Research Articles
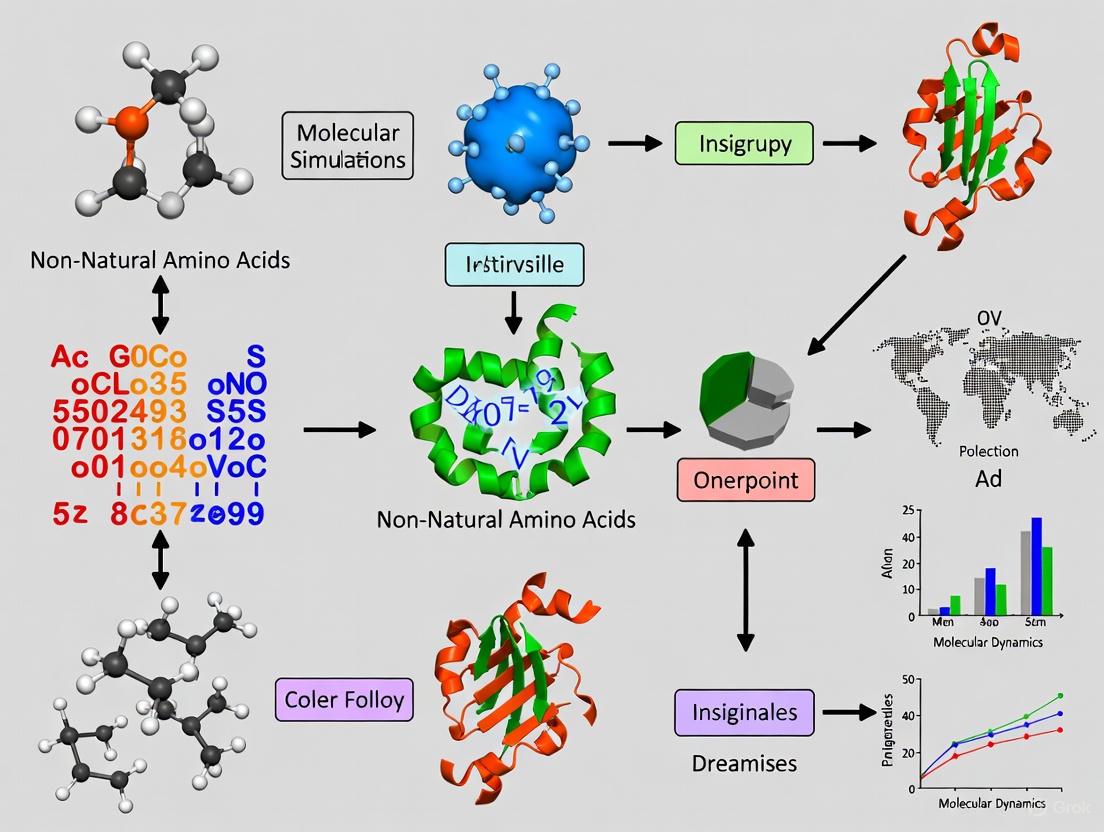
Navigating Non-Natural Amino Acids in Molecular Dynamics Folding Simulations: A Guide for Therapeutic Peptide Design
The incorporation of non-canonical amino acids (ncAAs) is a powerful strategy to enhance the stability, permeability, and binding affinity of therapeutic peptides.
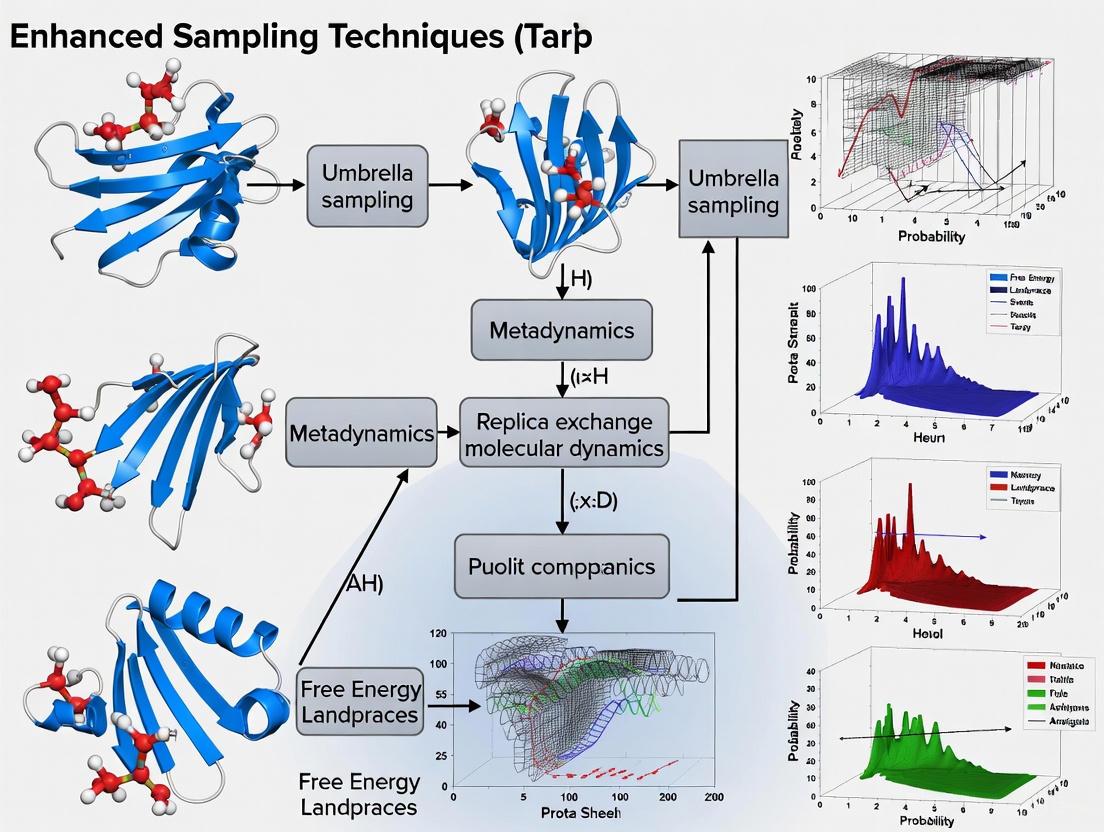
Breaking the Timescale Barrier: Enhanced Sampling Techniques for Simulating Rare Events in Protein Folding
This article provides a comprehensive overview of advanced computational methods designed to overcome the timescale limitations of molecular dynamics simulations in studying rare protein folding events.
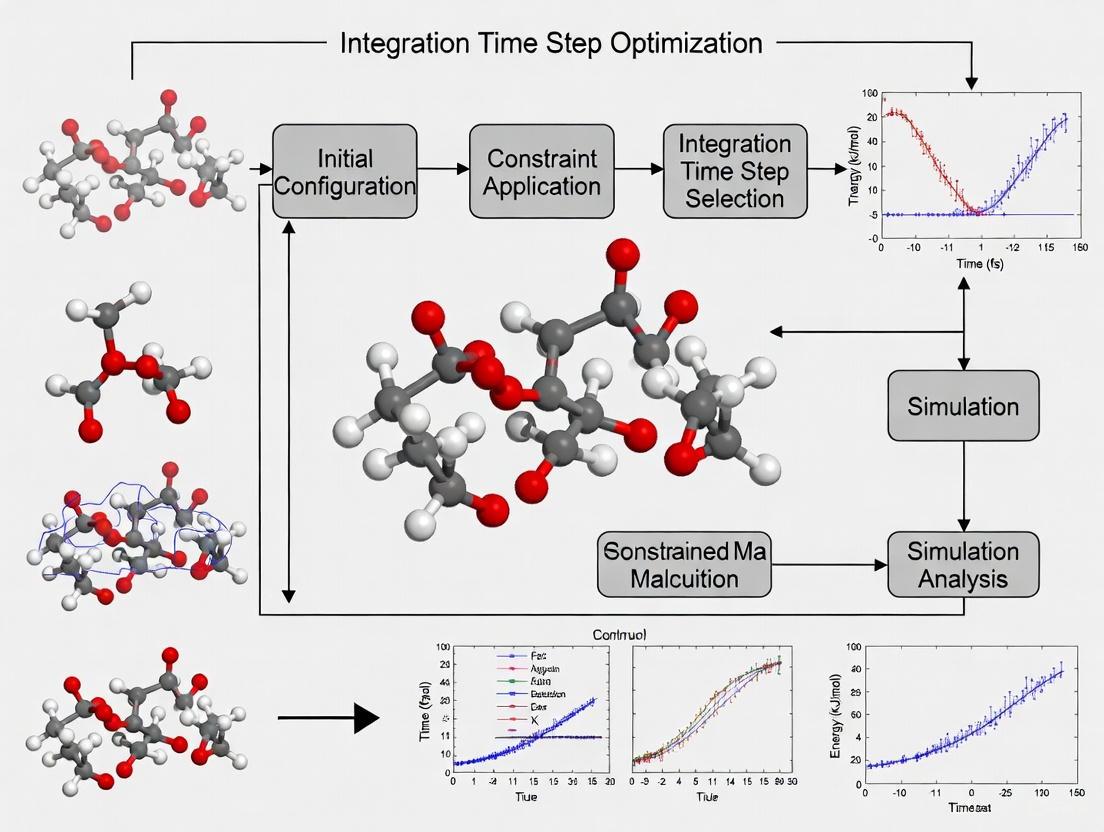
Optimizing Integration Time Steps in Constrained MD Simulations: A Guide for Biomedical Researchers
This article provides a comprehensive guide for researchers and drug development professionals on optimizing integration time steps in constrained Molecular Dynamics (MD) simulations.
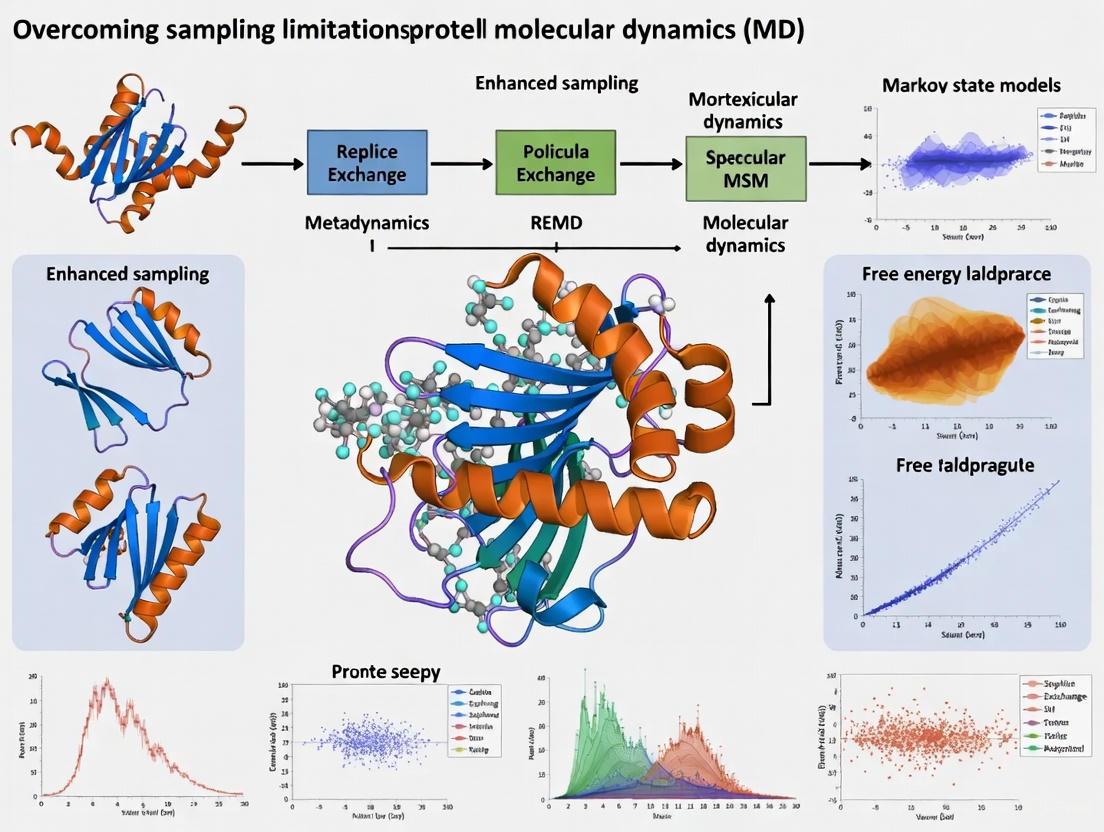
Beyond the Timescale Barrier: Modern Strategies to Overcome Sampling Limits in Small Protein Simulations
Molecular dynamics (MD) simulations are a cornerstone of structural biology and drug discovery, yet their utility is fundamentally constrained by the limited timescales accessible for sampling the conformational landscapes of...
GNEIMO Method: Revolutionizing Protein Folding and Refinement with Torsional Dynamics
This article explores the Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method, an advanced internal coordinate molecular dynamics (ICMD) technique transforming the study of protein folding and structure refinement.
Machine Learning Revolution: Next-Gen Coarse-Grained MD Parameters for Small Protein Folding
This comprehensive review explores the transformative impact of machine learning and advanced parameterization methods on coarse-grained molecular dynamics (CGMD) for small protein folding studies.
Beyond Static Structures: Integrating Machine Learning and Molecular Dynamics to Predict Dynamic Protein Ensembles
This article explores the integrated approach of machine learning (ML) and molecular dynamics (MD) for protein structure prediction, a paradigm shifting from static models to dynamic ensembles.
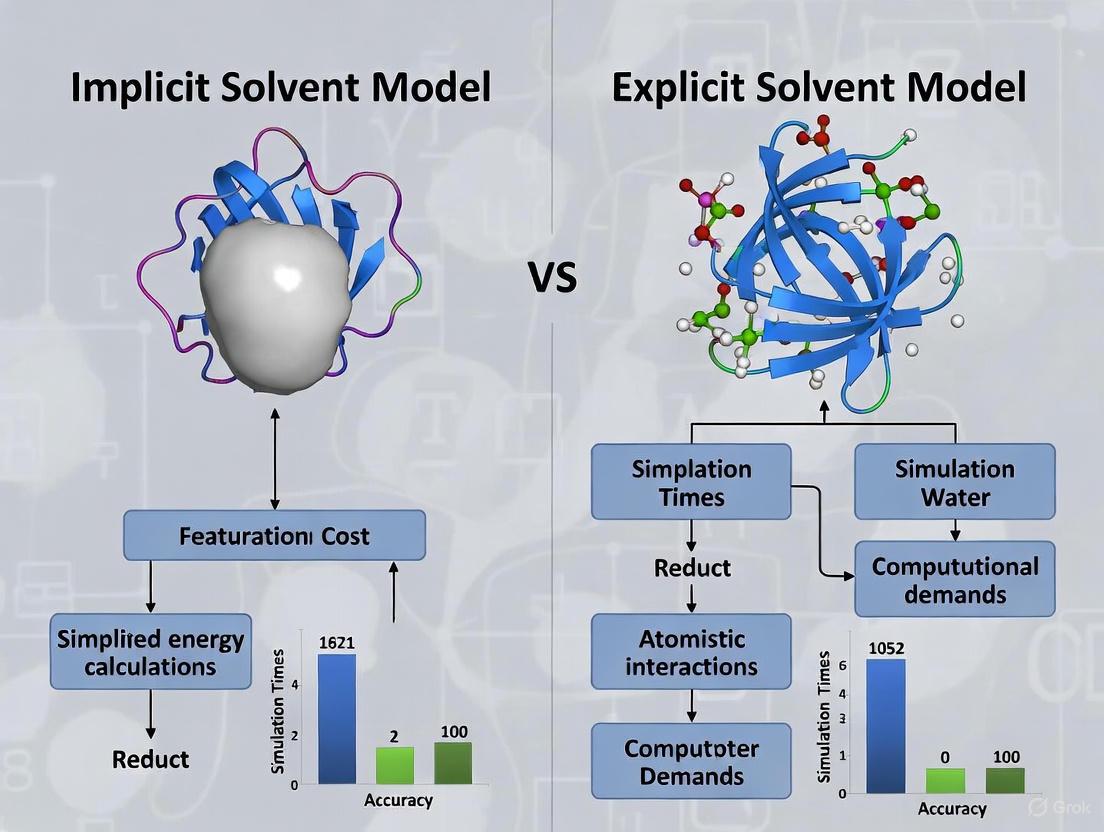
Implicit vs Explicit Solvent Models for Small Protein Simulations: A Practical Guide for Computational Researchers
This article provides a comprehensive analysis of implicit and explicit solvent models for molecular dynamics simulations of small proteins, targeting researchers and drug development professionals.
A Comprehensive Guide to All-Atom MD Simulation of Trp-Cage Folding: Protocols, Force Fields, and Validation
This article provides a comprehensive guide for implementing all-atom molecular dynamics (MD) simulations to study the folding of the Trp-cage miniprotein, a key model system in computational biophysics.
Low-Mass Molecular Dynamics: Accelerating Protein Folding Simulations for Biomedical Research
This article explores the low-mass molecular dynamics (MD) technique, a simple yet powerful method to dramatically enhance configurational sampling in protein folding simulations.