Constrained Molecular Dynamics for Small Protein Folding: Protocols, Applications, and Advancements in Computational Biophysics
This article provides a comprehensive overview of constrained molecular dynamics (MD) protocols for simulating small protein folding, a critical challenge in computational biophysics and structure-based drug discovery.
Constrained Molecular Dynamics for Small Protein Folding: Protocols, Applications, and Advancements in Computational Biophysics
Abstract
This article provides a comprehensive overview of constrained molecular dynamics (MD) protocols for simulating small protein folding, a critical challenge in computational biophysics and structure-based drug discovery. We explore the foundational principles that make constrained MD a computationally efficient alternative to all-atom MD, enabling enhanced sampling of near-native structures. The article details methodological implementations, including all-torsion and hierarchical 'freeze and thaw' clustering schemes, and their application to proteins with diverse secondary structures like α-helices, β-turns, and mixed motifs. We further discuss troubleshooting and optimization strategies to overcome sampling limitations and energy barriers, and present a comparative analysis with other enhanced sampling techniques like accelerated MD (aMD) and their validation against experimental data. This resource is tailored for researchers and drug development professionals seeking to leverage computational methods for protein structure prediction and refinement.
The Principles and Necessity of Constrained MD in Protein Folding
The Core Scientific Challenge
The "protein folding problem" is the grand challenge of predicting the precise three-dimensional (3D) structure of a protein from its one-dimensional amino acid sequence alone [1] [2]. This problem is foundational to modern biology because a protein's structure is the primary determinant of its function. Misfolded proteins are implicated in a range of diseases, including Alzheimer's and Parkinson's, and a detailed understanding of native protein structures is invaluable for drug discovery and understanding cellular processes [3].
The problem is framed by two seminal concepts. Anfinsen's dogma, for which Christian Anfinsen won the Nobel Prize in Chemistry in 1972, posits that a protein's native structure is the one that is thermodynamically most stable under its physiological conditions, meaning all the information required for folding is encoded in its amino acid sequence [1] [2]. However, this theoretical possibility collides with Levinthal's paradox, which highlights the computational infeasibility of this process. Cyrus Levinthal noted that a protein has an astronomically large number of possible conformations (on the order of 10^300); if it were to randomly sample all of them to find the native state, it would take longer than the age of the universe. Yet, in nature, proteins fold spontaneously on timescales of milliseconds to seconds [1] [2]. This paradox underscores the immense difficulty of computationally predicting protein structure.
Computational Approaches and Associated Timescales
Computational methods for studying protein folding must navigate the trade-off between atomic-level detail, which is computationally expensive, and the need to simulate processes that occur over long periods. The table below summarizes the key methodologies and their characteristics.
Table 1: Computational Methods for Protein Folding
| Method | Key Features | Representative Simulated Folding Times | Key Challenges |
|---|---|---|---|
| All-Atom Molecular Dynamics (MD) | Models all atoms with classical force fields; explicit or implicit solvent; high spatial and temporal resolution [4]. | TRP-cage (20 residues): ~4 µs [4]Villin headpiece (35 residues): ~1–10 µs [4]WW domain (mutant): <15 µs [4] | Extreme computational cost; limited sampling; force field inaccuracies can destabilize native states [4]. |
| Constrained MD (e.g., GNEIMO) | Fixes bond lengths/angles, uses torsional degrees of freedom; larger time steps (e.g., 5 fs); faster than all-atom MD [5]. | Simulations of small proteins (e.g., Trp-cage) achieved in ~20 ns/replica using replica exchange [5]. | Efficient conformational search; hierarchical "freeze-and-thaw" of rigid bodies can enhance native state sampling [5]. |
| Machine-Learned Coarse-Grained (CG) Models | Groups atoms into "beads"; learned from all-atom data; highly computationally efficient [6]. | Several orders of magnitude faster than all-atom MD; enables folding/unfolding simulations of proteins like villin and engrailed homeodomain (1ENH, 54 residues) [6]. | Reproducing correct multi-body interactions and thermodynamics; transferability across diverse protein sequences [6]. |
| Deep Learning AI (e.g., AlphaFold) | End-to-end deep learning trained on known structures/sequences; predicts static structure without simulating folding pathway [2]. | Predicts structure in days (using ~100-200 GPUs for training), bypassing the need for dynamical simulation [2]. | Does not model the folding process, intermediates, or dynamics; initially limited accessibility for commercial use [7] [8]. |
The fundamental challenge for dynamical simulation methods like all-atom MD is the vast difference between the femtosecond timestep required for numerical integration and the microsecond-to-second timescale of actual folding events [4]. This 10^9 to 10^15 gap makes direct simulation prohibitively expensive for all but the smallest, fastest-folding proteins.
Table 2: Experimentally Derived Folding Times for Model Systems
| Protein | Size (Residues) | Experimentally Derived Folding Time |
|---|---|---|
| TRP-cage | 20 | ~4 µs [4] |
| Villin Headpiece | 35 | ~1–10 µs [4] |
| WW Domain (mutant) | ~35 | <15 µs [4] |
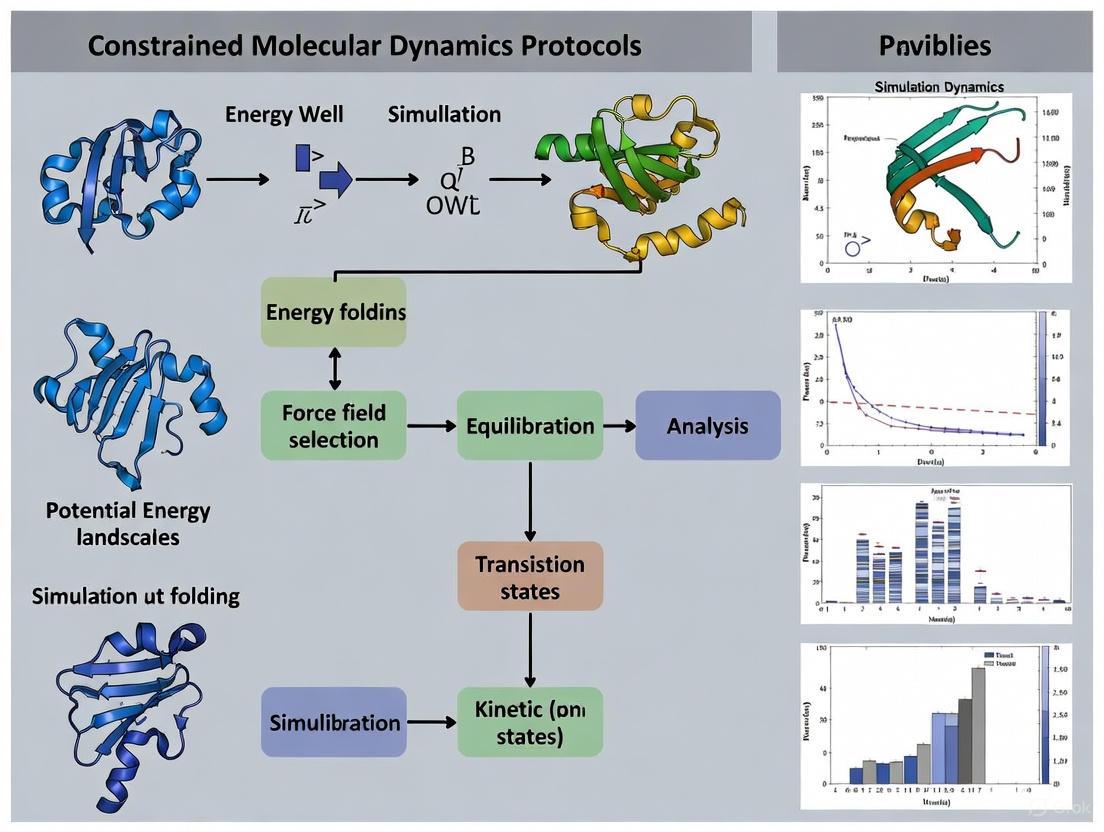
Constrained MD Protocols for Small Protein Folding
Constrained Molecular Dynamics (MD), also known as torsion-angle MD, offers a protocol to enhance conformational sampling for protein folding studies. The following section details a specific methodology as applied to small proteins like the Trp-cage miniprotein.
The following diagram illustrates the hierarchical constrained MD protocol for protein folding.
Detailed Experimental Methodology
1. System Preparation:
- Begin with an extended conformation of the protein's amino acid sequence [5].
- Perform energy minimization using a conjugate gradient algorithm until the force gradient converges to a predefined threshold (e.g., 10⁻² Kcal/mol/Å) to remove steric clashes [5].
2. Define the Constrained MD Model:
- Apply holonomic constraints to fix all bond lengths and bond angles. The molecule is treated as a collection of rigid bodies (clusters) connected by flexible torsional hinges [5].
- Choose a clustering scheme:
- All-Torsion Model: All torsional degrees of freedom are active [5].
- Hierarchical Clustering: Partially formed secondary structure regions identified in preliminary simulations are "frozen" into rigid clusters. Only the torsional degrees of freedom connecting these clusters are sampled, which aligns with zipping-and-assembly folding models and can accelerate convergence [5].
3. Simulation Parameters (using GNEIMO method):
- Force Field: Use an all-atom force field parameterization such as parm99 (AMBER99) [5].
- Solvation Model: Employ an implicit solvent model like the Generalized-Born Surface Area (GB/SA) to significantly reduce computational cost. Typical parameters include an interior dielectric of 1.75 for the solute and an exterior dielectric of 78.3 for water [5].
- Non-bonded Interactions: Apply a cutoff (e.g., 20 Å) with a smooth switching function to zero [5].
- Integration: Use an internal coordinate integrator (e.g., Lobatto integrator) with a timestep of 5 fs, made possible by the constrained model [5].
4. Enhanced Sampling via Replica Exchange:
- To overcome energy barriers and achieve adequate sampling, employ a replica exchange molecular dynamics (REMD) protocol [5].
- Set up multiple replicas (e.g., 8 replicas for a small protein) spanning a temperature range (e.g., 325K to 500K) [5].
- Attempt exchanges between neighboring replicas at regular intervals (e.g., every 2 ps) based on the Metropolis criterion [5].
- Note: The reduced number of degrees of freedom in constrained MD reduces the number of required replicas compared to all-atom REMD by approximately a factor of three [5].
5. Trajectory Analysis:
- Principal Component Analysis (PCA): Project the simulation trajectories onto the first two principal components to visualize the conformational landscape and population density [5].
- Clustering: Use algorithms like K-means to group similar conformations and identify dominant structural states. The population percentage of a cluster is the fraction of total conformations belonging to it [5].
- Native-likeness Metrics: Calculate the Root Mean Square Deviation (RMSD) of alpha-carbon atoms from the known native structure and compute the Fraction of Native Contacts (Q) to assess prediction accuracy and identify folded states [6].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Constrained MD Folding Studies
| Item | Function / Rationale | Example Specifications |
|---|---|---|
| All-Atom Force Field | Provides the empirical potential energy function governing atomic interactions; critical for accuracy. | AMBER (parm99) [5], CHARMM, OPLS/AA [4]. |
| Implicit Solvation Model | Mimics the effect of solvent (water) without explicit water molecules, drastically reducing compute cost. | Generalized-Born Surface Area (GB/SA) [5]. |
| Constrained MD Software | Software capable of performing MD in internal coordinates with rigid body clusters. | GNEIMO method [5]. |
| Replica Exchange Wrapper | Manages the parallel simulations at different temperatures and coordinate exchange attempts. | Custom or integrated scripts (e.g., in GNEIMO) [5]. |
| Trajectory Analysis Suite | Used for processing simulation output, calculating metrics (RMSD, Q), and visualizing results. | Tools for Principal Component Analysis (PCA), K-means clustering [5]. |
| Model Protein Systems | Small, well-characterized, fast-folding proteins for method development and validation. | TRP-cage (20 residues) [4] [5], Villin Headpiece (35 residues) [4], WW Domain [4]. |
The protein folding challenge, once hindered by Levinthal's paradox, is being addressed by a multi-faceted computational arsenal. While all-atom MD remains the gold standard for detailed dynamical insight, its extreme computational cost has driven the development of efficient alternatives like constrained MD. These methods, particularly when enhanced with replica exchange and hierarchical clustering, provide a powerful protocol for simulating the folding of small proteins and enriching the sampling of native-like states [5].
The field is now being transformed by AI systems like AlphaFold, which have demonstrated remarkable accuracy in predicting static protein structures from sequence, bypassing the timescale problem entirely [9] [2]. However, for understanding folding pathways, kinetics, and the role of energy landscapes, dynamical simulation methods remain essential. The future lies in integrating these approaches, with emerging technologies like machine-learned coarse-grained models promising to deliver the transferability and speed of AI with the dynamical insight of physics-based simulations [6]. The continued development of open-source AI models, such as OpenFold3 and Boltz-1, will also be crucial for broadening access and fueling further innovation in basic research and drug discovery [7] [8].
Reducing Degrees of Freedom with Constrained MD
Constrained Molecular Dynamics (MD) is an advanced simulation technique that addresses a fundamental bottleneck in all-atom Cartesian MD simulations: their prohibitively long computational times for studying processes like protein folding. By incorporating holonomic constraints directly into the molecular model, constrained MD reduces the number of degrees of freedom by approximately an order of magnitude, transforming the system into a collection of rigid bodies ('clusters') connected by flexible torsional hinges [5]. This approach fundamentally differs from all-atom MD by using torsional angle coordinates as the primary degrees of freedom instead of atomic Cartesian coordinates [5].
The computational advantage emerges from two key factors: the drastic reduction in system degrees of freedom and the elimination of high-frequency vibrational modes associated with bond stretching. This dual benefit enables stable dynamics with significantly larger integration time steps (up to 5 femtoseconds), leading to a substantial decrease in computational cost for exploring long-timescale biological processes such as protein folding [5]. For protein folding research, this method provides an alternate MD tool that enhances conformational search towards "native-like" structures while maintaining atomic-level insights [5] [10].
Methodological Framework
Core Computational Algorithm
The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method provides the mathematical foundation for efficient constrained MD simulations. This algorithm solves the coupled equations of motion in internal coordinates with O(N) computational cost, in contrast to conventional O(N³) techniques, making it practical for large protein systems [5]. The framework enables hierarchical "freeze and thaw" clustering schemes, allowing researchers to dynamically adjust which parts of the protein are treated as rigid bodies and which torsional degrees of freedom are sampled during simulation [5].
Table 1: Key Components of the Constrained MD Framework
| Component | Description | Function in Simulation |
|---|---|---|
| Holonomic Constraints | Fixed bond lengths and bond angles within rigid clusters | Eliminates high-frequency vibrations, enables larger time steps |
| Torsional Hinges | Flexible connections between rigid clusters | Maintains essential conformational flexibility |
| Spatial Operator Algebra | Mathematical framework for multibody dynamics | Enables O(N) computational scaling |
| Replica Exchange Method | Parallel sampling at multiple temperatures | Enhances conformational sampling efficiency |
Experimental Implementation Protocol
The following protocol outlines the standard methodology for protein folding studies using constrained MD:
System Preparation:
- Start with an extended conformation of the peptide/protein sequence [5]
- Perform conjugate gradient minimization with a convergence criterion of 10⁻² kcal/mol/Å in force gradient [5]
- Apply the parm99 forcefield (part of AMBER99) for energy calculations [5]
- Utilize implicit solvation with the Generalized-Born Surface Area (GB/SA) OBC model [5]
Simulation Parameters:
- Set GB/SA interior dielectric value to 1.75 for the solute and exterior dielectric constant to 78.3 for aqueous solvent [5]
- Use a solvent probe radius of 1.4Å for nonpolar solvation energy calculations [5]
- Apply a 20Å cutoff for non-bonded forces with smooth switching [5]
- Employ the Lobatto integrator with an integration step size of 5 fs [5]
Replica Exchange Configuration:
- Configure 8 replicas across a temperature range of 325K to 500K (in 25K steps) [5]
- Attempt temperature exchanges between replicas every 2ps [5]
- Run simulations for up to 20ns per replica [5]
Hierarchical Clustering:
- Identify partially formed structural regions during initial simulation trajectory
- Freeze these regions as rigid clusters in subsequent hierarchical simulations
- Sample only the torsional degrees of freedom connecting these clusters
Performance Analysis and Applications
Quantitative Assessment of Efficiency
Constrained MD demonstrates significant advantages over traditional all-atom MD across multiple performance metrics, particularly for small protein folding applications.
Table 2: Performance Comparison of MD Methods for Protein Folding
| Parameter | All-Atom MD | Constrained MD | Advantage Factor |
|---|---|---|---|
| Degrees of Freedom | ~3N (Atomic Cartesian) | ~N (Torsional) | ~3x reduction [5] |
| Integration Time Step | 1-2 fs | 5 fs | 2.5-5x increase [5] |
| Replica Count | Proportional to √(3N) | Proportional to √(N) | ~3x reduction [5] |
| Computational Scaling | O(N) to O(N²) | O(N) with GNEIMO | More efficient for large systems [5] |
| Native-State Enrichment | Baseline | Increased enrichment | Wider conformational search [5] |
Application to Model Protein Systems
The constrained MD method has been successfully validated across multiple protein systems with diverse structural motifs:
α-Helical Peptides:
- Polyalanine (Ala20): Simulations with GB/SA solvation at 300K achieved helical formation without elevated temperatures previously required in earlier studies [5]. Helicity was quantified by measuring the fraction of residues with backbone torsional angles within 20° of ideal α-helical angles (φ = -57°, ψ = -47°) [5].
- WALP16 Transmembrane Peptide: Employed a membrane-mimetic environment with external dielectric constant of 40.0, demonstrating the method's adaptability to different biological environments [5].
β-Sheet and Mixed Motif Proteins:
- Beta Hairpin (1E0Q): A β-turn motif derived from ubiquitin successfully folded using the all-torsion constrained MD approach [5] [10].
- Trp-cage Miniprotein: This 20-residue mixed-motif protein folds in approximately 4µs experimentally and serves as an important model system [4]. Hierarchical constrained MD simulations, where partially formed helical regions were frozen, demonstrated better sampling of near-native structures than all-torsion simulations, consistent with the zipping-and-assembly folding model [5].
Research Reagent Solutions
Table 3: Essential Computational Tools for Constrained MD Simulations
| Research Reagent | Type | Function in Constrained MD |
|---|---|---|
| GNEIMO Method | Software Algorithm | Solves constrained dynamics equations with O(N) computational scaling [5] |
| AMBER parm99 | Force Field | Provides potential energy parameters for molecular interactions [5] |
| GB/SA OBC Model | Implicit Solvation | Computes solvation effects without explicit water molecules [5] |
| Replica Exchange Method | Sampling Enhancement | Facilitates escape from local energy minima through temperature cycling [5] |
| Lobatto Integrator | Numerical Integration | Propagates dynamics with stable integration at 5fs time steps [5] |
| Principal Component Analysis | Trajectory Analysis | Identifies essential dynamics and conformational populations [5] |
Concluding Remarks
Constrained molecular dynamics represents a computationally efficient alternative to all-atom MD for protein folding studies, particularly for small proteins with diverse structural motifs. The method's key innovation lies in reducing the system's degrees of freedom through holonomic constraints while maintaining sufficient flexibility to sample biologically relevant conformational spaces. The hierarchical "freeze and thaw" approach extends this methodology by enabling dynamic adjustment of rigid clusters during simulation, aligning with theoretical folding models like zipping-and-assembly [5].
For the drug development researcher, constrained MD offers a practical tool for studying folding pathways and intermediate states that may be relevant to molecular recognition and binding events. The enhanced sampling of near-native structures combined with significantly reduced computational cost makes this method particularly valuable for rapid assessment of protein folding behavior in early-stage research programs.
Molecular dynamics (MD) simulation is a pivotal tool in structural biology, providing atomic-level details of protein folding and dynamics. However, the computational cost of all-atom simulations remains a significant barrier, particularly for processes occurring on biologically relevant timescales. Constrained MD protocols address this challenge by reducing the system's degrees of freedom, enabling enhanced conformational sampling with greater computational efficiency. This application note details the key advantages, quantitative performance metrics, and detailed protocols for implementing constrained MD in small protein folding research, providing researchers and drug development professionals with practical guidance for leveraging these methods.
Fundamental Principles and Key Advantages
Constrained MD methods operate on the principle of imposing holonomic constraints on bond lengths and bond angles, effectively reducing the number of computational degrees of freedom by approximately an order of magnitude compared to all-atom Cartesian MD [5]. This reduction enables two primary advantages: significant increases in integration time steps and enhanced exploration of conformational space.
The computational efficiency stems from the elimination of high-frequency vibrational modes, which normally limit time steps to 1-2 femtoseconds in all-atom MD. With constraints, stable dynamics can be achieved with time steps of 4-5 femtoseconds or larger [5]. This translates directly to the ability to simulate longer biological timescales with equivalent computational resources.
For conformational sampling, the constrained approach provides enhanced exploration of torsional space. Studies demonstrate that constrained MD exhibits wider conformational search characteristics than all-atom MD, with increased enrichment of near-native structures for protein folding applications [5]. The fixed covalent geometry and correction to torsional potential in constrained MD leads to more effective barrier crossing in the energy landscape.
Table 1: Key Algorithmic Comparisons in Constrained Molecular Dynamics
| Algorithm | Constraint Method | Computational Efficiency | Accuracy | Primary Applications |
|---|---|---|---|---|
| ILVES-PC | Newton's method for bond constraints | Up to 76× faster than SHAKE (multi-threaded) [11] | Higher than P-LINCS [11] | Polymer and protein dynamics |
| SHAKE | Nonlinear Gauss-Seidel method | Reference baseline | Default in many MD packages [11] | General biomolecular simulations |
| P-LINCS | Linear constraint solver | Faster than SHAKE | Lower than ILVES-PC [11] | Large biomolecular systems |
| GNEIMO | Torsional angle constraints | 10× fewer degrees of freedom [5] | Comparable to all-atom with enhanced sampling [5] | Protein folding, domain motions |
Quantitative Performance Data
Recent advances in constraint algorithms demonstrate substantial improvements in both computational efficiency and sampling effectiveness. The ILVES-PC algorithm, which solves nonlinear constraint equations using Newton's method rather than the nonlinear Gauss-Seidel approach of SHAKE, shows speedups of up to 4.9× in single-threaded executions and up to 76× in shared-memory multi-threaded executions when integrated into GROMACS [11]. This performance advantage grows with system size and parallelization.
In protein folding applications, constrained MD replica exchange methods have proven particularly effective. The reduced degrees of freedom in constrained models decrease the number of replicas required for efficient conformational sampling by approximately a factor of three compared to all-atom models [5]. This directly reduces computational requirements while maintaining thorough sampling of the folding landscape.
For small protein folding, constrained MD simulations have successfully folded various motifs including α-helices (polyalanine, WALP16), β-turns (1E0Q), and mixed motif proteins (Trp-cage) [5]. These simulations demonstrate that hierarchical constrained MD simulations, where partially formed secondary structure regions are frozen, show better sampling of near-native structures than all-torsion constrained MD, aligning with zipping-and-assembly folding models [5].
Table 2: Performance Metrics for Constrained MD in Protein Folding Applications
| System | Method | Time Step | Sampling Efficiency | Folding Accuracy |
|---|---|---|---|---|
| Polyalanine (Ala20) | All-torsion constrained MD with GB/SA | 5 fs [5] | High helicity formation at 300K [5] | Native-like α-helical structures |
| Trp-cage | Hierarchical constrained MD | 5 fs [5] | Wider conformational search than all-atom MD [5] | Near-native structures enriched |
| Chignolin (2RVD) | Machine-learned CG model | N/A | Comparable to all-atom MD [6] | Correct folded state prediction |
| Villin headpiece (1YRF) | Machine-learned CG model | N/A | Folding/unfolding transitions [6] | Native-like structures |
Experimental Protocols
Constrained MD with Replica Exchange for Protein Folding
Application: Folding of small proteins with various secondary structural motifs Primary Reference: [5]
Protocol Steps:
- System Preparation:
- Start from an extended conformation of the peptide/protein sequence
- Perform conjugate gradient minimization with convergence criterion of 10⁻² kcal/mol/Å in force gradient
Force Field and Solvation:
- Utilize parm99 forcefield (AMBER99) with implicit solvation GB/SA OBC model
- Set GB/SA interior dielectric value to 1.75 for solute and exterior dielectric constant of 78.3 for solvent
- Use solvent probe radius of 1.4Å for nonpolar solvation energy component
- Apply smooth non-bond force switching at cutoff radius of 20Å
Constrained Dynamics Setup:
- Implement all torsional degrees of freedom or hierarchical clustering schemes
- Apply Lobatto integrator with integration step size of 5 fs
- Use replica exchange method with 8 replicas in temperature range 325K to 500K (25K steps)
- Exchange temperatures between replicas every 2ps
- Total simulation duration: up to 20ns per replica
Hierarchical Clustering:
- Identify partially formed secondary structure regions during simulation
- Freeze backbone torsions in these regions to form rigid clusters
- Sample only torsional degrees of freedom connecting these clusters
- Continue sampling with this adaptive scheme to enhance native structure convergence
ILVES-PC Implementation for Enhanced Constraint Satisfaction
Application: Accurate and efficient constraint satisfaction in polymer/protein dynamics Primary Reference: [11]
Protocol Steps:
- Algorithm Selection:
- Implement Newton's method for solving nonlinear constraint equations
- Replace traditional SHAKE or P-LINCS algorithms in MD packages
Linear System Solution:
- Apply specialized linear solver exploiting protein structure
- Solve necessary linear systems with computational cost proportional to number of constraints
Integration with MD Packages:
- Incorporate into GROMACS or other MD software
- Maintain same system of differential algebraic equations as SHAKE
- Achieve higher accuracy through precise numerical solution
Parallelization:
- Leverage shared-memory multi-threading capabilities
- Distribute constraint solving across available computational resources
Constrained MD Folding Workflow
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Item | Function/Application | Implementation Notes |
|---|---|---|
| GROMACS | MD simulation package | ILVES-PC integration for constraint satisfaction [11] |
| AMBER99 | Force field parameters | Compatible with constrained MD GB/SA simulations [5] |
| GB/SA OBC model | Implicit solvation | Dielectric constant 78.3 (water), 40.0 (membrane) [5] |
| Lobatto integrator | Constrained dynamics integration | Enables 5 fs time steps in constrained MD [5] |
| Replica Exchange | Enhanced sampling method | Requires fewer replicas than all-atom MD [5] |
| NEIMO/GNEIMO | Constrained dynamics algorithms | O(N) computational cost for solving equations of motion [5] |
Advanced Applications and Integration
Integration with Enhanced Sampling Techniques
Constrained MD methods effectively combine with advanced sampling approaches to address complex biological questions. True reaction coordinates (tRCs) that control both conformational changes and energy relaxation can be identified through energy flow theory and the generalized work functional method [12]. Biasing these tRCs in enhanced sampling simulations has demonstrated remarkable acceleration of processes like flap opening and ligand unbinding in HIV-1 protease, accelerating processes with experimental lifetimes of 8.9×10⁵ seconds to 200 picoseconds in simulation [12].
The combination of constrained dynamics with replica exchange methods has proven particularly powerful. The reduced dimensionality of constrained models decreases the number of replicas required for efficient temperature-based sampling, reducing computational costs while maintaining thorough phase space exploration [5]. This hybrid approach enables studies of folding pathways and metastable states that would be prohibitively expensive with all-atom methods.
Machine-Learned Coarse-Grained Models
Recent advancements incorporate machine learning with coarse-grained modeling to create transferable force fields that maintain accuracy while dramatically improving computational efficiency. These models demonstrate the feasibility of universal, computationally efficient CG models for proteins that are several orders of magnitude faster than all-atom models while successfully predicting metastable states of folded, unfolded, and intermediate structures [6].
Multiscale Simulation Approaches
Constrained MD protocols represent a powerful approach for balancing computational efficiency with physical accuracy in protein folding research. The key advantages of increased time steps, reduced degrees of freedom, and enhanced conformational sampling make these methods particularly valuable for studying folding pathways and metastable states. Integration with modern enhanced sampling techniques and machine-learned coarse-grained models further extends their applicability to biologically relevant systems and timescales. As algorithm development continues, constrained MD approaches will remain essential tools for researchers and drug development professionals seeking to understand protein dynamics and folding mechanisms.
Molecular dynamics (MD) simulations are an indispensable tool in computational chemistry and structural biology, predicting the motion of every atom in a molecular system over time based on interatomic interactions [13]. These simulations provide unparalleled atomic-level insight into biomolecular processes, including protein folding, conformational change, and ligand binding [13]. However, the substantial computational cost of all-atom MD simulations significantly restricts the accessible timescales for many biologically relevant processes [5] [14].
Constrained MD has emerged as an alternative approach that addresses these limitations by reducing the system's number of degrees of freedom, enabling longer timescale simulations [5]. This application note provides a detailed comparison of these methodologies, focusing on their theoretical foundations, practical implementation, and application to small protein folding research.
Fundamental Methodological Differences
The core distinction between all-atom and constrained MD lies in their treatment of molecular degrees of freedom and integration techniques.
All-Atom Molecular Dynamics
In all-atom Cartesian MD, the system is modeled with all atoms moving freely in three-dimensional space. The equations of motion are numerically integrated using Newton's laws, where forces are derived from a molecular mechanics force field that includes bonded and non-bonded interaction terms [13] [15]. The potential energy function typically includes:
- Bonded interactions: Bond stretching, angle bending, and torsion potentials
- Non-bonded interactions: Lennard-Jones potential for van der Waals forces and Coulomb's law for electrostatic interactions [15]
A significant limitation of all-atom MD is the requirement for very small integration time steps (typically 1-2 femtoseconds) to accurately capture the fastest vibrational motions, particularly bond stretching [16] [17]. This fundamentally restricts the physical timescales that can be simulated with available computational resources.
Constrained Molecular Dynamics
Constrained MD, also known as internal coordinate MD or torsion angle MD, incorporates holonomic constraints directly into the molecular model [5]. The molecule is represented as a collection of rigid bodies ("clusters") connected by flexible torsional hinges, with fixed bond lengths and bond angles within each cluster. This approach reduces the number of degrees of freedom by approximately an order of magnitude compared to all-atom models [5].
The elimination of high-frequency bond vibrations allows for larger integration time steps (up to 5 femtoseconds or more) and decreased computational cost per time step [5]. Specialized algorithms such as SHAKE, RATTLE, or the more efficient NEIMO (Newton-Euler Inverse Mass Operator) method are used to solve the coupled equations of motion in internal coordinates [5] [11].
Table 1: Fundamental Characteristics of All-Atom MD vs. Constrained MD
| Characteristic | All-Atom MD | Constrained MD |
|---|---|---|
| Degrees of Freedom | 3N (where N = number of atoms) | Approximately N/10 (torsional degrees only) |
| Typical Time Step | 1-2 femtoseconds | 3-5 femtoseconds (5+ fs achievable) |
| High-Frequency Motions | Explicitly simulated | Eliminated via constraints |
| Computational Scaling | O(N) to O(N²) for force calculations | O(N) with NEIMO algorithm |
| Energy Conservation | Good with small time steps | Stable with larger time steps |
Quantitative Comparison of Performance Metrics
Research studies directly comparing these methodologies provide insight into their relative performance for protein folding applications.
Computational Efficiency
Constrained MD demonstrates significant advantages in computational efficiency. The reduced number of degrees of freedom combined with the ability to use larger time steps results in an overall decrease in computational cost [5]. Furthermore, the number of replicas required in replica exchange simulations—a common enhanced sampling technique—is proportional to the square root of the number of degrees of freedom. Since constrained MD has approximately one-tenth the degrees of freedom of all-atom models, the number of required replicas is reduced by approximately a factor of three [5].
Conformational Sampling Efficiency
Studies on small proteins with various secondary structural motifs (α-helix, β-turn, and mixed motifs) demonstrate that constrained MD replica exchange methods exhibit wider conformational search than all-atom MD with increased enrichment of near-native structures [5] [10]. For the Trp-cage miniprotein, "hierarchical" constrained MD simulations, where partially formed helical regions were frozen, showed better sampling of near-native structures than all-torsion constrained MD simulations [5]. This hierarchical approach aligns with the zipping-and-assembly folding model proposed by Dill and coworkers [5].
Table 2: Performance Comparison for Protein Folding Applications
| Performance Metric | All-Atom MD | Constrained MD | Experimental Context |
|---|---|---|---|
| Sampling Diversity | Moderate | Wider conformational search [5] | Replica exchange simulations of Trp-cage, polyalanine [5] |
| Near-Native Enrichment | Standard | Increased enrichment [5] | Folding simulations starting from extended conformations [5] |
| Replica Exchange Efficiency | Requires more replicas | Fewer replicas needed (∼1/3) [5] | Temperature range 325K-500K for small proteins [5] |
| Hierarchical Sampling | Not applicable | Better near-native sampling [5] | Freezing helical regions in Trp-cage [5] |
Experimental Protocols for Protein Folding Studies
Constrained MD Protocol for Small Protein Folding
The following protocol outlines a generalized approach for folding small proteins using constrained MD, based on methodologies successfully applied to systems such as polyalanine, WALP16, and Trp-cage [5]:
System Preparation
- Start from an extended conformation of the peptide/protein sequence
- Perform conjugate gradient minimization with a convergence criterion of 10⁻² kcal/mol/Å in force gradient
Simulation Parameters
- Use parm99 forcefield (part of AMBER99) or other modern forcefields
- Employ implicit solvation (e.g., GB/SA OBC model) with interior dielectric of 1.75 and exterior dielectric of 78.3 for water
- Set solvent probe radius to 1.4Å for nonpolar solvation energy
- Apply non-bond force cutoff of 20Å with smooth switching
- Use Lobatto integrator with time step of 3-5 femtoseconds
Enhanced Sampling
- Implement replica exchange method with 6-8 replicas
- Use temperature range of 325K-500K (adjust based on system)
- Attempt exchanges between replicas every 2ps
- Total simulation duration: up to 20ns per replica
Hierarchical Clustering (Optional)
- Identify partially formed secondary structure elements during simulation
- "Freeze" these regions as rigid clusters while sampling only connecting torsions
- Implement "freeze and thaw" dynamics to adaptively adjust flexible regions
Trajectory Analysis
- Perform principal component analysis (PCA) using Cα atom coordinates
- Use K-means clustering algorithm to identify structurally similar subsets
- Compute population percentages of different conformational clusters
- Generate representative structures by averaging snapshots from each cluster
Workflow Visualization
The following diagram illustrates the key decision points and methodological considerations when selecting between all-atom and constrained MD approaches for protein folding studies:
MD Methodology Selection Workflow
Research Reagent Solutions
Successful implementation of constrained MD simulations requires specific computational "reagents" and tools. The following table details essential components and their functions:
Table 3: Essential Research Reagent Solutions for Constrained MD
| Reagent/Tool | Function | Implementation Examples |
|---|---|---|
| Constraint Algorithms | Impose bond length/angle constraints to enable larger time steps | SHAKE [5], RATTLE [5], ILVES-PC [11] |
| Internal Coordinate Solvers | Solve equations of motion in reduced coordinate space | NEIMO [5], GNEIMO [5] |
| Implicit Solvation Models | Account for solvent effects without explicit water molecules | GB/SA [5], GBSW [16] |
| Enhanced Sampling Methods | Improve conformational sampling efficiency | Replica Exchange [5], Metadynamics [14] |
| Force Fields | Define empirical energy functions for molecular interactions | AMBER [5] [16], CHARMM [5] |
| Hierarchical Clustering Schemes | Freeze/thaw structural elements to match folding mechanisms | "Freeze and thaw" GNEIMO [5] |
Applications to Small Protein Folding
Constrained MD has proven particularly valuable for studying the folding of small proteins and peptides with various structural motifs:
α-Helical Peptides
Studies on polyalanine and WALP16 (a transmembrane peptide) demonstrate that constrained MD with implicit solvent can successfully fold these α-helical structures. For WALP16, an external dielectric constant of 40.0 effectively represents the membrane environment [5].
Mixed Motif Proteins
The Trp-cage miniprotein (20 residues) represents a more challenging system with mixed structural motifs. Constrained MD simulations successfully sample near-native structures, with hierarchical approaches (freezing partially formed helical regions) providing superior results [5] [16].
Oxidative Folding
Specialized constrained MD approaches can model disulfide bond formation during folding. The CYR residue type (cysteine with deprotonated thiol) enables simulation of oxidative folding pathways, relevant for peptides like guanylin and proguanylin [18].
Limitations and Complementary Methods
While constrained MD offers significant advantages for protein folding studies, several limitations should be considered:
- Fixed bond geometries may neglect important structural fluctuations in certain applications
- Implicit solvent models may not fully capture specific solvent effects [16]
- Implementation complexity is higher than standard all-atom MD
- May not be suitable for processes involving bond breaking/formation without specialized extensions [18]
Alternative approaches include Monte Carlo methods, which can provide extensive thermodynamic characterization of folding [16], and emerging machine learning methods like BioMD, which show promise for simulating long-timescale biomolecular dynamics [14].
Constrained MD represents a powerful methodology for studying small protein folding, offering enhanced conformational sampling and reduced computational cost compared to all-atom MD. The ability to implement hierarchical "freeze and thaw" schemes provides particular advantage for investigating zipping-and-assembly folding mechanisms. When selected appropriately for the research question and system characteristics, constrained MD serves as a valuable tool in the computational structural biologist's toolkit, enabling insights into protein folding landscapes that might otherwise be computationally inaccessible.
The study of small protein folding is a critical area of research in structural biology and drug discovery. Understanding how amino acid sequences encode specific three-dimensional structures provides fundamental insights into biological function and enables the design of novel therapeutic agents. This application note details constrained molecular dynamics (MD) protocols for simulating the folding of small proteins, with a specific focus on systems rich in α-helix, β-turn, and mixed structural motifs. Constrained MD methods offer a computationally efficient alternative to all-atom MD by reducing the number of degrees of freedom, enabling enhanced conformational sampling and the study of folding dynamics on biologically relevant timescales [5]. These protocols are particularly valuable for investigating folding mechanisms, predicting metastable states, and understanding the energy landscapes of proteins that may access multiple folded conformations, including the emerging class of fold-switching proteins [19] [20].
Background and Significance
Proteins populate a spectrum of stability, from single, highly stable folds to intrinsically disordered ensembles. Fold-switching proteins represent a fascinating intermediate, featuring energy landscapes with multiple minima corresponding to distinct, biologically active native-like conformations [20]. These proteins challenge the classical "one sequence–one structure" paradigm and play critical regulatory roles across all kingdoms of life. For instance, the human chemokine XCL1 interconverts between a monomeric chemokine fold and a dimeric fold with a completely reregistered hydrogen-bonding network, enabling it to perform dual signaling and pathogen-response functions [20]. Accurately simulating the folding landscapes of such systems, including their alternative folds and the transitions between them, requires advanced sampling techniques that can capture multiple metastable states.
Computational studies have demonstrated that coarse-grained models and constrained MD approaches can successfully predict folding/unfolding transitions, fluctuations of disordered regions, and relative folding free energies, achieving performance comparable to all-atom MD but at a fraction of the computational cost [5] [6]. The development of transferable, machine-learned coarse-grained force fields now enables extrapolative molecular dynamics on new protein sequences not used during model parameterization, opening new avenues for the predictive simulation of diverse protein folds [6].
Application Notes: Constrained MD for Structural Motifs
α-Helical Peptide Folding
Application: Constrained MD is highly effective for studying the folding of α-helical peptides such as polyalanine and transmembrane peptides like WALP16. The reduced number of degrees of freedom allows for efficient sampling of the helical conformational space.
Key Findings from Literature:
- Polyalanine (Ala20): Constrained MD replica exchange simulations using the GB/SA implicit solvation model successfully fold Ala20 at 300 K. A residue is considered helical if its backbone torsional angles (φ, ψ) are within 20° of ideal α-helical angles (-57°, -47°). Simulations demonstrate a rapid increase in helicity, reaching a stable fraction of residues in helical conformation [5].
- WALP16: This transmembrane peptide (Ace-GWW(LA)5WWA-Nme) is folded in a membrane-mimetic environment simulated using a GB/SA exterior dielectric constant of 40.0. Constrained MD accurately captures the stabilization of the helical structure in this hydrophobic environment [5].
Performance Data: Table 1: Constrained MD Performance on α-Helical Peptides
| Peptide | Sequence Length | Environment (Dielectric) | Helicity Definition (φ, ψ tolerance) | Simulation Success |
|---|---|---|---|---|
| Polyalanine (Ala20) | 20 residues | Water (78.3) | (-57°, -47°) ± 20° | Correctly folds to stable helix [5] |
| WALP16 | 16 residues | Membrane (40.0) | (-57°, -47°) ± 20° | Stabilizes helical structure in membrane [5] |
β-Turn and β-Hairpin Folding
Application: β-Turns are common reverse motifs that often serve as nucleation sites for the formation of more extensive β-sheets and β-hairpins. Constrained MD facilitates the sampling of the conformational transitions involved in turn formation and strand association.
Key Findings from Literature:
- A β-hairpin peptide derived from ubiquitin (PDB: 1E0Q) has been used as a model system for constrained MD folding studies. The method successfully samples the formation of the hairpin's hydrogen-bonded network and native-like structure [5].
- The folding of β-sheets is inherently more complex than α-helices due to non-local interactions where β-strands distant in sequence come together. This can lead to slower folding rates and a higher propensity for misfolding, making enhanced sampling techniques like replica exchange particularly valuable [21].
Mixed-Motif Protein Folding
Application: Proteins containing both α-helical and β-sheet elements represent a significant computational challenge. Constrained MD has been validated on several small, fast-folding mixed-motif proteins.
Key Findings from Literature:
- Trp-cage (PDB: 2JOF): This 20-residue miniprotein contains a C-terminal polyproline helix and a salt bridge. "Hierarchical" constrained MD simulations, where partially formed helical regions are frozen based on initial trajectory analysis, demonstrate improved sampling of near-native structures compared to all-torsion simulations. This approach aligns with the zipping-and-assembly folding model [5].
- Villin Headpiece (PDB: 1YRF) & BBA (PDB: 1FME): Machine-learned coarse-grained models, which share principles with constrained dynamics, successfully predict the folded states and folding/unfolding transitions of these mixed-motif proteins. The model stabilizes folded states with a high fraction of native contacts (Q ~1) and low Cα root-mean-square deviation (RMSD) [6].
- Chignolin (PDB: 2RVD): For this small β-hairpin, coarse-grained models can capture not only the native state but also a specific misfolded state with a similar free energy, as observed in all-atom reference simulations [6].
Performance Data: Table 2: Constrained MD and Coarse-Grained Model Performance on Mixed-Motif Proteins
| Protein (PDB ID) | Key Structural Features | Simulation Approach | Key Outcome |
|---|---|---|---|
| Trp-cage (2JOF) | C-terminal polyproline helix, salt bridge | Hierarchical Constrained MD | Better native-state sampling vs. all-torsion MD [5] |
| Villin Headpiece (1YRF) | Three-helix bundle | Machine-Learned Coarse-Grained MD | Predicts folded state with high native contacts (Q~1) [6] |
| BBA (1FME) | Helical and anti-parallel β-sheet | Machine-Learned Coarse-Grained MD | Folds to native-like state (local free energy min.) [6] |
| Chignolin (2RVD) | β-hairpin | Machine-Learned Coarse-Grained MD | Captures native state and a specific misfolded state [6] |
Experimental Protocols
Generalized Constrained MD Protocol for Protein Folding
This protocol utilizes the Generalized NEIMO (GNEIMO) method for constrained MD simulation, incorporating replica exchange for enhanced sampling [5].
1. System Preparation:
- Initial Conformation: Begin with an extended conformation of the protein sequence.
- Force Field and Solvation: Use the parm99 forcefield (AMBER99) with an implicit solvation model, such as the Generalized-Born Surface Area (GB/SA) OBC model.
- Set the GB/SA interior dielectric to 1.75 and the exterior dielectric to 78.3 for aqueous environments (use 40.0 for membrane environments).
- Use a solvent probe radius of 1.4 Å for nonpolar solvation energy and a non-bonded force cutoff of 20 Å.
2. Energy Minimization:
- Perform conjugate gradient minimization on the initial structure with a convergence criterion of 10⁻² kcal/mol/Å in force gradient.
3. Constrained MD Replica Exchange Simulation:
- Dynamics Engine: Conduct simulations using the GNEIMO constrained MD algorithm.
- Integration: Use a Lobatto integrator with a time step of 5 fs.
- Replica Exchange Parameters:
- Utilize 6-8 replicas for small peptides (e.g., 8 replicas for a temperature range of 325 K to 500 K in 25 K increments).
- Attempt exchanges between neighboring replicas every 2 ps.
- Run simulations for a total duration of up to 20 ns per replica to achieve convergence.
4. Hierarchical Clustering (Optional, for complex motifs):
- Analyze initial all-torsion simulation trajectories to identify partially formed stable secondary structures (e.g., helical regions).
- "Freeze" the torsional degrees of freedom within these identified regions into rigid clusters.
- Repeat the replica exchange simulation, sampling only the torsional degrees of freedom connecting these rigid clusters. This "freeze and thaw" scheme can accelerate convergence to the native state [5].
5. Trajectory Analysis:
- Secondary Structure Analysis: Calculate the fraction of residues in native helical or β-sheet conformation over the trajectory.
- Principal Component Analysis (PCA): Project simulation trajectories onto the first two principal components from the Cα atom covariance matrix to visualize the free energy landscape and conformational populations.
- Clustering: Use algorithms like K-means to cluster trajectories into structurally similar subsets and determine the population percentage of each cluster.
Workflow Visualization
The following diagram illustrates the logical workflow for the constrained MD protein folding protocol:
Energy Landscape of Fold-Switching Proteins
Fold-switching proteins exhibit a complex energy landscape with multiple minima, as depicted below:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools and Resources for Constrained MD Protein Folding
| Item Name | Function/Application | Specifications/Notes |
|---|---|---|
| GNEIMO MD Package | Engine for performing constrained MD simulations. | Implements Newton-Euler Inverse Mass Operator; allows "freeze and thaw" of torsional degrees of freedom [5]. |
| AMBER99 Forcefield | Provides potential energy functions for the protein. | Often used with parm99 parameters; compatible with constrained MD and GB/SA solvation [5]. |
| GB/SA Implicit Solvent | Models solvent effects without explicit water molecules. | OBC model; interior dielectric=1.75, exterior=78.3 (water) or 40.0 (membrane); accelerates sampling [5]. |
| Replica Exchange Wrapper | Manages parallel MD simulations at different temperatures. | Enables conformational swapping; typically 6-8 replicas for constrained MD; reduces required replicas vs. all-atom MD [5]. |
| Protein Data Bank (PDB) | Source of initial protein structures and validation targets. | Repository of experimentally solved structures; used to verify simulation results and design target folds [22] [19]. |
| Principal Component Analysis (PCA) | Analyzes trajectory data to identify major conformational motions. | Uses covariance matrix of Cα atoms; projects trajectories onto principal components to visualize free energy landscapes [5]. |
Implementing Constrained MD Protocols: From Theory to Practice
All-Tion Constrained MD and Replica Exchange Method Implementations
Molecular dynamics (MD) simulations are a cornerstone of modern computational biology, providing atomic-level insights into protein folding mechanisms. However, the timescales required for folding events often remain a formidable challenge for conventional all-atom MD. Constrained MD methods address this bottleneck by reducing the system's degrees of freedom, typically by fixing bond lengths and angles and focusing sampling on the crucial torsional degrees of freedom [5] [23]. This approach, often coupled with enhanced sampling techniques like the Replica Exchange Method (REM), enables more efficient exploration of the protein conformational landscape [5] [24].
This Application Note details the implementation of all-torsion constrained MD simulations within a replica exchange framework, providing a structured protocol for studying small protein folding. We contextualize these methods within a broader thesis on constrained MD protocols, emphasizing their utility for researchers and drug development professionals investigating protein dynamics and folding pathways.
Theoretical Background and Key Concepts
All-Torsion Constrained Molecular Dynamics
In all-atom Cartesian MD, the system's motion is simulated using all atomic coordinates, which includes high-frequency vibrations of bonds and angles. Constrained MD, also known as torsional dynamics or internal coordinate MD, employs a different molecular model. The protein is treated as a collection of rigid bodies (clusters) connected by flexible torsional hinges [5] [23].
- Reduced Degrees of Freedom: By imposing holonomic constraints on bond lengths and bond angles, the number of degrees of freedom is reduced by approximately an order of magnitude compared to all-atom MD [5].
- Increased Time Step: The elimination of high-frequency motions allows for a significant increase in the integration time step, typically to 5 femtoseconds or more, compared to the 1-2 femtoseconds common in all-atom MD [5].
- Computational Efficiency: The reduced degrees of freedom and larger time steps collectively lead to a substantial decrease in computational cost, enabling the simulation of longer biological timescales [5].
The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method is an advanced algorithm for solving the equations of motion in internal coordinates with O(N) computational cost, making it practical for larger protein systems [5] [23].
Replica Exchange Molecular Dynamics (REMD)
REMD is an enhanced sampling technique designed to overcome kinetic traps and sample the conformational space more thoroughly. The method involves running multiple simultaneous MD simulations (replicas) of the same system at different temperatures or with different Hamiltonians [25] [24].
Periodically, exchanges between neighboring replicas are attempted based on a Metropolis criterion. For temperature REMD, the probability of exchanging replicas i (at temperature T1) and j (at temperature T2) is:
[P(1 \leftrightarrow 2)=\min\left(1,\exp\left[ \left(\frac{1}{kB T1} - \frac{1}{kB T2}\right)(U1 - U2) \right] \right)]
where U1 and U2 are the potential energies of the two replicas, and kB is Boltzmann's constant [25]. This process allows conformations to diffuse across temperatures, ensuring that low-temperature replicas can escape local energy minima while maintaining the correct Boltzmann distribution [24].
The combination of constrained MD with REMD is particularly powerful. The reduced number of degrees of freedom in constrained MD lowers the number of replicas required for efficient sampling, as the number of replicas scales with the square root of the system's degrees of freedom [5].
Experimental Protocols and Workflows
Workflow for Constrained MD Replica Exchange Simulations
The following diagram illustrates the integrated workflow for setting up and running a constrained MD replica exchange simulation, from system preparation to analysis.
Detailed Step-by-Step Protocol
This protocol is adapted from studies on folding small proteins like polyalanine, beta-hairpins, and the Trp-cage miniprotein [5].
System Preparation
- Initial Structure: Begin with an extended conformation of the peptide or protein sequence. This ensures no native bias at the start of the simulation [5].
- Force Field: Select an appropriate all-atom force field. The AMBER parm99 and AMBER99SB*-ILDN force fields have been successfully used in constrained MD folding studies [5] [16].
- Solvation Model: Use an implicit solvation model to reduce computational cost. The Generalized-Born Surface Area (GB/SA) model is commonly employed, with parameters such as an interior dielectric of 1.75 for the solute and an exterior dielectric of 78.3 for water [5]. A solvent probe radius of 1.4 Å is typical for the nonpolar solvation term.
Energy Minimization
- Perform conjugate gradient minimization on the initial extended structure to remove any steric clashes.
- A convergence criterion of 10⁻² kcal/mol/Å for the force gradient is sufficient for subsequent dynamics [5].
Constrained MD and REMD Parameters
- Constrained Dynamics: Use the GNEIMO method or an equivalent constrained dynamics algorithm. Set all torsional degrees of freedom as flexible unless applying a hierarchical scheme.
- Integrator and Time Step: Use a Lobatto integrator with a time step of 5 fs [5].
- Non-bonded Interactions: Apply a cutoff for non-bonded forces (e.g., 20 Å), smoothly switching them off.
- REMD Setup:
- Number of Replicas: For small proteins, 6-8 replicas are often adequate due to the reduced dimensionality of constrained MD [5].
- Temperature Range: A range from 325 K to 500 K, with exchanges attempted every 2 ps, has been used successfully for peptides like Trp-cage [5].
- Exchange Attempts: Follow a scheme where odd-numbered replica pairs (0-1, 2-3...) attempt exchange on odd attempts, and even-numbered pairs (1-2, 3-4...) attempt exchange on even attempts to maintain detailed balance [25].
Hierarchical Clustering (Advanced Protocol)
- For proteins with known or predicted secondary structure, a "freeze and thaw" protocol can be applied. After an initial all-torsion simulation, partially formed secondary structure elements (e.g., helical regions) can be frozen into rigid clusters. Subsequent simulations then sample only the torsional degrees of freedom connecting these rigid bodies, which can lead to faster convergence to the native state by reducing the conformational search space [5] [23].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 1: Key computational tools and parameters for constrained MD-REMD simulations.
| Item | Function/Description | Example Usage/Note |
|---|---|---|
| GNEIMO MD Package | Software for performing constrained MD simulations with efficient O(N) spatial operator algebra. | Enables all-torsion and hierarchical "freeze and thaw" simulations [5] [23]. |
| AMBER Force Fields | Parametrized potential functions for calculating molecular energies and forces. | AMBER parm99 and AMBER99SB*-ILDN are widely used and tested for protein folding [5] [16]. |
| GB/SA Implicit Solvent | An efficient solvation model that approximates the electrostatic and non-polar effects of water. | Reduces computational cost; GB/OBC model with a 1.4 Å probe radius is standard [5]. |
| Lobatto Integrator | Numerical integrator for solving the equations of motion in constrained dynamics. | Allows for stable dynamics with a 5 fs time step [5]. |
| Replica Exchange Framework | Software infrastructure for managing multiple MD replicas, attempts, and configuration swaps. | Implemented in major MD packages like GROMACS and AMBER; requires MPI for parallelism [25] [24]. |
| Principal Component Analysis (PCA) | Dimensionality reduction technique to identify essential motions from simulation trajectories. | Used to project conformational sampling onto principal components for analysis [5]. |
Data Presentation and Analysis
Quantitative Performance Comparison
The efficacy of constrained MD-REMD is demonstrated by its ability to sample near-native structures more efficiently than all-atom MD for several small protein motifs.
Table 2: Performance of constrained MD-REMD in folding small proteins and peptides.
| Protein/Peptide | Secondary Structure | Key Simulation Parameters | Key Results and Performance |
|---|---|---|---|
| Polyalanine (Ala₂₀) | α-helix | 6 replicas, 300 K, GB/SA water (ε=78.3) [5]. | Achieved stable helix formation; simulations at 300K did not require elevated temperatures for folding [5]. |
| WALP16 | Transmembrane α-helix | 8 replicas, GB/SA membrane (ε=40.0) [5]. | Correctly folded in membrane-mimetic environment; demonstrated applicability to different dielectric environments [5]. |
| Beta-hairpin (1E0Q) | β-turn | 8 replicas, 325K-500K temperature range [5]. | Sampled near-native structures effectively, showing method's utility beyond helical peptides [5]. |
| Trp-cage | Mixed (α-helix, 3₁₀-helix) | 8 replicas, 325K-500K, 5 fs timestep, 20ns/replica [5]. | All-torsion REMD exhibited wider conformational search and increased enrichment of near-native structures vs. all-atom MD [5]. |
| Trp-cage (Hierarchical) | Mixed | Initial all-torsion, then freezing helical clusters [5]. | Improved sampling of near-native states vs. all-torsion alone, consistent with zipping-and-assembly folding models [5]. |
Analysis Methodologies
- Measuring Structural Formation: For α-helical peptides like polyalanine, the fraction of residues in a helical conformation can be tracked by counting residues whose backbone dihedral angles (φ, ψ) are within a threshold (e.g., 20°) of ideal α-helical angles (-57°, -47°) [5]. The helicity is the fraction of helical residues over the total.
- Conformational Clustering and Free Energy Landscapes: Use algorithms like K-means clustering to group structurally similar conformations from the simulation trajectory. The population percentage of each cluster indicates the relative stability of different states [5]. Projecting the trajectory onto the first two principal components from Principal Component Analysis (PCA) allows for visualization of the conformational landscape and the density of sampled states [5].
- Validation with Experimental Data: For the Trp-cage miniprotein, which has a known NMR structure, the Root Mean Square Deviation (RMSD) of the simulated structures from the native conformation is a critical metric for assessing the accuracy of the folding simulation [16].
Discussion and Outlook
The integration of all-torsion constrained MD with the replica exchange method provides a powerful and computationally efficient platform for simulating the folding of small proteins. The key advantage lies in its strategic reduction of the conformational search space, which leads to faster convergence and enhanced sampling of biologically relevant native-like states [5]. The hierarchical clustering approach, which leverages prior knowledge or dynamically identified structural elements, further refines this strategy and aligns well with modern folding theories like the "zipping-and-assembly" model [5].
While constrained MD with implicit solvent is highly effective, the field of biomolecular simulation is rapidly evolving. Emerging AI-driven methods, such as BioEmu and AI2BMD, promise to revolutionize the scale and accuracy of protein dynamics simulations. BioEmu is a generative AI system that can simulate protein equilibrium ensembles with 1 kcal/mol accuracy using a single GPU, achieving a speedup of 4–5 orders of magnitude for equilibrium distributions [26]. Similarly, AI2BMD uses a machine learning force field and a protein fragmentation scheme to perform ab initio-quality simulations of large proteins (>10,000 atoms) efficiently, demonstrating accurate protein folding and unfolding processes [27]. These methods represent a significant shift toward highly scalable and thermodynamically accurate simulations that could soon complement or extend the capabilities of traditional physics-based methods like constrained MD.
For researchers today, the constrained MD-REMD protocol remains a robust and accessible method for studying protein folding mechanisms, particularly for systems where atomic-level detail and torsional dynamics are of primary interest.
The Hierarchical 'Freeze and Thaw' Clustering Scheme
The Hierarchical 'Freeze and Thaw' Clustering Scheme is a specialized computational protocol within the Generalized Newton-Euler Inverse Mass Operator (GNEIMO) constrained molecular dynamics (MD) framework. It is designed to enhance the conformational sampling of proteins during folding simulations and structure refinement by strategically reducing the number of degrees of freedom [5] [28]. This method allows researchers to "freeze" parts of a protein—treating them as rigid bodies—while "thawing" other regions to remain flexible and sample torsional angles [28]. By dynamically controlling which parts of the protein are flexible, this scheme enables a more efficient exploration of the conformational landscape compared to all-atom or all-torsion MD, leading to faster convergence to native-like structures and providing insights into protein folding pathways, in agreement with the zipping-and-assembly model [5].
Theoretical Background and Principles
Conventional all-atom Cartesian MD simulations are computationally expensive for studying protein folding due to the large number of degrees of freedom and the requirement for small integration time steps [5] [28]. The GNEIMO method addresses this by applying holonomic constraints to high-frequency bond vibrations and angles, modeling the protein as a collection of rigid bodies (clusters) connected by flexible torsional hinges [5] [28]. This reduces the system's degrees of freedom by approximately an order of magnitude and allows for larger integration time steps (e.g., 5 fs), significantly decreasing computational cost [5].
The hierarchical "freeze and thaw" scheme builds upon this foundation by allowing strategic, dynamic control over the rigidity of protein segments:
- Freezing: Secondary structure elements (e.g., pre-formed α-helices or β-sheets) are treated as single rigid bodies. Their internal covalent geometry is fixed, and they move as a single unit [5] [28].
- Thawing: The flexible loops and torsional angles connecting these rigid clusters remain active and are sampled during the simulation [28].
- Hierarchical Clustering: This creates a dynamic model where the level of flexibility can be adjusted based on the protein's predicted or evolving structure, guiding the conformational search along low-frequency, functionally relevant motions [5].
Application Notes
Quantitative Performance in Protein Folding and Refinement
The hierarchical scheme has been quantitatively validated in folding small proteins and refining low-resolution homology models. The table below summarizes key performance metrics.
Table 1: Performance Metrics of the Hierarchical 'Freeze and Thaw' Scheme
| Application / Protein | Key Metric | Reported Performance | Comparative Context |
|---|---|---|---|
| General Folding (e.g., Trp-cage) [5] | Sampling of near-native structures | Better sampling than all-torsion constrained MD | Wider conformational search and increased enrichment of near-native structures vs. all-atom MD |
| Structure Refinement (8 proteins) [28] | Improvement in backbone RMSD to native | ~2 Å improvement | Starting from low-resolution decoys (2-5 Å RMSD) |
| Computational Efficiency [5] | Integration Time Step | 5 fs | Enabled by constrained dynamics; larger than typical all-atom MD time steps |
| Replica Exchange Sampling [5] | Number of Replicas Required | Reduced by a factor of ~3 | Due to fewer degrees of freedom vs. all-atom MD |
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details the key software, force fields, and solvation models required to implement the GNEIMO method with the hierarchical clustering scheme.
Table 2: Key Research Reagents and Computational Tools for GNEIMO Simulations
| Item | Function / Description | Example Products / Formulations |
|---|---|---|
| Simulation Software | Implements the GNEIMO algorithm and the "freeze and thaw" clustering framework. | GNEIMO simulation package [5] [28] |
| Force Field | Provides parameters for potential energy calculations (bonded and non-bonded terms). | AMBER parm99/AMBER99 forcefield [5] [28] |
| Implicit Solvent Model | Mimics the presence of a solvent environment, reducing computational cost. | Generalized-Born/Surface Area (GB/SA) OBC model [5] [28] |
| Enhanced Sampling Method | Accelerates exploration of conformational space by running multiple simulations at different temperatures. | Replica Exchange Molecular Dynamics (REXMD) [5] [28] |
| Analysis Tools | Used for processing simulation trajectories to assess structural convergence and quality. | Principal Component Analysis (PCA), K-means clustering, RMSD calculations [5] |
Experimental Protocol
This section provides a detailed, step-by-step protocol for setting up and running a hierarchical constrained MD simulation for protein structure refinement using the GNEIMO method.
System Setup and Initial Minimization
- Initial Structure: Begin with a protein structure in an extended conformation or a low-resolution decoy model [5] [28].
- Force Field and Solvation: Apply the AMBER99 forcefield and the GB/SA OBC implicit solvation model. Use an interior dielectric of 1.5-1.75 for the solute and an exterior dielectric of 78.3 for water. Set a non-bonded force cutoff of 20 Å [5] [28].
- Energy Minimization: Perform conjugate gradient minimization on the initial structure. A typical protocol involves 1000 steps of steepest descent followed by 1000 steps of conjugate gradient minimization, with a convergence criterion of 10⁻² kcal/mol/Å for the force gradient [5] [28].
Defining the Clustering Scheme
- Identify Structural Motifs: Analyze the initial structure to identify stable secondary structural elements, such as α-helices or β-sheets. This can be done visually or using secondary structure prediction tools [5] [28].
- Apply "Freeze and Thaw":
- For a mixed motif protein (e.g., α/β), treat the well-defined α-helical or β-sheet regions as rigid bodies. The backbone atoms within these motifs are "frozen" into a cluster [5] [28].
- The side chains of these clusters can remain flexible (all-torsion) or be frozen based on the desired level of sampling [28].
- The remaining regions of the protein (e.g., loops, termini) are "thawed," meaning all torsional degrees of freedom are active [28].
Running Constrained Replica Exchange MD
- Simulation Parameters:
- Replica Exchange Setup:
- Production Simulation:
Post-Simulation Analysis
- Conformational Clustering: Use algorithms like K-means on the combined trajectory from all replicas to group structurally similar conformations. Generate a representative structure for each cluster by averaging the coordinates of multiple snapshots [5].
- Principal Component Analysis (PCA): Perform PCA on the Cα atom coordinates to project the high-dimensional trajectory onto the essential dynamics space defined by the first two principal components. Visualize the projection as a 2D free-energy landscape or population density map [5].
- RMSD and Native-likeness Assessment: Calculate the backbone RMSD of the cluster representatives and sampled structures against the known experimental native structure. Monitor the fraction of native contacts or secondary structure content to assess the quality of the refined models [5].
Workflow and Data Interpretation
The following diagram illustrates the logical workflow of a typical hierarchical "freeze and thaw" simulation for protein structure refinement.
Hierarchical Clustering MD Workflow
Interpretation Guidelines
- Convergence: A successful simulation is indicated by the population of a few dominant clusters in the PCA space, with one cluster having a low RMSD to the native state [5].
- Refinement Success: An improvement of ~2 Å in backbone RMSD from the starting decoy to the representative refined structure is considered a successful application of the protocol [28].
- Folding Pathways: The hierarchical scheme often samples conformations that support the "zipping-and-assembly" folding model, where secondary structures form first and then assemble into the tertiary structure [5].
Troubleshooting and Optimization
- Lack of Convergence: If the simulation does not converge to native-like structures, consider increasing the simulation time per replica or adjusting the temperature range for replica exchange. Alternatively, re-evaluate the initial clustering scheme; it may be beneficial to "thaw" a previously frozen region to allow for structural adjustments [5] [28].
- Structural Instability: Ensure that the initial energy minimization has converged properly. Using a softened potential or a simulated annealing protocol before the main production run can help "swell" the initial decoy and escape deep energy minima [28].
- Computational Cost: For larger proteins, the reduction in computational cost achieved by constrained dynamics is significant. However, if resources are limited, one can start with a more aggressive clustering scheme (larger frozen blocks) and gradually thaw regions in subsequent hierarchical simulations [5].
Molecular dynamics (MD) simulation is a cornerstone computational technique for studying protein folding, but its application is often limited by the extreme computational cost of simulating biologically relevant timescales. Constrained molecular dynamics (MD) provides an alternative tool for protein structure prediction and refinement by reducing the number of degrees of freedom in the system, enabling longer timescale simulations and enhanced conformational sampling [5]. This approach addresses a fundamental bottleneck of all-atom Cartesian MD simulations, where folding processes can occur on timescales of microseconds or longer, making them computationally prohibitive for many research applications [5].
The core principle of constrained MD involves imposing holonomic bond length and bond angle constraints directly into the molecular model, effectively reducing the number of degrees of freedom by approximately an order of magnitude compared to all-atom Cartesian MD models [5]. In this approach, the molecule is modeled as a collection of rigid bodies termed 'clusters' connected by flexible hinges, with torsional angles serving as the primary degrees of freedom. This reduction in dimensionality, combined with the elimination of high-frequency vibrational modes, allows for a significant increase in integration time step size—typically over an order of magnitude larger than in all-atom MD [5]. For researchers investigating small protein folding, constrained MD methods offer a balanced approach that maintains atomic detail while expanding the accessible simulation timescales, making them particularly valuable for studying folding mechanisms and native-state dynamics.
Theoretical Framework and Constraint Selection
Mathematical Foundation of Constrained Dynamics
Constrained MD simulations utilize internal coordinate systems, primarily torsional angles, as degrees of freedom instead of atomic Cartesian coordinates. The equations of motion in these internal coordinates become coupled, and solving them for accelerations requires inverting a dense mass matrix with computational complexity that traditionally scaled with the third power of the number of degrees of freedom [5]. Advanced algorithms like the Newton-Euler Inverse Mass Operator (NEIMO) method have been adapted from Spatial Operator Algebra mathematical frameworks to solve these equations of motion with O(N) computational cost, making constrained MD practical for larger protein systems [5].
The Generalized NEIMO (GNEIMO) method extends this approach by providing a hierarchical framework that allows researchers to "freeze and thaw" torsional degrees of freedom as appropriate for the specific problem being studied [5]. This flexibility enables the creation of dynamic models ranging from all-torsion simulations, where all torsional angles are flexible, to partially constrained models where specific secondary structure elements are treated as rigid bodies while sampling only the torsions connecting these clusters. The ability to transition between these models during simulation provides a powerful approach for enhancing sampling efficiency while maintaining physical accuracy.
Strategic Selection of Constraint Types
Choosing appropriate constraints is critical for balancing computational efficiency with biological accuracy. The selection should be guided by the specific research objectives, protein characteristics, and available computational resources. The following table summarizes the primary constraint types available in constrained MD frameworks:
Table: Types of Constraints in Molecular Dynamics Simulations
| Constraint Type | Mathematical Basis | Computational Advantage | Typical Applications |
|---|---|---|---|
| Bond Length | Holonomic constraints fixing atom-atom distances | Eliminates fastest vibrational modes, allows ~5 fs time steps | General purpose MD with moderate acceleration |
| Bond Angle | Holonomic constraints fixing angle between three atoms | Further reduces high-frequency motions | Specialized applications requiring additional acceleration |
| Torsional (All-atom) | Internal coordinates with fixed bond lengths and angles | Reduces DOF by ~10x, enables largest time steps (5-10 fs) | Enhanced conformational sampling, folding studies |
| Hierarchical Clustering | Selected torsions frozen based on structural features | Directed sampling, reduced conformational space | Investigating specific folding pathways, zipping-assembly mechanisms |
For protein folding studies, all-torsion constrained MD provides the most comprehensive conformational sampling, while hierarchical clustering schemes offer more targeted approaches that can accelerate convergence to native-like structures [5]. Research has demonstrated that hierarchical constrained MD simulations, where partially formed helical regions are frozen based on initial trajectory analysis, show better sampling of near-native structures than all-torsion constrained MD simulations alone [5]. This approach aligns with the zipping-and-assembly folding model and can be particularly effective for proteins with mixed structural motifs.
Experimental Protocol: Constrained MD for Small Protein Folding
System Preparation and Initialization
The following step-by-step protocol outlines the application of constrained MD with replica exchange for folding small proteins, based on established methodologies that have successfully folded polyalanine, β-turn peptides, and mixed-motif proteins like Trp-cage [5]:
Initial Structure Preparation: Begin with an extended conformation of your target peptide or protein sequence. Extended structures ensure no inherent bias toward native conformations and allow the simulation to explore the complete folding pathway.
Energy Minimization: Perform conjugate gradient minimization on the initial structure with a convergence criterion of 10⁻² kcal/mol/Å in force gradient. This step removes any steric clashes and prepares the system for stable dynamics.
Force Field and Solvation Selection: Employ the parm99 forcefield (part of AMBER99) with implicit solvation using the Generalized-Born Surface Area (GB/SA) OBC model [5]. Key parameters include:
- GB/SA interior dielectric value: 1.75 for the solute
- GB/SA exterior dielectric constant: 78.3 for aqueous environments (adjust to 40.0 for membrane environments)
- Solvent probe radius: 1.4Å for the nonpolar solvation energy component
- Non-bond forces cutoff: 20Å with smooth switching
Dynamics Configuration: Configure the constrained MD simulation using:
- Integration algorithm: Lobatto integrator
- Integration step size: 5 fs (made possible by constraint implementation)
- Temperature coupling: Appropriate for your system (300K for folding with GB/SA forces)
- Constraint model: Select all-torsion or hierarchical clustering based on research goals
Replica Exchange Enhancement
To overcome potential energy barriers and enhance conformational sampling, implement the replica exchange method alongside constrained MD:
Replica Temperature Distribution: For small proteins, typically 8 replicas distributed between 325K and 500K in steps of 25K provide sufficient sampling [5]. The number of replicas scales with the square root of the number of degrees of freedom, and constrained MD requires approximately one-third the replicas of all-atom MD due to reduced dimensionality.
Exchange Attempt Frequency: Attempt exchanges between neighboring replicas every 2ps to maintain detailed balance while allowing sufficient decorrelation between exchange attempts.
Simulation Duration: Run simulations for up to 20ns per replica, though this may vary based on protein size and complexity. Monitor convergence through RMSD and secondary structure metrics.
Hierarchical Clustering Implementation
For proteins exhibiting partial secondary structure formation, implement hierarchical constraint strategies:
Initial All-Torsion Phase: Begin with all-torsion constrained MD to allow natural formation of secondary structure elements.
Cluster Identification: Analyze the initial trajectory segment to identify regions with persistent secondary structure using criteria such as:
- Backbone torsional angles within 20° of ideal α-helical angles (φ = -57°, ψ = -47°)
- Hydrogen bonding patterns consistent with secondary structure
- Spatial proximity of sequentially distant residues
Constraint Application: Freeze the identified structured regions into rigid clusters while maintaining flexibility in connecting regions and disordered segments.
Iterative Refinement: Periodically reassess clustering assignments and adjust constraints as the simulation progresses and new structures stabilize.
Diagram: Hierarchical Constraint Workflow for Protein Folding
Analysis and Validation Methods
Principal Component Analysis
Principal Component Analysis (PCA) serves as a powerful method for analyzing the high-dimensional data generated by constrained MD simulations. When applied to MD trajectories, PCA identifies the dominant modes of motion or conformational changes that contribute most to the system's overall variability [29]. The technique operates by constructing a covariance matrix of the atomic coordinates (typically Cα atoms) from simulation snapshots, then diagonalizing this matrix to extract eigenvectors (principal components) and eigenvalues (representing the variance along each component) [29].
To implement PCA for constrained MD analysis:
Trajectory Preparation: Extract snapshots of your protein structure at regular intervals throughout the simulation trajectory. Align all structures to a reference frame to remove global rotation and translation.
Covariance Matrix Construction: Build a covariance matrix using the Cartesian coordinates of Cα atoms after removal of translational and rotational motions.
Dimensionality Reduction: Diagonalize the covariance matrix to obtain eigenvectors and eigenvalues. The eigenvectors represent the principal components (directions of maximal variance), while the eigenvalues indicate the magnitude of variance along each component.
Projection and Visualization: Project the original high-dimensional trajectory onto the first two or three principal components to create a low-dimensional representation of the conformational landscape. Create free energy landscapes by calculating:
ΔG = -kBT ln(P(s))
where P(s) is the probability distribution along the principal components [29].
Collective Motion Analysis: Examine the structural changes associated with each principal component to identify collective motions relevant to folding, such as helix formation, hydrophobic collapse, or tertiary structure packing.
Validation Metrics for Folded Structures
Validating the quality of sampled conformations is essential for assessing simulation success. The following table outlines key metrics for evaluating constrained MD results:
Table: Validation Metrics for Protein Folding Simulations
| Metric | Calculation Method | Interpretation | Target Values |
|---|---|---|---|
| RMSD to Native | Root-mean-square deviation of Cα atoms after alignment | Measures global structural similarity to experimental structure | <2.0Å for native-like states |
| Fraction of Native Contacts | Q = Σ exp[-(rij/λ)²] / Ncontacts | Quantifies formation of native-like interactions | >0.7 for well-folded structures |
| Secondary Structure Content | DSSP or torsional angle analysis | Tracks formation of α-helices, β-sheets | Match experimental predictions |
| Radius of Gyration | Rg = √(Σ mi ri² / Σ mi) | Measures chain compactness | Consistent with native state |
| Principal Component Variance | Cumulative sum of eigenvalues from PCA | Assesses sampling completeness | >70% in first 3-5 components |
For proteins with known native structures, the fraction of native contacts (Q) provides a particularly robust metric, as it is less sensitive to overall structural alignment than RMSD and more directly reflects the formation of specific stabilizing interactions [5]. Combining multiple validation metrics offers the most comprehensive assessment of simulation quality.
Research Reagent Solutions
Successful implementation of constrained MD simulations requires both computational tools and methodological components. The following table outlines essential "research reagents" for conducting constrained MD studies:
Table: Essential Research Reagents for Constrained MD Simulations
| Reagent Category | Specific Tools/Components | Function | Implementation Notes |
|---|---|---|---|
| Constrained MD Software | GNEIMO simulator [5] | Performs internal coordinate MD with flexible constraint schemes | Supports "freeze and thaw" hierarchical clustering |
| Force Fields | AMBER parm99 [5] | Defines energy terms and parameters for molecular interactions | Compatible with GB/SA implicit solvation |
| Solvation Models | GB/SA OBC [5] | Implicitly models solvent effects without explicit water | Adjust dielectric constant for membrane environments |
| Sampling Enhancers | Replica Exchange MD [5] | Accelerates barrier crossing through temperature cycling | Requires fewer replicas than all-atom MD |
| Analysis Frameworks | Principal Component Analysis [29] | Identifies dominant conformational motions from trajectories | Projects high-D data onto essential subspaces |
| Validation Databases | PDB structures, experimental folding data [5] | Provides reference for native states and folding mechanisms | Essential for method benchmarking |
Diagram: Constrained MD Protocol Sequence
Constrained molecular dynamics provides a powerful framework for studying small protein folding with significantly enhanced computational efficiency compared to all-atom Cartesian MD. The strategic application of constraints—particularly through hierarchical "freeze and thaw" approaches that target specific structural elements—enables broader conformational sampling and increased enrichment of near-native structures [5]. When combined with enhanced sampling techniques like replica exchange and robust analysis methods such as principal component analysis, constrained MD offers researchers an effective tool for investigating protein folding mechanisms, predicting native structures, and characterizing folding landscapes. The continued development and application of these methods will further advance our understanding of protein folding principles and facilitate drug discovery efforts targeting protein dynamics and stability.
The protein folding problem remains one of the most fundamental challenges in structural biology and computational biophysics. Among the various model systems developed to study this phenomenon, the 20-residue Trp-cage miniprotein has emerged as a particularly valuable paradigm for investigating folding mechanisms due to its small size and rapid folding kinetics. This application note examines the folding of Trp-cage and other fast-folding proteins within the context of constrained molecular dynamics (MD) protocols, providing researchers with detailed methodologies and quantitative frameworks for studying folding dynamics. The Trp-cage protein (sequence: NLYIQ WLKDG GPSSG RPPPS) contains an N-terminal α-helix (residues 2-8), a 3₁₀-helix (residues 11-14), and a C-terminal polyproline motif, forming a hydrophobic core that cages the central Trp6 residue—a structural feature critical for its stability and folding behavior [30] [31].
Biological Significance and Structural Features
Trp-cage represents an ideal model system for protein folding studies due to its computationally tractable size and well-characterized structural elements. The folded architecture stabilizes its native state through multiple interactions, including a salt bridge between Asp9 and Arg16, hydrophobic clustering around the Trp6 indole ring, and specific secondary structure elements that cooperatively form the functional fold [30] [32]. Experimental studies indicate that TC5b, a stabilized Trp-cage variant, folds in approximately 4 μs at room temperature, making it amenable to both experimental characterization and detailed molecular simulations [31].
The stability of individual structural elements varies significantly within the Trp-cage architecture. While the α-helix and hydrophobic core demonstrate considerable stability, the 3₁₀-helix represents a more fragile structural component that unfolds at temperatures significantly below the global melting transition of the protein [31]. This differential stability creates a hierarchical folding landscape that can be exploited to study distinct aspects of the folding process.
Table 1: Key Structural Elements and Their Roles in Trp-cage Folding
| Structural Element | Residue Range | Key Interactions | Role in Folding |
|---|---|---|---|
| N-terminal α-helix | 2-8 | Backbone hydrogen bonds | Early folding nucleation |
| 3₁₀-helix | 11-14 | Backbone hydrogen bonds | Late stabilization, independent folding |
| Polyproline region | 17-19 | Proline rigidity | Structural constraint for core formation |
| Hydrophobic core | Around Trp6 | Aromatic and aliphatic sidechains | Driving force for collapse |
| Salt bridge | Asp9-Arg16 | Electrostatic interactions | Stabilization of native state |
Computational Methodologies
Constrained Molecular Dynamics Approaches
Constrained MD methods provide a powerful alternative to all-atom Cartesian MD simulations by significantly reducing the system's degrees of freedom. These approaches treat molecules as collections of rigid bodies connected by flexible torsional hinges, fixing bond lengths and angles through holonomic constraints. This reduction in degrees of freedom allows for larger integration time steps (typically 5 fs compared to 1-2 fs in all-atom MD) and enhanced conformational sampling efficiency [5].
The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method implements constrained MD using a hierarchical framework that enables "freeze-and-thaw" capabilities for specific torsional degrees of freedom. This approach scales with O(N) computational cost compared to O(N³) for conventional constrained dynamics methods, making it applicable to larger protein systems [5]. The method partitions proteins into dynamic clusters, allowing researchers to selectively rigidify structured regions while maintaining flexibility in functionally important hinges or disordered segments.
Advanced Sampling Techniques
Replica exchange molecular dynamics (REMD) significantly enhances the sampling efficiency of constrained MD simulations by running parallel simulations at different temperatures and periodically exchanging configurations between replicas. The reduced degrees of freedom in constrained models decrease the number of replicas required—approximately one-third of that needed for all-atom simulations—while maintaining effective sampling [5]. For Trp-cage folding studies, typical temperature ranges span 325K to 500K using 6-8 replicas, with exchange attempts every 2 ps.
Figure 1: Constrained MD Workflow for Protein Folding Studies
Action-Derived Molecular Dynamics (ADMD)
ADMD provides an alternative pathway-finding approach by converting the initial value problem of classical dynamics into a boundary value problem using the least action principle. This method generates classical low-potential-energy trajectories connecting defined initial and final states, making it particularly valuable for studying rare events like protein folding [30]. In practice, ADMD simulations discretize the atomic trajectory into numerous steps (typically 2000 for Trp-cage) and optimize the Passerone-Parrinello action to ensure total energy conservation along the pathway [30].
Machine Learning Force Fields
Recent advances in artificial intelligence have enabled the development of ab initio biomolecular dynamics systems such as AI2BMD, which uses machine learning force fields trained on density functional theory data to achieve quantum-chemical accuracy with significantly reduced computational cost [27]. This approach employs a protein fragmentation scheme, breaking large proteins into manageable dipeptide units for accurate energy and force calculations before reassembling the complete structure. For Trp-cage (281 atoms), AI2BMD achieves a 17,500-fold speed increase compared to conventional DFT calculations while maintaining high accuracy [27].
Experimental Protocols
Constrained MD with Replica Exchange for Trp-cage Folding
Protocol Objective: To fold Trp-cage from an extended conformation to its native state using constrained MD with replica exchange enhanced sampling.
Initial Structure Preparation:
- Build an extended conformation of Trp-cage using sequence: NLYIQ WLKDG GPSSG RPPPS
- Perform conjugate gradient minimization with convergence criterion of 10⁻² kcal/mol/Å force gradient
- Use the AMBER parm99 force field with GB/SA implicit solvation
- Set GB/SA interior dielectric to 1.75 and exterior dielectric to 78.3
- Apply a nonpolar solvation energy term with solvent probe radius of 1.4Å
- Implement a 20Å cutoff for non-bonded interactions with smooth switching
Constrained MD Simulation Parameters:
- Employ the GNEIMO constrained dynamics algorithm
- Set integration time step to 5 fs using Lobatto integrator
- Define temperature range of 325K to 500K for 8 replicas (6 replicas for simpler systems)
- Attempt replica exchanges every 2 ps
- Run simulations for 20 ns per replica
- Apply either all-torsion constraints or hierarchical clustering schemes
Hierarchical Clustering Scheme for Trp-cage:
- Identify and rigidify partially formed helical regions during simulation
- Sample only torsional degrees of freedom connecting rigid clusters
- Adapt clustering based on emerging secondary structure elements
Analysis Methods:
- Calculate root-mean-square deviation (RMSD) from native NMR structure (PDB: 1L2Y)
- Monitor native contact formation using a 4.5Å heavy atom cutoff
- Track hydrogen bond formation with donor-acceptor distance < 3.5Å and angle > 120°
- Quantify secondary structure content using DSSP or backbone dihedral analysis
- Perform principal component analysis on Cα coordinates to visualize conformational space
Multi-Probe Kinetic Characterization
Protocol Objective: To experimentally characterize Trp-cage folding kinetics using temperature-jump relaxation with multiple spectroscopic probes.
Sample Preparation:
- Prepare 13C-labeled TC10b variant (DAYAQWLKDGGPSSGRPPPS) with 13C=O labeled alanines
- Dissolve in appropriate buffer to final concentration of 1-2 mM for IR studies
- Confirm folding stability via circular dichroism spectroscopy measuring molar ellipticity at 222nm
Temperature-Jump Experimental Setup:
- Implement nanosecond laser-induced T-jump apparatus
- Monitor relaxation kinetics at multiple IR frequencies:
- 1612 cm⁻¹ for 13C-labeled α-helix signal
- 1580 cm⁻¹ for Asp sidechain (salt bridge formation)
- 1664 cm⁻¹ for 3₁₀-helix and disordered structures
- Collect data at multiple final temperatures (10-40°C)
- Perform single-exponential and double-exponential fitting of relaxation curves
Data Analysis:
- Extract relaxation rate constants at each probe frequency
- Compare temperature dependence of different structural elements
- Identify correlated and uncorrelated folding events
- Map temporal hierarchy of structure formation
Table 2: Key Experimental Findings from Trp-cage Folding Studies
| Experimental Observation | Structural Interpretation | Methodology | Reference |
|---|---|---|---|
| 4 μs folding time at 300K | Two-state folding behavior | T-jump fluorescence | [31] |
| Identical relaxation at 1580 cm⁻¹ and 1612 cm⁻¹ | Concurrent α-helix and cage formation | Multi-probe IR spectroscopy | [31] |
| Biphasic kinetics at 1664 cm⁻¹ | Independent 3₁₀-helix unfolding | Multi-probe IR spectroscopy | [31] |
| Fast phase (100s of ns) at 1664 cm⁻¹ | Local unfolding of 3₁₀-helix | T-jump IR spectroscopy | [31] |
| Hydrophobic contact before polyproline docking | Sequential folding mechanism | ADMD simulations | [30] |
Key Research Findings
Folding Pathway and Mechanisms
Computational and experimental studies of Trp-cage folding have revealed a detailed mechanistic pathway with distinct stages. ADMD simulations show that folding initiates with contact formation between Tyr3 and Trp6 side chains concurrent with N-terminal α-helix formation [30]. This early intermediate possesses approximately 40% of native contacts and 30% of native hydrogen bonds. The folding process continues with docking of the C-terminal polyproline segment onto the aromatic rings of Trp6 and Tyr3, facilitating hydrophobic core formation. Final stages involve slower structural adjustments to achieve the precise native configuration [30].
The salt bridge between Asp9 and Arg16 plays a complex role in Trp-cage folding. While some simulations suggest early salt bridge formation as a prerequisite for folding [32], experimental φ-value analysis indicates that this interaction likely forms on the downhill side of the major folding barrier [31]. This apparent discrepancy highlights the importance of integrating multiple computational and experimental approaches to develop a comprehensive folding mechanism.
Hierarchical Folding Dynamics
Trp-cage exhibits hierarchical folding dynamics with varying stability across its structural elements. The 3₁₀-helix represents the most fragile structural component, unfolding at temperatures significantly below the global melting transition of the protein [31]. This element demonstrates independent folding and unfolding kinetics on the hundreds of nanoseconds timescale, comparable to short α-helices [31]. In contrast, the α-helix and hydrophobic core display strongly cooperative folding behavior, forming concurrently at identical rates as measured by multi-probe infrared spectroscopy [31].
Figure 2: Hierarchical Folding Pathway of Trp-cage
Quantitative Folding Analysis
Constrained MD simulations of Trp-cage folding demonstrate enhanced sampling of near-native structures compared to all-atom MD approaches. Principal component analysis of constrained MD trajectories reveals broader conformational sampling and increased enrichment of native-like structures [5]. Hierarchical clustering schemes, where partially formed helical regions are treated as rigid bodies, further improve sampling efficiency and accelerate convergence to the native state [5].
Advanced machine learning force fields like AI2BMD achieve remarkable accuracy in protein folding simulations, with potential energy mean absolute errors of 0.045 kcal mol⁻¹ and force errors of 0.078 kcal mol⁻¹ Å⁻¹ compared to density functional theory calculations [27]. This represents a two-order-of-magnitude improvement over conventional molecular mechanics force fields, enabling more reliable prediction of folding pathways and thermodynamics.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for Folding Studies
| Tool/Reagent | Type | Function/Application | Specifications |
|---|---|---|---|
| AMBER parm99 Force Field | Computational | Potential energy calculations | Includes GB/SA implicit solvation parameters |
| GNEIMO Constrained MD | Software | Enhanced conformational sampling | O(N) computational scaling, hierarchical clustering |
| 13C-TC10b Peptide | Experimental | Multi-probe folding kinetics | 13C=O labeled alanines for specific IR signatures |
| GB/SA Implicit Solvent | Computational | Efficient solvation modeling | Interior dielectric 1.75, exterior dielectric 78.3 |
| T-Jump Spectrometer | Experimental | Microsecond kinetics measurement | Multi-wavelength IR detection capability |
| AI2BMD Potential | Computational | Ab initio accuracy ML force field | ViSNet architecture, fragmentation approach |
The folding of Trp-cage and other fast-folding proteins provides fundamental insights into protein folding mechanisms and represents an ideal test system for developing advanced computational methodologies. Constrained molecular dynamics protocols offer significant advantages for folding studies through reduced degrees of freedom, enhanced sampling efficiency, and ability to implement hierarchical clustering schemes that mirror the zipping-and-assembly mechanism of protein folding. The integration of computational approaches with multi-probe experimental techniques enables researchers to dissect complex folding pathways with unprecedented temporal and structural resolution. These advanced protocols continue to expand our understanding of protein folding while providing valuable tools for protein engineering and therapeutic development targeting misfolding diseases.
Integration with Implicit Solvent Models for Speed and Efficiency
The integration of implicit solvent models with constrained molecular dynamics (MD) represents a powerful strategy for accelerating the computational study of protein folding. This protocol details the application of these combined methods for the folding of small proteins, a context where achieving sufficient conformational sampling is a significant challenge. Explicit solvent MD simulations, while highly accurate, are often prohibitively expensive for studying folding, as these processes can occur on microsecond to millisecond timescales [5]. By replacing explicit water molecules with a continuum representation, implicit solvent models eliminate the need to simulate thousands of solvent degrees of freedom, thereby "pre-equilibrating" the solvent and drastically reducing computational cost [33]. When this is combined with constrained MD methods—which reduce the number of solute degrees of freedom by fixing bond lengths and angles—the resulting framework enables stable dynamics with larger integration time steps and enhanced conformational sampling [5] [10]. This Application Note provides a detailed guide for researchers aiming to apply these techniques to fold small proteins, complete with benchmarked protocols, quantitative accuracy assessments, and ready-to-use visualization workflows.
Background and Rationale
Implicit Solvation Theory
Implicit solvation, or continuum solvation, replaces the explicit solvent environment with a dielectric medium characterized by macroscopic properties like the dielectric constant [34]. The primary goal is to compute the solvation free energy (ΔG_solv), which is the free energy change associated with transferring a solute molecule from a vacuum into the solvent [35] [36].
The solvation free energy is typically partitioned into polar (electrostatic) and non-polar components [35] [36]: ΔGsolv = ΔGele + ΔG_np
The polar component (ΔGele) represents the cost of polarizing the solvent by the solute's charge distribution. The non-polar component (ΔGnp) accounts for the energy cost of forming a cavity in the solvent to accommodate the solute and the van der Waals interactions between the solute and solvent [35]. The non-polar term is often modeled as being proportional to the Solvent-Accessible Surface Area (SASA) [35] [34].
Table 1: Common Implicit Solvent Models for Biomolecular Simulations
| Model | Description | Key Features | Typical Applications |
|---|---|---|---|
| Generalized Born (GB) | A fast approximation to the Poisson-Boltzmann equation [33] [34]. | Computes ΔG_ele via a pairwise summation over atoms. High computational efficiency [35] [37]. | High-throughput MD simulations, protein folding studies, structure refinement [33] [5]. |
| Poisson-Boltzmann (PB) | A more rigorous, formally derived continuum electrostatic model [33] [34]. | Solves a partial differential equation for the electrostatic potential. Provides a global solution for electrostatics [33] [37]. | Visualization, analysis of electrostatic potentials, diffusion simulations, accuracy benchmarking [33] [37]. |
| SASA (SA) | Models non-polar solvation or the entire solvation energy [35] [34]. | ΔG_solv is a sum of atom-specific parameters multiplied by their SASA. Computationally very fast [35]. | Often used in conjunction with GB or PB models (GBSA/PBSA) to provide a complete solvation energy estimate [34] [37]. |
Constrained Molecular Dynamics
Constrained MD, also known as internal coordinate MD or torsion angle MD, is an approach that reduces the number of degrees of freedom in a system [5]. In this model, the molecule is treated as a collection of rigid bodies ("clusters") connected by flexible torsional hinges. By fixing bond lengths and bond angles using holonomic constraints, the number of degrees of freedom is reduced by approximately an order of magnitude compared to all-atom Cartesian MD [5]. This reduction allows for the use of larger integration time steps (e.g., 5 fs compared to 2 fs in Cartesian MD) and eliminates high-frequency motions, leading to a significant decrease in computational cost and an enhancement of conformational sampling [5] [10].
Quantitative Performance and Accuracy
The combination of implicit solvent and constrained MD offers substantial computational advantages. Studies have shown that constrained MD models can achieve a wider conformational search and increased enrichment of near-native structures compared to all-atom MD [10]. The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) constrained MD method, for instance, has been successfully coupled with the GB/SA implicit solvent model and replica exchange methods to fold various small peptides and proteins, including polyalanine, WALP16, and Trp-cage [5].
Table 2: Accuracy Comparison of Common Implicit Solvent Models for Small Molecules and Proteins [37]
| Implicit Solvent Model | Correlation with Explicit Solvent (Ligands) | Correlation with Explicit Solvent (Proteins) | Correlation with Explicit Solvent (Desolvation Energies) | Key Characteristics |
|---|---|---|---|---|
| APBS (PB) | 0.953 - 0.966 | 0.65 - 0.99 | High | High accuracy, suitable for benchmarking; computationally slower than GB [37]. |
| GBNSR6 (GB) | 0.953 - 0.966 | 0.65 - 0.99 | High | High accuracy for desolvation energies; a fast approximation to PB [37]. |
| DISOLV (S-GB) | 0.953 - 0.966 | 0.65 - 0.99 | Moderate | Fast, implemented in docking and post-processing workflows [37]. |
| DISOLV (PCM) | 0.953 - 0.966 | 0.65 - 0.99 | Moderate | High numerical accuracy; more computationally intensive than GB [37]. |
| DISOLV (COSMO) | 0.953 - 0.966 | 0.65 - 0.99 | Moderate | Conductor-like screening model; performance varies with implementation [37]. |
Despite their speed, users must be aware of the limitations and potential sources of error in implicit solvent models. These include the assumption of a linear and local dielectric response, which can break down near highly charged groups; ambiguity in dielectric interface definitions; the neglect of specific ion-solute interactions; and the use of a mean-field treatment of ions, which ignores ion correlations [33]. Furthermore, implicit models typically do not account for specific, directional interactions like hydrogen bonds between solute and solvent, nor do they capture the viscous damping effect of explicit water [34].
Experimental Protocols
Protocol 1: All-Torsion Folding with Replica Exchange
This protocol outlines the procedure for folding a small protein from an extended conformation using all-torsion constrained MD coupled with a replica exchange sampling strategy, as demonstrated for peptides like polyalanine and Trp-cage [5].
System Preparation
- Start with an extended conformation of the protein sequence.
- Assign force field parameters (e.g., AMBER parm99/ff14SB [5] [18] or CHARMM [5]) and partial charges.
- Pre-process the structure using a tool like PDB2PQR to assign correct titration states, add missing atoms, and optimize hydrogen bonding networks to minimize structure-based errors [33].
Energy Minimization
- Perform conjugate gradient energy minimization on the initial structure until the force gradient converges to a criterion of ≤ 10⁻² kcal/(mol·Å) [5].
Simulation Parameter Setup
- Solvation Model: Apply an implicit solvent model. For general aqueous folding, use the GB/SA (OBC) model with an interior dielectric of 1.0-1.75 for the solute and an exterior dielectric of 78.3 for water [5]. For membrane environments, adjust the exterior dielectric accordingly (e.g., ~40) [5].
- Non-polar Term: Set a solvent probe radius of 1.4 Å for the SASA-based non-polar solvation energy component [5].
- Dynamics: Use the constrained MD (GNEIMO) method with all torsional degrees of freedom flexible.
- Integrator: Use a Lobatto integrator with a time step of 5 fs [5].
- Non-bonded Interactions: Apply a cutoff of 20 Å for non-bonded forces, smoothly switched off [5].
Replica Exchange Setup
- The number of replicas scales with the square root of the number of degrees of freedom. For small proteins, 6-8 replicas are often sufficient [5].
- Set up a temperature distribution for the replicas, for example, from 325 K to 500 K in steps of 25 K [5].
- Configure replica exchanges to be attempted every 2 ps [5].
Production Simulation
- Run the replica exchange simulation for a duration sufficient to observe folding (e.g., up to 20 ns per replica for small peptides) [5].
- Monitor convergence by tracking metrics like the fraction of native contacts, radius of gyration, and Root Mean Square Deviation (RMSD) from the native state.
Protocol 2: Hierarchical Clustering for Folded Motifs
This protocol uses a "freeze and thaw" strategy to enhance the sampling of near-native states by treating pre-formed secondary structural elements as rigid bodies, aligning with the zipping-and-assembly folding model [5] [38].
Initial Sampling and Analysis
- First, perform an all-torsion constrained MD simulation (as in Protocol 1) for a short period.
- Analyze the resulting trajectory to identify regions that have formed stable secondary structure motifs (e.g., α-helices or β-hairpins).
Define Rigid Clusters
- Based on the analysis, define these stable secondary structure regions as rigid clusters within the constrained MD framework. The backbone torsions within these clusters are "frozen."
Refinement Simulation
- Run a new constrained MD simulation where only the torsional degrees of freedom connecting the rigid clusters are sampled. This hierarchical clustering reduces the conformational space to be searched.
- Maintain the same solvent, integrator, and replica exchange parameters as in Protocol 1.
Analysis and Validation
- Compare the population and structural quality (e.g., RMSD) of near-native states from the hierarchical simulation against the all-torsion simulation. Hierarchical clustering has been shown to yield better sampling of the native basin for proteins like Trp-cage [5].
- Use Principal Component Analysis (PCA) and clustering algorithms (e.g., K-means) to analyze and visualize the conformational landscape sampled [5].
Workflow Visualization
The following diagram illustrates the integrated computational workflow for protein folding using constrained MD and implicit solvation, incorporating the hierarchical clustering strategy.
Constrained MD Folding Workflow
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Item | Function/Description | Example Resources |
|---|---|---|
| Force Fields | Provides parameters for potential energy calculations. | AMBER (parm99, ff14SB) [5] [18], CHARMM [5], MMFF94 [37] |
| Implicit Solvent Models | Computes solvation free energy without explicit water. | Generalized Born (GB/OBC) [5] [35], Poisson-Boltzmann (APBS) [33] [37] |
| Constrained MD Software | Performs molecular dynamics with reduced degrees of freedom. | GNEIMO method [5], Internal Coordinate MD [5] |
| Structure Pre-processor | Prepares and optimizes protein structures for simulation. | PDB2PQR (adds missing atoms, assigns protonation states) [33] |
| Replica Exchange | Enhanced sampling technique to overcome energy barriers. | Implemented in various MD packages [5] |
| Analysis Software | Analyzes trajectories and quantifies results. | PCA, K-means clustering, RMSD/fraction native contact calculators [5] |
Overcoming Limitations and Optimizing Constrained MD Simulations
Identifying and Mitigating Sampling Inadequacies
In the field of computational structural biology, sampling inadequacies represent a fundamental challenge, particularly in the study of protein folding. These inadequacies refer to the inability of computational simulations to sufficiently explore the vast conformational landscape that a protein chain can occupy, potentially missing crucial intermediate states, misfolded structures, or the native functional state itself. Within the specific context of constrained molecular dynamics (cMD) protocols for small protein folding research, this problem manifests as limited access to biologically relevant timescales and an incomplete representation of conformational diversity. cMD methods, which reduce computational complexity by fixing certain molecular degrees of freedom (e.g., bond lengths and angles), offer a strategic approach to extend simulation timescales [5]. However, they introduce unique sampling artifacts that must be identified and systematically mitigated to ensure research validity. This Application Note details protocols for recognizing these limitations and provides experimentally validated methodologies to overcome them, enabling more accurate and predictive protein folding simulations.
Understanding Sampling Inadequacies in Constrained MD
Constrained MD simulations accelerate conformational sampling by reducing the system's degrees of freedom. Unlike all-atom Molecular Dynamics (MD), which models every atom and requires femtosecond timesteps, cMD treats parts of the molecule as rigid bodies connected by flexible torsional hinges. This reduces the number of degrees of freedom by approximately an order of magnitude, allowing for larger integration time steps (e.g., 5 fs) and longer simulation times [5]. The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method is one such constrained MD approach that enables efficient simulation of larger proteins [5].
However, this computational efficiency comes with inherent sampling risks:
- Limited Conformational Exploration: Fixed geometric parameters can restrict the natural flexibility of secondary structural elements and side chains, potentially overlooking alternative low-energy states.
- Kinetic Trapping: The reduced degrees of freedom may cause the simulation to become trapped in local energy minima, with insufficient perturbation to escape to global minima.
- Coarse-to-Fine Mapping Artifacts: When using multiscale protocols that reverse-map coarse-grained (CG) equilibrated structures to all-atom (AA) models for production simulations, artifacts can be introduced. For instance, in membrane protein studies, this hybrid protocol has been shown to artificially increase lipid density and decrease hydration in channel pores because lipids entering during CG simulation remain trapped in subsequent AA simulations due to kinetic mismatches [39].
Table 1: Common Sampling Inadequacies in Constrained MD Protocols
| Inadequacy Type | Molecular Manifestation | Impact on Folding Studies |
|---|---|---|
| Incomplete Backbone Sampling | Limited ϕ/ψ dihedral angle exploration | Failure to identify stable secondary structures or folding intermediates |
| Kinetic Trapping | Prolonged residence in meta-stable states | Overestimation of energy barriers; inaccurate folding pathways |
| Reverse-Mapping Artifacts | Non-physical lipid packing or solvent dehydration in refined models [39] | Biased protein-lipid interactions; erroneous pore hydration states |
| Ensemble Under-representation | Narrow structural distribution around initial conditions | Poor agreement with experimental ensemble measurements (NMR, cryo-EM) |
Protocols for Identifying Sampling Inadequacies
Quantitative Metrics for Sampling Assessment
Researchers should employ these key metrics to evaluate sampling completeness:
1. Root Mean Square Deviation (RMSD) Convergence Analysis:
- Protocol: Calculate the backbone RMSD of all sampled structures relative to both the starting structure and the predicted native state. Plot RMSD as a function of simulation time or Monte Carlo step count.
- Adequacy Threshold: Sampling is considered converged when the moving average of RMSD over a significant portion of the simulation (e.g., last 25%) shows no directional trend and fluctuates around a stable mean value with bounded variance.
- Data Interpretation: Persistent drift indicates ongoing exploration, while failure to return to low-RMSD states suggests possible kinetic trapping.
2. Principal Component Analysis (PCA) of Trajectories:
- Protocol: Perform PCA on the Cartesian coordinates of Cα atoms from all simulation snapshots. Project the entire trajectory onto the first two or three principal components, which represent the largest collective motions.
- Adequacy Threshold: Adequate sampling is indicated by a contiguous, multi-modal density distribution in the PCA projection space, where each basin corresponds to a structurally distinct state, and transitions between them are observed [5].
- Data Interpretation: Isolated, disconnected clusters suggest insufficient sampling of transition pathways. A single dense cluster may indicate kinetic trapping.
3. Free Energy Landscape Calculation:
- Protocol: Construct free energy surfaces by projecting trajectories onto collective variables (e.g., RMSD, radius of gyration, native contacts). Calculate free energy as G = -kBT ln P(RMSD, Rg), where P is the probability distribution.
- Adequacy Threshold: A well-sampled landscape shows clear, deep free energy minima connected by saddle points representing transition states.
- Data Interpretation: The presence of multiple folding intermediates and barriers can be identified. Landscapes with single, poorly defined minima warrant extended sampling.
Experimental Validation of Computational Ensembles
Computational sampling must be validated against experimental data that inherently report on ensemble averages:
A. NMR Restraint Satisfaction:
- Protocol: Calculate nuclear Overhauser effect (NOE)-derived distance restraints and J-couplings from computational ensembles. Compare with experimental NMR data.
- Inadequacy Indicator: Systematic violations of NOE distances or inability to reproduce J-couplings indicates missing conformational states in the simulation ensemble [40].
B. X-ray Crystallographic Density Agreement:
- Protocol: Compare simulated ensembles with experimental electron density maps. Compute real-space correlation coefficients (RSCC) and real-space R-values (RSR).
- Inadequacy Indicator: Low RSCC values or poor fit to electron density, particularly in flexible "altloc" regions, suggests inadequate sampling of alternative conformations present in the crystal [40].
C. Protein Stability Measurements:
- Protocol: Compare computational stability predictions (e.g., folding free energy ΔGfold) with experimental measurements from chemical or thermal denaturation assays monitored by circular dichroism (CD) or fluorescence spectroscopy.
- Inadequacy Indicator: Systematic deviations between predicted and measured stability for designed protein variants indicates inaccuracies in the energy landscape sampling [41] [42].
Experimentally Validated Mitigation Strategies
Enhanced Sampling Protocols
The following protocols have demonstrated success in mitigating sampling inadequacies for small protein folds:
Protocol 1: Replica Exchange with Hierarchical Clustering (For cMD)
This protocol enhances sampling of near-native structures by employing a "freeze and thaw" approach for secondary structure elements [5].
Step 1: System Preparation
- Start from an extended conformation of the protein sequence.
- Perform conjugate gradient minimization with a convergence criterion of 10⁻² kcal/mol/Å force gradient.
- Use the AMBER parm99 forcefield with implicit Generalized-Born Surface Area (GB/SA) solvation (OBC model). Set GB/SA interior dielectric to 1.75 and exterior dielectric to 78.3 for aqueous environments.
Step 2: Replica Exchange Setup
- Configure 8 replicas across a temperature range (e.g., 325K to 500K in 25K increments). The reduced degrees of freedom in cMD cuts the required replicas by approximately one-third compared to all-atom MD.
- Use the GNEIMO constrained MD algorithm with all torsional degrees of freedom.
Step 3: Hierarchical Sampling
- Perform initial all-torsion folding simulation.
- Identify partially formed secondary structure regions (e.g., helices) in trajectory analysis.
- "Freeze" these regions as rigid clusters and sample only the connecting torsional degrees of freedom in subsequent simulations.
- This zipping-and-assembly approach mirrors natural folding pathways.
Step 4: Analysis
- Employ Principal Component Analysis and k-means clustering to analyze conformational diversity.
- Validate against known native structures and experimental stability data.
Protocol 2: FoldArchitect for Shape Diversity Sampling (For De Novo Design)
This computational platform enables massively parallel design of proteins to explore shape diversity within given folds, overcoming limited natural sampling [41].
Step 1: Fold Definition
- Define the target fold topology (e.g., 3-helical bundle, ferredoxin, thioredoxin).
- Divide the fold into segments comprising two secondary structure elements with a loop connector.
Step 2: Incremental Folding with Diversity Sampling
- For each segment, dynamically vary secondary structure lengths and loop types during folding trajectories.
- Introduce loose distance constraints to bias sampling toward well-formed beta-sheets for strand-containing folds.
- Automatically generate strand pairing between neighbors with specified directionality.
Step 3: Sequence Design
- Option A: Sample rotamers based on solvent exposure, starting with low repulsive terms to find optimal sidechain interactions.
- Option B: Introduce residue "pair-motifs" (side-chain pairings from natural protein interfaces) to design protein cores before rotamer-based design.
Step 4: Experimental Validation
- Express and purify designs in E. coli.
- Assess monodispersity via size exclusion chromatography.
- Verify secondary structure formation using circular dichroism spectroscopy.
- Validate stability through chemical or thermal denaturation assays.
Table 2: Key Parameters for Enhanced Sampling Protocols
| Parameter | Replica Exchange with Hierarchical Clustering | FoldArchitect Shape Sampling |
|---|---|---|
| Force Field | AMBER parm99/GB/SA OBC solvation [5] | Rosetta-based energy functions [41] |
| Solvation Model | Implicit GB/SA | Implicit or explicit depending on protocol |
| Sampling Dimensions | Temperature (325-500K), torsional angles | Secondary structure length, loop conformation, strand alignment |
| Simulation Time | ~20ns per replica [5] | Variable; optimized for high-throughput (30,000+ designs) [41] |
| Experimental Validation | Comparison to native state geometry [5] | Yeast surface display, SEC, CD, stability assays [41] |
| Throughput | Medium (8 replicas for small proteins) | High (thousands of designs) |
Multiscale and Monte Carlo Approaches
Multiscale AA CG Protocol: This approach couples the accuracy of all-atom (AA) models with the sampling efficiency of coarse-grained (CG) simulations. The Simultaneous Lengthscale Multiple State (SLMS) method iteratively improves a CG potential using AA simulations, achieving high accuracy and efficiency simultaneously. The protocol involves: (1) Running AA simulations on selected CG structures, (2) Deriving an improved CG potential using the force-matching method, (3) Running new CG simulations with the updated potential, and (4) Iterating until convergence [43].
All-Atom Monte Carlo Sampling: As an alternative to MD, Monte Carlo (MC) simulations with all-atom force fields and implicit solvent can sample conformational landscapes without inherent timescale limitations. The protocol includes: (1) Using AMBER99SB*-ILDN force field with GB/SA implicit solvent, (2) Performing 200 million MC steps across a temperature range (330K-410K), (3) Employing combined displacement, rotation, and dihedral moves, and (4) Reconstructing free energy landscapes and identifying transition states [16]. This approach has successfully characterized the folding of Trp-cage, Villin headpiece, and WW-domain proteins.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Reagent/Software | Function/Application | Usage Notes |
|---|---|---|
| GNEIMO Constrained MD [5] | O(N) computational method for torsional dynamics | Enables hierarchical "freeze and thaw" of secondary structure elements |
| FoldArchitect Pipeline [41] | Massively parallel protein design with shape diversity sampling | Incorporates protein folding rules for structural features like helix-connecting loops |
| AMBER99SB*-ILDN [16] | All-atom force field for protein simulations | Compatible with both MD and MC protocols; parameterized for implicit solvent |
| GB/SA OBC Solvent Model [5] [16] | Implicit solvation for accelerated sampling | Uses a solvent probe radius of 1.4Å for nonpolar solvation energy component |
| Rosetta Design Suite [41] | De novo protein design and structure prediction | Knowledge-guided energy functions with Monte Carlo sampling |
| Yeast Surface Display [41] | High-throughput stability screening | Enables evaluation of 30,000+ designs via protease susceptibility assays |
Workflow Diagrams
Constrained MD Enhanced Sampling Workflow
Sampling Inadequacy Identification Protocol
The systematic identification and mitigation of sampling inadequacies is crucial for advancing small protein folding research using constrained molecular dynamics protocols. The methodologies presented here—including hierarchical replica exchange, multiscale sampling, and experimental validation—provide researchers with a comprehensive framework to enhance conformational sampling. By implementing these protocols and rigorously validating computational ensembles against experimental data, scientists can overcome fundamental sampling limitations, leading to more accurate predictions of protein structure, stability, and function. These advances are particularly valuable for drug development applications where understanding conformational dynamics can inform targeted therapeutic design.
Constrained Molecular Dynamics (MD) is a powerful computational technique that enhances the efficiency of protein folding simulations by reducing the system's number of degrees of freedom. By imposing constraints on fast-moving bonds and angles, it allows for significantly larger integration time steps compared to all-atom Cartesian MD [5]. This approach is particularly valuable for studying the folding of small proteins, where accessing biologically relevant timescales is computationally prohibitive with conventional methods. The strategic selection of parameters, specifically the integration time step and the use of enhanced sampling potentials, is critical for achieving physically accurate results within a feasible simulation time. This document provides detailed application notes and protocols for parameter selection, framed within the context of constrained MD protocols for small protein folding research.
Key Concepts and Quantitative Data
The Rationale for Constraints and Time Step Selection
In all-atom Cartesian MD, the presence of high-frequency bond vibrations (e.g., C-H bonds) severely limits the integration time step to typically 1-2 femtoseconds (fs) to maintain numerical stability and energy conservation [5]. Constrained MD addresses this bottleneck by fixing these fast degrees of freedom, often treating collections of atoms as rigid bodies connected by flexible torsional hinges. This reduces the number of degrees of freedom by approximately an order of magnitude and eliminates the highest frequency modes, enabling a substantial increase in the time step [5]. The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method, an example of a constrained MD algorithm, allows for stable dynamics with time steps as large as 5 fs [5]. A recent algorithm, ILVES-PC, further underscores the critical importance of accurate constraint solving for achieving efficient and physically sound simulations [11].
Table 1: Comparison of Simulation Parameters Between All-Atom and Constrained MD
| Parameter | All-Atom MD | Constrained MD |
|---|---|---|
| Typical Time Step | 1 - 2 fs | 3 - 5 fs [5] |
| Degrees of Freedom | ~3N (N = number of atoms) | ~N/10 (Torsional DOFs) [5] |
| Computational Scaling | Favorable for large systems | O(N) with efficient algorithms like NEIMO [5] |
| Key Limitation | High-frequency bond vibrations | Dense, coupled equations of motion |
Boost Potentials and Enhanced Sampling
While constraints improve efficiency, protein folding can still be hampered by high free energy barriers. "Boost potentials," or enhanced sampling methods, are used to accelerate barrier crossing. Within constrained MD, the Replica Exchange Method (REM) is highly effective [5]. Also known as Parallel Tempering, this method involves running multiple simultaneous simulations (replicas) of the same system at different temperatures. Periodically, exchanges between neighboring temperatures are attempted according to a Metropolis criterion, allowing conformations trapped in local energy minima at low temperatures to escape by visiting higher temperatures.
A key advantage of using REM with constrained MD is that the reduced number of degrees of freedom lowers the number of replicas required for efficient sampling, cutting down the computational cost by approximately a factor of three [5].
Table 2: Enhanced Sampling and Solvation Parameters for Protein Folding with Constrained MD
| Parameter Category | Recommended Setting | Function and Rationale |
|---|---|---|
| Replica Exchange Temperatures | 325K to 500K (in steps of 25K) [5] | Enables sampling over energy barriers; 8 replicas sufficient for small proteins. |
| Exchange Attempt Frequency | Every 2 ps [5] | Balances conformational mixing with computational overhead. |
| Implicit Solvent Model | GB/SA OBC (Onufriev, Bashford, Case) Model [5] | Approximates solvent effects efficiently, crucial for folding. |
| GB/SA Dielectric Constant (Interior) | 1.75 [5] | Represents the low dielectric of the protein interior. |
| GB/SA Dielectric Constant (Exterior) | 78.3 (Water), 40.0 (Membrane) [5] | Represents the solvent environment. |
| Non-bonded Cutoff | 20 Å (with a switch function) [5] | Manages computational cost of long-range interactions. |
Experimental Protocols
Protocol 1: All-Torsion Constrained MD Folding Simulation
This protocol outlines the procedure for folding a small protein from an extended conformation using all-torsion constrained MD coupled with replica exchange [5].
1. System Preparation:
- Initial Conformation: Start with an extended conformation of the protein sequence, generated using a molecular builder tool.
- Force Field Selection: Use a standard all-atom force field such as AMBER parm99 or a more modern equivalent [5].
- Solvation: Employ an implicit solvation model. The Generalized-Born Surface Area (GB/SA) model with the OBC correction is recommended [5]. Set the solute interior dielectric to 1.75 and the exterior dielectric to 78.3 for aqueous simulations. Use a solvent probe radius of 1.4 Å.
2. Energy Minimization:
- Perform conjugate gradient minimization on the initial extended structure.
- Use a convergence criterion of 10⁻² kcal/(mol·Å) for the force gradient.
3. Constrained MD Simulation Setup:
- Dynamics Algorithm: Use a constrained MD integrator such as the Lobatto integrator within the GNEIMO framework [5].
- Integration Time Step: Set to 5 fs.
- Constraint Tolerance: Ensure the algorithm (e.g., SHAKE, ILVES-PC) satisfies constraints within a tight tolerance [11].
- Non-bonded Interactions: Apply a cutoff of 20 Å for non-bonded interactions, with a smoothing switch function to avoid discontinuities.
4. Replica Exchange Configuration:
- Set up 6-8 replicas depending on protein size.
- Distribute temperatures exponentially between 325K and 500K.
- Attempt exchanges between neighboring replicas every 2 ps.
5. Production Run and Analysis:
- Run the simulation for a minimum of 20 ns per replica for small peptides. Monitor the fraction of native contacts (Q) and Cα root-mean-square deviation (RMSD) to track folding.
- Use Principal Component Analysis (PCA) and clustering algorithms (e.g., K-means) to analyze the resulting conformational landscape [5].
Protocol 2: Hierarchical Constrained MD Simulation
This protocol uses a "freeze and thaw" strategy to enhance sampling of near-native states by treating pre-formed secondary structure elements as rigid bodies, aligning with the zipping-and-assembly folding model [5].
1. Initial Sampling and Analysis:
- Perform a short all-torsion constrained MD simulation (as in Protocol 1).
- Analyze the trajectory to identify regions that have formed stable secondary structure (e.g., via backbone dihedral angles or hydrogen bonding patterns).
2. Cluster Definition:
- "Freeze" the identified stable regions (e.g., an α-helix or β-hairpin) by defining them as rigid clusters. The atoms within a cluster have fixed relative positions.
- The "thawed" flexible regions remain as torsional degrees of freedom connecting these rigid clusters.
3. Hierarchical Production Simulation:
- Run a new constrained MD simulation using this hybrid model. The dynamics will now sample only the torsional degrees of freedom between the rigid clusters, dramatically reducing the conformational space.
- This hierarchical approach has been shown to better sample near-native structures for proteins like Trp-cage by focusing the search on the assembly of pre-formed elements [5].
Workflow for Hierarchical Constrained MD Folding
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Constrained MD
| Reagent / Resource | Function and Application |
|---|---|
| Constrained MD Software (GNEIMO) | Implements the constrained MD formalism with an O(N) solver, enabling "freeze and thaw" dynamics for large proteins [5]. |
| ILVES-PC Algorithm | A high-accuracy constraint solver for proteins, providing speed and precision improvements over traditional methods like SHAKE [11]. |
| GB/SA Implicit Solvent | An efficient solvation model that eliminates the need for explicit water molecules, drastically reducing computational cost in folding simulations [5]. |
| Machine-Learned Coarse-Grained (CG) Model | A transferable, bottom-up CG force field that can serve as a powerful and fast prior for identifying folding basins or generating initial structures for refined constrained MD studies [6]. |
| Replica Exchange Framework | A software infrastructure for running parallel tempering simulations, essential for overcoming free energy barriers during protein folding [5]. |
| Analysis Tools (PCA, Clustering) | Software for trajectory analysis, such as Principal Component Analysis and K-means clustering, used to identify metastable states and project the free energy landscape [5]. |
Visualization of the Folding Landscape
The analysis of constrained MD trajectories often involves projecting the high-dimensional data onto a low-dimensional space to visualize the free energy landscape.
Workflow for Free Energy Landscape Analysis
Refining Structures with Post-Prediction MD Simulations
The advent of highly accurate protein structure prediction tools, such as AlphaFold2, has revolutionized structural biology by providing high-quality static models of proteins [44] [45]. However, these predicted structures are static and do not capture the dynamic conformational ensembles that proteins adopt in solution, which are crucial for understanding function, allosteric regulation, and facilitating drug discovery [45] [46]. Molecular dynamics (MD) simulations serve as a powerful complementary technique that can refine these static models by sampling their conformational landscape, assessing their stability, and identifying potential functional states [4] [45].
For the specific challenge of studying small protein folding, constrained MD methods offer a computationally efficient strategy for post-prediction refinement [5]. These methods reduce the number of simulated degrees of freedom, enabling longer timescale simulations that can more effectively probe folding pathways and the stability of predicted folds. This application note details protocols for employing constrained MD simulations to refine and validate protein structures, particularly those generated by AI-based prediction tools.
Key Concepts and Quantitative Comparisons
Constrained MD for Enhanced Sampling
Constrained MD, also known as internal coordinate MD or torsion angle MD, differs from standard all-atom Cartesian MD by treating molecules as collections of rigid bodies connected by flexible torsional hinges [5] [47]. This approach fixes bond lengths and bond angles using hard holonomic constraints, drastically reducing the number of degrees of freedom and allowing for larger integration time steps (e.g., 5 fs compared to 1-2 fs in Cartesian MD) [5]. The key advantage for folding studies is the enhanced conformational sampling efficiency, as the simulation can explore relevant torsional space more rapidly.
Table 1: Comparison of MD Simulation Approaches for Protein Folding
| Feature | All-Atom Cartesian MD | Constrained MD (All-Torsion) | Hierarchical Constrained MD |
|---|---|---|---|
| Degrees of Freedom | 3N (N = number of atoms) [5] | ~N/10 (Torsional angles only) [5] | Variable (User-defined rigid clusters) [5] |
| Typical Time Step | 1-2 fs [5] | 3-5 fs [5] | 3-5 fs [5] |
| Computational Cost | High (Microseconds computationally demanding) [4] | Lower (Order of magnitude reduction in DOFs) [5] | Variable (Adaptable to system and question) [5] |
| Primary Application | High-resolution dynamics, local interactions [4] | Enhanced conformational sampling, folding pathways [5] | Sampling assembly of pre-formed elements, zipping-and-assembly mechanisms [5] |
Analysis Methods for Folding Simulations
The trajectories generated from MD simulations require robust analysis methods to extract meaningful insights into folding mechanisms and dynamics.
Table 2: Key Analytical Methods for Folding Simulations
| Method | Description | Application in Folding Studies |
|---|---|---|
| Principal Component Analysis (PCA) | Identifies the main collective motions by diagonalizing the covariance matrix of atomic coordinates [5]. | Differentiates folding pathways and visualizes the free energy landscape along dominant reaction coordinates [5]. |
| Dynamic Cross-Correlation (DCC) | Calculates the correlation coefficient between atomic fluctuations, C(i,j), ranging from -1 (anti-correlated) to 1 (correlated) [48] [46]. | Identifies networks of residues moving together, revealing allosteric communication and coordinated motions during folding [48] [46]. |
| Graph Theory Network Analysis | Represents protein as a network; nodes are residues, edges represent significant dynamic correlations [46]. | Identifies key residues for signal propagation (high centrality) and communities of residues (modularity) involved in folding steps [46]. |
Experimental Protocols
The following diagram illustrates the comprehensive workflow for refining a predicted protein structure using constrained MD simulations, from initial setup to final analysis.
Protocol 1: System Setup and Constrained MD Simulation
This protocol uses the GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method as an example of a constrained MD approach [5].
Initial Structure Preparation:
- Obtain the initial protein structure from a prediction server like AlphaFold2 or ColabFold [45]. Place the structure in a PDB file.
- Solvation: Solvate the protein in an implicit solvent model, such as the Generalized-Born Surface Area (GB/SA) model, using an external dielectric constant of 78.3 to represent water [5]. For membrane proteins, a lower dielectric (e.g., 40.0) may be used [5].
- Ionization and Neutralization: Add ions to neutralize the system's charge and mimic physiological ionic strength.
Energy Minimization:
- Perform conjugate gradient minimization on the solvated system to remove bad steric clashes. A convergence criterion of 10⁻² kcal/(mol·Å) for the force gradient is typically sufficient [5].
Constrained MD Simulation with Replica Exchange:
- Define Constraints: Set all bond lengths and bond angles as constrained, leaving only torsional angles as flexible degrees of freedom [5] [47].
- Force Field and Parameters: Use a modern force field (e.g., AMBER parm99). Set up GB/SA solvation parameters: interior dielectric = 1.75, exterior dielectric = 78.3, solvent probe radius = 1.4 Å, and a non-bonded force cutoff of 20 Å [5].
- Integrator and Time Step: Use an integrator like the Lobatto integrator with a time step of 5 fs [5].
- Replica Exchange Setup: To enhance sampling, run 6-8 replicas of the system across a temperature range (e.g., 325 K to 500 K). Exchange temperatures between replicas every 2 ps [5].
- Simulation Duration: Run production simulation for a sufficient duration to observe folding/unfolding events (e.g., 20 ns per replica for small proteins) [5].
Protocol 2: Hierarchical Clustering for Complex Folding
For proteins suspected to fold via a "zipping-and-assembly" mechanism, a hierarchical constrained MD approach can be more efficient [5].
Preliminary All-Torsion Simulation: First, run a short all-torsion constrained MD simulation (as in Protocol 1) to identify regions that form stable secondary structures (e.g., α-helices) early in the folding process.
Define Rigid Clusters: Analyze the preliminary trajectory to identify residues forming persistent secondary structure elements. Define these regions as rigid clusters within the simulation software.
Freeze and Thaw Simulation: Run a new constrained MD simulation where the torsions within the identified stable clusters are "frozen" (treated as rigid bodies). Only the torsional degrees of freedom connecting these clusters and in the remaining flexible regions are sampled. This strategy accelerates the sampling of the assembly of pre-formed elements [5].
Protocol 3: Trajectory Analysis and Validation
Principal Component Analysis (PCA) and Clustering:
- Construct a covariance matrix from the Cα atomic coordinates of the trajectory [5].
- Diagonalize the matrix to obtain the eigenvectors (principal components, PCs) and eigenvalues.
- Project the trajectory onto the first two PCs to visualize the free energy landscape and identify major conformational clusters using an algorithm like k-means [5].
Dynamic Cross-Correlation and Graph Analysis:
- Calculate the cross-correlation matrix C(i,j) for Cα atoms using the Pearson correlation coefficient of their fluctuations [48] [46].
- Build a dynamical network model by representing residues as nodes and creating edges between residues with |C(i,j)| > 0.5 (or a chosen threshold) [46].
- Analyze the network to identify residues with high "betweenness centrality," which are potential key players in the folding pathway and allosteric communication [46].
The logical flow of this analysis pipeline, from raw simulation data to mechanistic insight, is shown below.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Software for Constrained MD
| Item Name | Function/Description | Example Software/Package |
|---|---|---|
| Structure Prediction Tool | Generates initial atomic coordinates from an amino acid sequence. | AlphaFold2, ColabFold [45] |
| Constrained MD Simulator | Performs molecular dynamics using internal coordinates, enforcing constraints. | GNEIMO [5] |
| General MD Software | Performs standard all-atom and/or constrained MD simulations; used for setup, equilibration, and analysis. | GROMACS [48], AMBER [5], CHARMM [47] |
| Implicit Solvent Model | Computationally efficient method to simulate solvent effects without explicit water molecules. | Generalized-Born Surface Area (GB/SA) [5] |
| Trajectory Analysis Suite | Tool for calculating dynamics cross-correlation, PCA, and other essential metrics. | Bio3D (R package) [48], GROMACS analysis tools [48] |
| Network Analysis Tool | Constructs and analyzes graph theory-based networks from MD trajectories. | Custom scripts using libraries like NetworkX, Bio3D [46] |
| Visualization Software | Visualizes 3D structures, trajectories, and dynamic networks. | PyMOL, VMD, UCSF Chimera |
Addressing Force Field Biases and Their Impact on Folding Pathways
Molecular dynamics (MD) simulations are a powerful tool for studying protein folding pathways at atomic resolution. However, a significant challenge in this field is the presence of force field biases, where the underlying energy functions inaccurately represent molecular interactions, leading to incorrect predictions of protein native states, folding mechanisms, and kinetics. For researchers employing constrained MD protocols, recognizing, quantifying, and correcting for these biases is essential for obtaining biologically relevant results. This Application Note examines the nature of force field biases, provides protocols for their identification, and outlines strategies to mitigate their impact on simulated folding pathways, with a specific focus on studies of small proteins.
Quantifying Force Field Bias: Key Evidence
Empirical studies have demonstrated that force field biases can significantly alter simulation outcomes. The table below summarizes quantitative findings from key investigations.
Table 1: Documented Force Field Biases in Protein Folding Simulations
| Protein Studied | Force Field | Observed Bias | Energetic Impact / Consequence | Citation |
|---|---|---|---|---|
| Human Pin1 WW Domain | CHARMM22 with CMAP | Misfolded helical states favored over native β-sheet state | Helical states more stable by 4.4–8.1 kcal/mol; failure to fold in 10-μs simulations [49] | |
| Villin Headpiece | Multiple Force Fields | Altered folding pathways and unfolded state properties | Correct folding rate & native structure, but different mechanisms dependent on force field [50] | |
| Coarse-grained TIA-1 | Martini (v3) | Excessively compact conformations in multi-domain protein | Poor agreement with SAXS data; required force field adjustment [51] |
These findings underscore a critical insight: a force field's ability to correctly predict a native structure and folding rate does not guarantee an accurate depiction of the folding pathway or the free energy landscape [50]. Biases can favor non-native secondary structures, as seen with the unwarranted stability of helices in a β-sheet protein, or distort the ensemble of unfolded and intermediate states [49].
Experimental Protocols for Identifying and Mitigating Bias
Protocol 1: Free Energy Calculation Using Deactivated Morphing
This protocol is designed to quantitatively assess a force field's preference for a misfolded state versus the native state, as applied to the Pin1 WW domain [49].
- Step 1: Simulation Trajectory Analysis. Perform multiple long-timescale MD folding simulations starting from different initial conformations (e.g., extended and heat-denatured states). Use cluster analysis to identify persistently populated non-native states.
- Step 2: Reference State Selection. Select the experimentally determined native structure (e.g., PDB: 2F21) and the predominant misfolded structures (e.g., helixU, helixL, helixV) identified in Step 1 as reference conformations.
- Step 3: Free Energy Calculation Path. For each pair of native and misfolded states, calculate the free energy difference (ΔG) by following a thermodynamic path using the Deactivated Morphing (DM) method. The path involves several stages, as shown in the workflow below:
Diagram 1: Deactivated morphing free energy calculation workflow.
- Step 4: Free Energy Difference Calculation. The free energy difference ΔG between the unrestrained ensembles of states A and B is the sum of the free energy changes for each step in the path. Error analysis should be performed using block averaging over the simulation data [49].
Protocol 2: Constrained MD with Replica Exchange for Enhanced Sampling
Constrained MD, which reduces computational cost by fixing bond lengths and angles and using torsional degrees of freedom, can be combined with replica exchange to improve sampling of native-like states [5].
- Step 1: System Setup. Begin with an extended conformation of the protein. Apply constraints to define the molecular model. In all-torsion constrained MD, all bond lengths and angles are fixed, and only torsional degrees of freedom are sampled.
- Step 2: Force Field and Solvation. Use an appropriate force field (e.g., AMBER parm99) with an implicit solvation model (e.g., GB/SA) to reduce computational expense.
- Step 3: Replica Exchange Simulation. Set up a replica exchange simulation with 6-8 replicas spanning a temperature range (e.g., 325K to 500K). Due to the reduced degrees of freedom in constrained MD, fewer replicas are required compared to all-atom MD. Perform dynamics using an internal coordinate integrator with a larger timestep (e.g., 5 fs). Attempt exchanges between neighboring replicas at regular intervals (e.g., every 2 ps).
- Step 4: Hierarchical Clustering (Optional). To accelerate sampling, a "freeze and thaw" strategy can be employed. After an initial all-torsion simulation, secondary structure motifs that form can be frozen into rigid clusters, and sampling is focused on the flexible torsions connecting them. This hierarchical approach aligns with zipping-and-assembly folding models [5].
Protocol 3: Integrating Simulations with Small-Angle Scattering Data
For multi-domain proteins or complex systems, biases can lead to incorrect global conformations. This protocol uses experimental data to refine and validate simulation ensembles [51].
- Step 1: Generate Initial Ensemble. Run coarse-grained MD simulations (e.g., using the Martini force field) of the multi-domain protein, applying an elastic network within folded domains to maintain their integrity while allowing linkers to be flexible.
- Step 2: Compare with SAXS Data. Calculate theoretical SAXS curves from the simulation trajectories after backmapping to all-atom structures. Compare these with experimentally measured SAXS data.
- Step 3: Force Field Refinement. If a systematic discrepancy is found (e.g., simulated ensembles are too compact), adjust force field parameters. For the Martini force field, tuning the protein-water interaction strength has been shown to improve agreement with SAXS data [51].
- Step 4: Ensemble Reweighting. Use a Bayesian/Maximum Entropy (BME) approach to reweight the simulation ensemble to achieve maximum consistency with the experimental SAXS data. This provides a refined structural ensemble that integrates computational and experimental information.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Force Fields for Bias-Aware Folding Simulations
| Tool Name | Type | Primary Function in Protocol | Key Consideration |
|---|---|---|---|
| NAMD | MD Software | Production all-atom MD simulations [49] | Supports multiple force fields; used in deactivated morphing studies. |
| GROMACS | MD Software | All-atom and coarse-grained MD; constraint algorithms [51] [11] | High performance; integrates with PLUMED for analysis. |
| CHARMM22/CMAP | All-Atom Force Field | Energy calculation for proteins [49] | Known to exhibit helical bias for some β-sheet proteins. |
| Martini | Coarse-Grained FF | Simulating larger systems and longer timescales [51] | May produce overly compact conformations; requires validation/tuning. |
| GB/SA Implicit Solvent | Solvation Model | Solvation effects in constrained MD [5] | Reduces computational cost; requires careful parameterization. |
| ILVES-PC | Constraint Algorithm | Imposing bond constraints in all-atom MD [11] | Offers greater speed and accuracy vs. SHAKE/P-LINCS for proteins. |
| PLUMED | Analysis & Enhanced Sampling | Calculating collective variables (e.g., Rg) [51] | Essential for analyzing pathways and constructing free energy surfaces. |
Force field biases represent a significant source of uncertainty in protein folding simulations. The protocols outlined here provide a structured approach for researchers to proactively diagnose these biases through free energy calculations, mitigate sampling issues via constrained MD and replica exchange, and validate simulation ensembles against experimental data. For the field of constrained MD, the development of more accurate force fields and constraint algorithms remains critical. By systematically addressing force field biases, scientists can enhance the reliability of their folding pathway predictions, thereby strengthening the value of computational studies in fundamental research and drug development.
Balancing Computational Cost with Sampling Accuracy
In molecular dynamics (MD) simulations of protein folding, the core challenge is to achieve a sufficient level of sampling accuracy while operating within practical computational budgets. Accurate sampling is essential for capturing the rare events and complex energy landscapes that characterize protein folding, but it often requires simulations at microsecond to millisecond timescales, which are prohibitively expensive for most research groups. This document outlines application notes and protocols that leverage constrained dynamics and advanced sampling techniques to optimize this balance, enabling more efficient and reliable studies of small protein folding. The methodologies presented are framed within a broader thesis on employing constrained MD protocols to make such investigations more computationally tractable without sacrificing the physical rigor required for meaningful biological insights.
Quantitative Comparison of Computational Methods
Selecting an appropriate method requires a clear understanding of its computational cost and predictive accuracy. The following table summarizes these factors for several prominent physics-based and AI-assisted approaches.
Table 1: Comparison of Computational Methods for Protein Studies
| Method | Primary Application | Reported Accuracy | Relative Computational Cost | Key Trade-offs |
|---|---|---|---|---|
| QresFEP-2 (Hybrid-Topology FEP) [52] | Protein stability changes upon mutation | Excellent correlation with experimental stability data (ΔΔG) | High (but highest efficiency among FEP protocols) [52] | Accuracy comes at a high computational cost; less suitable for very high-throughput virtual screening. |
| MM/PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) [53] | Binding free energy calculations | Often lacks sufficient accuracy for subtle single-point mutations [52] | Medium | Faster than FEP but may sacrifice accuracy for speed; useful for initial screening. |
| FragFold (AI-based prediction) [54] | Predicting inhibitory protein fragments | >50% experimental validation of predicted binders [54] | Low (leveraging pre-computed models) | Does not provide a full dynamical trajectory; reliant on the accuracy of the underlying AI model (AlphaFold). |
| Conventional MD (Explicit Solvent) [55] | Conformational sampling & folding pathways | Theoretically high, but limited by achievable timescales | Very High to Prohibitive | Directly simulates dynamics but may not reach biologically relevant timescales for folding. |
Detailed Experimental Protocols
Protocol 1: Hybrid-Topology Free Energy Perturbation (QresFEP-2)
Application: Accurately predicting the change in protein thermodynamic stability (ΔΔG) resulting from single-point mutations for protein engineering and drug design [52].
Materials and Equipment:
- Software: QresFEP-2, integrated with the molecular dynamics (MD) software Q [52].
- Hardware: Computer cluster for production runs. Initial setup can be performed on a desktop workstation (e.g., 4-core CPU, 16 GB memory) [55].
- Initial Input: Protein structure coordinate file (PDB format).
Procedure:
- System Preparation:
- Obtain the initial protein structure from the Protein Data Bank (PDB) or generate a homology model.
- Pre-process the structure to remove external water molecules and separate ligand coordinates if present [55].
- Convert the PDB file to the required molecular geometry format (
.gro) and generate a topology file (.top) using a command likepdb2gmx[55].
Define Simulation Box and Solvation:
- Place the protein in the center of a periodic boundary box (e.g., cubic, dodecahedron) with a defined distance (e.g., 1.4 nm) from the protein to the box edge using
editconf[55]. - Solvate the protein in the box using the
solvatecommand, which adds explicit water molecules and updates the topology file [55]. - Neutralize the system's net charge by adding counter ions (e.g., Na+, Cl-) using the
genioncommand [55].
- Place the protein in the center of a periodic boundary box (e.g., cubic, dodecahedron) with a defined distance (e.g., 1.4 nm) from the protein to the box edge using
QresFEP-2 Specific Setup:
- Hybrid Topology Construction: The protocol automatically generates a hybrid topology for the mutated residue. This topology combines a single-topology representation for the conserved backbone atoms with a dual-topology representation for the changing side-chain atoms [52].
- Restraint Application: Dynamically apply restraints between topologically equivalent atoms in the wild-type and mutant side chains to ensure sufficient phase-space overlap and prevent "flapping," where atoms incorrectly overlap with non-equivalent neighbors [52].
Production FEP Simulation:
- Run the FEP simulation using the Q software. The protocol performs MD sampling along the alchemical pathway that transforms the wild-type side chain into the mutant side chain.
- The simulation uses a spherical boundary condition to maximize computational efficiency [52].
Analysis:
- Analyze the output to calculate the relative free energy change (ΔΔG) associated with the mutation. The robustness of QresFEP-2 has been validated on large datasets, including a comprehensive scan of the Gβ1 protein domain involving over 400 mutations [52].
The following workflow diagram illustrates the key stages of the QresFEP-2 protocol.
Protocol 2: Standard MD Setup for Protein Folding Studies
Application: Simulating the folding pathway and conformational dynamics of a small protein using explicit solvent MD.
Materials and Equipment:
- Software: GROMACS MD suite, Rasmol (for visualization), text editor (e.g., Gedit) [55].
- Hardware: A desktop PC for initial setup and a supercomputer cluster for production MD runs [55].
- Initial Input: Protein structure file (PDB format), force field (e.g.,
ffG53A7), parameter files (.mdp) [55].
Procedure:
- Obtain and Prepare Protein Coordinates: Download and visually inspect the protein PDB file. Remove unnecessary molecules and add missing hydrogen atoms using
pdb2gmx, which also generates the topology [55]. - Setup Periodic Boundary Conditions: Define a simulation box around the protein using
editconfto place the protein centrally within a defined box type (e.g., cubic) [55]. - Solvate the System: Fill the box with water molecules using the
solvatecommand [55]. - Neutralize the System: Add ions to neutralize the system's net charge using the
geniontool [55]. - Energy Minimization: Run an energy minimization step to remove any steric clashes or unrealistic geometry in the initial structure.
- Equilibration: Perform two equilibration phases:
- NVT Ensemble: Equilibrate the system at constant Number of particles, Volume, and Temperature (NVT) for ~100-500 ps.
- NPT Ensemble: Equilibrate the system at constant Number of particles, Pressure, and Temperature (NPT) for ~100-500 ps.
- Production MD: Launch the final, unrestrained production simulation. For protein folding, this may require microsecond-to-millisecond timescales, necessitating high-performance computing resources [55].
- Trajectory Analysis: Analyze the resulting trajectory for metrics such as Root Mean Square Deviation (RMSD), Radius of Gyration (Rg), and formation of secondary structural elements.
The standard workflow for setting up and running a classical MD simulation is summarized below.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Materials for Constrained MD Protocols
| Item Name | Function/Application | Usage Notes |
|---|---|---|
| GROMACS MD Suite [55] | A robust, open-source software package for performing MD simulations. | Supports almost all major force fields. Ideal for standard MD setup, simulation, and analysis [55]. |
| Q Software with QresFEP-2 [52] | MD software integrating the hybrid-topology FEP protocol for free energy calculations. | Uses spherical boundary conditions for enhanced computational efficiency in FEP simulations [52]. |
| AlphaFold & FragFold [54] | AI systems for predicting protein structures (AlphaFold) and inhibitory fragments (FragFold). | Provides initial structural models and identifies potential binding fragments, informing targets for simulation [54]. |
| Force Field (e.g., ffG53A7) [55] | A set of empirical parameters describing the interatomic forces within the molecular system. | Critical for simulation accuracy. The choice (e.g., ffG53A7 for explicit solvent) is made during topology generation [55]. |
| Molecular Geometry File (.gro) [55] | A file containing the coordinates of all atoms in the system with continuous numbering. | Generated from the initial PDB file and used throughout the simulation process [55]. |
| Molecular Topology File (.top) [55] | A file describing the molecular structure, including bonds, angles, and force field parameters. | Created and updated during system setup to include protein, solvent, and ions [55]. |
| Parameter File (.mdp) [55] | A file containing all the settings and parameters for the MD run (e.g., timestep, temperature, pressure control). | Defines the conditions of each simulation step (minimization, equilibration, production) [55]. |
Benchmarking Performance: Validation Against Experiments and Other Methods
Validating Near-Native Structures Against NMR and X-ray Crystallography Data
The determination of accurate three-dimensional protein structures is fundamental to structural biology and drug development. For researchers employing computational methods like constrained molecular dynamics (MD) to fold small proteins, validating the resulting "near-native" structures against experimental data is a critical step. Such validation ensures computational models are biologically relevant and trustworthy for downstream applications, such as structure-based drug design [56]. This application note details protocols for validating protein structures using data from the two primary experimental techniques: X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy. The content is framed within the context of using constrained MD for protein folding, a method that enhances conformational sampling by reducing the number of degrees of freedom and allowing for larger integration time steps [5].
A fundamental understanding of the two primary structure determination techniques is a prerequisite for meaningful validation.
X-ray Crystallography provides a single, high-resolution "snapshot" of the protein structure in a crystalline state. The quality of a crystal structure is often described by its resolution (in Ångströms), where a lower value indicates greater detail. Key validation metrics include the R-factor and R-free, which measure the agreement between the experimental diffraction data and the data calculated from the atomic model [56] [57] [58].
NMR Spectroscopy typically yields an ensemble of structures in solution, all of which are consistent with the experimental data. Key metrics here include the number of restraints per residue (e.g., from Nuclear Overhauser Effect (NOE) measurements) and the root-mean-square deviation (RMSD) among the ensemble members, which is a measure of precision, though not necessarily of accuracy [59] [58].
For both methods, geometric quality checks are essential. These include analyzing Ramachandran plots for backbone dihedral angles and calculating clashscores to assess how well atoms are packed [56] [58]. It is crucial to recognize that an NMR ensemble's precision (low RMSD) does not guarantee its accuracy, and a high-resolution crystal structure might miss functionally important dynamics [56] [58].
Table 1: Key Validation Metrics for X-ray and NMR Structures
| Validation Aspect | X-ray Crystallography | NMR Spectroscopy |
|---|---|---|
| Primary Experimental Data | X-ray diffraction intensities [57] | Chemical shifts, NOEs, residual dipolar couplings [60] [58] |
| Primary Output | A single model [56] | An ensemble of models [59] |
| Key Numerical Metrics | Resolution, R-factor, R-free [58] | Restraints per residue, restraint violations, ensemble RMSD [58] |
| Standard Geometric Checks | Ramachandran plot, clashscore, side-chain rotamer outliers [56] [58] | Ramachandran plot, clashscore, side-chain rotamer outliers [58] |
| Comparative Accuracy | Typically more precise; can be too rigid in loop regions [58] | Captures solution-state dynamics; can be too floppy overall [58] |
Validation Parameters and Benchmarking Data
When validating a computational model, it is vital to compare its properties against the expected ranges derived from high-quality experimental structures. Systematic comparisons of NMR and X-ray structures of the same proteins provide invaluable benchmark data.
One study of 109 non-redundant protein pairs revealed that the average Cα root-mean-square deviation (RMSD) between NMR and X-ray structures ranges from approximately 1.5 Å to 2.5 Å [59]. This provides a realistic expected deviation for a "near-native" model. The study further found that beta-strands generally match better between the two techniques than helices and loops, and that hydrophobic residues buried in the protein core exhibit higher conformational similarity than hydrophilic residues [59].
The validation of ligands or small molecules bound to proteins requires special attention. The quality of small molecules in the Protein Data Bank (PDB) is highly variable, and incorrect ligand placement can misguide drug discovery efforts. Therefore, careful inspection of the electron density map around a ligand is crucial [56].
Table 2: Benchmark Values from Comparative NMR and X-ray Studies
| Structural Element | Observation in NMR vs. X-ray Comparison | Implication for Validation |
|---|---|---|
| Overall Protein Fold | Average global Cα RMSD of 1.5-2.5 Å [59] | A near-native model should fall within or near this range. |
| Secondary Structure | Beta-strands show better average match than helices and loops [59] | Pay close attention to the geometry of helical and loop regions. |
| Residue Environment | Hydrophobic/core residues are more similar than hydrophilic/surface residues [59] | Higher deviations in solvent-exposed loops may be acceptable. |
| Side Chains | Buried side chains seldom adopt different orientations [59] | Mismatches in the protein core are a sign of a poor model. |
Experimental Protocols for Structure Validation
Protocol 1: Validating Against X-ray Crystallographic Data
This protocol is used to validate a computationally derived structure against an existing crystal structure or to assess the quality of a new crystal structure itself.
Assess Global and Local Quality Metrics:
- Check the resolution of the crystal structure. Structures at resolutions better than 2.0 Å are generally considered high-quality, while those between 2.0-3.0 Å are medium-resolution [57].
- Examine the R-factor and R-free. A reliable structure typically has an R-free value below 25%, and it should be close to the R-factor to avoid over-fitting [58].
Analyze Electron Density Maps:
- Visually inspect the 2Fo-Fc and Fo-Fc electron density maps in a program like COOT [56].
- Ensure the atomic model fits the electron density well, with continuous density for the main chain and well-defined density for side chains.
- For ligand validation, confirm the ligand's density is unambiguous and its chemistry is correct [56].
Run Geometric Validation Software:
Figure 1: Workflow for validating a protein structure using X-ray crystallographic data.
Protocol 2: Validating Against NMR Data and the ANSURR Method
This protocol is for validating a computational model using NMR data, such as chemical shifts, or for assessing the quality of an NMR-derived structure ensemble.
Utilize Chemical Shift Data:
Compare to Structural Rigidity:
Apply the ANSURR Validation Method:
- The ANSURR (Accuracy of NMR Structures using RCI and Rigidity) method compares the RCI-derived rigidity (from experimental shifts) to the FIRST-derived rigidity (from the structure) [58].
- It produces two scores:
- A correlation score: Measures if rigid and flexible regions align (indicates correct secondary structure).
- An RMSD score: Measures if the overall rigidity is correct (sensitive to hydrogen bonding and side-chain packing) [58].
- Accurate models will have high scores for both measures.
Figure 2: Workflow for validating a protein structure using NMR data and the ANSURR method.
Application to Constrained MD for Protein Folding
Constrained MD methods, such as the Generalized NEIMO (GNEIMO) method, are powerful tools for studying protein folding. These methods fix bond lengths and angles, treating the molecule as a collection of rigid bodies connected by flexible torsional hinges, which drastically reduces the number of degrees of freedom and computational cost [5].
Validation against experimental data is essential to confirm that these accelerated simulations produce physically realistic results. For instance, constrained MD replica exchange simulations have been successfully used to fold small proteins like polyalanine and the Trp-cage miniprotein, and the resulting structures were validated by comparing their secondary structure content and overall topology to known native states [5]. Furthermore, a "hierarchical" constrained MD approach, where pre-formed secondary structure elements are frozen and sampling is focused on the connecting loops, has shown improved convergence to native-like structures, aligning with the zipping-and-assembly folding model [5]. The ANSURR method [58] is particularly well-suited for validating such MD-derived models against readily available NMR chemical shifts, providing a robust measure of accuracy that is independent of the force field used in the simulation.
The Scientist's Toolkit
Table 3: Essential Software and Resources for Structure Validation
| Tool Name | Type | Primary Function in Validation |
|---|---|---|
| MolProbity [56] | Software Suite | All-atom contact analysis, clashscore, Ramachandran plots, and side-chain rotamer evaluation. |
| COOT [56] | Molecular Graphics | Visual model building and fitting into electron density maps; integrates MolProbity validation. |
| PROCHECK [56] | Software | Comprehensive analysis of protein structure geometry, including Ramachandran plot quality. |
| ANSURR [58] | Web Server/Software | Validates NMR (or computational) structures by comparing chemical shift-derived rigidity (RCI) with structure-derived rigidity (FIRST). |
| FIRST [58] | Software | Performs rigidity analysis on a protein structure using mathematical graph theory. |
| PDB | Database | Primary repository for experimental structures; provides validation reports for deposited entries. |
| Cambridge Structural Database (CSD) [56] | Database | Reference for small-molecule geometry to validate ligands and ion coordination in macromolecular structures. |
Molecular dynamics (MD) simulations are an indispensable tool for studying protein folding, yet sampling the requisite timescales remains computationally challenging. Enhanced sampling methods have been developed to address this limitation. This application note provides a comparative analysis of two prominent techniques: Constrained MD and Accelerated MD (aMD), focusing on their application in small protein folding research. We detail their fundamental principles, provide structured protocols for their implementation, and discuss their respective advantages to guide researchers in selecting the appropriate method for their studies.
Theoretical Background and Key Differentiators
Constrained MD and aMD take fundamentally different approaches to enhance conformational sampling in molecular simulations.
Constrained Molecular Dynamics reduces the number of degrees of freedom in the system by imposing holonomic constraints on bond lengths and bond angles. The molecule is modeled as a collection of rigid bodies connected by flexible torsional hinges. This reduction, often by an order of magnitude, eliminates high-frequency motions, permitting larger integration time steps and decreasing computational cost. The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method is an efficient algorithm for solving the resulting equations of motion with O(N) computational cost. A key advanced application is "hierarchical clustering," where pre-formed secondary structure elements can be frozen as rigid bodies, and only the connecting torsional degrees of freedom are sampled, which can better align with zipping-and-assembly folding models [5].
In contrast, Accelerated MD (aMD) is a potential-energy-based method that applies a non-negative bias potential to the system's original potential energy surface. This bias effectively reduces the energy barriers separating low-energy states, thereby increasing the transition rates between conformational states and enhancing the sampling of rare events. The bias potential is typically applied to the dihedral potential energy and/or the total potential energy, leading to two common variants: dihedral-boost aMD and dual-boost aMD [62].
The table below summarizes the core differences in their approach and impact.
Table 1: Fundamental Comparison of Constrained MD and Accelerated MD
| Feature | Constrained MD | Accelerated MD (aMD) |
|---|---|---|
| Fundamental Principle | Reduces degrees of freedom by constraining bond lengths/angles. | Adds a non-negative bias potential to the true potential energy. |
| System Representation | System of rigid bodies connected by torsional hinges. | All-atom representation without simplification. |
| Effect on Energy Landscape | Explores the original, unmodified landscape in a reduced coordinate space. | Directly modifies the energy landscape by lowering barriers. |
| Energetics & Thermodynamics | Preserves the original potential; no direct reweighting needed. | Alters potential; requires complex reweighting to recover true kinetics/thermodynamics. |
| Computational Speed | Faster per-ns due to fewer degrees of freedom and larger timesteps (e.g., 5 fs). | Slower per-ns than constrained MD but much faster than conventional MD in crossing barriers. |
| Primary Sampling Enhancement | Achieved through more efficient exploration per unit computational time. | Achieved by promoting barrier crossing and sampling of higher-energy states. |
Application in Small Protein Folding: A Practical Comparison
Both methods have been successfully applied to study the folding of small proteins and peptides, demonstrating their utility in this domain.
Constrained MD has been effectively used in replica exchange simulations to fold various small peptides with implicit solvent, including α-helical peptides like polyalanine and WALP16, β-turn peptides, and the mixed-motif Trp-cage mini-protein. Studies show that constrained MD replica exchange exhibits a wider conformational search than all-atom MD with increased enrichment of near-native structures. The hierarchical "freeze and thaw" approach, where partially formed helical regions are frozen, has been shown to produce better sampling of near-native structures for Trp-cage, aligning with the zipping-and-assembly folding model [5].
aMD is also noted as a powerful enhanced sampling technique for quantitatively characterizing protein conformational landscapes, including folding. It is particularly useful for sampling the underlying conformational landscape and identifying intermediate and metastable states, which are essential for understanding folding mechanisms [62].
Table 2: Practical Application in Small Protein Folding
| Aspect | Constrained MD | Accelerated MD (aMD) |
|---|---|---|
| Proven Efficacy | Folding of α-helices, β-turns, and Trp-cage mini-protein [5]. | Sampling of conformational landscapes, intermediates, and rare events [62]. |
| Sampling Breadth | Wider conformational search and enrichment of near-native structures [5]. | Enhanced sampling of high-energy states and barrier crossing. |
| Integration with RE | Highly effective; reduced dofs cut required replicas by ~factor of three [5]. | Possible and commonly used (RE-aMD). |
| Handling of Disulfides | Specialized protocols exist for oxidative folding (e.g., using CYR residues) [18]. | Standard aMD does not explicitly model disulfide formation. |
Detailed Experimental Protocols
Protocol for Constrained MD Replica Exchange Folding Simulation
This protocol outlines the steps for setting up and running a folding simulation for a small peptide using the Constrained MD (GNEIMO) method with a replica exchange sampling scheme, as derived from the literature [5].
I. Initial System Setup
- Starting Structure: Begin with an extended conformation of the peptide generated from its sequence.
- Force Field and Solvation: Use the parm99 forcefield (part of AMBER99) with an implicit solvation model, such as the Generalized-Born/Surface Area (GB/SA) OBC model.
- Set the GB/SA interior dielectric to 1.75 and the exterior dielectric to 78.3 for water (or 40.0 for a membrane environment).
- Use a solvent probe radius of 1.4 Å for the nonpolar solvation component.
- Apply a 20 Å cutoff for non-bonded interactions, with forces smoothly switched off.
II. Energy Minimization
- Perform conjugate gradient minimization on the extended structure until the force gradient converges to a value of 10⁻² kcal/mol/Å.
III. Constrained MD Replica Exchange Parameters
- Dynamics Integrator: Use a Lobatto integrator.
- Time Step: 5 fs.
- Replica Setup: Use 8 replicas for a system like the 20-residue Trp-cage. The number of replicas is proportional to the square root of the number of degrees of freedom; constrained MD's reduced dofs can cut the required replicas by a factor of three compared to all-atom MD.
- Temperature Distribution: Distribute replicas across a temperature range, for example, from 325 K to 500 K in steps of 25 K.
- Replica Exchange Attempts: Attempt exchanges between neighboring replicas every 2 ps.
- Simulation Duration: Run each replica for up to 20 ns.
IV. Hierarchical Clustering (Advanced Application)
- After an initial all-torsion simulation, analyze the trajectory for partially formed secondary structures.
- Define rigid clusters based on these regions (e.g., helical segments).
- Run a subsequent simulation where the torsions within these clusters are "frozen," and only the torsions connecting them are sampled.
Protocol for Oxidative Folding with Constrained MD
For peptides containing disulfide bonds, such as guanylin, a specialized protocol can model disulfide formation during folding [18].
I. Cysteine Residue Modeling
- Define a new residue type, "CYR," which represents a cysteine with a deprotonated, neutral thiol group (-S). This mimics reactive species like thiyl radicals or sulfenates that can form disulfide bonds.
II. Simulation Setup
- Use the CYR residue type for all cysteines in the input structure.
- Employ the AMBER ff14SB force field.
- Run a series of standard constrained MD simulations. When two CYR sulfur atoms come into close proximity, they can form a disulfide bond in a barrierless fashion within this model.
III. Analysis
- Analyze the resulting trajectories to identify the distribution of different disulfide-bonded isomers.
General Protocol for Accelerated MD (aMD) Simulation
This is a generalized protocol for setting up an aMD simulation. Specific parameters depend on the software and system.
I. System Preparation
- Obtain Coordinates: Start with a PDB file of the protein.
- Solvate and Neutralize: Use tools like
solvateandgenionin GROMACS to place the protein in a solvent box (e.g., TIP3P water) and add ions to neutralize the system's charge [63].
II. Preprocessing for aMD
- Equilibration: Perform a short conventional MD simulation to equilibrate the solvent and ions around the protein.
- Calculate aMD Parameters: From the equilibrated conventional MD, calculate the average dihedral and total potential energies required to set the aMD boost potential.
III. aMD Production Run
- Run the production aMD simulation using the calculated boost parameters. The modified potential energy, ( V'(r) ), is given by: ( V'(r) = V(r) + \Delta V(r) ), where ( \Delta V(r) ) is the bias potential that is added when ( V(r) ) is below a predefined energy threshold, ( E ).
IV. Post-Processing and Analysis
- Reweighting: Apply reweighting algorithms (e.g., the Maclaurin series expansion or other techniques) to the aMD trajectory to recover the unbiased canonical ensemble and kinetics, which is a non-trivial step.
Workflow Visualization
The following diagram illustrates the logical workflow and key decision points when choosing between and applying these simulation methods.
Diagram 1: Decision workflow for choosing a simulation method.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Software, Tools, and Analytical Methods
| Category | Item/Solution | Function/Description |
|---|---|---|
| Software & Suites | GROMACS | A robust, open-source MD simulation suite supporting many force fields; ideal for aMD and conventional MD [63]. |
| GNEIMO | A specialized package for performing Constrained MD simulations, supporting hierarchical clustering and replica exchange [5]. | |
| AMBER | A suite of biomolecular simulation programs that includes support for force fields like parm99 and GB/SA implicit solvent used in constrained MD studies [5]. | |
| Force Fields | AMBER parm99/ff14SB | Force fields providing parameters for potential energy calculations; used in constrained MD and oxidative folding studies [5] [18]. |
| CHARMM | All-atom force fields, including modifications used in specialized simulations like those on the Anton supercomputer [62]. | |
| Solvation Models | GB/SA (OBC) | An implicit solvation model that approximates the solvent environment, significantly reducing computational cost [5]. |
| TIP3P | A common 3-site model for explicit water molecules, used in detailed all-atom simulations [62]. | |
| Analysis & Visualization | Principal Component Analysis (PCA) | A dimensionality reduction technique to project high-dimensional simulation data onto a low-dimensional space for analyzing collective motions [5] [62]. |
| Clustering (K-means, HDBSCAN) | Algorithms to group similar conformations from a trajectory into discrete states, aiding in the identification of metastable states [5] [62]. | |
| VADAR / Ramachandran Plots | Tools for analyzing the stereochemical quality and secondary structure content of protein models [64]. | |
| Specialized Hardware | Anton Supercomputer | A special-purpose machine designed for extremely long-scale MD simulations, microseconds and beyond [62]. |
| Computer Clusters (with GPUs) | General-purpose high-performance computing systems essential for running most MD simulations within a reasonable time [63]. |
In the field of structural biology, predicting the three-dimensional structure of proteins from their amino acid sequence remains a fundamental challenge. Two distinct computational approaches have emerged as powerful tools: physics-based Constrained Molecular Dynamics (MD) and artificial intelligence (AI)-based prediction tools like AlphaFold2 and Robetta. Constrained MD simulations incorporate physical principles and force fields to model protein folding pathways and dynamics by reducing the system's degrees of freedom [5]. In contrast, AI-based methods leverage deep learning architectures trained on vast datasets of known protein structures to predict static protein folds with remarkable speed and accuracy [65] [66]. This application note provides a comparative analysis of these methodologies, detailing their theoretical bases, respective protocols, and practical applications, with a specific focus on folding small proteins.
The core distinction between these methods lies in their foundational principles: Constrained MD is a physics-based simulation approach, while AI tools are data-driven predictors.
Constrained Molecular Dynamics simplifies the traditional all-atom MD simulation by fixing bond lengths and angles, treating the molecule as a collection of rigid bodies connected by flexible torsional hinges. This reduction in degrees of freedom allows for larger integration time steps (e.g., 5 fs) and a significant decrease in computational cost, enabling longer timescale simulations of folding events [5]. The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method implements this approach with O(N) computational cost, making it feasible for larger proteins [5]. Its strength lies in sampling folding pathways and near-native conformational ensembles.
AI-Based Prediction Tools like AlphaFold2 and Robetta rely on deep learning models. AlphaFold2 uses a novel neural network architecture (Evoformer) that processes multiple sequence alignments (MSAs) and incorporates physical and geometric constraints to directly predict the 3D coordinates of all heavy atoms [66]. Robetta, which incorporates RoseTTAFold, is a three-track deep neural network that similarly uses patterns in amino acid sequences and residue interactions for structure prediction [67] [68]. These methods excel at providing highly accurate static structures quickly but offer limited direct insight into folding dynamics or the energy landscape.
Table 1: Core Characteristics of Constrained MD and AI-Based Prediction Tools
| Feature | Constrained MD | AlphaFold2 | Robetta (RoseTTAFold) |
|---|---|---|---|
| Fundamental Principle | Physics-based simulation (Newtonian mechanics) [5] | AI/Deep Learning (Evoformer network) [66] | AI/Deep Learning (Three-track neural network) [67] |
| Primary Output | Folding pathways, conformational ensembles, dynamics [5] | Static, single 3D structure [66] | Static, single 3D structure [67] |
| Computational Cost | High (but lower than all-atom MD) [5] | Low to Moderate (per prediction) [66] | Low to Moderate (per prediction) [67] |
| Typical Simulation/Prediction Time | Nanoseconds to microseconds [5] | Minutes to hours (depending on sequence length) [66] | Minutes to hours [67] |
| Key Strength | Studies kinetics, thermodynamics, and folding mechanisms; refines structures [5] [67] | High accuracy for single, stable folds; high throughput [67] [66] | High accuracy; good performance even with limited homology [67] [68] |
| Key Limitation | Computationally expensive; limited by force field accuracy [5] | Limited conformational diversity; static output [69] [70] | Static output; performance can vary [67] |
Table 2: Comparative Performance in Practical Applications
| Application / Metric | Constrained MD | AlphaFold2 | Robetta |
|---|---|---|---|
| Small Protein Folding (e.g., Trp-cage) | Successfully folds with Replica Exchange; samples near-native states [5] | Highly accurate for most single-domain proteins [71] [66] | Accurately predicts structures of small peptides [67] |
| Refinement of Predicted Models | Effective for compacting and refining initial models (e.g., from I-TASSER) [67] | Output is typically used as a final model | Output is typically used as a final model |
| Handling Intrinsically Disordered Regions | Can simulate flexibility and disorder [5] | Lower confidence (low pLDDT) in disordered regions [70] | Lower confidence in disordered regions [70] |
| Domain Orientation in Multi-domain Proteins | Can theoretically sample domain motions | Prone to severe deviations from experimental domain packing [69] | Performance can vary; may suffer similar issues |
| Confidence Metric | Energetic and convergence analysis | pLDDT (per-residue), PAE (inter-residue) [69] [66] | Predicted TM-score, other internal metrics [67] |
Experimental and Computational Protocols
Constrained MD Protocol for Protein Folding
This protocol outlines the use of the GNEIMO constrained MD method with a replica exchange sampling strategy for folding small proteins, as applied to systems like the Trp-cage protein [5].
A. Initial System Setup
- Starting Conformation: Begin with an extended conformation of the protein sequence.
- Energy Minimization: Perform conjugate gradient minimization until the force gradient converges to 10⁻² kcal/mol/Å.
- Force Field and Solvation: Use the parm99 forcefield (part of AMBER99) with an implicit solvation model, such as the Generalized-Born Surface Area (GB/SA) model. Typical parameters include:
- GB/SA interior dielectric: 1.75
- GB/SA exterior dielectric: 78.3 (for water)
- Solvent probe radius: 1.4 Å
- Non-bonded force cutoff: 20 Å (with a smooth switching function) [5].
B. Dynamics Configuration
- Clustering Scheme: Define rigid clusters based on the molecular topology. For "all-torsion" dynamics, all torsional degrees of freedom are allowed to flex. For "hierarchical" dynamics, pre-formed secondary structure regions can be frozen as rigid clusters to enhance sampling efficiency [5].
- Integrator: Use the Lobatto integrator for dynamics.
- Time Step: Set to 5 fs, enabled by the constrained degrees of freedom.
- Simulation Temperature: Couple the system to a thermal bath (e.g., 300 K). For replica exchange, set up a temperature ladder.
C. Enhanced Sampling: Replica Exchange
- Replica Setup: Typically, 6-8 replicas are sufficient for small proteins due to the reduced degrees of freedom. A temperature range of 325 K to 500 K (in steps of 25 K) has been used successfully [5].
- Exchange Attempts: Attempt exchanges between neighboring replicas every 2 ps.
- Total Simulation Time: Run simulations for up to 20 ns per replica to achieve adequate sampling of the folded state [5].
D. Trajectory Analysis
- Native-like Structure Detection: Monitor the Root Mean Square Deviation (RMSD) of the backbone atoms from the known native structure.
- Secondary Structure Formation: Calculate the fraction of residues in helical or beta-sheet conformations based on backbone dihedral angles.
- Conformational Clustering: Use algorithms like K-means to cluster trajectories into structurally similar families and determine the population of native-like clusters [5].
- Principal Component Analysis (PCA): Project the simulation trajectories onto the first two principal components to visualize the conformational landscape and population densities [5].
AI-Based Structure Prediction Protocol
This protocol describes the standard procedure for predicting a protein structure using AlphaFold2 and Robetta. The example of the Hepatitis C virus core protein (HCVcp) illustrates a practical application [67].
A. Input Preparation
- Target Sequence: Obtain the amino acid sequence of the target protein in FASTA format.
B. Running AlphaFold2
- Multiple Sequence Alignment (MSA): The AlphaFold2 algorithm automatically searches databases (e.g., with HHblits or JackHMMER) to construct an MSA for the input sequence. This is a critical step for accurate prediction [66].
- Structure Prediction: The sequence and MSA are processed through the Evoformer neural network architecture, followed by the structure module, which iteratively refines the 3D coordinates. This is often done with multiple recycles (e.g., 3 cycles) to improve accuracy [66].
- Output: The model generates a PDB file containing the predicted atomic coordinates and a per-residue confidence score (pLDDT).
C. Running Robetta
- Sequence Submission: Submit the target amino acid sequence to the Robetta web server (https://robetta.bakerlab.org/).
- Template-Based vs. De Novo Prediction: The server automatically determines whether to use a template-based modeling approach (if a suitable structural homolog is found) or a de novo prediction using the RoseTTAFold algorithm.
- Output: The server returns one or more predicted models, often accompanied by confidence scores like the predicted TM-score.
D. Model Validation and Analysis
- Confidence Scores: Inspect the pLDDT scores from AlphaFold2 (scores < 70 indicate low confidence, often in flexible loops or disordered regions) and the Predicted Aligned Error (PAE) to assess inter-domain confidence [69] [66].
- Steric and Stereochemical Quality: Use tools like MolProbity within molecular visualization software (e.g., MOE, PyMOL) to check for clashes, and analyze phi-psi torsion angles in Ramachandran plots [67].
- Comparison with Experimental Data: If available, validate the model against experimental data such as NMR chemical shifts or mutagenesis studies.
Integrated and Specialized Applications
Synergistic Use of AI and Constrained MD
A powerful approach is to use AI-predicted structures as starting points for Constrained MD simulations to refine and validate the models. A study on HCVcp demonstrated this: structures predicted by AF2, Robetta, and other tools were subsequently refined using MD simulations. The MD simulations compacted the initial models, improved their stereochemical quality (as measured by ERRAT and Ramachandran plots), and led to theoretically more accurate and reliable structural models [67].
Specialized Applications
- Studying Folding Mechanisms: Constrained MD with hierarchical clustering, where partially formed native secondary structure elements are frozen, has been used to validate the "zipping-and-assembly" folding model for proteins like Trp-cage [5].
- Predicting Functional Peptides and Inhibitors: Advanced AI tools like FragFold build upon AlphaFold to predict small protein fragments that can bind to and inhibit target proteins. This method computationally fragments a protein and uses a pre-computed MSA to efficiently model how these fragments bind, enabling the discovery of genetically encodable inhibitors [54].
- Modeling Conformational Ensembles: Newer methods like FiveFold address the limitation of single-structure prediction by combining outputs from five different algorithms (including AF2 and RoseTTAFold) to generate an ensemble of plausible conformations. This is particularly useful for studying intrinsically disordered proteins and conformational diversity relevant to drug discovery [70].
The Scientist's Toolkit: Essential Research Reagents and Computational Tools
Table 3: Key Software Tools and Resources
| Tool / Resource Name | Type | Primary Function | Access / Reference |
|---|---|---|---|
| GNEIMO | Software Package | Performs Constrained MD simulations [5] | Research Code / PMC3124276 [5] |
| AlphaFold2 | AI Prediction Tool | Predicts protein structures from sequence [66] | Colab Notebook / Jumper et al., 2021 [66] |
| Robetta Server | AI Prediction Tool | Predicts protein structures (RoseTTAFold) [67] | https://robetta.bakerlab.org/ [67] |
| Molecular Operating Environment (MOE) | Software Suite | Homology modeling, visualization, and analysis [67] | Commercial Software (Chemical Computing Group) |
| FiveFold Methodology | Ensemble Method | Generates conformational ensembles from multiple AI tools [70] | Yang et al., 2025 (Scientific Reports) [70] |
| FragFold | Specialized AI Tool | Predicts inhibitory protein fragments [54] | Savinov et al., 2025 (PNAS) [54] |
| GB/SA Implicit Solvent | Solvation Model | Models solvent effects without explicit water molecules [5] | Onufriev et al., 2004 [5] |
Critical Limitations and Future Directions
Both methodologies have constraints that researchers must consider.
Limitations of Constrained MD:
- Its accuracy is inherently dependent on the quality of the underlying force field and the solvation model [5].
- While more efficient than all-atom MD, it remains computationally demanding for folding large proteins or simulating processes on millisecond timescales [5].
Limitations of AI Tools:
- They predominantly predict single, static structures and struggle to capture functional dynamics, multiple conformational states, and allosteric mechanisms [65] [70].
- Predictions for multi-domain proteins can show severe deviations from experimental structures in the relative orientation of domains, even when confidence metrics like PAE appear moderate [69].
- Performance can be poor for intrinsically disordered proteins, proteins with few homologous sequences, or unusual folds not well-represented in training data [69] [70].
Emerging Solutions: The future lies in hybrid approaches and next-generation AI. Tools like BioEmu are emerging as generative AI systems that can simulate protein equilibrium ensembles, capturing conformational changes and thermodynamics with high accuracy and a massive speedup over traditional MD [26]. Integrating these dynamic AI models with the physical realism of MD simulations promises a more comprehensive understanding of protein folding and function.
Evaluating Free Energy Profiles and Convergence
Within the framework of constrained molecular dynamics (MD) protocols for small protein folding, the accurate calculation and evaluation of free energy profiles is paramount. Free energy landscapes provide the fundamental link between a protein's atomic structure and its thermodynamic and kinetic properties. Free-energy-based simulations are increasingly supplying the narratives about the structures, dynamics, and biological mechanisms that constitute the fabric of protein science [72]. For researchers and drug development professionals, assessing whether a simulation has produced a converged free energy profile is a critical step in validating the reliability of computational predictions for understanding folding pathways, identifying metastable states, and guiding therapeutic design.
This application note details the protocols for computing free energy profiles in protein folding simulations and establishes a framework for rigorously evaluating their convergence. We focus on methodologies applicable to small proteins, emphasizing how constrained MD protocols can be leveraged to obtain statistically robust and thermodynamically meaningful results.
Free Energy Calculation Methods in Protein Folding
The "correct" folded structure of a protein is not merely one of minimal potential energy but one of minimal free energy, which balances potential energy with entropy [73]. At a finite temperature, the native state is a free energy minimum, and simulations must capture this balance. A variety of rigorous, physics-based methods are employed to compute these free energy landscapes.
Table 1: Key Free Energy Calculation Methods for Protein Folding
| Method | Fundamental Principle | Key Applications in Folding | Considerations for Convergence |
|---|---|---|---|
| Replica-Exchange MD (REMD) | Parallel simulations at multiple temperatures swap configurations, overcoming energy barriers and enhancing sampling [74]. | Efficiently exploring conformational space and characterizing folding/unfolding dynamics from an arbitrary extended conformation [74]. | Convergence requires sufficient replica swaps and simulation time to ensure random walks in temperature space and conformation space. |
| Alchemical Free Energy Calculations | Uses unphysical pathways to mutate, create, or annihilate atoms; includes equilibrium (FEP, TI) and non-equilibrium (based on Jarzynski/Crooks) methods [75]. | Calculating relative binding free energies (RBFE) and protein folding ΔΔG values due to mutations [75]. | Highly dependent on sufficient sampling along the alchemical parameter (λ) and the careful treatment of changing net charges. |
| Metadynamics / Infrequent Metadynamics | An external history-dependent bias potential is added to collective variables (CVs) to discourage the system from revisiting sampled states [72]. | Enhancing sampling of rare events, computing ligand-protein binding affinities, and calculating unbinding rates [72]. | Convergence is reached when the free energy landscape is fully filled by the bias. Requires careful selection of CVs. |
| Action-Derived MD (ADMD) | Generates a dynamic folding pathway between defined start and end states, which can serve as a reaction coordinate [74]. | Investigating the dynamic folding pathway model of a protein between fixed extended and compact conformations [74]. | The pathway itself is a single trajectory; convergence is assessed by the stability of the resulting free energy profile when combined with methods like REMD. |
The selection of a method often depends on the specific scientific question. For instance, the combination of Action-Derived MD (ADMD) and Replica-Exchange MD (REMD) provides a powerful protocol where ADMD identifies a plausible reaction coordinate for the folding transition, and REMD subsequently calculates the free energy profile along that coordinate as a function of temperature [74]. In studies focused on the effects of mutations, alchemical methods are the gold standard for computing changes in folding free energy (ΔΔG) [75].
Protocols for Free Energy Profile Generation
This section outlines two detailed protocols for generating free energy profiles, incorporating best practices for setup and execution.
Protocol 1: Combined ADMD and REMD for Folding Pathway Analysis
This protocol is designed to define a reaction coordinate and obtain its temperature-dependent free energy profile without an a priori assumption of the folding pathway [74].
System Preparation:
- Obtain an initial extended conformation of the protein. Ensure all atoms are present and the topology is consistent with the chosen force field.
- Solvate the protein in an explicit water box, adding ions as necessary to neutralize the system.
Action-Derived MD (ADMD) Simulation:
- Objective: To generate a dynamic transition pathway between the extended conformation and a known compact (folded or intermediate) conformation.
- Execution: Run ADMD simulations. The algorithm will produce a trajectory connecting the two end-states.
- Output Analysis: The step index of the ADMD simulation trajectory is defined as the reaction coordinate for subsequent analysis.
Replica-Exchange MD (REMD) Sampling:
- Objective: To perform enhanced conformational sampling along the identified reaction coordinate.
- Setup: Set up a REMD simulation with 20-50 replicas spanning a temperature range that includes the protein's expected folding transition (e.g., 270-500 K). Use the same solvated system as in Step 1.
- Execution: Run the REMD simulation, ensuring frequent exchange attempts between neighboring replicas (e.g., every 1-2 ps). The simulation length must be sufficient for the protein to repeatedly fold and unfold at temperatures near the transition.
Free Energy Profile Construction:
- Data Processing: Project all saved conformations from the REMD simulation onto the ADMD-derived reaction coordinate.
- Calculation: Use the Weighted Histogram Analysis Method (WHAM) or similar techniques to compute the free energy as a function of the reaction coordinate at the temperature of interest. The formula for the free energy is ( G(X) = -k_B T \ln P(X) ), where ( P(X) ) is the probability density along the reaction coordinate ( X ).
Protocol 2: Alchemical Free Energy Calculation for Mutation Effects (ΔΔG)
This protocol uses the pmx toolbox to calculate the change in folding free energy due to a point mutation [75].
System Preparation:
- Start with the folded structure of the wild-type protein from PDB or a previous folded simulation.
- Use
pmxto generate hybrid structures and topologies for the wild-type and mutant proteins. This creates a single molecular representation that can alchemically interpolate between the two states.
Double-System/Single-Box Setup:
- To handle mutations that change the net charge of the protein, employ the double-system/single-box approach [75].
- In a single simulation box, place two copies of the protein system: one being the wild-type and the other being the mutant. This ensures the overall system remains charge-neutral throughout the alchemical transformation.
Alchemical Transformation Simulation:
- Objective: To compute the free energy change for mutating the wild-type to the mutant in the folded state.
- Execution: Run an alchemical simulation, often using a non-equilibrium method based on the Crooks Fluctuation Theorem. The Hamiltonian ( H ) is coupled to a parameter ( \lambda ), which navigates the system from wild-type (( \lambda = 0 )) to mutant (( \lambda = 1 )).
- Sampling: Perform multiple independent "work" simulations (forward and backward) to obtain sufficient statistics for the free energy calculation via the Crooks Gaussian Intersection (CGI) method.
The following workflow diagram illustrates the logical relationship and sequence of the two primary protocols described above:
Assessing Convergence of Free Energy Profiles
A free energy calculation is only as reliable as the evidence demonstrating its convergence. Below are essential metrics and checks, summarized in a table for easy reference.
Table 2: Key Metrics for Assessing Convergence of Free Energy Profiles
| Metric | Description | Application and Interpretation |
|---|---|---|
| Time-Series Analysis | Monitor the free energy or the key collective variables as a function of simulation time. | The profile is considered converged when these values oscillate around a stable mean with no discernible drift over a long portion of the simulation. |
| Statistical Precision | Calculate the standard error or confidence intervals for the free energy difference (e.g., ΔΔG or barrier height) using block averaging or bootstrapping. | A converged result will show small confidence intervals (e.g., < 0.5 kcal/mol) that do not shrink significantly with increased simulation time. |
| Forward/Backward Consistency | For non-equilibrium alchemical simulations, compare the work distributions from the forward and reverse directions [75]. | Well-overlapping work distributions and a small statistical error from the Crooks Gaussian Intersection (CGI) analysis indicate robustness. |
| Replica Mixing (REMD) | Analyze the replica exchange acceptance rates and the random walk of replicas through temperature space. | Acceptance rates of 20-30% and a linear relationship between replica index and temperature visit count suggest efficient sampling and convergence. |
| Convergence of Landscape Features | Track the stability of the locations and depths of free energy minima (folded, unfolded, intermediates) over time. | The positions and relative stabilities of these states should remain constant over the latter part of the simulation. |
A critical conceptual point is that a single, long MD simulation will not necessarily locate the global free energy minimum but will sample configurations with a probability proportional to the Boltzmann factor, ( \exp(-G(x)/k_B T) ) [73]. Convergence means the simulation has adequately sampled the relevant regions of configuration space to reflect this Boltzmann distribution accurately.
The Scientist's Toolkit: Research Reagent Solutions
The following table details essential software and methodological "reagents" required for implementing the protocols discussed in this note.
Table 3: Essential Research Reagents for Free Energy Protein Folding Studies
| Reagent / Tool | Type | Primary Function | Example Use Case |
|---|---|---|---|
pmx |
Software Package | Automates the generation of hybrid structures and topologies for alchemical free energy calculations [75]. | Enabling double-system/single-box setup for charge-changing mutations in Protocol 3.2. |
| Force Fields (e.g., CHARMM, AMBER) | Parameter Set | Defines the potential energy function for the atoms in the system, including bonds, angles, and non-bonded terms [72]. | Providing the physical model for all MD simulations; accuracy is critical for realistic folding. |
| REMD Simulation Packages (e.g., GROMACS, NAMD) | MD Engine | Software capable of running complex MD protocols, including temperature and Hamiltonian replica exchange. | Executing the temperature swaps and dynamics sampling required in Protocol 3.1. |
| Weighted Histogram Analysis Method (WHAM) | Analysis Algorithm | Computes the potential of mean force (free energy profile) from umbrella sampling or REMD simulation data. | Constructing the final free energy profile from projected REMD data in Protocol 3.1. |
| Collective Variables (CVs) | Mathematical Descriptor | Low-dimensional functions of atomic coordinates that describe the progress of a slow process (e.g., folding). | In metadynamics, CVs like root-mean-square deviation (RMSD) or radius of gyration (Rg) are biased to explore the landscape. |
| MELD (Modeling Employing Limited Data) | Accelerated MD Method | Harnesses external, often vague, structural information to guide and drastically accelerate conformational searching [72]. | Finding native structures of small proteins (up to 92-mers) starting from extended states more efficiently than brute-force MD. |
Conclusion
Constrained molecular dynamics has emerged as a powerful and computationally efficient protocol for simulating the folding of small proteins, consistently demonstrating an enhanced ability to sample near-native structures compared to traditional all-atom MD. By strategically reducing the system's degrees of freedom through methods like all-torsion constraints and hierarchical 'freeze and thaw' schemes, researchers can overcome significant time-scale barriers. When integrated with other techniques like replica exchange or used in a multi-stage refinement pipeline with AI predictions, constrained MD offers a robust framework for exploring protein energy landscapes and folding pathways. The ongoing development of more accurate force fields and hybrid methods promises to further solidify its role. For biomedical research, these advances directly fuel structure-based drug discovery by providing reliable protein models, enabling the identification of cryptic pockets and informing the design of novel therapeutics against previously intractable targets.
References
- 1. What is the protein folding problem? | AlphaFold [https://www.ebi.ac.uk/training/online/courses/alphafold/an-introductory-guide-to-its-strengths-and-limitations/what-is-the-protein-folding-problem/]
- 2. AlphaFold: a solution to a 50-year-old grand challenge in ... [https://deepmind.google/blog/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology/]
- 3. The Protein Folding Problem: The day AI unlocked a secret of ... [https://medicine.iu.edu/blogs/research-updates/the-protein-folding-problem-the-day-ai-unlocked-a-secret-of-life]
- 4. Challenges in protein folding simulations: Timescale, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3032381/]
- 5. Folding of Small Proteins Using Constrained Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3124276/]
- 6. Navigating protein landscapes with a machine-learned ... [https://www.nature.com/articles/s41557-025-01874-0]
- 7. Open-source protein structure AI aims to match AlphaFold [https://www.nature.com/articles/d41586-025-03546-y]
- 8. What's Hot in 2025: Protein Structure Prediction using AI [https://www.criver.com/eureka/whats-hot-2025-protein-structure-prediction-using-ai]
- 9. AlphaFold: Five Years of Impact [https://deepmind.google/blog/alphafold-five-years-of-impact/]
- 10. Folding of small proteins using constrained molecular dynamics - PubMed [https://pubmed.ncbi.nlm.nih.gov/21591767/]
- 11. Accurate and efficient constrained molecular dynamics of ... [https://www.sciencedirect.com/science/article/pii/S0010465523000875]
- 12. Enhanced sampling of protein conformational changes via ... [https://www.nature.com/articles/s41467-025-55983-y]
- 13. Molecular dynamics simulation for all - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6209097/]
- 14. BioMD: All-atom Generative Model for Biomolecular ... [https://arxiv.org/html/2509.02642v1]
- 15. Practical considerations for Molecular Dynamics - GitHub Pages [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/aio/index.html]
- 16. Sampling of the conformational landscape of small proteins ... [https://www.nature.com/articles/s41598-020-75239-7]
- 17. Uncertainty driven active learning of coarse grained free ... [https://www.nature.com/articles/s41524-023-01183-5]
- 18. Simple MD-based model for oxidative folding of peptides ... [https://www.nature.com/articles/s41598-017-09229-7]
- 19. Evolutionary selection of proteins with two folds [https://www.nature.com/articles/s41467-023-41237-2]
- 20. Fold-switching proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12629603/]
- 21. Supramolecular assembly of protein building blocks [https://nanoconvergencejournal.springeropen.com/articles/10.1186/s40580-021-00294-3]
- 22. Exploration of novel αβ-protein folds through de novo design [https://pmc.ncbi.nlm.nih.gov/articles/PMC10442233/]
- 23. Mapping Conformational Dynamics of Proteins Using ... [https://www.sciencedirect.com/science/article/pii/S0006349513001835]
- 24. Replica Exchange Molecular Dynamics [https://pmc.ncbi.nlm.nih.gov/articles/PMC6484850/]
- 25. Replica exchange - GROMACS 2024 documentation [https://manual.gromacs.org/documentation/2024.0/reference-manual/algorithms/replica-exchange.html]
- 26. BioEmu: AI‐Powered Revolution in Scalable Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12643044/]
- 27. Ab initio characterization of protein molecular dynamics ... [https://www.nature.com/articles/s41586-024-08127-z]
- 28. Structure Refinement of Protein Low Resolution Models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3377353/]
- 29. A review on description dynamics and conformational ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482524013301]
- 30. Dynamic Folding Pathway Models of the Trp-Cage Protein [https://pmc.ncbi.nlm.nih.gov/articles/PMC3707288/]
- 31. A Case Study of the Folding Mechanism of Trp-cage - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3331591/]
- 32. Dynamics and cooperativity of Trp-cage folding [https://www.sciencedirect.com/science/article/abs/pii/S0003986108002257]
- 33. Solvation model background — APBS 3.4.1 documentation [https://apbs.readthedocs.io/en/latest/background.html]
- 34. Implicit solvation [https://en.wikipedia.org/wiki/Implicit_solvation]
- 35. Design and application of implicit solvent models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4045398/]
- 36. Implicit Solvent Models and Their Applications in Biophysics [https://www.mdpi.com/2218-273X/15/9/1218]
- 37. Accuracy comparison of several common implicit solvent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5313374/]
- 38. Union of Geometric Constraint-Based Simulations with ... [https://www.sciencedirect.com/science/article/pii/S0006349509017986]
- 39. Importance of molecular dynamics equilibrium protocol on ... [https://www.sciencedirect.com/science/article/pii/S2667074722000374]
- 40. Inverse problems with experiment-guided AlphaFold [https://arxiv.org/html/2502.09372v1]
- 41. of structure and sequence space of Sampling small protein folds [https://www.nature.com/articles/s41467-022-34937-8?error=cookies_not_supported&code=0dc34039-b31b-4a49-a714-c4a52f5d705f]
- 42. Quantitative and scalable approaches for measuring protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9337709/]
- 43. Multiscale methods for protein folding simulations [https://www.sciencedirect.com/science/article/abs/pii/S1046202310001325]
- 44. Accurate prediction of protein folding mechanisms by simple ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10587348/]
- 45. Machine learning/molecular dynamic protein structure ... [https://www.nature.com/articles/s41598-022-13714-z]
- 46. Graph Theory Approaches for Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11853848/]
- 47. Constraint (computational chemistry) [https://en.wikipedia.org/wiki/Constraint_(computational_chemistry)]
- 48. A beginner's guide to molecular dynamics simulations and ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687920301488]
- 49. Force Field Bias in Protein Folding Simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2711430/]
- 50. How Robust Are Protein Folding Simulations with Respect ... [https://www.sciencedirect.com/science/article/pii/S0006349511004097]
- 51. Combining molecular dynamics simulations with small-angle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7205321/]
- 52. Accurate predictions of protein mutational effects ... [https://www.nature.com/articles/s42004-025-01771-0]
- 53. for MM/PBSA Protocol Simulations of Molecular Dynamics Proteins [https://www.academia.edu/143100958/Protocol_for_MM_PBSA_Molecular_Dynamics_Simulations_of_Proteins]
- 54. AI system predicts protein fragments that can bind to or ... [https://news.mit.edu/2025/ai-system-fragfold-predicts-protein-fragments-0220]
- 55. for Protocol Simulations of Molecular Dynamics Proteins [https://bio-protocol.org/en/bpdetail?id=2051&type=0]
- 56. X-ray crystallography: Assessment and validation of protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3138648/]
- 57. x Ray crystallography - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC1186895/]
- 58. A method for validating the accuracy of NMR protein ... [https://www.nature.com/articles/s41467-020-20177-1]
- 59. Systematic Comparison of Crystal and NMR Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3032220/]
- 60. Automated protein fold determination using a minimal NMR ... [https://pubmed.ncbi.nlm.nih.gov/12761394/]
- 61. High-throughput three-dimensional protein structure ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166900002263]
- 62. Mapping conformational landscape in protein folding [https://www.sciencedirect.com/science/article/abs/pii/S0301462225000018]
- 63. Protocol for Molecular Dynamics Simulations of Proteins [https://en.bio-protocol.org/en/bpdetail?id=2051&type=0]
- 64. A comparative study of computational modeling ... [https://www.nature.com/articles/s41598-025-16866-w]
- 65. Structural Biology in the AlphaFold Era: How Far Is Artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109453/]
- 66. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 67. Comparison, Analysis, and Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10525864/]
- 68. Comparison, Analysis, and Molecular Dynamics ... [https://pubmed.ncbi.nlm.nih.gov/37760106/]
- 69. Severe deviation in protein fold prediction by advanced AI [https://www.nature.com/articles/s41598-025-89516-w]
- 70. A comprehensive application of FiveFold for conformation ... [https://www.nature.com/articles/s41598-025-17022-0]
- 71. Benchmarking AlphaFold2 on peptide structure prediction [https://www.sciencedirect.com/science/article/pii/S0969212622004798]
- 72. Advances in free-energy-based simulations of protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4785060/]
- 73. newtonian mechanics - Molecular vs Dynamics minimization... energy [https://physics.stackexchange.com/questions/330981/molecular-dynamics-vs-energy-minimization-in-protein-folding]
- 74. Transition pathway and its free - energy : a protocol for profile ... protein [https://pubmed.ncbi.nlm.nih.gov/23917881/]
- 75. Implementing and Assessing an Alchemical Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8044032/]