Research Articles

Beyond the Breaking Point: Navigating System Strain in Molecular Energy Minimization for Drug Development
This comprehensive review explores the critical challenge of system strain in energy minimization processes essential to computational drug design.
Optimizing Energy Minimization: A Practical Guide to Adjusting emtol and nsteps for Robust Convergence in Molecular Dynamics
This article provides a comprehensive guide for researchers and scientists on optimizing the critical energy minimization parameters 'emtol' and 'nsteps' in molecular dynamics simulations, with a focus on applications in...
Convergence Failure in Drug Development: A Researcher's Guide to Diagnosis, Resolution, and Prevention
This article provides a comprehensive guide for researchers and drug development professionals facing the 'maximum number of steps reached without convergence' error.
Solvent Molecules and Ions: From Molecular Design to Drug Development Applications
This article provides a comprehensive exploration of solvent molecule insertion and ion placement, critical processes in drug development and materials science.
How to Fix Fmax Too High Error in GROMACS: A Comprehensive Troubleshooting Guide
This article provides a complete, step-by-step guide for researchers and scientists facing the common yet critical 'Fmax too high' error during energy minimization in GROMACS.
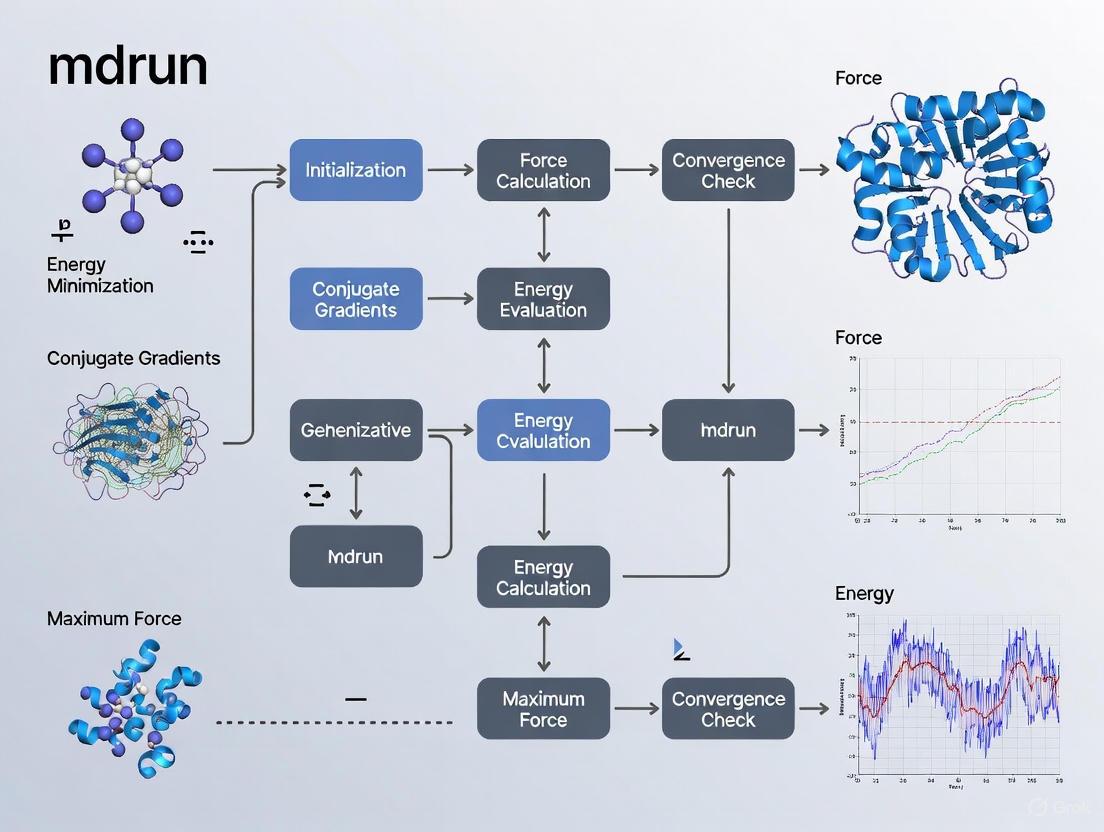
Mastering GROMACS mdrun for Energy Minimization: A Practical Guide for Biomedical Researchers
This comprehensive guide details the use of the GROMACS mdrun command for energy minimization, a critical step in preparing stable molecular dynamics simulations of biomolecules and drug candidates.
Energy Minimization for Membrane Protein Systems: Computational Strategies for Structure, Function, and Drug Discovery
This article provides a comprehensive overview of energy minimization strategies specifically for membrane protein systems, which are critical targets for over 50% of modern pharmaceuticals.
Solving 'Energy Minimization Stopped But Forces Not Converged': A Complete Guide for Computational Researchers
This comprehensive guide addresses the common but frustrating 'forces not converged' error during energy minimization in molecular dynamics simulations.
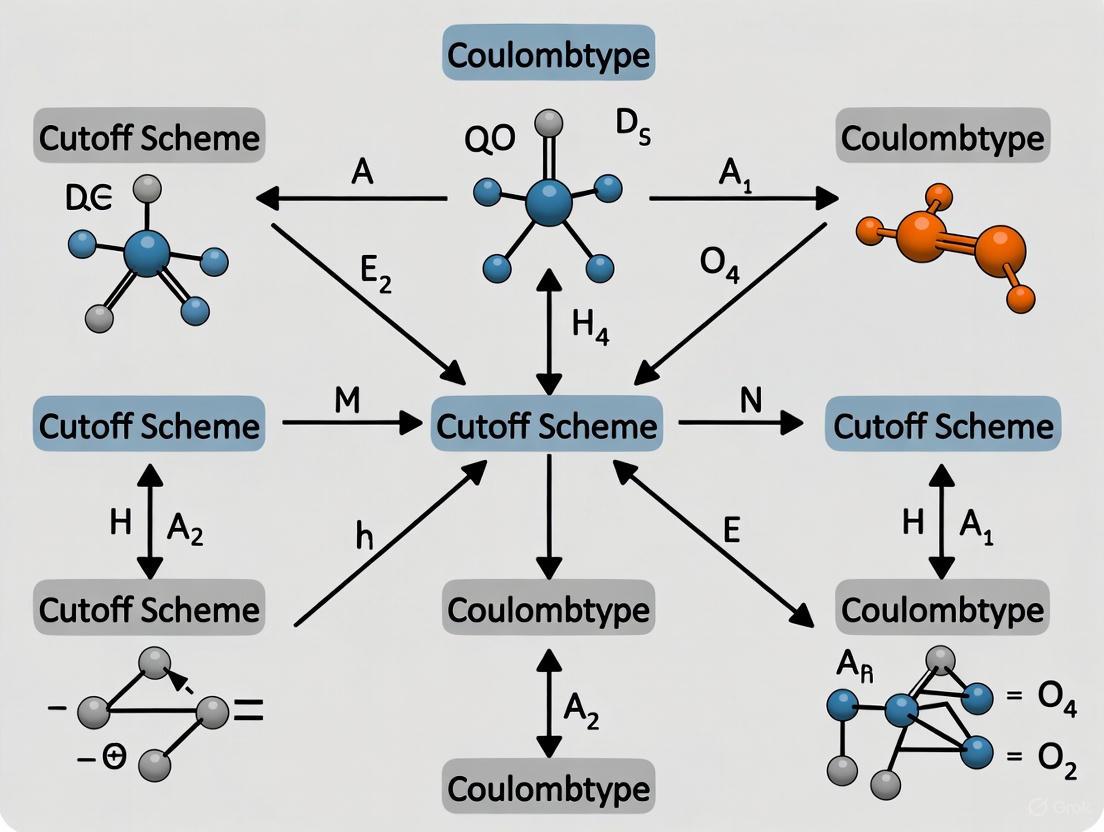
A Practical Guide to Electrostatic and Cutoff Scheme Selection for Energy Minimization in Biomolecular Simulation
This article provides a comprehensive, practical guide for researchers and scientists on configuring electrostatic and non-bonded interaction parameters for Energy Minimization (EM) in molecular dynamics.
Energy Minimization with Constraints and Restraints: Advanced Computational Strategies for Drug Discovery
This article provides a comprehensive exploration of energy minimization techniques incorporating constraints and restraints, tailored for the computational drug discovery pipeline.