Validating Solution Ensembles from Molecular Dynamics: A Comprehensive Guide for Biomedical Research
This article provides a comprehensive framework for researchers and drug development professionals to validate molecular dynamics (MD) simulations against experimental data.
Validating Solution Ensembles from Molecular Dynamics: A Comprehensive Guide for Biomedical Research
Abstract
This article provides a comprehensive framework for researchers and drug development professionals to validate molecular dynamics (MD) simulations against experimental data. Covering foundational concepts to advanced applications, it explores how techniques like Wide-Angle X-ray Scattering (WAXS) and Small-Angle X-ray Scattering (SAXS) can rigorously test structural ensembles. The content addresses critical methodological considerations for accurate profile calculation, troubleshooting common force field and sampling issues, and implementing robust validation protocols. By synthesizing current best practices, this guide enables more reliable interpretation of biomolecular dynamics and conformational heterogeneity, with direct implications for drug discovery and understanding disease mechanisms.
Understanding Biomolecular Solution Ensembles: Why Validation Matters
The Challenge of Biomolecular Dynamics in Solution
Understanding biomolecular dynamics in solution is fundamental to advancements in structural biology and rational drug design. The biological function of proteins, nucleic acids, and their complexes is intrinsically linked to their conformational behavior in an aqueous, solvated environment. Molecular dynamics (MD) simulations have emerged as a powerful computational microscope, enabling researchers to observe atomic-level motions and interactions that are difficult to capture experimentally. However, a significant challenge persists: accurately modeling the intricate balance of solute-solvent interactions and validating the resulting structural ensembles against experimental reality. This guide objectively compares the performance of predominant MD software tools within the critical context of validating solution ensembles, providing researchers with actionable data to select appropriate methodologies for their investigations.
Comparing Molecular Dynamics Software: Features and Performance
The selection of MD software significantly impacts the efficiency, accuracy, and scope of biomolecular simulations. The table below summarizes the core capabilities of major MD packages, highlighting their suitability for different aspects of solution-phase modeling [1].
Table 1: Feature Comparison of Molecular Dynamics Software
| Software | License | GPU Support | Implicit Solvent | QM/MM | Key Strengths |
|---|---|---|---|---|---|
| AMBER | Proprietary / Free | Yes | Yes | Yes | High-performance MD, comprehensive analysis tools, widely used for biomolecules [1]. |
| GROMACS | Open Source (GPL) | Yes | Yes | No | Extreme performance for MD on CPUs and GPUs, highly optimized for large systems [1]. |
| NAMD | Free / Source | Yes | Yes | Yes | Fast, parallel MD, excellent for large biomolecular complexes [1]. |
| CHARMM | Proprietary | Yes | Yes | Yes | Extensive force fields, versatile modeling capabilities [1]. |
| OpenMM | Open Source (MIT) | Yes | Yes | No | Highly flexible, scriptable with Python, cross-platform performance [1]. |
| Desmond | Proprietary | Yes | No | No | High performance, integrated with Schrödinger's GUI for setup and analysis [1]. |
| LAMMPS | Open Source (GPL) | Yes | Yes | Yes | Potentials for soft and solid-state materials, coarse-grain systems [1]. |
Performance Benchmarks on High-Performance Computing Systems
Theoretical features must be paired with practical performance. Benchmarking on Alliance clusters provides insight into computational efficiency, which is crucial for achieving sufficient sampling for ensemble validation [2].
Table 2: Example Simulation Performance and Resource Configuration
| Software | Hardware | Key Performance Configuration | Use Case |
|---|---|---|---|
| GROMACS | 1 GPU, 12 CPU cores | -nb gpu -pme gpu -update gpu -bonded cpu |
Standard protein-ligand simulation [2]. |
| AMBER (pmemd) | 1 GPU, 1 CPU core | Single GPU execution | Optimized for single GPU, ideal for smaller systems [2]. |
| NAMD 3 | 2 GPUs, 2 CPU cores | +idlepoll flag for GPU efficiency |
Large complexes (e.g., viral capsids) [2]. |
A key finding from performance evaluations is that using more CPUs does not always result in a faster simulation and can sometimes even slow it down. Computational efficiency, measured by comparing actual speedup to the ideal 100% efficient speedup (speed on 1 CPU × N), is critical. Low efficiency wastes valuable computational resources and can negatively impact a researcher's ability to run future jobs on shared clusters [2]. Furthermore, performance can be enhanced by employing hydrogen mass repartitioning, which allows for a 4 femtosecond time step, thereby extending the effective simulation time per computational day [2].
Experimental Protocols for Validating Solution Ensembles
Validation ensures that computational models produce physically realistic and biologically relevant ensembles. The following protocols are central to this process.
Protocol 1: Solvation Free Energy Calculations Using FlexiSol Benchmarking
Accurate solvation free energy prediction is a rigorous test for any MD force field and sampling protocol.
- Benchmark Set Selection: Utilize the FlexiSol dataset, a modern benchmark containing 824 experimental solvation energy and partition ratio data points for 1551 unique molecule-solvent pairs. It focuses on drug-like, flexible molecules (up to 141 atoms) and includes over 25,000 theoretical conformer/tautomer geometries [3] [4].
- Conformational Sampling: For each molecule, generate an exhaustive conformational ensemble. The FlexiSol study found that using either full Boltzmann-weighted ensembles or just the lowest-energy conformers yields similar accuracy, but random single-conformer selection significantly degrades performance, especially for larger, flexible systems [3].
- Simulation Setup:
- System Preparation: Solvate the solute in a box of explicit water molecules (e.g., TIP3P model) with sufficient padding.
- Force Field Selection: Choose an appropriate force field (e.g., AMBER, CHARMM, OPLS-AA).
- Simulation Run: Perform equilibrium MD followed by production MD to ensure proper sampling of solvent degrees of freedom.
- Free Energy Calculation: Use alchemical free energy methods, such as Free Energy Perturbation (FEP) or Thermodynamic Integration (TI), to compute the solvation free energy [3].
- Validation: Compare calculated solvation free energies against the experimental data in the FlexiSol set. A robust model should not systematically underestimate strong stabilizing interactions or overestimate weaker ones [3].
Protocol 2: MD Refinement of Predicted Protein Structures
MD simulation can be used to refine and validate protein structures predicted by deep learning and other computational tools [5].
- Structure Generation: Predict the initial protein structure using multiple algorithms (e.g., AlphaFold2, Robetta, trRosetta, I-TASSER, MOE-based homology modeling) [5] [6].
- Simulation Setup:
- Solvation: Place the predicted structure in an explicit solvent box.
- Neutralization: Add ions to neutralize the system charge.
- Equilibration: Gradually heat the system and equilibrate under constant temperature and pressure (NPT ensemble).
- Production MD and Analysis: Run a multi-nanosecond MD simulation. Monitor convergence by calculating the Root Mean Square Deviation (RMSD) of backbone atoms, Root Mean Square Fluctuation (RMSF) of Cα atoms, and the Radius of Gyration (Rg). A stable, compactly folded structure that converges indicates a high-quality model [5].
- Quality Assessment: Post-simulation, evaluate the refined model using quality assessment tools like ERRAT and phi-psi (Ramachandran) plot analysis [5].
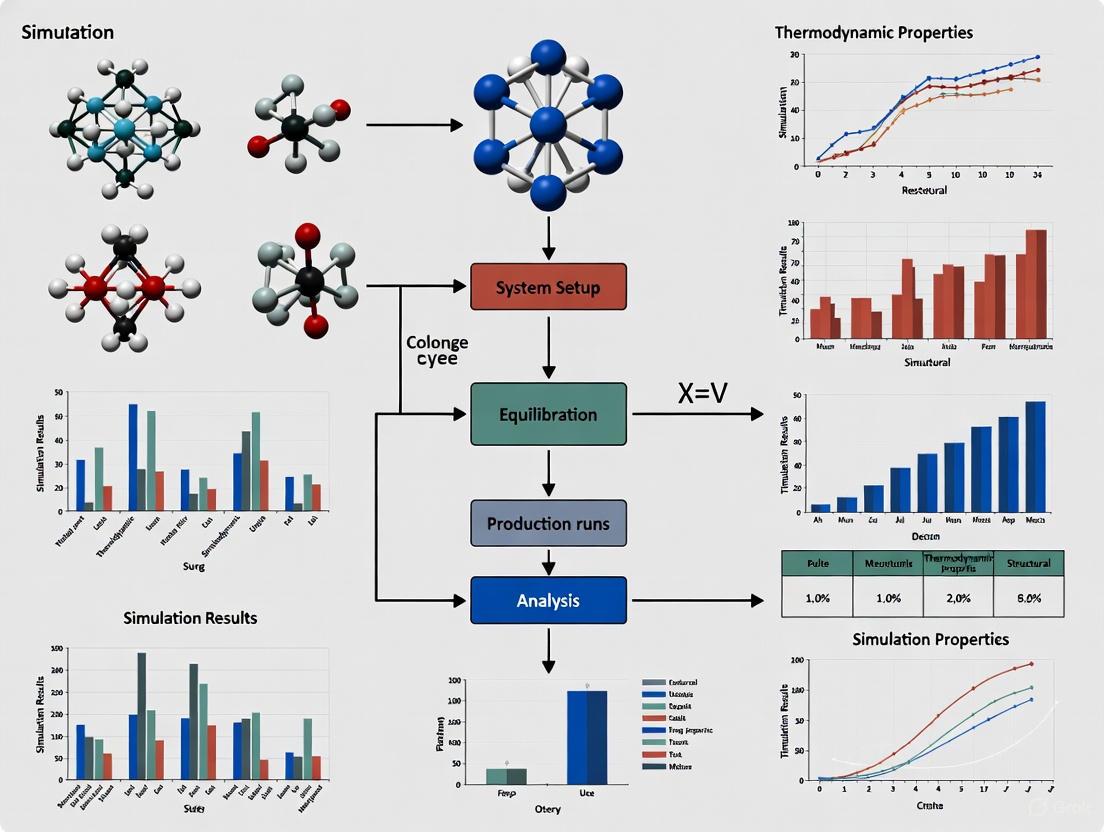
Visualizing Workflows for Validation and Simulation
The following diagrams, created with Graphviz, illustrate the logical flow of the key methodologies discussed.
Solution Ensemble Validation Workflow
Diagram 1: A high-level workflow for generating and validating a biomolecular solution ensemble through MD simulation and experimental comparison.
Molecular Dynamics Simulation Setup
Diagram 2: A detailed step-by-step protocol for setting up and running a standard molecular dynamics simulation.
Successful biomolecular dynamics research relies on a suite of software, hardware, and data resources.
Table 3: Essential Resources for Biomolecular Dynamics Research
| Resource Name | Type | Primary Function | Relevance to Solution Ensembles |
|---|---|---|---|
| FlexiSol Dataset | Data | Benchmark set for solvation models | Provides experimental solvation energies for flexible, drug-like molecules to validate simulations [3] [4]. |
| AMBER Tools | Software | MD simulation & analysis | A comprehensive suite for running simulations, particularly strong with nucleic acids and force field development [1]. |
| GROMACS | Software | High-performance MD engine | Enables extremely fast simulations, allowing for better sampling of conformational space [1] [2]. |
| GPU Cluster | Hardware | Parallel computing | Dramatically accelerates MD calculations, making nanosecond-to-microsecond timescales accessible [2]. |
| Force Fields (e.g., AMBER, CHARMM) | Parameter Set | Defines interatomic potentials | The physical model governing energy calculations; choice is critical for accuracy [1]. |
| VMD / PyMOL | Software | Trajectory visualization & analysis | Essential for visually inspecting simulations, analyzing interactions, and preparing figures [1] [5]. |
| AlphaFold2 | Software | Protein structure prediction | Provides high-quality initial structural models for proteins of unknown structure [5] [6]. |
| CP2K | Software | Quantum Chemistry/MD | Performs atomistic simulations using DFT, enabling QM/MM studies of chemical reactivity in solution [1]. |
WAXS and SAXS as Powerful Validation Tools
Small-Angle X-ray Scattering (SAXS) and Wide-Angle X-ray Scattering (WAXS) are complementary biophysical techniques that provide crucial structural information on biomolecules in solution under near-native conditions. SAXS typically probes structural correlations up to approximately 2 nanometers, offering insights into global parameters such as the radius of gyration, overall shape, and assembly state [7] [8]. In contrast, WAXS extends to higher scattering angles, accessing finer structural details including secondary structure elements and tertiary fold characteristics [7] [9]. For researchers focused on validating solution ensembles from molecular dynamics (MD) simulations, SAXS/WAXS data provides an invaluable experimental benchmark. The integration of these techniques with MD simulations has emerged as a powerful approach for deriving accurate structural ensembles, as computational methods alone cannot always distinguish between physically realistic and unrealistic models [9]. This guide compares the performance of different computational methods for interpreting SAXS/WAXS data within the context of MD validation, providing experimental protocols and resources for structural biologists and drug development professionals.
Table 1: Key Characteristics of SAXS and WAXS
| Feature | SAXS (Small-Angle X-ray Scattering) | WAXS (Wide-Angle X-ray Scattering) |
|---|---|---|
| Spatial Resolution | Low resolution (up to ~2 nm) [7] | Higher resolution (atomic to sub-nanometer) [7] |
| Primary Information | Global shape, radius of gyration, assembly state [7] [8] | Secondary structure, tertiary fold, finer structural details [7] |
| Typical q-range | Lower q-values (e.g., < 3 nm⁻¹) [7] | Higher q-values (e.g., extending beyond 15 nm⁻¹) [7] |
| Sensitivity | Overall conformation and large-scale transitions | Small conformational changes and local structural rearrangements [7] |
Computational Methods for Scattering Profile Prediction
The accurate calculation of SAXS/WAXS profiles from atomic coordinates is fundamental to their use as validation tools for MD simulations. Computational methods primarily differ in how they model the hydration layer surrounding biomolecules in solution, a significant contributor to the scattering profile [10]. These approaches can be broadly categorized into implicit-solvent models and explicit-solvent models, each with distinct strengths and limitations for validating molecular dynamics ensembles.
Implicit-Solvent Models
Implicit-solvent methods model the hydration layer as a continuous shell of uniform scattering density surrounding the biomolecule. Popular programs like CRYSOL, Pepsi-SAXS, and FoXS use this approach and are freely available on web servers [10]. They generally optimize fits to experimental data using adjustable parameters: one controlling the total excluded volume (often a scaling factor for atomic radii, rSc), and another for the contrast of the hydration layer (δρ) relative to the bulk solvent [10]. A major advantage of these methods is their computational speed, making them suitable for rapid screening of structural models [10]. However, the use of adjustable parameters carries a risk of overfitting, where differences between a model and the true solution structure can be masked during parameter optimization [7] [10]. This is a significant consideration when validating dynamic MD ensembles, as these parameters might absorb the subtle effects of conformational flexibility.
Explicit-Solvent Models
Explicit-solvent models, particularly those using all-atom molecular dynamics (MD) simulations, represent the solute and surrounding solvent molecules in atomic detail. This approach eliminates the need for fitting parameters associated with the hydration layer or excluded solvent, thereby minimizing the risk of overfitting and providing a more rigorous test for MD-derived ensembles [7] [10]. Furthermore, explicit-solvent MD naturally incorporates thermal fluctuations of both the protein and solvent, which has been shown to significantly improve agreement with experimental WAXS data [7]. Benchmarking studies have demonstrated that MD simulations without adjustable parameters can yield fits to consensus SAXS profiles that are at least as good as those from implicit-solvent methods, with the added benefit of more accurately representing the physical nature of the hydration shell [10]. The primary trade-off is the substantially higher computational cost compared to implicit-solvent methods [10].
Table 2: Comparison of SAXS/WAXS Computational Prediction Methods
| Method Type | Representative Programs | Key Features | Advantages | Disadvantages |
|---|---|---|---|---|
| Implicit-Solvent | CRYSOL, Pepsi-SAXS, FoXS [10] | Models hydration as uniform shell; fits parameters for excluded volume (rSc) and hydration contrast (δρ) [10] |
Fast computation; widely accessible via web servers [10] | Risk of overfitting; less physically realistic solvent model [7] [10] |
| Explicit-Solvent | Explicit-solvent MD simulations [7] [10] | Uses atomistic water models; no fitting parameters for hydration layer [7] [10] | Minimizes overfitting; accounts for thermal fluctuations; more accurate hydration model [7] [10] | High computational cost [10] |
Experimental Protocols for Validation
Generating Consensus Scattering Profiles
A robust protocol for validating MD simulations begins with high-quality, reproducible experimental data. International round-robin exercises have established methods to generate consensus SAXS profiles by combining hundreds of independent measurements from multiple beamlines [10]. For example, one such study created consensus datasets for five globular proteins (xylose isomerase, urate oxidase, xylanase, lysozyme, and ribonuclease A) by merging 171 SAXS profiles from 12 beamlines using a maximum likelihood method [10]. This process involves:
- Sample Preparation: Using a common protein source and standard buffers for all measurements to minimize variability.
- Data Collection: Collecting data using both in-line size-exclusion chromatography (SEC) and batch modes to eliminate contributions from aggregates or interparticle interference.
- Data Merging: Carefully scaling and merging datasets from different instrument configurations to create a consensus profile with improved statistical precision across a wide q-range [10].
These consensus profiles provide a gold standard for benchmarking computational methods and validating the solution ensembles generated by MD simulations.
Calculating Profiles from MD Simulations
The following protocol, adapted from studies validating solution ensembles with WAXS, outlines the key steps for calculating scattering profiles from explicit-solvent MD simulations [7]:
- MD Simulation Trajectory: Perform an unrestrained, explicit-solvent MD simulation of the biomolecule of interest.
- Spatial Envelope Definition: For each simulation frame, define a fixed spatial envelope around the solute. This envelope must be large enough to encompass all conformational states of the protein and its complete solvation layer, ensuring that water molecules outside the envelope exhibit bulk-like correlations [7].
- Scattering Intensity Calculation: Compute the excess scattering intensity,
I(q), using the formula:I(q) = IA(q) - IB(q)whereIA(q)is the scattering intensity from the solute-solvent system (within the envelope), andIB(q)is the scattering intensity from a pure-solvent system [7]. - Averaging: Perform an ensemble average over multiple simulation frames and a rotational average over all orientations of the solute to replicate the experimental conditions of a solution [7].
This explicit-solvent approach directly uses the simulated atomic positions without fitting parameters related to solvation, providing a stringent validation metric [7]. The entire workflow for integrating MD simulations with experimental validation is summarized in the diagram below.
Successful application of SAXS/WAXS as a validation tool requires access to specific instrumentation, software, and sample preparation materials. The following table details key resources used in the featured experiments and the broader field.
Table 3: Key Research Reagent Solutions for SAXS/WAXS and MD Validation
| Category | Item | Function / Application | Example from Literature |
|---|---|---|---|
| Instrumentation | High-Brilliance X-ray Source | Increases flux for weakly scattering biological samples; enables laboratory-based measurements that previously required synchrotrons [11]. | Liquid gallium metal-jet X-ray source [11] |
| Large-Area Pixel Array Detector | Increases speed and sensitivity of data collection via single-photon counting and a large active area [11]. | Detector with 5X larger active area [11] | |
| Software & Databases | CRYSOL, Pepsi-SAXS, FoXS | Web-server programs for predicting SAXS profiles from atomic structures using implicit-solvent models [10]. | Used for benchmarking against consensus data [10] |
| Molecular Dynamics Software | Software for running explicit-solvent MD simulations (e.g., AMBER, GROMACS). | Used to generate conformational ensembles for WAXS validation [7] [12] | |
| SASBDB (Small Angle Scattering Biological Data Bank) | Public repository for SAS data; source of consensus profiles for benchmarking [10]. | Consensus profiles for 5 proteins [10] | |
| Sample Preparation | In-line Size Exclusion Chromatography (SEC) | Purifies the sample immediately before measurement to remove aggregates and ensure monodispersity [10]. | Used in round-robin study to generate clean consensus data [10] |
| Standardized Buffers | Using common, precisely prepared buffers is critical for accurate background subtraction and reproducibility across experiments [10]. | Essential for multi-beamline consensus study [10] |
SAXS and WAXS have firmly established themselves as powerful and indispensable tools for validating solution ensembles derived from molecular dynamics simulations. While implicit-solvent methods offer speed and accessibility for initial comparisons, explicit-solvent MD simulations provide a more rigorous, physically realistic foundation for validation by accurately modeling the hydration layer and incorporating thermal fluctuations without overfitting risks. The development of consensus scattering profiles through community-wide efforts now provides benchmark datasets of exceptional quality for testing computational models. As both experimental techniques and simulation methodologies continue to advance, their synergistic combination will undoubtedly yield ever more precise and dynamic views of biomolecular structure and function in solution, directly impacting fields ranging from fundamental biophysics to rational drug design.
The Critical Role of Explicit Solvent Models
In molecular dynamics (MD) research, the choice of how to represent the solvent environment is foundational, influencing the predictive power of simulations studying biomolecular folding, binding, and chemical reactivity. This choice predominantly divides into two philosophies: explicit and implicit solvent models. Explicit models treat solvent molecules as individual entities, while implicit models approximate the solvent as a continuous, polarizable medium [13]. The central thesis of this guide is that while implicit models offer computational efficiency, explicit solvent models are critical for validating solution ensembles because they capture specific, atomistic solute-solvent interactions that continuum models cannot. These interactions—including hydrogen bonding, dipole-dipole interactions, and the hydrophobic effect—are vital for reproducing experimentally observed structures, dynamics, and free energies [14] [15]. The failure to capture these effects can lead to significant inaccuracies; for instance, implicit models are known to struggle with charged species and can produce solvation free energies that deviate from experiment by as much as 10 kcal/mol, rendering high-level quantum chemical results useless [14]. This guide provides a objective comparison of these approaches, supported by current experimental data and methodologies, to inform researchers and drug development professionals.
Model Comparison: Explicit vs. Implicit Solvents
The table below summarizes the core characteristics, performance metrics, and ideal use cases for explicit and implicit solvent models.
Table 1: Quantitative Comparison of Explicit and Implicit Solvent Models
| Feature | Explicit Solvent Models | Implicit Solvent Models (GB/PSA, PCM) |
|---|---|---|
| Fundamental Approach | Atomistic representation of individual solvent molecules [13] | Continuum dielectric medium representing an average solvent environment [13] |
| Computational Cost | High (adds 10³-10⁶ extra atoms, scales O(N²)–O(N³)) [14] | Low (dramatically reduces degrees of freedom) [16] |
| Sampling Speed | Slower (high solvent viscosity slows conformational transitions) [16] | Faster (solvent viscosity can be turned off) [16] |
| Treatment of Electrostatics | Explicit Coulombic interactions; captures local polarization and shielding [15] | Approximate (e.g., Generalized Born, Poisson-Boltzmann); captures long-range bulk effects [17] [16] |
| Key Strengths | Captures specific solute-solvent interactions (H-bonding), solvent structure, and entropy [14] [15] | Computational efficiency; well-defined potential energy surfaces for geometry optimization [14] [16] |
| Known Limitations | Computationally prohibitive for long-time/size scales; slow sampling [14] | Poor treatment of specific interactions (H-bonding), charged species, and entropy [14] [18] |
| Accuracy (Solvation Free Energy) | High (considered the "gold standard" for ΔGsolv) [19] | Variable; can show substantial discrepancies (>10 kcal/mol) for proteins/complexes [18] |
| Ideal Application Domains | Validation of solution ensembles, binding studies, processes with specific solvent interactions [15] [20] | High-throughput screening, long timescale folding studies, gas-phase reaction modeling with estimated solvation [16] |
Performance Benchmarking: A Data-Driven View
Independent benchmarks provide a clear picture of how these models perform in practice. A 2023 benchmark study on glycosaminoglycans (GAGs) highlighted the importance of model choice for biologically relevant systems. The study compared structural descriptors like the Radius of Gyration (Rg) and End-to-End Distance (EED) for a heparin decasaccharide across different solvent models [15]. The results demonstrate that explicit solvent models, particularly more advanced ones like OPC and TIP4PEw, can produce significantly different—and often more reliable—structural ensembles compared to implicit models.
Table 2: Structural Properties of a Heparin Decasaccharide in Different Solvent Models Data sourced from a 5 µs MD simulation benchmark study [15]
| Solvent Model | Type | Average Rg (Å) | Average EED (Å) |
|---|---|---|---|
| TIP3P | Explicit | 16.2 | 48.5 |
| OPC | Explicit | 15.8 | 45.1 |
| TIP4PEw | Explicit | 15.9 | 46.3 |
| SPC/E | Explicit | 16.1 | 47.8 |
| IGB=1 | Implicit | 14.5 | 35.2 |
| IGB=2 | Implicit | 14.8 | 36.0 |
| IGB=5 | Implicit | 15.0 | 37.1 |
| IGB=7 | Implicit | 14.7 | 35.8 |
| IGB=8 | Implicit | 14.9 | 36.5 |
Furthermore, a broader accuracy comparison on small molecules, proteins, and protein-ligand complexes revealed that while implicit models like PCM, COSMO, and various Generalized Born (GB) implementations show high correlation with experimental hydration energies for small molecules, this agreement deteriorates for larger systems. The study found that estimated protein solvation energies and protein-ligand binding desolvation energies can show substantial discrepancies of up to 10 kcal/mol compared to explicit solvent references [18]. This error margin is critical, as reliable prediction of inhibition activity requires a binding energy error not exceeding 1 kcal/mol [18].
Experimental Protocols for Solvent Model Validation
Protocol 1: Traditional MD Benchmarking with Explicit Solvent
This protocol is the established standard for generating reference data to validate implicit models or faster explicit ones [15].
- System Setup: The solute (e.g., protein, drug molecule) is parameterized with an appropriate force field (e.g., AMBER, CHARMM). It is then solvated in a periodic box of explicit water molecules (e.g., TIP3P, OPC) with a minimum distance between the solute and box edge (e.g., 8.0 Å). The system is neutralized with counterions (e.g., Na⁺, Cl⁻) [15].
- Energy Minimization: The system undergoes sequential energy minimization steps to remove bad contacts. This typically involves initial minimization with harmonic restraints on solute atoms, followed by a full minimization without restraints [15].
- System Equilibration: The minimized system is gently heated to the target temperature (e.g., 300 K) over 10-50 ps with restraints on solute atoms. This is followed by a longer equilibration (e.g., 50-100 ps) in the isothermal-isobaric (NPT) ensemble to stabilize the system's temperature and pressure [15].
- Production MD: An unrestrained MD simulation is run in the NPT ensemble for a time sufficient to achieve convergence of the properties of interest (e.g., µs-scale for some biomolecular properties). Standard settings include a 2 fs time step, a cutoff for nonbonded interactions (e.g., 8 Å), and the use of the Particle Mesh Ewald (PME) method for long-range electrostatics [15].
- Analysis: The resulting trajectory is analyzed for key properties, which are then used as a benchmark. These include structural metrics (e.g., Radius of Gyration, End-to-End Distance), dynamic properties, and free energy estimates derived from the trajectory (e.g., via MM/GBSA or similar) [15].
Protocol 2: Machine Learning Potentials with Active Learning for Explicit Solvent
This modern protocol, exemplified by Duarte and co-workers, uses machine learning (ML) to make explicit solvent accuracy accessible for studying chemical reactions [20].
- Initial Data Generation: A small, diverse set of configurations is generated. This includes:
- Substrates only in the gas phase or implicit solvent.
- Clusters of the substrate with a small number of explicit solvent molecules (e.g., 2, 33) to capture specific interactions.
- Clusters of solvent molecules only [20].
- Active Learning Loop:
- Train ML Potential: An initial ML potential (e.g., Atomic Cluster Expansion model) is trained on the current dataset.
- Run ML/MD: The trained model is used to run molecular dynamics simulations.
- Selector Evaluation: A descriptor-based selector (e.g., based on Smooth Overlap of Atomic Positions (SOAP)) evaluates simulation structures to identify regions of chemical space not well-represented in the training data.
- Quantum Chemical Calculation: The selected under-represented structures are passed to a high-level quantum chemical method (e.g., DFT) for accurate energy and force calculations.
- Expand Training Set: These new, accurately labeled structures are added to the training dataset. The loop repeats until the ML potential's predictions are converged and accurate [20].
- Production Simulation & Validation: The final, trained ML potential is used to run extensive biased sampling MD (e.g., metadynamics) to compute free energy surfaces and reaction rates. The results are validated against available experimental data [20].
The workflow for this protocol is illustrated below.
The Scientist's Toolkit: Essential Research Reagents and Solutions
This table details key computational "reagents" essential for working with explicit solvent models.
Table 3: Key Reagents for Explicit Solvent Molecular Dynamics
| Research Reagent | Function/Description | Examples |
|---|---|---|
| Explicit Water Models | Parametrized models representing water molecules, differing in complexity and accuracy. | TIP3P [15], SPC/E [15], TIP4P [15], TIP4PEw [15], OPC [15] |
| Force Fields | Empirical potentials describing intra- and inter-molecular interactions. | AMBER [15], CHARMM, GLYCAM06 (for carbohydrates) [15] |
| Molecular Dynamics Engines | Software to perform the numerical integration of Newton's equations of motion. | AMBER [15], GROMACS, NAMD, OpenMM |
| Machine Learning Potentials | Surrogate models trained on QM data to provide quantum accuracy at near-classical cost. | Atomic Cluster Expansion (ACE) [20], NequIP [14], SchNet [20] |
| Active Learning Selectors | Algorithms to identify and select new data points for training ML potentials. | Descriptor-based (e.g., SOAP) [20], Uncertainty-based (e.g., Query-by-Committee) [20] |
| Enhanced Sampling Methods | Techniques to accelerate the sampling of rare events (e.g., barrier crossing) in MD. | Metadynamics [14], Umbrella Sampling [20] |
The critical role of explicit solvent models in validating solution ensembles is undeniable. While implicit solvents are invaluable tools for high-throughput screening and systems where computational cost is the primary constraint, the data shows they can introduce significant inaccuracies in the description of complex biomolecular systems and chemical reactions. Explicit solvents, through their atomistic treatment of solute-solvent interactions, provide a more rigorous and reliable foundation for generating and validating solution ensembles. The emergence of machine learning potentials, trained via active learning on explicit solvent quantum chemical data, is a transformative development. This approach is beginning to bridge the accuracy-efficiency gap, promising to make the predictive power of explicit solvent models more accessible for routine use in drug discovery and molecular science [14] [20]. For research where quantitative accuracy is paramount, explicit solvent models remain the indispensable gold standard.
Molecular dynamics (MD) simulation has emerged as a transformative methodology that extends structural biology beyond static snapshots, enabling researchers to capture the thermal fluctuations and dynamic processes fundamental to biological function. Unlike crystallography or cryo-EM that provide stationary structural information, MD simulations reveal how biomolecules move, fluctuate, and undergo conformational changes in full atomic detail at extraordinarily fine temporal resolution [21]. This capability is particularly valuable for understanding allosteric regulation, drug binding, and functional mechanisms that depend entirely on molecular motion.
The growing impact of MD simulations in molecular biology and drug discovery is particularly noticeable in neuroscience and pharmaceutical development, where simulations have proven valuable in deciphering functional mechanisms of proteins, uncovering structural bases for disease, and optimizing therapeutic compounds [21]. This guide provides a comprehensive comparison of MD approaches specifically for characterizing thermal fluctuations, validating dynamical properties against experimental data, and selecting appropriate computational strategies for research objectives centered on molecular mobility and conformational ensembles.
Comparative Analysis of Molecular Dynamics Software
The selection of MD software significantly influences the efficiency, scale, and accuracy of simulations capturing thermal fluctuations. The table below provides a systematic comparison of predominant software packages used for molecular mechanics modeling, highlighting their specific capabilities for probing molecular dynamics [1].
Table 1: Comparison of Molecular Dynamics Software Platforms
| Software | MD Method | GPU Support | Enhanced Sampling | QM/MM | Implicit Solvent | License | Primary Applications |
|---|---|---|---|---|---|---|---|
| AMBER | Yes | Yes | Yes (REMD) | Yes | Yes | Proprietary/Open Source | Biomolecular simulations, drug discovery |
| GROMACS | Yes | Yes | Yes (REMD) | No | Yes | Open Source | High-performance MD of biomolecules |
| NAMD | Yes | Yes | Yes | No | Yes | Free Academic | Large biomolecular systems |
| CHARMM | Yes | Yes | Yes | Yes | Yes | Proprietary | Comprehensive biomolecular modeling |
| Desmond | Yes | Yes | Yes | No | No | Proprietary/Free | Drug discovery, molecular modeling |
| OpenMM | Yes | Yes | Yes (REMD) | No | Yes | Open Source | Customizable simulation algorithms |
| LAMMPS | Yes | Yes | Yes | Yes | Yes | Open Source | Soft materials, polymers, coarse-grain |
Each platform offers distinct advantages for specific aspects of thermal fluctuation analysis. GROMACS and OpenMM provide exceptional performance for large-scale biomolecular simulations due to sophisticated GPU acceleration [1]. AMBER and CHARMM offer extensive force fields and enhanced sampling methods like replica exchange MD (REMD) crucial for adequate conformational sampling [1]. For researchers focusing on drug discovery, Desmond integrates well with visualization and analysis tools commonly used in pharmaceutical development [1].
Experimental Validation of Thermal Fluctuations
Case Study: EF-SAM Domain as a Calcium and Thermal Sensor
Advanced MD simulations have revealed that the EF-SAM domain of STIM1 functions as a sophisticated dual sensor for both calcium concentration and temperature changes. Conventional and enhanced sampling MD simulations demonstrated that the hidden EF-hand (hEF) subdomain exhibits prompt and extended unfolding at slightly elevated temperatures, exposing a highly conserved hydrophobic Phe108 residue [22]. This study revealed an intricate interplay between calcium and temperature sensing, with both the canonical EF-hand (cEF) and hEF subdomains displaying significantly higher thermal stability in their calcium-loaded forms compared to calcium-free states [22].
The simulations further identified a modular architecture for the EF-SAM domain comprising three functional units: a thermal sensor (hEF), a calcium sensor (cEF), and a stabilizing domain (SAM). The SAM domain unexpectedly displayed high thermal stability compared to the EF-hands and appears to act as a stabilizer for the latter [22]. These findings provided crucial atomic-level insights into the mechanism of temperature-dependent regulation of STIM1, explaining how this protein can activate in response to thermal stimuli independent of endoplasmic reticulum calcium depletion.
Table 2: Quantitative Analysis of Thermal Fluctuation Methodologies
| Methodology | System Size | Time Scale | Spatial Resolution | Key Measurable Parameters | Validation Approaches |
|---|---|---|---|---|---|
| All-Atom MD | 10^4-10^6 atoms | ns-μs | Atomic (0.1-1 Å) | RMSD, RMSF, dihedral distributions | NMR relaxation, B-factors |
| Coarse-Grained MD | 10^5-10^8 atoms | μs-ms | Residue (5-10 Å) | Domain motions, large-scale fluctuations | Light scattering, FRET |
| Enhanced Sampling (REMD) | 10^4-10^5 atoms | Effectively ms-s | Atomic | Free energy landscapes, transition pathways | Single-molecule experiments |
| ab Initio MD | 10^2-10^4 atoms | ps-ns | Electronic | Bond vibrations, charge transfer | Spectroscopic data |
Internal Standard-Assisted Workflow for Thermal Stability Assessment
A novel internal standard-assisted ab initio MD simulation strategy enables direct and comparative evaluation of thermal stability and decomposition mechanisms, effectively mitigating temperature fluctuation issues and empirical limitations in traditional simulations [23]. This approach involves selecting both an object molecule and a reference molecule, subjecting them to parallel MD simulations, and analyzing the resulting decomposition events to provide mechanistic insights into relative stability [23]. The method has been validated using five pairs of energetic molecules with different structures and experimental thermal stability, demonstrating that the molecule decomposing first with higher frequency in each pair consistently corresponds to lower decomposition barriers [23].
Internal Standard-Assisted MD Workflow
Comparative Methods for Thermal Property Prediction
For thermodynamic properties like thermal conductivity, both Equilibrium MD (EMD) and Reverse Non-Equilibrium MD (RNEMD) methods have been systematically compared. In studies of n-decane at sub/supercritical pressures, EMD methods demonstrated better prediction accuracy than RNEMD methods when using the same united atom (UA) force field models [24]. United atom force field models significantly outperformed all-atom force field models for predicting thermal conductivity, with the SKS model showing the best prediction accuracy among all tested force field models [24]. The overall averaged absolute relative deviation (AARD) of EMD simulations with the SKS force field model was only 2.05% compared to experimental data [24].
Experimental Protocols for Validation
Protocol 1: Red Blood Cell Membrane Fluctuation Analysis
This protocol outlines a method for analyzing thermal shape fluctuations of red blood cells through comparison between experiments and coarse-grained molecular dynamics simulations [25].
Sample Preparation: Isolate red blood cells from fresh blood samples using standard centrifugation procedures. Suspend in appropriate physiological buffer.
Experimental Imaging: Record fluctuations of 2D equatorial contours of red blood cells using fast phase-contrast video microscopy at 37°C.
Image Analysis: Process video data to extract membrane contour coordinates with temporal resolution sufficient to capture thermal fluctuation frequencies.
Coarse-Grained Simulation Setup: Construct molecular model of red blood cell membrane with resolution appropriate for capturing bending modulus and elastic properties.
Parameter Matching: Adjust simulation parameters until fluctuation spectrum matches experimental measurements within statistical error.
Validation: Compare simulation-predicted fluctuation amplitudes across multiple wavevectors with experimental data to validate model accuracy.
Protocol 2: Internal Standard-Assisted Thermal Stability Assessment
This protocol describes the novel internal standard approach for comparative thermal stability evaluation of molecular systems [23].
Reference Selection: Identify and select appropriate reference molecule with known thermal decomposition properties similar to target molecules.
System Preparation: Construct simulation boxes containing both target and reference molecules with identical simulation parameters and conditions.
Parallel MD Simulations: Perform ab initio molecular dynamics simulations at multiple temperatures above decomposition thresholds.
Event Monitoring: Track decomposition events through bond breaking patterns, radical formation, or product molecule generation.
Frequency Analysis: Calculate and compare decomposition event frequencies between target and reference molecules across multiple simulation replicates.
Barrier Estimation: Correlate decomposition frequencies with known energy barriers to establish predictive capability for new molecules.
Protocol 3: Temperature-Dependent Domain Unfolding Analysis
This protocol details the approach for investigating thermal stability of specific protein domains, as demonstrated in STIM1 EF-SAM studies [22].
System Setup: Construct simulation system with protein solvated in appropriate physiological buffer using explicit water molecules.
Temperature Control: Implement gradual temperature ramping or multiple fixed-temperature simulations to probe unfolding transitions.
Order Parameter Definition: Establish quantitative metrics for domain stability, such as root-mean-square deviation (RMSD) of specific subdomains, radius of gyration, or contact maintenance.
Enhanced Sampling: Apply replica exchange molecular dynamics or metadynamics to adequately sample unfolding transitions within feasible simulation timeframes.
Hydrophobic Exposure Monitoring: Track solvent-accessible surface area of key hydrophobic residues to identify initiation sites for unfolding.
Experimental Correlation: Compare simulation-predicted unfolding temperatures and pathways with experimental circular dichroism or fluorescence thermal shift assays.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Computational Tools for Thermal Fluctuation Studies
| Reagent/Solution | Function/Application | Example Implementation |
|---|---|---|
| CHARMM Force Field | All-atom empirical energy function | Protein-ligand binding affinity calculations |
| AMBER Force Field | Biomolecular parameter set | Nucleic acid and protein simulations |
| OPLS-AA | All-atom optimized potentials | Liquid simulations and solvation free energies |
| GROMOS | Unified atom parameter set | Lipid membrane and membrane protein systems |
| SKS United Atom | Coarse-grained hydrocarbon model | Thermal conductivity prediction for n-alkanes [24] |
| TraPPE-UA | Transferable potentials for phase equilibria | Fluid phase behavior and transport properties |
| TIP3P/TIP4P Water | Explicit water models | Solvation dynamics and hydrophobic effects |
| CHARMM-GUI | Web-based simulation input generator | Membrane system assembly and parameterization |
Visualization of Thermal Fluctuation Analysis Workflow
The following diagram illustrates the integrated computational and experimental workflow for validating thermal fluctuations through molecular dynamics simulations, highlighting the cyclical nature of model refinement based on experimental validation.
Thermal Fluctuation Analysis Workflow
The integration of molecular dynamics simulations with experimental validation represents a powerful framework for advancing beyond static structural analysis to capture the essential thermal fluctuations governing molecular function. The comparative approaches outlined in this guide provide researchers with methodologies to select appropriate simulation strategies, validate dynamical properties against experimental data, and implement robust protocols for characterizing molecular flexibility and stability. As force fields continue to improve and computational resources expand, MD simulations will play an increasingly central role in bridging the gap between structural snapshots and functional dynamics across diverse research domains from drug discovery to materials science.
Overfitting Risks in Implicit Solvent Methods
Implicit solvent models are indispensable tools in biomolecular simulations, dramatically reducing computational cost by representing solvent as a continuum dielectric medium rather than explicit molecules [26]. This efficiency, however, comes with inherent validation challenges. Within the context of validating solution ensembles from molecular dynamics (MD) research, a critical yet often overlooked risk emerges: overfitting. This occurs when a model's parameters are tuned to reproduce limited experimental data without capturing the underlying physical principles, compromising transferability and predictive power for systems beyond the training set.
The fundamental risk stems from the parameterization process itself. As noted in recent biophysical literature, "Parameterization remains a challenge, as the accuracy of these models depends strongly on the choice of atomic radii, dielectric constants, and empirical coefficients" [26]. When these parameters are adjusted to optimize agreement with specific experimental measurements, they may inadvertently incorporate not only genuine physical effects but also noise or system-specific idiosyncrasies. This problem becomes particularly acute when integrating implicit solvent methods with experimental data for validating solution ensembles of biomolecules like intrinsically disordered proteins (IDPs), where the heterogeneous nature of conformational states creates ample opportunity for over-parameterization [27].
Quantifying Overfitting: Comparative Analysis of Solvation Methods
Table 1: Comparative Performance and Overfitting Indicators Across Solvation Methods
| Method Category | Representative Examples | Parameterization Flexibility | Primary Overfitting Risks | Typical Validation Approaches |
|---|---|---|---|---|
| Continuum Implicit Models | Poisson-Boltzmann (PB), Generalized Born (GB), Polarizable Continuum Model (PCM) | Medium: Atomic radii, dielectric constants, surface tension parameters [26] | Over-optimization to specific solute classes; inadequate transferability [26] | Solvation free energy predictions; comparison with explicit solvent references [26] |
| Machine Learning Implicit Models | LSNN, Katzberger & Riniker GNN, ML-corrected PB/GB [19] | High: Network architecture, weights, features [19] | Force-matching without energy reference; limited training data diversity [19] | Alchemical free energy calculations; conformational sampling comparison [19] |
| Explicit Solvent | TIP3P, TIP4P, SPC water models [28] | Low: Pre-defined force field parameters [29] | Limited due to physical constraints; force field parameterization bias [29] | Direct experimental comparison (WAXS, SAXS, NMR) [29] [27] |
The table above reveals a crucial trade-off: methods with higher parameterization flexibility (like ML-based approaches) offer potentially greater accuracy but present increased overfitting risks without proper validation protocols. Traditional continuum models face challenges in parameter transferability, while explicit solvent methods, though computationally demanding, provide a more physically constrained benchmark for validation.
Table 2: Experimental Validation Metrics for Implicit Solvent Models in Biomolecular Simulations
| Experimental Technique | Validated Molecular Properties | Implicit Solvent Performance Limitations | Explicit Solvent Reference Standard |
|---|---|---|---|
| Wide-Angle X-ray Scattering (WAXS) | Global shape, radius of gyration, structural fluctuations [29] | May miss specific solvent-mediated structural details [29] | Quantitative agreement with profiles using single fitting parameter [29] |
| Small-Angle X-ray Scattering (SAXS) | Ensemble dimensions, molecular form factors [27] | Difficulty capturing accurate solvent density near solute surface [27] | Combined with NMR for maximum entropy reweighting [27] |
| NMR Spectroscopy | Chemical shifts, J-couplings, residual dipolar couplings, relaxation parameters [27] | Limited accuracy for solvent-exposed regions without specific parameterization [27] | Primary data for integrative structural biology of IDPs [27] |
Methodological Frameworks: Protocols for Robust Validation
Maximum Entropy Reweighting for Force Field Assessment
Recent advances in validating solution ensembles employ maximum entropy reweighting to integrate experimental data with molecular dynamics simulations. This approach is particularly valuable for assessing implicit solvent methods while mitigating overfitting risks:
Experimental Integration Protocol:
- Perform long-timescale MD simulations using the implicit solvent model of interest (e.g., 30μs for IDP systems) [27]
- Calculate experimental observables from simulation trajectories using forward models for NMR chemical shifts, SAXS profiles, and other relevant data [27]
- Apply maximum entropy reweighting with minimal free parameters (typically just the effective ensemble size via Kish ratio) [27]
- Compare ensemble properties before and after reweighting to assess initial force field quality [27]
- Validate against orthogonal data not used in the reweighting process [27]
This methodology "facilitates the integration of MD simulations with extensive experimental datasets and demonstrates progress towards the calculation of accurate, force-field independent conformational ensembles" [27]. The approach minimizes subjective decisions about restraint strengths that could otherwise introduce bias.
Advanced Machine Learning with Physical Constraints
Machine learning approaches to implicit solvation face unique overfitting challenges, particularly the "arbitrary constant problem" in energy predictions. The recently developed LSNN (λ-Solvation Neural Network) addresses this through a modified training strategy:
LSNN Training Workflow:
- Architecture Selection: Employ graph neural networks (GNNs) that incorporate molecular topology [19]
- Multi-term Loss Function: Implement a combined optimization targeting:
- Training Data Diversity: Utilize large-scale datasets (~300,000 small molecules) to ensure broad chemical space coverage [19]
- Physical Constraints: Embed fundamental physical principles into the network architecture [19]
This approach ensures that "solvation free energies can be meaningfully compared across chemical species" rather than just achieving accurate forces for conformational sampling [19].
Table 3: Essential Computational Tools for Implicit Solvent Validation
| Tool/Resource | Category | Primary Function in Validation | Key Applications |
|---|---|---|---|
| Open Molecules 2025 (OMol25) | Dataset [30] | Provides high-accuracy quantum chemical reference data for biomolecules, electrolytes, and metal complexes [30] | Training and benchmarking ML-based solvation models [30] |
| Maximum Entropy Reweighting Protocol | Algorithm [27] | Integrates experimental data with MD simulations while minimizing overfitting through Kish ratio optimization [27] | Determining accurate conformational ensembles of IDPs [27] |
| LSNN (λ-Solvation Neural Network) | Machine Learning Model [19] | Predicts solvation properties with conserved free energies across chemical space [19] | Free energy calculations for drug discovery applications [19] |
| WAXS/SAXS Calculation Tools | Experimental Comparison [29] | Computes theoretical scattering profiles from structural models for direct experimental validation [29] | Validating solution ensembles from implicit solvent simulations [29] |
| General Solubility Equation (GSE) | Analytical Model [31] | Provides baseline prediction for solubility using logP and melting point [31] | Benchmarking MD-based solubility predictions [31] |
The validation of implicit solvent methods requires careful attention to overfitting risks, particularly as machine learning approaches expand the parameterization space. The most effective strategies combine multiple validation sources—including explicit solvent benchmarks, diverse experimental data, and physical constraints—to ensure models capture genuine physical behavior rather than idiosyncrasies of training data. The emerging paradigm emphasizes integration over replacement: implicit solvent models refined through experimental integration and multi-scale validation provide the most promising path toward both computational efficiency and physical accuracy. As noted in recent research, "hybridization as a best practice, meaning continuum cores refined by improved physics, such as multipolar water, ML correctors with uncertainty quantification and active learning" [26] represents the future of reliable solvation modeling. For researchers validating solution ensembles, this approach provides a robust framework for leveraging the speed of implicit solvent methods while maintaining confidence in the resulting conformational distributions.
Calculating Experimental Observables from MD Trajectories
Explicit-Solvent SAXS/WAXS Calculation Methods
Small- and Wide-Angle X-ray Scattering (SAXS/WAXS) has evolved into a powerful technique for investigating biomolecular structures and dynamics in near-native solution conditions [32] [9]. The interpretation of experimental scattering data requires accurate computational methods to calculate theoretical profiles from structural models. Traditional implicit-solvent approaches rely on parameterized corrections for hydration layers and excluded solvent effects, introducing free parameters that must be fit to experimental data [33] [34]. In contrast, explicit-solvent methods leverage molecular dynamics (MD) simulations to physically model solvent structure and interactions, eliminating fitting ambiguities and providing a more rigorous foundation for validating solution ensembles from molecular dynamics research [7] [33].
This comparison guide examines state-of-the-art explicit-solvent approaches for SAXS/WAXS calculation, focusing on their implementation, performance, and application in biomolecular research. By objectively evaluating these methods against alternatives and presenting supporting experimental data, we provide researchers with a foundation for selecting appropriate computational tools for interpreting scattering data.
Theoretical Foundation: Explicit vs. Implicit Solvent Models
Limitations of Implicit-Solvent Approaches
Implicit-solvent methods model hydration effects through simplified approximations, typically employing a homogeneous excess electron density layer around the solute [33] [34]. Popular implementations such as CRYSOL, FoXS, and PepsiSAXS require fitting parameters for the hydration layer density and excluded solvent volume [35] [34]. While computationally efficient, these approaches face significant limitations:
- Parameter ambiguity: The optimal hydration layer density (typically 10-15% of bulk water density) is not easily measurable and may differ between solutes [7] [33]
- Overfitting risk: Multiple fitting parameters can absorb subtle conformational differences, reducing sensitivity to actual structural changes [7] [9]
- Limited physical accuracy: Simplified solvent models struggle to capture specific solute-solvent interactions, particularly for highly charged biomolecules like RNA [35] [33]
- Wide-angle limitations: Implicit methods become increasingly inaccurate at wider angles where solvent structure contributions are significant [7] [33]
Explicit-Solvent Fundamentals
Explicit-solvent methods compute scattering profiles directly from atomistic MD simulations of the solute in aqueous solution [7] [34]. The theoretical foundation follows the experimental contrast method:
[I(q) = I{\text{sample}}(q) - I{\text{solvent}}(q)]
where (I{\text{sample}}(q)) is computed from the solute-solvent system and (I{\text{solvent}}(q)) from a pure-solvent simulation [7] [33]. This approach:
- Physically models hydration: Explicit water molecules naturally form structured hydration layers with density variations [7] [34]
- Eliminates solvent parameters: No fitting is required for hydration layer density or excluded solvent effects [7] [34]
- Captures thermal fluctuations: MD simulations naturally include conformational dynamics and their impact on scattering [7] [9]
- Accurately models ions: Explicit ions account for specific ion effects in buffer solutions [35]
The computational workflow involves constructing a spatial envelope around the solute that encompasses the hydration layer, then computing scattering intensities from the electron densities within this envelope for both sample and pure-solvent systems [7] [34].
Figure 1: Explicit-solvent SAXS/WAXS calculation workflow. The method requires parallel MD simulations of the solvated biomolecule and pure solvent, followed by scattering calculation within a constructed spatial envelope and solvent subtraction.
Comparative Analysis of Calculation Methods
Software Tools and Methodologies
Table 1: Comparison of SAXS/WAXS Calculation Methods and Software
| Method/Software | Solvent Treatment | Key Features | Computational Demand | Applicable q-Range |
|---|---|---|---|---|
| WAXSiS [7] [34] | Explicit solvent with spatial envelope | Web server automation; No solvent fitting parameters; Accounts for thermal fluctuations | High (MD required) | Full SAXS/WAXS range |
| Capriqorn [35] | Explicit solvent with geometric selection | Frame-by-frame spectra; User-defined sphere radius and shell width | High (MD required) | Full SAXS/WAXS range |
| CRYSOL [35] [34] | Implicit solvent with hydration layer | Spherical harmonics expansion; Fast computation; Fitted hydration parameters | Low | Primarily SAXS regime |
| PLUMED [35] | Implicit or coarse-grained | On-the-fly calculation for enhanced sampling; Coarse-grained representation available | Medium | SAXS regime |
Performance Comparison and Experimental Validation
Table 2: Quantitative Performance Comparison for RNA GAC System (Adapted from [35])
| Method | Rg from Guinier Fit (Å) | Computation Time | Agreement with Experiment | Solvent Parameters Fitted |
|---|---|---|---|---|
| WAXSiS | 22.4 ± 0.3 | ~Hours (MD dependent) | Excellent | None (only scale and offset) |
| Capriqorn | 22.6 ± 0.4 | ~Hours (MD dependent) | Excellent | None (only scale and offset) |
| CRYSOL 3 | 21.9 ± 0.2 | Minutes | Good | Hydration layer density |
| CRYSOL 2 | 22.1 ± 0.2 | Minutes | Fair | Hydration layer density |
| PLUMED (CG) | 22.7 ± 0.5 | Minutes | Fair | Excluded volume parameters |
Studies validating explicit-solvent methods against experimental data demonstrate their superior accuracy. For example, research on five different proteins showed that explicit-solvent MD simulations achieved excellent agreement with experimental WAXS profiles across small and wide angles using only a single fitting parameter for experimental uncertainties [7]. The explicit treatment eliminated free parameters associated with the solvation layer or excluded solvent, minimizing overfitting risks [7].
Notably, explicit-solvent methods correctly capture the increased radius of gyration (Rg) due to the hydration layer without empirical adjustments. MD simulations naturally reproduce the ~1-2% increase in Rg observed experimentally, whereas implicit methods must fit this effect through hydration layer parameters [34]. This physical accuracy makes explicit-solvent approaches particularly valuable for detecting subtle conformational changes that might be obscured by parameter fitting in implicit methods [7] [9].
Experimental Protocols and Implementation
WAXSiS Web Server Protocol
The WAXSiS server provides automated explicit-solvent SAXS/WAXS calculations through the following methodology [7] [34]:
System Preparation:
- Input protein or nucleic acid structure (PDB format)
- Server automatically solvates the system in explicit water
- Adds ions to achieve physiological concentration (e.g., 150 mM NaCl)
Molecular Dynamics Simulation:
- Runs explicit-solvent MD simulation (typically 20-500 ps depending on system size)
- Applies position-restraining potentials (1000 kJ/mol/nm²) to backbone/heavy atoms
- Samples thermal fluctuations of side chains, water, and counterions
- Runs separate pure-solvent simulation for background subtraction
Spatial Envelope Construction:
- Builds envelope from icosphere subdivided recursively
- Adjusts vertices to maintain minimum distance (default 7Å) from all solute atoms
- Uses identical envelope across all simulation frames
Scattering Calculation:
- Computes scattering amplitudes using atomic form factors with electron-withdrawing corrections for water
- Evaluates orientational average using spiral method for vector distribution
- Applies buffer density matching (default 334 e/nm³ for pure water)
Experimental Data Fitting (if provided):
- Fits only scale factor (f) and constant offset (c): Ifit(q) = fIexp(q) + c
- No fitting of hydration layer or excluded solvent parameters
GROMACS-SWAXS Implementation
For researchers implementing explicit-solvent calculations locally, the GROMACS-SWAXS extension provides comprehensive tools [33]:
Simulation Requirements:
- All-atom MD simulation with explicit water (typically 20-100 ns)
- Compatible force fields (AMBER, CHARMM, OPLS-AA)
- Appropriate ion concentrations matching experimental conditions
- Parallel pure-solvent simulation with identical buffer composition
Envelope Selection Criteria:
- Envelope distance: Typically 5-7Å from solute surface
- Must encompass entire first hydration shell
- Should maintain consistent volume across conformational ensemble
Convergence Considerations:
- Number of frames required depends on solute-solvent contrast
- Small biomolecules and IDPs require more frames due to lower contrast
- Reference values: ~1000 frames for globular proteins, ~5000 for IDPs [33]
Figure 2: Spatial envelope concept in explicit-solvent calculations. The envelope encompasses the solute and structured hydration layer while excluding bulk solvent, enabling accurate computation of scattering contrast.
Table 3: Essential Computational Tools for Explicit-Solvent SAXS/WAXS Research
| Tool/Resource | Function | Implementation | Access |
|---|---|---|---|
| WAXSiS Web Server [34] | Automated explicit-solvent calculations | Full pipeline from structure to scattering profile | Web interface (http://waxsis.uni-goettingen.de) |
| GROMACS-SWAXS [33] | MD simulations with integrated scattering calculations | GROMACS extension for trajectory analysis | Local installation (https://gitlab.com/cbjh/gromacs-swaxs) |
| Capriqorn [35] | Explicit-solvent trajectory analysis | Standalone software for MD trajectories | Local installation |
| AMBER/CHARMM | Force fields for biomolecular MD | Parameter sets for proteins, nucleic acids, lipids | Academic licensing |
| OPC Water Model [35] | Accurate water structure prediction | Four-point water model compatible with ion parameters | MD software implementation |
Applications in Solution Ensemble Validation
Explicit-solvent SAXS/WAXS methods have proven particularly valuable for validating solution ensembles from molecular dynamics simulations across multiple biological systems:
RNA Structural Dynamics
Studies on the 57-nucleotide GTPase-associated center (GAC) RNA demonstrated that explicit-solvent methods accurately capture scattering profiles under varying ionic conditions [35]. The calculations revealed solvent effects significant even at relatively low scattering vectors, highlighting the importance of explicit solvent modeling for nucleic acids that profoundly influence their hydration environment [35].
Protein Conformational Transitions
Research on periplasmic binding proteins, aspartate carbamoyltransferase, and nuclear exportins demonstrated that explicit-solvent SWAXS-driven MD can refine structures against experimental data without prior knowledge of reaction paths [36]. The combination accelerates conformational transitions in MD simulations while reducing force-field bias [36].
Sensitivity to Structural Fluctuations
Explicit-solvent calculations have revealed the remarkable sensitivity of WAXS profiles to minor conformational rearrangements. Free MD simulations up to one microsecond demonstrated detectable scattering changes from increased loop flexibility or Rg variations as small as 1% [7]. This sensitivity enables detection of subtle structural changes inaccessible to implicit solvent methods.
Explicit-solvent SAXS/WAXS calculation methods represent a significant advancement for validating solution ensembles from molecular dynamics research. By physically modeling solvent structure and interactions through MD simulations, these approaches eliminate fitting ambiguities associated with implicit solvent methods and provide more rigorous validation of structural models.
While computationally demanding, explicit-solvent methods offer unparalleled accuracy across the entire scattering range, particularly for wide angles where solvent contributions become dominant. The development of user-friendly implementations like the WAXSiS web server and GROMACS-SWAXS has made these powerful tools accessible to non-specialists, enabling broader adoption in structural biology research.
As MD force fields continue to improve and computational resources expand, explicit-solvent approaches are poised to become standard tools for interpreting solution scattering data, ultimately enhancing our understanding of biomolecular structure and dynamics in physiologically relevant environments.
Advanced Ensemble Refinement Techniques (BioEn, EROS)
The accurate structural characterization of biomolecules is fundamental to advancing our understanding of their biological functions and for rational drug design. However, biomolecules are inherently dynamic, often sampling multiple conformational states that are crucial for their function. Traditional single-structure approaches in structural biology fail to capture this complexity, potentially leading to oversimplified or misleading models. This limitation is particularly pronounced for highly flexible systems such as intrinsically disordered proteins, multi-domain proteins with flexible linkers, and large RNA molecules [37] [38].
Ensemble refinement has emerged as a powerful methodology that bridges this gap by combining molecular simulations with experimental data to generate structural ensembles that represent the dynamic nature of biomolecules. These techniques address the fundamental challenge that most experimental measurements in structural biology are ensemble-averaged, where the observed signal comes from the average across many molecules in different conformational states [38]. By refining ensembles against experimental data, researchers can obtain more faithful descriptions of the true conformational diversity present in biological systems.
The Bayesian inference of ensembles (BioEn) and ensemble refinement of SAXS (EROS) methods represent sophisticated approaches in this domain, enabling researchers to infer detailed structural models for biomolecules exhibiting significant dynamics by combining input from experiment and simulation in a balanced manner [39]. These methods are particularly valuable for validating solution ensembles derived from molecular dynamics simulations, as they provide a statistical framework for assessing whether computational ensembles accurately reflect experimental reality.
Theoretical Foundations and Methodological Frameworks
The Bayesian Inference of Ensembles (BioEn) Method
The BioEn method provides a general Bayesian framework for ensemble refinement, building upon maximum-entropy principles [39] [40]. In this approach, the posterior probability is maximized as a function of the statistical weights ( w_\alpha ) assigned to each ensemble member:
[ P(w|\text{data}) \propto P(\text{data}|w)P(w) ]
where ( P(\text{data}|w) ) is the likelihood function and ( P(w) ) is the prior given by:
[ P(w) = \exp\left(\theta S\right) ]
with ( S ) being the relative entropy:
[ S = -\sum{\alpha=1}^N w\alpha \ln\left(\frac{w\alpha}{w\alpha^0}\right) ]
Here, ( w_\alpha^0 ) are the reference weights (typically from the simulation force field), and the parameter ( \theta ) expresses confidence in the reference ensemble [39]. Large values of ( \theta ) indicate high confidence in the prior ensemble, resulting in optimal weights closer to the reference weights.
For uncorrelated Gaussian errors in experimental measurements, the negative log-posterior becomes:
[ L = \frac{1}{2}\sum{i=1}^M \frac{\left(\langle yi \rangle - Yi\right)^2}{\sigmai^2} - \theta S ]
where ( \langle yi \rangle = \sum{\alpha=1}^N w\alpha y{i\alpha} ) is the ensemble-averaged calculated value of observable ( i ), ( Yi ) is the experimental measurement, and ( \sigmai ) encompasses uncertainties from both experiment and calculation [39].
Ensemble Refinement of SAXS (EROS)
The EROS method is a special case of the more general BioEn framework, specifically adapted for refining ensembles against Small-Angle X-ray Scattering data [39] [38]. While sharing the same theoretical foundation based on maximum entropy principles, EROS focuses particularly on SAXS data, which provides ensemble-averaged information about macromolecular size and shape [39].
In EROS, the objective is to minimize the function ( \chi^2 - \theta S ), where ( \chi^2 ) represents the discrepancy between calculated and experimental SAXS profiles, and ( S ) is the negative Kullback-Leibler divergence [39]. The BioEn and EROS formulations differ mainly by a factor of ( \frac{1}{2} ) scaling the ( \chi^2 ) term, which is equivalent to a trivial rescaling of the confidence parameter ( \theta ).
Optimization Strategies for Large-Scale Problems
Ensemble refinement by reweighting presents computationally challenging optimization problems, particularly when dealing with large numbers of structures and experimental data points. To address this, researchers have developed efficient numerical methods:
Table 1: Optimization Methods for Ensemble Refinement
| Method | Optimization Variables | Dimensionality | Key Features |
|---|---|---|---|
| Log-Weights Optimization | Log-weights ( g\mu = \ln w\mu ) | N-1 variables | Transforms constrained problem to unconstrained; ensures weight positivity [39] |
| Generalized Forces Optimization | Generalized forces ( \lambda_i ) | M variables | Solves dual problem; efficient when M << N [39] |
The log-weights method introduces variables ( g\alpha = \ln w\alpha ), which are determined up to an additive constant that cancels in normalization [39]. This transformation converts the constrained optimization problem into an unconstrained one, enabling the use of efficient gradient-based optimization algorithms.
The generalized forces method leverages the analytical solution for the optimal weights:
[ w\alpha = \frac{w\alpha^0 \exp\left(-\sumi \lambdai y_{i\alpha}\right)}{Z} ]
where ( Z ) is the normalization constant, and optimizes directly with respect to the generalized forces ( \lambda_i ) [39]. This approach is particularly efficient when the number of experimental data points (M) is much smaller than the number of structures (N).
Comparative Analysis: BioEn vs. EROS
Technical Implementation and Algorithmic Differences
While BioEn and EROS share common theoretical roots in maximum entropy principles, they differ in their specific implementations and applications:
Table 2: Comparison of BioEn and EROS Methods
| Feature | BioEn | EROS |
|---|---|---|
| Theoretical Basis | General Bayesian framework [39] | Maximum entropy principle [39] |
| Data Compatibility | Multiple data types (NMR, SAXS, DEER, etc.) [39] | Originally developed for SAXS [39] |
| Mathematical Formulation | Minimizes ( L = \frac{1}{2}\chi^2 - \theta S ) [39] | Minimizes ( \chi^2 - \theta S ) [39] |
| Reference Implementation | BioEn package | EROS implementation |
| Optimization Methods | Log-weights and generalized forces [39] | Similar optimization approaches |
BioEn represents a generalization of EROS, with the key mathematical difference being a scaling factor in the ( \chi^2 ) term [39]. This difference is equivalent to a rescaling of the confidence parameter ( \theta ), making the two methods closely related rather than fundamentally distinct.
Performance Characteristics and Computational Efficiency
Both BioEn and EROS have been demonstrated to be robust, accurate, and efficient methods for ensemble refinement [39]. The computational efficiency of these methods depends on the relationship between the number of structures in the ensemble (N) and the number of experimental data points (M):
- For systems where M << N, the generalized forces optimization method is typically more efficient
- For systems where N is not excessively large, the log-weights optimization method provides robust performance
The unique solution guaranteed by the convexity of the optimization problem ensures that both methods converge to the same result given equivalent input data and parameters [39]. Performance benchmarks using synthetic data have demonstrated that both methods can handle realistic numbers of structures (hundreds of thousands) and experimental data points (thousands) efficiently [39].
Experimental Protocols and Applications
Protocol for Reweighting MD Simulations Using BioEn
Application Example: Reweighting of Ala-5 Peptide Ensemble Using NMR J-Couplings [39]
Generate Reference Ensemble: Perform all-atom molecular dynamics simulation of the disordered peptide Ala-5 using the AMBER99SB*-ildn-q force field to generate an initial structural ensemble.
Calculate Observables: For each structure in the ensemble, calculate the theoretical values of NMR J-couplings that will be compared with experimental data.
Set Confidence Parameter: Choose an appropriate value for the confidence parameter ( \theta ) based on the reliability of the force field and the quality of the experimental data.
Optimize Weights: Solve the optimization problem using either the log-weights or generalized forces method to obtain the optimal weights ( w_\alpha ) that maximize the posterior probability.
Validate Results: Assess the quality of the refined ensemble by examining:
- Agreement with experimental data (reduced ( \chi^2 ))
- Changes in conformational populations
- Physical plausibility of the resulting ensemble
In the Ala-5 case study, after reweighting, researchers observed a consistent increase in the population of polyproline-II conformations and a decrease of α-helical-like conformations, demonstrating how ensemble refinement can reveal structural insights that might be missed in conventional analysis [39].
Protocol for Cryo-EM Ensemble Refinement Using Metainference
Application Example: RNA Structure Refinement Using Cryo-EM Data [37]
Initial Model Preparation: Inspect the deposited cryo-EM RNA structure, identify mismodeled regions, and rebuild problematic sections using appropriate tools (e.g., DeepFoldRNA for missing fragments, MD simulations with restraints to correct mispaired helices).
Set Up Metainference Simulation: Initialize a multi-replica MD simulation (minimum 8 replicas for the group II intron ribozyme case) with restraints based on the cryo-EM density map.
Run Ensemble Refinement: Perform production simulation while enforcing agreement between the experimental density map and the ensemble average computed across replicas.
Release Restraints: After initial equilibration period (e.g., 5 ns), release artificial helical restraints to allow the ensemble to naturally sample conformations compatible with the experimental data.
Analyse Results: Collect trajectories, analyze conformational diversity, and validate against experimental data.
In the group II intron ribozyme application, this approach revealed that a single structure could not simultaneously be compatible with the experimental data and expected RNA helical geometry, resulting in mismodeling of flexible regions [37]. Ensemble refinement correctly identified stable helices that were misrepresented in the single-structure approach.
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for Ensemble Refinement
| Tool/Resource | Type | Function | Application Context |
|---|---|---|---|
| AMBER99SB*-ildn-q | Molecular mechanics force field | Provides prior structural ensemble for BioEn reweighting [39] | Protein ensemble refinement |
| GROMACS | Molecular dynamics software | Generates reference ensembles for reweighting | MD simulation production |
| BioEn Package | Software library | Implements Bayesian ensemble refinement algorithm [39] | Reweighting MD ensembles |
| PLUMED | Plugin for enhanced sampling | Implements metainference for on-the-fly refinement [37] | Integrative MD simulations |
| ModelSEED | Metabolic network reconstruction | Drafts model reconstructions in SBML format [41] | Metabolic engineering applications |
| KEGG Pathway | Metabolic pathway database | Reference metabolic pathways for comparative analysis [41] | Network reconstruction |
| MetaCyc | Metabolic pathway database | Organism-specific metabolic network diagrams [41] | Pathway analysis |
| SBML | Model representation format | Standard format for exchanging metabolic models [41] | Data interoperability |
Workflow Visualization
Ensemble Refinement Workflow Diagram
Ensemble refinement methods like BioEn and EROS represent sophisticated approaches for validating solution ensembles from molecular dynamics research. By combining the physical realism of molecular simulations with the empirical constraints of experimental data, these methods provide a balanced approach to modeling biomolecular dynamics. The Bayesian framework underlying BioEn offers a principled approach to managing the trade-offs between force field accuracy and experimental agreement.
As structural biology continues to recognize the importance of conformational heterogeneity, and as experimental techniques provide increasingly diverse data types, ensemble refinement methods will play an increasingly crucial role in bridging the gap between simulation and experiment [38]. Future developments will likely focus on improving scalability for larger systems, integrating more diverse data types, and developing more sophisticated prior distributions that better capture our knowledge of biomolecular physics.
For researchers in drug development, these methods offer powerful approaches for characterizing the dynamic landscapes of therapeutic targets, potentially revealing cryptic binding sites and allosteric mechanisms that would be invisible in single-structure approaches. The continued refinement and application of these techniques will undoubtedly enhance our ability to connect structural dynamics to biological function and therapeutic intervention.
Handling Solvent Contributions and Buffer Subtraction
In molecular dynamics (MD) simulations, accurately modeling solvent contributions and performing correct buffer subtraction are critical for validating solution ensembles against experimental data. These factors directly influence the interpretation of biomolecular structure, dynamics, and interactions. Implicit and explicit solvent models offer distinct trade-offs between computational efficiency and physical accuracy, while proper buffer subtraction protocols ensure that calculated scattering intensities meaningfully compare with experimental measurements. This guide objectively compares predominant methodologies, evaluates their performance, and provides detailed experimental protocols for researchers and drug development professionals working within the broader thesis of validating solution ensembles.
Comparative Analysis of Solvent Models in MD Simulations
Solvent models in computational chemistry account for the behavior of solvated condensed phases, falling primarily into explicit, implicit, and hybrid categories [13]. Each approach offers distinct advantages and limitations for modeling biomolecular systems.
Explicit solvent models treat solvent molecules as individual entities with defined coordinates and degrees of freedom [13]. These models provide a physically realistic, spatially resolved description of the solvent, capturing specific solute-solvent interactions, hydrogen bonding fluctuations, and solvent ordering effects. Popular explicit water models include TIP3P, SPC/E, TIP4P, TIP4PEw, OPC, and TIP5P, which differ in their computational complexity and accuracy in reproducing water properties [15].
Implicit solvent models (also called continuum models) replace explicit solvent molecules with a polarizable medium characterized by properties such as dielectric constant [13]. These models significantly reduce computational cost by eliminating solvent degrees of freedom and are implemented in various forms, including Poisson-Boltzmann (PB), Generalized Born (GB), and Solvent-Accessible Surface Area (SASA) methods [17]. The free energy of solvation in these models is typically decomposed into cavity formation (ΔGcav), van der Waals (ΔGvdW), and electrostatic (ΔGele) components [17].
Hybrid models combine aspects of both explicit and implicit approaches, such as QM/MM methods where a quantum mechanics core is surrounded by molecular mechanics water molecules and an implicit solvent outer layer [13].
Table 1: Comparison of Explicit vs. Implicit Solvent Models for Biomolecular Simulations
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Computational Cost | High (more degrees of freedom) | Low (fewer degrees of freedom) |
| Spatial Resolution | Atomistic detail of solvent molecules | Homogeneous continuum approximation |
| Solvation Structure | Captures specific hydrogen bonding and solvent ordering | Misses local solvent density fluctuations |
| Electrostatic Treatment | Explicit dipole reorientation | Mean-field dielectric response |
| Common Implementations | TIP3P, TIP4P, SPC/E, TIP5P [15] | GB, PB, SASA, COSMO [13] [17] |
| Best Applications | Accurate solvation shell modeling; WAXS validation [7] | High-throughput screening; large conformational sampling |
Quantitative Performance Benchmarking
Accuracy in Glycosaminoglycan Simulations
A 2023 benchmark study compared five implicit (IGB=1,2,5,7,8) and six explicit (TIP3P, SPC/E, TIP4P, TIP4PEw, OPC, TIP5P) solvent models for simulating heparin dp10, a glycosaminoglycan [15]. The table below summarizes key structural parameters compared to experimental references:
Table 2: Solvent Model Performance for Heparin dp10 Simulations (5 μs MD) [15]
| Solvent Model | Radius of Gyration (Å) | End-to-End Distance (Å) | Deviation from Reference |
|---|---|---|---|
| Experimental Reference | ~16.5 | ~46.2 | - |
| TIP3P | 16.3 | 45.9 | Minimal |
| OPC | 16.4 | 46.1 | Minimal |
| TIP4PEw | 16.2 | 45.7 | Minimal |
| SPC/E | 16.1 | 45.5 | Moderate |
| IGB=2 (GB) | 15.8 | 44.2 | Significant |
| IGB=8 (GB) | 17.1 | 48.3 | Significant |
The study revealed that while TIP3P remains a reasonable choice for glycosaminoglycan simulations, more modern water models like OPC and TIP4PEw can provide improved performance, particularly for modeling chain flexibility and ionic interactions [15].
Impact on Wide-Angle X-ray Scattering (WAXS) Validation
Explicit solvent MD simulations demonstrate particular advantage for calculating accurate WAXS profiles. Research shows that using explicit solvation eliminates free parameters associated with the solvation layer or excluded solvent, minimizing overfitting risks [7] [42]. With only a single fitting parameter to account for experimental uncertainties in buffer subtraction and dark currents, explicit solvent MD achieves excellent agreement with experimental WAXS profiles across both small and wide angles [7].
Notably, the influence of different water models (TIP3P vs. TIP4P) and protein force fields on calculated WAXS profiles was found to be insignificant up to q ≈ 15 nm⁻¹, suggesting that the choice of explicit solvent representation matters more than minor force field variations [7]. Incorporating thermal fluctuations through MD simulations significantly improved agreement with experimental data compared to static models, demonstrating the importance of protein dynamics in interpreting WAXS profiles [7].
Experimental Protocols for Buffer Subtraction and WAXS Validation
Explicit-Solvent WAXS Calculation Protocol
The following methodology enables accurate calculation of WAXS profiles from MD simulations with proper buffer subtraction [7]:
System Preparation:
- Solvate the biomolecule in an explicit water box with appropriate dimensions (minimum 8-10 Å padding)
- Add ions to neutralize system charge and match experimental buffer conditions
- Energy minimize and equilibrate the system using standard MD protocols
Production Simulation:
- Run unrestrained MD simulations (microsecond-scale for protein folding/unfolding processes)
- Maintain constant temperature and pressure (NPT ensemble) using thermostats and barostats
- Save trajectories at appropriate intervals for analysis (e.g., every 100 ps)
Scattering Intensity Calculation:
- Compute the instantaneous scattering intensity for each saved snapshot using: I(q) = ⟨|Ã(q)|²⟩' - ⟨|B̃(q)|²⟩' [7] where Ã(q) and B̃(q) are Fourier transforms of the electron densities of the solution and pure solvent systems, respectively
- The ensemble average ⟨···⟩' includes averages over solute rotations and conformational fluctuations
Spatial Envelope Implementation:
- Construct a constant spatial envelope around the solute that encompasses all conformational states and the solvation layer
- Ensure the envelope distance from solute atoms is sufficient to capture bulk solvent behavior
- This approach reduces computational cost and statistical noise while allowing calculation from heterogeneous ensembles
Single-Parameter Fitting:
- Apply a single scaling parameter to account for experimental uncertainties in buffer subtraction and dark currents
- Avoid fitting parameters related to solvation layer density or excluded volume
AI-Enhanced Ab Initio Biomolecular Dynamics
Recent advances introduce artificial intelligence-based ab initio biomolecular dynamics (AI2BMD) that simulate full-atom biomolecules with quantum chemical accuracy at dramatically reduced computational cost [43]. The protocol involves:
Protein Fragmentation: Split proteins into overlapping dipeptide units covering all 20 amino acid types
Machine Learning Force Field Training: Train ViSNet models on comprehensive datasets generated with density functional theory (DFT) calculations
Energy and Force Calculation: Use the AI2BMD potential to compute energies and forces with ab initio accuracy
Explicit Solvent Integration: Combine with polarizable solvent models (e.g., AMOEBA) for realistic solvation
This approach achieves potential energy mean absolute errors of 0.045 kcal mol⁻¹ compared to 3.198 kcal mol⁻¹ for classical molecular mechanics force fields, while reducing computation time by several orders of magnitude compared to conventional DFT [43].
Figure 1: Workflow for Validating Solution Ensembles with Explicit Solvent MD and WAXS
Research Reagent Solutions for Solvent Modeling
Table 3: Essential Research Reagents and Computational Tools for Solvent Modeling
| Reagent/Tool | Type | Function | Example Applications |
|---|---|---|---|
| TIP3P Water Model | Explicit solvent | Three-site water model with fixed point charges | General biomolecular simulations; balance of accuracy and efficiency [15] |
| OPC Water Model | Explicit solvent | Optimized four-point water model with improved accuracy | Glycosaminoglycan simulations; properties requiring precise water geometry [15] |
| Generalized Born (GB) Models | Implicit solvent | Approximates Poisson-Boltzmann electrostatics with speed | High-throughput screening; protein folding studies [17] |
| AMOEBA Force Field | Polarizable force field | Accounts for electronic polarization effects | Accurate ion solvation dynamics; combined with AI2BMD [43] |
| ViSNet Potential | Machine learning force field | Provides ab initio accuracy with quantum chemical data | AI2BMD for protein folding/unfolding; accurate free energy calculations [43] |
| SASA Methods | Implicit solvent | Models nonpolar solvation via solvent-accessible surface area | Protein-ligand binding; membrane systems [17] |
Practical Guidelines for Method Selection
When to Use Explicit vs. Implicit Solvent
Choose explicit solvent models when:
- Validating against experimental WAXS/SAXS data [7]
- Studying specific solvent-mediated interactions (e.g., hydrogen bonding networks)
- Modeling systems with highly ordered water molecules (e.g., ion channels)
- Simulating interface phenomena (e.g., ice-water interfaces in freezing studies) [44]
- Investigating solvent structure around highly charged molecules like glycosaminoglycans [15]
Choose implicit solvent models when:
- Computational resources are limited and system size is large
- Sampling large conformational spaces (e.g., protein folding landscapes)
- Performing high-throughput screening of ligand binding
- Solvent acts primarily as a dielectric continuum without specific interactions
Recommendations for Buffer Subtraction in Scattering Experiments
For accurate buffer subtraction in WAXS validation:
- Use explicit solvent MD simulations to eliminate free parameters associated with solvation layers [7]
- Employ a spatial envelope approach to reduce computational cost while maintaining accuracy [7]
- Incorporate thermal fluctuations through MD simulations rather than relying on static structures [7]
- Apply only a single fitting parameter to account for experimental uncertainties rather than adjusting multiple solvation parameters [7]
- Ensure simulation boxes are sufficiently large to capture bulk water behavior beyond the solvation shell
The demonstrated sensitivity of WAXS profiles to minor conformational changes (e.g., <1% increase in radius of gyration) makes the explicit solvent MD approach particularly valuable for detecting subtle structural rearrangements in biomolecular solutions [7] [42].
SWAXS-AMDE and Other Modern Computational Tools
In structural biology, the synergy between computational simulations and experimental data is paramount for accurately depicting biomolecular conformations in their native solution states. Small- and Wide-Angle X-ray Scattering (SWAXS) has emerged as a powerful technique to probe the structure and dynamics of biological macromolecules. However, interpreting scattering data requires sophisticated computational tools to calculate theoretical profiles from atomic models and compare them with experiments. This guide objectively compares modern software for integrating SWAXS with Molecular Dynamics (MD), with a specific focus on validating solution ensembles from MD research. The accuracy of these ensembles is critical for meaningful biological insights, particularly for flexible systems like multi-domain proteins and Intrinsically Disordered Proteins (IDPs) [45] [46]. We evaluate tools based on their computational efficiency, treatment of solvent effects, number of free parameters, and ability to avoid overfitting, providing a clear framework for scientists to select the appropriate tool for their research.
Comparative Analysis of Modern Computational Tools
The following table summarizes the key features and performance metrics of prominent SWAXS computation tools.
Table 1: Comparison of Modern Computational Tools for SWAXS Analysis
| Tool Name | Solvent Treatment Method | Key Fitting Parameters | Computational Efficiency | Key Advantages & Validation |
|---|---|---|---|---|
| SWAXS-AMDE [47] | Explicit-solvent, atomic detail | None (Accounts for hydration density changes) | N/A (Post-processing of trajectories) | Generalizable model; Works with AMBER, GROMACS, NAMD, OpenMM; Accounts for solute thermal fluctuations, crucial for IDPs. |
| DENSS (denss.pdb2mrc.py) [48] | Implicit solvent (adjusted atomic volumes) | Bulk solvent density, Mean hydration shell contrast | Comparable to leading software; Up to 10x faster for large molecules | Reduced parameters; Eliminates excluded volume fitting; High-quality fits to public SWAXS profiles. |
| Explicit-Solvent MD (Grishaev et al.) [7] | Explicit-solvent MD simulations | Single parameter (for buffer subtraction/experimental uncertainties) | High computational cost (explicit solvent) | Minimizes overfitting risk; Excellent agreement with experiment up to q≈15 nm⁻¹; Highly sensitive to minor conformational changes. |
| CRYSOL [49] | Implicit solvent (continuum model) | Excess solvent density, Excluded volume, Atomic radius scaling | Fast | Widely used; Can be applied to nucleic acids; Frozen solute assumption limits accuracy for flexible molecules. |
| WAXSiS [47] | Explicit-solvent, atomic detail | None | N/A (Post-processing of trajectories) | Accounts for solvent in atomic detail and thermal fluctuations of the solute. |
| SWAXS-driven MD [36] | Explicit-solvent MD | None (Experimental data used as restraint) | High computational cost | Directly refines structures against SWAXS data without foreknowledge of reaction paths; Reduces force-field bias. |
A critical metric for comparing these tools is their quantitative agreement with experimental data. The table below summarizes reported performance from independent studies.
Table 2: Summary of Experimental Validation and Key Applications
| Tool / Method | Reported Accuracy / Agreement | Primary Biomolecules Tested | Key Application Context |
|---|---|---|---|
| SWAXS-AMDE [47] | Good agreement with SAXS data for EK polyampholyte sequences. | Intrinsically Disordered Proteins (IDPs), Polyampholytes | Validating force fields; Testing generalizability of simulation methods. |
| DENSS [48] | High-quality fits to eight public SWAXS profiles; Significantly more accurate than leading software with optimization disabled. | Biological Macromolecules (proteins) | Comparing atomic models to experimental SWAXS data; Density modeling. |
| Explicit-Solvent MD [7] | Excellent agreement with experimental WAXS profiles at small and wide angles. | Proteins (5 different types) | Validating solution ensembles from MD; Studying protein dynamics. |
| HREMD with Explicit Solvent [45] | Excellent agreement with SAXS/SANS and NMR (χ² defined in Eq. 5) for IDPs. | Intrinsically Disordered Proteins (IDPs) | Generating unbiased, accurate ensembles for IDPs. |
| Coarse-Grained MD (Martini) [46] | Too compact without tuning; Good agreement after strengthening protein-water interactions. | Multi-domain protein (TIA-1) | Studying conformation and relative domain orientation in flexible proteins. |
Detailed Methodologies for Key Tools
SWAXS-AMDE (Small and Wide Angle X-ray Scattering for All Molecular Dynamics Engines)
SWAXS-AMDE is an open-source scattering model designed for post-processing trajectories from various MD engines, including AMBER, GROMACS, NAMD, and OpenMM [47]. Its core function is to calculate background-subtracted scattering profiles by accounting for electron density changes in atomic detail.
Experimental Protocol:
- System Setup and Simulation: Run an all-atom, explicit-solvent MD simulation using your chosen engine and force field.
- Trajectory Output: Save molecular trajectory frames at regular intervals in a format compatible with SWAXS-AMDE (e.g., via binary trajectory files and topology files).
- Scattering Calculation: Input the trajectory and topology files into SWAXS-AMDE. The software calculates the scattering intensity I(q) using the equation:
I(q) = < |Ṽ_solute(q) + δṼ_hydration(q)|² >_ens
where
Ṽ_solute(q)is the Fourier transform of the solute's electron density,δṼ_hydration(q)is the transform of the electron density change of the hydration layer relative to bulk solvent, and< >_ensdenotes the average over all solute conformations and orientations [47]. - Comparison with Experiment: The final computed profile is directly compared to the experimental SWAXS data without using free parameters for fitting, serving as a stringent test of the MD-derived ensemble's accuracy.
Explicit-Solvent MD with a Spatial Envelope
This methodology, detailed by Grishaev et al., calculates SWAXS profiles directly from explicit-solvent MD simulations, effectively eliminating free parameters associated with solvation [7].
Experimental Protocol:
- MD Simulation: Perform an explicit-solvent MD simulation of the biomolecule and a separate simulation of pure solvent.
- Define a Spatial Envelope: Construct a fixed envelope around the solute that is large enough to encompass all its conformational states and a sufficiently thick hydration layer (see diagram below). This envelope must remain constant during analysis.
- Compute Intensity: For each simulation frame and orientation, calculate the scattering amplitude
Ã(q)andB̃(q)as Fourier transforms of the electron densities within the envelope for the solution (A(r)) and pure solvent (B(r)), respectively. - Calculate Excess Scattering: The key quantity is
D(q) = <|Ã(q)|²>_(ω) - <|B̃(q)|²>_(ω), where< >_(ω)is the average over solute and solvent fluctuations at a fixed solute orientation. The final excess scattering intensityI(q)is the orientational average ofD(q)[7]. - Parameter Fitting: A single fitting parameter is adjusted to account for experimental uncertainties in buffer subtraction and dark currents, rather than for solvation effects.
The following diagram illustrates the workflow for calculating SWAXS profiles from explicit-solvent MD simulations.
DENSS (denss.pdb2mrc.py)
The DENSS algorithm employs a different approach by generating high-resolution electron density maps from atomic models to predict solution scattering profiles [48].
Experimental Protocol:
- Input Atomic Model: Provide the atomic coordinates of the biomolecular structure.
- Calculate Adjusted Atomic Volumes: The algorithm computes unique adjusted atomic volumes directly from the coordinates to account for the excluded volume of bulk solvent, eliminating this as a free parameter.
- Generate Electron Density Map: Create a high-resolution electron density map from the atomic model.
- Implicit Hydration Model: Apply an implicit model of the hydration shell using the form factor of water.
- Parameter Optimization (Optional): Two parameters—the bulk solvent density and the mean hydration shell contrast—can be optimized to best fit the experimental data. However, the default values are already close to the true solution, and disabling optimization still yields highly accurate results compared to other software [48].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key computational "reagents" and resources essential for working with SWAXS data and MD simulations.
Table 3: Key Research Reagent Solutions in SWAXS-Guided MD Research
| Item Name | Function / Role | Example Use Case |
|---|---|---|
| Explicit Solvent Models (e.g., TIP4P/2005s, OPC) [47] [45] | Simulate water molecules with realistic interactions; crucial for accurate scattering profile calculation. | Used in explicit-solvent MD simulations to model the hydration layer and bulk solvent without free parameters. |
| Optimized Force Fields (e.g., AMBER ff19SB, a99SB-disp) [47] [45] | Define potential energy functions for proteins; critical for accurate conformational sampling. | Generating realistic solution ensembles of both folded proteins and IDPs for comparison with SWAXS data. |
| Enhanced Sampling Methods (e.g., HREMD) [45] | Accelerate conformational sampling in MD simulations to overcome energy barriers. | Generating unbiased and converged structural ensembles for IDPs, which are consistent with SAXS/SANS data. |
| Coarse-Grained Force Fields (e.g., Martini) [46] | Reduce computational cost by grouping atoms into beads; enables longer timescale simulations. | Simulating large, flexible systems like multi-domain proteins; often requires tuning (e.g., protein-water interaction strength). |
| Bayesian/Maximum Entropy (BME) Reweighting [46] | Integrate simulation ensembles with experimental data by reweighting trajectory frames. | Refining initial MD ensembles (e.g., from coarse-grained simulations) to achieve full consistency with SAXS data. |
The evolution of computational tools for SWAXS analysis marks a significant advancement in our ability to validate and refine molecular dynamics ensembles. Tools like SWAXS-AMDE and explicit-solvent MD methods have set a new standard by minimizing free parameters, thereby reducing the risk of overfitting and providing more trustworthy validation of simulation models [7] [47]. The trend is moving away from simplistic implicit solvent models towards methods that either explicitly model solvent molecules or intelligently incorporate solvent effects with fewer adjustable parameters. For the future, the integration of SWAXS data directly as restraints in MD simulations (SWAXS-driven MD) shows promise for guiding simulations toward experimentally relevant conformations [36]. Furthermore, the critical importance of enhanced sampling techniques, such as HREMD, for achieving converged and accurate ensembles of flexible molecules like IDPs cannot be overstated [45]. As these tools continue to mature and become more integrated into workflows, they will undoubtedly play a central role in accelerating drug development by providing more reliable, atomic-resolution insights into the dynamic behavior of therapeutic targets in solution.
Molecular dynamics (MD) simulations have become an indispensable tool in computational structural biology, providing atomic-level insights into the conformational dynamics of biomolecules. This capability is crucial for drug discovery, where understanding the dynamic interactions between a drug and its target can inform the optimization of therapeutic candidates [50]. While MD has long been applied to studying folded proteins with stable three-dimensional structures, its application to intrinsically disordered proteins (IDPs) presents unique challenges and opportunities. IDPs, which lack a fixed tertiary structure and exist as dynamic conformational ensembles, play critical roles in cellular signaling and regulation, and their dysfunction is linked to numerous diseases [51].
Validating the structural ensembles generated by MD simulations against experimental data remains a central challenge in the field, particularly for IDPs where traditional high-resolution structural methods are insufficient. This guide compares the application of MD simulations to both folded proteins and IDPs, with a specific focus on validation methodologies within the broader thesis of ensemble verification. We examine case studies, experimental protocols, and performance metrics to provide researchers with a practical framework for assessing and applying these computational approaches in drug discovery pipelines.
Case Study Comparison: Folded Proteins vs. Intrinsically Disordered Proteins
Case Study 1: Drug Solubility Prediction Using Folded Protein Dynamics
A 2025 study demonstrated the power of MD simulations for predicting critical pharmaceutical properties, specifically the aqueous solubility of drug compounds. Researchers conducted MD simulations on 211 diverse drugs and extracted ten dynamic properties that influence solubility. Through machine learning analysis, they identified seven key MD-derived properties that effectively predicted solubility: logP, Solvent Accessible Surface Area (SASA), Coulombic and Lennard-Jones interaction energies (Coulombic_t, LJ), Estimated Solvation Free Energy (DGSolv), Root Mean Square Deviation (RMSD), and the Average number of solvents in the Solvation Shell (AvgShell) [52].
The MD simulations were performed using the GROMACS 5.1.1 software package in the isothermal-isobaric (NPT) ensemble with the GROMOS 54a7 force field. The simulation protocol involved placing molecules in a cubic box with 4 nm dimensions, solvating with SPC water model, and energy minimization using the steepest descent algorithm. This was followed by equilibration and production runs with the particle mesh Ewald method for electrostatic interactions [52]. When these properties were used as features in ensemble machine learning algorithms, the Gradient Boosting algorithm achieved exceptional predictive performance with an R² of 0.87 and RMSE of 0.537 on the test set, demonstrating that MD-derived properties from folded protein simulations possess comparable predictive power to models based on structural descriptors alone [52].
Case Study 2: Force Field Validation for IDP Conformational Ensembles
In contrast to folded proteins, IDPs require specialized force fields and validation approaches. A 2025 force field validation study focused on COR15A, an IDP on the verge of folding, evaluated 20 different MD models for their ability to capture structural and dynamic aspects of disordered proteins [51]. The researchers employed a rigorous two-step validation protocol: (1) initial validation of short 200 ns simulations against small-angle X-ray scattering (SAXS) data, followed by (2) detailed evaluation of the six best-performing models through extended 1.2 μs MD simulations against nuclear magnetic resonance (NMR) data, including a single-point mutant with slightly increased helicity [51].
The results revealed significant differences in force field performance. Only DES-amber and ff99SBws successfully captured the subtle helicity differences between wild-type COR15A and its mutant, though ff99SBws overestimated helicity. Notably, only DES-amber adequately reproduced the COR15A dynamics as assessed by NMR relaxation times at different magnetic field strengths [51]. This study highlights the critical importance of force field selection for IDP simulations and identifies remaining discrepancies that necessitate further force field development.
Performance Comparison Table
Table 1: Comparative performance metrics for MD simulations of folded proteins versus IDPs
| Performance Metric | Folded Protein Study | IDP Force Field Study |
|---|---|---|
| System Size | 211 drug compounds | COR15A protein (1 system, multiple force fields) |
| Simulation Duration | Not specified (standard MD) | 200 ns initial + 1.2 μs extended |
| Key Validation Methods | Machine learning prediction of solubility | SAXS and NMR validation |
| Best Performing Method | Gradient Boosting algorithm | DES-amber force field |
| Quantitative Performance | R² = 0.87, RMSE = 0.537 | Captured helicity differences and NMR relaxation |
| Primary Application | Drug solubility prediction | Force field validation for disordered proteins |
Emerging Integrative Approaches and Validation Frameworks
AlphaFold-Metainference for Disordered Protein Ensembles
A groundbreaking approach called AlphaFold-Metainference has been developed to address the challenge of predicting structural ensembles for disordered proteins. This method integrates deep learning-predicted distances with molecular dynamics simulations to construct structural ensembles of both ordered and disordered proteins [53]. The approach uses AlphaFold-predicted inter-residue distances as structural restraints in MD simulations through the maximum entropy principle within the metainference framework, generating ensembles consistent with both predicted distances and experimental data.
Validation against SAXS data for 11 highly disordered proteins demonstrated that AlphaFold-Metainference generated structural ensembles in better agreement with experimental data compared to individual AlphaFold structures or CALVADOS-2 ensembles [53]. The method successfully reproduced the radius of gyration (Rg) values derived from SAXS experiments and showed particular improvement for proteins with scaling exponents ν that deviate from the Flory value for random coils (ν = 0.5). When applied to partially disordered proteins like TDP-43 (associated with amyotrophic lateral sclerosis), ataxin-3 (linked to Machado–Joseph disease), and the human prion protein, AlphaFold-Metainference effectively captured the structural characteristics of both ordered and disordered domains within these multifunctional proteins [53].
Integrated Workflows in Targeted Drug Discovery
The integration of MD with other computational methods has shown significant promise in drug discovery pipelines. A 2025 study on SARS-CoV-2 papain-like protease (PLpro) inhibitors combined MD, molecular docking, and machine learning to identify potential drug candidates from existing FDA-approved drugs [54]. Researchers performed long-timescale MD simulations on PLpro-ligand complexes at two known binding sites, followed by structural clustering to capture representative structures. These ensembles were then used for molecular docking screens of over 1,100 FDA-approved drugs.
The workflow involved training a random forest model on docking scores from representative conformations, which achieved 76.4% accuracy through leave-one-out cross-validation [54]. This integrated approach identified five promising drug repurposing candidates for COVID-19 treatment, demonstrating the power of combining MD-generated structural ensembles with machine learning for rapid therapeutic development against emerging viral targets. The study highlighted that utilizing multiple conformational states from MD simulations, rather than single crystal structures, provides more biologically relevant results for virtual screening [54].
Table 2: Research Reagent Solutions for Ensemble Validation Studies
| Research Reagent | Type/Category | Function in Validation |
|---|---|---|
| GROMACS | MD Software | Molecular dynamics simulation engine [52] |
| GROMOS 54a7 | Force Field | Parameterization for folded protein simulations [52] |
| DES-amber | Force Field | Specialized parameterization for IDP simulations [51] |
| SAXS | Experimental Technique | Validation of global structural properties and dimensions [51] [53] |
| NMR Spectroscopy | Experimental Technique | Validation of local structure and dynamics [51] |
| AlphaFold-Metainference | Computational Method | Ensemble generation using AI-predicted restraints [53] |
| CAMShift | Computational Tool | Back-calculation of NMR chemical shifts from structures [53] |
Experimental Protocols for Ensemble Validation
MD Simulation Protocol for Folded Protein-Ligand Systems
The protocol for simulating folded protein-ligand systems, as applied in drug solubility studies, involves these critical steps [52]:
System Setup: Begin with the protein structure from databases like RCSB PDB. For ligand-containing systems, generate initial coordinates using tools like smina and sample different protonation states and tautomers using Schrödinger tools. Place the molecular system in a cubic simulation box with dimensions approximately 4×4×4 nm.
Solvation and Ionization: Solvate the system using explicit water models (SPC water model is commonly used). Add ions to neutralize the system and achieve physiological salt concentrations.
Energy Minimization: Perform energy minimization using the steepest descent algorithm to remove steric clashes and unfavorable contacts.
Equilibration: Conduct equilibration runs in the NPT (isothermal-isobaric) ensemble with position restraints on heavy atoms to relax the solvent around the solute.
Production Simulation: Run production MD simulations in the NPT ensemble using software like GROMACS. For electrostatic interactions, employ the particle mesh Ewald method. Maintain constant temperature and pressure using appropriate thermostats and barostats.
Property Extraction: Calculate key properties from trajectories, including SASA, interaction energies (Coulombic and Lennard-Jones), solvation free energies, RMSD, and solvation shell characteristics.
Validation Protocol for IDP Structural Ensembles
The validation of IDP structural ensembles requires specialized approaches [51] [53]:
Multi-Scale Simulation Approach: Begin with shorter simulations (200 ns) of multiple force fields for initial screening against SAXS data. Select top-performing force fields for extended simulations (1.2 μs) for detailed validation.
Experimental Data Integration: Validate against multiple experimental techniques:
- SAXS Data: Compare theoretical scattering profiles calculated from MD ensembles with experimental SAXS data. Calculate the radius of gyration (Rg) and pairwise distance distributions for quantitative comparison.
- NMR Data: Validate against chemical shifts, residual dipolar couplings, and relaxation data. Use tools like CamShift for back-calculation of chemical shifts from structures.
- NMR Diffusion Measurements: Utilize label-free information about inter-residue distance distributions in disordered states.
Mutational Validation: Include single-point mutants with known structural perturbations (e.g., slightly increased helicity) to test the sensitivity of the force field to subtle sequence changes.
Quantitative Metrics: Employ statistical measures such as Kullback-Leibler divergence to quantify agreement between experimental and simulation-derived distance distributions.
Figure 1: Ensemble Validation Workflow. This diagram illustrates the iterative process of generating and validating structural ensembles from MD simulations against experimental data.
The application of molecular dynamics simulations has expanded significantly from folded proteins to intrinsically disordered proteins, driven by advances in force fields, validation methodologies, and integrative computational approaches. For folded proteins, MD simulations provide valuable dynamic properties that can predict critical drug characteristics like solubility with remarkable accuracy, as demonstrated by the machine learning model achieving R² = 0.87 [52]. For IDPs, specialized force fields like DES-amber and innovative approaches like AlphaFold-Metainference enable the generation of validated structural ensembles that capture the inherent heterogeneity of these dynamic systems [51] [53].
Robust validation against multiple experimental techniques—particularly SAXS and NMR for IDPs—remains essential for generating biologically relevant ensembles. The emerging trend of integrating MD with machine learning, molecular docking, and deep learning methods creates powerful pipelines for drug discovery and structural biology [53] [54]. As these methodologies continue to evolve, they will further bridge the gap between static structural snapshots and the dynamic reality of biomolecular function in solution, ultimately enhancing our ability to develop therapeutics for a wide range of diseases.
Figure 2: MD Integration in Drug Discovery. This diagram shows how MD simulations integrate with other computational methods in modern drug discovery pipelines.
Optimizing Force Fields and Overcoming Sampling Limitations
In molecular dynamics (MD) research, the validation of solution ensembles—ensuring that simulations accurately reflect the structural and dynamic properties of molecules in solution—is a central challenge. The choice of force field, the mathematical model that describes the potential energy of a system of particles, is fundamental to this process. An accurate force field is crucial for modeling the balance between a molecule's affinity for its solvent environment and its inherent conformational preferences. This balance directly determines the reliability of simulated solution ensembles when compared against experimental data.
Among the most widely used force fields in biomolecular simulation are AMBER (Assisted Model Building with Energy Refinement) and CHARMM (Chemistry at HARvard Macromolecular Mechanics). This guide provides an objective comparison of these force fields and their specialized variants, focusing on their performance in reproducing experimental observables. We summarize quantitative benchmarking data, detail key experimental protocols used for validation, and provide resources to inform selection for specific research applications, particularly in drug development.
Force Field Philosophies and Parameterization
The CHARMM and AMBER force fields, while sharing a similar functional form for the potential energy function, are built upon distinct philosophical approaches to parameterization. These differences originate in how they model key interactions, leading to variations in performance when simulating different molecular systems.
The general potential energy function for CHARMM, which is similar in form to that used by AMBER, is given by:
[ \begin{aligned} V = &\sum{bonds}k{b}(b-b{0})^{2} + \sum{angles}k{\theta}(\theta-\theta{0})^{2} + \sum{dihedrals}k{\phi}[1+\cos(n\phi-\delta)] \ &+ \sum{impropers}k{\omega}(\omega-\omega{0})^{2} + \sum{Urey-Bradley}k{u}(u-u{0})^{2} \ &+ \sum{nonbonded}\left(\epsilon{ij}\left[\left({\frac{R{min{ij}}}{r{ij}}}\right)^{12}-2\left({\frac{R{min{ij}}}{r{ij}}}\right)^{6}\right]+{\frac{q{i}q{j}}{\epsilon{r}r{ij}}}\right) \end{aligned} ]
This equation includes terms for bonds, angles, dihedrals, impropers, Urey-Bradley interactions, and nonbonded (van der Waals and electrostatic) interactions [55].
A key difference lies in the assignment of atomic partial charges. The CHARMM General Force Field (CGenFF) assigns charges to best represent the Coulombic interaction of an atom with a proximal TIP3P water molecule, evaluated at the HF/6-31G(d) level of quantum mechanical (QM) theory. This approach aims to explicitly capture polarization effects of the condensed phase. In contrast, the Generalized AMBER Force Field (GAFF) uses the AM1-BCC charge model, which is optimized to reproduce the gas-phase electrostatic surface potential (ESP) calculated at the HF/6-31G* level. The AMBER philosophy presumes that the overestimated gas-phase dipole moment fortuitously accounts for condensed-phase polarization [56].
These foundational differences mean that the force fields may perform differently when simulating specific chemical environments, such as the solvation of particular functional groups or the behavior of intrinsically disordered proteins.
Table 1: Overview of Force Field Philosophies and Scope
| Feature | CHARMM/CGenFF | AMBER/GAFF |
|---|---|---|
| Charge Model | Target: HF/6-31G(d) interaction with TIP3P water [56] | AM1-BCC to reproduce HF/6-31G* ESP [56] |
| Polarization Treatment | Aims to explicitly model condensed-phase effects [56] | Relies on fortuitous cancellation with gas-phase parameters [56] |
| Biomolecular Coverage | Proteins, nucleic acids, lipids, carbohydrates, small molecules (CGenFF) [55] | Proteins, nucleic acids, small molecules (GAFF) |
| Specialized IDP Variants | CHARMM36m, CHARMM36IDPSFF, CHARMM36m2021s3p [57] | a99sb-ildn, a99SB-4P-ew, a19SB-OPC [57] |
| Common Water Models | TIP3P, mTIP3P [57] | TIP3P, SPC/E, OPC, TIP4P-ew |
Performance Comparison and Experimental Data
Independent benchmarking studies provide critical insights into the strengths and limitations of various force fields. The performance can vary significantly based on the system being studied, such as ordered proteins, intrinsically disordered proteins (IDPs), or small drug-like molecules in solution.
Hydration Free Energy of Drug-like Molecules
The hydration free energy (HFE) is a critical property for drug design, quantifying a molecule's affinity for an aqueous environment. A 2024 study analyzed the HFE prediction accuracy of CGenFF and GAFF for over 600 molecules from the FreeSolv database. The investigation linked systematic errors to specific functional groups [56]:
- Nitro-groups: Molecules with nitro-groups were over-solubilized in CGenFF and under-solubilized in GAFF.
- Amine-groups: Molecules with amine-groups were under-solubilized in both force fields, but the error was more pronounced in CGenFF.
- Carboxyl groups: Molecules with carboxyl groups were more over-solubilized in GAFF than in CGenFF.
This study highlights that despite similar overall accuracy, the functional group-specific performance of general force fields can vary, informing their application in drug discovery projects [56].
Characterization of Intrinsically Disordered Proteins
IDPs are a major challenge for force fields parameterized for stable, globular proteins. A 2023 benchmark evaluated 13 force fields (including AMBER and CHARMM variants) on the intrinsically disordered R2 region of the FUS-LC domain, which is implicated in ALS. The study used multiple measures—radius of gyration (Rg), secondary structure propensity (SSP), and intra-peptide contact maps—to provide a comprehensive evaluation [57].
Table 2: Benchmarking Scores for Selected Force Fields on the R2-FUS-LC IDP [57]
| Force Field | Final Score | Rg Score | SSP Score | Contact Map Score |
|---|---|---|---|---|
| c36m2021s3p (CHARMM) | 1.00 | 1.00 | 0.74 | 0.89 |
| a99sb-4P-ew (AMBER) | 0.79 | 0.71 | 0.68 | 0.83 |
| a19SB-OPC (AMBER) | 0.74 | 0.82 | 0.60 | 0.54 |
| c36ms3p (CHARMM) | 0.69 | 0.66 | 0.53 | 0.59 |
| c36m3pm (CHARMM) | 0.17 | 0.17 | 0.12 | 0.98 |
The study concluded that CHARMM36m2021s3p with the mTIP3P water model was the most balanced force field, capable of generating diverse conformations compatible with experimental data. It also noted a general tendency for AMBER force fields to generate more compact conformations with more non-native contacts compared to CHARMM force fields. Furthermore, the mTIP3P water model was found to be computationally more efficient than the four-site water models often paired with top-ranked AMBER force fields [57].
Experimental Protocols for Force Field Validation
To ensure the reliability of simulation results, researchers must employ rigorous experimental protocols for validation. The following sections detail common methodologies used to benchmark force field performance.
Absolute Hydration Free Energy Calculations
The HFE is the free energy change for transferring a solute from the gas phase into a dilute aqueous solution. This property is sensitive to solute-solvent interactions and is a key benchmark for force field accuracy.
Protocol Overview:
- System Setup: The solute molecule is placed in a cubic box of explicit water molecules (e.g., TIP3P), with a minimum distance of 14 Å between the solute and the box edge. Periodic boundary conditions are applied [56].
- Alchemical Transformation: The solute is "annihilated" (its interactions are turned off) both in vacuum and in the aqueous phase using an alchemical pathway. This is typically implemented with a hybrid Hamiltonian, ( H(\lambda) = \lambda H0 + (1-\lambda) H1 ), where (\lambda) is a coupling parameter that progresses from 0 to 1. At (\lambda=0), the solute is fully interacting, and at (\lambda=1), it is non-interacting [56].
- Free Energy Calculation: The free energy change for the annihilation process is computed in both vacuum ((\Delta G{vac})) and solvent ((\Delta G{solvent})). The HFE is then obtained using the formula: ( \Delta G{hydr} = \Delta G{vac} - \Delta G_{solvent} ) The free energy terms can be computed using methods such as Free Energy Perturbation (FEP), Thermodynamic Integration (TI), or the Bennett Acceptance Ratio (BAR) [56].
- Analysis: The computed HFEs are compared against experimental reference data to evaluate force field accuracy.
Validation of Intrinsically Disordered Protein Ensembles
Validating force fields for IDPs requires comparing simulation ensembles against multiple experimental measures that probe both global and local structural properties.
Protocol Overview:
- System Preparation: Construct a model of the IDP sequence. For aggregation-prone regions like R2-FUS-LC, a trimer of peptides may be used. The system is solvated in an appropriate water model and neutralized with ions [57].
- Molecular Dynamics Simulation: Run multiple, relatively long (e.g., 500 ns) replica simulations to sample a diverse set of conformations. A total aggregate simulation time of several microseconds is often targeted [57].
- Conformational Analysis:
- Radius of Gyration (Rg): Calculated for the trimer and individual peptides to assess global compactness. The Rg distribution from simulation is compared to values derived from experimental structures (e.g., from NMR or cryo-EM) and theoretical models for the unfolded state [57].
- Secondary Structure Propensity (SSP): The propensity to form elements like β-strands is calculated from the simulation trajectory and compared to experimental data or the known cross-β structure of fibrils [57].
- Contact Map: The frequency of specific intra- and inter-peptide contacts is calculated and compared to reference structures to assess the formation of native-like interactions [57].
- Scoring and Ranking: A final score can be computed by combining the scores from the Rg, SSP, and contact map analyses to provide a quantitative ranking of force field performance [57].
Success in molecular dynamics simulations relies on a combination of software, force fields, and computational resources.
Table 3: Essential Resources for Molecular Dynamics Research
| Resource | Type | Function | Example |
|---|---|---|---|
| Simulation Software | Software Package | Performs energy minimization, molecular dynamics, and analysis. | CHARMM [58], AMBER [2], GROMACS [2], NAMD [59], OpenMM [56] |
| System Building Tools | Cyberinfrastructure | Automates the setup of complex simulation systems, including membranes and multicomponent assemblies. | CHARMM-GUI [60] |
| General Force Fields | Parameter Set | Provides parameters for small, drug-like molecules. | CGenFF [56], GAFF [56] |
| Specialized Force Fields | Parameter Set | Optimized for specific challenges, such as simulating intrinsically disordered proteins. | CHARMM36m [57], CHARMM36IDPSFF [57], a99SB-4P-ew [57] |
| Water Models | Parameter Set | Defines the solvent environment and its interactions with solutes. | TIP3P [56], mTIP3P [57], OPC [57] |
| High-Performance Computing | Hardware | Provides the computational power needed for nanoseconds-to-microseconds of simulation time. | GPU Clusters [59], NVIDIA RTX 4090/6000 Ada [59] |
Selecting between AMBER and CHARMM force fields is not a matter of identifying a universally superior option, but rather of choosing the most appropriate tool for a specific research problem. The experimental data summarized here shows that:
- For simulating small drug-like molecules, both CGenFF and GAFF show good overall performance for Hydration Free Energy prediction, but researchers should be aware of functional group-specific biases, such as the under-solvation of amines in CGenFF or over-solvation of carboxylates in GAFF [56].
- For studying intrinsically disordered proteins, specialized variants like CHARMM36m2021s3p have demonstrated a balanced ability to reproduce multiple experimental observables, while many AMBER force fields may produce overly compact conformations [57].
The validation of solution ensembles remains an ongoing effort. Best practices involve using multiple experimental measures—both global (e.g., Rg) and local (e.g., contact maps)—to comprehensively assess force field performance. As force fields continue to be refined and new specialized variants are developed, this comparative landscape will evolve, necessitating continuous benchmarking against high-quality experimental data.
The validation of solution ensembles in molecular dynamics (MD) research represents a cornerstone for reliable simulations in computational chemistry and drug development. The accuracy of these simulations hinges critically on the water model employed, as water dictates the solvation environment that influences biomolecular structure, dynamics, and interaction energetics. Water models are simplified mathematical representations that use an empirical potential energy function to describe water molecules and their interactions, balancing computational cost with accurate reproduction of water's physical-chemical properties [61]. This guide objectively compares two advanced water models, TIP4P-D and OPC, within the critical context of validating solution ensembles. Both models emerged from systematic efforts to overcome limitations in earlier representations, incorporating specific physical insights to better capture water's complex behavior, particularly regarding solvation effects crucial for biomolecular simulations.
Evolution of Water Models: From Simple to Specialized
The development of water models illustrates a continuous trade-off between computational efficiency and physical accuracy. Early rigid non-polarizable models like SPC, SPC/E, and TIP3P simplified water molecules as rigid entities with fixed point charges, ignoring polarization effects. These 3-site models placed charges on oxygen and hydrogen atomic nuclei, with Lennard-Jones (LJ) interactions centered on oxygen [62]. While computationally efficient and still widely used in biomolecular simulations, their fixed charge distributions and simplified electrostatics limit accuracy in reproducing certain thermodynamic properties and heterogeneous environments [61].
The introduction of 4-site models like TIP4P and its variants (TIP4P/Ew, TIP4P/2005) marked a significant advancement. These models incorporate an additional massless virtual site (M-site) located near the oxygen atom to better represent the molecular charge distribution. This M-site, carrying the negative charge, creates a more realistic electrostatic potential compared to 3-site models [62]. The TIP4P/2005 variant is often regarded as one of the best non-polarizable rigid models for reproducing various water properties, including the temperature of maximum density and phase behavior [61].
Further refinements led to 5-site models (e.g., TIP5P) that use two virtual sites to represent oxygen's lone electron pairs, creating a more tetrahedral charge distribution. While successful in capturing water's density anomaly, they offer limited improvement for other properties like dielectric constant and come with increased computational cost [62] [61].
The latest generation, including polarizable models (e.g., AMOEBA) and highly optimized fixed-charge models like OPC and TIP4P-D, addresses the critical limitation of fixed charge distributions. Polarizable models explicitly account for electronic polarization response to changing electrostatic environments, while optimized fixed-charge models incorporate polarization effects implicitly through parameter optimization [61]. TIP4P-D and OPC represent sophisticated 4-site models specifically parameterized to address critical shortcomings in biomolecular simulations, particularly concerning solvation thermodynamics and dispersion interactions.
Table 1: Classification and Evolution of Key Water Models
| Model Type | Key Features | Representative Examples | Primary Strengths | Common Applications |
|---|---|---|---|---|
| 3-Site Rigid | Fixed charges on O/H atoms; LJ center on O; Rigid geometry | SPC, SPC/E, TIP3P [62] | Computational efficiency; Good for bulk properties | Biomolecular simulation (default in many force fields) |
| 4-Site Rigid | Additional M-site for negative charge; Improved electrostatics | TIP4P, TIP4P/2005, TIP4P-Ew [62] | Better liquid structure & phase diagram; Improved dielectric properties | Accurate bulk water simulation; Phase behavior studies |
| 5-Site Rigid | Two virtual sites for lone pairs; Tetrahedral charge distribution | TIP5P, TIP5P-E [62] | Excellent density maximum reproduction | Modeling water's anomalous properties |
| Polarizable | Explicit induced dipoles; Environment-responsive charges | AMOEBA, SWM4-NDP [62] | Physically realistic response to ions & interfaces | Heterogeneous systems; Spectroscopy |
| Advanced 4-Site | Optimized for specific properties like dispersion or charge | TIP4P-D, OPC [63] | Superior solvation free energies; Balanced properties | Biomolecular binding; Solution ensemble validation |
Comparative Analysis: TIP4P-D vs. OPC
TIP4P-D Water Model
The TIP4P-D water model was specifically developed to address the underestimated dispersion interactions that plague many classical water models. This underestimation manifests as inaccuracies in solvation free energies of hydrophobic solutes and the stability of protein secondary structures in simulation. TIP4P-D modifies the Lennard-Jones parameters on the oxygen atom to enhance attractive dispersion forces while maintaining the TIP4P/2005 geometry and charge distribution [63]. This targeted enhancement allows TIP4P-D to more accurately capture the delicate balance between water-water and water-solute interactions that govern hydrophobic hydration, protein folding, and macromolecular association. Its parameterization placed significant emphasis on reproducing experimental hydration free energies for small molecules and protein folding equilibria, making it particularly suitable for simulating biological systems where these phenomena are critical [63].
OPC Water Model
The OPC (Optimal Point Charge) model represents a different optimization philosophy. Rather than targeting specific interaction deficiencies, OPC employed a comprehensive and unconstrained parameterization strategy. Starting from a 4-site geometry, OPC optimized all parameters (including geometry, charges, and LJ parameters) simultaneously against a extensive training set of experimental data encompassing liquid water properties. These properties included internal energy, density, dielectric constant, and self-diffusion coefficient, among others [63]. This holistic approach resulted in a model that achieves excellent across-the-board accuracy for bulk water properties while simultaneously improving performance for solvation thermodynamics. OPC is notable for its accurate reproduction of water's dielectric constant and diffusion coefficient, properties crucial for modeling electrostatic screening and dynamics in biological systems [63].
Table 2: Quantitative Comparison of TIP4P-D and OPC Water Models
| Property (at 298 K) | TIP4P-D | OPC | Experimental Value | Key Implications |
|---|---|---|---|---|
| Density (g/cm³) | ~0.997 [63] | ~0.997 [63] | 0.997 | Proper system packing & volume |
| Diffusion Coefficient (10⁻⁹ m²/s) | ~2.3 [62] | ~2.3 [62] | ~2.3 | Correct water dynamics & mobility |
| Dielectric Constant (ε) | ~78 [63] | ~78 [63] | 78.4 | Accurate electrostatic screening |
| Vaporization Enthalpy (kJ/mol) | ~44.0 [62] | ~44.0 [62] | 44.0 | Balanced water-water interactions |
| Hydration Free Energy Accuracy | Excellent for organics [63] | Excellent overall [63] | Benchmark data | Reliable solute solvation thermodynamics |
| Peptide/Protein Stability | Enhanced stability [63] | Balanced representation [63] | Experimental structures | Proper biomolecular folding & association |
| Dispersion Interactions | Explicitly enhanced [63] | Optimized implicitly [63] | Quantum mechanics | Correct hydrophobic & stacking interactions |
| Compatible Force Fields | AMBER, CHARMM, OPLS [63] | AMBER, CHARMM, OPLS [63] | - | Broad biomolecular application |
Experimental Protocols for Validation
Validating solution ensembles requires rigorous methodologies that probe both bulk water behavior and specific solvation effects. The following experimental protocols provide essential data for water model validation:
Bulk Property Characterization
Liquid Density Measurements at ambient conditions (298 K, 1 atm) provide the most fundamental validation. MD simulations typically employ NPT (constant Number of particles, Pressure, and Temperature) ensemble with at least 500 water molecules. The average density over 10-20 ns production run should match the experimental value of 0.997 g/cm³ within 1% [62] [64].
Dielectric Constant Determination requires calculating the collective dipole moment fluctuations during an NVT simulation: ε = 1 + (4π/3VkBT)(⟨M²⟩ - ⟨M⟩²), where M is the total dipole moment vector, V is volume, T is temperature. Simulations should extend for 20+ ns to ensure adequate sampling of long-range correlations [61] [64].
Diffusion Coefficient Calculation uses the Einstein relation from mean-squared displacement in an NVT simulation: D = (1/6t)limt→∞⟨|rᵢ(t) - rᵢ(0)|²⟩, where rᵢ(t) is the position of molecule i at time t. Statistical precision requires averaging over all water molecules and multiple independent trajectories [64].
Solvation Thermodynamics Protocols
Small Molecule Hydration Free Energy calculations employ thermodynamic integration (TI) or free energy perturbation (FEP) methods. The protocol involves gradually decoupling the solute from its environment through a series of intermediate states (λ-values). For each λ, equilibrium sampling (1-5 ns per window) collects energy differences. The hydration free energy is integrated over the complete pathway, with calculations performed for diverse compounds (alkanes, alcohols, ions) to test transferability [65].
Ionic Solvation Free Energy determination follows similar alchemical pathways but requires careful treatment of long-range electrostatics and proper correction for finite-size effects. Comparison to experimental data validates ion-water interactions critical for biomolecular electrostatics [66].
Biomolecular Validation Experiments
Protein Folding Stability assessment involves simulating model peptides (e.g., α-helices, β-hairpins) and monitoring secondary structure persistence using DSSP or similar metrics. Multiple independent replicas (5-10) of 100-500 ns duration provide statistics on folded state stability. Comparison to NMR or CD spectroscopy data validates the balance between protein-water and protein-protein interactions [63].
Hydrogen Bond Network Dynamics can be probed through Terahertz (THz) Absorption Spectroscopy coupled with MD simulation. The hydrogen bond lifetime (τₕ₋bond) correlates with the center frequency of the hydrogen bond network's THz absorption peak. MD simulations calculate the hydrogen bond population correlation function C(t) = ⟨h(0)h(t)⟩/⟨h⟩, where h(t)=1 if a specific donor-acceptor pair is hydrogen-bonded at time t, else 0. The integral of C(t) provides τₕ₋bond, which shows a linear relationship with THz absorption peak frequency [64]. Temperature-dependent studies (278-318 K) reveal how water models respond to thermal perturbations, with superior models reproducing the experimental red-shift in absorption with increasing temperature [64].
Successful implementation and validation of water models in solution ensemble simulations requires specific computational tools and resources. The following table details essential components of the methodological toolkit:
Table 3: Essential Research Reagents and Computational Tools for Water Model Validation
| Tool/Resource | Function/Purpose | Application Context | Key Features |
|---|---|---|---|
| Molecular Dynamics Software | Engine for running simulations and sampling configurations | All simulation stages | GROMACS [67] [65], AMBER [65], NAMD [63], LAMMPS [63] |
| Topology/Parameter Generators | Create force field parameters for molecules in selected water model | System setup | Sobtop [67], AuToFF [63], acpype, tleap |
| Force Field Databases | Provide parameters for biomolecules compatible with specific water models | System setup | GAFF/GAFF2 [63] [66], AMBER force fields [66], CHARMM [63], OPLS-AA [63] |
| Free Energy Calculation Tools | Compute solvation thermodynamics and binding affinities | Validation & production | gmx_MMPBSA [65] [66], FEP, TI implementations |
| Analysis Suites | Process trajectory data to extract structural and dynamic properties | Post-simulation analysis | GROMACS tools [65], VMD, MDAnalysis, in-house scripts |
| Ion Parameters | Specialized parameters for ions compatible with specific water models | Electrolyte solution simulation | Merz/IOD parameters [63] [66], HaiyangZhang parameters [63] |
| Validation Datasets | Experimental reference data for benchmarking | Validation | Hydration free energies [63], protein stability data [63], THz spectra [64] |
Within the critical framework of validating solution ensembles for molecular dynamics research, both TIP4P-D and OPC water models represent significant advancements over earlier representations. The selection between them should be guided by specific research priorities and the nature of the system under investigation. For studies where accurate dispersion interactions are paramount—such as hydrophobic hydration, protein-ligand binding with significant non-polar components, or nucleic acid simulations—TIP4P-D offers targeted excellence. Conversely, when comprehensive accuracy across diverse bulk and solvation properties is required for heterogeneous biomolecular systems, OPC provides outstanding balanced performance. Both models substantially improve upon older 3-site and earlier 4-site models in reproducing experimental solution ensembles, thereby enhancing the reliability of MD simulations in drug development and molecular sciences. Their continued validation and refinement against expanding experimental datasets will further solidify their role in advancing predictive computational biology.
Addressing Sampling Challenges in Heterogeneous Systems
Molecular dynamics (MD) simulations provide a powerful "virtual molecular microscope" for investigating the structural and dynamic properties of biomolecular systems at atomistic resolution [68]. However, when studying heterogeneous systems—such as lipid nanoparticles (LNPs) for drug delivery, partially disordered proteins, or complex polymer electrolytes—researchers face a significant challenge: the limited sampling of rare events and conformational diversity inherent to such systems [69] [70]. The sampling problem represents one of two fundamental limitations in MD simulations, alongside the accuracy problem of force fields [68]. This guide objectively compares multiple computational strategies for addressing sampling challenges, with a specific focus on validating the resulting solution ensembles against experimental data.
The core issue stems from the vast temporal and spatial scales of biomolecular processes. While experimental data represents averages over ~10^17-10^19 molecules, MD simulations typically track just one molecule at a time [70]. For heterogeneous systems, this sampling limitation is exacerbated by their inherent structural diversity and the complex energy landscapes they navigate. Consequently, comparing simulation results with experimental observations requires careful consideration of whether the simulated ensemble adequately represents the true heterogeneity of the system.
Comparative Analysis of Sampling Methods
Table 1: Comparison of Enhanced Sampling Methods for Heterogeneous Systems
| Method | Key Mechanism | Computational Cost | Best for Heterogeneous Systems | Key Limitations |
|---|---|---|---|---|
| Multiple Independent Simulations | Statistical improvement via parallel sampling [68] [70] | Moderate to High (multiple replicates) | Capturing divergent pathways and denatured state ensembles [70] | No guidance along specific coordinates; purely statistical |
| Enhanced Sampling (Umbrella Sampling, Metadynamics) | Accelerates exploration along predefined Collective Variables (CVs) [69] | High per simulation (but fewer simulations needed) | Focusing on specific transitions (e.g., endosomal escape in LNPs) [69] | Quality depends on CV selection; risk of missing important dimensions [69] |
| Coarse-Grained (CG-MD) | Reduces degrees of freedom for larger systems [69] | Lower per simulation (enables longer timescales) | Initial self-assembly processes of LNPs; large-scale conformational changes [69] | Loss of atomic detail; potential accuracy trade-offs |
| Machine Learning-Guided Sampling | Identifies important features automatically [69] [52] | High initial training cost | Discovering unexpected collective variables in complex transitions [69] | Requires substantial data for training; model dependency |
Each method presents distinct trade-offs between computational efficiency, system size limitations, and the risk of introducing bias. For heterogeneous systems specifically, multiple independent simulations have demonstrated particular value by revealing how similar observable properties can emerge from diverse conformational pathways [70]. This approach helps overcome the fundamental limitation of single-molecule observation in MD simulations compared to the ensemble averages obtained experimentally [70].
Experimental Validation Protocols for Solution Ensembles
Validating MD-derived ensembles requires comparison with experimentally measurable properties. The following protocols represent key methodologies for benchmarking simulations of heterogeneous systems.
Wide-Angle X-ray Scattering (WAXS) Validation
Protocol Objective: To quantitatively compare MD-simulated conformational ensembles with experimental WAXS data, providing a robust validation method sensitive to minor structural fluctuations [7].
Table 2: Key Steps in Explicit-Solvent WAXS Validation Protocol
| Step | Description | Critical Parameters | Rationale |
|---|---|---|---|
| 1. System Preparation | Solvate protein in explicit water box with sufficient padding [7] | 10Å beyond protein atoms; TIP4P-EW water model [68] | Realistic solvent distribution and solvation layer structure |
| 2. MD Simulation | Perform unrestrained MD simulations under experimental conditions [7] | Fixed orientation for averaging; sufficient sampling time [7] | Capture thermal fluctuations essential for accurate WAXS profiles |
| 3. Scattering Calculation | Compute IA(q) and IB(q) from explicit solvent trajectories [7] | Spatial envelope enclosing all conformational states [7] | Eliminates free parameters for solvation layer and excluded solvent |
| 4. Buffer Subtraction | Calculate excess intensity I(q) = IA(q) - IB(q) [7] | Single fitting parameter for experimental uncertainties [7] | Minimizes risk of overfitting compared to implicit solvent methods |
| 5. Ensemble Comparison | Compare simulated and experimental WAXS profiles | Sensitivity to 1% changes in radius of gyration [7] | Detects minor conformational rearrangements and flexibility changes |
This explicit-solvent approach eliminates free parameters typically associated with modeling solvation layers in implicit solvent methods, thereby reducing overfitting risks and providing more rigorous validation [7]. The method demonstrates high sensitivity, detecting increased loop flexibility and radius of gyration changes as small as 1% [7].
Multiple Trajectory Analysis Framework
Protocol Objective: To systematically compare multiple unfolding trajectories and characterize denatured state ensembles, addressing the sampling limitations of single simulations [70].
Implementation Steps:
- Execute multiple independent simulations (typically 3-11 replicates) under identical conditions [70]
- Segment each trajectory into unfolding process and denatured state segments [70]
- Apply complementary analysis methods:
Key Insight: For proteins like BPTI, barnase, and CI2, this approach revealed that while unfolding pathways often diverged, the denatured state ensembles showed similar conformational properties despite different unfolding routes [70]. This highlights the danger of assuming similar observed properties imply similar conformations—a critical consideration for heterogeneous systems [70].
Integrated Workflow for Ensemble Validation
The following diagram illustrates a comprehensive workflow integrating multiple sampling methods with experimental validation for heterogeneous systems:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools and Resources for Ensemble Validation
| Tool/Resource | Function | Application in Heterogeneous Systems |
|---|---|---|
| GROMACS [52] | Molecular dynamics simulation package | High-performance MD for proteins and nanoparticles |
| AMBER [68] | Molecular dynamics simulation package | Specialized for biomolecular systems |
| NAMD [68] | Molecular dynamics simulation package | Scalable for large systems |
| Martini Coarse-Grained Force Field [69] | Reduced-resolution modeling | Self-assembly of lipid nanoparticles (LNPs) [69] |
| Constant pH Molecular Dynamics (CpHMD) [69] | Environment-dependent protonation states | Ionizable lipids in LNPs with pH-dependent behavior [69] |
| Explicit Solvent WAXS Calculator [7] | Calculates theoretical scattering profiles | Quantitative comparison with experimental WAXS data [7] |
| NMR Chemical Shift Predictors [68] | Calculates NMR observables from structures | Validation against solution NMR data [71] |
Addressing sampling challenges in heterogeneous systems requires a multi-faceted approach that combines complementary sampling strategies with rigorous experimental validation. The comparative analysis presented here demonstrates that no single method universally outperforms others; rather, each offers distinct advantages for specific aspects of the sampling problem. For reliable results, researchers should prioritize methods that directly address the specific heterogeneity in their systems—whether through multiple independent trajectories to capture diverse pathways, enhanced sampling to accelerate specific transitions, or coarse-grained models to access larger scales.
The critical importance of validation against experimental data cannot be overstated. As demonstrated in the protocols outlined here, approaches like explicit-solvent WAXS calculations and multiple trajectory analysis provide quantitative benchmarks for assessing the representativeness of simulated ensembles. By integrating these computational and experimental strategies, researchers can significantly improve the reliability of molecular dynamics studies for complex, heterogeneous systems across drug delivery, materials science, and structural biology applications.
Software-Specific Considerations (AMBER, GOMACS, NAMD)
This guide objectively compares the performance of AMBER, GROMACS, and NAMD, key molecular dynamics (MD) software packages. Performance data for current hardware, detailed experimental methodologies, and essential computational tools are provided to inform research aimed at validating solution ensembles.
Performance Comparison of MD Software
Molecular dynamics simulations are computationally intensive. The choice of software and hardware significantly impacts research efficiency. The following sections compare performance across different GPUs and system sizes.
AMBER Performance
AMBER's pmemd.cuda engine is highly optimized for single-GPU acceleration. Performance varies significantly with GPU architecture and the size of the molecular system.
Table: AMBER 24 Performance (ns/day) on NVIDIA GPUs for Various Systems [72]
| GPU | STMV (1.07M atoms) | Cellulose (408k atoms) | Factor IX (90.9k atoms) | DHFR (23.6k atoms) |
|---|---|---|---|---|
| RTX 5090 | 109.75 | 153.30 | 494.45 | 1632.97 |
| B200 SXM | 114.16 | 161.19 | 427.26 | 1447.75 |
| RTX 6000 Ada | 70.97 | 114.99 | 442.91 | 1666.84 |
| GH200 Superchip | 101.31 | 152.40 | 206.06 | 1322.17 |
| H100 PCIe | 74.50 | 113.81 | 385.12 | 1500.37 |
Independent benchmarks from an HPC center show a similar performance hierarchy for AMBER22/24, noting that the cost-to-performance ratio of the data center A100 GPU was less favorable than the A40 for AMBER simulations [73].
GROMACS Performance
GROMACS is known for its high performance and scalability on both CPU and GPU. Recent versions have focused on offloading more work to the GPU. Key performance improvements in GROMACS 2025 include [74] [75]:
- GPU-by-default: The
mdrun-updatestep now automatically runs on the GPU, significantly boosting performance with a single MPI rank [74]. - CUDA Graphs: Reduces CPU and GPU scheduling overheads, especially beneficial for smaller systems. Enabled via the
GMX_CUDA_GRAPH=1environment variable [74] [76]. - Increased default coupling intervals: The default temperature and pressure coupling intervals have been increased, improving performance for GPU and parallel runs [74].
Optimal configuration often involves using a single MPI rank per GPU (-ntmpi 1) and setting the number of OpenMP threads to the number of CPU cores (-ntomp) [76]. Using a powerful GPU often leads to CPU underutilization, which is acceptable as the goal is to offload maximum work to the GPU [76].
NAMD Performance
NAMD is designed for parallel simulation of large biomolecular systems, supporting multi-GPU configurations.
Table: NAMD Performance (ns/day) on a 456K Atom System (Her1-Her1 Membrane) [77]
| GPU Configuration | 1x GPU | 2x GPUs | 4x GPUs |
|---|---|---|---|
| RTX 6000 Ada | 21.21 | 35.97 | 63.90 |
| RTX A5500 | 16.39 | 29.32 | 51.84 |
| RTX A4500 | 13.00 | 23.23 | 41.29 |
For a single GPU, the consumer-grade RTX 4090 delivered 19.87 ns/day, offering a high value for its cost, though it lacks the scalability of professional GPUs [77]. Benchmarks on OpenBenchmarking.org for a 327,506-atom ATPase system show a wide range of performance, with top-tier results around 32.2 ns/day and median performance around 2.1 ns/day [78].
Hardware configuration matters; single-CPU workstations (e.g., Intel Xeon W9-3495X) often outperform dual-CPU servers (e.g., dual Intel Xeon 8490H) due to bottlenecks in CPU interconnect [77].
Experimental Protocols for Benchmarking
The performance data presented was generated using standardized benchmarks and methodologies to ensure reproducibility and fair comparison.
AMBER Benchmarking Protocol
The following methodology was used for generating the AMBER 24 performance data [72]:
- Software Version: AMBER 24 with AmberTools 25.
- Benchmark Systems: A standard set of molecular systems from the AMBER benchmark suite was used, ranging from the 1,067,095-atom Satellite Tobacco Mosaic Virus (STMV) to the 23,558-atom Dihydrofolate Reductase (DHFR, JAC).
- Simulation Parameters: Both NPT and NVE ensembles were tested with timesteps of 2fs or 4fs, as appropriate for the system.
- Execution: Each benchmark was run on a single GPU, even in multi-GPU systems. The
pmemd.cudaengine was used for all tests. - Measurement: Performance is reported in nanoseconds simulated per day (ns/day), calculated by the AMBER benchmark suite.
NAMD Benchmarking Protocol
The NAMD benchmarks were conducted as follows [77]:
- Software Version: NAMD 2.14.
- Benchmark System: A 456,000-atom Her1-Her1 membrane simulation.
- Hardware Platform: Benchmarks were run on an Intel Xeon W9-3495X workstation.
- GPU Scaling: Tests were performed with 1, 2, and 4 GPUs to assess multi-GPU scaling performance.
- Performance Metric: Results are reported in nanoseconds per day (ns/day).
Workflow for Validating Solution Ensembles
Integrating simulation and experimental data is crucial for validating the structural ensembles of proteins, especially for disordered systems.
Validating Structural Ensembles
This workflow illustrates the integration of computational and experimental approaches for determining a protein's structural ensemble, as demonstrated in AlphaFold-Metainference studies [53].
- Start: The process begins with a Protein Sequence.
- AlphaFold Prediction: AlphaFold predicts a distribution of inter-residue distances (a distogram) from the sequence, which provides information for both ordered and disordered regions [53].
- MD Simulation: The AlphaFold-predicted distances are used as structural restraints in Molecular Dynamics (MD) Simulation (e.g., using AMBER, GROMACS, or NAMD) to generate a structural ensemble [53].
- Experimental Data: In parallel, label-free experimental data, such as Wide-Angle X-ray Scattering (WAXS) or Small-Angle X-ray Scattering (SAXS) profiles, are collected for the protein in solution. WAXS is highly sensitive to minor conformational rearrangements and internal protein dynamics [29].
- Ensemble Validation: The structural ensemble from the simulation is used to calculate a theoretical scattering profile. This profile is quantitatively compared against the experimental data to validate the ensemble [29] [53].
- Final Output: A Validated Solution Ensemble that is consistent with both the deep learning predictions and the experimental data is obtained [53].
The Scientist's Toolkit: Essential Research Reagents & Materials
This table details key computational and experimental "reagents" essential for conducting and validating molecular dynamics simulations of solution ensembles.
Table: Essential Reagents for Solution Ensemble Validation
| Item | Function in Research |
|---|---|
| MD Software (AMBER, GROMACS, NAMD) | The core engine for performing molecular dynamics simulations to generate structural ensembles and dynamics data [79]. |
| High-Performance GPU (e.g., NVIDIA RTX 5090/6000 Ada) | Accelerates computation, dramatically reducing simulation time from years to days for complex biological systems [79] [72]. |
| AlphaFold-Metainference | A method that uses AlphaFold-predicted distances as restraints in MD simulations to generate structural ensembles for disordered and ordered proteins [53]. |
| WAXS/SAXS Experimental Data | Provides label-free, ensemble-averaged structural information against which computational ensembles are validated, ensuring accuracy and biological relevance [29] [53]. |
| Biomolecular Force Field | Defines the potential energy functions and parameters governing atomic interactions, forming the physical basis for the MD simulation [29]. |
| Benchmarking Suite | Standardized tests (e.g., provided with AMBER) to evaluate hardware performance and optimize resource allocation for specific research needs [72]. |
Rigorous Validation Against Experimental Data
Quantitative Comparison Metrics for SAXS/WAXS Profiles
Small-Angle and Wide-Angle X-ray Scattering (SAXS and WAXS) have emerged as indispensable tools for characterizing nanoscale and atomic-level structures across diverse scientific fields, from materials science to structural biology. For researchers engaged in validating solution ensembles from molecular dynamics (MD) simulations, establishing robust, quantitative metrics for comparing experimental and computational scattering profiles is paramount [49] [29]. This guide provides a systematic overview of the core metrics, experimental protocols, and analytical frameworks essential for objective performance comparison between MD-derived ensembles and experimental SAXS/WAXS data, with a particular focus on applications in pharmaceutical and biomolecular research.
Core Quantitative Metrics for Profile Comparison
The quantitative comparison between experimental and theoretical scattering profiles relies on specific mathematical metrics that assess different aspects of similarity and deviation.
Table 1: Key Quantitative Metrics for SAXS/WAXS Profile Comparison
| Metric | Formula | Application Context | Interpretation & Advantage |
|---|---|---|---|
| Chi-Squared (χ²) [29] | ( \chi^2 = \frac{1}{N} \sum{i=1}^{N} \frac{[I{exp}(qi) - I{calc}(qi)]^2}{\sigmai^2} ) | General goodness-of-fit for full profiles across the entire q-range. | Quantifies the agreement relative to experimental errors (σᵢ). Lower values indicate better fit. Directly assesses model accuracy against raw data. |
| Linear Combination Fitting (LCF) R-Factor [80] | ( R = \frac{\sum |I{exp}(q) - I{calc}(q)|}{\sum |I_{exp}(q)|} ) | Quantifying species composition in a mixture by fitting with reference spectra. | Provides a normalized measure of the residual. Useful for quantifying the fractional contribution of different structural components or conformations. |
| Radius of Gyration (Rg) | Derived from the Guinier approximation: ( I(q) ≈ I(0)exp(-\frac{1}{3}q^2Rg^2) ) for ( q \cdot Rg < 1.3 ) [81] [82]. | Assessing global particle size and compactness in solution, primarily from SAXS. | A model-free parameter. Discrepancies between experimental and MD-derived Rg indicate issues with the overall compactness of the simulated ensemble. |
| Porod Exponent | Determined from the slope at high-q in a log(I) vs. log(q) plot: ( I(q) ∝ q^{-P} ) [81]. | Inferring surface and internal structure (e.g., mass fractal dimension, surface smoothness). | P ≈ 4 for smooth surfaces; P < 3 for mass fractals; P > 4 for diffuse interfaces. Reveals information on the particle's internal structure and solvation. |
Beyond these standard metrics, the use of intensity difference curves is a powerful qualitative and quantitative tool, particularly for visualizing where in the q-range a computational model succeeds or fails to reproduce experimental data [49]. This involves plotting ( \Delta I(q) = I{exp}(q) - I{calc}(q) ), which can pinpoint specific structural deficiencies.
Experimental Protocols for High-Quality Data
The reliability of any quantitative comparison is contingent on the quality of the experimental scattering data. Adherence to rigorous measurement and correction protocols is non-negotiable.
SAXS/WAXS Measurement Methodology
A robust experimental protocol involves several critical stages, from sample preparation to data collection. The following workflow outlines the key steps for obtaining data suitable for quantitative validation of MD ensembles.
Title: SAXS/WAXS Experimental Workflow
- Sample Preparation: For biomolecules like proteins or nucleic acids, extensive dialysis into a matched buffer is crucial to minimize interparticle interactions and ensure a uniform solvent background [49]. For nanomaterials and polymers, protocols must ensure colloidal stability and monodispersity [82]. Centrifugation or filtration is often employed to remove large aggregates that could dominate the scattering signal [82].
- Data Collection: Measurements are performed on both the sample and its matched buffer. The sample-to-detector distance is set to access the desired q-range: a longer distance (e.g., 1-4 m) for SAXS to probe larger structures, and a shorter distance (e.g., 0.4-0.5 m) for WAXS to access atomic-level details [49]. The q-range must be sufficient to fully cover the Porod regime to avoid over/underestimation of structural parameters [81]. Multiple short exposures are recommended to screen for and mitigate radiation damage.
- Primary Data Processing: This involves the subtraction of the buffer scattering from the sample scattering, followed by absolute scale calibration, often using water as a standard [49]. This step yields the purified, normalized scattering profile ( I(q) ) ready for analysis.
Advanced and Coupled Techniques
For complex or polydisperse systems, in-line fractionation techniques coupled to SAXS are invaluable:
- Size-Exclusion Chromatography SAXS (SEC-SAXS): Effectively "polishes" the sample on-the-fly, separating monomers from aggregates or oligomers immediately before measurement [82].
- Asymmetrical-Flow Field-Flow Fractionation SAXS (AF4-SAXS): Particularly powerful for broadly distributed nanoparticles, like lipid nanoparticles (LNPs), as it separates particles by hydrodynamic size and provides a quantitative, size-resolved structural characterization [82].
Computational Validation Using Molecular Dynamics
Integrating SAXS/WAXS with Molecular Dynamics (MD) simulations creates a powerful cycle for validating and refining structural models of molecules in solution.
Calculating Theoretical Scattering Profiles
The primary method for validating MD ensembles involves calculating theoretical scattering profiles from simulation snapshots and comparing them directly to experiment. The most rigorous approaches use explicit-solvent MD simulations, which model the solvent molecules directly, thereby eliminating free parameters associated with modeling the solvation layer and providing a more physically realistic representation of the system [29]. From the simulation trajectories, hundreds or thousands of snapshots are extracted in PDB format. The theoretical scattering profile ( I{calc}(q) ) for each snapshot is then computed using programs like CRYSOL, which calculates the solution scattering pattern by considering the hydration shell and counterions [49]. Finally, the calculated profiles from the ensemble of snapshots are averaged and compared to the experimental ( I{exp}(q) ) using the quantitative metrics in Table 1.
Metrics in Practice: Validating MD with WAXS
The agreement between MD simulations and WAXS data is highly sensitive to the structural details and dynamics of the biomolecule. Studies have shown that incorporating full thermal fluctuations from MD simulations significantly improves agreement with experimental WAXS data compared to using static, high-resolution crystal structures alone [29]. Furthermore, WAXS profiles are exquisitely sensitive to minor conformational rearrangements, such as increased loop flexibility or an increase of the radius of gyration by less than 1% [29]. This high sensitivity makes WAXS a stringent test for MD-derived solution ensembles. The use of a single fitting parameter to account for experimental uncertainties in buffer subtraction, in conjunction with explicit-solvent calculations, minimizes the risk of overfitting and leads to robust validation [29].
Essential Research Toolkit
Successful execution of SAXS/WAXS experiments for MD validation requires a suite of specialized tools and resources.
Table 2: Essential Research Reagents and Solutions
| Item | Function / Rationale |
|---|---|
| Matched Buffer Solution | Serves as the background for scattering subtraction. Its precise chemical matching to the sample solvent is critical for accurate background subtraction [49]. |
| Absolute Calibrant (e.g., Pure H₂O) | Used to calibrate the scattering intensity to absolute units, enabling direct comparison of intensities between different experiments and instruments [49] [82]. |
| Standard Samples (e.g., BSA Protein) | Well-characterized reference materials used for instrument alignment, validation of data collection protocols, and testing data analysis workflows [82]. |
Table 3: Key Software Packages for Data Analysis
| Software | Primary Function | Key Features |
|---|---|---|
| CRYSOL [49] | Calculation of SAXS/WAXS profiles from atomic models. | Accounts for hydration shell; fits experimental data by adjusting the excluded volume and contrast of the hydration layer. |
| SASfit / SasView [83] | Model fitting of scattering data from non-atomic systems. | Extensive library of form factors (spheres, cylinders, etc.) and structure factors for complex morphologies. |
| BioXTAS RAW | Integrated SAXS data processing and analysis. | Handles data reduction, Guinier analysis, P(r) calculation, and basic modeling in a single pipeline. |
| MD Simulation Suites (e.g., AMBER) [49] | Generation of solution ensembles for validation. | Produces the trajectories of atomic coordinates from which theoretical scattering profiles are calculated. |
Detecting Subtle Conformational Changes and Flexibility
Molecular dynamics (MD) simulations provide an atomic-level view of protein dynamics, capturing motions and conformational changes that are central to biological function but often difficult to observe experimentally. Unlike static structural models, MD simulations can reveal transient states, flexible regions, and the pathways connecting different conformations, offering critical insights for understanding biological mechanisms and facilitating drug discovery [84] [85]. The central thesis of modern biomolecular simulation research asserts that validating dynamical ensembles against experimental data is paramount for establishing the physiological relevance of computational models. This guide objectively compares the performance of predominant MD software in capturing subtle conformational changes, providing researchers with the experimental data and methodologies needed to select appropriate tools for studying protein flexibility.
Comparative Analysis of Molecular Dynamics Software
The selection of MD software involves balancing computational efficiency, feature availability, and the specific biological questions under investigation. The table below summarizes the key characteristics of major MD packages.
Table 1: Key Features of Major Molecular Dynamics Software Packages
| Software | License | GPU Acceleration | Strengths and Specializations | Notable Force Fields |
|---|---|---|---|---|
| GROMACS | Free open source (GPL) | Yes | High performance, excellent parallel scaling, extensive analysis tools [1] [86] | GROMOS, AMBER, CHARMM, Martini (coarse-grained) [1] |
| AMBER | Proprietary (free academic use) | Yes | Accurate force fields, particularly for proteins & nucleic acids; advanced free energy methods [1] [86] | AMBER force fields, GAFF [1] |
| NAMD | Proprietary (free academic use) | Yes | Excellent scalability on parallel systems, tight VMD integration for visualization [1] [86] | CHARMM, AMBER, GROMOS [1] |
| CHARMM | Proprietary, commercial | Yes | Comprehensive all-atom additive force field, extensive functionality [1] | CHARMM force fields [1] |
| OpenMM | Free open source (MIT) | Yes | High flexibility, Python scriptable, cross-platform [1] [84] | Supports multiple force fields [1] |
| LAMMPS | Free open source (GPL) | Yes | Broad potentials for soft/solid-state materials & coarse-grain systems [1] | Various specialized force fields [1] |
| Desmond | Proprietary, commercial/gratis | Yes | High performance MD, user-friendly GUI [1] | OPLS, AMBER, CHARMM [1] |
Performance Benchmarks
Practical performance directly impacts research throughput, especially for large systems or long timescales. Recent benchmarking data provides critical insights for resource planning and software selection.
Table 2: AMBER 24 Performance Benchmarks (ns/day) Across GPU Architectures [72]
| GPU Model | STMV (1M atoms) | Cellulose (408k atoms) | Factor IX (90k atoms) | DHFR (23k atoms) | Myoglobin (2.5k atoms) |
|---|---|---|---|---|---|
| NVIDIA RTX 5090 | 109.75 | 169.45 | 529.22 | 1655.19 | 1151.95 |
| NVIDIA RTX 5080 | 63.17 | 105.96 | 394.81 | 1513.55 | 871.89 |
| NVIDIA RTX PRO 6000 Max-Q | 97.44 | 149.84 | 475.04 | 1464.14 | 940.57 |
| NVIDIA RTX PRO 4500 Blackwell | 54.17 | 88.41 | 389.54 | 1481.61 | 924.67 |
| NVIDIA H100 PCIe | 74.50 | 125.82 | 410.77 | 1532.08 | 1094.57 |
Performance data reveals several critical patterns for hardware selection. Larger systems like STMV (1 million atoms) show the greatest performance spread across GPU tiers, with high-end consumer cards like the RTX 5090 delivering nearly double the throughput of mid-range professional cards [72]. Smaller systems demonstrate less performance variation, suggesting that cost-effective options like the RTX PRO 4500 Blackwell provide excellent value for typical simulation sizes [72]. Notably, AMBER simulations primarily utilize GPU resources, with CPU performance having minimal impact on simulation throughput [72].
Comparative discussions among researchers highlight nuanced trade-offs: "GROMACS stands out for its speed, versatility, and open-source nature" but "visualization is not its strongest suit," while "NAMD, especially when paired with VMD, offers much better support for visual analysis" [86]. AMBER is frequently noted for its accurate force fields, though licensing can present barriers for commercial applications [86]. Performance characteristics also vary with simulation type, as one researcher notes: "For calculations like MMGBSA, MMPBSA and FEP, Amber and Desmond are more user-friendly" than GROMACS [86].
Experimental Protocols for Ensemble Validation
Standard MD Protocol for Conformational Analysis
Robust MD protocols require careful system preparation, equilibration, and production phases to generate physically meaningful ensembles.
Diagram 1: MD Simulation and Validation Workflow
System Setup: Begin with a protein structure from crystallography, NMR, or AlphaFold prediction. Process the structure using pdbfixer to repair missing residues, atoms, and termini, and assign standard protonation states at pH 7.0 [84]. Prepare the system using the tleap module in AMBER or similar tools in other packages, adding hydrogens and generating force field parameters.
Solvation and Ions: Solvate the system with explicit water molecules (TIP3P, TIP4P) using a minimum 1.0 nm padding around the protein [84]. Add ions to neutralize system charge and achieve physiological concentration (0.15 M NaCl) [84].
Energy Minimization: Perform steepest descent minimization (5,000-10,000 steps) to remove steric clashes, followed by conjugate gradient minimization. Apply position restraints on protein heavy atoms (force constant 1000 kJ/mol·nm²) during initial minimization [2].
Equilibration: Conduct NVT equilibration for 100-500 ps using a Berendsen or Langevin thermostat (300 K, coupling constant 1.0 ps⁻¹) with continued position restraints [84]. Follow with NPT equilibration for 100-500 ps using a Monte Carlo or Parrinello-Rahman barostat (1 atm, coupling every 25 steps) [84]. Gradually release position restraints during this phase.
Production MD: Run unrestrained production simulation using a 2-4 fs timestep with hydrogen mass repartitioning enabled [2] [84]. Use Langevin dynamics for temperature coupling (300 K, γ = 1.0 ps⁻¹) and a Monte Carlo barostat for pressure control (1 atm) [84]. For comprehensive conformational sampling, simulate for at least 100 ns - 1 μs, depending on system size and the timescales of relevant dynamics.
Enhanced Sampling Considerations: For complex conformational transitions, implement replica exchange molecular dynamics (REMD) or weighted ensemble (WE) sampling. WE sampling uses progress coordinates from time-lagged independent component analysis (TICA) to efficiently explore conformational space [84].
Trajectory Analysis Methods
Root Mean Square Deviation (RMSD): Calculate backbone RMSD relative to the initial structure to assess overall stability and convergence. Systems are typically considered equilibrated when RMSD stabilizes (e.g., ~1.5-3.0 Å for well-folded proteins) [87].
Root Mean Square Fluctuation (RMSF): Compute per-residue RMSF to identify flexible regions and compare with crystallographic B-factors [88] [87]. Higher fluctuations often occur in loop regions, with specific patterns depending on bound nucleotide and conformational state [87].
Principal Component Analysis (PCA): Perform PCA on Cα coordinates to identify essential dynamics and collective motions. Project trajectories onto the first few principal components to visualize conformational landscapes and transitions between states [89].
Interaction Analysis: Calculate interaction fingerprints throughout trajectories to monitor specific protein-ligand or intra-protein contacts, hydrogen bonds, and salt bridges [85].
Validation Against Experimental Data
Crystallographic B-Factors: Compare RMSF profiles with experimental B-factors after converting using the relationship: RMSF ≈ √(3B/8π²) [88]. Note that B-factors capture both dynamic flexibility and static disorder, while MD primarily reflects dynamics.
NMR Ensembles: Compare coordinate variances in MD trajectories with uncertainties in NMR-derived structural ensembles. Both capture peptide bond unit motions, though potential force field deficiencies may create similar artifacts in both MD and NMR refinement [88].
Ensemble-Refinement Methods: Integrate experimental data directly as restraints during simulation to generate ensembles consistent with both computational and experimental data [88].
Advanced Approaches and Emerging Methodologies
Machine Learning-Enhanced Sampling
Recent advances integrate machine learning with MD to overcome timescale limitations. Weighted ensemble sampling using WESTPA enables efficient exploration of conformational space by running multiple replicas with periodic resampling based on progress coordinates [84]. Latent diffusion models like aSAM (atomistic structural autoencoder model) generate structural ensembles at reduced computational cost by learning from MD data, accurately sampling side-chain and backbone torsion angle distributions [89]. Temperature-conditioned models (aSAMt) capture temperature-dependent ensemble properties and generalize beyond training temperatures, enhancing exploration of conformational landscapes [89].
Coarse-Grained Methods for Extended Timescales
For large-scale conformational changes or initial flexibility screening, coarse-grained methods offer significant speed advantages. CABS-flex provides rapid simulations of protein flexibility with a computational speed advantage of three to four orders of magnitude over all-atom MD while maintaining competitive accuracy [90]. The method combines coarse-grained simulation with all-atom reconstruction, offering multiple flexibility modes (Flexible, Rigid, Rigid-pLDDT, Unleashed) to balance restraint levels with sampling requirements [90].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool/Category | Specific Examples | Function and Application |
|---|---|---|
| MD Simulation Engines | AMBER, GROMACS, NAMD, OpenMM, CHARMM [1] | Core software for performing molecular dynamics simulations with different algorithms and optimizations |
| Force Fields | AMBER, CHARMM, GROMOS, OPLS-AA [1] [84] | Mathematical representations of interatomic potentials that determine simulation accuracy |
| Visualization & Analysis | VMD, CPPTRAJ, MDAnalysis, PyTraj [85] | Trajectory examination, measurement, and feature extraction |
| System Preparation | PDBFixer, tleap, CHARMM-GUI, MOE [1] [84] | Structure repair, solvation, ionization, and parameter generation |
| Enhanced Sampling | WESTPA, PLUMED, MetaDynamics [84] | Accelerate rare events and improve conformational space sampling |
| Validation Datasets | ATLAS, mdCATH [84] [89] | Reference MD trajectories and experimental data for method benchmarking |
| Specialized Hardware | NVIDIA RTX 5090, H100, GH200 Superchip [72] | GPU accelerators for computationally intensive MD calculations |
| Coarse-Grained Tools | CABS-flex 3.0, MARTINI [90] | Rapid flexibility screening and large-scale conformational sampling |
Selecting the appropriate MD software requires careful consideration of research goals, system characteristics, and available computational resources. GROMACS excels in raw performance for typical biomolecular systems, while AMBER provides sophisticated force fields and advanced sampling methods crucial for studying subtle conformational changes. NAMD offers superior visualization integration, and OpenMM enables exceptional flexibility through Python scripting. Validation against experimental data remains essential for establishing the biological relevance of MD-derived ensembles, with RMSF, PCA, and interaction analysis providing complementary views of protein dynamics. Emerging methodologies, particularly machine learning-enhanced sampling and standardized benchmarking frameworks, promise to accelerate progress in simulating and validating conformational ensembles, ultimately enhancing our understanding of protein function in health and disease.
Benchmarking Different Force Fields and Water Models
The validation of solution ensembles in molecular dynamics (MD) research hinges on the careful selection of force fields and water models, which collectively determine the accuracy of simulating biomolecular processes. As MD simulations become increasingly integral to drug development and molecular sciences, researchers face a challenging landscape of competing models, each with distinct performance characteristics across various systems. This guide provides an objective comparison of different force fields and water models based on recent benchmarking studies, offering experimental data and methodologies to inform model selection for specific research applications. By synthesizing evidence from systematic evaluations across protein systems, polyamide membranes, and glycosaminoglycan complexes, we establish a framework for validating solution ensembles that balances computational efficiency with physical accuracy.
The critical importance of force field and water model selection stems from their profound influence on simulation outcomes. Molecular dynamics serves as a computational microscope for studying atomic-level processes, but its predictive power depends entirely on the underlying potential functions [84]. Classical force fields approximate atomic interactions through parameterized potential functions, while water models explicitly represent solvent molecules with varying degrees of complexity. Recent advances include machine learning force fields that promise ab initio accuracy with significantly reduced computational costs [43], yet their generalizability remains challenging. Within this context, rigorous benchmarking against experimental data provides the essential foundation for validating solution ensembles in MD research.
Performance Comparison of Water Models
Structural Accuracy Across Temperature Ranges
Comprehensive evaluations of water models reveal significant differences in their ability to reproduce experimental structural properties. A systematic assessment of 44 classical water potential models compared their performance against neutron and X-ray diffraction data across a wide temperature range [91]. The study calculated radial distribution functions and total scattering structure factors, identifying clear performance trends among model categories.
Table 1: Performance of Water Model Types for Structural Prediction
| Model Type | Representative Models | Structural Accuracy | Computational Efficiency | Best Applications |
|---|---|---|---|---|
| Three-site | OPC3, OPTI-3T, TIP3P, SPC/E | Good to excellent | High | Large biomolecular systems, high-throughput screening |
| Four-site | TIP4P-type variants | Best overall agreement | Moderate | Accurate solvation studies, parameter development |
| Flexible/polarizable | SPC/ε, FBA/ε | Moderate | Low | Dielectric properties, specific cluster analyses |
| Five-site | TIP5P | Variable | Low | Specialized structural studies |
Notably, models with more than four interaction sites, as well as flexible or polarizable models with higher computational requirements, did not provide significant advantages in accurately describing water structure [91]. This finding challenges the assumption that increased complexity necessarily improves performance. Recent three-site models like OPC3 and OPTI-3T demonstrated considerable progress in structural prediction, though the best agreement with experimental data across the entire temperature range was achieved with four-site, TIP4P-type models [91].
Performance in Specific Biomolecular Contexts
Water model performance varies significantly across different biomolecular systems, necessitating context-specific selection. In protein-glycosaminoglycan (GAG) complexes, systematic evaluation of explicit water models (TIP3P, SPC/E, TIP4P, TIP4PEw, OPC, TIP5P) and implicit solvent models revealed substantial differences in binding pose characterization [92]. TIP5P and OPC water models provided the best agreement with experimental data for both local and global structural features of heparin in MD simulations [92].
For polyamide membrane simulations, water model selection significantly influenced predictions of water permeability and membrane properties [93]. The TIP4P model demonstrated superior performance in hydrated membrane simulations when combined with the GAFF force field, accurately predicting experimental pure water permeability within a 95% confidence interval [93]. This context-dependence underscores the importance of matching water models to specific research applications rather than seeking a universal solution.
Table 2: Water Model Performance in Specialized Applications
| Application Domain | Best Performing Models | Key Metrics | Experimental Validation |
|---|---|---|---|
| Protein-GAG complexes | TIP5P, OPC | Binding pose stability, structural features | NMR data, crystal structures |
| Polyamide membranes | TIP4P, TIP3P | Water permeability, pore size distribution | Experimental permeability measurements |
| Bulk water properties | TIP4P-type, OPC3 | Radial distribution functions, structure factors | Neutron and X-ray diffraction |
| Water clusters | SPC/ε | Electronic structure representation, complexity measures | Information-theoretic analysis |
Force Field Evaluation and Comparison
Performance Across Biomolecular Systems
Force field selection profoundly impacts simulation outcomes across diverse biomolecular contexts. Benchmarking studies have evaluated numerous force fields against experimental data to establish objective performance criteria.
In polyamide membrane simulations, six force fields (PCFF, CVFF, SwissParam, CGenFF, GAFF, DREIDING) were systematically compared for their ability to predict membrane properties [93]. The evaluation encompassed dry and hydrated states, as well as non-equilibrium reverse osmosis conditions. CVFF, SwissParam, and CGenFF force fields performed best in dry states, accurately predicting experimental Young's modulus, density, porosity, and pore size distribution [93]. For hydrated membranes, PCFF and CGenFF provided the most accurate predictions of water diffusion coefficients, with PCFF achieving the closest agreement with experimental pure water permeability data [93].
For protein simulations, the AMBER14 force field combined with TIP3P-FB water model has been employed in standardized benchmarking frameworks, demonstrating robust performance across a diverse set of nine proteins ranging from 10 to 224 residues [84]. This combination effectively captured folding complexities and various topological challenges, establishing it as a reliable choice for protein dynamics studies.
Emerging Machine Learning Force Fields
Machine learning force fields (MLFFs) represent a paradigm shift in molecular simulations, offering ab initio accuracy with significantly reduced computational costs. AI2BMD, an artificial intelligence-based ab initio biomolecular dynamics system, demonstrates the potential of this approach [43]. By using a protein fragmentation scheme and machine learning force field, AI2BMD achieves generalizable ab initio accuracy for energy and force calculations for various proteins comprising more than 10,000 atoms [43].
Performance evaluations reveal that AI2BMD reduces computational time by several orders of magnitude compared to density functional theory while maintaining high accuracy [43]. For energy calculations, AI2BMD achieved a mean absolute error of 0.045 kcal mol⁻¹ compared to 3.198 kcal mol⁻¹ for classical molecular mechanics force fields [43]. Similarly, for force calculations, AI2BMD demonstrated a MAE of 0.078 kcal mol⁻¹ Å⁻¹ versus 8.125 kcal mol⁻¹ Å⁻¹ for classical approaches [43]. This dramatic improvement in accuracy positions MLFFs as compelling alternatives for future biomolecular simulations, though their generalizability across diverse molecular systems requires further validation.
Methodologies for Systematic Benchmarking
Standardized Evaluation Frameworks
The rapid evolution of MD methods has outpaced the development of standardized validation tools, hindering objective comparisons between simulation approaches. To address this challenge, recent initiatives have introduced modular benchmarking frameworks that systematically evaluate MD methods using enhanced sampling analysis [84]. These frameworks employ weighted ensemble (WE) sampling via the Weighted Ensemble Simulation Toolkit with Parallelization and Analysis (WESTPA), enabling efficient exploration of protein conformational space.
A key innovation in these frameworks is the use of progress coordinates derived from Time-lagged Independent Component Analysis (TICA), which allows for fast and efficient sampling of conformational states [84]. The frameworks include flexible, lightweight propagator interfaces that support arbitrary simulation engines, accommodating both classical force fields and machine learning-based models. Comprehensive evaluation suites compute more than 19 different metrics and visualizations across various domains, providing multidimensional assessment of force field performance [84].
Table 3: Key Metrics for Force Field and Water Model Evaluation
| Metric Category | Specific Metrics | Experimental Reference | Computational Method |
|---|---|---|---|
| Structural fidelity | Radial distribution functions, Rg distributions | X-ray/neutron diffraction | MD simulations with explicit solvent |
| Energetic accuracy | Potential energy MAE, force MAE | DFT calculations, ab initio data | MLFF comparisons |
| Dynamic properties | Self-diffusion coefficient, dielectric constant | Experimental measurements | Equilibrium MD simulations |
| Thermodynamic properties | Density, heat of vaporization | Experimental measurements | NPT simulations |
| Sampling efficiency | Conformational space coverage, rare event frequency | Enhanced sampling data | Weighted ensemble simulations |
Information-Theoretic Analysis
Beyond traditional metrics, information-theoretic measures provide complementary tools for force field evaluation. Comprehensive analyses employ five fundamental descriptors—Shannon entropy, Fisher information, disequilibrium, LMC complexity, and Fisher-Shannon complexity—calculated in both position and momentum spaces to quantify electronic delocalizability, localization, uniformity, and structural sophistication [94].
This approach has revealed distinct scaling behaviors that correlate with experimental accuracy across water clusters of increasing size (1, 3, 5, 7, 9, and 11 molecules) [94]. The SPC/ε model demonstrates superior electronic structure representation with optimal entropy-information balance and enhanced complexity measures, while TIP3P shows excessive localization and reduced complexity that worsen with increasing cluster size [94]. These information-theoretic measures establish quantitative relationships between force field parameters and experimentally observable properties, offering insights beyond traditional structural comparisons.
Integrated Workflows for Force Field Validation
Multi-scale Validation Approach
Robust force field validation requires integrated workflows that assess performance across multiple spatial and temporal scales. These workflows begin with cluster-level analyses using information-theoretic measures to evaluate electronic structure representation, progress to bulk property validation against experimental physicochemical data, and culminate in biomolecular system assessments comparing structural and dynamic properties against experimental observations [94].
For water models, this approach has established clear connections between cluster-level properties and bulk behavior. The transferability from clusters to bulk properties is demonstrated through systematic convergence of information-theoretic measures toward bulk-like behavior with increasing cluster size [94]. Similarly, for biomolecular force fields, validation workflows assess performance from dipeptide-level accuracy to full-protein dynamics, ensuring consistent behavior across scales.
Enhanced Sampling for Rare Events
Comprehensive force field validation requires adequate sampling of rare conformational events, which traditional MD struggles to capture within feasible computational timeframes. Enhanced sampling strategies, particularly weighted ensemble (WE) simulations, address this challenge by running multiple replicas of a system and periodically resampling them based on user-defined metrics of conformational space coverage [84].
This approach allocates computational resources adaptively, increasing the likelihood of observing rare events within tractable timeframes and making WE a powerful tool for capturing critical transitions in complex systems [84]. When integrated with standardized benchmarking frameworks, WE sampling enables consistent evaluation of force fields across their ability to reproduce not just stable states but also transition pathways and rare conformational changes.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Computational Tools for Force Field Benchmarking
| Tool Name | Type | Primary Function | Application Context |
|---|---|---|---|
| WESTPA 2.0 | Software toolkit | Weighted ensemble sampling | Enhanced conformational sampling |
| OpenMM | Simulation engine | MD simulation with GPU acceleration | Force field implementation and testing |
| AMBER14 | Force field | Biomolecular parameter set | Protein dynamics simulations |
| GLYCAM06 | Force field | Carbohydrate parameter set | Glycosaminoglycan simulations |
| TIP3P-FB | Water model | Three-site water representation | Balanced accuracy and efficiency |
| TIP4P-type | Water model | Four-site water representation | High-accuracy structural studies |
| OPC | Water model | Four-site optimized potential | Superior dielectric properties |
| AI2BMD | ML framework | Ab initio accuracy MD | Reference calculations |
| PLOT | Analysis tool | Information-theoretic measures | Electronic structure evaluation |
Benchmarking studies consistently demonstrate that force field and water model performance is highly context-dependent, with no single universal solution emerging across all application domains. For general biomolecular simulations, four-site TIP4P-type water models provide the best balance of structural accuracy and computational efficiency, while recent three-site models like OPC3 offer compelling alternatives for specific applications [91]. Among force fields, AMBER14 for proteins and GLYCAM06 for carbohydrates, combined with appropriate water models, deliver robust performance across diverse systems [84] [92].
Machine learning force fields represent a transformative development, offering ab initio accuracy with dramatically reduced computational costs [43]. As these methods mature and address current limitations in generalizability, they promise to redefine the standards for force field validation and application. Nevertheless, classical force fields remain essential workhorses for most biomolecular simulations, particularly when selected through systematic benchmarking against experimental data relevant to specific research contexts.
The establishment of standardized benchmarking frameworks with comprehensive metric suites enables more rigorous and reproducible force field evaluations [84]. By adopting these frameworks and the methodologies outlined in this guide, researchers can make informed decisions in force field selection, advancing the validation of solution ensembles in molecular dynamics research and enhancing the reliability of computational predictions in drug development and molecular sciences.
Validation Across Diverse Biomolecular Systems
Validating the structural ensembles generated by Molecular Dynamics (MD) simulations against experimental data is a critical step in computational biology. It ensures that the simulated dynamics accurately reflect the behavior of biomolecules in solution, which is fundamental for reliable applications in drug design and mechanistic studies. Without rigorous validation, simulation results may represent computational artifacts rather than physiologically relevant states [70]. This guide objectively compares the performance of various validation methodologies and analysis tools, providing a framework for researchers to assess the accuracy of their MD ensembles.
Comparative Performance of Validation Methods
The table below summarizes the core validation methods used to compare MD simulation ensembles with experimental data, highlighting their applications, key outcome measures, and performance insights.
| Validation Method | Biomolecular System | Key Measured Parameters | Validation Outcome / Performance Insight |
|---|---|---|---|
| Wide-Angle X-Ray Scattering (WAXS) [29] | Five different proteins in solution | Scattering profile agreement (q ≈ 15 nm⁻¹); Radius of Gyration changes (< 1%); Loop flexibility. | Excellent agreement with experiment when using explicit solvent and incorporating thermal fluctuations. Highly sensitive to minor conformational changes. |
| X-Ray/Neutron Diffraction [95] | Dioleoylphosphatidylcholine (DOPC) lipid bilayer | Bilayer thickness; Area per lipid; Structure factors; Scattering-density profiles. | United-atom (GROMACS) and all-atom (CHARMM22/27) force fields did not reproduce experimental data within error, though CHARMM27 showed improvement. |
| Analysis of Multiple Trajectories [70] | BPTI, CI2, Barnase (unfolding studies) | Cα Root-Mean-Square Deviation (RMSD); Property similarity (e.g., SASA, Rg); Conformational clustering. | Unfolding pathways diverged in conformational space but often shared similar physical properties in the denatured state, supporting a "funnel" description of folding. |
Experimental Protocols for Ensemble Validation
Protocol 1: Validation Using Wide-Angle X-Ray Scattering (WAXS)
This protocol uses WAXS data to validate solution ensembles from MD simulations with minimal fitting, thus reducing overfitting risks [29].
- Simulation Setup: Perform explicit-solvent MD simulations of the biomolecule(s) of interest. The study utilized free MD simulations up to one microsecond.
- Theoretical Scattering Calculation: Calculate the theoretical WAXS profile directly from the MD simulation trajectories. This employs explicit solvation, eliminating free parameters typically associated with modeling the solvation layer or excluded solvent.
- Data Comparison: Compare the calculated WAXS profiles to the experimental data. The study used only a single fitting parameter to account for uncertainties in buffer subtraction and detector dark currents.
- Sensitivity Analysis: Assess the sensitivity of the profile by analyzing simulations with increasing protein flexibility and through minor conformational rearrangements.
Protocol 2: Diffraction-Based Validation for Lipid Bilayers
This model-independent method rigorously compares simulated and experimental bilayer structures by analyzing data in an identical manner [95].
- System Simulation: Perform multi-nanosecond MD simulations of the lipid bilayer (e.g., DOPC at 66% RH).
- Reciprocal Space Analysis:
- Determine the discrete and continuous structure factors (
F(s)) from the simulation. - This mimics the experimentalist's analysis of diffraction data and avoids the approximations of simple binning procedures.
- Determine the discrete and continuous structure factors (
- Real Space Analysis:
- Perform a Fourier reconstruction of the discrete structure factors to obtain the overall transbilayer scattering-density profile.
- Quantitative Comparison: Compare the simulation-derived structure factors and density profiles with experimental results. The comparison includes bilayer thickness, area per lipid, and individual molecular-component distributions, with careful accounting of experimental and simulation uncertainties.
Protocol 3: Comparative Analysis of Multiple Unfolding Trajectories
This suite of methods compares multiple MD unfolding trajectories to derive general insights about pathways and denatured states, facilitating comparison with ensemble-based experiments [70].
- Trajectory Generation: Perform multiple, independent high-temperature MD simulations of protein unfolding.
- Structure-Based Analysis:
- Calculate the Cα Root-Mean-Squared Deviation (RMSD) between all pairs of structures across different trajectories to quantify conformational similarity.
- Perform clustering analysis to identify dominant conformational states populated across different runs.
- Property-Based Analysis:
- Track time-dependent physical properties (e.g., radius of gyration, solvent-accessible surface area, secondary structure content) for each trajectory.
- Compare the evolution of these "property spaces" to determine if different conformational pathways yield similar experimental observables.
- Data Interpretation: Synthesize findings from both structural and property-based analyses to characterize the breadth of the unfolding pathway and the nature of the denatured state ensemble.
Workflow Visualization
The following diagram illustrates the logical workflow for validating MD ensembles using the protocols described above, integrating both simulation and experimental components.
Diagram Title: Workflow for MD Ensemble Validation
The Scientist's Toolkit: Essential Research Reagents and Solutions
The table below details key computational tools and resources essential for conducting and analyzing MD simulations and their validation.
| Tool / Resource | Function / Purpose | Relevance to Validation |
|---|---|---|
| FastMDAnalysis [96] | Software for automated analysis of MD trajectories via CLI or Python API. | Streamlines calculation of key validation metrics like RMSD, Rg, and SASA; generates publication-quality figures. |
| MDWiZ [97] | An online platform for converting file formats and force fields between different MD software packages. | Crucial for preparing simulation data for analysis with different tools and for reproducing results across software. |
| GROMACS & NAMD [95] | High-performance MD simulation software packages. | Used to produce the simulation data that is subsequently validated against experimental evidence. |
| CHARMM & GROMACS Force Fields [95] | Empirical parameter sets defining interatomic interactions for MD simulations. | Their accuracy is directly tested during validation; newer versions (e.g., CHARMM27) show improved performance. |
| MedCalc-Bench [98] | A benchmark for evaluating Large Language Models (LLMs) on medical calculation tasks. | Represents the broader trend of developing specialized benchmarks to validate computational models in life sciences. |
Molecular dynamics (MD) simulation has emerged as a powerful computational technique for studying the physical movements of atoms and molecules over time, providing unparalleled atomic-scale insights into biological and materials systems [99]. However, the predictive power and reliability of MD simulations are fundamentally dependent on rigorous validation against experimental data. Without such validation, simulation results risk being computational artifacts rather than representations of physical reality. This comparison guide examines the methodologies and performance of integrating multiple experimental data sources to validate solution ensembles derived from molecular dynamics research, with particular emphasis on wide-angle X-ray scattering (WAXS), advanced computational hardware, and specialized software tools.
The importance of this integration lies in the complementary strengths and limitations of computational and experimental approaches. While MD simulations can provide atomic-level temporal resolution of molecular processes, they operate within timescales and force field approximations that require experimental verification [99]. Conversely, experimental techniques often yield ensemble-averaged structural information but lack the atomic-level dynamical details that simulations can provide. This guide objectively compares the performance of various validation approaches, computational tools, and hardware configurations to assist researchers in selecting optimal strategies for their specific validation challenges.
Methodologies for Experimental Validation of MD Simulations
Wide-Angle X-Ray Scattering (WAXS) Validation
WAXS has become an increasingly valuable technique for validating MD simulations due to technical advances in light sources and detectors [7]. The methodology involves calculating theoretical WAXS profiles from MD trajectories and comparing them directly with experimental data. The key advantage of this approach is its sensitivity to minor conformational rearrangements – simulations demonstrate that WAXS profiles are highly sensitive to increased flexibility of protein loops or increases of the radius of gyration by less than 1% [7].
The experimental protocol for WAXS validation involves several critical steps. First, MD simulations are performed on the system of interest using explicit solvent models to ensure realistic solvation effects. The scattering intensity I(q) is calculated from the MD trajectories using the equation: I(q) = ⟨|Ã(q)|²⟩' - ⟨|B̃(q)|²⟩', where Ã(q) and B̃(q) are the Fourier transforms of the instantaneous electron densities of the solution and pure solvent systems, respectively, and the ensemble average ⟨⋯⟩' includes averages over solute and solvent fluctuations as well as orientations of the solute [7]. This explicit-solvent formulation eliminates free parameters associated with the solvation layer and excluded solvent, minimizing the risk of overfitting. Only a single fitting parameter is typically required to account for experimental uncertainties related to buffer subtraction and dark currents [7].
The workflow for WAXS validation of MD simulations can be visualized as follows:
Grain Growth and Materials Validation
In materials science, MD simulations of processes like grain growth in polycrystalline materials provide additional validation opportunities. The methodology involves using experimental data as the initial microstructure for MD simulations, enabling direct comparison of simulation outcomes with experimental observations [100]. This approach has been successfully applied to study the relationship between grain boundary curvature and velocity in impurity-free systems, contributing evidence to the reported absence of correlation between velocity and curvature during grain growth in polycrystals [100].
The experimental protocol for grain growth validation includes developing bidirectional methods for converting data between voxelized and atomic structures. MD grain growth simulations are then performed using these experimentally-derived structures, and the results are compared with actual grain growth observations. This methodology confirms that certain phenomena observed experimentally are not related to solutes, precipitates, processing route, or characterization method, but rather inherent features of the 3D grain boundary network [100].
Multi-Technique Integration Framework
The most robust validation approach integrates multiple experimental data sources to create a comprehensive validation framework. This multi-technique methodology leverages the complementary strengths of various experimental approaches to address different aspects of MD validation:
- Structural validation: Using techniques like WAXS, NMR, and cryo-EM to validate structural ensembles
- Dynamic validation: Employing techniques like fluorescence resonance energy transfer and hydrogen-deuterium exchange to validate dynamic properties
- Thermodynamic validation: Utilizing isothermal titration calorimetry and surface plasmon resonance to validate binding energies and thermodynamic parameters
The integration of these diverse data sources provides a more complete validation picture than any single technique can offer independently, significantly increasing confidence in simulation results.
Performance Comparison of Validation Methodologies
Quantitative Validation Performance Metrics
Table 1: Performance Comparison of MD Validation Methodologies
| Validation Method | Spatial Resolution | Temporal Resolution | Key Strengths | Key Limitations |
|---|---|---|---|---|
| WAXS/SAS | 1-2 nm (global structure) | Milliseconds to seconds | Sensitive to global shape and flexibility; minimal sample requirements | Ensemble-averaged; limited structural details |
| NMR Spectroscopy | Atomic-level (local environment) | Picoseconds to seconds | Atomic-level structural and dynamic information | Limited to smaller proteins; complex analysis |
| Cryo-EM | 3-5 Å (single-particle) | Static snapshots | Visualizes large complexes; no size limitation | Sample preparation challenges; static information |
| FRET | 2-8 nm (inter-atomic distances) | Nanoseconds to milliseconds | Sensitive distance measurements in solution | Requires labeling; limited structural details |
| HDX-MS | 5-20 residues (local dynamics) | Seconds to minutes | Probes backbone dynamics and solvent accessibility | Limited spatial resolution; complex interpretation |
Hardware Performance for Validation Workflows
The computational demands of MD simulations required for experimental validation necessitate high-performance hardware. Different MD software packages show varying performance across hardware configurations:
Table 2: GPU Performance for AMBER 24 (ns/day) on Various Hardware Configurations [72]
| GPU Model | STMV (1M atoms) | Cellulose (408K atoms) | Factor IX (91K atoms) | DHFR (24K atoms) | Myoglobin GB (2.5K atoms) |
|---|---|---|---|---|---|
| RTX 5090 | 109.75 | 169.45 | 529.22 | 1655.19 | 1151.95 |
| RTX 5080 | 63.17 | 105.96 | 394.81 | 1513.55 | 871.89 |
| RTX PRO 6000 Blackwell | 97.44 | 149.84 | 475.04 | 1464.14 | 940.57 |
| B200 SXM | 114.16 | 182.32 | 473.74 | 1513.28 | 1020.24 |
| H100 PCIe | 74.50 | 125.82 | 410.77 | 1532.08 | 1094.57 |
| RTX 6000 Ada | 70.97 | 123.98 | 489.93 | 1697.34 | 1016.00 |
For GROMACS and NAMD simulations, which are also commonly used in validation workflows, the NVIDIA RTX 4090 and RTX 6000 Ada have proven to be excellent choices, with the RTX 4090 offering substantial CUDA cores (16,384) and 24 GB of GDDR6X VRAM for handling large datasets typical in molecular simulations [101].
Software Tools for Experimental Data Integration
Table 3: Software Tools for Integrating Experimental Data with MD Simulations
| Software Tool | Primary Function | Supported Experimental Data Types | Key Features |
|---|---|---|---|
| AMBER | MD simulation engine | WAXS/SAS, NMR, FRET | PMEMD GPU acceleration; explicit solvent simulations |
| GROMACS | MD simulation package | WAXS, NMR, HDX | High performance; multi-GPU support |
| NAMD | MD simulation program | Cryo-EM, SAS | Scalable parallelization; visualization integration |
| WAXSiS | WAXS profile calculation | WAXS | Web server for profile calculation from structures |
| SASSIE | SAS analysis | Small-angle scattering | Generation and analysis of structural ensembles |
Experimental Protocols for Key Validation Approaches
WAXS Experimental Protocol for MD Validation
The integration of WAXS experimental data with MD simulations follows a detailed protocol to ensure accurate validation:
Sample Preparation: Protein or other biomolecular samples are prepared in appropriate buffers at concentrations typically ranging from 1-10 mg/mL. The buffer composition must match exactly between experimental samples and simulation conditions.
Data Collection: WAXS experiments are performed at synchrotron beamlines with high-precision detectors. Data is collected at multiple concentrations to extrapolate to infinite dilution and eliminate interparticle interference effects. The momentum transfer range typically extends to q ≈ 15-20 nm⁻¹ to capture both small-angle and wide-angle information.
Data Processing: Experimental data is processed using standard procedures including background subtraction, normalization, and buffer subtraction. The excess scattering intensity I(q) is calculated according to Equation 1, accounting for the solvent scattering contribution.
MD Simulation Parameters: Simulations are performed using explicit solvent models with sufficient system size to avoid artifacts. Typical production runs range from 100 ns to 1 μs, with frames saved at 1-100 ps intervals for subsequent analysis.
Theoretical Profile Calculation: WAXS profiles are calculated from MD trajectories using explicit-solvent methods that incorporate the excluded solvent and solvation layer without additional fitting parameters. The spatial envelope method is employed to reduce computational cost and statistical noise.
Quantitative Comparison: Theoretical and experimental profiles are compared using χ² analysis or similar metrics. The sensitivity of the comparison is enhanced by incorporating thermal fluctuations from the MD simulations.
Multi-Scale Validation Protocol for Complex Systems
For complex systems such as calcium aluminosilicate hydrate (CASH) in cementitious materials or polymers for oil displacement, a multi-scale validation protocol is employed:
Atomic-Level Validation: MD simulations of CASH gels are validated against nuclear magnetic resonance data for atomic structure and position, providing insights into the role of Al in the molecular structure, dynamics, and mechanical behavior [99].
Mesoscale Validation: For oil-displacement polymers, MD simulations are validated against rheological measurements to optimize polymer properties and design novel polymers with specific oil-displacement characteristics [102].
Macroscale Validation: Mechanical properties predicted from MD simulations are validated against bulk mechanical testing, creating connections between atomic-scale interactions and macroscopic material performance.
The workflow for this multi-technique validation approach is visualized below:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Materials for MD Validation Studies
| Item | Function | Application Examples | Technical Specifications |
|---|---|---|---|
| Synchrotron Beam Access | WAXS/SAXS data collection | Protein structural validation | High-brilliance X-ray source; q-range 0.1-20 nm⁻¹ |
| NVIDIA RTX 5090 GPU | MD simulation acceleration | Large system simulations | 16,384 CUDA cores; 32 GB GDDR6X memory |
| NVIDIA RTX 6000 Ada | MD simulation for complex systems | CASH and polymer simulations | 18,176 CUDA cores; 48 GB GDDR6 memory |
| Explicit Solvent Force Fields | Realistic solvation in MD | WAXS profile calculation | CHARMM36, AMBER19, OPLS-AA |
| Buffer Matching Kits | Consistent experimental conditions | Sample preparation for WAXS | pH 3-10 range; various ionic strengths |
| BIZON Z5 Workstation | High-performance MD simulations | Multi-GPU simulation setups | 4-8 GPU capacity; advanced cooling systems |
| AMBER Software Suite | MD simulations and analysis | Biomolecular simulations | PMEMD engine; CUDA acceleration |
| GROMACS Package | MD simulations | High-throughput validation | MPI parallelization; multi-GPU support |
The integration of multiple experimental data sources for validating molecular dynamics simulations has matured into a sophisticated methodology that significantly enhances the reliability of computational predictions. Through rigorous comparison of validation approaches, several best practices emerge. First, WAXS validation with explicit solvent simulations provides exceptional sensitivity to conformational changes while minimizing overfitting risks. Second, hardware selection should align with specific simulation requirements, with high-memory GPUs like the RTX 6000 Ada offering advantages for large systems, while cost-effective options like the RTX 5090 provide excellent performance for standard simulations. Third, multi-technique validation approaches that combine structural, dynamic, and thermodynamic data provide the most comprehensive assessment of simulation accuracy.
The continuing advancement of both experimental techniques and computational power promises even more robust validation methodologies in the future. Emerging trends include the integration of machine learning with MD simulations, the development of multi-scale simulation methodologies, and increased emphasis on validating complex systems in materials science and drug development. By adhering to the rigorous comparison protocols outlined in this guide, researchers can ensure their molecular dynamics simulations provide accurate, reliable insights into molecular behavior across diverse scientific domains.
Conclusion
Validating molecular dynamics simulations against experimental data like WAXS and SAXS has emerged as a powerful paradigm for obtaining accurate solution ensembles of biomolecules. The integration of explicit-solvent simulations with advanced scattering calculations enables researchers to move beyond static structures and capture the essential dynamics governing biological function. As force fields continue to improve and computational methods become more sophisticated, this approach will play an increasingly crucial role in drug discovery—particularly for challenging targets like intrinsically disordered proteins involved in neurodegenerative diseases and membraneless organelle formation. Future directions will likely involve more sophisticated multi-modal data integration, machine learning-enhanced sampling, and the development of truly generalizable force fields capable of accurately describing both folded and disordered states, ultimately enabling more predictive computational models in biomedical research.
References
- 1. Comparison of software for molecular mechanics modeling [https://en.wikipedia.org/wiki/Comparison_of_software_for_molecular_mechanics_modeling]
- 2. Running Molecular Dynamics on Alliance clusters with AMBER [https://computecanada.github.io/molmodsim-amber-md-lesson/26-MD_Software_Benchmarks/index.html]
- 3. A diverse and chemically relevant solvation model ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12581188/]
- 4. A diverse and chemically relevant solvation model benchmark ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d5sc06406f]
- 5. Comparison, Analysis, and Molecular Dynamics ... [https://www.mdpi.com/2306-5354/10/9/1004]
- 6. A comparative study of computational modeling ... [https://www.nature.com/articles/s41598-025-16866-w]
- 7. Validating Solution Ensembles from Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4104061/]
- 8. A practical guide to small angle X-ray scattering (SAXS) of ... [https://www.sciencedirect.com/science/article/pii/S0014579315007279]
- 9. Interpreting solution X-ray scattering data using molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X17300891]
- 10. Benchmarking predictive methods for small-angle X-ray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11364021/]
- 11. Next Generation SAXS/WAXS System [https://biopacificmip.org/platform/instrumentation/next-generation-saxswaxs-system]
- 12. Understanding nucleic acid structural changes by ... [https://www.osti.gov/pages/biblio/1327123]
- 13. Solvent model [https://en.wikipedia.org/wiki/Solvent_model]
- 14. Machine Learning for Explicit Solvent Molecular Dynamics [https://corinwagen.github.io/public/blog/20230720_ml_aimd.html]
- 15. Solvent Model Benchmark for Molecular Dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10091405/]
- 16. Implicit Solvent Models in Molecular Dynamics Simulations [https://www.sciencedirect.com/science/article/abs/pii/S1574140008000078]
- 17. Design and application of implicit solvent models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4045398/]
- 18. Accuracy comparison of several common implicit solvent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5313374/]
- 19. Extending machine learning model for implicit solvation to ... [https://arxiv.org/html/2510.20103v1]
- 20. Modelling chemical processes in explicit solvents with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11271496/]
- 21. Molecular Dynamics Simulation for All [https://www.sciencedirect.com/science/article/pii/S0896627318306846]
- 22. Molecular dynamics simulations reveal the hidden EF-hand of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10400917/]
- 23. Internal Standard-Assisted Ab Initio MD Simulation for ... [http://www.sciencedirect.com/science/article/pii/S266664722500048X]
- 24. Comparison of atomic simulation methods for computing ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732221022029]
- 25. Red blood cell thermal fluctuations: comparison between ... [https://pubs.rsc.org/en/content/articlelanding/2009/sm/b910422d]
- 26. Implicit Solvent Models and Their Applications in Biophysics [https://www.mdpi.com/2218-273X/15/9/1218]
- 27. Determining accurate conformational ensembles of ... [https://www.nature.com/articles/s41467-025-64098-3]
- 28. Machine-Learned Potentials for Solvation Modeling [https://arxiv.org/html/2505.22402v3]
- 29. Validating Solution Ensembles from Molecular Dynamics ... [https://www.sciencedirect.com/science/article/pii/S0006349514006109]
- 30. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 31. Machine learning analysis of molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12290097/]
- 32. Interpreting SAXS/WAXS Data with Explicit-Solvent ... [https://pubmed.ncbi.nlm.nih.gov/33582993/]
- 33. Predicting solution scattering patterns with explicit-solvent ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687922003603]
- 34. WAXSiS: a web server for the calculation of SAXS/WAXS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4489308/]
- 35. Comparing state-of-the-art approaches to back-calculate ... [https://link.springer.com/article/10.1140/epjb/s10051-021-00186-9]
- 36. Interpretation of Solution X-Ray Scattering by Explicit- ... [https://www.sciencedirect.com/science/article/pii/S0006349515004038]
- 37. Ensemble refinement of mismodeled cryo-EM RNA ... [https://www.nature.com/articles/s41467-025-59769-0]
- 38. The emerging role of physical modeling in the future ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X17301033]
- 39. Efficient Ensemble Refinement by Reweighting - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6727217/]
- 40. Integrative modelling of biomolecular dynamics [https://arxiv.org/html/2510.01108v1]
- 41. Computational Tools for Metabolic Engineering - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3361690/]
- 42. Validating solution ensembles from molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/25028885/]
- 43. Ab initio characterization of protein molecular dynamics ... [https://www.nature.com/articles/s41586-024-08127-z]
- 44. Molecular Dynamics Simulation to Uncover the ... [https://www.sciencedirect.com/science/article/abs/pii/S0022354921000071]
- 45. Full structural ensembles of intrinsically disordered ... [https://www.nature.com/articles/s42003-021-01759-1]
- 46. Combining molecular dynamics simulations with small-angle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7205321/]
- 47. The Protein Force Field Plays a Crucial Role in Obtaining ... [https://arxiv.org/html/2508.18570]
- 48. Fitting high-resolution electron density maps from atomic ... [https://www.sciencedirect.com/science/article/pii/S0006349523006707]
- 49. Understanding nucleic acid structural changes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4884185/]
- 50. The role of molecular dynamics simulations in drug discovery [https://cresset-group.com/solutions/by-discovery-phase/target-validation/molecular-dynamics-simulations/]
- 51. Molecular Dynamics of the Intrinsically Disordered Protein ... [https://pubmed.ncbi.nlm.nih.gov/40907012/]
- 52. Machine learning analysis of molecular dynamics ... [https://www.nature.com/articles/s41598-025-11392-1]
- 53. AlphaFold prediction of structural ensembles of disordered ... [https://www.nature.com/articles/s41467-025-56572-9]
- 54. Integrating Molecular Dynamics, Molecular Docking, and ... [https://www.mdpi.com/1420-3049/30/14/2985]
- 55. CHARMM [https://en.wikipedia.org/wiki/CHARMM]
- 56. Exploring the limits of the generalized CHARMM and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11275216/]
- 57. Benchmarking of force fields to characterize the intrinsically ... [https://www.nature.com/articles/s41598-023-40801-6]
- 58. CHARMM: The Biomolecular Simulation Program - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2810661/]
- 59. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOoo156gqqG_mbKY1JFjoRx-4TFHVPym35Qg5gtF-8iI_wC_DmlGZ]
- 60. CHARMM-GUI Multicomponent Assembler for modeling ... [https://www.nature.com/articles/s41467-024-49700-4]
- 61. 分子模拟中的水模型有哪些? [https://www.huasuankeji.com/news/?p=363836]
- 62. 如何模拟H2O?从简单模型到极化效应的迭代优化! [https://www.huasuankeji.com/news/?p=340206]
- 63. 快速入门— AuToFF-Manual documentation [https://autoff.readthedocs.io/en/latest/User%20Guide.html]
- 64. 水的氢键网络动力学与其太赫兹频谱的关系 [https://wulixb.iphy.ac.cn/article/doi/10.7498/aps.70.20211731]
- 65. gmx_MMPBSA.py的安装及使用--只翻译部分内容,具体可 ... [https://blog.csdn.net/lixiang_92/article/details/125853041]
- 66. gmx_MMPBSA输入文件——待完善_mpbsa是什么 [https://blog.csdn.net/yin1331102028yin/article/details/149295474]
- 67. Sobtop [http://sobereva.com/soft/Sobtop/]
- 68. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 69. Challenges and opportunities in computational studies for ... [https://www.nature.com/articles/s44386-025-00024-3]
- 70. Analysis methods for comparison of multiple molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283699928437]
- 71. Structure-Based Experimental Datasets for Benchmarking ... [https://arxiv.org/abs/2303.11056]
- 72. AMBER 24 NVIDIA GPU Benchmarks | B200, RTX PRO ... [https://www.exxactcorp.com/blog/molecular-dynamics/amber-molecular-dynamics-nvidia-gpu-benchmarks]
- 73. AMBER performance on NHR@FAU GPU systems [https://hpc.fau.de/2025/05/21/amber-performance-on-nhrfau-gpu-systems/]
- 74. Performance improvements - GROMACS 2025-rc ... [https://manual.gromacs.org/2025-rc/release-notes/2023/major/performance.html]
- 75. Performance improvements - GROMACS 2025.0-beta ... [https://manual.gromacs.org/2025.0-beta/release-notes/2021/major/performance.html]
- 76. Optimizing CPU/GPU efficiency and performance in ... [https://gromacs.bioexcel.eu/t/optimizing-cpu-gpu-efficiency-and-performance-in-gromacs-simulations/11509]
- 77. NAMD GPU Benchmarks and Hardware Recommendations [https://www.exxactcorp.com/blog/benchmarks/namd-gpu-benchmarks-and-hardware-recommendations]
- 78. NAMD Benchmark [https://openbenchmarking.org/test/pts/namd&eval=d03c92614e18f4e121d1fd9fd6893158013f0df5]
- 79. Best CPU, GPU, RAM for Molecular Dynamics in 2024 [ Updated ] [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics]
- 80. Can laboratory-based XAFS compete with XRD and ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0323678]
- 81. A comprehensive small angle X-ray scattering analysis on ... [https://www.sciencedirect.com/science/article/abs/pii/S0032386122000970]
- 82. Quantitative size-resolved characterization of mRNA ... [https://www.nature.com/articles/s41598-023-42274-z]
- 83. Using in-situ small-angle scattering to reveal the structure ... [https://www.nature.com/articles/s41467-025-65010-9]
- 84. A Standardized Benchmark for Machine-Learned ... [https://arxiv.org/html/2510.17187v1]
- 85. From Data to Knowledge: Systematic Review of Tools for Automatic... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8960308/]
- 86. Choosing the best tool for MD simulations: GROMACS ... [https://www.linkedin.com/posts/omarariasgaguancela_gromacs-vs-amber-vs-namd-which-one-do-you-activity-7339451262129606656-xgBd]
- 87. Molecular dynamics simulations reveal differences in the ... [https://www.nature.com/articles/s41598-024-66763-x]
- 88. Patterns in Protein Flexibility: A Comparison of NMR ... [https://pubmed.ncbi.nlm.nih.gov/33803249/]
- 89. Deep generative modeling of temperature-dependent ... [https://www.nature.com/articles/s42004-025-01737-2]
- 90. CABS-flex 3.0: an online tool for simulating protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12230700/]
- 91. Comparison of water models for structure prediction [https://www.sciencedirect.com/science/article/pii/S0167732225011523]
- 92. Benchmarking Water Models in Molecular Dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10934818/]
- 93. Benchmarking forcefields for molecular dynamics ... [https://www.sciencedirect.com/science/article/abs/pii/S0376738824010871]
- 94. Information-Theoretic Analysis of Selected Water Force ... [https://www.mdpi.com/1099-4300/27/10/1073]
- 95. Experimental Validation of Molecular Dynamics Simulations of Lipid Bilayers: A New Approach - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1305157/]
- 96. GitHub - aai-research-lab/FastMDAnalysis: Software for automated... [https://github.com/aai-research-lab/fastmdanalysis]
- 97. MDWiZ: a platform for the automated translation of molecular ... [https://pubmed.ncbi.nlm.nih.gov/24434017/]
- 98. ncbi-nlp/MedCalc-Bench [https://github.com/ncbi-nlp/MedCalc-Bench]
- 99. A Review of the Progress in Molecular Dynamics ... [https://www.mdpi.com/2313-4321/10/4/132]
- 100. Comparing molecular dynamics simulations of grain ... [https://www.sciencedirect.com/science/article/abs/pii/S1359646224004640]
- 101. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOorGYfBQh98tg8OavMsrpVqL9UO8xjr90SmxfDDM0TsGHlv_K5Sx]
- 102. Frontier advances in molecular dynamics simulations for the ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra00059a?page=search]