Research Articles
Expectation-Maximization Algorithms in Biomolecular Systems: From Foundational Theory to Advanced Drug Discovery Applications
This article provides a comprehensive exploration of Expectation-Maximization (EM) algorithms and their critical role in computational biology and drug discovery.
Beyond the Minimum: A Practical Guide to Validating Molecular Structures with Dynamics Simulations
This article provides a comprehensive guide for researchers and drug development professionals on validating energy-minimized molecular structures through Molecular Dynamics (MD) simulations.
Benchmarking Energy Minimization in Biomedical Systems: From Algorithms to Clinical Applications
This article provides a comprehensive framework for benchmarking energy minimization parameters across diverse biomedical systems, tailored for researchers and drug development professionals.
Beyond Outliers: A Practical Guide to Validating Minimized Protein Structures with Ramachandran Plots
This article provides a comprehensive guide for researchers and drug development professionals on utilizing the Ramachandran plot for rigorous validation of energy-minimized protein structures.
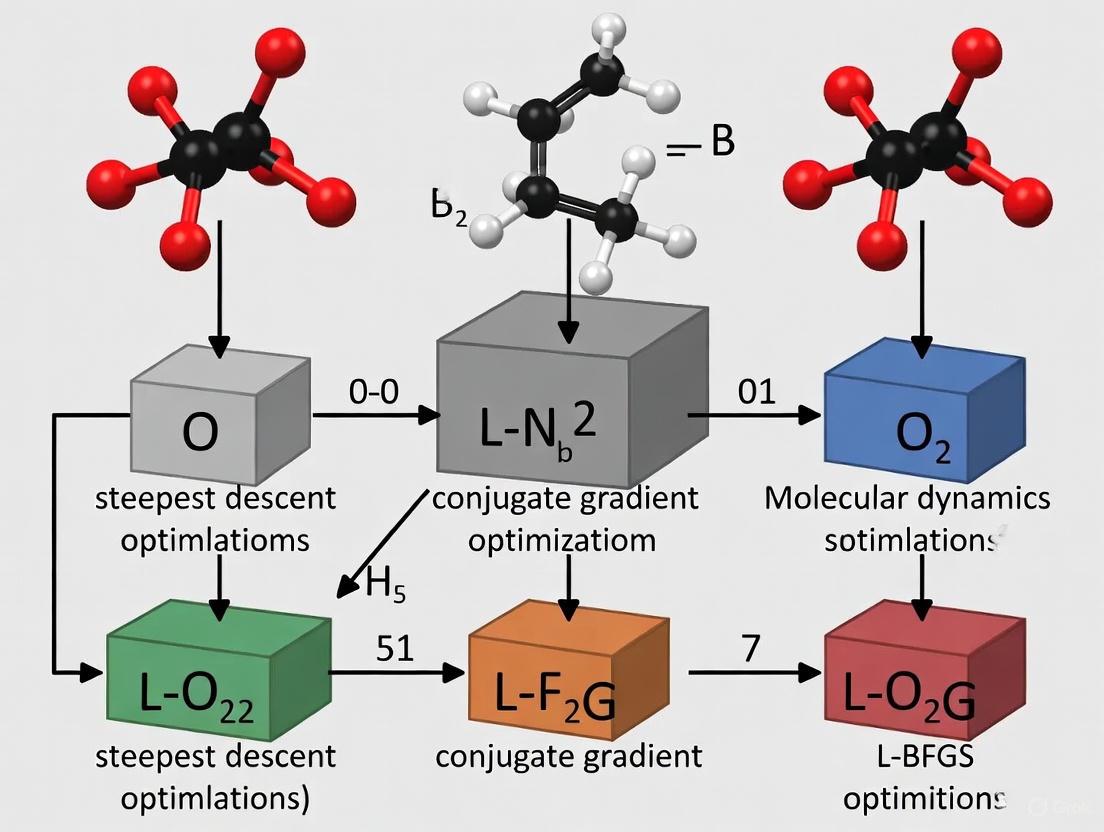
Steepest Descent vs. Conjugate Gradient vs. L-BFGS: A Performance Guide for Biomedical Researchers
This article provides a comprehensive comparison of three fundamental optimization algorithms—Steepest Descent, Conjugate Gradient, and L-BFGS—tailored for researchers and professionals in drug development and bioinformatics.
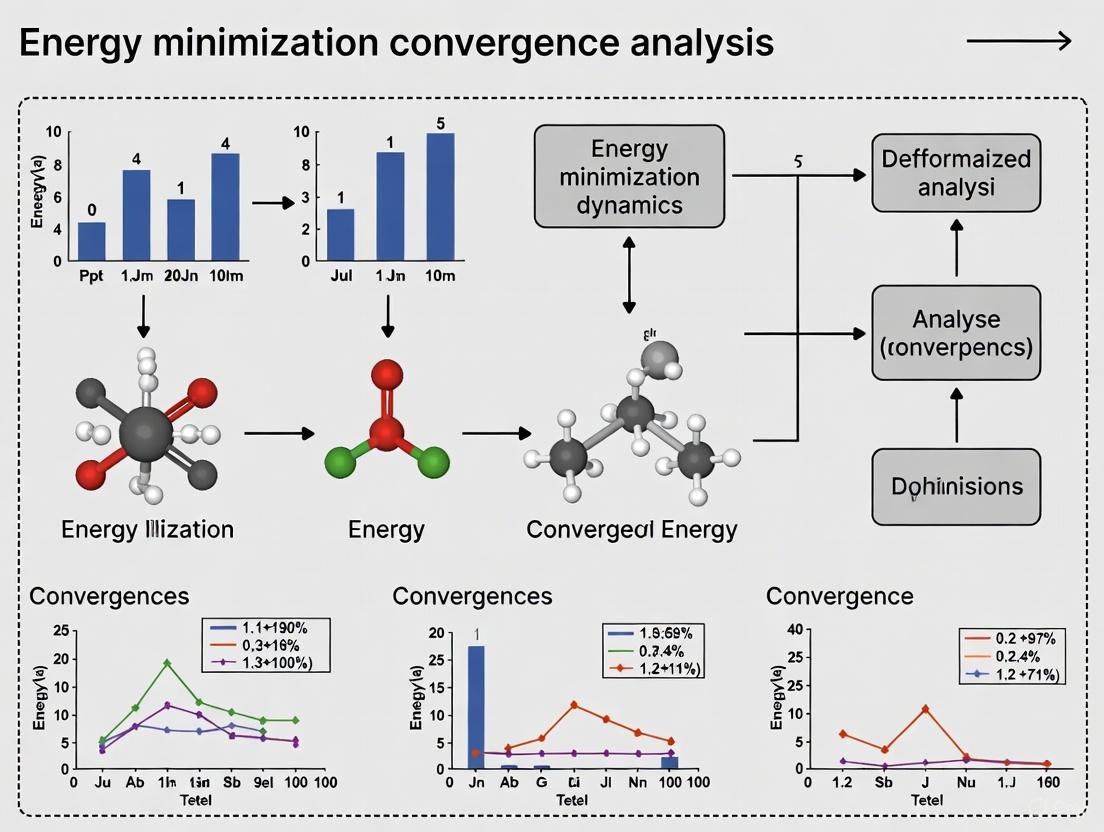
Energy Minimization Convergence Analysis: Techniques for Robust Biomolecular Simulations in Drug Development
This article provides a comprehensive analysis of energy minimization convergence techniques, a critical prerequisite for stable and accurate biomolecular simulations in pharmaceutical research.
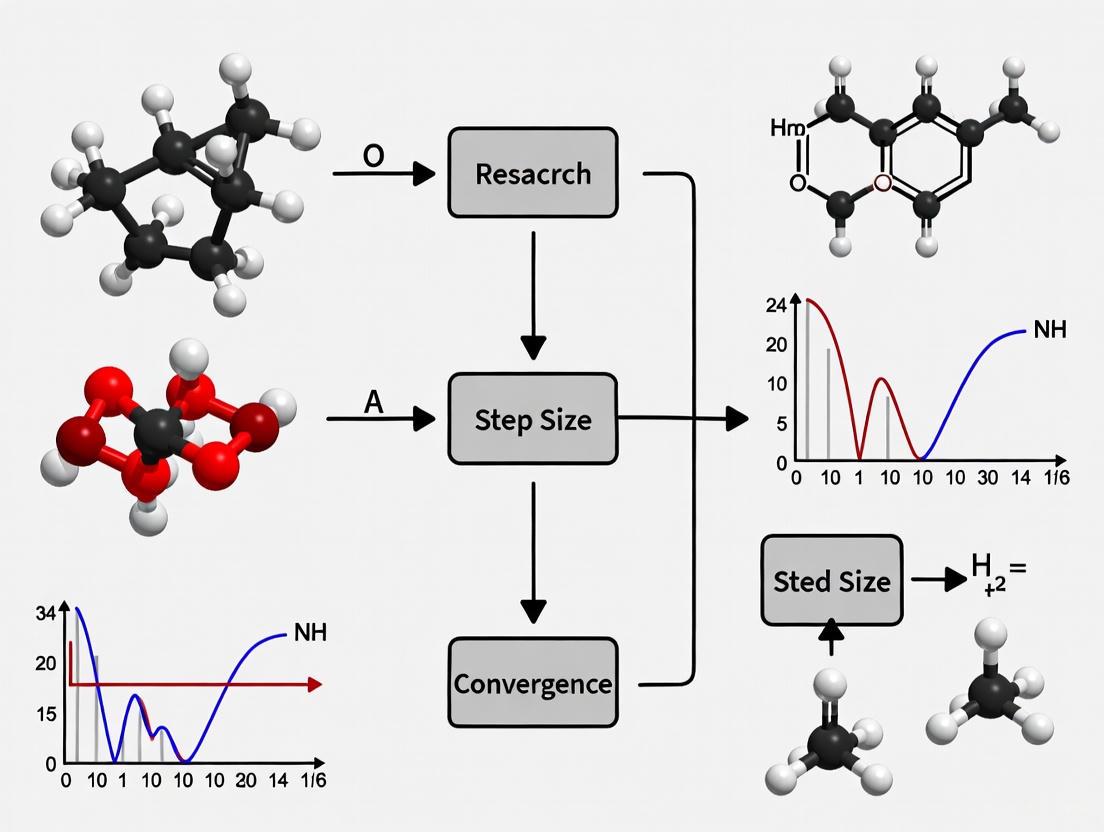
Step Size Control in Steepest Descent: Achieving Robust Convergence in Biomedical Optimization
This article provides a comprehensive analysis of step size adaptation strategies for the steepest descent method, focusing on applications in drug discovery and clinical research.

Validating Energy Minimization: Best Practices for Accurate Potential Energy Predictions in Drug Discovery
This article provides a comprehensive framework for validating energy minimization protocols and the potential energy values they generate, with a specific focus on applications in biomedical research and drug development.
Pre-EM Structural Validation: A Comprehensive Guide to Checking for Structural Issues and Missing Atoms
This article provides a systematic framework for researchers, scientists, and drug development professionals to validate atomic structures prior to Electron Microscopy (EM) analysis.

Resolving High Energy from Atomic Overlaps: A Computational Guide for Biomolecular Modeling
This article provides a comprehensive guide for researchers and scientists in drug development on identifying, troubleshooting, and resolving high-energy states caused by atomic overlaps in molecular models.