Navigating Protein Folding: From Free Energy Landscape Theory to Advanced Simulations in Drug Discovery
This article provides a comprehensive overview of the free energy landscape theory for protein folding simulations, a cornerstone concept in computational biophysics.
Navigating Protein Folding: From Free Energy Landscape Theory to Advanced Simulations in Drug Discovery
Abstract
This article provides a comprehensive overview of the free energy landscape theory for protein folding simulations, a cornerstone concept in computational biophysics. Tailored for researchers and drug development professionals, it explores the foundational principles of funneled landscapes that guide proteins to their native states. The content delves into cutting-edge methodologies, including machine-learned coarse-grained models and all-atom molecular dynamics, highlighting their application in predicting folding mechanisms, simulating disordered proteins, and characterizing misfolding. It further addresses practical challenges in simulation setup and analysis, compares the performance of different computational approaches against experimental data, and discusses the transformative impact of these tools in accelerating structure-based drug discovery and the design of therapeutic interventions.
The Funneled Landscape: Core Principles of Protein Folding and Misfolding
The Levinthal paradox highlights the apparent impossibility of protein folding, noting that a random conformational search would require cosmological timescales to find the native structure, contrary to the millisecond-to-second timescales observed experimentally. This paradox is resolved by energy landscape theory, which reveals that natural proteins have evolved minimally frustrated, funneled landscapes that direct the folding process. This in-depth technical guide explores the theoretical foundations of funneled landscapes, details experimental and computational methodologies for their study, and presents quantitative data on folding thermodynamics and kinetics. Framed within the context of free energy landscape theory for protein folding simulations research, this review provides researchers, scientists, and drug development professionals with a comprehensive understanding of the principles enabling efficient protein folding and their implications for biomolecular engineering and therapeutic design.
The protein folding problem represents one of the most fundamental challenges in structural biology: understanding how a one-dimensional amino acid sequence spontaneously folds into a unique, functional three-dimensional structure. In 1969, Cyrus Levinthal noted that a protein sampling possible conformations at random would require timescales exceeding the age of the universe to find its native structure, creating an apparent paradox given that proteins typically fold on timescales of milliseconds to seconds [1]. This paradox, while based on an oversimplified model of the folding process, highlighted the need for a theoretical framework to explain the rapid, reliable folding observed in nature.
The solution emerged from energy landscape theory, which reconceptualizes protein folding not as a random search but as a directed process on a minimally frustrated, funneled energy landscape. This framework, first proposed by Bryngelson and Wolynes, recognizes that natural proteins are not random heteropolymers but have been evolutionarily selected for folding efficiency [1]. The funneled landscape picture has since been validated through both experimental studies and advanced computational simulations, providing a unified understanding of how proteins navigate the vast conformational space to achieve their native structures.
Theoretical Framework: Energy Landscape Theory
The Levinthal Paradox and Its Resolution
Levinthal's paradox arises from considering the astronomical number of possible conformations available to a polypeptide chain. For a typical protein of 100 amino acids, assuming just three possible configurations per residue, the total conformational space exceeds 10^100 states. Even sampling at nanosecond rates, a random search would require timescales far exceeding the age of the universe, contradicting experimental observations of rapid folding [1].
The resolution lies in recognizing that the energy landscape is not flat but funneled. Evolution has selected protein sequences with minimally frustrated landscapes, meaning that stabilizing interactions are consistently reinforced toward the native structure rather than competing with one another. This organizational principle creates a bias toward the native state, dramatically reducing the effective search space [1]. As the protein folds, it loses conformational entropy but gains stabilizing energy, creating a downhill trajectory toward the native state.
Principles of Funneled Energy Landscapes
Energy landscape theory introduces several key concepts that enable rapid folding:
Minimal Frustration: In minimally frustrated systems, favorable native interactions dominate over non-native ones. This contrasts with random heteropolymers or strongly frustrated systems where competing interactions create deep kinetic traps [1].
Funnel Topography: The energy landscape resembles a funnel, with the native state at the bottom. The width represents conformational entropy, while the depth represents energy. As the protein folds, it moves downward and inward toward the native state [1].
Thermodynamic Phase Transitions: Minimally frustrated sequences exhibit two key temperatures: the folding transition temperature (TF) where folding occurs, and the glass transition temperature (Tg) below which the system would become trapped in metastable states. Easy-to-fold sequences have high TF/Tg ratios [1].
The following diagram illustrates the key concepts of a funneled energy landscape compared to a frustrated landscape:
Table 1: Key Characteristics of Funneled vs. Frustrated Energy Landscapes
| Characteristic | Funneled Landscape | Frustrated Landscape |
|---|---|---|
| Frustration Level | Minimal | High |
| TF/TG Ratio | High | Low |
| Kinetic Traps | Few | Many |
| Folding Mechanism | Directed, nucleation-like | Random search, glassy dynamics |
| Thermodynamic Control | Yes | No |
| Evolutionary Optimization | Yes | No |
Experimental Methodologies for Studying Folding Landscapes
High-Throughput Stability Measurements
Recent advances in high-throughput experimental methods have enabled large-scale analysis of protein folding stability. The cDNA display proteolysis method represents a breakthrough in scalability, allowing measurement of thermodynamic folding stability for up to 900,000 protein domains in a single experiment [2]. This method combines cell-free molecular biology with next-generation sequencing to quantify protease resistance as a proxy for folding stability.
Experimental Protocol: cDNA Display Proteolysis
- Library Preparation: Synthetic DNA oligonucleotide pools are designed, with each oligonucleotide encoding one test protein variant.
- Transcription and Translation: The DNA library is transcribed and translated using cell-free cDNA display, producing proteins covalently attached to their cDNA at the C-terminus.
- Protease Incubation: Protein-cDNA complexes are incubated with varying concentrations of protease (typically trypsin or chymotrypsin).
- Reaction Quenching and Pull-Down: Reactions are quenched, and intact (protease-resistant) proteins are isolated using an N-terminal pull-down tag.
- Sequencing and Analysis: The relative abundance of surviving proteins is quantified by deep sequencing, and folding stabilities (ΔG) are calculated using a Bayesian model of protease cleavage kinetics [2].
This method has demonstrated high consistency with traditional stability measurements (Pearson correlations > 0.75 for 1,188 variants of 10 proteins), validating its accuracy while achieving unprecedented scale [2].
Quantitative Analysis of Folding Landscapes
Large-scale stability measurements have revealed fundamental principles governing folding landscapes:
- Environmental Influences: The effect of amino acid substitutions on stability depends strongly on the local structural environment, with buried residues typically showing larger stability changes than surface-exposed ones [2].
- Thermodynamic Couplings: Unexpected interactions between distal sites can significantly influence stability, revealing long-range couplings in protein structures.
- Evolutionary Divergence: Natural protein sequences often deviate from optimal stability, suggesting competing evolutionary pressures beyond folding efficiency alone [2].
Table 2: Representative Folding Stabilities from Large-Scale Analysis [2]
| Protein Domain | Length (aa) | Wild-type ΔG (kcal/mol) | Most Stabilizing Mutation | ΔΔG (kcal/mol) |
|---|---|---|---|---|
| Domain A | 40 | -4.2 | V23I | -1.8 |
| Domain B | 52 | -5.1 | A45F | -2.1 |
| Domain C | 61 | -3.8 | L12M | -1.5 |
| Domain D | 68 | -4.7 | F34W | -2.3 |
| Domain E | 72 | -5.3 | K29R | -1.2 |
Computational Approaches and Simulation Methodologies
All-Atom Molecular Dynamics Simulations
All-atom molecular dynamics (MD) simulations represent the gold standard for detailed computational studies of protein folding. By numerically solving Newton's equations of motion for all atoms in the system, MD can theoretically simulate the complete folding process with atomic resolution. Recent advances have enabled millisecond-scale simulations using specialized hardware like Anton, providing unprecedented insights into folding mechanisms [3].
Simulation Protocol: All-Atom Folding Simulations
- System Preparation: The protein sequence is placed in an extended conformation within a simulation box containing explicit water molecules and ions.
- Force Field Selection: Appropriate force fields are selected (e.g., ff99SB/ff14SB for proteins with explicit or implicit solvent models) [3].
- Equilibration: The system undergoes energy minimization and gradual heating to the target temperature (typically 300 K).
- Production Simulation: Extended MD simulations are performed, often using enhanced sampling techniques like replica exchange molecular dynamics (REMD) to improve conformational sampling.
- Trajectory Analysis: Simulations are analyzed using order parameters such as Cα root-mean-square deviation (RMSD), fraction of native contacts (Q), and radius of gyration to characterize folding pathways and kinetics [3].
Studies have demonstrated successful folding to native-like structures (Cα RMSD < 2 Å) for 16 of 17 diverse proteins using implicit solvent models on GPU hardware, achieving simulation rates of ~1 μs/day [3].
Machine-Learned Coarse-Grained Models
While all-atom MD provides detailed information, its computational cost limits applications to small proteins and shorter timescales. Coarse-grained (CG) models address this limitation by representing multiple atoms with single interaction sites, dramatically increasing computational efficiency. Recent machine learning approaches have enabled the development of transferable CG force fields that maintain accuracy while achieving speedups of several orders of magnitude over all-atom simulations [4].
Methodology: Development of Machine-Learned CG Models
- Training Data Generation: Diverse sets of all-atom explicit solvent simulations are performed for small proteins with varied structures.
- Neural Network Training: Deep learning models (e.g., CGSchNet) are trained using the variational force-matching approach to reproduce the equilibrium distribution of all-atom reference simulations.
- Model Validation: The trained CG model is tested on unseen protein sequences to assess transferability and predictive accuracy.
- Production Simulations: The validated model is used for extensive sampling of folding landscapes, often employing parallel-tempering to ensure convergence [4].
These machine-learned CG models successfully predict metastable states of folded, unfolded, and intermediate structures, fluctuations of intrinsically disordered proteins, and relative folding free energies of protein mutants [4].
The following diagram illustrates the workflow for developing and applying machine-learned coarse-grained models:
Table 3: Performance Comparison of Protein Simulation Methods [4] [3]
| Method | Resolution | Speed | Accuracy | Applications |
|---|---|---|---|---|
| All-Atom MD | Atomic | 1 μs/day (GPU) | High (0.5-2 Å RMSD) | Detailed mechanism studies |
| Machine-Learned CG | Coarse-grained | 100-1000 μs/day | Medium-high (1-3 Å RMSD) | Folding landscapes, mutant effects |
| Structure-Based Models | Cα or backbone | ~ms/day | System-dependent | Native basin dynamics |
| Implicit Solvent MD | Atomic | 5-10 μs/day | Variable | Extended folding simulations |
Deep Learning Structure Prediction
Recent breakthroughs in deep learning have revolutionized protein structure prediction, with methods like AlphaFold achieving accuracy competitive with experimental structures. While these approaches primarily predict final structures rather than folding pathways, they provide insights into the sequence-structure relationships that underlie funneled landscapes [5].
AlphaFold incorporates physical and biological knowledge about protein structure through novel neural network architectures that jointly embed evolutionary information from multiple sequence alignments with geometric constraints. The system uses an iterative refinement process that progressively improves structural accuracy, demonstrating how evolutionary constraints shape foldable landscapes [5].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Tools for Protein Folding Studies
| Tool/Resource | Function | Application Examples |
|---|---|---|
| cDNA Display Proteolysis | High-throughput stability measurement | Quantifying ΔG for 900,000 variants [2] |
| GB-Neck2 Implicit Solvent | Accelerated MD simulations | All-atom folding simulations on GPU hardware [3] |
| Machine-Learned CG Models | Efficient landscape sampling | Predicting metastable states and folding mechanisms [4] |
| AlphaFold | Structure prediction from sequence | Assessing native state geometry and confidence [5] |
| GoFold | Educational visualization of CMO problems | Template selection and contact map analysis [6] |
Discussion and Research Implications
The framework of funneled energy landscapes has transformed our understanding of protein folding, resolving the Levinthal paradox by revealing how evolution has shaped sequences for foldability. The minimal frustration principle explains how proteins can navigate vast conformational spaces efficiently, with funnels guiding the search for the native state.
Key insights from this paradigm include:
Evolutionary Optimization: Natural protein sequences represent exceptional instances of minimally frustrated heteropolymers, contrasting with the rugged landscapes of random sequences [1].
Landscape Topography Determines Mechanism: The specific topography of a protein's folding funnel dictates whether folding follows nucleation-collapse, diffusion-collision, or other mechanisms, with different proteins exhibiting varying degrees of landscape roughness [1] [3].
Thermodynamic and Kinetic Trade-offs: While minimal frustration generally promotes folding speed, some natural proteins may retain controlled frustration to enable functional dynamics or alloster regulation [2].
Implications for Design: Understanding funneled landscapes enables computational protein design, with recent methods successfully creating novel foldable sequences by optimizing for funneled landscapes [2].
The integration of theoretical concepts, high-throughput experiments, and advanced simulations continues to advance our understanding of protein folding, with applications in drug discovery, biomolecular engineering, and therapeutic development. As methods like cDNA display proteolysis and machine-learned force fields mature, we anticipate increasingly accurate predictions of folding stability and mechanisms for arbitrary protein sequences.
The Levinthal paradox, once considered a fundamental challenge to understanding protein folding, has been resolved through the framework of funneled energy landscapes. Natural proteins fold rapidly not despite the vast conformational space, but because their evolutionary-optimized landscapes direct the search for the native state. This paradigm, supported by both theoretical analysis and experimental evidence, has profound implications for computational structural biology, protein design, and therapeutic development.
Continued advances in high-throughput experimentation and machine learning approaches promise to further elucidate the quantitative principles governing folding landscapes, potentially enabling accurate prediction of folding pathways and stability for arbitrary protein sequences. This capability would transform fields from protein engineering to drug discovery, where understanding stability and folding is often critical for developing effective therapeutics.
The free energy landscape paradigm provides a powerful conceptual and quantitative framework for understanding protein folding, binding, and function. This technical guide details the core components of this framework: the order parameter Q, quantifying structural nativeness, and the free energy f(Q), defining the thermodynamic landscape. Within the broader thesis of energy landscape theory for protein folding simulations, we establish that the explicit construction and quantification of f(Q) enables researchers to distinguish between ordered and intrinsically disordered proteins, rationalize folding mechanisms, and predict folding kinetics. This whitepaper provides researchers, scientists, and drug development professionals with in-depth methodologies, quantitative benchmarks, and visualization tools essential for applying these concepts in computational and experimental research.
The free energy landscape is the cornerstone concept in modern protein folding theory, resolving the Levinthal paradox by positing that folding is not a random search but a directed process on a globally funneled energy surface [7]. This funneled topography ensures that as the protein approaches its native structure, its free energy decreases, biasing the conformational search toward the functional state [8] [9].
Quantifying this landscape requires moving beyond qualitative descriptions to precise, measurable definitions. This guide focuses on two central pillars of this quantification:
- The Order Parameter (Q): A reaction coordinate that measures progress along the folding reaction.
- The Free Energy (f): A configuration-dependent thermodynamic function that defines the landscape's topography.
The synergy between Q and f(Q) allows for the reduction of a high-dimensional conformational space into an interpretable and quantifiable landscape, enabling the prediction of folding stability, kinetics, and mechanisms.
Defining the Order Parameter Q
Conceptual Foundation and Mathematical Definition
The order parameter Q, the fraction of native contacts, is a structural metric that quantifies how similar a given protein configuration is to its experimentally determined native structure. It serves as a powerful reaction coordinate because it smoothly connects the unfolded state (low Q) to the folded state (high Q) [9] [7].
Mathematically, for any protein conformation, Q is defined as:
[ Q = \frac{\text{Number of native contacts present in the configuration}}{\text{Total number of native contacts in the native structure}} ]
A native contact is typically defined for a pair of amino acid residues that are within a specific cutoff distance (e.g., 6 Å) in the native state but are not adjacent in the protein sequence (separated by at least three other residues) [9]. The value of Q is normalized between 0 and 1, where Q ≈ 0 represents the fully unfolded ensemble and Q ≈ 1 represents the native state.
Computational Protocol and Best Practices
Protocol: Calculating Q from a Molecular Dynamics Trajectory
Generate the Native Contact Map:
- Using the native protein structure (e.g., from PDB), identify all pairs of non-bonded heavy atoms or Cα atoms from residues i and j where |i-j| > 3 and the interatomic distance is less than a chosen cutoff (e.g., 6.0 Å) [9].
- This list of atom pairs defines the total universe of native contacts, N_total.
Analyze Simulation Snapshots:
- For each saved configuration (r) from an MD simulation, check the same list of atom pairs.
- A native contact is considered "formed" if the distance between the atom pair is less than the same cutoff (or a slightly larger one to account for thermal fluctuations).
Compute Q for Each Snapshot:
- For a given configuration, count the number of native contacts that are formed, N_formed(r).
- Calculate ( Q(\mathbf{r}) = N{\text{formed}}(\mathbf{r}) / N{\text{total}} ).
Table 1: Quantitative Characteristics of Q for Model Proteins
| Protein | Structure | Folded State (Q) | Unfolded State (Q) | Transition State (Qts) |
|---|---|---|---|---|
| HP-35 | α-helical | ~0.89 [8] | ~0.20 [8] | Not Specified |
| WW Domain | β-sheet | High Q | Low Q | Not Specified |
| Generic Two-State Folder | Mixed | ~1.0 | ~0.0 | ~0.6 [9] |
The following diagram illustrates the workflow for calculating the order parameter Q and its role in characterizing protein states.
Defining the Free Energy f and the Landscape f(Q)
The Solvent-Averaged Effective Energy f
The true free energy landscape for a protein in solution is defined not by the gas-phase potential energy but by the solvent-averaged effective energy, denoted as f. This quantity is unambiguously defined for any individual protein conformation r as [8] [7]:
[ f(\mathbf{r}) = Eu(\mathbf{r}) + G{\text{solv}}(\mathbf{r}) ]
Here:
- ( E_u(\mathbf{r}) ) is the gas-phase potential energy of the protein conformation r, computed directly from the force field used in the simulation.
- ( G_{\text{solv}}(\mathbf{r}) ) is the solvation free energy for the protein conformation r, representing the free energy cost of transferring the protein from a vacuum into the solvent.
This definition explicitly averages over the solvent degrees of freedom, making f(r) a genuine thermodynamic free energy that governs the protein's statistical properties. The probability of observing a configuration r is proportional to ( \exp(-\beta f(\mathbf{r})) ), where ( \beta = 1/k_B T ) [7].
Distinction Between f(Q) and F(Q)
A critical distinction must be made between two different free energy quantities:
- ( f(Q) ) - The Free Energy Landscape: This is the average of ( f(\mathbf{r}) ) over all configurations sharing the same Q value, ( f(Q) = \langle f(\mathbf{r}) \rangle_{Q(\mathbf{r})=Q} ). It represents the internal energy + solvation free energy of the system at a specific Q and lacks configurational entropy. This landscape is expected to be globally funneled, showing a steady decrease in energy as Q increases [7].
- ( F(Q) ) - The Free Energy Profile: This is derived from the probability distribution ( P(Q) ) of the order parameter, ( F(Q) = -kB T \log P(Q) ). It includes the effects of configurational entropy (( S{\text{config}}(Q) )). The relationship is ( F(Q) = f(Q) - T S_{\text{config}}(Q) ) [7].
For a typical two-state folding protein, f(Q) displays a monotonic downhill slope, while F(Q) shows a characteristic double-well shape with a barrier separating the unfolded and folded states.
Protocol for Calculating f(Q)
Protocol: Constructing the Free Energy Landscape f(Q)
Perform Molecular Dynamics Sampling:
- Run an MD simulation (using GROMACS, AMBER, NAMD, etc.) that adequately samples the folded, unfolded, and transition states of the protein.
Compute f(r) for Sampled Configurations:
Compute Q(r) for the Same Configurations:
- Follow the protocol in Section 2.2.
Construct the f(Q) Landscape:
- Bin the data along the Q-axis (e.g., from 0 to 1 in steps of 0.01).
- For each bin, calculate the average value of ( f(\mathbf{r}) ) for all structures whose Q value falls within that bin.
- Plot the average ( f(Q) ) versus Q.
Table 2: Quantitative Free Energy Landscape Slopes for Selected Proteins
| Protein | Class | Landscape Slope (kcal/mol) | Functional Implication |
|---|---|---|---|
| HP-35 [7] | Ordered (α-helical) | ~ -50 | Steep funnel ensures fast, reliable folding. |
| WW Domain [7] | Ordered (β-sheet) | ~ -50 | Steep funnel ensures fast, reliable folding. |
| pKID (Isolated) [7] | Intrinsically Disordered | ~ -24 | Shallowness explains conformational disorder. |
| pKID (Bound to KIX) [7] | Upon Binding | ~ -54 | Partner binding creates a steep folding funnel. |
The diagram below illustrates the computational pipeline for constructing the free energy landscape f(Q) and its relationship to the more common free energy profile F(Q).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Free Energy Landscape Analysis
| Research Reagent | Function in Analysis | Exemplary Software/Packages |
|---|---|---|
| Specialized MD Hardware/Software | Generates high-dimensional folding/unfolding trajectory data. | Anton Supercomputer [8], GPU-accelerated GROMACS/NAMD [8] |
| Molecular Dynamics Force Fields | Provides parameters for calculating the gas-phase potential energy ( E_u(\mathbf{r}) ). | CHARMM, AMBER, OPLS |
| Implicit Solvation Models | Enables efficient calculation of the solvation free energy ( G_{\text{solv}}(\mathbf{r}) ) for each snapshot. | Integral-Equation Theory [8], Generalized Born (GB) models |
| Analysis & Clustering Tools | Computes order parameters (Q, RMSD, Rg) and performs dimensionality reduction and clustering. | MDTraj, LOOS, MDAnalysis, MSMBuilder |
| Markov State Model (MSM) Software | Constructs kinetic models from simulation data to interpret folding pathways and rates. | PyEMMA, MSMBuilder |
Advanced Applications: From Order Parameters to Kinetic Models
The combination of Q and f enables sophisticated analyses beyond static landscape characterization. The funneledness of the landscape can be directly assessed by plotting f versus Q, which for well-designed proteins shows a strong negative correlation (e.g., Pearson correlation R ≈ 0.86 for HP-35 [8]). This funneledness is a hallmark of minimally frustrated sequences evolved for rapid and reliable folding [9].
Furthermore, the discretization of conformational space using both geometric (Q) and thermodynamic (f) order parameters can lead to improved Markov State Models (MSMs) [8]. These models provide a quantitative understanding of folding kinetics and mechanisms. Advanced methods, such as time-lagged independent component analysis (TICA), can also be used to identify optimal linear combinations of order parameters (like Q and the total number of contacts) that best describe the slow folding dynamics, providing improved estimates of folding rates [10].
The rigorous definition and computational implementation of the order parameter Q and the free energy f(Q) are foundational to a quantitative protein folding free energy landscape theory. This guide has detailed the protocols for their calculation, distinguished key concepts, and provided quantitative benchmarks. The explicit construction of f(Q) empowers researchers to move beyond qualitative pictures to a numerical framework that can differentiate protein classes, predict the effects of mutations on stability and kinetics, and elucidate the mechanisms of coupled folding and binding. As simulation capabilities and analytical methods continue to advance, this framework will remain central to bridging the gap between atomistic simulations and experimental observables in fundamental research and drug development.
The concept of the free energy landscape is fundamental to understanding how proteins navigate their vast conformational space to achieve a unique, functional three-dimensional structure. This paradigm resolves Levinthal's paradox, which posited that a random conformational search would take astronomical timescales, by demonstrating that evolution has selected for sequences with "funneled" landscapes that make folding both rapid and reliable [1]. These landscapes are not random; for naturally occurring proteins, they are shaped by the principle of minimal frustration, meaning that the native structure is optimized to have consistently stabilizing interactions, unlike random heteropolymer sequences that are plagued by conflicting interactions ("frustration") leading to a rough, glassy energy landscape with many deep metastable minima [1].
The folding process can be described as a navigation across this free energy landscape. For ordered proteins, this landscape often resembles a smooth, steep funnel, guiding the protein efficiently toward its native state. In contrast, Intrinsically Disordered Proteins (IDPs) exhibit characteristically different landscapes, which are often shallower or even multifunneled, explaining their inherent flexibility and lack of a stable structure in the unbound state [11]. The quantitative study of these landscapes provides profound insights not only into basic protein biophysics but also into practical applications in drug discovery, where understanding and predicting binding affinities is paramount [12] [13].
Quantitative Comparison of Landscapes: Ordered Proteins vs. IDPs
The steepness of a folding funnel is a quantitative measure of the energetic bias driving a protein toward its native state. Recent advances in computational methods, employing fully atomistic molecular dynamics simulations and explicit characterization of free energy, allow for the direct calculation of this landscape slope [7].
Defining the Landscape Slope
The free energy landscape, ( f(\mathbf{r}) ), is defined for a protein configuration, ( \mathbf{r} ), as the sum of the gas-phase potential energy and the solvation free energy, ( f(\mathbf{r}) = Eu(\mathbf{r}) + G{solv}(\mathbf{r}) ) [7]. To visualize and quantify this high-dimensional landscape, it is reduced using an order parameter ( Q ), typically the fraction of native contacts. The resulting reduced landscape, ( f(Q) ), represents the average free energy of all configurations with a given ( Q ) value. The global slope of ( f(Q) ) from the unfolded (( Q \sim 0 )) to the native state (( Q \sim 1 )) provides a metric for the funnel's steepness and the folding drive [7].
Table 1: Measured Free Energy Landscape Slopes for Representative Proteins
| Protein | Structural Class | Native State | Landscape Slope (kcal/mol) |
|---|---|---|---|
| HP-35 | Ordered Protein | α-helical | ~ -50 [7] |
| WW Domain | Ordered Protein | β-sheet | ~ -50 [7] |
| pKID (free) | Intrinsically Disordered Protein (IDP) | Disordered | ~ -24 [7] |
| pKID (bound to KIX) | Coupled Folding and Binding | Folded upon binding | ~ -54 [7] |
Interpreting the Quantitative Data
As summarized in Table 1, the data reveals a clear distinction. Ordered proteins like HP-35 and the WW domain exhibit steep folding funnels with slopes of approximately -50 kcal/mol. This signifies that for every 10% of native contacts formed, the free energy of the system decreases by about 5 kcal/mol, creating a strong thermodynamic driving force for folding [7].
In stark contrast, the free form of the IDP pKID has a significantly shallower landscape with a slope of -24 kcal/mol. This reduced bias means that the energetic gain from forming native-like contacts is insufficient to overcome the chain's conformational entropy, resulting in a disordered ensemble under native conditions [7]. The dramatic transformation upon binding to its partner, KIX, is a key finding: the landscape becomes steep (-54 kcal/mol), demonstrating that intermolecular interactions, primarily from hydrophobic residues, provide the necessary energetic bias to guide the disordered chain into a unique, folded structure [7].
Methodologies for Characterizing Protein Folding Landscapes
Molecular Dynamics and Free Energy Landscape Construction
The explicit construction of a free energy landscape, as performed in the cited studies, involves a rigorous computational protocol [7]:
- Sampling: Perform extensive all-atom molecular dynamics (MD) simulations that encompass both the folded and unfolded states of the protein. For instance, the study on HP-35 utilized a ~400 μs folding-unfolding trajectory [7].
- Configuration Analysis: For each sampled configuration ( \mathbf{r} ), calculate the order parameter ( Q(\mathbf{r}) ) (e.g., fraction of native contacts) and the total free energy ( f(\mathbf{r}) = Eu(\mathbf{r}) + G{solv}(\mathbf{r}) ). The gas-phase energy ( Eu ) is derived from the force field, while the solvation free energy ( G{solv} ) can be computed using methods like the molecular integral-equation theory [7].
- Averaging and Landscape Generation: Average the free energy ( f(\mathbf{r}) ) over all configurations sharing the same ( Q ) value to obtain the reduced one-dimensional landscape, ( f(Q) ). This ( f(Q) ) versus ( Q ) plot quantitatively defines the funnel's characteristics.
It is critical to distinguish the free energy landscape ( f(Q) ) from the free energy profile ( F(Q) = -k_B T \ln P(Q) ), where ( P(Q) ) is the probability distribution from equilibrium sampling. While ( f(Q) ) shows a globally funneled slope, ( F(Q) ) for a two-state folder displays a characteristic barrier between the unfolded and folded minima [7].
Advanced Sampling and Free Energy Calculations
To overcome the limitations of standard MD in sampling rare events like folding or binding, advanced methods are employed in drug discovery and landscape studies:
- Alchemical Transformations: Methods like Free Energy Perturbation (FEP) and Thermodynamic Integration (TI) use a coupling parameter ( \lambda ) to interpolate between two states (e.g., a bound and unbound ligand). The free energy difference is calculated by integrating the derivative ( \langle \partial V\lambda / \partial \lambda \rangle\lambda ) along ( \lambda ) [12]. These are widely used for calculating relative binding free energies between similar compounds [12] [13].
- Path-Based Methods: These methods use collective variables (CVs) to describe the progression along a reaction pathway, such as binding. A key result is the potential of mean force (PMF), which is the free energy profile along the selected CV [12]. Sophisticated CVs like Path Collective Variables (PCVs) can map a protein-ligand binding process onto a curvilinear pathway for more accurate calculation of absolute binding free energies [12].
- Nonequilibrium Switching (NES): This emerging approach runs many short, independent, and bidirectional transitions between states far from equilibrium. The collective statistics from these fast, parallel simulations allow for efficient and scalable free energy calculations, offering a potential 5-10x higher throughput than traditional equilibrium methods [13].
The Scientist's Toolkit: Essential Reagents and Computational Methods
Table 2: Key Research Reagents and Computational Tools for Free Energy Landscape Studies
| Item / Method | Type | Primary Function in Research |
|---|---|---|
| All-Atom Molecular Dynamics (MD) | Computational Method | Simulates the physical movements of atoms over time, providing trajectories for analyzing folding pathways and constructing energy landscapes [7] [12]. |
| Molecular Integral-Equation Theory | Computational Solver | Calculates the solvation free energy ( G_{solv}(\mathbf{r}) ) for individual protein configurations, a critical component of the free energy landscape ( f(\mathbf{r}) ) [7]. |
| Collective Variables (CVs) | Computational Descriptor | Low-dimensional parameters (e.g., RMSD, native contacts, Path CVs) that describe the progress of a reaction; used in enhanced sampling to compute free energy landscapes [12]. |
| Path Collective Variables (PCVs) | Advanced CV Type | Defines a progression variable S(x) and a path deviation Z(x) relative to a pre-defined pathway, enabling efficient sampling of complex transitions like binding to flexible targets [12]. |
| Nonequilibrium Switching (NES) | Enhanced Sampling Method | Performs many fast, independent transitions between states to calculate free energy differences with high throughput and scalability, ideal for drug candidate ranking [13]. |
| Free Energy Perturbation (FEP) | Alchemical Method | Computes free energy differences between two states by sampling using a hybrid Hamiltonian; widely used for relative binding free energy calculations in lead optimization [12] [13]. |
| Potential of Mean Force (PMF) | Computational Result | The free energy profile along a chosen collective variable; the key output of path-based simulations that reveals barriers and stable states along a reaction pathway [12]. |
Experimental Protocols for Key Studies
Protocol: Quantifying Landscape Slopes for Ordered and Disordered Proteins
This protocol is derived from the methodology used to generate the quantitative data in Table 1 [7].
- System Preparation: Construct the initial atomic coordinates for the protein of interest (e.g., HP-35, WW domain, pKID) in a simulation box. Solvate the protein with explicit water molecules and add ions to neutralize the system.
- Molecular Dynamics Simulation: Run a long-timescale, all-atom MD simulation using a force field (e.g., CHARMM, AMBER) at physiological temperature and pressure. The simulation must be sufficiently long to capture multiple folding and unfolding events. For pKID-KIX binding, simulate the complex.
- Trajectory Analysis:
- For each saved configuration: Calculate the fraction of native contacts ( Q ).
- For each saved configuration: Compute the free energy ( f(\mathbf{r}) = Eu(\mathbf{r}) + G{solv}(\mathbf{r}) ). ( Eu ) is obtained from the force field's potential energy function. ( G{solv} ) is computed using a method like the 3D-RISM molecular integral-equation theory.
- Construct ( f(Q) ): Group configurations by their ( Q ) value (e.g., in bins of 0.01). For each bin, calculate the average ( f ). Plot the average ( f ) versus ( Q ) to generate the reduced one-dimensional landscape.
- Calculate Slope: Perform a linear regression on the ( f(Q) ) versus ( Q ) plot over the range Q = 0 to Q = 1. The slope of this linear fit is reported as the quantitative measure of the landscape's steepness.
Protocol: Semiautomatic Pipeline for Binding Free Energy Using Path CVs
This protocol outlines a modern path-based approach for calculating absolute binding free energies, relevant for studying IDP folding upon binding [12].
- Define End-States: Identify the initial (unbound, disordered IDP and separate partner) and final (bound, folded complex) states.
- Generate a Reference Path: Create a discrete set of configurations (a "string") that connects the unbound and bound states. This can be done through preliminary steering MD simulations or by interpolation.
- Set Up Path Collective Variables: Define the PCVs, S(x) and Z(x), using the reference path. S(x) measures progress along the path, and Z(x) measures the deviation from it.
- Run Enhanced Sampling Simulation: Perform a meta-dynamics or other biased simulation using S(x) as the collective variable. This biases the simulation to explore the entire binding pathway and efficiently overcome barriers.
- Compute Potential of Mean Force and ( \Delta Gb ): Re-weight the simulation data to obtain the PMF along S(x). The difference in the PMF between the bound and unbound states provides an estimate of the standard binding free energy, ( \Delta Gb ).
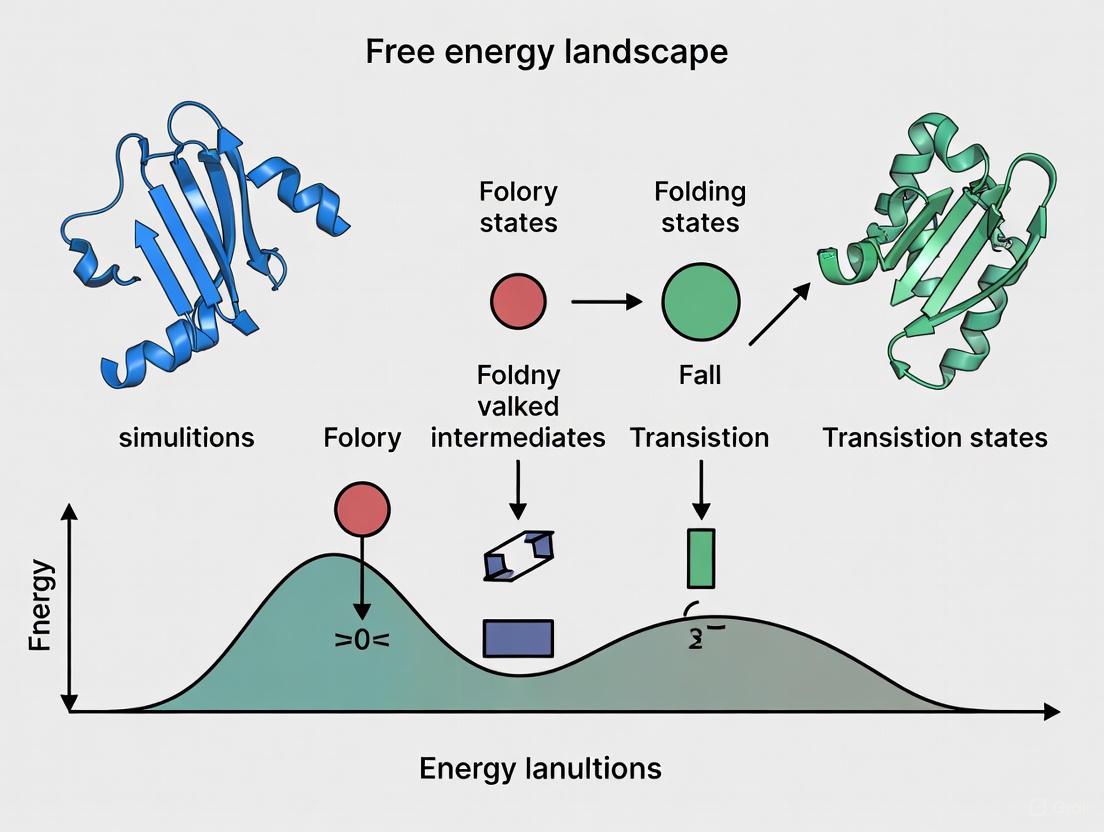
The proper folding of proteins into their unique three-dimensional structures is fundamental to biological function. The energy landscape theory provides a powerful conceptual framework for understanding this process, visualizing protein folding as a funnel-shaped journey from a high-entropy unfolded state to a low-energy native conformation [14] [15]. This landscape encodes the relative stabilities of different states—native, partially folded intermediates, unfolded, misfolded, and aggregated—as well as the energy barriers separating them [14]. In a perfectly funneled landscape, the native state is reached efficiently because stabilizing contacts are mutually supportive and "minimally frustrated" [14].
However, the landscape for protein folding is often rugged, with kinetic traps that can lead to misfolding and aggregation. These off-pathway events are particularly relevant to human diseases, as the accumulation of misfolded proteins into insoluble amyloid fibrils is a hallmark of numerous neurodegenerative disorders, including Alzheimer's, Parkinson's, and prion diseases [14] [15]. The misfolding and aggregation landscape is characterized by multiple competing conformations separated by substantial kinetic barriers, making it significantly rougher than the landscape for native folding [14]. Understanding the topological features of these landscapes—the arrangement of energy basins, barriers, and pathways—is therefore crucial for elucidating the molecular mechanisms of protein misfolding diseases and developing therapeutic strategies.
Fundamental Principles of Protein Energy Landscapes
Theoretical Foundations and Historical Development
The conceptual foundation of protein folding was established by Anfinsen's thermodynamic hypothesis, which demonstrated that the native structure represents the most thermodynamically stable conformation under physiological conditions and is determined solely by the amino acid sequence [15]. This principle, however, left unresolved questions about folding pathways, highlighted by Levinthal's paradox which noted the impossibility of random conformational sampling [15].
The development of energy landscape theory in the 1990s addressed this paradox by framing folding as a funnel-guided process where native states occupy energy minima [15]. According to this view, the ruggedness of the folding landscape—arising from partially folded states and misfolded conformations—accounts for the heterogeneity and complexity observed in folding pathways [15]. The funnel analogy illustrates how high-energy, high-entropy unfolded states at the top of the funnel fold along various paths down to the low-energy, low-entropy native conformation [14].
Contrasting Native Folding and Misfolding Landscapes
A critical insight from landscape theory is the fundamental difference between landscapes for native folding versus misfolding and aggregation. While native folding follows a minimally frustrated landscape toward a single stable state, aggregation landscapes typically feature multiple competing pathways with significant kinetic barriers [14].
Table 1: Key Differences Between Native Folding and Misfolding Landscapes
| Characteristic | Native Folding Landscape | Misfolding/Aggregation Landscape |
|---|---|---|
| Topology | Smooth, funneled | Rough, multi-funneled |
| Pathways | Convergent, directed | Divergent, competing |
| Stabilizing Interactions | Native contacts, minimally frustrated | Non-native contacts, frustrated |
| Kinetic Barriers | Generally lower | Substantial, rate-limiting |
| Final State | Unique native conformation | Multiple aggregate polymorphs |
This distinction explains why misfolding and aggregation are complex processes involving rare, transient intermediates and multiple pathways [14]. In the case of prion protein (PrP), for instance, the native structure may be kinetically trapped by a ~20 kcal/mol barrier separating it from a more thermodynamically stable, β-rich oligomeric state [14].
Experimental Methods for Mapping Energy Landscapes
Single-Molecule Force Spectroscopy
Single-molecule techniques, particularly force spectroscopy, have revolutionized the study of protein energy landscapes by enabling observation of rare transient events and heterogeneous pathways that are obscured in ensemble measurements [14]. These methods capture the statistical mechanics of structural fluctuations, allowing researchers to measure energy landscapes in unprecedented detail [14].
Experimental Protocol: Single-molecule force spectroscopy (SMFS) using optical tweezers:
- Protein immobilization: PrP monomers or dimers are covalently tethered between microspheres [14]
- Force application: Dual-beam optical tweezers apply precisely controlled forces [14]
- Data collection: Force-extension curves are generated by ramping force up and down [14]
- Transition identification: Abrupt changes ("rips") in force-extension curves indicate structural transitions [14]
- Landscape reconstruction: Statistical analysis of transitions reveals energy barriers and intermediate states [14]
In studies of prion protein (PrP), this approach revealed that isolated monomers frequently sample various misfolded conformations off the native folding pathway, though none were thermodynamically stable [14]. However, when studying PrP dimers—the smallest form of oligomer—researchers observed stable misfolded structures involving approximately 240 amino acids, compared to 104 in the monomer structure [14]. This demonstrates how intermolecular interactions stabilize misfolded states that are unstable in monomers.
Kinetic Analysis of Folding Pathways
Stopped-flow kinetic experiments provide crucial information about folding mechanisms and the presence of intermediates. Recent studies of engineered metamorphic proteins (B4 and Sb3) that share highly similar sequences but adopt distinct topologies reveal how topology dictates folding mechanisms at an early stage [16].
Experimental Protocol: Stopped-flow (un)folding kinetics:
- Rapid dilution: Protein is rapidly diluted into solutions containing varying denaturant concentrations (e.g., GdnHCl) [16]
- Fluorescence monitoring: Time course of emission fluorescence is recorded [16]
- Data fitting: Observed rates (k˅obs) are fitted to exponential equations [16]
- Chevron plot analysis: Logarithm of k˅obs plotted against denaturant concentration [16]
- Mechanism determination: Curve shape (V-shaped vs. roll-over) reveals two-state versus three-state folding [16]
For metamorphic proteins B4 and Sb3, which share nearly identical sequences but different topologies, kinetic analyses revealed that B4 follows a two-state folding mechanism, while Sb3 involves formation of an intermediate species [16]. This demonstrates that folding landscapes are primarily shaped by final native structures rather than sequence composition alone [16].
Table 2: Kinetic Parameters for Metamorphic Proteins B4 and Sb3
| Parameter | B4 Protein | Sb3 Protein |
|---|---|---|
| Folding Mechanism | Two-state | Three-state with intermediate |
| Unfolding Rate (kᵤ) | 35.9 ± 1.4 s⁻¹ | 12.4 ± 1.2 s⁻¹ |
| Stabilization Response | V-shaped chevron retained with 0.3M Na₂SO₄ | Roll-over effect revealing intermediate with 0.3M Na₂SO₄ |
| pH Sensitivity | Significant outside pH 4.5-7.0 | Minimal sensitivity to pH changes |
| Parent Protein | GB1 (ku = 0.57 ± 0.02 s⁻¹) | S6 (ku = 0.0003 ± 0.00004 s⁻¹) |
Computational Approaches to Landscape Characterization
Machine-Learned Coarse-Grained Models
Recent advances in deep learning have enabled the development of transferable coarse-grained (CG) models that capture the essential physics of protein folding while being computationally efficient. These models combine deep-learning methods with large training sets of all-atom protein simulations to create force fields with chemical transferability, capable of extrapolative molecular dynamics on new sequences [4].
Methodology: Machine-learned coarse-grained force field development:
- Training data generation: All-atom explicit solvent simulations of small proteins with diverse folded structures [4]
- Model architecture: CGSchNet framework using neural networks to represent many-body interactions [4]
- Parameter optimization: Variational force-matching approach to reproduce all-atom distributions [4]
- Validation: Testing on unseen proteins with low sequence similarity (<40%) to training set [4]
- Simulation: Parallel-tempering and Langevin dynamics for converged sampling [4]
This approach has successfully predicted metastable states of folded, unfolded, and intermediate structures, fluctuations of intrinsically disordered proteins, and relative folding free energies of protein mutants, while being several orders of magnitude faster than all-atom models [4]. The model accurately reproduces folding landscapes for various test proteins, including chignolin, TRPcage, BBA, and villin headpiece [4].
Nested Sampling for Rugged Landscapes
Nested sampling is a Bayesian technique designed to explore probability distributions where the posterior mass is localized in an exponentially small area of parameter space, making it particularly suited for protein folding landscapes characterized by first-order phase transitions [17].
Algorithm: Parallel Nested Sampling:
- Initialization: Sample K points ({θ₁...θₖ}) uniformly from the prior distribution [17]
- Likelihood calculation: Compute likelihoods {L(θ₁),...,L(θₖ)} for all points [17]
- Point removal: Identify and remove the point with smallest likelihood, saving it as (L₁, X₁) [17]
- Point replacement: Generate a new point sampled uniformly from {θ: L(θ) > L₁} [17]
- Iteration: Repeat steps 3-4, generating (L₂, X₂), (L₃, X₃), etc. [17]
- Evidence calculation: Estimate evidence Z = ∫₀¹ L(X)dX via numerical integration [17]
This approach allows efficient calculation of free energies and thermodynamic observables at any temperature through simple post-processing of the output, without requiring new simulations at each temperature [17]. When applied to protein folding with Gō-like force fields, it has yielded large efficiency gains over other sampling techniques, including parallel tempering [17].
The Role of Landscapes in Misfolding Diseases
Prion Protein Misfolding
The prion protein (PrP) provides a compelling example of how landscape topology influences disease. The cellular isoform (PrPᶜ) can undergo conformational conversion to a pathological scrapie isoform (PrPˢᶜ) that aggregates and causes neurodegenerative diseases [14]. Single-molecule studies reveal that while isolated PrP monomers sample various misfolded conformations, none are thermodynamically stable, consistent with the view that misfolded PrP is stable only within aggregates [14].
The energy landscape for PrP features a kinetic trap—a ~20 kcal/mol barrier separating the native state from a more thermodynamically stable, β-rich oligomeric state [14]. Mutations or template aggregates (PrPˢᶜ) may enhance aggregation by stabilizing the misfolding transition state, suggesting therapeutic approaches that block binding to PrPˢᶜ or kinetically trap intermediates [14].
Therapeutic Strategies Targeting Energy Landscapes
Understanding energy landscape topology enables novel therapeutic approaches for misfolding diseases:
- Kinetic stabilizers: Small molecules that increase barriers to unfolding or misfolding [14]
- Transition-state analogs: Compounds that block the binding to pathological aggregates like PrPˢᶜ [14]
- Proteostasis network modulators: Agents that enhance cellular folding capacity or clearance of misfolded proteins [15]
- Chaperone modulators: Drugs that regulate molecular chaperones to assist proper folding or prevent aggregation [15]
These strategies aim to manipulate the energy landscape to favor native folding over pathological aggregation, either by stabilizing native states, increasing barriers to misfolding, or redirecting misfolded species to productive pathways [14] [15].
Research Toolkit: Essential Methods and Reagents
Table 3: Research Reagent Solutions for Protein Folding Studies
| Reagent/Resource | Function/Application | Technical Specifications |
|---|---|---|
| PrP Dimers (Tethered) | Study early aggregation events | Covalently linked monomers; increases effective local concentration [14] |
| Engineered Metamorphic Proteins (B4, Sb3) | Compare folding mechanisms with minimal sequence differences | Highly similar sequences (differ only at position 5) but distinct topologies [16] |
| Guanidine Hydrochloride (GdnHCl) | Chemical denaturant for folding/unfolding experiments | Varying concentrations to modulate protein stability; used in stopped-flow kinetics [16] |
| Sodium Sulfate (Na₂SO₄) | Stabilizing salt for marginal stability proteins | 0.3 M concentration to reveal hidden intermediates [16] |
| Coarse-Grained Model (CGSchNet) | Machine-learned force field for efficient simulation | Trained on diverse all-atom simulations; transferable to new sequences [4] |
The energy landscape perspective provides a unifying framework for understanding how proteins navigate from native folding to pathological aggregation. Key insights emerge from this approach: (1) Landscape topology—dictated more by native structure than sequence alone—determines folding mechanisms and susceptibility to misfolding; (2) Kinetic barriers play a crucial role in protecting against aggregation, as the aggregated form is often thermodynamically more stable but kinetically inaccessible; and (3) Experimental and computational advances—from single-molecule spectroscopy to machine-learned coarse-grained models—are providing unprecedented resolution of these landscapes.
For therapeutic development, targeting the topological features of energy landscapes offers promising strategies for misfolding diseases. By mapping the precise contours of these landscapes—identifying critical intermediates, transition states, and barriers—researchers can design interventions that kinetically stabilize native folds, block pathological transitions, or redirect misfolding trajectories toward harmless outcomes. As these approaches mature, energy landscape theory will continue to bridge fundamental biophysical principles with therapeutic innovation for protein misfolding diseases.
Simulation Tools and Techniques: From All-Atom MD to Machine-Learned Coarse-Graining
All-atom molecular dynamics (MD) simulations represent the gold standard for investigating protein folding and biomolecular dynamics at atomic resolution. By explicitly representing every atom in a system and employing physics-based force fields, all-atom MD provides unparalleled insights into folding pathways, intermediate states, and the underlying free energy landscapes that govern protein behavior. However, this methodological rigor comes with extraordinary computational costs that severely limit accessible timescales and system sizes, presenting a fundamental challenge for studying biologically relevant folding processes. This technical review examines the theoretical foundations, current capabilities, and computational burdens of all-atom MD within the context of free energy landscape theory for protein folding, while exploring emerging methodologies that seek to overcome these limitations through enhanced sampling algorithms and machine learning approaches.
All-atom molecular dynamics has established itself as the premier computational technique for studying protein folding dynamics with full atomic detail. Unlike simplified or coarse-grained models that reduce computational demands through abstraction, all-atom MD simulations explicitly represent every atom in the protein-solvent system and numerically integrate Newton's equations of motion using detailed empirical force fields [18]. This approach provides several fundamental advantages: it captures specific atomic interactions including hydrogen bonding, solvation effects, and side-chain packing; enables direct comparison with experimental structural biology techniques; and offers the potential to predict how mutations and small molecules impact folding pathways and stability [19] [20].
The theoretical framework for understanding protein folding has been profoundly shaped by the energy landscape theory and the principle of minimal frustration, which posits that naturally occurring protein sequences have evolved to have landscapes biased toward the native structure [19]. Within this framework, all-atom MD serves as a crucial computational microscope for characterizing the detailed topography of these folding landscapes, including the presence and properties of folding intermediates, transition states, and alternative misfolded states [19]. The combination of all-atom simulation with free energy landscape theory has revolutionized our understanding of how proteins navigate their conformational space to achieve functional native states.
Methodological Foundations: Force Fields and Sampling Algorithms
Modern Force Field Families and Their Parameterization
The accuracy of all-atom MD simulations fundamentally depends on the quality of the force field—the mathematical representation of interatomic interactions. Current force field development focuses on achieving a balanced description of diverse protein states, from folded domains to intrinsically disordered regions, while maintaining thermodynamic transferability [20]. The four major force field families—AMBER, CHARMM, OPLS, and GROMOS—have undergone extensive parameterization against experimental data (crystallography, spectroscopy) and ab initio quantum mechanical calculations [20].
Recent refinements have addressed critical limitations in modeling protein-water interactions and backbone torsional sampling. For instance, the amber ff03w-sc variant incorporates selective upscaling of protein-water interactions, while ff99SBws-STQ′ includes targeted improvements to glutamine torsional parameters to correct overestimated helicity in polyglutamine tracts [20]. These developments highlight the ongoing effort to balance the stabilization of folded proteins with accurate description of disordered ensembles, a central challenge in force field design [20].
Table 1: Major Force Field Families and Their Key Characteristics
| Force Field Family | Key Strengths | Common Pairings | Recent Refinements |
|---|---|---|---|
| AMBER | Excellent for proteins and nucleic acids; widely used in academic research | TIP3P, TIP4P water models | ff03w-sc, ff99SBws-STQ′ with improved protein-water balance [20] |
| CHARMM | Balanced treatment of lipids, proteins, and carbohydrates | Modified TIP3P with additional LJ parameters on hydrogen atoms | charmm36m with enhanced IDP description [20] |
| OPLS | Strong emphasis on liquid state properties and solvation | TIP4P water models | Continued parameter optimization for protein-ligand systems |
| GROMOS | Unified atom approach reduces computational cost | SPC water model | Parameter sets optimized for specific biomolecular classes |
Enhanced Sampling Techniques for Free Energy Landscape Characterization
Due to the high energy barriers separating conformational states in protein folding, standard MD simulations often become trapped in local minima, making comprehensive sampling of free energy landscapes computationally prohibitive. Numerous enhanced sampling methods have been developed to address this limitation:
Replica Exchange Molecular Dynamics (REMD) utilizes multiple parallel simulations at different temperatures, allowing periodic exchange of configurations between replicas to overcome energy barriers more efficiently [19]. Metadynamics employs a history-dependent bias potential that discourages the system from revisiting previously sampled states, effectively filling free energy basins to promote exploration of new regions [19]. Accelerated MD modifies the potential energy surface to reduce barrier heights while preserving the relative stability of low-energy states [19].
These techniques, when combined with careful choice of collective variables that capture the essential motions of folding, enable the construction of detailed free energy landscapes from which folding mechanisms, intermediate states, and kinetic properties can be derived [19].
Computational Costs: The Fundamental Limitation of All-Atom MD
Quantitative Assessment of Computational Demands
The computational expense of all-atom MD simulations scales dramatically with both system size and simulation timescale, creating fundamental limitations for studying large proteins and slow folding processes. Specialized supercomputers like ANTON have pushed the boundaries of what is feasible, enabling millisecond-scale simulations for small proteins (<100 amino acids) [19]. However, the median protein length in eukaryotes is 532 amino acids [19], with folding times ranging from microseconds to tens of minutes—far beyond the reach of current all-atom MD for most systems of biological interest.
Table 2: Computational Requirements for All-Atom MD Protein Folding Simulations
| System Size (Residues) | Typical Atom Count | Simulation Timescale | Computational Resources Required | Limitations and Challenges |
|---|---|---|---|---|
| Small Proteins (<100 aa) | ~10,000-50,000 atoms | Microseconds to milliseconds | Specialized hardware (ANTON), distributed computing (Folding@home) [19] | Limited to fast-folding proteins; insufficient for proteins with slow folding intermediates |
| Medium Proteins (100-300 aa) | ~50,000-200,000 atoms | Nanoseconds to microseconds | High-performance computing clusters; GPU-accelerated MD | Often unable to observe complete folding events; limited sampling of rare transitions |
| Large Multidomain Proteins (>300 aa) | >200,000 atoms | Nanoseconds | Leadership-class supercomputing facilities | Primarily limited to stability and local dynamics of already-folded states; cannot simulate folding from unfolded states |
| Virus Capsids & Complexes | Millions of atoms | Nanoseconds | Exascale computing resources [21] | Focused on global dynamics and assembly rather than folding pathways |
Case Studies: Computational Costs in Practice
Recent simulations illustrate both the capabilities and limitations of all-atom MD. Studies of folded protein stability using refined force fields like ff99SBws have demonstrated the ability to maintain structural integrity of systems like Ubiquitin and Villin HP35 over microsecond timescales [20]. However, even these extensive simulations represent only a fraction of the complete folding process for many proteins. All-atom simulations of intact virus capsids, comprising millions of atoms, have provided unprecedented insights into collective motions and assembly principles but remain restricted to nanosecond timescales that capture local fluctuations rather than large-scale conformational changes [21].
The computational burden stems from multiple factors: the need for femtosecond-level integration steps to capture atomic vibrations, the calculation of pairwise interactions between thousands of atoms, and the explicit representation of solvent molecules that typically constitute the majority of atoms in the system. These factors collectively make all-atom MD simulations of protein folding among the most computationally demanding tasks in modern scientific computing.
Emerging Solutions: Bridging the Gap Between Accuracy and Feasibility
Machine Learning Approaches and Generative Models
Recent advances in machine learning offer promising pathways to overcome the timescale limitations of conventional all-atom MD. The BioMD framework represents a groundbreaking approach as the first all-atom generative model for simulating long-timescale protein-ligand dynamics [22]. Using a hierarchical framework of forecasting and interpolation, BioMD successfully generates ligand unbinding paths for 97.1% of protein-ligand systems within ten attempts, demonstrating the ability to explore critical pathways that would be prohibitively expensive with conventional MD [22]. These machine learning approaches learn the essential dynamics from existing simulation data and experimental constraints, enabling the generation of physically plausible trajectories at a fraction of the computational cost.
Multiscale and Hybrid Modeling Strategies
Integrating all-atom MD with coarse-grained models provides another powerful strategy for balancing atomic detail with computational efficiency. Coarse-grained models reduce complexity by representing multiple atoms with single interaction sites, dramatically decreasing the number of degrees of freedom [18]. The MARTINI force field and UNICORN model of biological macromolecules are examples of coarse-grained approaches that can access microsecond to millisecond timescales for large systems [18]. Hybrid methodologies that combine all-atom resolution in regions of interest with coarse-grained representations elsewhere enable targeted investigation of specific folding events while maintaining manageable computational demands.
Diagram 1: Multiscale computational approaches to protein folding. Selection of methodology depends on the specific research question, system size, and required temporal resolution.
Experimental Protocols: Best Practices for All-Atom Folding Simulations
Standard Protocol for Folded Protein Stability Assessment
Based on recent force field validation studies [20], the following protocol represents current best practices for assessing protein stability using all-atom MD:
System Preparation: Obtain initial coordinates from experimental structures (e.g., PDB IDs: 1D3Z for Ubiquitin, 2F4K for Villin HP35). Add missing hydrogen atoms using standard protonation states at physiological pH.
Solvation and Ion Addition: Place the protein in an appropriate water box (minimum 10Å padding from protein surface). Add ions to neutralize system charge and achieve physiological concentration (e.g., 150mM NaCl).
Energy Minimization: Perform steepest descent minimization (5,000 steps) to remove steric clashes and bad contacts.
Equilibration: Conduct gradual heating from 0K to target temperature (e.g., 300K) over 100ps with position restraints on protein heavy atoms (force constant of 1000 kJ/mol/nm²). Follow with pressure equilibration (1 bar) for 100ps with maintained restraints.
Production Simulation: Run unrestrained MD for multiple independent replicas (minimum 2.5μs each) using a modern balanced force field (e.g., ff99SBws or ff03w-sc) paired with an appropriate water model (TIP4P2005 for ff99SBws).
Analysis: Calculate backbone RMSD, radius of gyration, and per-residue RMSF. Monitor secondary structure evolution (DSSP) and native contact formation (Q fraction).
Key Research Reagents and Computational Tools
Table 3: Essential Research Tools for All-Atom MD Protein Folding Studies
| Tool Category | Specific Examples | Primary Function | Application in Protein Folding |
|---|---|---|---|
| Force Fields | AMBER ff99SBws, ff03w-sc, CHARMM36m | Define interatomic potentials | Determine accuracy in modeling folded stability and disordered ensembles [20] |
| Water Models | TIP3P, TIP4P2005, OPC | Represent solvent interactions | Critical for balancing protein-water and protein-protein interactions [20] |
| Specialized Hardware | ANTON supercomputer, GPU clusters | Enable long timescales | Facilitate microsecond-millisecond simulations of folding [19] |
| Sampling Algorithms | Replica Exchange MD, Metadynamics | Enhance conformational sampling | Overcome energy barriers to explore free energy landscapes [19] |
| Analysis Software | MDTraj, VMD, CPPTRAJ | Process trajectory data | Quantify order parameters, structural metrics, and free energies |
All-atom molecular dynamics remains the gold standard for computational studies of protein folding due to its atomic resolution and rigorous physical foundations. However, the extraordinary computational costs associated with this approach present fundamental limitations that restrict its application to small proteins and relatively short timescales. The ongoing development of balanced force fields that accurately capture both folded and disordered states, combined with emerging machine learning approaches like BioMD [22], promises to expand the accessible territory of all-atom simulations. Furthermore, multiscale strategies that strategically combine all-atom detail with coarse-grained models offer a practical pathway to study large biomolecular systems while retaining essential atomic-level information.
As hardware advances continue with exascale computing coming online [21], and algorithmic innovations improve sampling efficiency, all-atom MD will increasingly bridge the gap between theoretical free energy landscape concepts and experimental observations of protein folding. This convergence will enhance our fundamental understanding of folding mechanisms while strengthening the predictive power of computational methods for addressing biomedical challenges including misfolding diseases and rational drug design.
The Rise of Machine-Learned Coarse-Grained (CG) Models for Transferable and Fast Sampling
The most popular and universally predictive protein simulation models employ all-atom molecular dynamics (MD), but they come at extreme computational cost, creating a significant bottleneck for studying biologically relevant phenomena at appropriate time and length scales. The development of a universal, computationally efficient coarse-grained (CG) model with similar prediction performance has represented a long-standing challenge in computational biophysics for over five decades [4] [23]. While traditional CG approaches have shown success in specific systems, they have consistently fallen short of achieving the chemical transferability necessary to predict conformational landscapes of arbitrary proteins not used during parameterization [23]. The core challenge has centered on efficiently modeling the multi-body interaction terms essential for realistically representing protein thermodynamics and implicit solvation effects [4].
Recent advances in deep learning methods have fundamentally changed this landscape. By combining these methods with large and diverse training sets of all-atom protein simulations, researchers have now developed bottom-up CG force fields with genuine chemical transferability [4]. These models can be used for extrapolative molecular dynamics on new sequences not used during model parameterization, successfully predicting metastable states of folded, unfolded and intermediate structures, the fluctuations of intrinsically disordered proteins, and relative folding free energies of protein mutants—all while being several orders of magnitude faster than all-atom models [4] [24]. This breakthrough showcases the feasibility of a universal and computationally efficient machine-learned CG model for proteins, opening new possibilities for applications in drug discovery and protein engineering [24].
Theoretical Foundation: Energy Landscape Theory and CG Modeling
The Energy Landscape Perspective
Energy landscape theory provides the fundamental framework for understanding protein folding and dynamics. According to this theory, the spontaneous folding of monomeric globular proteins represents the simplest form of biological self-organization, generally involving only one molecule at a time working without sophisticated biological machinery [25]. The Principle of Minimal Frustration states that natural protein sequences have evolved to have energy landscapes that are smoothly "funneled" toward the native state, with native interactions being sufficiently strong compared to non-native interactions [25]. This funneled landscape contrasts sharply with the rugged landscapes of typical random heteropolymers, where the global ground state is nearly degenerate with other very different structures [25].
This theoretical framework justifies the search for simplified CG models that can capture the essential features of these funneled landscapes. The goal is to create models that maintain the thermodynamic consistency of all-atom models while dramatically increasing computational efficiency. From a statistical mechanics perspective, bottom-up coarse-graining aims to model the effective CG free energy surface defined by:
U(R) = -β^(-1) ln ∫ δ(R - M(r)) exp(-βu(r)) dr [23]
where M maps all-atom configurations r to their CG counterparts R = M(r), u is the reference all-atom energy, and β is the inverse temperature [23]. Intuitively, this equation defines how particles at the CG resolution should interact to preserve the thermodynamics of the original all-atom system.
The Coarse-Graining Challenge
The central challenge in CG modeling lies in the fact that when atomistic details are integrated out, the resulting CG representation naturally gives rise to multi-body interactions between the CG beads [26]. These interactions are not straightforward to model using physical intuition or traditional force field parameterization approaches. While classical all-atom force fields can model most non-bonded interactions as a sum of two-body terms, CG force fields require more complex multi-body terms to correctly represent protein thermodynamics and implicit solvation effects [4].
Previous CG models have exhibited significant limitations in this regard. Structure-based models rely on known native structures to explore free energy landscapes [4]. The Martini force field effectively models intermolecular interactions but inaccurately models intramolecular protein dynamics [4]. Other approaches like UNRES or AWSEM often fail to capture alternative metastable states [4]. The inability of these models to achieve true chemical transferability has hindered their widespread adoption for predictive simulations of novel protein sequences.
Machine Learning Framework for CG Models
Neural Network Architectures for CG Force Fields
The CGSchNet model represents a breakthrough in applying graph neural networks to learn the effective interactions between particles in coarse-grained protein simulations [24]. This architecture is particularly well-suited to molecular systems because it can naturally handle the graph-like structure of molecules, where nodes represent CG sites and edges represent their interactions. The model is trained to learn a CG potential U(x; θ) parameterized by θ that minimizes the force-matching loss function:
L(R; θ) = 1/(3nM) ∑_(c=1)^M ‖ΞF(r_c) + ∇U(Ξr_c; θ)‖² [27]
where Ξ is the mapping from all-atom to CG coordinates, F(r_c) are the all-atom forces, and the sum is over M configurations in the training dataset [27]. This variational force-matching approach ensures that the CG model reproduces the forces of the reference all-atom system.
To reduce the conformational space that must be learned and prevent poor exploration during CG simulation, the model incorporates a prior energy function that enforces basic physics constraints [27]. This prior typically includes bonded and repulsive terms to prevent chain rupture and bead clashes, along with dihedral terms to enforce proper chirality and prevent mirror images of native structures [27]. The neural network thus performs delta-learning between the all-atom forces and the prior forces, significantly reducing the complexity of the learning problem.
Training Data and Model Transferability
A critical factor in achieving transferability is the composition of the training dataset. The CGSchNet model was trained on a large and diverse set of all-atom explicit solvent simulations of small proteins with diverse folded structures, as well as many combinations of dimers of mono- and dipeptides [4]. This diverse training enables the model to learn generalizable interactions rather than memorizing specific sequences or folds.
The model demonstrates remarkable transferability by successfully simulating proteins with low (16-40%) sequence similarity to any sequence in the training or validation datasets [4]. This represents a significant advancement over previous CG models, which typically required homologous sequences in the training set to produce accurate results. The ability to extrapolate to novel sequences suggests that the model has learned fundamental physical principles of protein interactions rather than merely memorizing training examples.
Table 1: Key Components of Machine-Learned CG Force Fields
| Component | Function | Implementation in CGSchNet |
|---|---|---|
| Mapping Function | Defines how atomistic coordinates are reduced to CG sites | Linear mapping Ξ to Cα atoms or other backbone coordinates |
| Prior Potential | Enforces basic physical constraints and reduces conformational space | Bonded, repulsive, and dihedral terms to prevent chain rupture and maintain chirality |
| Neural Network Potential | Learns multi-body interactions from atomistic data | Graph neural network (SchNet architecture) trained via variational force-matching |
| Training Data | Provides reference for learning effective interactions | Diverse set of all-atom MD simulations of proteins with various folds |
Experimental Framework and Validation
Model Training and Simulation Protocol
The development of a transferable CG model requires careful attention to training methodologies and simulation protocols. The training process begins with the collection of extensive all-atom reference data. For example, in related work, researchers built a unique dataset of unbiased all-atom MD simulations totaling approximately 9 milliseconds for twelve different proteins with diverse secondary structure elements [27]. This dataset included proteins ranging from 10 to 80 amino acids with α-helical, β-sheet, and mixed α/β structures.
Once trained, the CG model can be used for molecular dynamics simulations using Langevin dynamics or advanced sampling techniques like parallel tempering to ensure converged sampling of the equilibrium distribution [4]. These simulations typically use a coarse-grained representation where each amino acid is represented by one or a few beads, dramatically reducing the number of degrees of freedom compared to all-atom representations.
Diagram 1: Workflow for Training Machine-Learned Coarse-Grained Force Fields. The process begins with all-atom MD simulations, which are mapped to CG representations to generate reference forces. A neural network potential is then trained via force matching to create a transferable CG force field for efficient MD simulations.
Validation Metrics and Performance
The performance of machine-learned CG models is validated using multiple metrics that assess both structural and thermodynamic accuracy. Key validation metrics include:
- Free energy surfaces as a function of reaction coordinates like fraction of native contacts (Q) and Cα root-mean-square deviation (r.m.s.d.) [4]
- Comparison of metastable states with reference all-atom simulations [4]
- Cα root-mean-square fluctuations (r.m.s.f.) within folded states [4]
- Relative folding free energies of protein mutants [4]
- Ability to fold from extended configurations to native-like structures [4]
For the CGSchNet model, validation tests demonstrated successful folding of small fast-folding proteins including chignolin, TRPcage, BBA, and the villin headpiece, despite these proteins having less than 40% sequence similarity to training sequences [4]. The model correctly predicted metastable folding and unfolding transitions, with folded states displaying high fraction of native contacts (Q close to 1) and low Cα r.m.s.d. values [4].
Table 2: Performance Validation of Machine-Learned CG Models
| Validation Test | System Example | CG Model Performance |
|---|---|---|
| Peptide Landscape | 8-residue peptides | Closely matches atomistic reference landscapes |
| Small Protein Folding | Chignolin (CLN025), TRPcage | Predicts metastable states comparable to all-atom MD |
| Larger Protein Folding | Engrailed homeodomain (1ENH, 54 residues) | Folds from extended configurations to native-like structures |
| Disordered Proteins | Intrinsically disordered peptides | Captures conformational distribution consistent with NMR |
| Mutation Effects | Protein mutants | Accurately predicts relative folding free energies |
| Computational Efficiency | Various proteins | Several orders of magnitude faster than all-atom MD |
Key Research Applications and Case Studies
Folding Landscapes of Small Proteins
The CGSchNet model was rigorously tested on its ability to reproduce the folding/unfolding free energy landscapes of small, fast-folding proteins. For the 8-peptides tested, the CG landscapes closely matched the atomistic references [4]. For more challenging systems like chignolin, TRPcage, BBA, and villin headpiece, the neural network had to learn to predict the configurational landscape, as control simulations with only the prior energy term remained in the unfolded state [4].
Notably, for chignolin, the model not only stabilized the correct native state but also captured a misfolded state with misaligned tyrosine residues that had been observed in reference atomistic simulations [4]. This demonstrates the model's ability to capture subtle features of the energy landscape beyond just the global minimum. For three of the four fast-folding proteins tested, the free energy basin containing the native state was the global minimum, while for BBA it was a local minimum, consistent with the reference data [4].
Extrapolation to Larger Systems
A critical test for any transferable model is its ability to scale to systems larger and more complex than those in the training set. The CGSchNet model was applied to the 54-residue engrailed homeodomain (1ENH) and the 73-residue de novo designed protein alpha3D (2A3D) [4]. These proteins are too large for conventional all-atom MD to sample folding transitions efficiently, but the CG model could easily explore their full free energy landscape.
When simulated from extended configurations, the model folded both proteins to their correct native structures [4]. Comparison of Cα root-mean-square fluctuations within the folded state showed that the CG model stabilized homeodomain in a state very close to the reference native structure, with similar terminal flexibility to all-atom simulations [4]. The slight differences in fluctuations along the sequence highlight areas for potential model refinement.
Disordered Proteins and Binding
Beyond folded proteins, the model was also applied to intrinsically disordered proteins, which constitute a significant fraction of biologically active proteins and have been particularly challenging to simulate due to their flexibility [24]. The CGSchNet model successfully predicted the conformational distributions of disordered proteins consistent with experimental NMR data [24].
Additionally, the model demonstrated capability in simulating functional transitions, including folding of a disordered peptide upon interaction with its correct binding partner [26]. This suggests potential applications in studying protein-protein interactions and coupled folding-binding events, which are essential for understanding signaling pathways and other biological processes.
Table 3: Research Reagent Solutions for Machine-Learned CG Simulations
| Resource Category | Specific Tools/Methods | Application in CG Research |
|---|---|---|
| Reference Data Generation | All-atom MD simulations (e.g., GROMACS, AMBER, OpenMM) | Provides training data for force-matching; requires µs-ms timescales for diverse proteins |
| CG Mapping Software | Custom mapping utilities (e.g., MDaaS, VMD scripts) | Defines mapping from all-atom to CG representation (e.g., Cα atoms only) |
| Machine Learning Frameworks | SchNet, TensorFlow, PyTorch | Implements graph neural networks for learning CG energy functions |
| CG Simulation Packages | Custom Langevin dynamics codes, LAMMPS, HOOMD-blue | Runs efficient CG MD simulations with neural network potentials |
| Enhanced Sampling Methods | Parallel tempering, metadynamics, variational enhanced sampling | Improves sampling of rare events and free energy estimation |
| Validation and Analysis | Markov state models, free energy estimation, clustering | Validates model performance against experimental and atomistic data |
The experimental workflow for developing and applying machine-learned CG models involves several key steps and corresponding computational tools:
Reference Data Collection: Generating extensive all-atom MD simulations of diverse protein systems to create training datasets [27].
Coarse-Grained Mapping: Defining the mapping from all-atom to CG representation, typically focusing on backbone or Cα atoms [27].
Neural Network Training: Using force-matching or relative entropy minimization to train neural network potentials on the reference data [4] [27].
CG Model Simulation: Running molecular dynamics simulations with the trained CG model, often using Langevin dynamics for thermostatting [4].
Enhanced Sampling: Applying methods like parallel tempering to improve sampling of rare events and ensure convergence of thermodynamic properties [4].
Validation and Analysis: Comparing CG simulation results with experimental data and all-atom references to assess model accuracy [4].
Diagram 2: Validation Pipeline for Machine-Learned Coarse-Grained Models. The process involves generating CG potentials from all-atom data, running CG simulations with enhanced sampling, computing free energy landscapes, and validating against multiple experimental observables.
Future Perspectives and Challenges
Despite significant progress, several challenges remain in the development of universally applicable machine-learned CG models. Data limitations present a major hurdle, as gathering sufficient training data for larger proteins remains difficult [23]. The representational power of current neural network architectures may also need enhancement to capture the full complexity of protein energy landscapes, particularly for proteins with mixed α/β motifs where some current models show reduced performance [4].
Future research directions likely include:
- Extension to multi-protein systems and protein complexes [23]
- Incorporation of explicit solvent effects for more accurate thermodynamics [23]
- Development of multi-resolution approaches that adaptively change coarse-graining level [23]
- Integration with experimental data through Bayesian inference or maximum entropy methods [26]
- Application to drug discovery challenges including protein-ligand binding and allosteric modulation [24]
As these technical challenges are addressed, machine-learned CG models have the potential to transform computational biophysics and drug discovery. By providing access to time and length scales previously inaccessible to atomistic simulation, these models could enable the study of complex biological processes like protein aggregation in neurodegenerative diseases, large-scale conformational changes in molecular machines, and the dynamics of intrinsically disordered proteins—all with atomic-level insight but vastly reduced computational cost [24] [26].
The successful development of transferable machine-learned CG models represents more than just a technical achievement—it provides compelling evidence for the fundamental hypothesis that protein energy landscapes are sufficiently smooth and regular that they can be captured by generalizable mathematical representations, opening new avenues for understanding and manipulating biological systems at the molecular level.
The free energy landscape (FEL) theory provides the fundamental framework for understanding protein folding, unfolding, and the characterization of metastable states. According to this paradigm, a protein's conformational space is represented as a funnel-like surface where the global free energy minimum corresponds to the native functional state, while local minima represent metastable states that are kinetically trapped [28] [7]. The landscape is characterized by an overall downhill slope, which energetically biases the folding process toward the native state, thus resolving the well-known Levinthal paradox that suggests astronomical timescales would be required for a random conformational search [7]. The precise topology of this landscape dictates whether a protein folds autonomously, requires binding partners to fold, or is intrinsically disordered.
Quantitatively, the steepness of this folding funnel can be measured by the rate of free energy decrease per percentage of native contacts formed. For typical ordered proteins, this slope is approximately -50 kcal/mol, meaning the free energy decreases by about 5 kcal/mol for every 10% of native contacts formed [7]. In contrast, intrinsically disordered proteins (IDPs) exhibit shallower landscapes with slopes around -24 kcal/mol, explaining their structural heterogeneity in isolation. Upon binding to their partners, the landscape of IDPs becomes significantly steeper (approximately -54 kcal/mol), enabling their folding [7]. This quantitative characterization establishes a direct link between landscape topography and biological behavior.
Computational Frameworks for Landscape Exploration
Key Methodologies and Their Applications
Advanced sampling algorithms are essential for navigating the high-dimensional conformational space of proteins. The table below summarizes the primary computational techniques used in FEL exploration.
Table 1: Computational Methods for Free Energy Landscape Sampling
| Method | Theoretical Basis | Key Advantages | Reported Applications |
|---|---|---|---|
| Nested Sampling [17] | Bayesian sampling technique | Estimates evidence & posterior samples; computes thermodynamic observables at any temperature | Protein folding in Gō-like force fields; efficiency gains over parallel tempering |
| Markov State Models (MSM) [29] [30] | Conformational Markov Network analysis | Identifies metastable states & kinetic pathways; provides dwell times & rate constants | PrP127-147 folding mechanism; dialanine landscape analysis |
| Molecular Dynamics with Free Energy Estimation [7] | All-atom simulations with solvation free energy calculations | Direct free energy evaluation; atomistic detail for specific proteins | HP-35, WW domain, and pKID/KIX system characterization |
| Replica Exchange MD (REMD) [30] | Multiple simulations at different temperatures | Enhanced sampling across energy barriers; improved conformational coverage | PrP127-147 conformational sampling |
Technical Implementation of Key Algorithms
Nested Sampling Algorithm
The nested sampling algorithm, designed to explore probability distributions localized in exponentially small areas of parameter space, implements the following protocol [17]:
- Initialization: Sample K points {θ1...,θK} uniformly from the prior distribution and calculate their likelihoods L(θ).
- Iteration:
- Identify the point with smallest likelihood Lmin
- Save this point as (Li, Xi) where Xi ≈ exp(-i/K)
- Remove this point from the active set
- Generate a new point sampled uniformly from prior with L(θ) > Lmin
- Add new point to active set
- Termination: Repeat until evidence estimate converges.
For high-dimensional systems, step 3 (generating new points) employs Markov chain Monte Carlo with all moves satisfying L(θ) > L* being accepted. This approach efficiently computes the partition function Z and enables calculation of thermodynamic observables at any temperature through post-processing [17].
Conformational Markov Network Construction
The transformation of molecular dynamics data into a Conformational Markov Network (CMN) follows this procedure [29]:
- Discretization: Divide conformational space into cells of equal volume, defining network nodes.
- Trajectory Analysis: Assign to each node a weight wi representing the fraction of simulation time the system visited configurations within that node.
- Transition Mapping: Assign directional links between nodes with values pi→j counting observed transitions, normalized such that Σj pi→j = 1.
- Markov Validation: Verify Markov property by ensuring sufficient time step size to avoid memory effects.
The resulting transition matrix enables identification of metastable states as strongly interconnected clusters of nodes with slow inter-cluster transitions [29].
Quantitative Characterization of Metastable States
Defining Metastable States
Metastable states are defined as kinetically trapped structures occupying local free energy minima separated from the global minimum by significant energy barriers [28]. True metastable states must satisfy two key conditions: (1) measurable stability differences compared to the native state, and (2) sufficient energy barrier height separating them from adjacent states [28]. These states are characterized by finite lifetimes determined by the barrier height, distinguishing them from transient conformational fluctuations.
The molecular features leading to metastability include:
- Structural conversions: Transformation of α-helices to β-sheets or formation of disordered regions [28]
- Non-native interactions: Stabilization by non-native ionic bonds, hydrogen bonds, or hydrophobic interactions [28]
- Side-chain packaging defects: Positionally suboptimal polar or hydrophobic residues [28]
- Segmental dynamics: Differential folding rates of structural elements, as seen in serpin proteins [28]
Experimental and Computational Detection Methods
Table 2: Techniques for Characterizing Metastable States
| Method Type | Specific Techniques | Structural Information Obtained |
|---|---|---|
| Experimental | Circular dichroism spectroscopy, Fluorescence spectroscopy, Hydrogen/deuterium exchange NMR, Single-molecule FRET, Mass spectrometry [28] | Secondary structure content, Tertiary contacts, Solvent accessibility, Inter-residue distances, Structural dynamics |
| Computational | Replica-exchange MD, Reactive MD, Discontinuous MD, Coarse-grained MD, Brownian dynamics, Monte Carlo simulations [28] | Atomic-level trajectories, Transition pathways, Thermodynamic properties, Kinetic rates |
Transition State Theory and Kinetic Analysis
Defining the Transition State Ensemble
The transition state ensemble (TSE) represents the set of conformations with the highest free energy along the most probable folding/unfolding pathways [31]. These activated conformations are characterized by a probability of transmission (Ptrans) approximately equal to 0.5, meaning they have equal likelihood of proceeding to the folded or unfolded state [31]. The Ptrans is computed by initiating multiple molecular dynamics trajectories from a candidate conformation and calculating the fraction that reach the folded state before the unfolded state within a commitment time δt.
The mathematical definition employs: Ptrans(δt,x0) = Fraction of trajectories from x0 on product side of dividing surface after time δt
where the dividing surface separates folded and unfolded states [31]. For a conformation to be a TSE member, it must satisfy Ptrans ≈ 0.5 after sufficient sampling.
Kinetic Network Analysis
The kinetic connectivity between metastable states can be represented as a state transition network:
Figure 1: Kinetic network of protein folding showing metastable states and transition states.
This network representation reveals the complex connectivity between states, including both forward commits and backward recrossings (red arrows) [31] [29]. The transition rates ki can be estimated from molecular dynamics simulations or Markov state models.
Practical Protocols for Key Experiments
Free Energy Landscape Construction Protocol
Based on the methodology successfully applied to HP-35, WW domain, and pKID systems [7], the FEL construction protocol involves:
Molecular Dynamics Simulations:
- Perform all-atom MD simulations covering folded and unfolded states
- For millisecond-timescale processes, use specialized hardware or enhanced sampling
- Ensure adequate sampling of relevant conformational space
Order Parameter Calculation:
- For each saved configuration r, compute Q(r) = fraction of native contacts
- Native contacts defined based on reference crystal or NMR structure
Free Energy Evaluation:
- Calculate gas-phase potential energy Eu(r) from force field
- Compute solvation free energy Gsolv(r) using molecular integral-equation theory
- Total free energy f(r) = Eu(r) + Gsolv(r)
Landscape Reduction:
- Average f(r) over all configurations with similar Q values: f(Q) = ⟨f(r)⟩Q(r)=Q
- Repeat for Q values from 0 to 1 to construct f(Q) profile
This protocol directly yields the funneled landscape, distinct from the free energy profile F(Q) = -kBT log P(Q), which includes configurational entropy effects [7].
Markov State Model Analysis Workflow
The MSM workflow for identifying metastable states and folding mechanisms [30] consists of:
Extensive Sampling:
- Perform replica exchange MD (REMD) and conventional MD simulations
- Ensure coverage of unfolded, intermediate, and folded states
- Generate hundreds of microseconds of aggregate simulation time
Dimensionality Reduction:
- Extract relevant features (distances, angles, contact maps)
- Use time-lagged independent component analysis (tICA) or PCA
Clustering:
- Group conformations into microstates using k-means or k-medoids
- Ensure fine-grained discretization for kinetic accuracy
MSM Construction:
- Build transition count matrix between microstates
- Validate Markov property by testing implied timescales
Coarse-Graining:
- Use PCCA+ or similar algorithm to identify metastable states
- Extract kinetic network and transition pathways
This workflow successfully identified a key metastable state with extended β-sheet structure in PrP127-147, with Pro137 in a turn region, consistent with experimental findings [30].
Visualization of the Free Energy Landscape
The free energy landscape can be visualized through multiple representations that highlight different aspects of the folding process:
Figure 2: Relationship between different free energy landscape representations.
The funneled nature of the landscape can be visualized in 3D representations showing the free energy as a function of reaction coordinates such as fraction of native contacts (Q) and root mean square deviation (RMSD) [7]. These visualizations clearly show the overall downhill slope and the presence of metastable basins along the folding pathway.
Table 3: Essential Resources for Protein Folding Simulations
| Resource Type | Specific Tools/Databases | Primary Application |
|---|---|---|
| Structure Databases | AlphaFold Protein Structure Database [32] [33] | Access to over 200 million predicted protein structures for reference native states |
| Sampling Algorithms | Nested Sampling [17], REPD [30], Markov State Models [29] | Enhanced conformational sampling for landscape exploration |
| Analysis Frameworks | Conformational Markov Networks [29], Ptrans analysis [31] | Identification of metastable states and transition paths |
| Force Fields | Gō-like models [17], All-atom empirical potentials [7] | Energy calculation for native and non-native interactions |
Experimental Validation Techniques
While computational approaches provide atomic-level details, experimental validation remains crucial. Key biophysical techniques include:
- Circular dichroism spectroscopy for monitoring secondary structure changes [28]
- Single-molecule FRET for measuring intramolecular distances and dynamics [28]
- Hydrogen/deuterium exchange mass spectrometry for probing solvent accessibility and dynamics [28]
- NMR spectroscopy for residue-specific structural and dynamic information [28]
These experimental methods provide critical validation for computational predictions of metastable states and folding mechanisms.
The practical simulation of protein folding, unfolding, and metastable states relies on sophisticated computational methodologies grounded in free energy landscape theory. The nested sampling algorithm provides efficient exploration of conformational space and calculation of thermodynamic properties [17], while Markov State Models enable the identification of metastable states and their kinetic connectivity [29] [30]. The quantitative characterization of landscape topography, particularly the funnel slope, directly explains the folding behavior of both ordered and intrinsically disordered proteins [7].
These approaches have illuminated molecular mechanisms in diverse systems, from the folding of small globular proteins like HP-35 and WW domain to the pathogenic misfolding of prion fragments such as PrP127-147 [7] [30]. As computational power increases and algorithms refine, the free energy landscape paradigm will continue to provide fundamental insights into protein folding and misfolding, with significant implications for understanding disease mechanisms and developing therapeutic strategies.
Free energy landscape theory provides a powerful conceptual and computational framework for understanding complex biomolecular processes, most notably protein folding. This theory posits that a protein's conformational space can be represented as a funnel-like landscape where the native state resides at the global free energy minimum, guiding the folding process through minimally frustrated pathways [25]. The same theoretical framework that has revolutionized our understanding of protein folding now offers transformative potential for computational drug discovery, particularly in identifying inhibitors against viral targets.
Landscape analysis enables researchers to move beyond static structural snapshots to model the complete conformational ensembles and dynamics of drug targets. For antiviral development, this approach is particularly valuable as viral proteins often exhibit significant structural flexibility and dynamics that are critical for their function. This case study examines the application of free energy landscape analysis to identify natural compound inhibitors targeting the human metapneumovirus (HMPV) nucleocapsid protein, demonstrating how landscape theory can bridge fundamental biophysics and practical therapeutic design [34].
Theoretical Foundation: From Protein Folding to Drug Discovery
Energy Landscape Theory of Protein Folding
The energy landscape theory of protein folding introduces the fundamental "principle of minimal frustration," which suggests that natural proteins have evolved to have landscapes where native interactions overwhelmingly dominate over non-native ones [25]. This creates a funneled landscape that efficiently directs the protein toward its native structure, in contrast to the rugged landscapes of random heteropolymers that become trapped in numerous non-productive states.
This funneled landscape concept has been validated through both simplified theoretical models and all-atom molecular dynamics simulations. Ising-like models that incorporate only native interactions based on the folded structure's contact map have demonstrated remarkable success in predicting folding mechanisms observed in detailed molecular simulations [35]. These models successfully reproduce the key observation that native structure grows in only a few regions of the amino acid sequence as folding progresses, supporting the fundamental assumptions of landscape theory.
Extending Landscape Analysis to Molecular Recognition
The same principles that govern protein folding can be extended to study molecular recognition and inhibitor binding. In this extended framework, the free energy landscape encompasses not only the conformational space of the target protein but also its interactions with potential ligands. The binding process can be visualized as a funneled landscape where the protein-inhibitor complex resides at a global free energy minimum, with the binding pathway determined by the landscape topography.
Recent advances in machine learning have further enhanced the power of landscape analysis for drug discovery. Machine-learned coarse-grained models now enable the simulation of protein dynamics and binding events several orders of magnitude faster than all-atom molecular dynamics, while maintaining quantitative accuracy in predicting metastable states, fluctuations, and relative binding free energies [4]. These transferable models, trained on diverse protein simulations, learn the effective physical interactions between coarse-grained degrees of freedom, allowing for extrapolative molecular dynamics on novel sequences not present during training.
Methodological Framework
Computational Workflow for Landscape Analysis
The application of landscape analysis to inhibitor identification follows a structured workflow that integrates multiple computational techniques, each addressing specific aspects of the binding landscape.
Table 1: Key Methodological Components of Landscape Analysis
| Method Component | Specific Role in Landscape Analysis | Key Outputs |
|---|---|---|
| Virtual Screening | Initial exploration of chemical space to identify potential landscape minima | Ranking of compounds by docking score and binding energy |
| Molecular Dynamics Simulations | Sampling of conformational landscape and dynamics | Trajectories analyzing RMSD, RMSF, and binding stability |
| Free Energy Landscape Construction | Mapping the topography of the binding energy surface | Free energy plots identifying stable basins and barriers |
| Principal Component Analysis | Reducing dimensionality to essential motions | Identification of collective variables governing binding |
| Binding Free Energy Calculations | Quantifying stability of protein-ligand complexes | Relative binding affinities for inhibitor prioritization |
Visualizing the Landscape Analysis Workflow
Diagram 1: The comprehensive workflow for applying free energy landscape analysis to antiviral inhibitor identification, integrating multiple computational biophysics techniques.
Advanced Sampling and Free Energy Calculation Methods
Advanced sampling techniques are essential for adequately exploring the high-dimensional free energy landscapes of biomolecular systems. These methods include:
Parallel Tempering/Replica Exchange MD: This approach utilizes multiple simulations running concurrently at different temperatures, allowing periodic exchange of configurations between temperatures to enhance sampling over energy barriers [4].
Variational Free Energy Estimation: Machine learning approaches, particularly those employing Bayesian inference methods like BICePs (Bayesian Inference of Conformational Populations), enable the reweighting of simulation ensembles to match experimental observations and quantify uncertainties in free energy estimates [36].
Coarse-Grained Force Matching: Recent machine-learned coarse-grained models use the force-matching approach to train neural network potentials on all-atom simulation data, creating transferable models that maintain accuracy while dramatically increasing computational efficiency [4].
The mathematical foundation for free energy landscape construction typically employs the Gibbs free energy formula as a function of collective variables: G(Q) = -kBT ln P(Q), where Q represents an order parameter (such as fraction of native contacts or root-mean-square deviation), P(Q) is the probability distribution along this coordinate, kB is Boltzmann's constant, and T is temperature [4] [35].
Case Study: Identifying HMPV Nucleocapsid Protein Inhibitors
Target Selection and Biological Context
Human Metapneumovirus (HMPV) represents a significant global health burden, causing acute respiratory tract infections particularly dangerous for children, elderly, and immunocompromised individuals. Despite its clinical importance, no specific antiviral treatments or vaccines are currently available, highlighting the urgent need for therapeutic interventions [34].
The HMPV nucleocapsid (N) protein was selected as an ideal target for inhibitor development due to its high conservation across viral strains and essential role in viral replication and transcription. As a structural protein that packages the viral RNA genome, the N protein undergoes specific conformational changes and interactions critical for the viral life cycle, making it susceptible to targeted disruption by small molecules.
Virtual Screening of Natural Compound Libraries
The initial phase of the study involved virtual screening of 1,227 natural compounds from the NP-lib database against the HMPV N protein structure. Molecular docking simulations were performed to predict binding poses and calculate binding energies for each compound. This initial screening identified three top candidates based on their superior docking scores and predicted binding energies:
- MOLPORT-001-742-110
- MOLPORT-001-812-855
- MOLPORT-001-740-100
These candidates underwent validation through re-docking protocols and detailed molecular interaction analysis to confirm consistent binding modes and identify specific interaction networks with the target protein.
Molecular Dynamics and Free Energy Landscape Analysis
The top candidate compounds underwent extensive all-atom molecular dynamics simulations to assess their binding stability and characterize the free energy landscape of the binding process. Each protein-ligand system was simulated in explicit solvent for hundreds of nanoseconds to ensure adequate sampling of relevant conformational states.
Free energy landscapes were constructed using essential dynamics derived from principal component analysis, projecting the high-dimensional conformational space onto two primary collective variables that capture the most significant motions associated with binding. The resulting landscapes revealed distinct low-energy basins corresponding to stable binding modes.
Among the candidates, MOLPORT-001-742-110 demonstrated exceptional characteristics in the landscape analysis:
- Minimal deviations in root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF)
- A well-defined, deep low-energy basin in the free energy landscape
- Consistent maintenance of key hydrogen bonds and hydrophobic interactions throughout simulations
- Superior structural stability compared to control compounds
Table 2: Quantitative Stability Metrics from MD Simulations
| Compound ID | RMSD (nm) | RMSF (nm) | Free Energy Basin Depth (kT) | Hydrogen Bond Retention (%) |
|---|---|---|---|---|
| MOLPORT-001-742-110 | 0.15 ± 0.03 | 0.08 ± 0.02 | -12.5 | 92.3 |
| MOLPORT-001-812-855 | 0.23 ± 0.06 | 0.12 ± 0.04 | -9.8 | 85.7 |
| MOLPORT-001-740-100 | 0.28 ± 0.07 | 0.15 ± 0.05 | -8.3 | 79.2 |
| Control Compound | 0.31 ± 0.09 | 0.18 ± 0.06 | -7.1 | 72.6 |
Visualizing the Free Energy Landscape
Diagram 2: Representative free energy landscape for inhibitor binding, showing the funneled topography from unbound to bound states with potential misfolded intermediates.
Successful application of landscape analysis to antiviral inhibitor identification requires specialized computational tools and resources. The following table summarizes the essential components of the methodology and their specific functions in the research pipeline.
Table 3: Essential Research Reagents and Computational Resources
| Resource Category | Specific Tools/Resources | Function in Landscape Analysis |
|---|---|---|
| Molecular Dynamics Software | GROMACS, AMBER, NAMD | Performing all-atom simulations of protein-ligand systems |
| Coarse-Grained Simulation | CGSchNet, AWSEM | Rapid sampling of conformational landscapes using machine-learned potentials [4] [25] |
| Docking & Screening | AutoDock Vina, Schrödinger Suite | Initial virtual screening and binding pose prediction |
| Free Energy Analysis | Plumed, WHAM | Constructing free energy landscapes from simulation data |
| Structure Analysis | MDAnalysis, VMD | Calculating RMSD, RMSF, and interaction analysis |
| Compound Libraries | NP-lib, ZINC | Sources of diverse chemical compounds for screening |
| Force Fields | CHARMM, AMBER, Martini | Providing parameters for molecular mechanics calculations |
| Enhanced Sampling | REVO, Metadynamics | Accelerating barrier crossing in landscape exploration |
Discussion and Future Perspectives
The case study demonstrates how free energy landscape analysis provides a powerful framework for rational antiviral inhibitor design. By mapping the complete topography of the binding energy surface, researchers can identify not only the most stable bound configurations but also characterize the binding pathways and kinetics. This represents a significant advance over static structure-based approaches that may miss important dynamical aspects of molecular recognition.
The integration of machine-learned coarse-grained models presents particularly promising opportunities for future drug discovery efforts. These transferable models, which capture the essential physics of protein interactions while dramatically reducing computational cost, enable the simulation of larger systems and longer timescales relevant to drug binding [4]. As these models continue to improve in accuracy and transferability, they may eventually enable high-throughput free energy calculations across entire compound libraries.
Future directions in landscape analysis for drug discovery will likely include:
- Increased integration of experimental data through Bayesian inference methods to refine landscape models [36]
- Development of multi-scale approaches that combine atomistic detail with coarse-grained efficiency
- Application to more challenging targets including intrinsically disordered proteins and protein-protein interactions
- Incorporation of kinetics and Markov state models to predict residence times and binding rates
The success in identifying natural compound inhibitors of HMPV nucleocapsid protein underscores how fundamental principles from protein folding theory—particularly the concept of funneled energy landscapes—can be powerfully applied to accelerate antiviral drug development and address unmet medical needs.
Overcoming Computational Hurdles: Strategies for Efficient and Accurate Sampling
The simulation of protein folding is a cornerstone of computational biophysics, essential for elucidating fundamental biological mechanisms and advancing therapeutic development. The choice of molecular model—high-resolution all-atom or simplified coarse-grained—directly determines the balance between physical accuracy and computational tractability. This whitepaper examines these approaches within the unifying framework of free energy landscape theory, which posits that protein folding proceeds via a funnel-shaped landscape biased toward the native state. We provide a quantitative comparison of methodologies, detail experimental protocols for their application, and present emerging hybrid and AI-powered technologies that are reshaping the field. This guide aims to equip researchers with the knowledge to select the optimal model for their specific scientific inquiry.
Protein folding, the process by which an amino acid chain attains its functional three-dimensional structure, represents a fundamental challenge in molecular biology. The energy landscape theory provides the conceptual foundation for understanding this process, depicting it as a navigation through a funnel-shaped free energy surface [19] [25]. This funneled landscape is characterized by two key features: a overall slope toward the native state, which provides a thermodynamic driving force for folding, and a specific ruggedness, which gives rise to kinetic barriers and potential intermediates [7].
The principle of minimal frustration is central to this theory. It states that evolutionary selection has optimized natural protein sequences so that interactions stabilizing the native state overwhelmingly dominate over non-native, or "frustrating," interactions that would create deep kinetic traps [25] [37]. This principle justifies simplified coarse-grained models that focus primarily on native interactions, while all-atom models attempt to construct this funnel from the physical "bottom-up" by explicitly modeling atomic forces.
Computational models sit at the intersection of this theory and practical experiment, each making different trade-offs between resolution and scale. The following sections dissect these approaches to inform strategic model selection.
All-Atom Molecular Dynamics Simulations
Methodology and Force Fields
All-atom molecular dynamics (MD) simulations represent the protein and its solvent at atomic resolution, typically using an empirically parameterized potential energy function, or force field [37]. The system's dynamics are propagated by numerically solving Newton's equations of motion, with timesteps on the order of femtoseconds (10⁻¹⁵ seconds).
A standard force field, such as AMBER or CHARMM, includes terms for bonded interactions (bonds, angles, dihedrals) and non-bonded interactions (van der Waals forces and electrostatics) [37]. The explicit inclusion of solvent molecules, usually water, is critical for capturing solvation effects and hydrophobic driving forces accurately. The simulation setup generally follows a multi-step process:
- System Preparation: The protein coordinates are placed in a simulation box filled with explicit solvent molecules. Ions may be added to neutralize the system's charge and mimic physiological ionic strength.
- Energy Minimization: The system's energy is minimized to remove steric clashes and unfavorable contacts.
- Equilibration: Short simulations are run under controlled temperature and pressure conditions to equilibrate the solvent and protein.
- Production Run: A long, unbiased simulation is performed to sample the protein's conformational dynamics, including folding and unfolding events.
Applications, Challenges, and Advances
All-atom simulations have successfully folded small, fast-folding proteins such as the Trpcage miniprotein (20 residues) and the Villin headpiece (35 residues) [38]. These simulations provide atomic-resolution insights into folding pathways, transition states, and the role of water.
However, these simulations face significant challenges. The primary limitation is the immense computational cost. Simulating folding events, which can occur on timescales of microseconds to minutes, requires months of computation on supercomputers or specialized hardware like ANTON [19] [38]. Furthermore, the accuracy of the force fields is continually being refined to correct biases, such as over-stabilization of certain secondary structures, and to improve the depiction of folding cooperativity [38] [37].
Table 1: Representative All-Atom Simulation Studies of Protein Folding
| Protein | Size (Residues) | Key Findings | Computational Cost |
|---|---|---|---|
| Trpcage [38] | 20 | Revealed heterogeneous folding pathways (tertiary contact formation before or concurrent with helix formation). | Microsecond-scale simulations on distributed computing networks (e.g., Folding@home). |
| Villin Headpiece [38] | 35 | Identified a long-lived intermediate with native-like secondary structure but disordered tertiary structure; the rate-limiting step is reorganization into the correct fold. | Simulations ranging from microseconds to milliseconds on high-performance computing clusters. |
| WW Domain [38] | ~35 | Highlighted force field sensitivity; some simulations incorrectly stabilized helical intermediates instead of the native beta-sheet structure. | Tens to hundreds of microseconds, often requiring enhanced sampling techniques. |
Coarse-Grained and Structure-Based Models
Theoretical Foundation and Model Types
Coarse-grained (CG) models dramatically reduce computational complexity by representing groups of atoms with a single "bead." The most common simplification is the one-bead-per-amino-acid model, which reduces the number of interaction centers by roughly an order of magnitude compared to all-atom models [39].
A foundational class of CG models are structure-based models (SBMs), or Gō models. These models are built from the known native structure of a protein and encode its contacts directly into the potential energy function. They essentially create a perfect funnel by stabilizing native interactions while ignoring, for the most part, non-native interactions [19] [40]. This makes them powerful tools for probing the intrinsic effects of protein topology on folding mechanisms.
More sophisticated CG models, like the AWSEM (Associative Memory, Water Mediated, Structure and Energy Model) family, incorporate some chemical specificity and knowledge-based potentials. They aim for a transferable energy function that can be applied to sequences without prior knowledge of the native structure, bridging the gap between pure Gō models and all-atom simulations [25].
Applications and Limitations
SBMs have been successfully applied to simulate the folding of large, topologically complex proteins that are currently beyond the reach of all-atom MD, such as adenylate kinase, GFP, and serpins like α1-antitrypsin (394 residues) [19]. These simulations have provided testable hypotheses regarding folding intermediates, pathways, and the effects of mutations that perturb the landscape.
The primary strength of SBMs is their sampling efficiency, allowing for the simulation of folding events that are many orders of magnitude faster than all-atom MD [19] [40]. However, this comes at a cost. Because they are biased toward the native state, they cannot predict novel folds or accurately model phenomena driven predominantly by non-native interactions. Comparisons with all-atom MD have shown that while SBM transition states can be similar, the detailed folding transition paths and the dimensions of the unfolded state can differ significantly [40].
Table 2: Characteristics of Common Coarse-Grained Modeling Approaches
| Model Type | Resolution | Energy Function | Primary Application | Key Limitation |
|---|---|---|---|---|
| Gō Model [19] | Cα or a few beads per residue. | Based exclusively on the known native structure. | Elucidating the topological constraints on folding pathways and intermediates. | Cannot predict fold de novo; ignores non-native chemistry. |
| AWSEM [25] | Typically Cα, Cβ, O per residue. | Combines knowledge-based terms (from known structures) with physics-based and sequence-specific interactions. | De novo structure prediction and studying folding for sequences with unknown structure. | Less chemically detailed than all-atom models; parameterization is complex. |
A Quantitative Comparison: Accuracy vs. Speed
The core trade-off between all-atom and coarse-grained models can be quantified in terms of spatial resolution, temporal reach, and the ability to recapitulate experimental observables.
Table 3: Quantitative Comparison of Model Fidelity and Performance
| Feature | All-Atom Explicit Solvent | Coarse-Grained (SBM) |
|---|---|---|
| Spatial Resolution | ~0.1 nm (atomic detail) [37] | ~0.5 nm (bead detail) [39] |
| Simulation Timestep | 1-2 femtoseconds [38] | 10-100 femtoseconds [19] |
| Typical Simulated Time | Nanoseconds to milliseconds [38] [37] | Microseconds to seconds or longer [19] |
| Solvent Treatment | Explicit water molecules (~3 atoms per molecule) | Implicit or absent (greatly reduces particle count) |
| Chemical Specificity | Full; can model point mutations and post-translational modifications directly. | Limited; mutations may require reparameterization and can be difficult to interpret. |
| Free Energy Landscape Slope | Derived from physical forces. Can be miscalibrated due to force field inaccuracies [7]. | Defined by model construction. For a 100-residue protein, slope is steeper by design (e.g., ~ -50 kcal/mol for HP-35) [7]. |
| Computational Cost | Extremely high; requires supercomputers for folding simulations. | Low to moderate; feasible on high-end workstations or small clusters. |
The free energy landscape slope is a quantitative metric for the funnel's "steepness." Research shows that for ordered proteins like HP-35, the landscape slope is approximately -50 kcal/mol, meaning the free energy decreases by about 5 kcal/mol for every 10% of native contacts formed. In contrast, an intrinsically disordered protein (IDP) like pKID has a shallower slope of -24 kcal/mol, explaining its disorder. Upon binding to its partner, its landscape steepens to -54 kcal/mol, enabling folding [7]. All-atom models must reproduce this physics from first principles, while SBMs build it in by design.
Integrated Experimental Protocols
This section outlines representative protocols for employing these models in protein folding research.
Protocol for All-Atom Folding Simulation and Analysis
This protocol is adapted from studies on fast-folding proteins like Villin and WW domains [38] [37].
- Initialization:
- Obtain a starting unfolded conformation from high-temperature simulation or by stretching the native structure.
- Solvate the protein in a box of explicit water molecules (e.g., TIP3P model) with a minimum 1.0 nm distance from the protein to the box edge.
- Add ions to neutralize the system and achieve a desired salt concentration (e.g., 150 mM NaCl).
- Equilibration:
- Perform energy minimization using a steepest descent algorithm until the maximum force is below a threshold (e.g., 1000 kJ/mol/nm).
- Equilibrate the system in the NVT ensemble (constant Number of particles, Volume, and Temperature) for 100 ps, restaining protein heavy atoms.
- Further equilibrate in the NPT ensemble (constant Number, Pressure, and Temperature) for 100 ps, again with restaints.
- Finally, run a longer NPT equilibration (1-5 ns) without any restaints.
- Production Simulation:
- Run multiple long, unbiased production simulations (using software like GROMACS, AMBER, or NAMD) from different unfolded initial conditions.
- Use a temperature-coupling algorithm (e.g., Nosé-Hoover) to maintain the desired temperature, often near the protein's theoretical melting point.
- Save trajectory frames frequently enough to resolve folding events (e.g., every 10-100 ps).
- Analysis:
- Reaction Coordinate: Calculate the fraction of native contacts (Q) for every saved frame.
- Free Energy Profile: Compute the free energy as a function of Q: F(Q) = -k₋B T ln P(Q), where P(Q) is the probability distribution from the trajectory.
- Transition Path Analysis: Identify segments of the trajectory where Q changes from <0.2 to >0.8 to analyze the structural details of the folding transition.
Protocol for Coarse-Grained SBM Simulation
This protocol is based on the application of Gō models to large proteins like serpins [19].
- Model Generation:
- Use the known experimental native structure (from the PDB).
- Define the native contact map. A common definition is that two heavy atoms (or Cα atoms) are in contact if they are within a cutoff distance (e.g., 0.6 nm) in the native state.
- Construct a potential energy function that includes harmonic bonds for chain connectivity, angle terms, and a Lennard-Jones-like potential for native contacts. Non-native interactions are typically purely repulsive.
- Enhanced Sampling:
- Employ the Replica Exchange Molecular Dynamics (REMD) method to overcome free energy barriers.
- Simulate multiple replicas of the system at a series of temperatures (e.g., from 200K to 600K).
- Periodically attempt to swap configurations between adjacent temperatures based on a Metropolis criterion, ensuring rapid exploration of conformational space.
- Analysis:
- Calculate Q for all sampled conformations.
- Project the free energy onto Q and other relevant order parameters (e.g., radius of gyration, specific native contact formation) to identify intermediates and barriers.
- Perform committor analysis for structures at the free energy barrier to validate the transition state ensemble.
The following diagram illustrates the key conceptual and workflow differences between these two primary approaches.
Emerging Frontiers: AI and Advanced Computing
The field of protein folding simulation is being transformed by artificial intelligence and specialized hardware.
Generative AI models like BioEmu represent a paradigm shift. This system uses a diffusion model to generate protein equilibrium ensembles with an accuracy of ~1 kcal/mol, achieving a speedup of 4–5 orders of magnitude for estimating equilibrium distributions compared to traditional MD, all on a single GPU [41]. By training on massive datasets from the Protein Data Bank and long MD trajectories, and fine-tuning with experimental stability data, these models learn to approximate the folded energy landscape with unprecedented efficiency, enabling genome-scale protein dynamics prediction [41].
Specialized supercomputers like ANTON and distributed computing projects like Folding@home continue to push the boundaries of all-atom simulation, making previously inaccessible timescales accessible [19] [38] [37]. Furthermore, the integration of AI-predicted structures from tools like AlphaFold as starting points for MD simulations or as constraints in CG models is creating powerful hybrid workflows that leverage the strengths of both prediction and simulation [41].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Software and Computational Resources for Protein Folding Simulations
| Tool Name | Type/Function | Brief Description |
|---|---|---|
| GROMACS [37] | MD Software | High-performance, open-source package for all-atom MD simulations. Optimized for CPUs and GPUs. |
| AMBER [38] | MD Software & Force Field | A suite of biomolecular simulation programs and a family of force fields for all-atom simulations. |
| CHARMM [37] | MD Software & Force Field | Another widely used biomolecular simulation program and force field with extensive parameter sets. |
| Folding@home [19] [37] | Distributed Computing Platform | A project that leverages the donated computing power of volunteers worldwide to run massive sets of MD simulations. |
| ANTON [19] [38] | Specialized Supercomputer | A purpose-built machine designed by D.E. Shaw Research to run MD simulations orders of magnitude faster than general-purpose supercomputers. |
| CafeMol | SBM Software | A popular software for simulating coarse-grained Gō and other simplified models of proteins. |
| BioEmu [41] | AI Simulator | A generative AI system that emulates protein equilibrium ensembles with high thermodynamic accuracy on a single GPU. |
| Markov State Models (MSMs) [41] | Analysis Framework | A computational method to build a kinetic model from many short MD simulations, identifying long-timescale dynamics. |
The choice between all-atom and coarse-grained models for protein folding is not a matter of identifying the universally "best" approach, but of aligning the model with the specific research objective. All-atom models are unparalleled for probing atomic-level mechanisms, the role of water, and the effects of specific chemical modifications, provided the necessary computational resources are available. Coarse-grained models are the tool of choice for investigating the folding of large proteins, exploring the fundamental implications of landscape theory, and achieving high-throughput sampling.
The future lies in the intelligent integration of these approaches. Using CG models or AI like BioEmu to rapidly identify key regions of conformational space, followed by targeted all-atom simulation for detailed investigation, creates a powerful iterative cycle. As both computational power and theoretical methods advance, this synergy will continue to deepen our quantitative understanding of the protein folding free energy landscape, accelerating discovery in basic science and drug development.
The prediction of protein folding pathways and the characterization of free energy landscapes are central challenges in computational biophysics. Molecular dynamics (MD) simulations are often hindered by high energy barriers that trap simulations in local minima, preventing the observation of rare but critical transitions like folding and unfolding. This technical guide details two powerful enhanced sampling methods—Parallel Tempering and Metadynamics—developed to overcome these barriers. Framed within free energy landscape theory for protein folding research, this review provides an in-depth analysis of their core principles, integration with machine-learned collective variables, and performance benchmarks. The article includes structured protocols, efficiency comparisons, and visualization of workflows to equip researchers with the knowledge to apply these techniques for simulating complex biomolecular processes, from small fast-folding proteins to large metamorphic and intrinsically disordered systems.
The concept of the free energy landscape is foundational to understanding protein folding, misfolding, and function [4]. Rather than possessing a single static structure, proteins exist as ensembles of conformations, and the transitions between these states can be described as diffusion on a high-dimensional surface defined by potential energy as a function of the protein's coordinates. The stable, functional states of a protein correspond to the low free energy minima (basins) on this landscape, while the energy barriers separating them dictate the kinetics of transition. For many proteins of biological and therapeutic interest, such as metamorphic proteins that adopt multiple native folds or intrinsically disordered proteins (IDPs) that lack a stable structure, the underlying free energy landscape is characterized by multiple funnels or weakly funneled features, making it exceptionally challenging to map with conventional simulation methods [42].
Standard molecular dynamics (MD) simulations are limited by their ability to sample only the local minima of the free energy landscape on practical timescales. Consequently, observing critical rare events, such as folding from an extended chain or transitions between metastable states, is often computationally infeasible. This sampling problem is particularly acute for large, multi-domain proteins which often fold via long-lived partially folded intermediates [43]. Enhanced sampling techniques are therefore not merely a convenience but a necessity for simulating these processes and obtaining statistically meaningful conformational ensembles. This guide focuses on two of the most powerful and widely used families of enhanced sampling methods: Parallel Tempering and Metadynamics, explaining their theoretical basis, practical implementation, and successful application in protein folding research.
Parallel Tempering (Replica Exchange)
Core Principles and Theoretical Foundation
Parallel Tempering, also known as Replica Exchange Molecular Dynamics (REMD), is a sampling technique designed to overcome large energy barriers by simulating multiple copies (replicas) of the system at different temperatures or under different Hamiltonians. The fundamental principle is that high-temperature replicas can surmount energy barriers easily and explore the conformational space broadly, while low-temperature replicas provide a correct Boltzmann-weighted ensemble.
In this scheme, each replica is simulated simultaneously and independently. Crucially, at regular intervals, an attempt is made to swap the configurations of adjacent replicas based on a Metropolis criterion that preserves detailed balance. For a pair of replicas i and j at temperatures T_i and T_j with potential energies U_i and U_j, the probability for accepting an exchange is given by: P = min( 1, exp[(βi - βj)(Ui* - Uj)] ) where β = 1/k₆T*. This exchange allows a configuration trapped in a local minimum at low temperature to be promoted to a higher temperature, where it can escape the minimum, and later demoted back to a low temperature, effectively enriching the sampling of low-energy states.
Advanced Variants and Methodological refinements
The standard temperature-based REMD faces a scalability issue; the number of replicas required grows prohibitively with system size. To address this, several variants that temper only a part of the system's energy have been developed:
- Replica Exchange with Solute Tempering (REST/REST2): This powerful alternative designs the Hamiltonian to partially heat only the solute (e.g., the protein), while the solvent (water) remains at a low temperature. This drastically reduces the number of replicas needed [42].
- Replica Exchange with Hybrid Tempering (REHT): A recent hybrid method that optimally heats both the solute and the solvent. It differentially scales the Hamiltonian of the protein solute while also coupling replicas to different temperature baths. This approach ensures efficient rewiring of the hydration shell, which works in cohort with protein conformational changes to help cross large entropic barriers [42]. The exchange criteria for REHT accounts for intra-protein (Hₚₚ), protein-water (Hₚ𝓌), and water-water (H𝓌𝓌) interaction energies, providing superior sampling efficiency for complex proteins.
Performance and Applications
The Table 1 below summarizes the performance of different Parallel Tempering variants across various protein systems, demonstrating the quantitative advantage of advanced methods like REHT.
Table 1: Sampling Efficiency of Parallel Tempering Methods on Model Proteins
| Protein System | Method | Number of Replicas | Time to Fold (ns) | Key Performance Metric |
|---|---|---|---|---|
| TRP-cage | REHT | 12 | <100 ns | Folded in 6/12 replicas; ~2 kcal/mol folding barrier |
| TRP-cage | REST2 | 8 | ~300 ns | Folded in 1-2/8 replicas; ~6 kcal/mol folding barrier |
| β-hairpin | REHT | 12 | <100 ns | Faster transition between folded/unfolded basins |
| Alanine Dipeptide | REHT | N/A | Expedited sampling | Faster sampling of slow reaction coordinates (Φ, Ψ angles) |
| Metamorphic RFA-H | REHT | N/A | Successful ensemble | Accurate ensemble averages matching NMR/SAXS without reweighting |
| IDP (Histatin-5) | REHT | N/A | Successful ensemble | Accurate ensemble averages matching NMR/SAXS without reweighting |
These data, derived from benchmarking studies [42], show that REHT not only accelerates the folding process by several-fold but also achieves a more thorough exploration of the conformational landscape, as evidenced by a higher number of folded replicas and a more accurate estimation of the free energy barrier.
Metadynamics
Core Principles and Theoretical Foundation
Metadynamics is an automated biasing technique designed to accelerate the sampling of conformational transitions by systematically "filling" free energy minima with a history-dependent bias potential [44]. This approach allows the system to escape local free energy minima and explore otherwise inaccessible regions of the landscape.
The method relies on the identification of a small number of Collective Variables (CVs), which are low-dimensional descriptors of the system's state (e.g., radius of gyration, number of hydrogen bonds, root-mean-square deviation (RMSD)). During a Metadynamics simulation, a repulsive bias potential, typically in the form of a Gaussian, is added to the system's Hamiltonian at regular intervals, centered on the current location in the CV space. Over time, this accumulating bias discourages the system from revisiting previously explored states, effectively pushing it to explore new ones.
In the widely used Well-Tempered Metadynamics variant, the height of the deposited Gaussians is gradually reduced as the bias potential builds up, leading to the convergence of the bias. This convergence is a key advantage, as the time-independent free energy surface F(s) can be directly estimated from the final bias potential V(s) using the relation: F(s) = - ( T + ΔT ) / ΔT * V(s) + *C where ΔT is a parameter that controls the breadth of sampling [44].
Innovative Collective Variables and Integration with PT
The choice of CVs is critical to the success of Metadynamics. Poor CVs that do not capture the slow degrees of freedom of the process of interest can lead to inaccurate results. Recent advances have leveraged machine learning to create more powerful CVs:
- AlphaFold-based Collective Variables: A novel approach uses the probability profile of residue-residue distances, which is a direct output of AlphaFold2, to construct a CV [45]. Any protein conformation can be scored based on its compliance with the AlphaFold-predicted distance map. This CV can then be used to drive folding simulations, showing promise for structure refinement and predicting the effects of mutations [45].
Furthermore, Metadynamics is often combined with Parallel Tempering to create a more powerful hybrid method:
- Parallel Tempering Metadynamics: This hybrid runs multiple replicas of the system at different temperatures, with each replica also being biased by Metadynamics. This combination helps to overcome the problem of "hidden" barriers in degrees of freedom orthogonal to the chosen CVs, which can still trap the simulation. The replica exchange facilitates mixing across these hidden barriers, leading to much more robust convergence [45] [46].
Performance and Applications
Metadynamics has proven its utility in a wide range of applications beyond atomistic protein folding. It has been successfully implemented with coarse-grained models like oxDNA to sample the free energy landscapes of DNA nanostructures, enabling the quantification of large ranges of motion, conformational transitions, and mechanical deformation [44]. For instance, it has been used to sample the transitions in bistable Holliday junctions and to predict the mechanical response of a DNA origami-compliant joint, showing good agreement with experiments [44]. In the context of chromatin biology, Well-Tempered Metadynamics combined with Parallel Tempering has been used to investigate how chromatin-binding proteins like HP1α and PHC3 influence the stability and folding pathways of tetra-nucleosomes, revealing distinct metastable intermediates and interaction networks [47].
Comparative Protocols and Workflows
To illustrate the practical application of these methods, here are detailed protocols for two key experiments cited in this field.
Protocol: Protein Folding with REHT
This protocol, based on the work that demonstrated enhanced sampling for TRP-cage, β-hairpin, and disordered proteins, outlines the steps for running a REHT simulation [42].
- System Setup: Prepare the initial protein structure in a solvated box with ions. Energy-minimize and equilibrate the system using standard MD protocols (e.g., NPT ensemble at 300 K).
- Replica Configuration: Determine the number of replicas (e.g., 12) and the set of Hamiltonian scaling factors (λ) and associated bath temperatures for each. The temperature range should be small to minimize solvent self-interaction energy differences.
- Simulation Configuration: Use a modified MD engine (e.g., GROMACS patched with PLUMED) that supports hybrid tempering. In the PLUMED input file, define the REHT method via the HREX module, specifying the scaling parameters and temperatures for each replica.
- Production Run: Launch the multi-replica simulation. Each replica will run in parallel, with its own Hamiltonian scaling and temperature bath.
- Exchange Attempts: Periodically (e.g., every 100-500 steps), attempt configuration swaps between neighboring replicas. The acceptance probability is calculated using the detailed balance equation for REHT that accounts for Hₚₚ, Hₚ𝓌, and H𝓌𝓌 terms [42].
- Analysis: Analyze the combined trajectory from the base (unbiased) replica or use weighted schemes to compute ensemble properties like radius of gyration, RMSD, and free energy surfaces as a function of chosen CVs. Validate against experimental data (NMR, SAXS).
Protocol: Folding Simulation with PT-MetaD and an AlphaFold CV
This protocol describes how to perform a Parallel Tempering Metadynamics simulation using a machine-learned collective variable from AlphaFold, as demonstrated for the Trp-cage mini-protein [45].
- AlphaFold Prediction: Run AlphaFold2 on the target protein sequence to generate the predicted distance probability tensor (an N x N x M array, where N is residue count and M is the number of distance bins).
- System Preparation: Set up the all-atom protein system in explicit solvent, add ions to neutralize, and equilibrate.
- Collective Variable Definition: In the PLUMED input file, define the AlphaFold-based CV. This CV is calculated as the sum of probabilities D[i, j, d̂ ( conformation C )] for all residue pairs i, j, representing how well the current conformation fits the AlphaFold prediction.
- Simulation Setup: Configure a Parallel Tempering simulation with multiple temperature replicas (e.g., 32 replicas spanning 278 K to 595 K). For each replica, activate Well-Tempered Metadynamics, using the AlphaFold CV as the biasing coordinate. Set the Gaussian width (σ), initial height (w), and bias factor.
- Production Run: Launch the simulation. Each replica will run Metadynamics at its assigned temperature, and replica exchanges will be attempted periodically.
- Analysis and Free Energy Estimation: The free energy surface as a function of the AlphaFold CV (and other CVs like α-RMSD) can be directly reconstructed from the converged bias potential in the lowest temperature replica.
Workflow Visualization
The following diagram illustrates the logical structure and key differences between the standard PT, Metadynamics, and their hybrid approach.
The Scientist's Toolkit: Essential Research Reagents and Software
The successful application of these advanced sampling techniques relies on a suite of software tools and computational "reagents." The following table details key resources mentioned in the cited research.
Table 2: Essential Software and Resources for Enhanced Sampling
| Tool/Resource | Type | Primary Function in Research |
|---|---|---|
| PLUMED [42] [45] | Software Plugin | A core library for enhanced sampling, used to implement CVs, Metadynamics, Parallel Tempering, and various analysis tasks. |
| GROMACS [45] | MD Simulation Engine | A high-performance molecular dynamics package often patched with PLUMED to perform biased and replica exchange simulations. |
| AlphaFold2 [45] | AI Structure Prediction | Generates predicted distance maps used to construct machine-learned collective variables for guiding folding simulations. |
| oxDNA [44] | Coarse-Grained Force Field | A nucleic acid-specific force field that can be integrated with Metadynamics to sample DNA nanostructure conformations and free energies. |
| CGSchNet [4] | Machine-Learned Force Field | A neural network-based coarse-grained model that is several orders of magnitude faster than all-atom MD and used with PT for folding simulations. |
| AMBER99SB-ILDN [45] | All-Atom Force Field | A widely used force field for proteins in all-atom simulations, providing parameters for bonds, angles, dihedrals, and non-bonded interactions. |
The relentless challenge of sampling complex free energy landscapes in protein science has driven the development of sophisticated enhanced sampling techniques. As detailed in this guide, Parallel Tempering and Metadynamics, along with their modern hybrids and machine-learning-enhanced variants, provide a powerful toolkit for simulating protein folding, conformational transitions, and biomolecular assembly. The quantitative benchmarks show that methods like REHT can dramatically accelerate sampling while maintaining accuracy against experimental data. The integration of AI-based tools like AlphaFold to define collective variables and machine-learned coarse-grained force fields represents the cutting edge, promising to further expand the scope and scale of systems that can be studied. For researchers in computational biophysics and drug development, mastering these techniques is key to unlocking a deeper, mechanistic understanding of protein function and dysfunction, ultimately accelerating the design of novel therapeutics.
In the study of protein folding, the free energy landscape provides a conceptual and quantitative framework for understanding how a polypeptide chain navigates its conformational space to attain its unique native structure [48]. Computer simulations, particularly molecular dynamics (MD), are indispensable tools for exploring these landscapes, offering insights into folding pathways, transition states, and intermediate ensembles [49]. However, the utility of any simulation is contingent upon the robustness of the free energy estimates it produces. These calculations are often hampered by a pervasive challenge: the presence of free energy barriers that create a wide gap between the biologically relevant timescales of folding and the much shorter timescales accessible by standard simulations [49]. This discrepancy can lead to inadequate sampling, where simulations become trapped in local energy minima, failing to provide a statistically representative view of the configurational ensemble. Consequently, validating convergence is not a mere supplementary step but a fundamental requirement for ensuring that computed free energies are reliable, reproducible, and physically meaningful. This guide details the core principles and practical methodologies for assessing convergence, providing researchers with a toolkit to bolster the credibility of their findings in protein folding research and drug development.
Theoretical Foundation: Free Energy Landscapes and Protein Folding
The energy landscape theory of protein folding imagines the process as a funnel, where the breadth represents the conformational entropy and the depth represents the stabilizing native interactions [48]. A perfectly funneled landscape is minimally frustrated, guiding the protein efficiently toward its native state [50]. The native state resides at a global free energy minimum, and the folding process can be viewed as a collapse in conformational entropy driven by the sequestration of hydrophobic residues and the formation of native contacts [51] [48].
The "reaction coordinate" or "progress variable" is central to quantifying movement on this landscape. Commonly used coordinates include the fraction of native contacts (Q), the radius of gyration (Rg), or root-mean-square deviation (RMSD) from the native structure. In simulations, the free energy, G, as a function of a reaction coordinate, q, is given by: G(q) = -kBT * ln P(q) where P(q) is the probability distribution of the system along q, kB is Boltzmann's constant, and T is the temperature [49]. Accurate calculation of G(q) requires that P(q) is sampled sufficiently, which is the core aim of convergence validation. Inadequate sampling results in a rough, poorly defined landscape replete with spurious minima and barriers that obscure the true folding mechanism.
Quantifiable Metrics for Assessing Convergence
Robust validation requires multiple, independent quantitative metrics. The following table summarizes key indicators and their interpretation.
Table 1: Quantitative Metrics for Validating Convergence in Free Energy Simulations
| Metric | Description | Interpretation of Convergence |
|---|---|---|
| Time-Series Stationarity | Statistical stability of the free energy estimate over simulation time. | The mean and variance of ΔG computed over sequential blocks of time show no monotonic trends and fluctuate around a stable value. |
| Statistical Inefficiency (g) | Integrated autocorrelation time of the energy or reaction coordinate; measures the correlation between samples. | A low g value indicates samples are largely independent, meaning effective sample size is large. |
| Overlap Statistics (Histogram) | Comparison of probability distributions P(q) from different simulation replicates or time segments. | High overlap (e.g., >90%) between histograms indicates consistent sampling of the same regions of phase space. |
| Bootstrap Confidence Intervals | Resampling with replacement from simulation data to estimate the distribution of the free energy. | Narrow, symmetric confidence intervals (e.g., <0.5 kcal/mol) around the mean ΔG estimate indicate high precision. |
Detailed Methodology for Key Metrics
Time-Series Stationarity Analysis:
- Procedure: Divide the total simulation time (or ensemble of simulations) into N sequential blocks (e.g., 10 blocks).
- For each block i, calculate the free energy difference (ΔGi) or the PMF along the reaction coordinate.
- Plot the block-averaged estimates (ΔGi) as a function of block index or simulation time.
- Convergence Criterion: The plot should oscillate randomly around the final mean value without any systematic drift. The standard deviation of these block averages should be small relative to the required precision (e.g., < 0.1 kcal/mol for binding affinity predictions) [52].
Statistical Inefficiency Calculation:
- Procedure: Track a key observable, such as the potential energy or reaction coordinate Q, throughout the simulation.
- Calculate the autocorrelation function C(t) for this time series.
- The statistical inefficiency, g, is defined as g = 1 + 2 * Σ C(t), integrated over the decay time.
- The effective sample size is then approximately Neff = Ntotal / g.
- Convergence Criterion: A sufficiently large N_eff (e.g., > 100 independent samples) for a reliable estimate of the mean and variance. A low g value (near 1) is ideal.
Enhanced Sampling Protocols for Achieving Convergence
Due to the high free energy barriers in protein folding, enhanced sampling techniques are often necessary. These methods accelerate the exploration of phase space and facilitate convergence.
Table 2: Enhanced Sampling Protocols for Protein Folding Simulations
| Method | Core Principle | Typical Workflow | Convergence Checks |
|---|---|---|---|
| Umbrella Sampling (US) | Restrains the system at specific windows along a pre-defined reaction coordinate using harmonic potentials. | 1. Define reaction coordinate (e.g., Q, Rg).2. Run independent simulations in each window.3. Reconstruct PMF using WHAM or MBAR. | Compare PMFs from first and second halves of production data; check for histogram overlap between adjacent windows. |
| Metadynamics | Progressively "fills" free energy minima with a history-dependent bias potential, forcing the system to explore new regions. | 1. Select collective variables (CVs).2. Deposit Gaussian potentials along the CVs during simulation.3. The free energy is the negative of the added bias. | Monitor the time evolution of the reconstructed free energy; it should fluctuate stably around a final profile once the landscape is filled. |
| Temperature Replica Exchange MD (T-REMD) | Runs multiple replicas at different temperatures; allows exchanges between replicas based on Metropolis criterion, enabling escape from local minima. | 1. Set up a temperature ladder.2. Run MD for each replica.3. Attempt periodic swaps between neighboring replicas.4. Analyze the lowest-temperature replica for folding thermodynamics. | Check for random walk of replicas through temperature space and high acceptance ratios (>20%). Ensure all relevant states are sampled in the target replica. |
The following workflow diagram illustrates a general protocol for setting up and validating a converged free energy simulation, integrating the enhanced sampling methods and validation metrics described above.
Free Energy Convergence Workflow
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Successful free energy simulation requires a suite of specialized software and force fields. The following table catalogs key resources.
Table 3: Essential Research Reagents and Computational Tools for Free Energy Simulations
| Category / Item | Specific Examples | Function and Application |
|---|---|---|
| Biomolecular Force Fields | CHARMM, AMBER, OPLS-AA | Provide the potential energy functions and parameters governing atomic interactions; critical for accurate modeling of protein energetics. |
| Simulation Engines | GROMACS, NAMD, OpenMM, DESMOND | High-performance software to perform molecular dynamics simulations, including support for enhanced sampling algorithms. |
| Free Energy Analysis Tools | WHAM, MBAR, plumed | Post-processing tools to calculate Potentials of Mean Force (PMF) and analyze free energies from simulation data. PLUMED also enables on-the-fly enhanced sampling. |
| Enhanced Sampling Suites | PLUMED, SSAGES | Libraries that integrate with major MD engines to implement a wide variety of enhanced sampling methods like metadynamics, umbrella sampling, and variationally enhanced sampling. |
| System Preparation Suites | CHARMM-GUI, AMBER tleap, MDWeb | Web servers and software tools for building complex simulation systems, including solvation, ion addition, and input file generation. |
In protein folding research, the journey from simulation trajectory to scientific insight is bridged by a rigorous demonstration of convergence. By employing a multi-faceted validation strategy—combining quantitative metrics like stationarity analysis and statistical inefficiency with powerful enhanced sampling protocols—researchers can ensure their free energy estimates are robust. This diligence is paramount for generating reliable predictions of folding mechanisms, stability, and molecular interactions that can confidently guide experimental efforts in structural biology and rational drug design.
The simulation of protein folding is a cornerstone of computational biology, enabling researchers to interpret experimental data, design novel experiments, and probe the effects of mutations on protein function and stability. While major progress has been made in understanding the folding of small proteins, research on larger, multidomain proteins—which constitute the majority of proteins in eukaryotes, bacteria, and archaea—remains a significant challenge [19]. The median protein length is 532 amino acids in eukaryotes, 365 in bacteria, and 329 in archaea, with folding times ranging from microseconds to tens of minutes [19]. Simulating the folding of these larger, slower-folding proteins using standard all-atom Molecular Dynamics (MD) is often computationally intractable, even with anticipated increases in computing power [19]. Large proteins frequently fold via long-lived, partially folded intermediates, which correspond to deep local energy minima on the folding landscape. Conventional MD simulations often become trapped in these minima, making it difficult to observe complete folding pathways [19]. This article examines optimized computational strategies, grounded in free energy landscape theory, to overcome these memory and runtime barriers for complex protein systems.
Free Energy Landscape Theory and Its Implications for Simulation
The energy landscape theory of protein folding provides a conceptual framework for understanding both the challenges and solutions for simulating large proteins. This theory posits that a protein's conformational space can be described by a funnel-shaped energy landscape, where the native, folded state resides at the global free energy minimum [19] [53]. The principle of minimal frustration suggests that naturally occurring protein sequences have evolved so that interactions stabilizing the native structure are highly optimized, with minimal conflict between competing interactions [19]. This results in a landscape that is globally funneled but still contains local ruggedness—manifested as local minima and barriers—which corresponds to partially folded intermediates and kinetic traps [19].
For large proteins, this landscape is inherently more complex. The larger number of residues and potential domain interactions leads to a higher-dimensional landscape with more numerous and deeper metastable states. This complexity directly translates into computational intractability for all-atom MD, as simulations must overcome significant entropic barriers and sample a vast conformational space. The funneled nature of the landscape, however, justifies the use of simplified, native-centric models that bias the simulation toward the native structure, thereby dramatically reducing the computational resources required to explore relevant folding pathways [19].
Strategic Approaches to Manage Memory and Runtime
Coarse-Grained (CG) Modeling
Coarse-grained (CG) modeling is a powerful strategy to reduce computational cost by representing groups of atoms with single "beads," thereby decreasing the number of interacting particles and the complexity of the potential energy surface. The development of a universal, computationally efficient CG model with predictive accuracy comparable to all-atom MD has been a long-standing challenge [4].
A breakthrough approach involves combining deep-learning methods with large and diverse training sets of all-atom protein simulations to develop a bottom-up CG force field that is chemically transferable. This means the model can perform extrapolative molecular dynamics on new protein sequences not present during the training phase [4]. This machine-learned CG model has been demonstrated to successfully predict metastable states of folded, unfolded, and intermediate structures, as well as the fluctuations of intrinsically disordered proteins and the relative folding free energies of protein mutants [4].
- Key Advantage: This approach is several orders of magnitude faster than all-atom models while maintaining high predictive accuracy for folding landscapes [4].
- Memory Benefit: By reducing the system's degrees of freedom, CG models require significantly less memory to store particle coordinates and interaction potentials.
Structure-Based Models (SBM) and Enhanced Sampling
Structure-based models (SBM), also known as Gō models, offer another highly efficient alternative. These models simplify the energy landscape by encoding the protein's native structure directly into the potential energy function, primarily favoring native contacts while largely ignoring non-native interactions [19]. This reduction in complexity makes rare folding events computationally accessible.
To further improve sampling efficiency, SBMs are often combined with enhanced sampling methods. A prominent example is the parallel-tempering (PT) method, also known as replica exchange molecular dynamics (REMD) [4] [19]. This technique runs multiple simulations of the same system at different temperatures (or with other modified Hamiltonian parameters) and periodically attempts to exchange configurations between them. This allows the system to escape deep local energy minima at low temperatures by temporarily visiting higher temperatures where barriers are easier to overcome.
- Runtime Benefit: PT simulations ensure converged sampling of the equilibrium distribution, which is crucial for obtaining accurate folding/unfolding free energy landscapes [4].
- Application: PT has been successfully used with both CG and all-atom-based methods to achieve converged folding landscapes for proteins that would otherwise be inaccessible to standard MD [4] [19].
Geometric Deep Learning and Graph Representations
Geometric deep learning represents a paradigm shift in analyzing biomolecular structures. In this approach, a protein's 3D structure is transformed into a graph representation, where nodes (representing amino acids or atoms) are connected by edges based on spatial proximity or specific chemical interactions [54]. Graph Neural Networks (GNNs) can then learn from these representations, compressing topological and biochemical information into embeddings useful for downstream prediction tasks [54].
Libraries like Graphein facilitate this process by providing turn-key tools for converting protein structures from databases like the PDB or AlphaFold into machine-learning-ready graph formats compatible with popular deep learning libraries such as DGL and PyTorch Geometric [55].
- Runtime & Memory Benefit: This approach can rapidly predict structural properties and dynamics without explicitly simulating physics-based trajectories, offering immense savings in computational time. The graph representation itself is a more memory-efficient abstraction of the protein structure.
- Function: While not a direct simulation method, it is a powerful tool for rapid analysis and prediction that can guide and complement more detailed simulations.
The following workflow diagram illustrates how these different strategies can be integrated into a cohesive computational pipeline for studying large protein systems.
Diagram 1: A unified workflow for optimizing large protein simulations. This diagram integrates coarse-graining, native-centric potentials, enhanced sampling, and deep learning to efficiently navigate the free energy landscape.
Quantitative Comparison of Simulation Methods
The choice of simulation method involves a direct trade-off between computational cost and the level of chemical detail. The following table summarizes the key characteristics of different approaches, highlighting their suitability for large systems.
Table 1: Performance and characteristics of protein simulation methods.
| Method | Typical System Size | Computational Cost | Relative Speed | Key Applications |
|---|---|---|---|---|
| All-Atom MD [19] | ~100 amino acids | Extremely High | 1x (Baseline) | Atomistic detail, folding mechanisms, ligand binding |
| Machine-Learned CG [4] | Small to Large proteins | Low | "Orders of magnitude faster" than all-atom MD | Folding landscapes, metastable states, mutant free energies |
| Structure-Based Models (SBM) [19] | Large proteins (e.g., 394-residue serpins) | Very Low | "Many orders of magnitude less" than conventional MD | Folding pathways, intermediate states, topology effects |
| All-Atom with Enhanced Sampling [19] | Larger than possible with standard MD | High (but more efficient than standard MD) | Faster than standard MD for equivalent sampling | Overcoming large barriers, probing rare events with atomistic detail |
Table 2: Analysis of performance gains from coarse-grained modeling.
| Metric | All-Atom MD | Machine-Learned Coarse-Grained Model |
|---|---|---|
| Computational Efficiency | Baseline | Several orders of magnitude improvement [4] |
| Simulation Transferability | System-specific; force fields are general but sampling is limited. | Chemically transferable; can simulate proteins with low (<40%) sequence similarity to training set [4] |
| Predictive Capability | High for small, fast-folding proteins. | Predicts metastable folded/unfolded states, intermediate structures, and relative folding free energies of mutants [4] |
Detailed Experimental and Computational Protocols
Protocol for Machine-Learned Coarse-Grained Simulation
This protocol outlines the steps for developing and deploying a transferable CG model as described in the recent literature [4].
Training Data Generation:
- Generate a diverse dataset of all-atom, explicit-solvent MD simulations of small proteins with varied folded structures.
- Include simulations of dimers of mono- and dipeptides to capture fundamental interaction data.
Model Training:
- Employ a deep-learning architecture, such as CGSchNet, designed for molecular systems.
- Use a variational force-matching approach to train the neural network potential. The objective is for the CG force field to reproduce the forces and distributions of the reference all-atom system.
Simulation of Unseen Proteins:
- Select target proteins with low sequence similarity (<40%) to any sequence in the training or validation datasets.
- Conduct extensive CG molecular dynamics simulations using the trained model. To ensure robust sampling, employ advanced techniques like parallel-tempering (PT).
Validation and Analysis:
- Compare the CG simulation results against reference all-atom simulations or experimental data where available.
- Standard metrics for comparison include:
- Free energy surfaces as a function of reaction coordinates (e.g., Fraction of Native Contacts Q, Cα Root-Mean-Square Deviation (RMSD)).
- Root-mean-square fluctuations (RMSF) of the folded state.
- Populations of metastable states and folding intermediates.
- Relative folding free energies for protein mutants.
Protocol for Folding Simulation Using Structure-Based Models
This protocol is based on established methods for simulating the folding of large proteins like serpins using native-centric approaches [19].
Model Construction:
- Obtain the native structure of the target protein from an experimental database (e.g., PDB).
- Construct a coarse-grained representation, typically using a single bead per amino acid (Cα model).
- Define the contact map of the native structure. A contact is typically formed if two Cα atoms are within a specified cutoff distance (e.g., 7-8 Å) in the native state.
Potential Energy Function:
- Define a potential energy function that strongly favors the formation of native contacts. Non-native interactions are usually repulsive or weakly attractive to prevent frustration.
- The total energy often includes terms for bond stretching, angle bending, and dihedral angles, all derived from the native structure.
Enhanced Sampling Execution:
- Due to the complex energy landscape of large proteins, implement a replica exchange molecular dynamics (REMD) scheme.
- Set up a temperature ladder with a sufficient number of replicas to ensure good exchange rates between adjacent temperatures.
- Run the simulation, allowing for periodic configuration swaps between replicas according to the Metropolis criterion.
Pathway and Intermediate Analysis:
- Analyze the resulting trajectories to identify folding pathways and populated intermediates.
- Use clustering algorithms to group structurally similar conformations.
- Construct free energy profiles projected onto relevant order parameters (e.g., Q, RMSD) to identify and characterize stable intermediates.
- Compare the computationally predicted intermediates with experimental data (e.g., from hydrogen-deuterium exchange or fluorescence spectroscopy) for validation.
Table 3: A toolkit of essential software and databases for large-scale protein folding studies.
| Tool / Resource | Type | Primary Function | Relevance to Large Systems |
|---|---|---|---|
| Graphein [55] | Python Library | Converts protein structures and interaction networks into graph representations for machine learning. | Facilitates geometric deep learning; enables rapid analysis of large complexes and networks without simulation. |
| CGSchNet [4] | Deep Learning Model | A neural network architecture for learning transferable coarse-grained force fields from all-atom data. | Provides a ready-made framework for developing highly efficient and accurate CG models for large proteins. |
| AlphaFold Structure Database [55] | Database | Provides high-accuracy predicted protein structures for nearly the entire human proteome. | Source of reliable native structures for proteins without experimental data, essential for building structure-based models. |
| GetContacts [55] | Computational Tool | Computes and analyzes intermolecular and intramolecular interactions within structures. | Helps define native contact maps for Structure-Based Models and analyze interaction networks in large complexes. |
| StringDB / BioGrid [55] | Database | Curated databases of protein-protein interactions. | Informs the construction of protein-protein interaction graphs, crucial for studying multi-protein assemblies. |
Benchmarking and Validation: Linking Simulations to Experiments and AI Prediction
Quantitative Comparison with Single-Molecule Experiments (FRET, Optical Tweezers)
The study of protein folding has been revolutionized by the ability to probe individual molecules, allowing researchers to dissect the complex pathways and heterogeneous behaviors that are obscured in ensemble-averaged measurements. Central to this revolution are two powerful techniques: single-molecule Förster resonance energy transfer (smFRET) and optical tweezers (OT). When framed within the context of free energy landscape theory, these methods provide complementary insights into the conformational states, transition barriers, and kinetic trajectories that define a protein's folding journey. smFRET functions as a nanoscale molecular ruler, measuring distances between specific sites on a protein to report on conformational changes. Optical tweezers employ focused laser light to apply and measure piconewton-scale forces, directly probing the mechanical unfolding and refolding pathways of proteins. Together, these techniques enable the direct experimental reconstruction of free energy landscapes, revealing not only the stable folded and unfolded basins but also the rare intermediate states and transition pathways that are fundamental to understanding protein folding mechanisms. This technical guide provides an in-depth comparison of these methodologies, their quantitative capabilities, experimental implementation, and their critical role in validating and informing computational models of protein folding landscapes.
Technical Foundations and Quantitative Capabilities
Core Physical Principles and Quantitative Ranges
Single-Molecule FRET relies on the distance-dependent non-radiative energy transfer between a donor fluorophore and an acceptor fluorophore. The FRET efficiency, (E), reports on the intervening distance, (R), through the relationship (E = [1 + (R/R0)^6]^{-1}), where (R0) is the Förster radius at which the energy transfer efficiency is 50% [56]. This relationship makes smFRET exquisitely sensitive to distance changes in the 1-10 nm range, perfectly suited for tracking conformational changes in proteins [57]. In practice, the FRET efficiency is calculated from the emission intensities as (E = IA/(IA + γID)), where (IA) and (I_D) are the background-corrected acceptor and donor intensities, and (γ) is a correction factor accounting for differences in quantum yields and detection efficiencies between the two channels [57].
Optical Tweezers operate on the principle of radiation pressure, where a tightly focused laser beam creates a potential well that can trap dielectric particles ranging from hundreds of nanometers to microns in size [58]. By attaching a protein between two trapped beads or one trapped bead and a surface, researchers can apply controlled forces and measure displacements with remarkable precision. Modern optical tweezers achieve force resolutions down to 0.1 pN and spatial resolutions as fine as 0.1 nm, enabling the detection of single base-pair steps in nucleic acid-processing proteins and sub-nanometer structural transitions in proteins [59].
Table 1: Quantitative Comparison of smFRET and Optical Tweezers
| Parameter | Single-Molecule FRET | Optical Tweezers |
|---|---|---|
| Distance Resolution | 1-10 nm range [57] | 0.1-1 nm (displacement) [59] |
| Force Capability | Not direct | 0.1-100 pN [58] |
| Temporal Resolution | 1-100 ms (cameras); 10-50 μs (SPADs) [57] | 0.1-1 ms [59] |
| Energy Resolution | ~0.1 kT (through fluctuations) | ~0.1 kT (direct) |
| Key Observables | Inter-dye distances, conformational dynamics, subpopulations [57] | End-to-end extension, unfolding/refolding forces, mechanical work [59] |
| Free Energy Reconstruction | From state populations and transition rates [60] | From force-extension relationships (Jarzynski equality, Crooks fluctuation theorem) |
Operational Modes and Experimental Configurations
smFRET experiments are typically conducted in two primary configurations. In surface-immobilized experiments, biomolecules are tethered to a passivated surface and observed using total internal reflection fluorescence (TIRF) microscopy to minimize background [60]. This approach allows observation of individual molecules for extended periods (1-30 seconds), ideal for studying slow conformational dynamics [57]. In freely-diffusing experiments, molecules diffuse through a confocal excitation volume, producing bursts of fluorescence as they pass through the detection zone. This method avoids potential surface perturbations and enables higher temporal resolution (10-50 μs) using single-photon avalanche diodes (SPADs) [57].
Optical tweezers experiments for protein folding typically employ either a single-bead trap (one bead trapped and attached to a surface) or a dual-bead trap (one bead in each of two optical traps) [59]. The dual-trap configuration is particularly powerful as it enables precise control over the force applied to the protein while minimizing instrument drift. More advanced configurations include angular optical traps that can measure torques and rotations, and combined optical tweezers-fluorescence microscopy systems that simultaneously monitor force and FRET signals [61].
Experimental Methodologies and Protocols
Sample Preparation and Labeling Strategies
smFRET Sample Preparation: Successful smFRET experiments require site-specific labeling of the protein of interest with donor and acceptor fluorophores. The most common approach utilizes cysteine mutations introduced at desired positions, followed by labeling with maleimide-functionalized dyes such as Cy3/Cy5, ATTO550/ATTO647N, or Alexa555/Alexa647 [56]. Key considerations for dye selection include brightness, photostability, spectral separation, and minimal environmental sensitivity. An optimal dye pair should have a Förster radius ((R_0)) matching the expected distance changes, typically in the 5-6 nm range [56].
Critical steps in sample preparation include:
- Surface passivation: Using PEG-coated surfaces to prevent non-specific adsorption of biomolecules [60].
- Oxygen scavenging systems: Employing enzymatic systems (e.g., protocatechuate dioxygenase with protocatechuic acid) or chemical additives (Trolox) to reduce photobleaching and suppress blinking [56].
- Bioconjugation: Site-specific attachment via cysteine chemistry, unnatural amino acids, or peptide tags for immobilization and labeling.
Optical Tweezers Sample Preparation: For optical tweezers experiments, proteins are typically tethered between micron-sized beads using DNA handles. This configuration separates the protein from the bead surfaces and minimizes non-specific interactions. A common strategy involves:
- DNA handle preparation: Modification of DNA fragments (~600-1000 bp) with biotin and digoxigenin at opposite ends.
- Protein labeling: Introduction of specific tags (e.g., HaloTag, SNAP-tag) or cysteine residues for conjugation to complementary DNA handles.
- Assembly: Formation of the bead-protein-bead assembly through specific interactions (streptavidin-biotin for biotinylated handles, anti-digoxigenin for digoxigeninated handles) [61].
Recent advances enable orthogonal labeling for combined optical tweezers and smFRET experiments. For example, a study on Hsp90 utilized thiol-maleimide chemistry for FRET pair attachment (maleimide-Atto 550 and maleimide-Atto 647N) alongside inverse electron demand Diels-Alder cycloaddition for DNA handle conjugation, enabling simultaneous mechanical and FRET measurements [61].
Instrumentation and Data Acquisition
smFRET Instrumentation: A basic smFRET setup with TIRF microscopy can be constructed from off-the-shelf components and typically includes:
- Lasers: 532 nm and 633 nm for excitation of common dye pairs
- Microscope: Inverted microscope with high-numerical-aperture objective (NA ≥ 1.4)
- Detectors: Electron-multiplying CCD (EMCCD) or scientific CMOS cameras for imaging
- Dichroic mirrors and filters: For spectral separation of donor and acceptor emissions
- Flow chamber: For sample introduction and immobilization [60]
Data acquisition involves recording movies of surface-immobilized molecules at frame rates appropriate for the dynamics of interest (typically 10-100 Hz). For freely-diffusing molecules, photon arrival times are recorded with microsecond resolution using SPAD detectors and time-correlated single photon counting (TCSPC) electronics [57].
Optical Tweezers Instrumentation: A dual-trap optical tweezers system typically consists of:
- Laser source: High-power infrared laser (1064 nm) to minimize photodamage
- Beam steering: Acousto-optic deflectors (AODs) or spatial light modulators (SLMs) for trap positioning
- Detection: Back-focal plane interferometry with quadrant photodiodes for precise position and force detection
- Piezo stage: For sample positioning and manipulation
- Force calibration: Typically performed through power spectrum analysis of bead position fluctuations [59]
Data acquisition involves recording bead positions at high sampling rates (10-100 kHz) while controlling trap positions or forces through feedback mechanisms.
Figure 1: Experimental Workflow for Single-Molecule Protein Folding Studies. This diagram illustrates the parallel pathways for smFRET and optical tweezers experiments, from initial sample preparation to final free energy landscape reconstruction.
Data Analysis and Free Energy Landscape Reconstruction
smFRET Data Processing and Analysis
The analysis of smFRET data involves multiple stages to extract quantitative information about protein dynamics:
Trace Selection and Validation: Identification of single-molecule traces showing anti-correlated donor and acceptor signals, proper single-step photobleaching, and sufficient signal-to-noise ratio. Traditional methods require manual curation, but recent advances in deep learning (e.g., DeepFRET software) enable automated trace classification with >95% accuracy, significantly accelerating this process [62].
FRET Efficiency Calculation: For each time point, the FRET efficiency is calculated as (E = IA/(IA + γI_D)), where the correction factor (γ) accounts for differences in quantum yields and detection efficiencies between channels [57].
State Identification and Kinetic Analysis: Idealization of FRET trajectories into discrete states using change-point algorithms (e.g., hidden Markov modeling) followed by transition rate extraction [60].
Free Energy Landscape Construction: From the equilibrium populations ((Pi)) of discrete states, the relative free energies can be calculated as (ΔGi = -kBT \ln(Pi)). Kinetic information from state transitions provides additional constraints on barrier heights between states.
Advanced analysis methods include multiparameter fluorescence detection, which incorporates fluorescence lifetimes and anisotropies to provide additional constraints on distance measurements and dye dynamics [57].
Optical Tweezers Data Analysis
Analysis of optical tweezers data focuses on extracting mechanical properties and reconstructing energy landscapes:
Force-Extension Analysis: Experimental force-extension curves are fitted to polymer elasticity models such as the worm-like chain (WLC) model to extract protein end-to-end distances and persistence lengths [59].
Work Calculation: The mechanical work performed during unfolding/refolding cycles is calculated from the integral (W = \int F\,dx).
Free Energy Reconstruction: Using non-equilibrium work relations such as the Jarzynski equality ((e^{-ΔG/kBT} = \langle e^{-W/kBT} \rangle)) or the Crooks fluctuation theorem, the equilibrium free energy landscape can be reconstructed from multiple non-equilibrium pulling trajectories [59].
Kinetic Analysis: Force-dependent transition rates can be extracted from unfolding/refolding force distributions and modeled within the framework of Bell's model or more sophisticated landscape models.
Table 2: Key Research Reagents and Materials for Single-Molecule Experiments
| Reagent/Material | Function | Examples/Specifications |
|---|---|---|
| Fluorescent Dyes | FRET donor/acceptor pairs | Cy3/Cy5, ATTO550/ATTO647N, Alexa555/Alexa647 [56] |
| Oxygen Scavengers | Reduce photobleaching | Protocatechuate dioxygenase (PCD)/protocatechuic acid (PCA), Trolox [56] |
| Surface Passivation | Prevent non-specific binding | PEG-biotin/PEG-silane, BSA-biotin [60] |
| DNA Handles | Tether proteins in OT | ~600-1000 bp DNA with biotin/digoxigenin modifications [61] |
| Functionalized Beads | Mechanical manipulation | Streptavidin-coated polystyrene beads [59] |
| Bioorthogonal Tags | Site-specific labeling | HaloTag, SNAP-tag, non-canonical amino acids [61] |
Integration with Free Energy Landscape Theory
Single-molecule experiments provide the critical connection between theoretical models of protein folding and experimental observations. According to energy landscape theory, protein folding can be described as a diffusion process on a funnel-shaped energy landscape where the native state resides at the global minimum [4]. Both smFRET and optical tweezers offer direct experimental access to key features of this landscape:
smFRET reveals the distribution of conformational states along specific reaction coordinates defined by the dye labeling positions. By monitoring transitions between these states, researchers can map the local topography of the energy landscape, including metastable intermediates and transition state barriers. The heterogeneity observed in single-molecule trajectories directly reflects the rough nature of the folding landscape, with different molecules following distinct pathways—a prediction of landscape theory that is invisible to ensemble techniques.
Optical Tweezers provide a direct measure of the mechanical work required to navigate the energy landscape along a force-defined reaction coordinate. The force-dependent unfolding and refolding rates follow Arrhenius-like behavior, enabling extraction of the distance to the transition state ((Δx^‡)) and the height of the energy barriers ((ΔG^‡)). Reconstruction of the full energy landscape from work measurements using Jarzynski's equality or Crooks' theorem provides a comprehensive picture of the folding thermodynamics.
Recent advances combine these techniques with coarse-grained molecular dynamics simulations parameterized using machine learning approaches. For example, a recently developed transferable coarse-grained model demonstrated the ability to predict metastable states of folded, unfolded, and intermediate structures, with performance comparable to all-atom molecular dynamics but at several orders of magnitude lower computational cost [4]. Such integrated approaches promise to bridge the gap between simulation and experiment, enabling atomic-level interpretation of single-molecule data while experimentally validating computational models.
Emerging Frontiers and Combined Approaches
The field of single-molecule studies is rapidly advancing through technical innovations and methodological integrations:
Combined Force and Fluorescence Measurements: The integration of optical tweezers with smFRET enables simultaneous monitoring of mechanical forces and internal conformational changes. This approach was recently demonstrated in a study of Hsp90, where researchers utilized orthogonal labeling strategies to attach both FRET pairs and DNA handles to the same protein, enabling correlation of mechanical unfolding transitions with internal distance changes [61].
Machine Learning-Enhanced Analysis: Deep learning approaches are revolutionizing data analysis in single-molecule biophysics. Tools like DeepFRET provide automated, objective classification of smFRET data with >95% accuracy, dramatically reducing analysis time and minimizing human bias [62]. Similar approaches are being developed for optical tweezers data to automatically detect transitions and classify unfolding pathways.
Advanced Coarse-Grained Simulations: Machine-learned coarse-grained models trained on diverse protein simulations now enable transferable force fields that can predict folding pathways, metastable states, and relative folding free energies for proteins not included in the training set [4]. These models provide a powerful bridge between all-atom simulations and single-molecule experiments.
High-Throughput Methodologies: Advances in instrumentation and data analysis are enabling high-throughput single-molecule studies, opening the door to systematic investigations of protein folding landscapes under varying conditions and for large sets of protein variants.
As these techniques continue to evolve and integrate with computational approaches, they promise to provide increasingly detailed and quantitative maps of protein energy landscapes, ultimately enabling predictive understanding of protein folding, stability, and function.
The prediction of protein folding rates, free energy landscapes, and native state recognition represents a critical benchmark for computational methods operating within the framework of free energy landscape theory. This theory posits that protein folding is a stochastic process guided by a funnel-shaped landscape, where the native state resides at the thermodynamic minimum [25] [63]. The "principle of minimal frustration" ensures that naturally evolved proteins have landscapes that are relatively smooth, minimizing kinetic traps and enabling efficient folding [25]. Assessing a model's predictive power across these interrelated phenomena—kinetics (folding rates), thermodynamics (free energies), and structural recognition (native state)—provides a rigorous test of its physical realism and its ability to capture the essential interplay between native topology, chain dynamics, and energetics that governs the folding process. This review synthesizes advances in physics-based and machine-learned models, evaluating their success in navigating the complex multi-scale challenges of protein folding simulation.
Core Predictive Tasks and Quantitative Metrics
Folding Rates
Protein folding rates ((kf)) quantify the kinetics of the transition from the unfolded to the native state. Within energy landscape theory, the rate is determined by the free energy barrier separating these states. Experimentally, rates are strongly correlated with native-state contact order, a measure of topological complexity defined as the average sequence separation between residues in spatial contact, divided by the total chain length [51] [64]. This relationship underscores the dominant role of native topology in determining folding kinetics. Simulation studies with Gō-like models have derived a unified scaling law for the folding rate at the transition temperature: (kF \sim \exp(-c \cdot RCO \cdot N^{0.6})), where (RCO) is the relative contact order and (N) is the number of residues [64]. This law couples the influence of native topology ((RCO)) with chain length ((N)), bridging theoretical predictions and experimental observations.
Free Energy Landscapes and Barriers
The free energy landscape is a central concept in protein folding, providing a unified framework for understanding both thermodynamics and kinetics [63]. The landscape is typically projected onto one or two reaction coordinates, such as the fraction of native contacts ((Q)) or the number of native residues ordered ((n)). The height of the free energy barrier ((\Delta G^\ddagger)) between the unfolded and native basins directly determines the folding rate ((kf \sim \exp(-\Delta G^\ddagger / kB T))). Physics-based models aim to reconstruct these landscapes from native structure information alone. For example, the WSME (Wako-Saitô-Muñoz-Eaton) model and its derivatives use statistical mechanics to calculate exact partition functions and rigorously determine free energy landscapes, transition states, and intermediates [65].
Native State Recognition
Native state recognition, or threading, is the most fundamental test of a structure prediction model. It assesses whether the model's energy function is minimized for the experimentally determined native structure compared to decoy or misfolded structures [25]. A successful model must not only identify the native state but also stabilize it as the global free energy minimum. This task evaluates the model's ability to embody the Principle of Minimal Frustration, where native interactions are sufficiently strong to overcome non-native frustrations that could trap the chain in non-functional states [25].
Table 1: Key Predictive Tasks in Protein Folding
| Predictive Task | Central Question | Key Quantitative Metric | Theoretical Basis in Landscape Theory |
|---|---|---|---|
| Folding Rates | How fast does the protein fold? | Folding rate constant ((k_f)), Correlation with Contact Order | Height of the free energy barrier on a funneled landscape |
| Free Energies | What is the stability of states along the folding pathway? | Free energy ((G)), Barrier height ((\Delta G^\ddagger)) | Direct mapping of the landscape from the partition function |
| Native State Recognition | Does the model select the correct native fold? | Energy/Z-score of native vs. decoys, pLDDT | "Minimal frustration" – the native state is the global energy minimum |
Established Physics-Based Models and Methodologies
Free Energy Functional Models
These models explicitly parameterize the free energy as a function of the degree of native structure formation. A seminal example uses a free energy function ((G)) that balances favorable native interactions against entropic costs [51]: [ G = - \gamma (A{buried} - A{unfolded}) + \alpha n + \beta \ln(L/L_0) ] Here, the first term represents the favorable energy from burying surface area upon forming native contacts, the second term is the entropic cost of ordering (n) residues, and the third term is the additional entropic penalty for closing a loop of length (L) between two ordered segments [51]. The parameters (\gamma), (\alpha), and (\beta) are tuned based on physical estimates. The model considers configurations with one or two contiguous native segments, and folding pathways are identified by allowing single residues to order or disorder. The transition state ensemble is rigorously defined as the highest free-energy configurations on the lowest free-energy pathways connecting the unfolded and native states [51].
Structure-Based Statistical Mechanical Models (WSME and WSME-L)
The WSME model and its extension, the WSME-L model, are Ising-like models that provide an exact analytical solution for the free energy landscape [65]. In the original WSME model, each residue is in either a native (1) or non-native (0) state. A native contact between residues (i) and (j) is formed only if all intervening residues are also native (i.e., (m{i,j} = \prod{k=i}^{j} m_k = 1)). This assumption enforces a strict coupling between local and non-local interactions but fails for multidomain proteins where discontinuous segments can fold independently.
The WSME-L model overcomes this by introducing "virtual linkers" that enable nonlocal interactions between distant residues (u) and (v) without requiring the intervening sequence to be folded [65]. The Hamiltonian for a model with one linker is: [ H^{(u,v)}({m}) = \sum{i=1}^{N-1} \sum{j=i+1}^{N} \epsilon{i,j} \left\lceil \frac{m{i,j} + m{i,j}^{(u,v)}}{2} \right\rceil ] where (m{i,j}^{(u,v)}) is 1 if residues (i) and (u), and (v) and (j), are connected by native stretches. The final model is an ensemble of such partition functions, each with a linker at a specific native contact, providing a comprehensive description of the landscape that successfully predicts complex multidomain folding mechanisms [65].
Gō-like Models and Molecular Dynamics
Gō-like models are coarse-grained molecular dynamics models where the energy function is designed to make the native structure the unique global minimum—a "perfect funnel" [64]. These models include attractive interactions only for native contacts (those present in the target structure) and repulsive interactions for non-native overlaps. Simulations with these models have been instrumental in validating the relationship between contact order and folding rates and in characterizing the structure of the denatured state and transition state ensemble for many small proteins [64]. The AWSEM model is an example of a more sophisticated Gō-like model that incorporates transferable contact potentials, allowing it to be applied to sequences without prior knowledge of the native structure [25].
Emerging Machine-Learned Coarse-Grained Models
The development of machine-learned coarse-grained (CG) models represents a paradigm shift. These models use deep learning, trained on large datasets of all-atom molecular dynamics simulations, to infer a potential of mean force that governs the CG degrees of freedom [4]. A key advance is the creation of transferable models that can be applied to protein sequences not seen during training.
One such model, CGSchNet, uses a deep neural network to model many-body interactions between CG sites (typically Cα atoms) [4]. The model is trained using a variational force-matching approach, where the goal is to match the forces on CG sites derived from all-atom reference simulations. The resulting force field can then be used to run molecular dynamics simulations on new proteins that are several orders of magnitude faster than all-atom simulations.
These models have demonstrated remarkable success in predicting folding/unfolding landscapes for small fast-folding proteins, populating metastable states (folded, unfolded, and intermediates), and reproducing the fluctuations of intrinsically disordered proteins [4]. Furthermore, they show promise in predicting relative folding free energies of protein mutants, a challenging test of thermodynamic accuracy.
Table 2: Comparison of Model Capabilities and Limitations
| Model Type | Key Features | Strengths | Limitations / Challenges |
|---|---|---|---|
| Free Energy Functional [51] | Analytic free energy function; Native contacts & entropy | Computationally efficient; Rigorous definition of pathways and TSE | Simplified configuration set; Relies on knowledge of native structure |
| WSME/WSME-L [65] | Ising-like; Exact analytical solution; Virtual linkers | Handles multi-domain folding; Can incorporate disulfide bonds | Still a native-centric model; Performance can vary with protein topology |
| Gō-like/AWSEM [25] [64] | Coarse-grained MD; Perfect funnel landscape; Can be transferable | Good for kinetics & TSE analysis; AWSEM is sequence-based | May struggle with alternative metastable states [4] |
| Machine-Learned CG [4] | Neural network potential; Trained on atomistic data | Transferable; Fast; Captures non-native states in principle | Training data dependent; Can be unstable; Explainability is limited |
Experimental Protocols for Model Validation
Protocol 1: φ-Value Analysis
Purpose: To validate the structural details of the predicted transition state ensemble (TSE) against experimental data. Principle: φ-values measure the extent to which a point mutation at a specific residue affects the free energy of the transition state ((\Delta \Delta G^\ddagger)) relative to its effect on the stability of the native state ((\Delta \Delta G_N)). A φ-value of 1 indicates the residue's native interactions are fully formed in the TSE, while a value of 0 indicates they are completely absent. Computational Procedure:
- Identify the ensemble of transition-state configurations from the model (e.g., the 100 lowest free-energy transition-state configurations) [51].
- Compute the change in free energy of this TSE ((\Delta \Delta G^\ddagger)) for an in-silico mutation that truncates a sidechain. This is often done by perturbing the contact energy ((\epsilon_{i,j})) for the mutated residue.
- Compute the change in free energy of the native state ((\Delta \Delta G_N)) for the same mutation.
- Calculate the predicted φ-value as: (\phi = \frac{\Delta \Delta G^\ddagger}{\Delta \Delta G_N}).
- Average this value over the entire transition-state ensemble and compare the profile of φ-values across the protein sequence with experimental data [51] [64].
Protocol 2: Folding Landscape Reconstruction
Purpose: To compute the equilibrium free energy landscape as a function of reaction coordinates and identify metastable states, barriers, and folding pathways. Procedure for WSME-L Model:
- Input: The native protein structure (e.g., from PDB) to identify all native contacts (\epsilon_{i,j}).
- Partition Function Calculation: Use the transfer matrix method to compute the exact partition function (Z_L(n)) as defined by the WSME-L Hamiltonian, which includes contributions from all possible virtual linkers [65].
- Free Energy Calculation: Calculate the free energy as a function of the order parameter (n) (fraction of native residues) using the standard relationship: (G(n) = -kB T \ln ZL(n)).
- Analysis: Plot the free energy profile (G(n)) or the 2D landscape (G(n, Q)). The global minimum should correspond to the native state ((n \approx 1)). The highest point on the lowest free energy pathway between unfolded ((n \approx 0)) and folded states defines the transition state.
Procedure for Machine-Learned CG Models:
- Sampling: Run extensive molecular dynamics simulations (often enhanced by parallel tempering/replica exchange) to ensure convergence of equilibrium properties [4].
- Reaction Coordinates: Calculate the fraction of native contacts ((Q)) and the Cα root-mean-square deviation (RMSD) from the native structure for each sampled configuration.
- Landscape Estimation: Construct a histogram of the sampled configurations in the (Q, RMSD) space and convert it to a free energy surface using (G(Q, RMSD) = -k_B T \ln P(Q, RMSD)), where (P) is the probability distribution.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Software and Data Resources for Protein Folding Prediction
| Tool/Resource | Type | Function in Research |
|---|---|---|
| AWSEM [25] | Coarse-grained Force Field | Transferable model for de novo structure prediction and folding pathway simulation. |
| WSME-L Model [65] | Analytical Statistical Model | Predicting detailed folding mechanisms, pathways, and free energy landscapes from a native structure. |
| CGSchNet [4] | Machine-Learned CG Model | Running fast, transferable molecular dynamics to predict landscapes and folding free energies. |
| AlphaFold Database [66] | Structure Repository | Source of high-accuracy predicted structures for initial coordinates or native contact definition. |
| Protein Data Bank (PDB) | Structure Repository | Source of experimental native structures for model parameterization, validation, and training. |
Visualizing Model Workflows and Relationships
The following diagrams illustrate the logical workflow of a free-energy functional model and the key relationships between different modeling paradigms.
Diagram 1: Workflow of a Free-Energy Functional Model. This chart outlines the key steps in a physics-based approach to predicting folding mechanisms, starting from the native structure and culminating in testable predictions [51].
Diagram 2: Convergence of Research Paradigms. This diagram shows how different research traditions are contributing to the ultimate goal of a universal predictive model for protein folding. The integration of physical principles with data-driven accuracy is a key trend in the field [4] [67].
The assessment of predictive power across folding rates, free energies, and native state recognition remains the cornerstone of progress in protein folding simulation. Physics-based models, grounded in energy landscape theory, have provided profound insights into the universal principles of folding, particularly the dominant role of native topology. The emergence of machine-learned, transferable coarse-grained models marks a significant advance, offering a powerful synthesis of physical reasoning and data-driven performance. However, challenges persist, especially in predicting the complex energy landscapes of large, multi-domain, and allosterically regulated proteins [68]. The future of the field lies in a continued synergy between these approaches—leveraging the interpretability and rigor of physics-based models with the speed and accuracy of machine learning—to achieve a truly universal and predictive understanding of protein folding.
The computational prediction of protein structure and dynamics relies on force fields to model molecular interactions. This review provides a comprehensive performance evaluation of three distinct approaches: the all-atom AMBER force field, the coarse-grained AWSEM model, and emerging machine-learned coarse-grained force fields. Framed within the context of free energy landscape theory, we examine how each methodology navigates the conformational space of proteins, the key validation experiments employed, and their respective computational trade-offs. The analysis indicates that while traditional force fields provide a solid physical foundation, machine-learned coarse-grained models are rapidly advancing, offering a promising path toward universal, transferable, and computationally efficient protein simulations.
The problem of predicting a protein's three-dimensional structure from its amino acid sequence remains a central challenge in computational biophysics. The "funneled" nature of protein energy landscapes provides the conceptual framework for this endeavor [25]. This theory posits that evolution has selected protein sequences whose energy landscapes are relatively smooth and funneled toward the native state, a concept known as the Principle of Minimal Frustration [25]. This contrasts with the rugged landscapes of random heteropolymers, which are riddled with deep kinetic traps. The funneled landscape allows proteins to fold reliably and predictably.
Computational simulations of protein folding aim to replicate this natural process by employing mathematical force fields. The core challenge is to develop models that are both physically accurate and computationally tractable. All-atom force fields like AMBER provide high fidelity but at extreme computational cost, limiting the timescales and system sizes that can be studied [4]. Coarse-grained models like AWSEM address this by reducing the number of degrees of freedom, often leveraging energy landscape theory to design funneled landscapes. Most recently, machine learning has been harnessed to learn transferable coarse-grained force fields from vast amounts of atomistic simulation data, promising to combine accuracy with efficiency [4] [69]. This review evaluates the cross-platform performance of these three distinct paradigms.
Theoretical Foundations and Methodologies
Free Energy Landscape Theory
Energy landscape theory reconceptualizes protein folding from a discrete path to a statistical journey over a continuous surface. The landscape is described as a funnel where the native state resides at the global free energy minimum. The steepness and roughness of this funnel determine the kinetics and thermodynamics of folding. Coarse-grained models are particularly well-suited to this theory, as their reduced resolution can smooth over atomic-scale roughness, making the overarching funnel more navigable for both proteins and algorithms [25]. Simulation methods aim to sample this landscape, identifying metastable states, transition states, and pathways.
The force fields evaluated in this review represent different philosophical and technical approaches to modeling the energy landscape.
- AMBER (All-Atom): AMBER is a classic all-atom force field. Its energy function is a sum of physically motivated terms including bond stretching, angle bending, torsional potentials, and non-bonded van der Waals and electrostatic interactions [70]. It is parameterized using a combination of quantum-chemical calculations and experimental data, providing a high-resolution, chemically specific representation.
- AWSEM (Coarse-Grained): The Associative Memory, Water Mediated, Structure and Energy Model (AWSEM) is a knowledge-based coarse-grained model that incorporates lessons from energy landscape theory [25]. It uses a combination of physical interactions (e.g., burial, electrostatics) and bioinformatically derived "associative memories" to bias the landscape toward native-like structures, ensuring it is funneled.
- Machine-Learned Force Fields (Coarse-Grained): These models, such as CGSchNet, use deep learning to approximate the effective CG free energy from all-atom simulation data [4] [69]. They employ a bottom-up approach, learning a many-body potential that is thermodynamically consistent with the reference all-atom model, as defined by the force-matching or relative entropy minimization frameworks [69].
Key Methodological Workflows
The development and application of these force fields follow distinct workflows, particularly in how they are parameterized and validated.
The following diagram illustrates the core methodology for developing a bottom-up machine-learned coarse-grained force field, a process that highlights the data-driven nature of this modern approach.
Diagram 1: Workflow for developing a machine-learned coarse-grained force field. The process begins with generating high-fidelity all-atom data, which is used to train a neural network potential. The resulting model is validated against independent data in an iterative cycle.
In contrast, the workflow for structure-based models like AWSEM often begins directly with the native structure to define the funnel, while all-atom force fields like AMBER rely on pre-defined physical parameterizations applied directly to the system of interest.
Performance Comparison and Benchmarking
Quantitative Performance Metrics
The table below summarizes the key performance characteristics of the three force field types across several critical metrics.
Table 1: Cross-Platform Performance Comparison of Protein Force Fields
| Performance Metric | AMBER (All-Atom) | AWSEM (Coarse-Grained) | ML-CG (e.g., CGSchNet) |
|---|---|---|---|
| Resolution | All-atom [70] | Coarse-grained (1+ beads per residue) [25] | Coarse-grained (Cα or backbone) [4] [69] |
| Computational Speed | ~1x (Baseline) [4] | Orders of magnitude faster than all-atom [25] | Orders of magnitude faster than all-atom [4] |
| Transferability | High (by design) [70] | Good for related folds [25] | Promising for low-similarity sequences [4] |
| Folding Landscape | Detailed, can be rugged | Funneled by construction [25] | Reproduces metastable states from AA data [4] |
| Key Strengths | High physical fidelity; Chemical specificity [70] | Captures folding nuclei; Good for large systems [25] [43] | High accuracy from data; Efficient [4] [69] |
| Key Limitations | Extreme computational cost; Slow dynamics [4] | Limited chemical specificity; Native-state bias [25] | Data-hungry; Training complexity [69] [23] |
Performance on Specific Biological Problems
Folding of Small Proteins and Peptides
Machine-learned coarse-grained models have demonstrated remarkable success in folding small, fast-folding proteins like chignolin, TRP-cage, and the villin headpiece. For instance, CGSchNet predicts metastable folded, unfolded, and intermediate states with structural metrics (fraction of native contacts Q, Cα RMSD) closely matching reference all-atom simulations [4]. Similarly, AWSEM has been extensively used to explore the principles of folding and to identify folding nuclei for a wide range of proteins, leveraging its funneled landscape to reliably find the native state [25]. AMBER can provide the gold-standard reference for these systems, but achieving converged folding/unfolding transitions requires immense computational resources, often only feasible for the smallest proteins on specialized supercomputers [4].
Conformational Dynamics of Larger Proteins
For larger proteins like the 54-residue engrailed homeodomain (1ENH) and the 73-residue alpha3D (2A3D), converged all-atom simulations of folding are often not practical. Here, coarse-grained models excel. The machine-learned CG model successfully folded both proteins to their correct native structures from extended configurations and reproduced the native-state fluctuations observed in shorter, stable all-atom simulations [4]. This demonstrates the potential of ML-CG models for extrapolative molecular dynamics on sequences not used during training.
Complex Phenomena: Multimerization and Misfolding
AWSEM has been applied to complex problems beyond single-domain folding, including multidomain protein misfolding and the initial stages of protein aggregation [25]. Its energy function can be adapted to study protein-protein association. Similarly, all-atom models like AMBER can be used to study protein-protein association, revealing, for example, that the process can be driven by enthalpy and opposed by entropy, involving water expulsion and side-chain conformational changes [71]. The application of machine-learned force fields to such complex, multi-protein phenomena remains a frontier area of research.
The Scientist's Toolkit: Essential Research Reagents and Methods
This section details key computational tools and methodological concepts essential for conducting research in this field.
Table 2: Key Reagents and Methodologies for Protein Folding Simulations
| Tool / Concept | Type | Function and Relevance |
|---|---|---|
| Molecular Dynamics (MD) | Algorithm | Core simulation engine for sampling conformational space using a given force field. |
| Variational Force-Matching | Training Method | A primary method for training ML force fields to be thermodynamically consistent with all-atom data without running CG simulations during training [69]. |
| Relative Entropy Minimization | Training Method | An alternative iterative method for training CG models by minimizing the divergence between CG and all-atom distributions [69]. |
| Parallel Tempering (Replica Exchange) | Sampling Method | Enhanced sampling technique crucial for overcoming free energy barriers and obtaining converged equilibrium distributions, especially in CG simulations [4]. |
| Markov State Models (MSMs) | Analysis Framework | Used to analyze long MD trajectories, identify metastable states, and model kinetics, often applied to all-atom simulation data [69]. |
| Root-Mean-Square Deviation (RMSD) | Metric | Measures the average distance between atoms of superimposed structures, used to quantify similarity to the native state. |
| Fraction of Native Contacts (Q) | Metric | Measures the fraction of native pairwise contacts formed in a given conformation, a key reaction coordinate for folding. |
Visualizing Free Energy Landscapes
A primary application of these simulation methods is the calculation of free energy landscapes, which provide a powerful visual and quantitative summary of protein folding thermodynamics and kinetics.
Diagram 2: A simplified schematic of a protein folding free energy landscape. The landscape is often projected onto reaction coordinates like Root-Mean-Square Deviation (RMSD) and the Fraction of Native Contacts (Q). The unfolding pathway (red) and folding pathway (blue) may differ, passing through intermediate (I) and transition (T) states. The funneled nature of the landscape is evident as the energy generally decreases toward the native state (N).
Different force fields yield different landscapes. Structure-based models like AWSEM produce strongly funneled landscapes by design. All-atom models like AMBER produce more detailed landscapes that can include subtle barriers and misfolded states. The goal of machine-learned force fields is to produce landscapes that match the all-atom reference at a fraction of the cost, as demonstrated by their ability to capture metastable states for proteins like chignolin and TRP-cage [4].
The evaluation of AMBER, AWSEM, and machine-learned coarse-grained force fields reveals a trade-off space defined by computational cost, resolution, and transferability. AMBER remains the benchmark for atomic-level accuracy but is prohibitively expensive for studying large-scale conformational changes. AWSEM and similar knowledge-based models provide powerful insights into folding principles and mechanisms for large systems by leveraging energy landscape theory. The emergence of machine-learned coarse-grained models represents a paradigm shift, demonstrating for the first time the feasibility of a transferable, bottom-up CG model that can approach the predictive accuracy of all-atom simulations while being orders of magnitude faster [4] [23].
Future developments in this field will likely focus on overcoming the remaining challenges. For machine-learned force fields, these include the insatiable demand for diverse training data and improving the robustness and physical interpretability of the learned potentials [69] [23]. The integration of physical constraints and multi-scale modeling approaches, combining the strengths of all-atom and coarse-grained descriptions, also holds great promise. As these methods mature, the vision of a universal, computationally efficient protein force field that can reliably predict structure, dynamics, and function for any sequence is steadily becoming a reality, with profound implications for basic research and drug development.
The emergence of deep learning systems like AlphaFold has revolutionized protein structure prediction, achieving accuracy competitive with experimental methods in many cases [33] [5]. However, these static structural predictions represent only one facet of the complex protein folding problem. This technical guide examines the synergistic integration of AI-based structure prediction with molecular dynamics (MD) simulations through the theoretical framework of free energy landscapes. We demonstrate how AlphaFold/OpenFold provide highly accurate initial structural models that serve as ideal starting points for dynamics simulations, which in turn elucidate conformational ensembles, folding pathways, and functional dynamics that lie beyond the capabilities of static prediction alone. This integrated approach enables researchers to construct comprehensive models of protein energy landscapes, bridging the gap between structure prediction and mechanistic understanding of folding processes and function.
The "protein folding problem" encompasses two fundamental challenges: predicting a protein's native three-dimensional structure from its amino acid sequence, and understanding the physical mechanism by which it folds to that structure [72]. Free energy landscape theory provides the essential conceptual framework, visualizing protein folding as a biased random walk on a funnel-shaped energy surface toward the native state [25] [7]. While historically treated as separate challenges, the integration of AI-based structure prediction with physics-based simulations now offers a unified approach to both problems.
AlphaFold and similar systems have demonstrated remarkable accuracy in structure prediction, achieving median backbone accuracy of 0.96 Å in CASP14 [5]. The system employs a novel neural network architecture that incorporates evolutionary, physical, and geometric constraints through Evoformer blocks and a structure module that enables end-to-end coordinate prediction [5]. However, as noted in recent analyses, "AlphaFold predicts a single structure and is subject to the same limitations as experimental techniques that resolve a single structure" [72]. Proteins are inherently dynamic systems that sample multiple conformational states, and understanding function often requires characterization of these ensembles rather than single structures.
This whitepaper provides a technical framework for integrating AI-predicted structures with dynamics simulations to construct comprehensive energy landscape models, enabling researchers to address biological questions that require understanding beyond static structures.
Theoretical Foundation: Free Energy Landscape Theory
The free energy landscape concept represents protein folding as a funnel-shaped surface where the depth corresponds to energy and the width represents conformational entropy [25] [7]. A perfectly funneled landscape has minimal energetic frustration, with no large barriers trapping misfolded intermediates. The landscape slope quantitatively characterizes the energetic bias toward the native state.
Quantitative Landscape Characterization
The folding free energy landscape can be quantitatively described using an order parameter Q, typically defined as the fraction of native contacts formed. The reduced free energy landscape f(Q) is obtained by averaging the full free energy function over configurations with specific Q values [7]. The landscape slope, calculated as the free energy decrease per unit change in Q, differentiates between ordered and intrinsically disordered proteins:
Table 1: Free Energy Landscape Slopes for Representative Protein Classes
| Protein | Class | Landscape Slope (kcal/mol) | Structural Consequence |
|---|---|---|---|
| HP-35 | α-helical | -50 | Stable, ordered protein |
| WW domain | β-sheet | -50 | Stable, ordered protein |
| pKID (isolated) | Intrinsically Disordered | -24 | Disordered in free state |
| pKID-KIX complex | Coupled folding/binding | -54 | Folds upon binding |
The steeper landscape slopes for ordered proteins (-50 kcal/mol) compared to intrinsically disordered proteins (-24 kcal/mol) quantitatively explain their different structural properties [7]. Upon binding to their partners, disordered proteins like pKID develop significantly steeper landscapes (-54 kcal/mol), enabling folding.
Funneled Landscape and Minimal Frustration
The Principle of Minimal Frustration dictates that natural proteins have evolved to have landscapes where native interactions are sufficiently strong compared to non-native interactions, creating a smoothly funneled landscape [25]. This principle underlies both natural protein folding and successful structure prediction methods, as both require consistent energetic biases toward native-like configurations.
Methodological Approaches: AI Prediction and Dynamics Simulations
AI-Based Structure Prediction Systems
AlphaFold employs a sophisticated neural network architecture with two main stages. The Evoformer block processes multiple sequence alignments and residue pair information through attention mechanisms, while the structure module generates atomic coordinates using an equivariant transformer that respects rotational and translational symmetry [5]. The system is trained with multiple losses including intermediate refinement cycles and produces per-residue confidence estimates (pLDDT) [33] [5].
OpenFold provides an open-source implementation that replicates AlphaFold's functionality, enabling community development and customization. Both systems can predict structures with atomic accuracy even when no homologous structures are available [5].
Molecular Dynamics Simulation Approaches
Molecular dynamics simulations numerically solve Newton's equations of motion for all atoms in the protein and solvent system, typically using femtosecond time steps [38]. MD provides extremely high-resolution spatial and temporal data on folding processes but faces challenges of timescale limitations and force field accuracy [38].
Table 2: Comparison of Simulation Methods for Protein Folding
| Method | Spatial Resolution | Typical Timescale | Key Applications | Limitations |
|---|---|---|---|---|
| All-atom MD with explicit solvent | Atomic | Nanoseconds to microseconds | Folding mechanisms, solvent effects | Computationally expensive, limited by timescales |
| Coarse-grained MD | Coarse-grained (multiple atoms per bead) | Microseconds to milliseconds | Aggregation studies, large systems | Loss of atomic detail |
| Enhanced sampling methods | Atomic | Effective milliseconds | Free energy calculations, rare events | Requires careful validation |
| 4D Diffusion models [73] | Atomic | Multiple time steps | Dynamic structure prediction | Emerging methodology |
Advanced sampling techniques like replica exchange molecular dynamics (REMD) accelerate conformational sampling by running multiple simulations at different temperatures and exchanging configurations. The free energy landscape construction method developed by [7] combines all-atom MD with molecular integral-equation theory to compute solvation free energies for individual configurations.
Integration Framework: Bridging Prediction and Dynamics
The integration of AI-predicted structures with dynamics simulations creates a powerful cycle for exploring protein energy landscapes. The following workflow diagram illustrates this complementary relationship:
AI-Simulation Integration Workflow
From Static Structures to Dynamic Ensembles
AlphaFold-predicted structures provide highly accurate starting configurations for dynamics simulations, bypassing the need for lengthy de novo folding simulations. These structures can be solvated in explicit solvent, energy-minimized, and used as initial conditions for MD simulations [72]. The pLDDT confidence scores help identify regions that may require special attention during simulation setup.
Molecular dynamics then explores the conformational landscape around the AI-predicted structure, capturing:
- Local flexibility and backbone dynamics
- Side-chain rearrangements
- Transient secondary structure elements
- Alternative conformational states
Recent advances include 4D diffusion models that generate dynamic protein structures across multiple time steps simultaneously, incorporating molecular dynamics simulation data to learn dynamic protein structures [73].
Validating and Refining AI Predictions
Dynamics simulations can validate AI predictions by assessing:
- Structural stability during simulation
- Agreement with experimental data (NMR order parameters, B-factors)
- Presence of conserved conformational transitions
Simulations can also refine AI predictions by relaxing strained geometries and optimizing side-chain packing, particularly in low-confidence regions identified by pLDDT scores.
Technical Protocols and Implementation
Free Energy Landscape Construction Protocol
Based on the methodology of [7], the following protocol constructs explicit folding free energy landscapes:
Perform MD Simulations: Run extensive all-atom molecular dynamics simulations covering both folded and unfolded states. For HP-35, this required ~400 μs of folding-unfolding trajectory [7].
Compute Order Parameters: For each saved configuration, calculate the fraction of native contacts Q using the native contact definition of [7].
Calculate Free Energy Components: For each configuration, compute:
- Gas-phase potential energy (E_u) from force field parameters
- Solvation free energy (G_solv) using molecular integral-equation theory
Construct Free Energy Landscape: Average f(r) = Eu(r) + Gsolv(r) over configurations with specific Q values to obtain f(Q)
Visualize Landscape: Create 3D representations showing free energy as a function of both Q and additional structural parameters (e.g., radius of gyration)
AI-Structure Initialized Simulation Protocol
Structure Prediction: Generate protein structure using AlphaFold/OpenFold with multiple sequence alignments from standard databases.
System Preparation:
- Solvate the structure in explicit solvent (TIP3P water)
- Add ions to physiological concentration (150 mM NaCl)
- Energy minimize using steepest descent algorithm
Equilibration:
- Run 100 ps simulation with position restraints on protein heavy atoms
- Gradually reduce restraint force constants
- Perform 10-100 ns unrestrained equilibration
Production Simulation:
- Run extended MD simulation (microsecond timescales for folding studies)
- Employ enhanced sampling if needed (temperature replica exchange, metadynamics)
Analysis:
- Calculate free energy surfaces using order parameters
- Identify metastable states and transition pathways
- Compare with experimental data for validation
Research Reagent Solutions
Table 3: Essential Computational Tools for Integrated Structure-Dynamics Studies
| Tool/Resource | Type | Function | Access |
|---|---|---|---|
| AlphaFold Protein Structure Database [33] | Database | Over 200 million predicted structures | Public access |
| AlphaFold Server [32] | Prediction server | Protein-ligand interaction predictions | Free for non-commercial research |
| OpenFold | Software | Open-source AlphaFold implementation | GitHub |
| GROMACS | Software | Molecular dynamics simulation | Open source |
| AMBER | Software | Molecular dynamics force fields & simulation | Commercial & academic |
| CHARMM | Software | Molecular dynamics force fields & simulation | Commercial & academic |
| PLUMED | Plugin | Enhanced sampling and free energy calculations | Open source |
| VMD | Software | Visualization and analysis of MD trajectories | Open source |
Applications and Case Studies
Intrinsically Disordered Proteins
The integrated approach is particularly valuable for studying intrinsically disordered proteins (IDPs) like pKID, which fold upon binding to their partner KIX [7]. AI prediction alone cannot capture the disorder-order transition, but combined with dynamics simulations, researchers can:
- Use AlphaFold to predict the folded structure of the complex
- Simulate the isolated IDP to characterize its disordered ensemble
- Perform binding simulations to observe coupled folding and binding
- Construct free energy landscapes for both isolated and bound states
This approach quantitatively explained why pKID is disordered in isolation (shallow landscape slope of -24 kcal/mol) but folds upon binding (steep slope of -54 kcal/mol) [7].
Protein Misfolding and Aggregation
Coarse-grained molecular dynamics simulations combined with AI-based structure prediction enable study of protein aggregation phenomena [74]. Deep learning strategies including genetic algorithms and reinforcement learning can generate peptides with tunable aggregation propensities, with coarse-grained MD used to evaluate solvent-accessible surface area and define aggregation propensity [74].
Conformational Switching and Allostery
For proteins that switch between multiple functional states, AlphaFold typically predicts only one conformation [72]. However, by leveraging sequence variations in multiple sequence alignments, researchers can sometimes trick AlphaFold into generating alternative conformations [72]. These structures can then serve as starting points for simulations that map the complete conformational landscape and transition pathways.
The field is rapidly advancing toward integrated models that simultaneously predict structure and dynamics. AlphaFold 3 has expanded capabilities to predict interactions with nucleic acids, small molecules, ions, and modified residues [32]. Four-dimensional diffusion models now generate dynamic protein structures across multiple time steps, incorporating MD simulation data to learn dynamic protein structures with both backbone and side-chain representations [73].
The emerging paradigm treats structure prediction and dynamics simulation not as competing approaches but as complementary components of a comprehensive framework for understanding protein energy landscapes. This integrated approach will be essential for addressing fundamental questions in structural biology and enabling novel applications in drug discovery and protein design.
As noted by [72], "The ability to predict such ensembles should reveal the mechanisms of protein folding and the different structural states that are important to a protein's function, whether they be the on/off states of a molecular switch or the different conformations of a catalytic cycle." The combination of AI-based structure prediction and dynamics simulations, grounded in free energy landscape theory, provides a powerful toolkit for realizing this vision.
Conclusion
Free energy landscape theory provides a unified and quantitative framework for understanding protein folding, stability, and misfolding. The integration of advanced simulation methodologies, particularly machine-learned coarse-grained models, has dramatically increased the scale and scope of problems that can be addressed, making the prediction of folding mechanisms for large proteins and the discovery of drugs targeting folding intermediates a tangible reality. The continued synergy between computational simulations—validated by single-molecule experiments—and AI-based structure prediction tools is paving the way for a new era in molecular biology. Future directions will focus on improving the accuracy of force fields for complex motifs, modeling larger biomolecular assemblies involved in disease, and ultimately enabling the de novo design of proteins and therapeutics with tailored folding landscapes, thereby deepening our impact on biomedical and clinical research.
References
- 1. Evolution, Energy Landscapes and the Paradoxes of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4472606/]
- 2. Mega-scale experimental analysis of protein folding ... [https://www.nature.com/articles/s41586-023-06328-6]
- 3. Folding Simulations for Proteins with Diverse Topologies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4195377/]
- 4. Navigating protein landscapes with a machine-learned ... [https://www.nature.com/articles/s41557-025-01874-0]
- 5. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 6. An interactive visualization tool for educational outreach in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10982686/]
- 7. Folding Free Energy Landscape of Ordered and ... [https://www.nature.com/articles/s41598-019-50825-6]
- 8. Examining a Thermodynamic Order Parameter of Protein ... [https://www.nature.com/articles/s41598-018-25406-8]
- 9. Quantifying Nonnative Interactions in the Protein-Folding Free ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4968343/]
- 10. Improved theoretical description of protein folding kinetics ... [https://pubmed.ncbi.nlm.nih.gov/15260647/]
- 11. Energy Landscapes of Protein Aggregation and ... [https://pubmed.ncbi.nlm.nih.gov/34358545/]
- 12. On Free Energy Calculations in Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12548091/]
- 13. Fast, scalable free energy prediction with nonequilibrium ... [https://www.drugtargetreview.com/article/190846/fast-scalable-free-energy-prediction-with-nonequilibrium-switching/]
- 14. Comparing the energy landscapes for native folding and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4981195/]
- 15. Navigating the landscape of protein folding and proteostasis [https://www.nature.com/articles/s41392-025-02439-w]
- 16. Topological determinants in protein folding dynamics [https://biologydirect.biomedcentral.com/articles/10.1186/s13062-025-00642-x]
- 17. Exploring the Energy Landscapes of Protein Folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3283771/]
- 18. Theory and Practice of Coarse-Grained Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8465211/]
- 19. Successes and challenges in simulating the folding of large ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6952611/]
- 20. Optimized protein-water interactions and torsional ... [https://www.nature.com/articles/s41467-025-65603-4]
- 21. Atomistic simulations of intact virus capsids [https://www.sciencedirect.com/science/article/pii/S0959440X25001009]
- 22. BioMD: All-atom Generative Model for Biomolecular ... [https://arxiv.org/abs/2509.02642]
- 23. Machine learned coarse-grained protein force-fields [https://www.sciencedirect.com/science/article/abs/pii/S0959440X23000076]
- 24. Advancing Protein Simulation with Machine Learning [https://www.fu-berlin.de/en/presse/informationen/fup/2025/fup_25_124_clementi-nature/index.html]
- 25. Learning To Fold Proteins Using Energy Landscape Theory [https://pmc.ncbi.nlm.nih.gov/articles/PMC4189132/]
- 26. Navigating protein landscapes with a machine-learned ... [https://communities.springernature.com/posts/navigating-protein-landscapes-with-a-machine-learned-transferable-coarse-grained-model]
- 27. Machine learning coarse-grained potentials of protein ... [https://www.nature.com/articles/s41467-023-41343-1]
- 28. The metastable states of proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7314396/]
- 29. Exploring the Free Energy Landscape: From Dynamics to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2694367/]
- 30. The folding mechanism and key metastable state ... [https://pubs.rsc.org/en/content/articlelanding/2017/cp/c7cp01521f]
- 31. Kinetic Definition of Protein Folding Transition State ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1479057/]
- 32. AlphaFold Protein Structure Database [https://deepmind.google/science/alphafold/]
- 33. AlphaFold Protein Structure Database [https://alphafold.ebi.ac.uk/]
- 34. Structural insights into natural compound inhibitors of the ... [https://pubmed.ncbi.nlm.nih.gov/41188391/]
- 35. Comparing a simple theoretical model for protein folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3816406/]
- 36. Quantifying the accuracy of protein simulation models [https://foldingathome.org/2025/09/04/quantifying-the-accuracy-of-protein-simulation-models/]
- 37. Atomistic molecular simulations of protein folding [https://www.sciencedirect.com/science/article/abs/pii/S0959440X11002041]
- 38. Challenges in protein folding simulations: Timescale, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3032381/]
- 39. Unfolding the secrets of proteins - Yale Engineering [https://engineering.yale.edu/news-and-events/news/unfolding-secrets-proteins]
- 40. A critical comparison of coarse - grained structure-based approaches... [https://pubmed.ncbi.nlm.nih.gov/28530269/]
- 41. BioEmu: AI‐Powered Revolution in Scalable Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12643044/]
- 42. High resolution ensemble description of metamorphic and ... [https://www.nature.com/articles/s41467-021-21105-7]
- 43. Successes and challenges in simulating the folding of ... [https://www.sciencedirect.com/science/article/pii/S0021925817495470]
- 44. Probing the Mechanical Properties of DNA Nanostructures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9245350/]
- 45. Collective Variable for Metadynamics Derived From ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9234394/]
- 46. [2503.09747] Parallel Tempered Metadynamics [https://arxiv.org/abs/2503.09747]
- 47. How chromatin-binding proteins direct distinct folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12324292/]
- 48. Folding funnel [https://en.wikipedia.org/wiki/Folding_funnel]
- 49. Strategies for the exploration of free energy landscapes [https://www.sciencedirect.com/science/article/pii/S2405428317300059]
- 50. The Folding Energy Landscape and Free ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4145804/]
- 51. Prediction of protein-folding mechanisms from free-energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC18029/]
- 52. Free Energy Simulations to Quantitatively Study ... [https://chemrxiv.org/engage/chemrxiv/article-details/6824aae950018ac7c5244e09]
- 53. Navigating the landscape of protein folding and proteostasis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550075/]
- 54. Graph representation learning for structural proteomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8786289/]
- 55. Graphein 1.4.0 [https://graphein.ai/]
- 56. A Practical Guide to Single Molecule FRET - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3769523/]
- 57. Single-molecule FRET [https://en.wikipedia.org/wiki/Single-molecule_FRET]
- 58. Optical tweezers in single-molecule experiments [https://link.springer.com/article/10.1140/epjp/s13360-020-00907-6]
- 59. Optical tweezers in single-molecule biophysics [https://www.nature.com/articles/s43586-021-00021-6]
- 60. A practical guide to single-molecule FRET [https://www.nature.com/articles/nmeth.1208]
- 61. One‐pot dual protein labeling for simultaneous mechanical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11915586/]
- 62. DeepFRET, a software for rapid and automated single ... [https://elifesciences.org/articles/60404]
- 63. Protein folding: bringing theory and experiment closer ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X03000071]
- 64. Roles of native topology and chain-length scaling in ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283601950375]
- 65. Accurate prediction of protein folding mechanisms by ... [https://www.nature.com/articles/s41467-023-41664-1]
- 66. Comprehensive analysis of experimental and AlphaFold 2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12149446/]
- 67. Challenges, Advances, and the Shift of Research Paradigms [https://www.sciencedirect.com/science/article/pii/S1672022923000657]
- 68. Challenging AlphaFold in predicting proteins with large- ... [https://www.nature.com/articles/s42004-025-01763-0]
- 69. Machine Learned Coarse-Grained Protein Force-Fields [https://pmc.ncbi.nlm.nih.gov/articles/PMC10023382/]
- 70. Comparison of protein force fields for molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/18446282/]
- 71. Article Exploring the Free-Energy Landscape and ... [https://www.sciencedirect.com/science/article/pii/S0006349520306019]
- 72. AlphaFold and protein folding: Not dead yet! The frontier is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11892350/]
- 73. 4D Diffusion for Dynamic Protein Structure Prediction with ... [https://arxiv.org/html/2408.12419v2]
- 74. Artificial intelligence-driven approaches for the rational ... [https://www.nature.com/articles/s44431-025-00005-6]