Research Articles
A Practical Guide to Replica Exchange Molecular Dynamics for Enhanced Peptide Sampling
This article provides a comprehensive resource for researchers and professionals on implementing Replica Exchange Molecular Dynamics (REMD) for studying peptides.
Constrained Molecular Dynamics for Small Protein Folding: Protocols, Applications, and Advancements in Computational Biophysics
This article provides a comprehensive overview of constrained molecular dynamics (MD) protocols for simulating small protein folding, a critical challenge in computational biophysics and structure-based drug discovery.
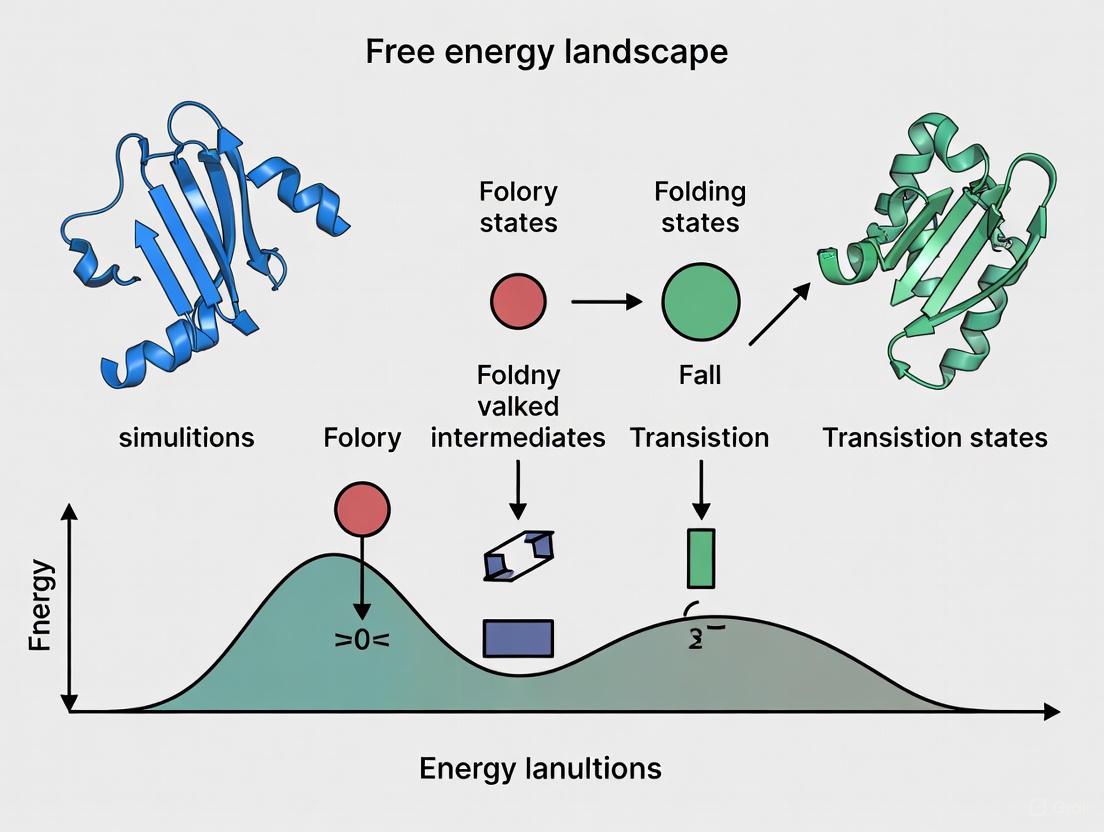
Navigating Protein Folding: From Free Energy Landscape Theory to Advanced Simulations in Drug Discovery
This article provides a comprehensive overview of the free energy landscape theory for protein folding simulations, a cornerstone concept in computational biophysics.
From Microseconds to AI: The Evolution of Protein Molecular Dynamics Simulations in Drug Discovery
This article traces the transformative journey of molecular dynamics (MD) simulations from their rudimentary beginnings to their current status as an indispensable computational microscope in life sciences.
Statistical Mechanics in Protein Folding: From Foundational Principles to Drug Discovery Applications
This article explores the pivotal role of statistical mechanics in simulating and understanding protein folding, a fundamental process with profound implications for health and disease.
Molecular Mechanics Force Fields Explained: How AMBER and CHARMM Power Biomolecular Simulations
This article provides a comprehensive guide for researchers and drug development professionals on the role of molecular mechanics force fields, specifically AMBER and CHARMM, in molecular dynamics (MD) simulations.
Resolving Levinthal's Paradox: How Molecular Dynamics and AI Are Decoding Protein Folding
This article explores the resolution of Levinthal's paradox—the apparent contradiction between the astronomical number of possible protein conformations and their rapid, reliable folding.
The Protein Folding Problem Solved? How AI, Quantum Computing, and New Algorithms Are Reshaping Biology and Drug Discovery
This article provides a comprehensive analysis of the modern state of the protein folding problem in computational biology, a grand challenge once considered intractable.
Benchmarking Ensemble Generation Methods for Disordered Proteins: From Molecular Simulations to Machine Learning
This article provides a comprehensive benchmark of computational methods for generating conformational ensembles of intrinsically disordered proteins (IDPs).
Protein Folding Unfolded: A Guide to Molecular Dynamics Simulations for Biomedical Research
This article provides a comprehensive guide to molecular dynamics (MD) simulations for studying protein folding, tailored for researchers and drug development professionals.