Research Articles
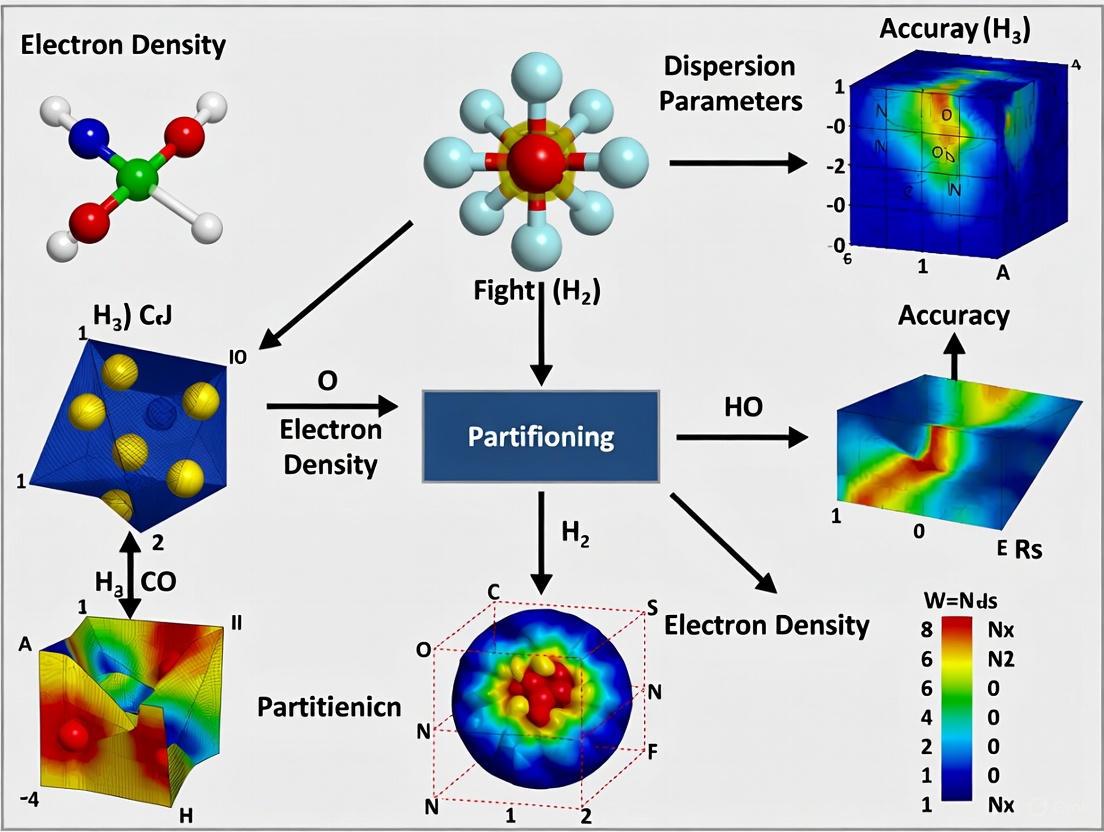
Electron Density Partitioning: A Critical Factor for Accurate Dispersion Parameters in Drug Discovery
Accurately modeling dispersion interactions is crucial for predicting molecular binding in drug design, yet the accuracy of these parameters is intrinsically linked to how a molecule's electron density is partitioned...
Quantum Mechanical Data in Force Field Validation: Enhancing Accuracy in Molecular Dynamics for Drug Discovery
This article explores the critical role of quantum mechanical (QM) data in validating and improving molecular mechanics force field parameters, which are foundational to reliable molecular dynamics simulations.
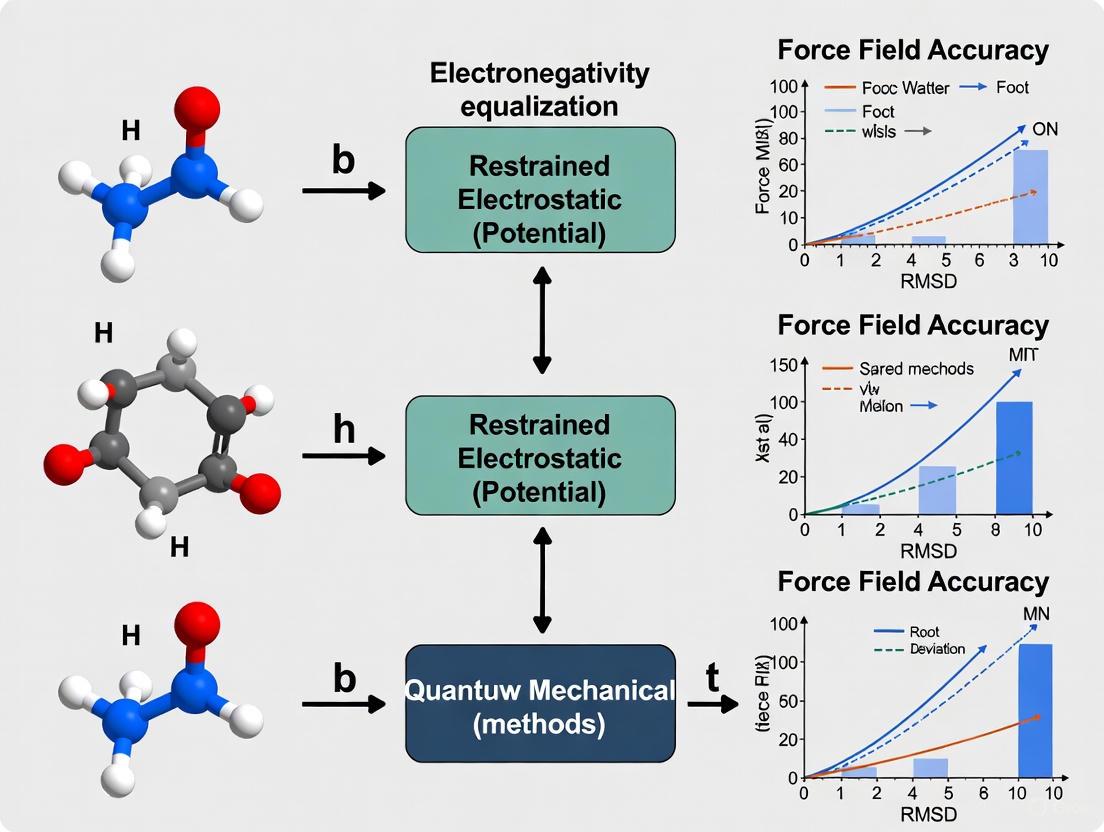
Partial Charge Methods and Force Field Accuracy: Foundations, Optimization, and Validation for Biomolecular Simulation
This article provides a comprehensive analysis of how methods for assigning atomic partial charges critically impact the accuracy of molecular force fields, with direct consequences for the reliability of molecular...

Force Field Errors in Drug Discovery: Navigating Accuracy and Validation in Molecular Simulations
This article examines the critical impact of force field accuracy on the reliability of molecular simulations in drug discovery.
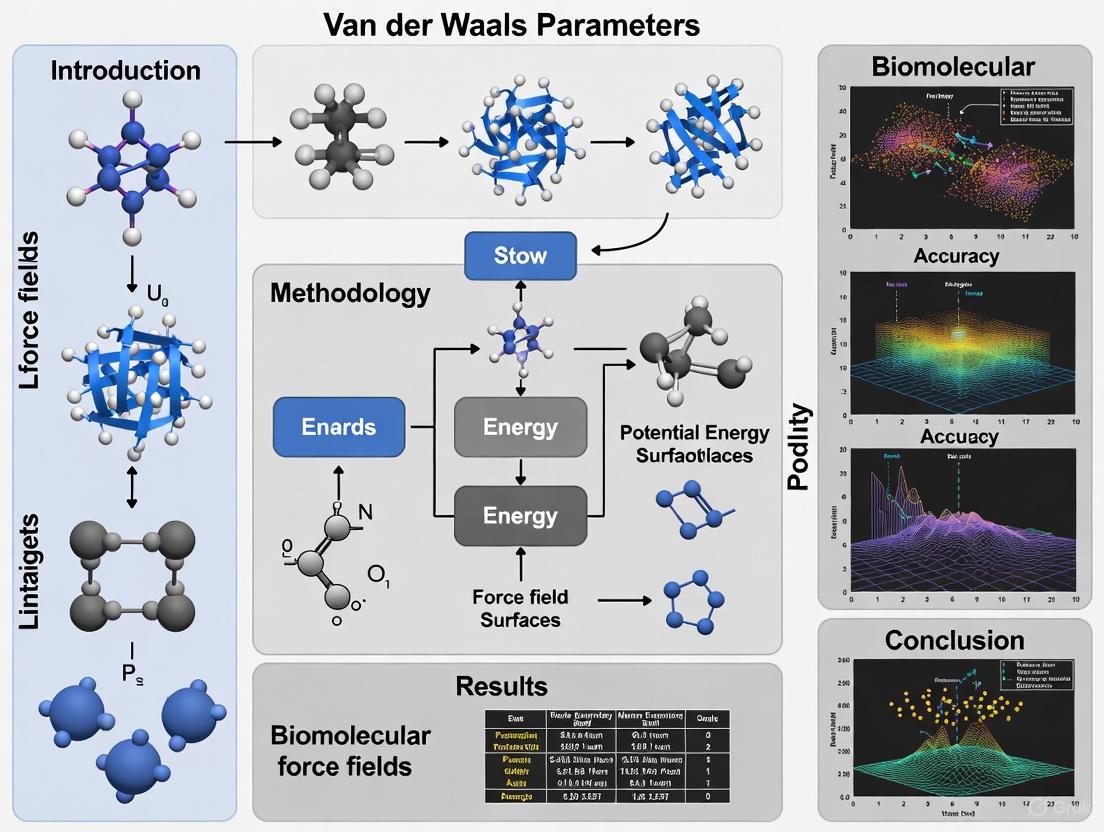
Beyond the Basics: Understanding and Addressing van der Waals Parameter Limitations in Biomolecular Force Fields
This article provides a comprehensive examination of van der Waals (vdW) parameters in biomolecular force fields, addressing critical challenges and modern solutions for researchers and drug development professionals.
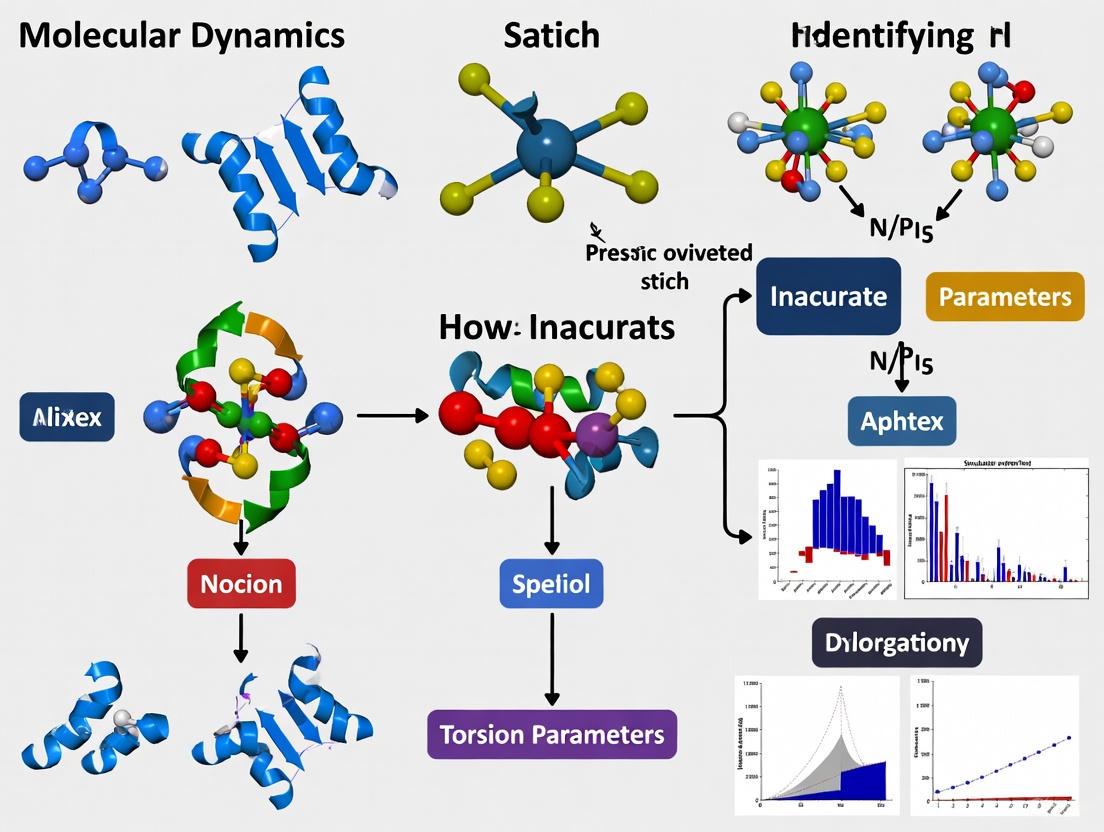
How to Identify and Fix Inaccurate Torsion Parameters in Molecular Dynamics Simulations
Accurate torsion parameters are fundamental to the reliability of Molecular Dynamics (MD) simulations in biomedical research, directly influencing predictions of molecular conformation, dynamics, and ligand binding.
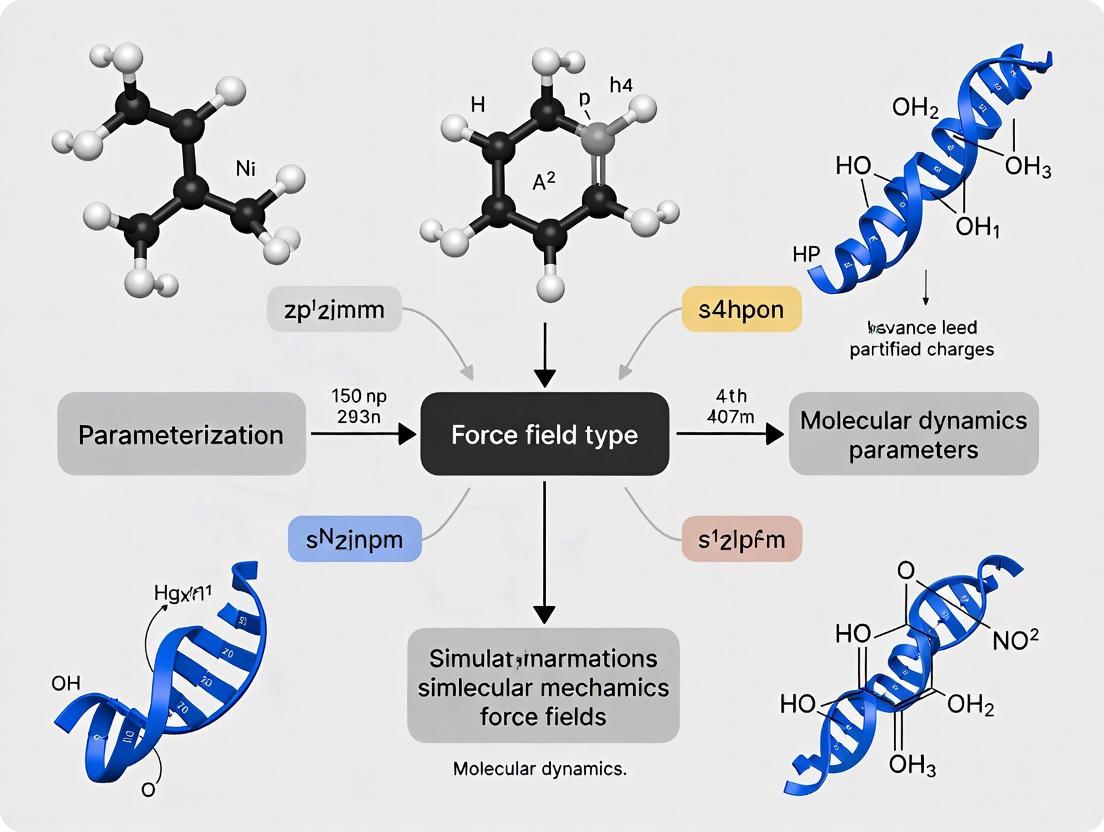
Common Sources of Error in Molecular Mechanics Force Fields: From Foundations to AI-Driven Solutions
This article provides a comprehensive analysis of the inherent limitations and common errors in molecular mechanics force fields, which are crucial for biomolecular simulation and computer-aided drug discovery.
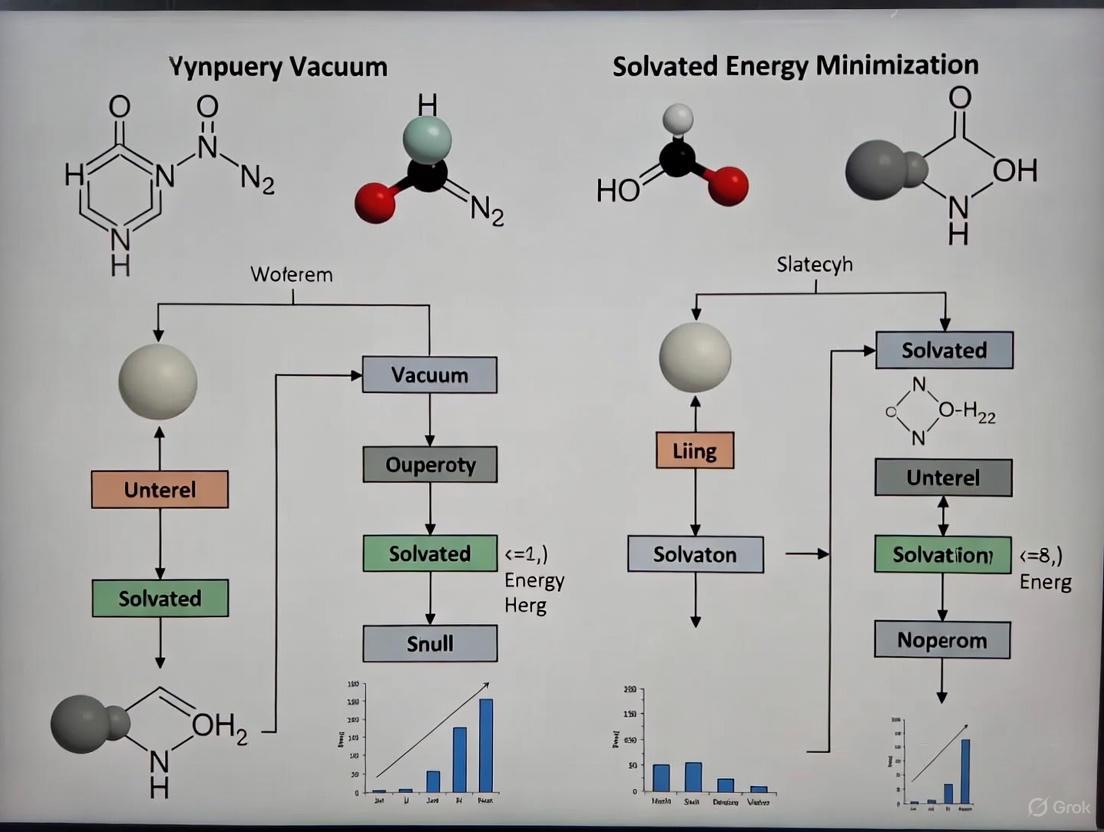
Vacuum vs. Solvated Energy Minimization: A Comprehensive Guide for Computational Researchers
This article provides a detailed comparative analysis of in vacuo and solvated energy minimization protocols for researchers and drug development professionals.
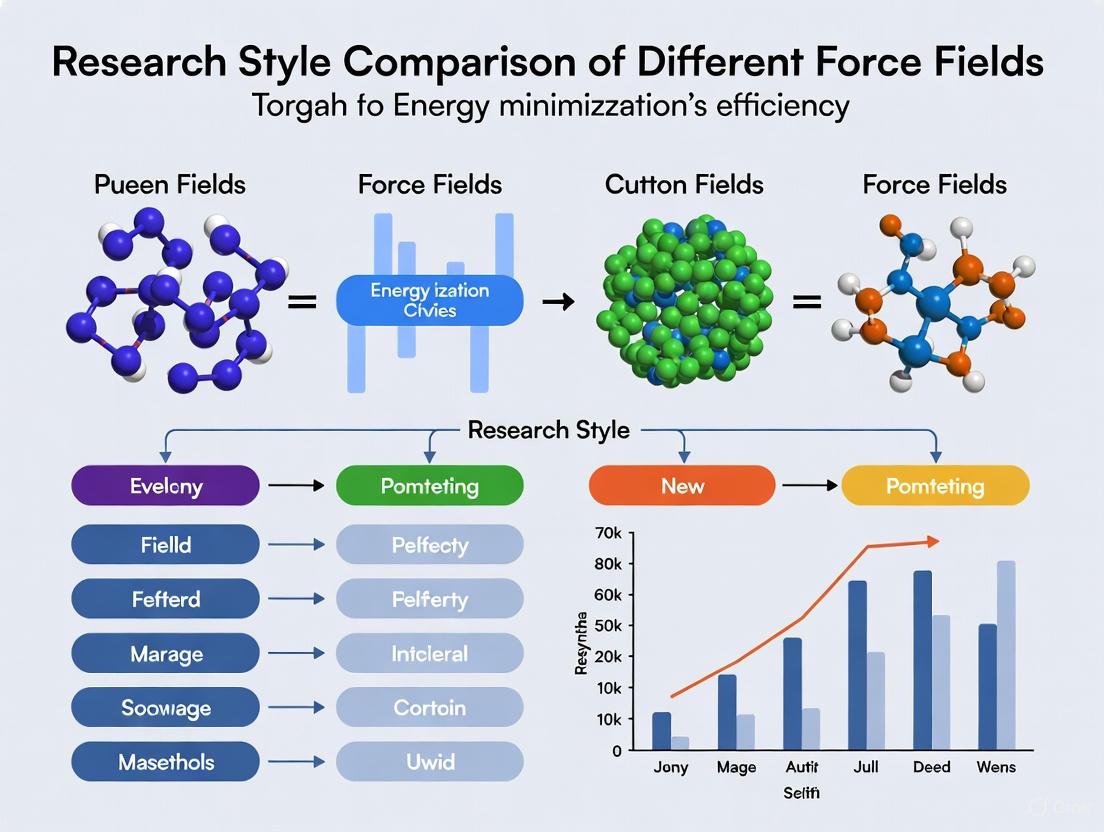
Force Field Efficiency Showdown: A Practical Guide to Energy Minimization for Biomolecular Simulation
Energy minimization is a critical first step in molecular dynamics simulations, yet the choice of force field significantly impacts both computational efficiency and result accuracy.
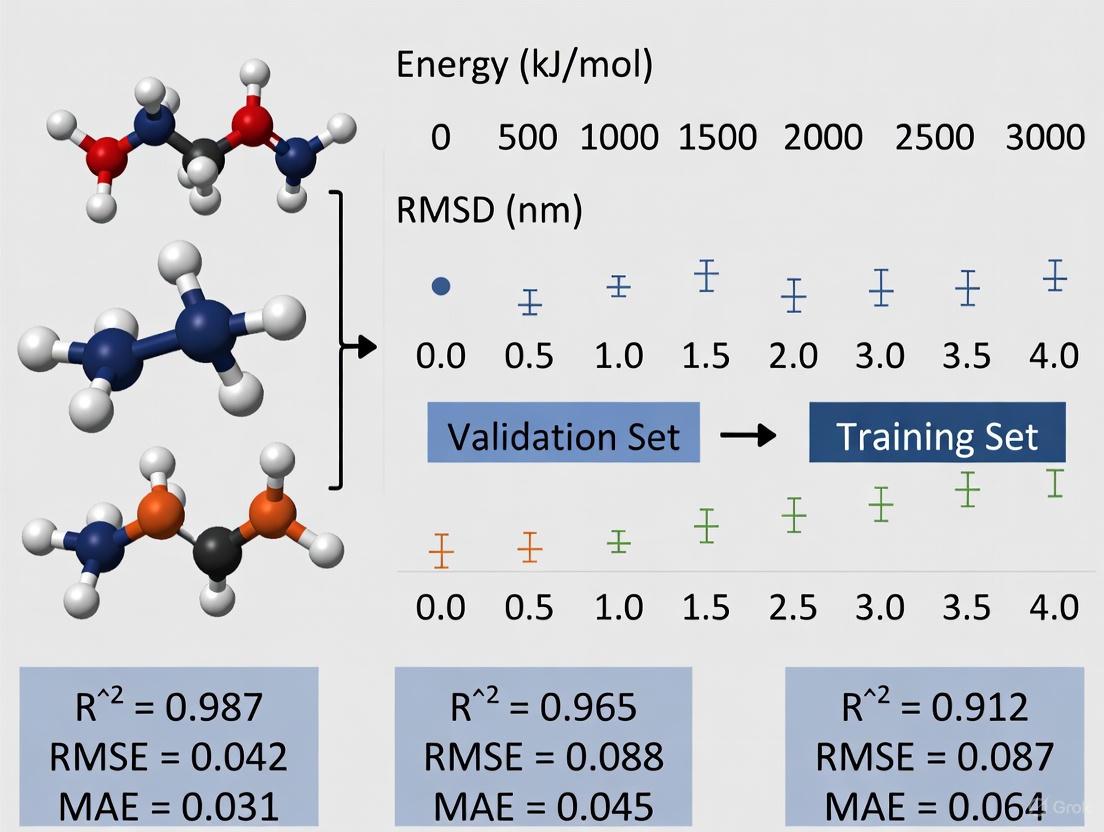
Validating Energy Minimization in Biomolecular Modeling: A Practical Guide to RMS Force Criteria
This article provides a comprehensive guide for researchers and drug development professionals on validating energy minimization procedures in biomolecular modeling, with a focus on root mean square (RMS) force as...