This article provides a detailed comparison of Molecular Dynamics (MD) and Monte Carlo (MC) simulation methods for researchers and professionals in computational biology and drug development. It explores the foundational principles of both stochastic (MC) and deterministic (MD) approaches, highlighting their unique strengths in sampling conformational space and simulating time evolution. The scope covers core methodologies, diverse applications in biomolecular simulation and drug design, strategies for troubleshooting sampling efficiency and system setup, and quantitative comparisons of performance and reliability. The review synthesizes these insights to offer practical guidance on method selection and discusses future directions for integrating these techniques in biomedical research.
Featured Articles
Latest Articles

This article provides a detailed comparison between Classical Molecular Dynamics (MD) and Reactive Force Fields, with a focus on ReaxFF, tailored for researchers and professionals in drug development and biomedical...
This article provides a comprehensive framework for researchers, scientists, and drug development professionals seeking to validate diffusion coefficients derived from Molecular Dynamics (MD) simulations against experimental data.
This article provides a systematic comparison of AMBER, CHARMM, and OPLS force fields, essential tools for molecular dynamics simulations in drug discovery and structural biology.
Molecular dynamics (MD) simulations have become an indispensable tool for predicting thermodynamic properties critical to drug design and materials science, such as binding free energy, entropy, and enthalpy.
This article provides a systematic comparison of explicit and implicit solvent models in molecular dynamics (MD) simulations, tailored for researchers and professionals in computational biophysics and drug development.
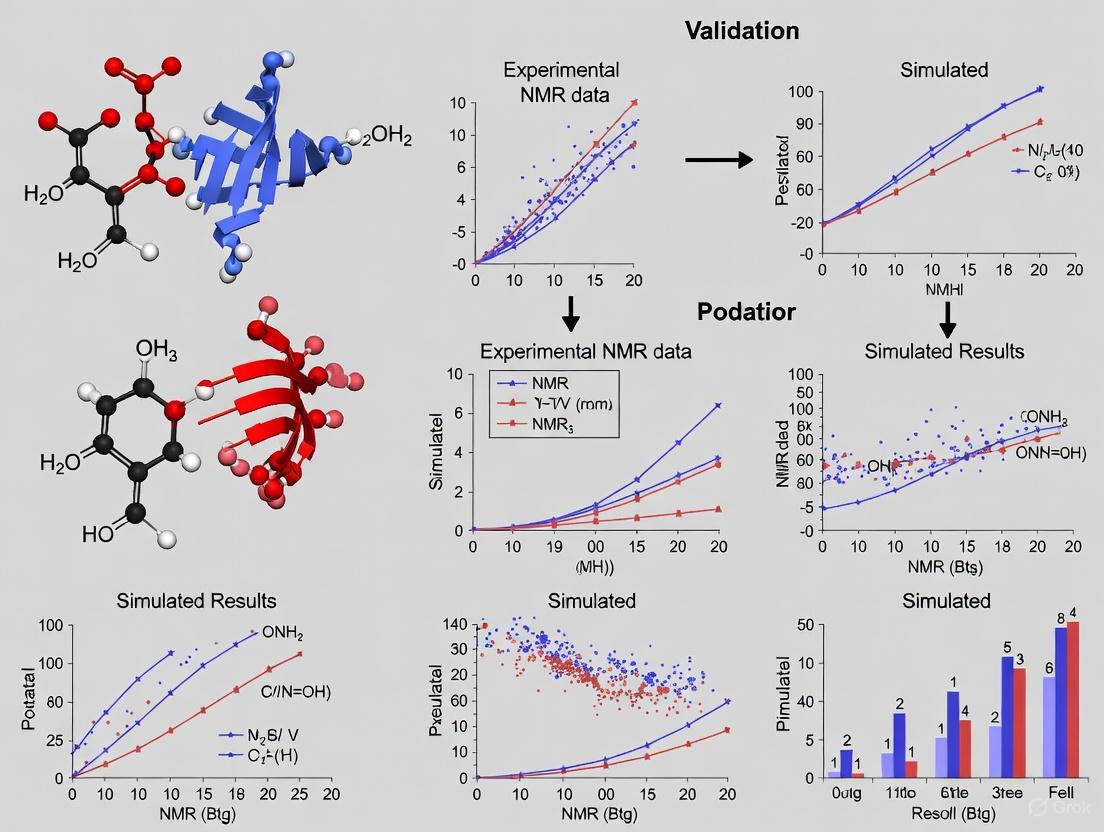
This article provides a comprehensive framework for validating Molecular Dynamics (MD) simulations using experimental Nuclear Magnetic Resonance (NMR) data, a critical synergy for advancing structural biology and rational drug design.
This guide provides a comprehensive framework for researchers and drug development professionals to optimize molecular dynamics (MD) simulations for large-scale biomedical systems.
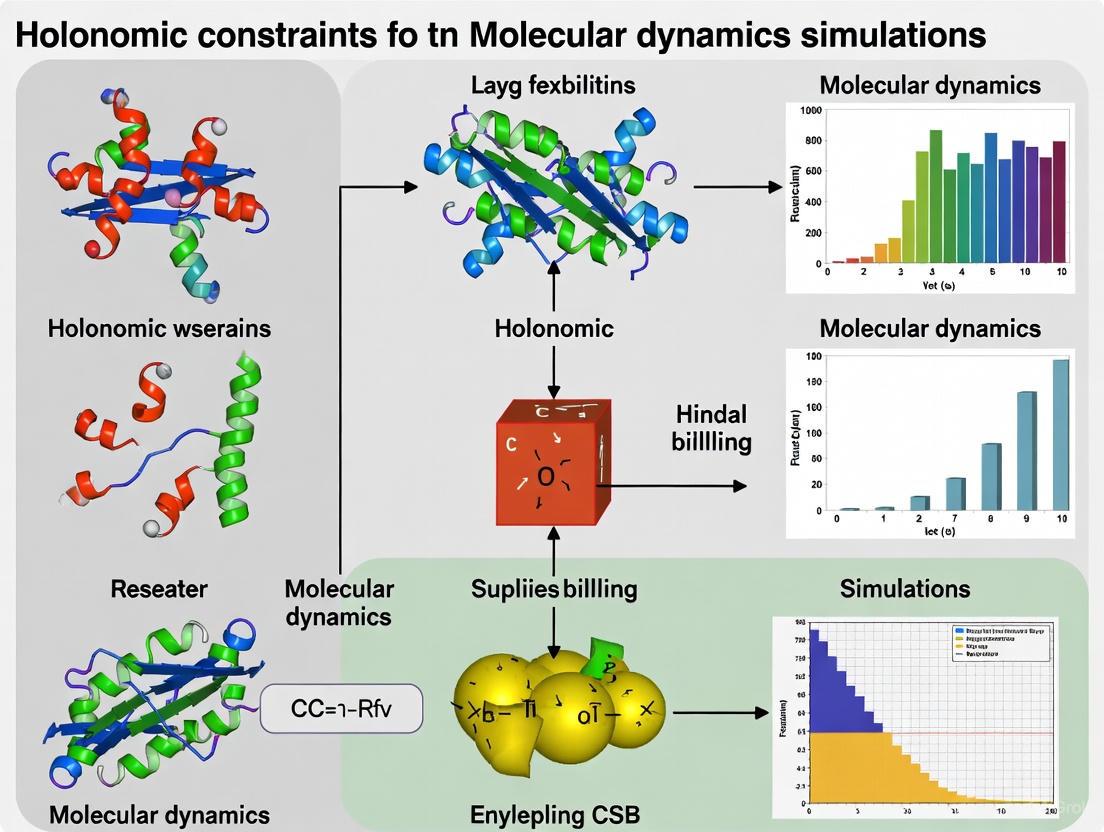
This comprehensive guide explores the fundamental principles and practical implementation of holonomic constraints in molecular dynamics simulations, specifically tailored for researchers and drug development professionals.
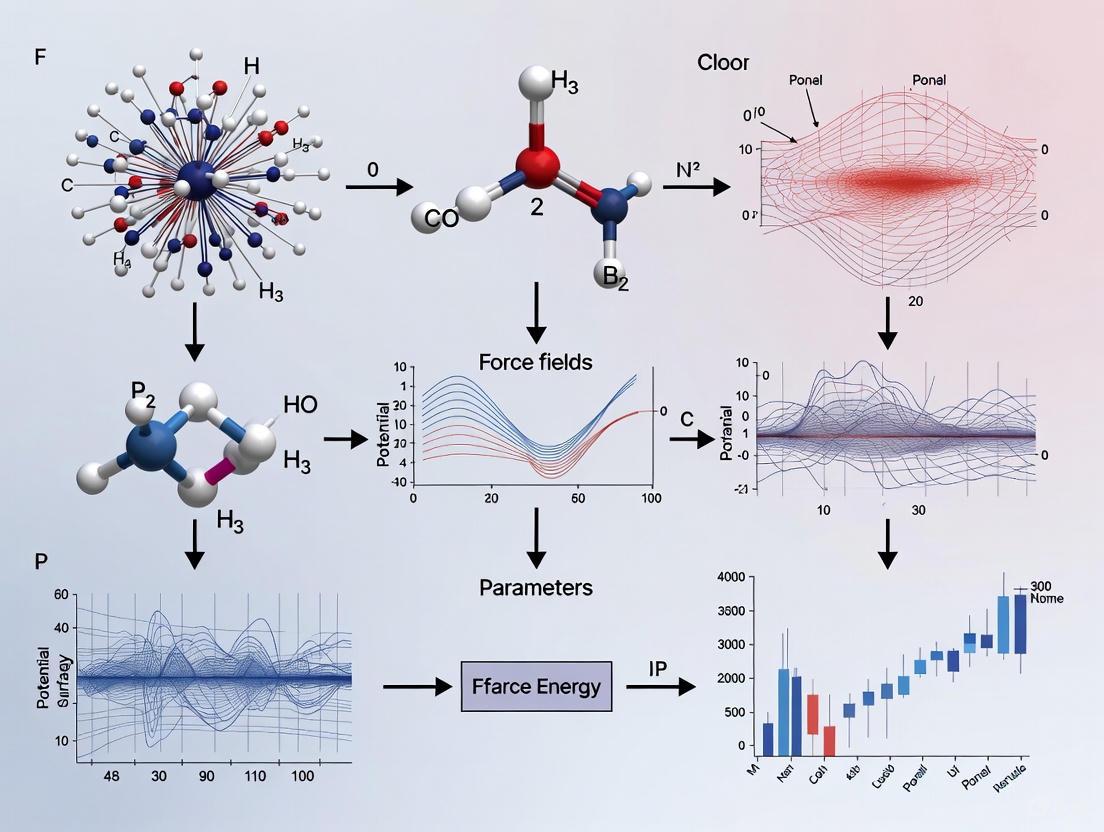
This article provides a comprehensive guide for researchers and drug development professionals on understanding, identifying, and overcoming inaccuracies in molecular force field parameters.
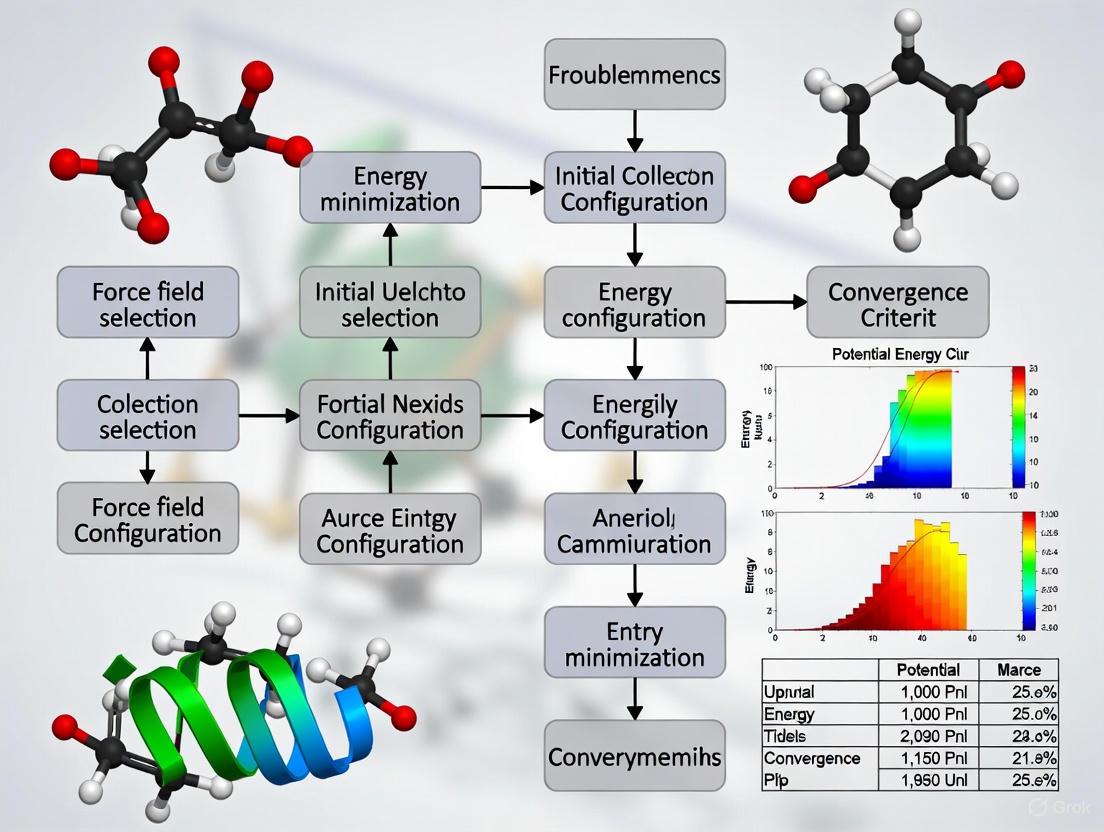
This article provides a comprehensive framework for understanding and resolving common energy minimization failures in Molecular Dynamics (MD) simulations, a critical step in computational drug discovery and biomolecular modeling.
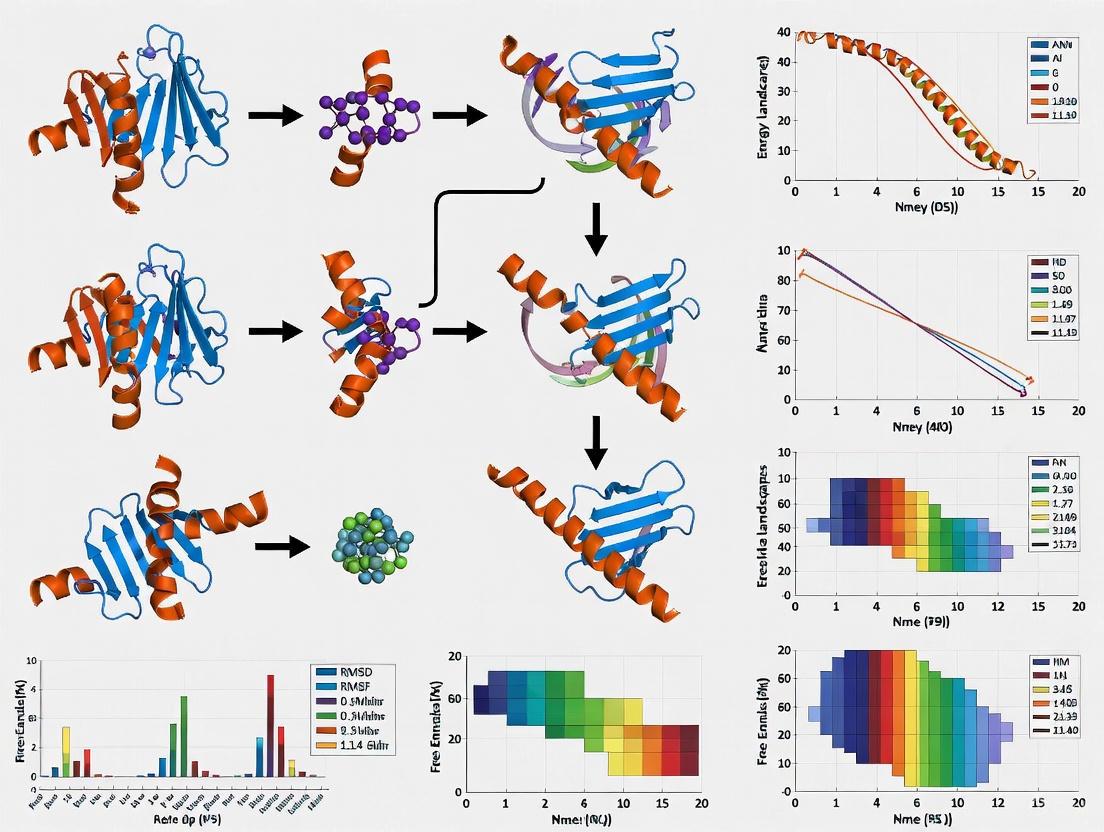
This article provides a comprehensive overview of the application of Molecular Dynamics (MD) simulations in studying biomolecular conformational changes, a critical process in understanding biological function and enabling structure-based drug...
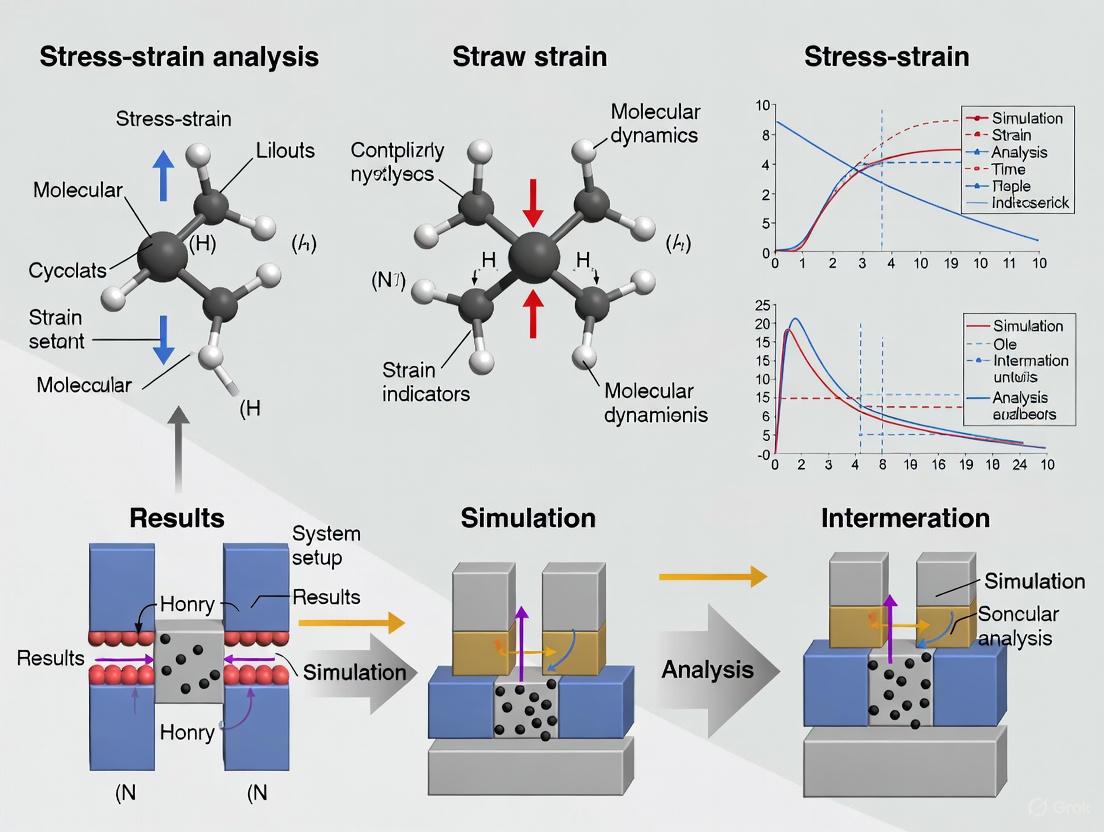
This article provides a comprehensive guide for researchers and scientists on performing stress-strain analysis using Molecular Dynamics (MD) simulations.
Recommended Articles
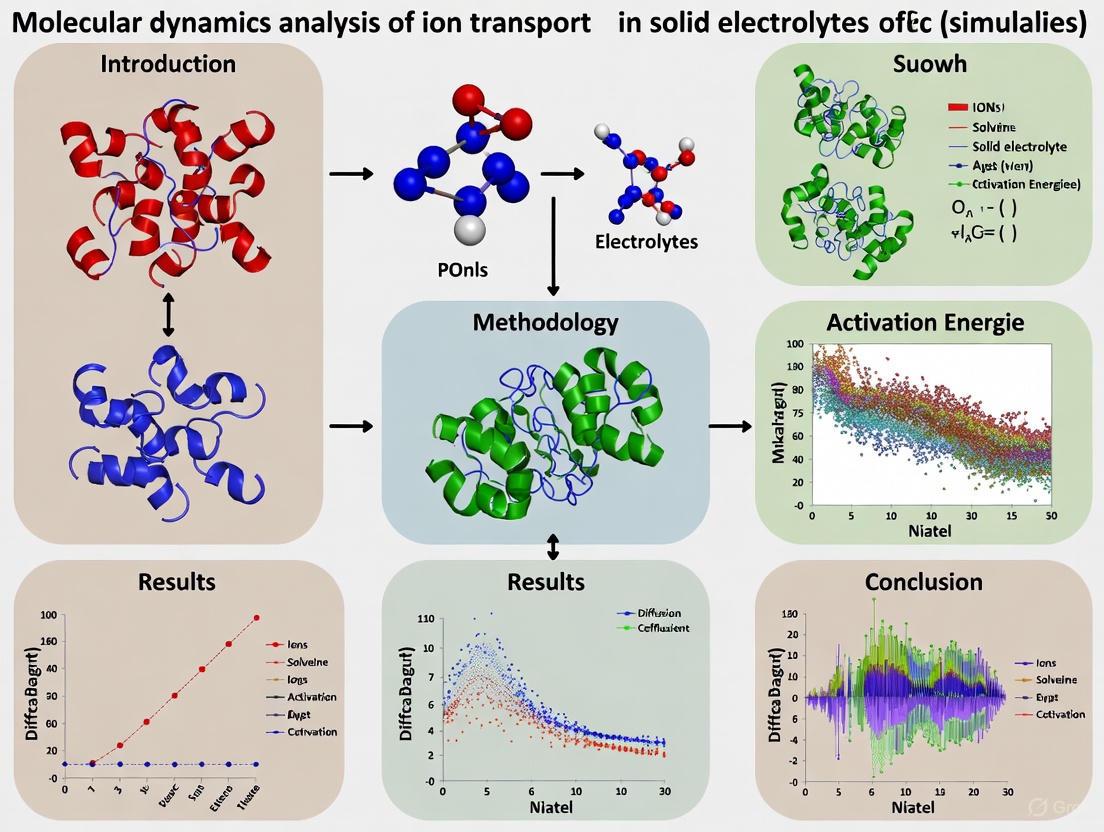
This article provides a comprehensive overview of how Molecular Dynamics (MD) simulations are revolutionizing the understanding and development of solid electrolytes for advanced battery technologies.
This article provides a comprehensive guide for researchers and scientists on performing stress-strain analysis using Molecular Dynamics (MD) simulations.
This article provides a systematic comparison of explicit and implicit solvent models in molecular dynamics (MD) simulations, tailored for researchers and professionals in computational biophysics and drug development.
This article provides a comprehensive overview of Molecular Dynamics (MD) simulations as a pivotal tool for predicting and understanding the mechanical properties of materials.
This article provides a comprehensive guide for researchers and drug development professionals on applying Molecular Dynamics (MD) simulations to refine protein homology models.
This article provides a comprehensive examination of molecular dynamics (MD) integration algorithms, exploring their foundational principles, methodological applications, optimization strategies, and validation frameworks. Tailored for researchers and drug development professionals, it synthesizes current technological advancements including quantum-AI integration, machine learning enhancement, and multi-omics data fusion. Through systematic comparison of classical, statistical, and deep learning-based approaches, we establish practical guidelines for algorithm selection based on dataset characteristics and computational requirements. The analysis addresses critical challenges in force field accuracy, computational scalability, and clinical translation while highlighting emerging opportunities in personalized cancer therapy and accelerated drug screening.

This article provides a comprehensive overview of Molecular Dynamics (MD) simulations, a computational technique that tracks atomic motion over time by solving Newton's equations of motion.
Replica-Exchange Molecular Dynamics (REMD) has emerged as a powerful computational technique to overcome the timescale limitations of conventional molecular dynamics, enabling the study of complex biomolecular processes like protein folding,...
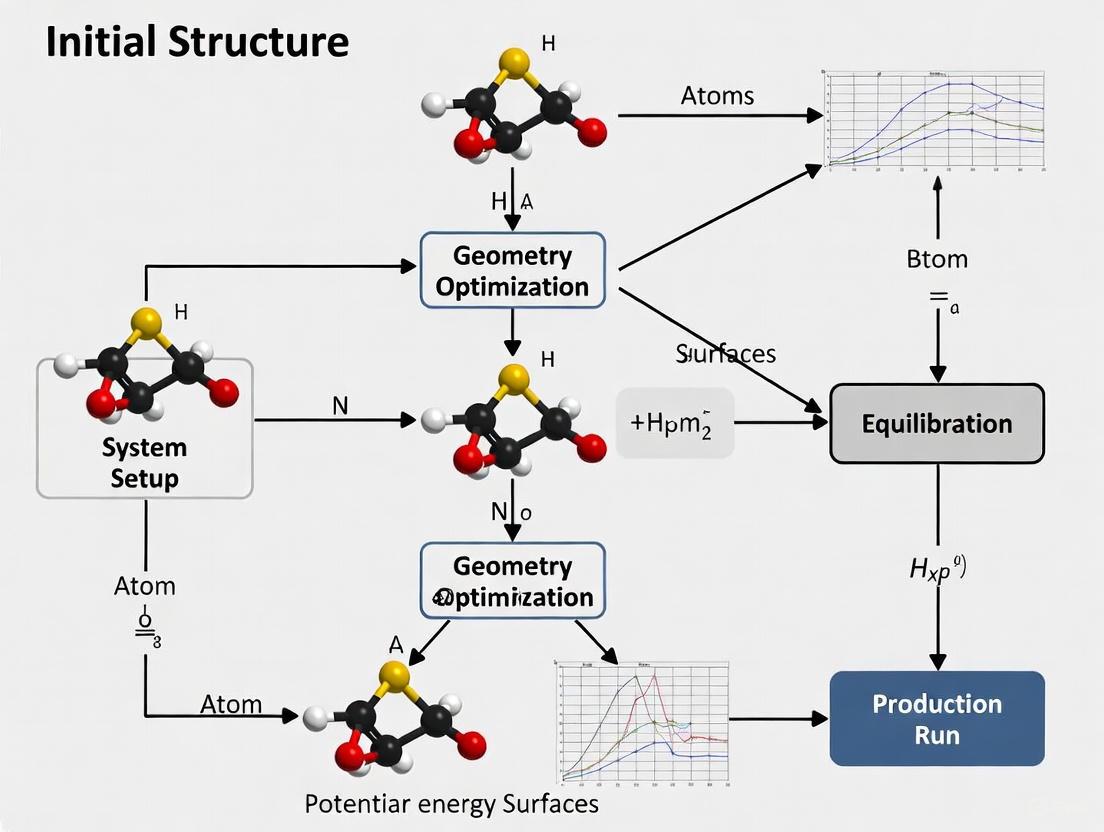
This guide provides a comprehensive framework for researchers and drug development professionals to prepare robust initial structures for Molecular Dynamics (MD) simulations.
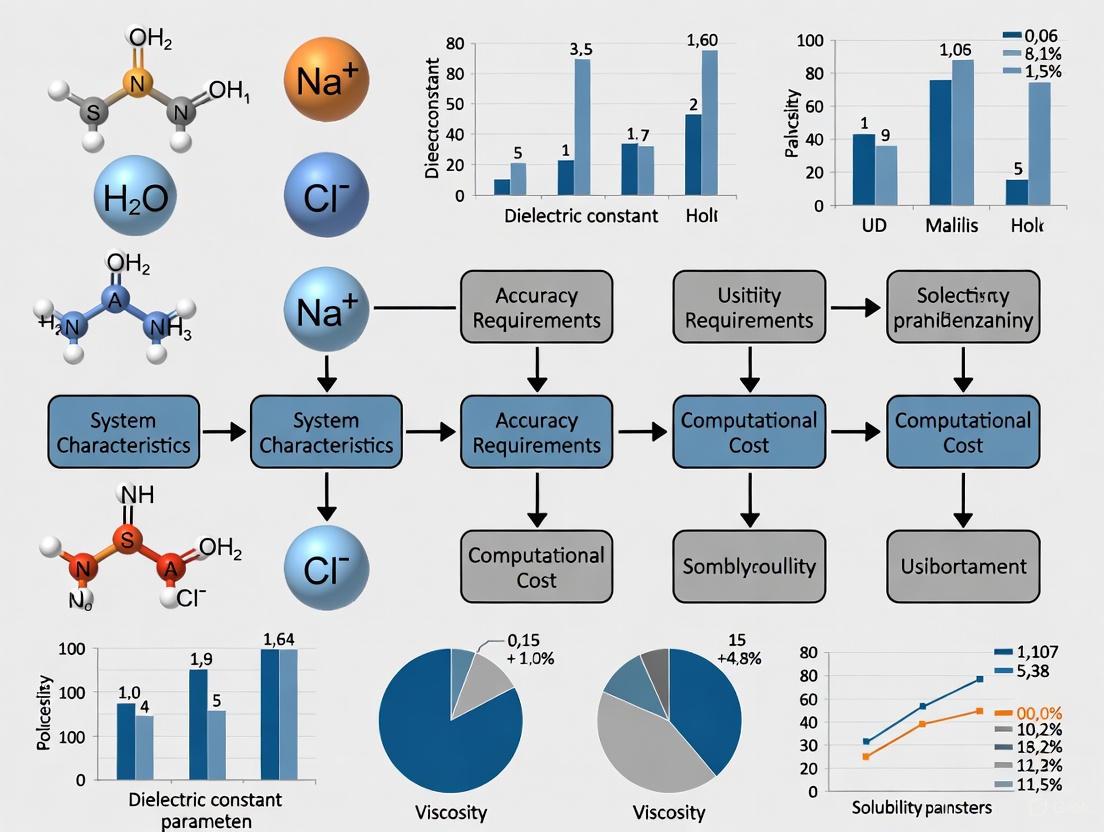
This article provides a comprehensive guide for researchers and drug development professionals on selecting appropriate solvent models for computational studies.
Molecular dynamics (MD) simulation has become an indispensable computational microscope, providing atomic-level insights into biomolecular processes that are often impossible to observe experimentally.
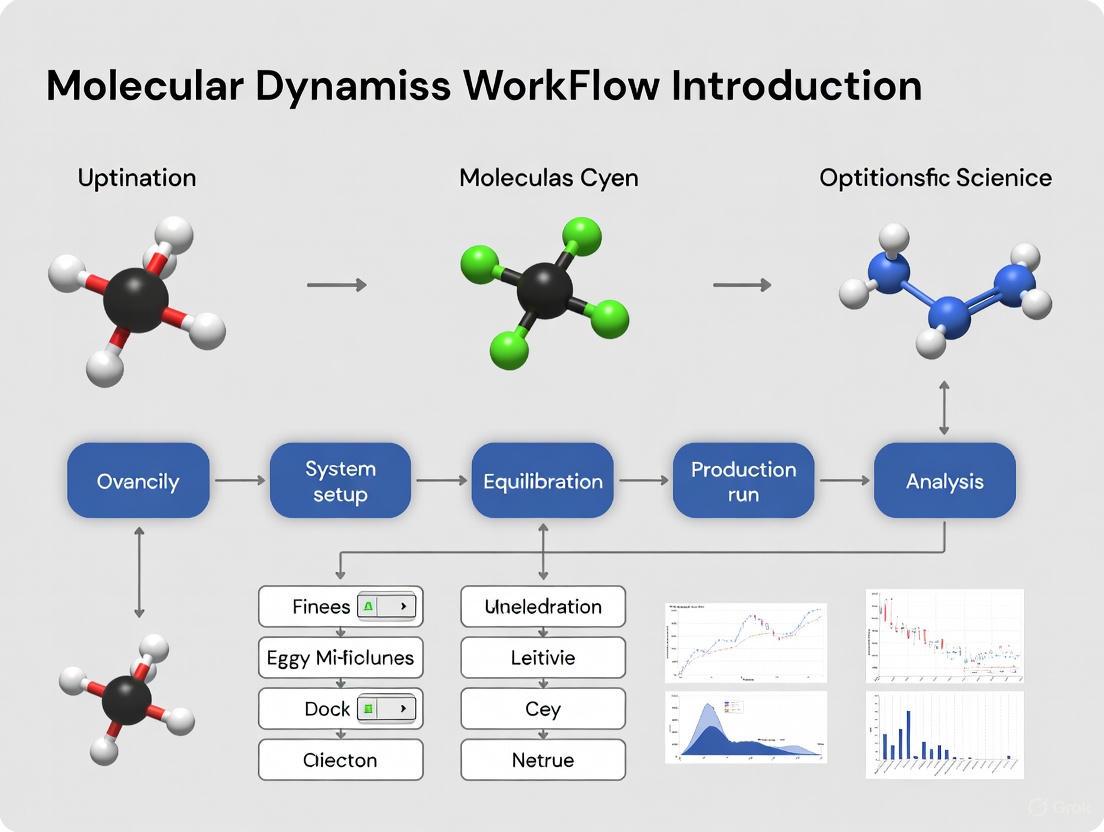
This article provides a comprehensive guide to molecular dynamics (MD) workflows, tailored for researchers and drug development professionals.
Stay at the Forefront of Molecular Science
Get exclusive content, research updates, and simulation tips delivered to your inbox