Explicit vs. Implicit Solvent Models in MD Simulations: A Comprehensive Guide for Computational Researchers
This article provides a systematic comparison of explicit and implicit solvent models in molecular dynamics (MD) simulations, tailored for researchers and professionals in computational biophysics and drug development.
Explicit vs. Implicit Solvent Models in MD Simulations: A Comprehensive Guide for Computational Researchers
Abstract
This article provides a systematic comparison of explicit and implicit solvent models in molecular dynamics (MD) simulations, tailored for researchers and professionals in computational biophysics and drug development. It covers foundational principles, key methodologies like Generalized Born and Poisson-Boltzmann, and practical applications in protein folding and ligand binding. The content addresses common limitations and troubleshooting strategies and presents rigorous validation and performance comparisons, including speedups and accuracy. Finally, it explores hybrid approaches and future directions, offering a balanced perspective for selecting the appropriate solvent model for specific research objectives.
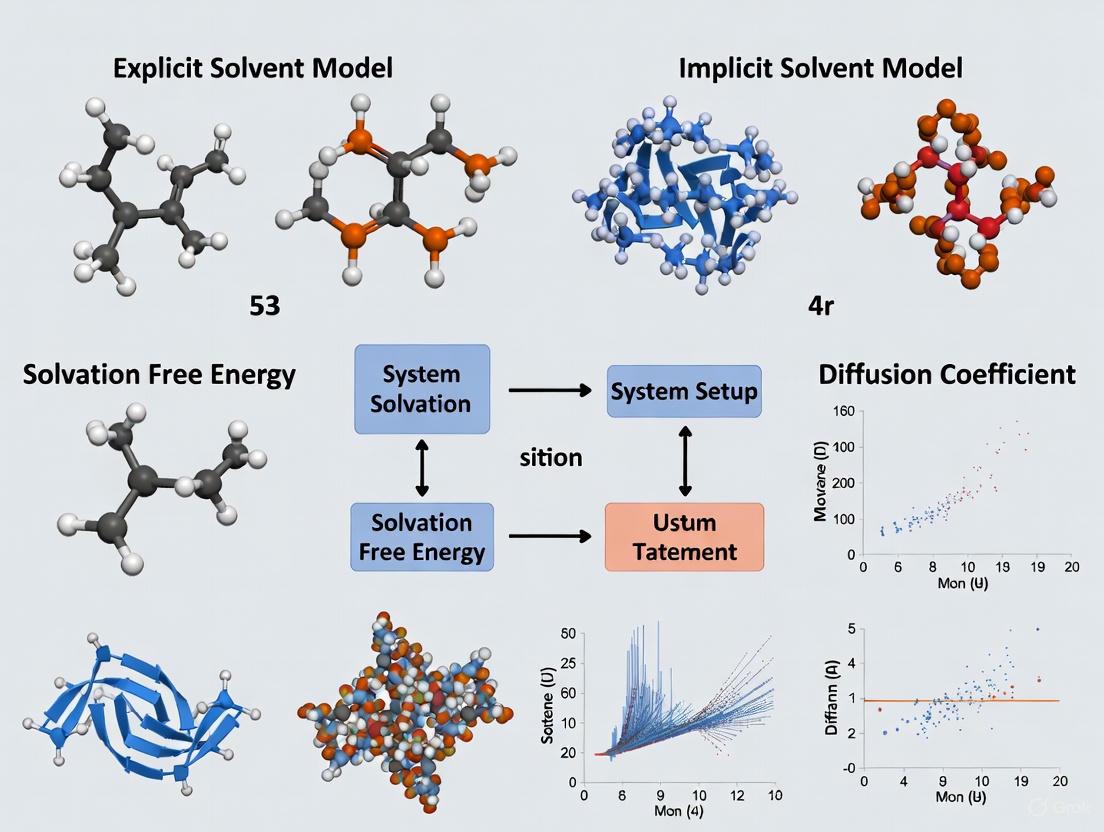
Understanding Solvent Models: From Explicit Water Molecules to Implicit Continuum
In molecular dynamics (MD) research, the choice of how to represent the solvent environment represents a fundamental compromise between computational accuracy and efficiency. Explicit solvent models treat each solvent molecule as a discrete particle, providing an atomistically detailed view of the solvation environment that captures specific molecular interactions, solvent structure, and dynamics with high fidelity. In contrast, implicit solvent models approximate the solvent as a continuous dielectric medium, offering significant computational acceleration at the potential cost of physical realism. This comparison guide objectively examines the performance characteristics, methodological foundations, and practical applications of explicit solvent models alongside emerging hybrid and machine learning approaches, providing researchers and drug development professionals with experimental data to inform their simulation strategies. The ongoing development of methods that bridge the explicit-implicit divide, particularly through machine learning potentials, promises to redefine the boundaries of what is computationally feasible in biomolecular simulation.
Fundamental Definitions and Theoretical Frameworks
Explicit and implicit solvent models operate on fundamentally different physical principles, each with distinct implications for simulation accuracy and computational demand. The core distinction lies in the representation of solvent molecules: explicit models maintain atomic-level detail, while implicit models use continuum approximations.
Explicit solvent models simulate each solvent molecule individually using classical force fields. In biomolecular simulations, the solute (e.g., protein, DNA, or ligand) is immersed in a box of explicit water molecules, typically employing three-site models like TIP3P or four-site models like TIP4P. These models naturally capture molecular details such as hydrogen bonding networks, water-bridged interactions, and solvent entropy contributions that arise from discrete molecular interactions. The particle mesh Ewald (PME) method is commonly employed to handle long-range electrostatic interactions in these explicitly solvated systems [1] [2].
Implicit solvent models, particularly Generalized Born (GB) models, replace discrete solvent molecules with a continuum representation characterized by a dielectric constant. The solvation free energy (ΔGsolv) is typically partitioned into polar (electrostatic) and non-polar components. The polar component is calculated using continuum electrostatics approaches such as Poisson-Boltzmann (PB) or Generalized Born (GB) approximations, while the non-polar contribution is often estimated based on solvent-accessible surface area (SASA) [1] [3]. This simplification dramatically reduces the number of particles in the simulation, but fails to capture specific solute-solvent interactions.
Table 1: Fundamental Characteristics of Explicit and Implicit Solvent Models
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Solvent Representation | Discrete molecules | Dielectric continuum |
| Computational Demand | High | Low |
| Key Physical Interactions | Hydrogen bonding, specific water networks, molecular crowding | Bulk electrostatic screening, surface tension effects |
| Common Implementations | TIP3P, TIP4P water models with PME electrostatics | Generalized Born (GB), Poisson-Boltzmann (PB) |
| Sampling Speed | Limited by solvent viscosity | Accelerated due to reduced friction |
| Handling of Solvent Entropy | Natural, through molecular motions | Approximated empirically |
Quantitative Performance Comparison: Accuracy vs. Efficiency
Systematic comparisons between explicit and implicit solvent approaches reveal a complex performance landscape where the optimal choice depends heavily on the specific biological process under investigation and the computational resources available. The trade-offs between physical accuracy and simulation efficiency follow predictable patterns across different classes of biomolecular systems.
Conformational Sampling Efficiency
The sampling efficiency advantage of implicit solvent models varies significantly depending on the type and scale of conformational change being simulated. A comprehensive comparative study evaluating the particle mesh Ewald (PME) explicit solvent method against a popular Generalized Born (GB) implicit solvent model found speedup factors ranging from approximately 1-fold for small conformational changes (dihedral angle flips) to between 1-100 fold for large conformational changes (nucleosome tail collapse, DNA unwrapping), with approximately 7-fold speedup for mixed cases (miniprotein folding) [1]. This study attributed the accelerated sampling primarily to reduction in solvent viscosity rather than differences in free-energy landscapes between the solvent models. When algorithmic differences are combined with reduced viscosity, the combined speedups were approximately 2-fold, 1-60 fold, and 50-fold for small, large, and mixed conformational changes, respectively [1].
Structural Accuracy and Validation
The ability to recapitulate experimental solvent structures provides a critical validation metric for explicit solvent simulations. Research on endoglucanase crystals compared MD-simulated water positions with those determined by joint X-ray and neutron diffraction. When harmonic restraints were applied to bias the protein toward its crystal structure, the recall of crystallographic waters was excellent—with 98% of top waters within 1.4Å of MD water density peaks. However, unrestrained simulations showed markedly poorer performance, with only 51-62% recall in the same range [2]. This demonstrates that while explicit solvent models can accurately reproduce experimental solvent structure, the accuracy is highly dependent on proper restraint of the biomolecular framework.
Table 2: Quantitative Performance Comparison of Solvent Models
| Performance Metric | Explicit Solvent (PME/TIP3P) | Implicit Solvent (GB) |
|---|---|---|
| Sampling Speed (Relative) | 1x (baseline) | 1-100x (system-dependent) |
| Crystallographic Water Recall | 98% (restrained) | Not applicable |
| Computational Cost for Small Systems | Lower due to PME overhead | Higher (algorithmic advantage) |
| Computational Cost for Large Systems | Higher (scales with solvent atoms) | Lower (algorithmic disadvantage) |
| Free Energy Landscape Accuracy | Gold standard for specific interactions | May differ substantially from explicit |
Experimental Protocols and Methodologies
Crystalline Molecular Dynamics Validation Protocol
The validation of explicit solvent models against experimental crystallographic data follows a rigorous protocol designed to maximize structural accuracy while maintaining computational feasibility:
System Preparation: The protein structure is derived from high-resolution joint X-ray and neutron diffraction data (e.g., 1.0Å X-ray, 1.5Å neutron), enabling experimentally assigned protonation states. Protonation states for exchangeable sites are modeled using neutron scattering density and chemical environment analysis [2].
Solvation and Ion Placement: The crystalline system is solvated within a 2×2×2 supercell with explicit solvent molecules. Counterions are added to neutralize the system, with additional ions included to match experimental mother liquor concentration (e.g., 50 mM). Alternative solvent models may include specific buffers like Tris-Cl [2].
Force Field Parameterization: Protein structures are parameterized using specialized force fields (e.g., amber99sb-ildn), with water modeled using TIP3P or other explicit water models. Ligands are parameterized using appropriate tools (e.g., GLYCAM for carbohydrates) [2].
Simulation with Restraints: Harmonic restraints are applied to protein non-hydrogen atoms to maintain the experimental crystal structure while allowing solvent dynamics. Simulations are typically run for tens of nanoseconds with coordinates recorded for analysis [2].
Water Density Analysis: Water scattering density maps are computed from MD trajectories using standard macromolecular crystallography methods. Peaks in MD water electron density are compared to crystallographic water positions with recall measured at various distance thresholds (0.5Å, 1.0Å, 1.4Å) [2].
This protocol demonstrates that with appropriate experimental restraints, explicit solvent models can successfully recover crystallographic water structures, establishing a benchmark for solvent model validation.
Machine Learning Potentials for Explicit Solvent Systems
Emerging machine learning approaches offer promising pathways to maintain explicit solvent accuracy while reducing computational cost. The active learning strategy for generating machine learning potentials (MLPs) for chemical processes in explicit solvents employs a sophisticated workflow:
Initial Data Generation: Create small training sets of configurations labeled with reference energies and forces. For reactions, initial training typically starts from transition state structures. Explicit solvent configurations are generated using cluster models with sufficient solvent molecules to cover the MLP's cut-off radius [4].
Active Learning Loop:
- Train initial MLP on the starting dataset
- Perform short MD simulations using the current MLP
- Evaluate structures using descriptor-based selectors (e.g., Smooth Overlap of Atomic Positions - SOAP)
- Select underrepresented structures for retraining
- Iteratively expand the training set based on chemical space coverage [4]
Selector Implementation: Descriptor-based selectors analyze the SOAP descriptor space to identify regions poorly represented in the training set, providing a general metric applicable across different MLP approaches at low computational cost compared to committee-based uncertainty estimation [4].
This strategy has been successfully applied to study Diels-Alder reactions in water and methanol, demonstrating the ability to obtain reaction rates in agreement with experimental data while capturing explicit solvent effects [4].
Emerging Paradigms: Machine Learning and Quantum Approaches
The distinction between explicit and implicit solvent modeling is being redefined by machine learning approaches that capture explicit solvent accuracy with reduced computational overhead. Neural network potentials (NNPs) trained on massive quantum chemical datasets represent a particularly promising development. Meta's Open Molecules 2025 (OMol25) dataset contains over 100 million calculations at the ωB97M-V/def2-TZVPD level of theory, spanning biomolecules, electrolytes, and metal complexes [5]. Models trained on this dataset, such as the equivariant Smooth Energy Network (eSEN) and Universal Model for Atoms (UMA), achieve accuracy comparable to high-level density functional theory while enabling simulations of large systems that were previously computationally prohibitive [5].
These ML-based approaches demonstrate surprising capability in predicting charge-related properties despite not explicitly modeling Coulombic physics. In benchmarks of reduction potential and electron affinity predictions, OMol25-trained NNPs performed comparably or better than low-cost DFT and semiempirical methods for organometallic species, though with more variable performance on main-group species [6]. This suggests that ML models can learn complex solvation effects implicitly from training data, potentially bypassing the need for explicit physics-based modeling of certain interactions.
Another innovative approach bridges the gap between explicit and implicit modeling through graph neural networks that incorporate alchemical variables. The λ-Solvation Neural Network (LSNN) extends traditional force-matching by incorporating derivatives with respect to electrostatic and steric coupling factors (λelec and λsteric), enabling accurate solvation free energy predictions while maintaining computational efficiency comparable to implicit solvent models [7].
Table 3: Essential Research Tools for Solvation Model Development and Validation
| Tool/Resource | Type | Function/Purpose | Example Implementations |
|---|---|---|---|
| Explicit Water Models | Force Field Parameters | Represent water molecules with atomic detail | TIP3P, TIP4P, OPC |
| Continuum Solvent Models | Implicit Solvation | Approximate solvent as dielectric continuum | Generalized Born (GB), Poisson-Boltzmann (PB) |
| Neural Network Potentials | Machine Learning Models | Learn potential energy surfaces from QM data | eSEN, UMA, ANI, MACE |
| Quantum Chemical Datasets | Training Data | Provide reference data for ML potential development | OMol25, SPICE, ANI-2x |
| Crystalline MD Protocols | Validation Methodology | Validate solvent models against experimental data | Joint X-ray/neutron refinement |
| Active Learning Frameworks | Sampling Algorithm | Efficiently explore chemical space for ML training | SOAP descriptor-based selectors |
Explicit solvent models remain the gold standard for simulating specific solute-solvent interactions, water-mediated processes, and accurately reproducing experimental solvent structures, particularly when validated against crystallographic data. The computational cost of these models continues to motivate the development of implicit solvent approaches for enhanced sampling and large-scale simulations. However, the emerging paradigm of machine learning potentials trained on extensive quantum chemical datasets promises to transcend the traditional explicit-implicit dichotomy, offering near-quantum accuracy with dramatically reduced computational cost. For researchers and drug development professionals, the optimal choice of solvent model depends critically on the specific scientific question, with explicit models providing unparalleled detail for mechanistic studies and ML-based approaches enabling previously intractable simulations of complex biomolecular processes in their native aqueous environments.
In computational chemistry and biophysics, accurately modeling the solvent environment is essential for realistic simulations of biomolecules, as most biological processes occur in aqueous solution [8] [3]. Two primary methodologies have emerged to address this challenge: explicit solvent models, which treat each solvent molecule as a discrete particle, and implicit solvent models, which replace the explicit solvent with a continuous, polarizable medium [9]. Implicit solvent models, the subject of this guide, offer a computationally efficient framework by approximating the average effect of the solvent on the solute [10]. This approach significantly reduces the number of particles in a simulation system, lowering computational cost and accelerating conformational sampling compared to explicit solvent simulations [11] [10]. The core idea of implicit solvation is to forgo the atomistic detail of individual solvent molecules in favor of a continuum representation described by macroscopic properties, primarily its dielectric constant [9]. This guide provides a comprehensive comparison of implicit solvent models against explicit and hybrid alternatives, detailing their theoretical foundations, performance data, methodological protocols, and practical applications in modern molecular research.
Theoretical Foundations of Implicit Solvation
The conceptual foundation of implicit solvent models rests on the partitioning of the solvation free energy into physically meaningful components [3]. Typically, the total solvation free energy (ΔG_solv) is decomposed into a polar (electrostatic) component and a non-polar component [12] [3].
The polar component (ΔGele) accounts for the interaction of the solute's charge distribution with the dielectric environment. This is typically computed by solving the Poisson-Boltzmann (PB) equation or its efficient approximation, the Generalized Born (GB) model [12] [3]. The non-polar component (ΔGnp) describes the free energy cost of forming a cavity in the solvent to accommodate the solute, along with van der Waals interactions [12]. This term is often modeled as being proportional to the Solvent-Accessible Surface Area (SASA) [8] [12]. Thus, the total solvation free energy can be approximated as: ΔGsolv ≈ ΔGGB + γ × SASA [7]
Where ΔG_GB is the polar Generalized Born energy and γ is a surface tension parameter [7]. This combination is widely known as the GBSA model. The following diagram illustrates the core conceptual relationship between these energy components and the overall solvation process.
Several popular implicit solvent models have been developed based on these principles. The Polarizable Continuum Model (PCM) and its variants, such as the Conductor-like Screening Model (COSMO) and the Solvation Model based on Density (SMD), are widely used in quantum chemistry calculations to describe solvation effects on electronic structure [8] [9] [3]. In classical molecular dynamics simulations, the GB model coupled with a SASA term (GBSA) is particularly popular due to its favorable balance of efficiency and accuracy [11] [10].
Performance Comparison: Implicit vs. Explicit Solvation
While implicit solvent models offer significant computational advantages, their accuracy in reproducing experimental data and explicit solvent benchmarks is a critical consideration. The table below summarizes key performance metrics for various solvation approaches, highlighting their respective strengths and limitations.
Table 1: Performance Comparison of Solvation Methods
| Solvation Method | Computational Cost | Key Accuracy Metrics | Best-Suited Applications |
|---|---|---|---|
| Implicit Solvent (GBSA) [11] [10] | Low to Moderate | RNA stem-loop folding: Stem region RMSD <2 Å, loop region RMSD ~4 Å [11] | Protein folding simulations [11]; Long-timescale conformational sampling [10] |
| Explicit Solvent (TIP3P) [7] | Very High | Considered the "gold standard" for solvation energy calculations [7] | Detailed study of specific solute-solvent interactions; Validation of other models [9] |
| Explicit Solvent (IRS Method) [8] | High | Predictive accuracy comparable to SMD; Superior to PB/GBSA [8] | Accurate solvation energy calculations where explicit water effects are critical [8] |
| Machine Learning (ML-PCM) [13] | Low (after training) | Mean Unsigned Error (MUE) of 0.40-0.52 kcal/mol for solvation free energies [13] | High-throughput screening; Fast and accurate solvation energy predictions [13] |
| Machine Learning (LSNN) [7] | Low (after training) | Free energy predictions with accuracy comparable to explicit solvent [7] | Drug discovery; Free energy calculations where both speed and accuracy are needed [7] |
The computational efficiency of implicit solvent models is their most significant advantage. By drastically reducing the number of particles in a simulation system, they lower computational cost and, due to the absence of solvent viscosity, can accelerate conformational sampling compared to explicit solvent simulations [11] [10]. However, this efficiency can come at the cost of limited accuracy in capturing specific, atomistic solvent effects. For instance, in RNA stem-loop folding simulations, while implicit solvents (GB-neck2) successfully folded stem regions with high accuracy (RMSD <2 Å), the modeling of loop structures was less precise (RMSD ~4 Å), indicating a limitation in capturing the fine structural details often stabilized by specific solvent interactions [11].
Quantitative accuracy for solvation free energy prediction has seen remarkable improvements with machine-learning-augmented implicit models. The ML-PCM model improves the accuracy of widely accepted continuum solvation models by almost an order of magnitude, achieving a Mean Unsigned Error (MUE) of 0.40-0.52 kcal/mol compared to experimental data [13]. Similarly, the graph neural network-based LSNN model achieves free energy predictions with accuracy comparable to explicit-solvent alchemical simulations but at a much lower computational cost [7].
Experimental Protocols and Methodologies
Protocol for Implicit Solvent MD Simulation (GBSA)
The following workflow outlines a typical protocol for running molecular dynamics simulations using an implicit solvent model, as applied in studies of RNA stem-loop folding [11]:
- System Preparation: Start with an initial solute structure (e.g., an extended RNA conformation).
- Force Field Selection: Employ a refined atomistic force field (e.g., the DESRES-RNA force field).
- Implicit Solvent Model Selection: Apply a modern Generalized Born model (e.g., GB-neck2 with parameters refined for nucleic acids).
- Energy Minimization: Perform initial energy minimization to remove steric clashes.
- Equilibration and Production MD: Run conventional MD simulations at the target temperature (e.g., near the predicted melting temperature).
- Analysis: Calculate Root Mean Square Deviation (RMSD) of the resulting structures (e.g., for stem and loop regions separately) against experimental reference structures [11].
Protocol for Explicit-Solvent IRS Solvation Energy Calculation
The Interaction-Reorganization Solvation (IRS) method is an explicit solvent approach for calculating molecular solvation energies, providing a useful benchmark for implicit model performance [8].
- Simulation Setup: Generate molecular dynamics simulations of the solute molecule in explicit solvent.
- Conformational Sampling: Create an extensive conformational space of solute conformations in solution.
- Energy Decomposition: Decompose the solvation free energy into two terms:
- Solute-Solvent Interaction Free Energy (ΔGint): Computed directly from MD simulations as the sum of electrostatic (Eele) and van der Waals (Evdw) interactions between solute and solvent atoms [8].
- Reorganization Free Energy (ΔGreo): Derived by fitting a polynomial expansion of the interaction term along with cavitation parameters (e.g., SASA) from a training set of experimental solvation energies [8].
- Validation: Test the fitted model on a separate validation set to confirm predictive accuracy [8].
Protocol for Machine Learning Model Training (ML-PCM)
Machine learning models like ML-PCM are trained to correct or enhance traditional implicit solvent models [13].
- Data Set Curation: Utilize a large data set of experimental solvation free energies for diverse molecules.
- Feature Selection: Use SCRF energy components and solvation free energies computed from a baseline continuum model (e.g., CPCM) as input features.
- Model Training: Train a neural network to map the input features to the experimental solvation free energies.
- Model Validation: Screen appropriately trained models via a rigorous post-validation strategy to select the best-performing one (e.g., with the lowest MUE) [13].
The Scientist's Toolkit: Essential Research Reagents & Solutions
Successful implementation and application of solvation models require a suite of computational tools and parameters. The following table details key resources mentioned in the cited research.
Table 2: Key Computational Reagents for Solvation Modeling
| Research Reagent / Parameter | Function / Purpose | Example Usage Context |
|---|---|---|
| Generalized Born (GB) Model [11] [10] [12] | Efficiently approximates the electrostatic component of solvation energy. | Core of GBSA implicit solvent for MD simulations [11] [10]. |
| Solvent-Accessible Surface Area (SASA) [8] [12] | Models the non-polar contribution to solvation energy (cavity formation + van der Waals). | Used in GBSA and as a descriptor in the IRS method [8]. |
| GB-neck2 Parameters [11] | A specific, refined set of GB parameters for nucleic acids. | Enables accurate folding of RNA hairpins in implicit solvent MD [11]. |
| DESRES-RNA Force Field [11] | An extensively refined atomistic force field for RNA molecules. | Provides accurate energetics for base stacking and pairing in simulations [11]. |
| AMBER-OL3 Force Field [11] | A standard classical force field for RNA systems in AMBER MD software. | The baseline force field upon which several refinements are built [11]. |
| Machine-Learned Potentials (MLPs) [14] | Surrogates for quantum chemistry methods, offering first-principles accuracy at reduced cost. | Used for solvation-aware atomistic modeling of complex systems [14]. |
| Graph Neural Network (GNN) [7] | A type of neural network architecture that operates on graph-structured data. | Core of the LSNN model for predicting solvation free energies [7]. |
| Alchemical Coupling Parameters (λelec, λsteric) [7] | Variables that scale interaction energies in free energy calculations. | Used in training the LSNN model to enable accurate absolute free energy comparisons [7]. |
Implicit solvent models represent a powerful compromise between computational efficiency and physical realism, enabling the study of biomolecular processes that would be prohibitively expensive with explicit solvent [10] [3]. The continuum medium approximation excels at capturing bulk solvent effects, particularly those related to polarity, making it suitable for protein folding, conformational sampling, and rapid screening [11] [10].
However, the choice between implicit and explicit models remains application-dependent. Implicit models are ideal for large-scale conformational searches, long-time dynamics, and applications requiring high computational throughput. Explicit models are necessary when investigating processes where atomistic details of solvent structure (e.g., specific hydrogen bonds, water bridges, or ion coordination) are critical [9].
The future of solvation modeling lies in hybrid approaches and machine learning augmentation. ML-corrected implicit models like ML-PCM and LSNN demonstrate that it is possible to achieve near-explicit solvent accuracy at implicit solvent speeds [13] [7]. Furthermore, integrating a few explicit solvent molecules within a continuum bath (the cluster-continuum approach) can capture specific interactions while maintaining much of the efficiency of pure implicit models [9]. As these hybrid and machine-learning techniques mature, they will further blur the lines between the two paradigms, offering researchers an increasingly powerful and accurate toolkit for modeling the complex role of solvation in molecular phenomena.
The free energy of solvation and the potential of mean force (PMF) are cornerstone concepts in computational biophysics and drug design. The free energy of solvation quantifies the thermodynamic work required to transfer a molecule from the gas phase into a solvent, a property critically important for predicting drug solubility, membrane permeability, and binding affinities [3]. The potential of mean force, in turn, provides a statistical mechanical description of the effective energy landscape along a reaction coordinate, accounting for the averaged influence of all other degrees of freedom, particularly the solvent [7].
The calculation of these properties in molecular dynamics (MD) simulations hinges on the treatment of the solvent environment. Explicit solvent models represent solvent molecules individually, offering high fidelity but at a great computational cost. Implicit solvent models treat the solvent as a continuous dielectric medium, offering speed but potentially sacrificing accuracy in capturing specific molecular details [3]. This guide provides an objective comparison of these approaches, detailing their theoretical foundations, performance, and practical applications in modern research.
Theoretical Foundations
Free Energy of Solvation
The solvation free energy (( \Delta G_{\text{solv}} )) is a fundamental thermodynamic quantity that can be partitioned into contributions from different physical interactions. A common framework separates it into polar (electrostatic) and non-polar components [3]:
[ \Delta G{\text{solv}} = \Delta G{\text{ele}} + \Delta G_{\text{np}} ]
The polar component (( \Delta G{\text{ele}} )) arises from the interaction between the solute's charge distribution and the dielectric solvent environment. The non-polar component (( \Delta G{\text{np}} )) encompasses the free energy cost of forming a cavity in the solvent to accommodate the solute and the van der Waals interactions between the solute and solvent. This non-polar term is often further decomposed [3]:
[ \Delta G{\text{solv}} = \Delta G{\text{cav}} + \Delta G{\text{ele}} + \Delta G{\text{vdW}} ]
where ( \Delta G{\text{cav}} ) is the cavity formation energy and ( \Delta G{\text{vdW}} ) represents the van der Waals dispersion interactions.
Potential of Mean Force (PMF)
The Potential of Mean Force provides a statistical description of the effective energy along a chosen reaction coordinate. Formally, for a solute configuration ( \vec{r}A ), the solvation PMF (( \Delta W(\vec{r}A, T) )) is defined by integrating out all solvent degrees of freedom (( \vec{r}_S )) [15]:
[ \exp(-\beta \Delta W(\vec{r}A, T)) = \frac{\int \exp(-\beta (U{AS}(\vec{r}A, \vec{r}S) + US(\vec{r}S))) d\vec{r}S}{\int \exp(-\beta US(\vec{r}S)) d\vec{r}S} ]
Here, ( U{AS} ) represents the solute-solvent interaction energy, ( US ) is the solvent-solvent potential, and ( \beta = 1/k_B T ). The PMF thus encapsulates the averaged influence of the solvent on the solute, and its gradient gives the mean force acting on the solute atoms.
Comparative Analysis: Explicit vs. Implicit Solvent Models
Table 1: Comparison of Explicit and Implicit Solvent Models for Free Energy Calculations.
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Fundamental Approach | Discrete representation of individual solvent molecules [3] | Continuum dielectric medium representing average solvent behavior [3] |
| Computational Demand | High; majority of computation spent on solvent-solvent interactions [7] | Low; no solvent degrees of freedom to simulate [3] |
| Solvation Free Energy Components | Emerges naturally from explicit solute-solvent interactions | Separated into polar (e.g., GB/PB) and non-polar (e.g., SASA) terms [7] [3] |
| Treatment of Solvent Entropy | Inherently included but difficult to quantify [15] | Implicitly included in the solvation potential [15] |
| Handling of Specific Solvent Effects | Excellent for specific interactions (e.g., hydrogen bonds, water bridges) [16] | Poor; cannot capture specific, local solvent structuring [3] |
| Performance in Solvation Free Energy Prediction | Generally higher accuracy but can be forcefield-dependent [17] [16] | Often lower agreement with experiment (e.g., RMSD ~15 kJ/mol for organic solvents) [17] |
| Performance in Partitioning (LogP) | Good, often due to error cancellation [16] | Varies; SMD model can show good agreement [17] |
Performance and Limitations
Studies indicate that for calculating solvation free energies in organic solvents, implicit models like Poisson-Boltzmann (PB) and Generalized Born (GB) can show poor agreement with both explicit solvent calculations and experimental data, with root-mean-square deviations of approximately 15 kJ/mol [17]. A key identified weakness is the prediction of the apolar contribution, which involves complex solvent entropy effects [17].
Explicit solvent models, while accurate, are computationally intensive. Their accuracy is also sensitive to the empirical forcefield used, particularly the fixed-charge models for electrostatic interactions, which do not account for geometry-dependent polarization [18] [16]. For properties like octanol-water transfer free energies (LogP), explicit solvent methods can achieve remarkable accuracy (e.g., mean unsigned error < 1 kcal/mol with the ABCG2 charge model), often benefiting from systematic error cancellation between different environments [16].
Experimental Protocols and Methodologies
Alchemical Free Energy Calculations with Explicit Solvent
Alchemical transformations are a standard explicit-solvent approach for calculating rigorous free energy differences. These methods leverage the fact that free energy is a state function, using an artificial pathway to connect the states of interest [18].
Core Protocol:
- Hamiltonian Definition: A hybrid Hamiltonian ( H(\vec{r}, \lambda) ) is defined as a linear combination of the Hamiltonians of the two end states (e.g., solute in solvent vs. solute in vacuum), controlled by an alchemical parameter ( \lambda ) that ranges from 0 to 1 [18]: [ H(\vec{r}, \lambda) = \lambda H1(\vec{r}) + (1-\lambda) H0(\vec{r}) ]
- Softcore Potentials: To avoid numerical singularities when atoms are decoupled, softcore potentials are used to modify the nonbonded interactions. A common form for the softcore Lennard-Jones potential is [18]: [ U(\lambda,r)=4\epsilon\lambda^n \left[ \left( \alpha{LJ}(1-\lambda)^m + \left( \frac{r}{\sigma} \right)^6 \right)^{-2} - \left( \alpha{LJ}(1-\lambda)^m + \left( \frac{r}{\sigma} \right)^6 \right)^{-1} \right] ] where ( \alpha_{LJ}, m, n ) are tunable softening parameters.
- Free Energy Estimation: The free energy difference is computed by sampling at multiple ( \lambda ) values. Using Thermodynamic Integration (TI), the free energy is calculated as [18]: [ \Delta G = \int0^1 \left\langle \frac{\partial H(\vec{r}, \lambda)}{\partial \lambda} \right\rangle\lambda d\lambda ]
Diagram 1: Alchemical free energy calculation workflow.
Machine Learning-Augmented Implicit Solvent Models
Recent advancements aim to overcome the accuracy limitations of traditional implicit models by using machine learning.
Core Protocol (LSNN Model):
- Model Architecture: A Graph Neural Network (GNN) is used to represent the implicit solvent potential. The model takes atomic coordinates, charges, and other atomistic features as input [7].
- Training Procedure: The key innovation is to move beyond simple force-matching. The model is trained by minimizing a loss function that includes derivatives with respect to both atomic coordinates and alchemical coupling parameters (( \lambda{\text{elec}} ) and ( \lambda{\text{steric}} )) [7]: [ \mathcal{L} = wF \left( \left\langle \frac{\partial U{\text{solv}}}{\partial \mathbf{r}i} \right\rangle - \frac{\partial f}{\partial \mathbf{r}i} \right)^2 + w{\text{elec}} \left( \left\langle \frac{\partial U{\text{solv}}}{\partial \lambda{\text{elec}}} \right\rangle - \frac{\partial f}{\partial \lambda{\text{elec}}} \right)^2 + w{\text{steric}} \left( \left\langle \frac{\partial U{\text{solv}}}{\partial \lambda{\text{steric}}} \right\rangle - \frac{\partial f}{\partial \lambda{\text{steric}}} \right)^2 ] This ensures the model learns a potential that is consistent with both structural forces and free energy changes, making it suitable for PMF calculations.
- Free Energy Prediction: The trained model can be used to rapidly compute solvation free energies and conformational landscapes, offering accuracy near explicit solvent levels at a fraction of the cost [7].
Diagram 2: Machine learning-implicit solvent model development.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Methods for Solvation Free Energy Calculations.
| Tool/Method | Type | Primary Function | Key Considerations |
|---|---|---|---|
| GBSA/MMPBSA [15] | Implicit Solvent Model | Estimates free energies from MD snapshots by combining Generalized Born/Poisson-Boltzmann electrostatics with a Surface Area term. | Computationally efficient but accuracy is limited by the SASA approximation for non-polar contributions [17]. |
| Thermodynamic Integration (TI) [18] | Explicit Solvent Method | Calculates free energy differences by integrating the ensemble-averaged derivative of the Hamiltonian along an alchemical path. | Considered a rigorous "gold standard" but requires significant sampling at multiple λ windows [18]. |
| Alchemical Transfer Method (ATM) [18] | Explicit Solvent Method | A non-alchemical alternative that uses coordinate transformation to interpolate between two physical end states. | Can be easier to set up and is compatible with ML potentials [18]. |
| ABCG2 Charge Model [16] | Fixed-Charge Parametrization | An empirical protocol for deriving atomic partial charges for small molecules in forcefields. | Outperforms older models (AM1/BCC) and can achieve accuracy near costly QM/MM methods for LogP [16]. |
| Machine Learned Potentials (MLPs) [18] [7] | Potential Energy Model | Learns a quantum-mechanically accurate potential energy surface from data, used as a replacement for empirical forcefields. | High accuracy but computationally expensive; requires specialized free energy methods [18]. |
| λ-LSNN [7] | ML-Augmented Implicit Model | A graph neural network trained to predict forces and free energy derivatives for fast and accurate solvation calculations. | Aims to bridge the speed of implicit models with the accuracy of explicit ones [7]. |
The choice between explicit and implicit solvent models for calculating solvation free energy and PMF involves a fundamental trade-off between computational efficiency and physical accuracy. Explicit solvent models remain the gold standard for reliability, particularly for processes where specific solvent structure is critical, but their high cost limits application in high-throughput screening. Implicit models offer the speed necessary for large-scale studies but have documented limitations in accuracy. The most promising future direction lies in hybrid approaches and machine learning, where the physical rigor of explicit solvents or the speed of implicit models is enhanced with data-driven corrections, steadily closing the gap between computational feasibility and quantitative accuracy.
In molecular dynamics (MD) simulations, the accurate description of the solvent environment is fundamental because biological processes occur in an aqueous medium. Implicit solvent models have emerged as a powerful alternative to explicit solvent representations, significantly reducing computational cost by treating the solvent as a continuum rather than simulating individual water molecules. This approach replaces explicit water molecules with a potential of mean force, allowing for more efficient conformational sampling. The three major classes of implicit solvent models—SASA (Solvent Accessible Surface Area), Generalized Born (GB), and Poisson-Boltzmann (PB)—each provide distinct methodologies for estimating solvation effects. When combined in methods like MM/PB(GB)SA, they become essential tools for predicting binding free energies in drug discovery, enabling researchers to accelerate cost-efficient binding free energy calculations for various protein systems, including soluble and membrane-bound targets [19] [12].
Theoretical Foundations of Implicit Solvation
Fundamental Principles and Energy Components
Implicit solvation models compute the free energy of solvation (ΔGsol), which represents the work required to transfer a solute molecule from a vacuum to the solvent. This free energy is traditionally decomposed into three components related to a theoretical process: creation of a cavity within the solvent to accommodate the solute molecule (ΔGcav), van der Waals interactions between solute and solvent (ΔGvdW), and electrostatic interactions (ΔGele) [12].
The overall solvation free energy can be expressed as: ΔGsol = ΔGcav + ΔGvdW + ΔGele
In practical implementation, these components are often grouped into polar and nonpolar contributions: ΔGsol = ΔGpol + ΔG_np
Where ΔGpol represents the electrostatic component (calculated by PB or GB methods), and ΔGnp represents the nonpolar component (typically calculated using SASA-based approaches) [20] [12].
Relationship Between Model Classes
The relationship between SASA, GB, and PB models can be understood through their complementary roles in calculating these energy components. SASA models primarily address the nonpolar contribution (ΔGnp), which includes both cavity formation and van der Waals interactions. GB and PB methods focus on the polar component (ΔGpol), which describes the electrostatic interactions between the solute and solvent continuum [12]. In many modern implementations, these approaches are combined—for example, in GB/SA or PB/SA methods—to provide a comprehensive description of solvation effects.
Solvent Accessible Surface Area (SASA) Models
Theoretical Basis and Algorithmic Implementation
The Solvent Accessible Surface Area (SASA) model quantifies the surface area of a biomolecule that is accessible to a solvent molecule. The concept was first introduced by Lee and Richards in 1971, and the canonical computational method was developed by Shrake and Rupley in 1973 [21] [22]. The fundamental principle assumes that the nonpolar solvation free energy is proportional to the molecular surface area: ΔG_np = γ × SASA
Where γ is the surface tension parameter, typically with identical values for all atoms [20].
The Shrake-Rupley algorithm implements this concept by creating a mesh of points equidistant from each atom at a distance equal to the van der Waals radius plus the probe radius (typically 1.4Å, approximating a water molecule). The algorithm then counts how many of these points are not buried within other atoms, with the accessible area being proportional to the number of exposed points [22] [23]. This represents a numerical "rolling ball" approach where a sphere of solvent is rolled along the molecular surface [21].
Advanced SASA Algorithms and Recent Developments
Several algorithms have been developed to improve the accuracy and efficiency of SASA calculations:
- LCPO (Linear Combinations of Pairwise Overlaps): An analytical approximation method that estimates SASA based on neighbor atoms, with computation complexity of O(N²) where N is the number of atoms [20].
- dSASA (differentiable SASA): A recently developed exact geometric method that calculates SASA analytically using Alpha Complex theory and inclusion-exclusion method, implemented on GPUs for accelerated performance [20].
- pwSASA (pairwise SASA): A fast pairwise approximation method suitable for GPU implementation, though with limited transferability to non-protein systems [20].
The table below compares the key characteristics of these SASA calculation methods:
Table 1: Comparison of SASA Calculation Algorithms
| Algorithm | Method Type | Accuracy | Speed | Derivatives Available | System Applicability |
|---|---|---|---|---|---|
| Shrake-Rupley | Numerical | High (reference) | Slow | No | Universal |
| LCPO | Analytical approximation | Moderate (1-3 Ų error) | Medium (O(N²)) | Yes | Universal |
| pwSASA | Pairwise approximation | Variable (R²: 0.6-0.9) | Fast | Yes | Proteins only |
| dSASA | Exact analytical | High (>98% accuracy) | Fast (GPU-accelerated) | Yes | Universal |
Applications and Limitations in Biomolecular Simulations
SASA-based models find extensive application in calculating binding free energies, protein folding studies, and predicting protein-ligand interactions. The nonpolar term calculated via SASA is frequently combined with polar solvation terms from PB or GB models in methods like MM/PBSA and MM/GBSA for binding affinity prediction [19]. Recent work has demonstrated that including an accurate SASA-based nonpolar term in GB/SA MD simulations produces more stable trajectories and better simulated melting temperatures compared to simulations without this term [20].
A significant limitation of traditional SASA models is their empirical nature and the assumption of a uniform surface tension parameter (γ) for all atoms. Additionally, early implementations lacked reliable atomic derivatives, making them unsuitable for molecular dynamics simulations. However, newer methods like dSASA have addressed this limitation by providing exact analytical derivatives [20].
Generalized Born (GB) Models
Theoretical Foundation and Mathematical Formulation
The Generalized Born (GB) model is an approximation to the Poisson-Boltzmann equation that provides a computationally efficient method for calculating electrostatic solvation energies. Introduced by Still et al., the GB method aims to approximate the solution of the PB equation with simpler functional forms [12]. The model is based on the Born formula for a single ion, which provides an analytical solution for the solvation free energy of a spherical ion with charge q and radius α:
ΔGsolv = -(1/2)(1/εint - 1/ε_ext) × (q²/α)
GB models extend this concept to complex molecules of arbitrary shape by defining an effective Born radius for each atom, which represents its degree of burial within the solute molecule. The generalized form of the equation for multiple atoms is [12]:
ΔGele = -(1/2)(1/εint - 1/εext) × ΣΣ (qi qj / fGB(rij, Ri, R_j))
Where fGB is a function that depends on the distance between atoms (rij) and their effective Born radii (Ri, Rj).
Parameterization and Modern Implementations
The accuracy of GB models heavily depends on the calculation of effective Born radii. Recent developments have focused on parametrizing GB screening parameters to provide better matching of effective radii to reference methods, with pairwise decomposition versions being ideal for GPU implementation with parallel computing [20]. The latest GB models for proteins and nucleic acids have shown significant improvement in achieving agreement with PB electrostatic free energies.
GB models are particularly valuable in molecular dynamics simulations due to their computational efficiency. When combined with SASA-based nonpolar terms in GB/SA approaches, they can produce stable trajectories and improve folding stabilities in protein folding studies [20]. The performance of GB models has been enhanced through GPU implementations, with recent versions achieving nearly 20-fold acceleration compared to CPU versions [20].
Poisson-Boltzmann (PB) Models
Mathematical Framework and Numerical Solutions
The Poisson-Boltzmann (PB) equation provides a more rigorous approach to modeling electrostatic interactions in implicit solvent. It is a differential equation that describes the electrostatic potential around a solute molecule with a dielectric discontinuity at the solute-solvent interface. The general form of the PB equation is [12]:
∇ · [ε(r)∇Φ(r)] = -ρ(r)/ε₀ - Σ ci^b zi q e^(-zi q Φ(r)/kB T)
Where:
- Φ(r) is the electrostatic potential
- ε(r) is the position-dependent dielectric constant
- ρ(r) is the fixed charge density of the solute
- c_i^b is the bulk concentration of ion i
- z_i is the valence of ion i
- q is the unit charge
- k_B is Boltzmann's constant
- T is the temperature
For systems where the exponential term is small, the equation can be linearized to [12]:
∇ · [ε(r)∇Φ(r)] = -ρ(r)/ε₀ + Σ (ci^b zi² q² Φ(r))/(k_B T)
Applications, Challenges, and Recent Advances
PB models are considered the gold standard for electrostatic calculations in implicit solvent modeling due to their theoretical rigor. They are widely used in binding free energy calculations, pKa prediction, and analysis of molecular interactions. However, several challenges limit their application in MD simulations: nonlinearity, dielectric jumps, charge singularity, and geometric complexity make PB solutions computationally demanding [24].
Recent advances include the development of more efficient numerical solvers and machine learning approaches. The second-order accurate MIBPB solver has demonstrated high accuracy, while PB-based machine learning (PBML) models have been developed to provide accurate electrostatic solvation free energies with significantly reduced computational cost [24]. These ML approaches can rapidly predict PB-quality electrostatic solvation energies for new biomolecules or conformations generated by molecular dynamics.
Comparative Analysis of Model Performance
Accuracy and Efficiency in Binding Free Energy Prediction
Comprehensive benchmarks comparing MM/PB(GB)SA methods have been conducted on both soluble and membrane protein systems. These studies evaluate different parameters including ligand charges, protein force fields, GB models, nonpolar optimization methods, and dielectric constants [19]. The results reveal several key insights into the relative performance of these implicit solvent approaches.
Table 2: Performance Comparison of Implicit Solvent Methods in Binding Free Energy Prediction
| Method | System Type | Accuracy (RMSE) | Computational Cost | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| MM/PBSA | Soluble proteins | 1.5-2.5 kcal/mol [19] | High | High accuracy for electrostatic interactions | Slow, computationally expensive |
| MM/GBSA | Soluble proteins | 1.6-2.7 kcal/mol [19] | Medium | Balanced speed/accuracy | Dependent on GB model parameterization |
| MM/PBSA | Membrane proteins | Case-dependent [19] | High | Handles dielectric heterogeneity | Requires membrane-specific parameters |
| MM/GBSA | Membrane proteins | Case-dependent [19] | Medium | Faster than PBSA for membranes | Membrane dielectric constant needs optimization |
| Docking | Both | >3.0 kcal/mol [19] | Low | Very fast | Lowest accuracy |
| FEP | Both | 1.4-2.3 kcal/mol [19] | Very High | Highest theoretical accuracy | Extremely computationally expensive |
Systematic Benchmarking Studies
A notable benchmarking study performed on 140 ligands bound to six soluble proteins and 37 ligands bound to three membrane proteins revealed that MM/PB(GB)SA shows competitive performance with the more rigorous Free Energy Perturbation (FEP) method, while docking scored significantly worse [19]. The study emphasized that parameters for MM/PB(GB)SA calculations need to be optimized for specific systems, with no universal parameter set guaranteeing accuracy across all applications.
For membrane protein systems—particularly important drug targets like GPCRs—specific considerations such as membrane dielectric constants significantly impact the accuracy of both PB and GB models [19]. The performance in these systems is case-dependent, requiring careful parameterization for reliable results.
Experimental Protocols and Methodologies
Standard Protocol for MM/PB(GB)SA Binding Free Energy Calculations
The typical workflow for MM/PB(GB)SA calculations involves multiple stages of system preparation, simulation, and energy analysis:
System Preparation
- Obtain protein-ligand complex structures from crystallography or docking
- Assign protonation states at physiological pH (e.g., pH 7.0)
- Generate ligand charges using appropriate methods (CGenFF, AM1-BCC, RESP-HF, RESP-DFT) [19]
Molecular Dynamics Simulation
- Perform explicit solvent MD simulation to generate conformational ensembles
- Extract snapshots at regular intervals for end-point free energy calculations
Energy Calculation
- Calculate molecular mechanics energy (ΔE_MM) including bond, angle, dihedral, electrostatic, and van der Waals contributions
- Compute polar solvation energy (ΔG_PB/GB) using PB or GB models
- Calculate nonpolar solvation energy (ΔG_SA) using SASA-based methods
- Optionally estimate entropy term (-TΔS), though this is often omitted due to high computational cost and large errors [19]
The binding free energy is then computed as: ΔGbind = ΔEMM + ΔGsolv - TΔS ΔGsolv = ΔGPB/GB + ΔGSA
Protocol for SASA-Based Binding Energy Estimation
For rapid binding affinity estimation using SASA-based models:
Structure Preparation
- Obtain or generate complex structures
- Identify binding interfaces
Buried Surface Area (BSA) Calculation
- Calculate solvent accessible surface area (SASA) for free components and complex
- Compute BSA as: BSA = SASAprotein + SASAligand - SASA_complex
Free Energy Calculation
- Apply empirical relationship: ΔG_binding = γ × BSA
- Use appropriate surface tension parameter (γ), typically in the range of -7 to -25 cal mol⁻¹ Å⁻² [21]
The free energy density γ varies depending on the system and methodology, with values of -15 ± 1.2 cal mol⁻¹ Å⁻² reported for hydrophobic residues, -7 cal mol⁻¹ Å⁻² when considering only ligand burial, and -25 cal mol⁻¹ Å⁻² when considering total hydrophobic burial [21].
Table 3: Essential Computational Tools for Implicit Solvent Simulations
| Tool/Resource | Type | Primary Function | Key Features | Applicable Models |
|---|---|---|---|---|
| AMBER | Software Suite | MD simulations with implicit solvent | GPU acceleration, MMPBSA.py, GB/SA implementation | SASA, GB, PB |
| APBS | Software Tool | Electrostatic calculations | Solves PB equation, PDB2PQR conversion | PB |
| MDTraj | Python Library | Trajectory analysis | Shrake-Rupley SASA implementation, efficient analysis | SASA |
| HYDROPRO | Software Tool | Hydrodynamic properties | Calculates hydrodynamic radius, diffusion constants | SASA-based properties |
| g_mmpbsa | GROMACS Tool | MM/PBSA calculations | Integration with GROMACS, automated binding energy | GB, PB, SASA |
| PDB2PQR | Software Tool | Structure preparation | Adds hydrogens, assigns charges, creates PQR files | PB |
| MMPBSA.py | AMBER Tool | Binding free energy | End-point free energy method, trajectory analysis | GB, PB, SASA |
| GSnet | Machine Learning Model | Property prediction | Graph neural network for SASA and pKa prediction | ML-enhanced SASA |
Emerging Trends and Future Directions
Machine Learning-Enhanced Implicit Solvent Models
Recent advances integrate machine learning with traditional implicit solvent models to overcome computational limitations. Graph neural networks like GSnet demonstrate remarkable capability in predicting molecular properties including SASA and residue-specific pKa values directly from 3D protein structures [25]. These ML approaches achieve accuracy comparable to simulation-based methods while enabling high-throughput applications across proteome-wide studies.
PB-based machine learning (PBML) models trained with accurate numerical solvers can provide PB-quality electrostatic solvation free energies for biomolecular structures with significantly reduced computational cost [24]. This integration of physical models with data-driven approaches represents a promising direction for maintaining accuracy while dramatically improving efficiency.
Advanced SASA Algorithms and GPU Acceleration
The development of exact analytical methods like dSASA implemented on GPUs addresses longstanding challenges in SASA calculation accuracy and efficiency [20]. These methods enable stable GB/SA MD simulations with inclusion of nonpolar terms, improving the performance of implicit solvent simulations for protein folding and binding studies. The GPU implementation of these algorithms achieves up to 20-fold acceleration compared to CPU versions, making implicit solvent simulations more practical for large biomolecular systems.
Application to Challenging Systems
While implicit solvent models have traditionally excelled with soluble proteins, recent research focuses on extending their applicability to more challenging systems like membrane proteins, nucleic acids, and glycans [19] [12]. For membrane proteins, careful optimization of membrane dielectric constants and other system-specific parameters has shown promising results in benchmarking studies [19]. The development of models that can handle the unique dielectric and solvation properties of these systems remains an active area of research.
SASA, Generalized Born, and Poisson-Boltzmann models represent complementary approaches within the implicit solvent modeling paradigm, each with distinct strengths and optimal application domains. SASA models provide efficient calculation of nonpolar solvation contributions, GB models offer a balanced approach for electrostatic interactions with moderate computational cost, and PB methods deliver high accuracy for electrostatic calculations at greater computational expense.
The choice between these models depends on the specific research requirements, balancing accuracy needs with computational resources. For high-throughput virtual screening, MM/GBSA with SASA-based nonpolar terms often provides the best compromise. For detailed mechanistic studies requiring high electrostatic accuracy, MM/PBSA remains preferable when computational resources permit. Recent advances in machine learning integration and GPU acceleration are blurring these traditional trade-offs, enabling faster simulations without sacrificing accuracy.
As implicit solvent models continue to evolve, their importance in drug discovery and biomolecular simulation is likely to grow, particularly through hybrid approaches that combine physical rigor with data-driven efficiency. These developments will further establish implicit solvent methods as indispensable tools for researchers studying biomolecular structure, function, and interactions.
The accurate modeling of solvent effects is a cornerstone of reliable molecular dynamics (MD) simulations, profoundly influencing the study of biomolecular structure, dynamics, and function. The computational landscape is dominated by two distinct paradigms: explicit solvent models, which treat each solvent molecule individually, and implicit solvent models, which average solvent effects into a continuous polarizable medium [3] [26]. Explicit models are considered the gold standard for realism but carry a prohibitive computational cost, as simulating the thousands of solvent molecules in a typical system requires significant resources [10]. Implicit solvent models emerged as an efficient alternative, dramatically reducing computational expense and accelerating conformational sampling by eliminating solvent viscosity [27] [10]. This guide traces the theoretical evolution from foundational continuum concepts to modern machine-learning-driven formulations, providing a comparative analysis of their performance, applications, and limitations within MD research.
Historical Development of Implicit Solvent Theory
The conceptual foundation of implicit solvation rests on early dielectric theories developed by Onsager and Debye, who first treated solvents as dielectric continua to estimate solvation energies from bulk properties like dielectric constant and molecular polarizability [3]. This established the core principle of partitioning the solvation free energy (( \Delta G_{solv} )) into physically distinct components.
The Born model, a seminal advance, provided an analytical solution for the electrostatic solvation energy of a spherical ion in a continuum dielectric [7]. Its simplicity laid the groundwork for more complex approximations. Subsequent developments introduced the Poisson-Boltzmann (PB) equation, a rigorous framework describing electrostatic interactions between a solute and a dielectric medium with ionic strength effects [3]. While accurate, solving the PB equation numerically remains computationally demanding for large systems.
To address this bottleneck, the Generalized Born (GB) model was developed as an efficient pairwise approximation to the PB formalism [11] [10]. GB models estimate the electrostatic solvation energy using an analytical formula, enabling rapid calculations for large biomolecules. Modern iterations like GB-neck and GB-neck2 have been parameterized to better reproduce PB solvation energies for proteins and nucleic acids [11].
Concurrently, advancements in quantum chemistry spurred the creation of models like the Polarizable Continuum Model (PCM) and the Conductor-like Screening Model (COSMO), which integrate solvation effects directly into electronic structure calculations [3] [28]. The most recent paradigm shift incorporates Machine Learning (ML). ML-based implicit solvent models, such as Graph Neural Network Implicit Solvent (GNNIS) models, are trained on data from explicit-solvent simulations to learn and predict the explicit-solvent effect, offering a new route to achieving high accuracy with the speed of a continuum model [28] [7].
Comparative Performance Analysis of Solvation Models
The performance of implicit solvent models is benchmarked against explicit solvent references and experimental data across various metrics. The following table summarizes key quantitative comparisons.
Table 1: Performance Comparison of Implicit vs. Explicit Solvent Models
| Performance Metric | Explicit Solvent (TIP3P, PME) | Generalized Born (GB) Implicit Solvent | Machine Learning Implicit Solvent (GNNIS) |
|---|---|---|---|
| Computational Speed | Baseline (1x) | 1x to 100x faster for conformational sampling [27] | Computational speedup comparable to implicit models, but with added training cost [7] |
| Solvation Free Energy Accuracy | High (reference) | Moderate; errors in non-polar component [7] | Near-explicit solvent accuracy for small molecules [7] |
| Conformational Sampling Efficiency | Limited by solvent viscosity | High efficiency due to low viscosity [27] [10] | High efficiency, learns from explicit solvent data [28] |
| Protein Folding Accuracy | Accurate, but computationally intensive | GB-neck2 with ff14SBonlysc folds diverse proteins, better balance than explicit TIP3P in some cases [11] | N/A |
| RNA Stem-Loop Folding | Achieved with specialized force fields | Successful folding of 26 RNA stem-loops to near-native structures (stem RMSD <2 Å) [11] | N/A |
| Handling of Explicit Solvent Effects | Naturally includes specific effects (e.g., H-bonds) | Poor; lacks atomic detail [26] [4] | Good; designed to capture explicit-solvent effects missing from continuum models [28] |
The table shows a clear trade-off. Implicit solvents like GB offer dramatic speedups, between 1-fold and 100-fold for large conformational changes, primarily due to reduced solvent viscosity [27]. This enables the folding of proteins and RNA stem-loops that is challenging with explicit solvents [11]. However, traditional implicit models fail to capture specific solute-solvent interactions, such as hydrogen bonds. ML-based models are emerging to bridge this accuracy gap, with GNNIS demonstrating an ability to reproduce experimental NMR and IR trends that are unattainable by state-of-the-art implicit models like SMD paired with static QM calculations [28].
Table 2: Comparison of Fundamental Solvation Model Types
| Characteristic | Explicit Solvent | Continuum Implicit Solvent (e.g., PB/GB) | Machine-Learned Implicit Solvent |
|---|---|---|---|
| Theoretical Basis | Newtonian mechanics for all atoms | Continuum electrostatics & empirical terms | Data-driven; trained on explicit solvent simulations |
| Solvent Representation | Individual molecules | Dielectric continuum | Mathematical approximation of mean forces |
| Computational Cost | Very High | Low | Low (after training) |
| Sampling Speed | Slow | Fast | Fast |
| Key Strengths | High realism, captures specific effects | Computational efficiency, fast sampling | Balances speed and accuracy for specific effects |
| Key Limitations | Extreme computational demand, slow dynamics | Poor for specific interactions, parameter sensitivity | Training data dependency, generalizability concerns |
Experimental Protocols and Workflows
Workflow for Developing Machine Learning Potentials for Solvation
The creation of robust MLPs for solvation employs an iterative active learning (AL) strategy to ensure data efficiency and comprehensive coverage of the chemical space [4]. The workflow, depicted in the diagram below, involves several key stages.
Diagram Title: Active Learning Workflow for ML Potentials
Detailed Methodology:
- Initial Data Generation: A small set of initial configurations is generated and labeled with reference energies and forces from quantum chemistry calculations. This typically includes:
- The solute in the gas phase or with an implicit solvent, with geometries sampled by randomly displacing atomic coordinates, often starting from a transition state for reactions.
- Cluster models containing the solute and a limited number of explicit solvent molecules placed at relevant positions, ensuring the solvent shell radius is at least as large as the MLP's cut-off radius [4].
- MLP Training and Validation: An initial MLP is trained on this dataset. Its stability is tested by running short MD simulations (e.g., starting at 2 fs and progressively increasing).
- Active Learning Loop: Structures from the MLP-driven MD are evaluated by a descriptor-based selector, such as Smooth Overlap of Atomic Positions (SOAP). This measures the similarity of new configurations to the existing training set.
- Uncertain/New Configuration: If a structure lies in an under-sampled region of the chemical space (high "uncertainty"), it is added to the training set.
- Well-Represented Configuration: If the structure is similar to existing training data, the MLP is considered reliable for that region.
- Iteration and Production: The process of re-training the MLP with the expanded dataset and running new MD continues until the MLP's predictions converge and no new significant configurations are found. The final, validated MLP is then used for production simulations [4].
Protocol for RNA Folding Simulations with Implicit Solvent
A specific application demonstrating the power of implicit solvent is the de novo folding of RNA stem-loops [11].
- Force Field: The DESRES-RNA force field (an extensive revision of the AMBER RNA force field) or the AMBER-OL3 force field is used.
- Solvation Model: The GB-neck2 implicit solvent model, parameterized for nucleic acids.
- System Preparation: Simulations start from the extended conformations of 26 different RNA stem-loops (10 to 36 residues), with and without bulges or internal loops.
- Simulation Type: Conventional molecular dynamics simulations are run, without enhanced sampling techniques.
- Analysis: Success is measured by the formation of all native base pairs in the stem regions and the calculation of the root mean square deviation (RMSD) relative to experimentally determined structures. Successful folding is defined as achieving RMSD values of <2 Å for stem regions and <5 Å for the entire molecule [11].
The Scientist's Toolkit: Essential Research Reagents and Solutions
This section details key computational tools and datasets referenced in the studies, essential for replicating and advancing research in this field.
Table 3: Key Research Reagents and Computational Solutions
| Item Name | Type | Primary Function | Relevant Context |
|---|---|---|---|
| OMol25 Dataset | Dataset | Provides over 100M high-accuracy quantum chemical calculations for training and benchmarking NNPs [29]. | Used to train Meta's Universal Model for Atoms (UMA); covers biomolecules, electrolytes, and metal complexes [29]. |
| GB-neck2 | Implicit Solvent Model | An optimized Generalized Born model for accurate calculation of solvation energies in biomolecular simulations [11]. | Successfully used in folding proteins and RNA stem-loops with the AMBER force field [11]. |
| DESRES-RNA Force Field | Molecular Force Field | An extensively refined AMBER force field for RNA that accurately reproduces energetics of base stacking and pairing [11]. | Combined with GB-neck2 or TIP4P-D water for successful RNA folding simulations [11]. |
| Graph Neural Network Implicit Solvent (GNNIS) | Machine Learning Model | A graph neural network that learns explicit-solvent effects from MD data to correct continuum model predictions [28]. | Transfers knowledge from MM to QM, applicable to various organic solvents without needing QM/MM training data [28]. |
| Open-Molman | Software Library | An open-source toolkit containing implementations of modern NNP architectures like eSEN and UMA [29]. | Provides pre-trained models for molecular modeling on datasets like OMol25 [29]. |
| AMBER | MD Software Package | A suite of biomolecular simulation programs that includes implementations of various force fields and implicit solvent models like GB-neck2 [11]. | Widely used for MD simulations of proteins and nucleic acids in explicit and implicit solvent [27] [11]. |
| LSNN (λ-Solvation Neural Network) | Machine Learning Model | A GNN trained to match forces and derivatives of alchemical variables, enabling accurate solvation free energy calculations [7]. | Overcomes the limitation of standard force-matching by ensuring energy predictions are suitable for absolute free energy comparisons [7]. |
The journey from the foundational Born model to modern analytical formulations illustrates a consistent drive to balance computational efficiency with physical accuracy in solvation modeling. While classical continuum models like PB and GB remain vital tools for large-scale conformational sampling, the frontier of the field is being reshaped by machine learning. ML-based implicit solvent models, trained on massive datasets like OMol25 or knowledge transferred from classical simulations, represent a paradigm shift. They promise to capture the explicit-solvent effects that have long been the Achilles' heel of continuum approaches, potentially offering a new generation of models that do not force a strict choice between accuracy and speed. For researchers in drug development and biophysics, this evolution opens the door to more reliable and ambitious simulations of biomolecular processes in their native aqueous environments.
Implementing Solvent Models: Methodologies and Practical Applications in Biomedicine
Implicit solvent models provide a computationally efficient framework for modeling solvation effects in molecular simulations by replacing the explicit simulation of solvent molecules with a continuum mean-field approximation. This approach drastically reduces the number of particles in a simulation system, lowering computational cost and accelerating conformational sampling. [7] [11] Within this paradigm, the Generalized Born Surface Area (GB/SA) and Poisson-Boltzmann Surface Area (PBSA) frameworks have emerged as two of the most popular methods for calculating binding affinities and solvation free energies in drug discovery and biomolecular research. [30] These end-point methods, notably Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) and Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA), are widely used for predicting protein-ligand binding free energies, helping to reduce costs and accelerate early-stage drug development. [31] [7] This guide provides a detailed, objective comparison of these frameworks, their performance, and their implementation within the AMBER and CHARMM software suites.
Theoretical Foundations of GB/SA and PBSA
Both MM/GBSA and MM/PBSA methods calculate the binding free energy (ΔG_bind) of a complex in solution as the difference between the free energy of the complex and its isolated receptor and ligand components. The general equation is:
ΔGbind = Gcomplex - (Greceptor + Gligand)
The free energy for each entity (complex, receptor, ligand) is estimated as a sum of molecular mechanics energy, polar solvation energy, and non-polar solvation energy:
G = EMM + Gsolv = (Eint + Eele + Evdw) + (Gpol + G_np) [30]
Here, EMM is the molecular mechanics gas-phase energy, comprising internal (Eint), electrostatic (Eele), and van der Waals (Evdw) components. Gsolv is the solvation free energy, decomposed into a polar (Gpol) contribution and a non-polar (G_np) contribution. [30]
The key difference between MM/GBSA and MM/PBSA lies in the treatment of the polar solvation term, G_pol, while the non-polar term is typically modeled using the solvent-accessible surface area (SASA).
- Polar Solvation (Gpol) in PBSA: The PBSA method uses the Poisson-Boltzmann (PB) equation to compute Gpol. The PB equation is an electrostatic continuum model that numerically solves for the electrostatic potential of a solute embedded in a dielectric continuum, providing a rigorous and theoretically sound solution. However, this numerical calculation is computationally demanding. [30] [32]
- Polar Solvation (Gpol) in GBSA: The GBSA method approximates the solution of the PB equation using the Generalized Born (GB) model. The GB model provides an analytical approximation for Gpol, which is significantly faster to compute but relies on empirical parameters and can be less accurate for certain systems, particularly those with complex electrostatic environments. [30] [32] [11]
- Non-Polar Solvation (Gnp): Both methods often use a SASA-based term for the non-polar contribution, which accounts for cavity formation and van der Waals interactions between the solute and solvent. The general form is Gnp = γ × SASA + b, where γ is a surface tension coefficient and b is a constant. [30] [33] It is worth noting that the specific parameters for this equation can differ between PB and GB calculations within the same software package. [34]
The following workflow diagram illustrates how these components are integrated in a typical binding free energy calculation.
Performance Comparison: Accuracy, Speed, and Applicability
The choice between MM/GBSA and MM/PBSA involves a trade-off between computational speed and accuracy, which can vary significantly depending on the system and parameters.
Quantitative Performance Data
The table below summarizes key findings from a recent head-to-head comparison study on the CB1 cannabinoid receptor and other systems.
| Performance Metric | MM/GBSA | MM/PBSA | System / Notes |
|---|---|---|---|
| Correlation with Experiment (Pearson's r) | 0.433 - 0.652 [31] | 0.100 - 0.486 [31] | CB1 receptor ligands (46 compounds) [31] |
| Calculation Speed | Faster [31] [32] | Slower [31] [32] | GB is an analytical approximation; PB requires numerical solving [31] |
| Result Agreement | Can show large differences [32] | Can show large differences [32] | Example: GB=-57.25 vs. PB=-2.53 kcal/mol for a protein-ligand complex [32] |
| SASA Calculation | Uses surften (e.g., 0.0072) & surfoff [34] |
Uses cavity_surften (e.g., 0.0378) & cavity_offset [34] |
Different non-polar models in AMBER can cause SASA value differences [34] |
| Solute Dielectric Constant (ε_in) | Improved correlations with larger values (ε=2,4) [31] | Improved correlations with larger values (ε=2,4) [31] | System-dependent; higher ε_in can better capture polarization [31] |
Critical Performance Factors
Several parameters critically influence the performance of both methods:
- Sampling and Starting Structures: Using ensembles from molecular dynamics (MD) simulations consistently provides better results than using single, energy-minimized structures. [31]
- Solute Dielectric Constant (εin): The choice of the internal dielectric constant is highly system-dependent. Studies on the CB1 receptor showed that improved correlations with experimental binding affinities were observed with larger εin values (2 or 4) compared to a value of 1. [31]
- Entropy Compensation: The incorporation of entropic terms (usually calculated via normal mode or quasi-harmonic analysis) is computationally expensive and often fails to improve, or can even degrade, the correlation with experimental data. [31]
- GB Model Variants: Within the GB framework, the choice of specific model (e.g., GBHCT, GBOBC1, GBOBC2, GBNeck, GBNeck2) can exert a varying effect on the results, and no single model is universally superior. [31]
- Non-Polar Model: The treatment of the non-polar solvation term is a significant source of variation. As noted in AMBER documentation, MM/PBSA and MM/GBSA can use different equations and parameters for this term, leading to different SASA values and final results. [34]
Software Implementation: AMBER vs. CHARMM
Implementation in AMBER
The AMBER software package provides extensive and well-documented support for both MM/PBSA and MM/GBSA calculations, primarily through the MMPBSA.py and gmx_MMPBSA tools. [31] [30]
- Force Field Compatibility: While AMBER force fields are native,
MMPBSA.pycan be used with CHARMM-generated topologies converted via thechambertool. However, caution is advised as this combination is less tested. [35] - GB Model Selection: AMBER offers multiple GB models (
igbkeyword).igb=5(GBOBC1) is often cited as a preferred model for its balance of accuracy and speed, though the optimal choice may be system-dependent. [31] [35] - PBSA Radii Setting: When using non-AMBER force fields, it is recommended to use
radiopt=0to employ the intrinsic atomic radii from the force field, as the optimized radii (radiopt=1) are parameterized specifically for AMBER atom types. [35] - SASA Algorithms: AMBER has evolved through several SASA algorithms:
- LCPO: A fast CPU-based algorithm, but can become a bottleneck in GPU-accelerated simulations due to CPU/GPU data transfer. [33]
- pwSASA: A pairwise approximation designed for GPUs, but with lower accuracy and parameters trained only on standard amino acids. [33]
- dSASA: A new, exact geometric method implemented on GPUs that offers high accuracy for proteins and nucleic acids, enabling stable and fast implicit solvent MD simulations. [33]
Implementation in CHARMM
CHARMM also features robust implementations of implicit solvation, with a different philosophical approach to certain parameters.
- Native GB Models: It is generally recommended to use CHARMM's own GB models (e.g.,
gbsw) for MM/GBSA calculations within the CHARMM software, as the parameters are optimized for the CHARMM force field. [36] - Force Field Specificity: The parameters for GB models, which include adjustable "shielding" parameters, are not easily transferable between force fields. Using AMBER's GB parameters with a CHARMM potential is unlikely to work well. [36]
- Limited Decomposition Tools: Historically, CHARMM has had limited modules for per-residue energy decomposition that incorporate entropy, which is a reason researchers might try to use AMBER's
MMPBSA.pywith CHARMM-generated structures. [36]
Experimental Protocols and Case Studies
Protocol for MM/PB(GB)SA in AMBER
A typical protocol for running a calculation in AMBER, as derived from published studies, involves the following steps: [31] [30]
- System Preparation: Model the protein-ligand complex, add missing atoms/residues, and assign protonation states. For ligands, parameters are often generated with Antechamber using the GAFF force field. [30]
- Molecular Dynamics Simulation: Solvate the system and run an explicit solvent MD simulation to generate an equilibrated trajectory. This step provides the conformational ensemble for the end-point calculation.
- Post-Processing:
- Use the
MMPBSA.pyscript to post-process the MD trajectory. - The script strips water molecules and ions and calculates the energies for the complex, receptor, and ligand across multiple snapshots.
- Input file example for running both GB and PB:
- Use the
- Binding Energy Calculation: The script automatically calculates the average binding free energy and its components.
Case Study: CB1 Cannabinoid Receptor Ligands
A 2024 study provides a robust benchmark for MM/PBSA and MM/GBSA performance on a pharmaceutically relevant target. [31]
- System: 46 CB1 receptor ligands (23 agonists and 23 antagonists).
- Methods: Structures were docked and then subjected to 30 ns MD production simulations using GROMACS. Binding free energies were calculated with
gmx_MMPBSAusing various parameters (solute dielectric, GB models, entropy). - Key Outcome: MM/GBSA consistently provided higher correlation with experimental binding affinities than MM/PBSA, while also being computationally faster. The study highlights that performance is highly individualized to the specific system and simulation parameters. [31]
The Scientist's Toolkit: Essential Research Reagents
The table below lists key computational "reagents" and tools essential for conducting MM/PB(GB)SA research.
| Tool / Reagent | Function / Purpose | Example or Note |
|---|---|---|
| MD Software | Generates the conformational ensemble for the bound and unbound states. | GROMACS [31], AMBER [30], CHARMM/NAMD [36] |
| MMPBSA Analysis Tool | The main engine for performing the end-point free energy calculations. | MMPBSA.py [30] [35] or gmx_MMPBSA [31] in AMBER |
| Implicit Solvent Model | Calculates the polar solvation free energy (G_pol). | Generalized Born (GB) or Poisson-Boltzmann (PB) models [30] |
| SASA Algorithm | Calculates the solvent-accessible surface area for the non-polar term (G_np). | LCPO [33], pwSASA [33], or the newer dSASA [33] |
| Force Field | Defines the molecular mechanics potential (E_MM). | AMBER (ff19SB, GAFF) [31] [30], CHARMM (C36) [36] [35] |
| Ligand Parameterizer | Generates force field parameters for small molecule ligands. | Antechamber (for GAFF) [30] |
| Dielectric Constant (ε_in) | Empirical parameter representing the solute's internal dielectric. | A value of 1, 2, or 4 is commonly tested [31] |
| Surface Tension (γ) | Empirical parameter scaling the SASA to energy. | Different values for PB (0.0378) and GB (0.0072) in AMBER [34] |
The field of implicit solvation continues to evolve. A prominent research direction is the integration of machine learning (ML) to develop more accurate and efficient solvation potentials. [7] For instance, novel Graph Neural Network (GNN)-based models like the λ-Solvation Neural Network (LSNN) are being trained not only on forces but also on derivatives of alchemical variables, enabling them to predict solvation free energies with near-explicit solvent accuracy while maintaining computational efficiency. [7] Furthermore, algorithmic improvements continue, such as the development of exact analytical methods like dSASA for more accurate and faster SASA calculations on GPUs. [33]
In conclusion, both GB/SA and PBSA frameworks are powerful tools for predicting binding affinities and modeling solvation. MM/GBSA is generally faster and can, in many cases, provide equal or better correlation with experiment than MM/PBSA. However, the performance is highly system- and parameter-dependent. Researchers must carefully select their parameters, including the dielectric constant, GB model, and non-polar treatment, and should not expect the two methods to yield numerically similar results. The ongoing integration of machine learning promises to further bridge the accuracy gap between implicit and explicit solvent models in the years to come.
Molecular dynamics (MD) simulation is a cornerstone technique in computational biophysics and drug discovery, providing atomic-level insights into the behavior of proteins, nucleic acids, and other biomolecules. A fundamental choice in setting up any MD simulation is how to represent the solvent environment. Explicit solvent models treat each water molecule as an individual entity with specific atomic interactions, while implicit solvent models (also known as continuum solvent models) represent the solvent as a continuous dielectric medium, dramatically reducing computational complexity [37] [3]. This guide provides a comprehensive comparison of these approaches, with specific focus on practical implementation of implicit solvent workflows, their performance advantages, and their limitations relative to explicit solvent simulations.
The theoretical foundation of implicit solvent models rests on partitioning solvation free energy into polar (electrostatic) and nonpolar components [3]. The polar component is typically calculated using approximations to the Poisson-Boltzmann equation, such as the Generalized Born (GB) method, while the nonpolar component accounts for cavity formation and van der Waals interactions, often estimated using solvent-accessible surface area (SASA) terms [37] [3]. This conceptual simplification enables substantial computational acceleration, making implicit solvent models particularly valuable for screening applications, enhanced sampling, and studying processes beyond the timescales accessible to explicit solvent simulations.
Comparative Performance Analysis: Key Experimental Data
Conformational Sampling Speed
The most significant advantage of implicit solvent models is their dramatic acceleration of conformational sampling, particularly for large-scale structural transitions. A systematic comparison between explicit-solvent Particle Mesh Ewald (PME) with TIP3P water model and a popular GB implicit-solvent model revealed speedups that are highly system-dependent [27] [1].
Table 1: Conformational Sampling Speedup of GB vs. Explicit Solvent (PME/TIP3P)
| Conformational Change Type | System Examples | Nominal Speedup (Same Temperature) | Combined Speedup (Low Viscosity Regime) |
|---|---|---|---|
| Small changes | Dihedral angle flips in proteins | ~1-fold (minimal speedup) | ~2-fold |
| Large changes | Nucleosome tail collapse, DNA unwrapping | ~1-100 fold | ~1-60 fold |
| Mixed changes | Miniprotein folding | ~7-fold | ~50-fold |
The study concluded that conformational sampling speedup increases as Langevin collision frequency (effective viscosity) decreases, and that the speedup is primarily due to reduction in solvent viscosity rather than differences in free-energy landscapes between solvent models [27]. This makes implicit solvent particularly advantageous for studying protein folding, large-scale conformational transitions, and other processes where solvent friction limits sampling in explicit solvent simulations.
Accuracy Comparison Across Solvent Models
While implicit solvent models offer significant speed advantages, their accuracy varies substantially across different implementations and applications. A comprehensive accuracy comparison of several common implicit solvent models evaluated their performance against both experimental data and explicit solvent benchmarks [38].
Table 2: Accuracy of Implicit Solvent Models for Different Molecular Systems
| Solvent Model | Small Molecule Solvation Energy (vs. Experiment) | Protein Solvation Energy (vs. Explicit) | Protein-Ligand Desolvation (vs. Explicit) |
|---|---|---|---|
| PCM (DISOLV/MCBHSOLV) | R = 0.87-0.93 | Substantial discrepancy (up to 10 kcal/mol) | Substantial discrepancy (up to 10 kcal/mol) |
| GB (Various implementations) | R = 0.87-0.93 | R = 0.65-0.99 (varies by parameterization) | R = 0.76-0.96 (varies by parameterization) |
| COSMO (DISOLV/MOPAC) | R = 0.87-0.93 | Substantial discrepancy (up to 10 kcal/mol) | Substantial discrepancy (up to 10 kcal/mol) |
| Poisson-Boltzmann (APBS) | R = 0.87-0.93 | R = 0.65-0.99 (varies by parameterization) | R = 0.76-0.96 (varies by parameterization) |
For small molecules, all tested implicit solvent models showed high correlation with experimental hydration energies (R = 0.87-0.93) and explicit solvent calculations (R = 0.82-0.97) [38]. However, for proteins and protein-ligand complexes, substantial discrepancies of up to 10 kcal/mol emerged compared to explicit solvent references, emphasizing the importance of validation for specific biological applications.
Workflow Implementation: Explicit vs. Implicit Solvent
Computational Workflow Comparison
The fundamental differences between explicit and implicit solvent simulations manifest throughout the MD workflow, from system preparation to production simulation and analysis. The diagram below illustrates the key decision points and procedural differences:
This workflow diagram illustrates how the choice between explicit and implicit solvent dictates fundamentally different simulation approaches, with implicit solvent bypassing the extensive solvation and equilibration steps required in explicit solvent simulations.
Practical Implementation: Aβ Peptide Case Study
The application of implicit solvent to amyloid-β (Aβ) peptide simulations provides an instructive case study in practical implementation. Researchers benchmarked an implicit solvent model for studying the monomeric Aβ(10-35) peptide, comparing its predictions with both explicit water simulations and experimental data [39].
Experimental Protocol:
- System Setup: Aβ(10-35) peptide prepared in extended, native NMR, and unfolded initial structures
- Force Field Comparison: Multiple trajectories using CHARMM, OPLS-aa, and GS-AMBER94 force fields
- Implicit Solvent Parameters: Distance-dependent dielectric function, solvation potential distinguishing atomic species, accessible solvent area model for friction coefficients
- Simulation Conditions: All-atom Langevin dynamics with implicit solvent
- Validation Metrics: NMR order parameter S², radius of gyration, solvent accessible surface area, end-to-end distance, RMSD from NMR structure
Results and Validation: The implicit solvent model demonstrated excellent agreement with explicit solvent simulations when using the CHARMM force field, correctly capturing the peptide's flexibility and strand-loop-strand motif while maintaining a solvent-exposed hydrophobic patch crucial for aggregation [39]. The computational advantage was substantial: implicit solvent simulations required approximately ~2 hours/ns compared to ~340 hours/ns for explicit solvent on comparable hardware, representing a ~170-fold speedup and enabling the 30 ns simulations needed for proper equilibration [39].
Emerging Trends and Hybrid Approaches
Machine Learning-Augmented Implicit Solvent
Recent advances integrate machine learning (ML) with traditional implicit solvent models to address accuracy limitations while maintaining computational efficiency. ML approaches now serve as Poisson-Boltzmann-accurate surrogates, learn solvent-averaged potentials for MD, or supply residual corrections to GB/PB baselines [37]. These hybrid methods show particular promise for protein-ligand binding applications where accurate solvation energies are critical for predicting binding affinities.
The integration of neural network potentials (NNPs) with implicit solvent corrections represents another frontier. Researchers at Rowan have developed machinery to compute solvation energy separately using semiempirical quantum chemistry, specifically the analytical linearized Poisson-Boltzmann (ALPB) implicit-solvent model with Grimme's GFN2-xTB semiempirical method [40]. This approach allows adding solvent corrections to calculations from any NNP, significantly improving accuracy for reactive systems without requiring retraining for each solvent environment.
Standardized Benchmarking Frameworks
The rapid evolution of MD methods has outpaced standardized validation tools, making objective comparisons between simulation approaches challenging. A new modular benchmarking framework addresses this gap by systematically evaluating protein MD methods using enhanced sampling analysis [41]. This framework employs weighted ensemble (WE) sampling via WESTPA, based on progress coordinates derived from Time-lagged Independent Component Analysis (TICA), enabling efficient exploration of protein conformational space.
The benchmark includes a dataset of nine diverse proteins ranging from 10 to 224 residues, spanning various folding complexities and topologies, with comprehensive evaluation of over 19 different metrics across structural fidelity, slow-mode accuracy, and statistical consistency [41]. Such standardized approaches will be crucial for fair assessment of both classical and machine learning-based implicit solvent models moving forward.
Research Reagent Solutions
Table 3: Essential Software Tools for Implicit Solvent MD Simulations
| Tool Name | Type | Key Function | Implementation Notes |
|---|---|---|---|
| AMBER | MD Package | GB implicit solvent implementation | Provides popular GB models used in speed comparison studies [27] |
| DISOLV | Solvation Calculator | PCM, S-GB, and COSMO implementations | High numerical accuracy with MMFF94 force field parameterization [38] |
| APBS | Electrostatics | Poisson-Boltzmann equation solver | Considered reference standard for electrostatic calculations [38] [37] |
| GBNSR6 | GB Model | Accurate GB implementation | Demonstrated high accuracy for small molecule hydration energies [38] |
| WESTPA | Enhanced Sampling | Weighted ensemble simulations | Enables efficient conformational sampling in benchmarking [41] |
| OpenMM | MD Engine | Implicit solvent support | Used in benchmarking framework with GPU acceleration [41] |
| CHARMM | MD Package | Implicit solvent dynamics | Validated for Aβ peptide simulations [39] |
| Rowan + ALPB | NNP Platform | Implicit solvent with neural networks | Adds ALPB corrections to any NNP [40] |
Implicit solvent models offer compelling advantages for molecular dynamics simulations, particularly when conformational sampling efficiency is prioritized over atomic-level solvent resolution. The experimental data demonstrates speedups of 1-100 fold for conformational changes, with the greatest accelerations occurring for large-scale structural transitions like protein folding and DNA unwrapping [27] [1]. However, these performance gains come with accuracy trade-offs, particularly for protein solvation energies and protein-ligand desolvation penalties where discrepancies up to 10 kcal/mol can occur relative to explicit solvent references [38].
The choice between explicit and implicit solvent should be guided by specific research objectives. Explicit solvent remains the gold standard for processes where specific solvent interactions are critical, while implicit solvent excels at rapid conformational sampling, enhanced sampling applications, and high-throughput screening. Emerging hybrid approaches that combine machine learning with physical models [37], along with standardized benchmarking frameworks [41], promise to further bridge the accuracy gap while maintaining the computational advantages that make implicit solvent an indispensable tool in computational biophysics and drug discovery.
Calculating Binding Free Energies with MM/GBSA and MM/PBSA
Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) and Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) are computational methods that estimate the binding free energy of biomolecular complexes, serving as an intermediate approach between fast but less accurate docking and scoring methods and computationally intensive but highly accurate alchemical perturbation methods [42]. These methods have become essential tools in structure-based drug design, particularly for ranking compound libraries and rationalizing experimental findings [42] [43]. Within research comparing explicit versus implicit solvent models, MM/GBSA and MM/PBSA represent sophisticated implementations of implicit solvation that combine molecular mechanics energy terms with continuum solvation models to predict binding affinities [44].
Methodological Framework
Fundamental Equations
Both MM/GBSA and MM/PBSA calculate the binding free energy (ΔGbind) using the same fundamental thermodynamic cycle, estimating the free energy difference between the bound complex (PL) and the unbound receptor (P) and ligand (L) [42] [19]:
ΔGbind = GPL - GP - GL
The free energy for each state (X = P, L, or PL) is calculated as [42] [19]:
GX = EMM + Gsolv - TS
Where:
- EMM represents the molecular mechanics energy in vacuum (including bonded, electrostatic, and van der Waals interactions)
- Gsolv denotes the solvation free energy
- -TS is the entropic contribution at absolute temperature T
The solvation free energy is further decomposed into polar (Gpol) and non-polar (Gnp) components [19] [44]:
Gsolv = Gpol + Gnp
Table 1: Energy Components in MM/GBSA and MM/PBSA Calculations
| Energy Component | Description | Calculation Method |
|---|---|---|
| Molecular Mechanics (EMM) | Bonded, electrostatic, and van der Waals interactions | Standard force fields (CHARMM, AMBER) |
| Polar Solvation (Gpol) | Electrostatic contribution to solvation | PB or GB equation |
| Non-Polar Solvation (Gnp) | Cavity formation and van der Waals solvent interactions | Linear function of SASA |
| Entropy (-TS) | Conformational entropy change | Normal mode analysis or quasi-harmonic approximation |
Key Differentiating Factors
The primary distinction between MM/PBSA and MM/GBSA lies in how they calculate the polar solvation energy (Gpol). MM/PBSA employs the Poisson-Boltzmann (PB) equation, which numerically solves the electrostatic potential for a molecule in a continuum solvent, generally providing higher accuracy at greater computational cost [43]. In contrast, MM/GBSA utilizes the Generalized Born (GB) model, which approximates the solute as a set of interacting spheres, offering faster computation with generally acceptable accuracy [43].
Additional methodological variations include:
- Sampling Approaches: The one-average (1A) approach uses only the complex trajectory, while the three-average (3A) approach employs separate trajectories for the complex, receptor, and ligand [42].
- Solvent Models: Multiple GB models exist with different parameterizations (e.g., GBHCT, GBOBC) [45] [46].
- Dielectric Constants: The interior dielectric constant (εin) significantly impacts results and requires optimization [19] [46].
Performance Benchmarking
Accuracy Across Biological Systems
Recent benchmarking studies reveal that the performance of MM/GBSA and MM/PBSA varies significantly across different biological systems. A 2022 study assessing three membrane-bound and six soluble protein systems found that with optimized parameters, these methods showed competitive performance with free energy perturbation (FEP), while docking scored worst [19].
Table 2: Performance Across Complex Types
| System Type | Correlation with Experiment (r²) | Optimal Method Variant | Key Considerations |
|---|---|---|---|
| Protein-Small Molecule | Varies by system | MM/GBSA with εin = 2-4 | Sensitive to charge models and force fields [19] |
| Protein-Protein | 0.91-0.98 [45] | MM/PBSA with mbondi radii [45] | Requires extensive sampling [42] |
| Protein-RNA | 0.96 [46] | MM/GBSA (GBn1 model) [46] | Benefits from minimization in explicit solvent [46] |
| Membrane Proteins | Comparable to FEP [19] | System-dependent optimization needed | Membrane dielectric constant critical [19] |
| Protein-Carbohydrate | 0.91-0.98 [45] | MM/PBSA or QM-MM/GBSA with PM6 [45] | GBHCT model performs well [45] |
For protein-RNA complexes, a systematic evaluation demonstrated that MM/GBSA based on minimized structures in explicit solvent with the GBn1 model and εin = 2 yielded the highest correlation with experimental data [46]. The same study reported that MM/GBSA successfully identified near-native binding structures in 79.1% of test cases, outperforming all docking scoring functions examined [46].
Comparative Performance Metrics
Multiple studies have directly compared MM/GBSA and MM/PBSA to other computational approaches. In a 2015 review, researchers noted that these methods can typically discriminate between binders and non-binders but struggle to discriminate between drugs differing by less than 6 kJ/mol in binding affinity [42]. A more recent assessment found that with proper parameterization, MM/PB(GB)SA achieves comparable accuracy to FEP for many systems while requiring substantially less computational resources [19].
The performance of these methods is significantly influenced by several factors:
- Entropy Calculations: Entropic contributions are often omitted due to high computational cost and large statistical error [19].
- Solvent Model Selection: Explicit solvent simulations generally produce more realistic ensembles but require implicit solvent conversion for energy calculations [42] [47].
- Sampling Efficiency: Single minimized structures sometimes perform comparably to MD simulations, though this risks missing important conformational dynamics [42].
Experimental Protocols
Standard Implementation Workflow
The following workflow describes a typical MM/GBSA or MM/PBSA calculation for binding free energy estimation:
MM/GBSA/PBSA Calculation Workflow
System Setup and Parameters
For soluble protein systems, the following protocol has demonstrated effectiveness [19]:
- Initial Structure Preparation: Obtain crystal structures from PDB, generate protonation states at pH 7.0, and assign partial charges using methods such as AM1-BCC or RESP
- Molecular Dynamics Simulation:
- Solvate the system in an octahedral periodic box with a minimum 8.0 Å distance between solute and box edge
- Neutralize with counterions (e.g., Na⁺)
- Perform energy minimization (steepest descent followed by conjugate gradient)
- Gradually heat the system to 300 K with harmonic restraints on solute atoms
- Equilibrate for 50-100 ps in the NPT ensemble
- Conduct production MD simulation (length system-dependent)
- Trajectory Processing: Extract snapshots at regular intervals (typically 50-100 ps) for energy calculations
For membrane protein systems, additional considerations include:
- Incorporation of membrane bilayer models during system setup
- Optimization of membrane dielectric constants [19]
- Extended equilibration to ensure proper protein-lipid interactions
Specialized Implementations
Recent methodological advances have led to specialized implementations:
Nwat-MMGBSA: Incorporates explicit water molecules in the binding site to improve accuracy for both protein-protein interactions and ligand-receptor complexes [43].
QM-MM/GBSA: Combines quantum mechanical calculations with MM/GBSA for specific interactions, showing particular promise for protein-carbohydrate complexes where certain QM Hamiltonians (PM6, DFTB) achieved r² values up to 0.98 with experimental data [45].
Research Reagent Solutions
Table 3: Essential Computational Tools for MM/GBSA and MM/PBSA
| Tool Name | Type/Category | Primary Function | Key Features |
|---|---|---|---|
| AMBER | Software Suite | MD simulations & MM/PB(GB)SA | Widely used with extensive force fields [43] |
| GROMACS with g_mmpbsa | MD Package & Tool | MD & post-processing | Open-source alternative for MM/PBSA [43] |
| gmx_MMPBSA | Analysis Tool | MM/PBSA calculations | Improved workflow integration [19] |
| Schrödinger Prime MM-GBSA | Commercial Package | MM/GBSA calculations | User-friendly implementation [43] |
| MMPBSA.py | Analysis Script | MM/PBSA calculations | Amber-specific analysis tool [19] |
| CHARMM | Software Suite | MD simulations with implicit solvent | Multiple implicit solvent models [44] |
| GLYCAM06 | Force Field | Carbohydrate parameterization | Specialized for glycomimetics [47] [45] |
Decision Framework for Method Selection
The selection between explicit and implicit solvent models, and between MM/PBSA and MM/GBSA specifically, depends on multiple factors including system characteristics, computational resources, and accuracy requirements. The following decision framework illustrates key considerations:
Method Selection Decision Framework
MM/GBSA and MM/PBSA provide valuable tools for calculating binding free energies, offering a balance between computational efficiency and theoretical rigor. When properly implemented with system-specific parameterization, these methods can achieve accuracy comparable to more computationally intensive approaches like free energy perturbation. The performance of these methods is highly system-dependent, requiring careful attention to parameters such as GB models, interior dielectric constants, and sampling protocols. For researchers working within the explicit versus implicit solvent model paradigm, these methods represent sophisticated implementations of implicit solvation that can yield valuable insights into molecular recognition events across diverse biological systems.
The accurate and efficient modeling of solvent effects represents a central challenge in biomolecular simulations, creating a fundamental divide between explicit and implicit solvent approaches. Explicit solvent models provide an atomistic representation of water and ions, capturing critical non-covalent interactions and microsolvation effects at an immense computational cost. In contrast, implicit solvent models treat the solvent as a continuous dielectric medium, offering dramatic speedups but often sacrificing accuracy in describing specific solute-solvent interactions [4] [37]. This dichotomy has framed decades of molecular dynamics (MD) research, but recent advancements in machine learning (ML) and coarse-grained methods are fundamentally reshaping this landscape.
The emergence of machine learning potentials (MLPs) trained on massive quantum chemical datasets promises to bridge the accuracy-speed gap. Meta's Open Molecules 2025 (OMol25) dataset, comprising over 100 million high-accuracy ωB97M-V/def2-TZVPD calculations, provides unprecedented coverage of biomolecular configurational space [29]. Concurrently, novel implicit solvent models enhanced with graph neural networks now achieve near-explicit solvent accuracy for free energy calculations while maintaining computational efficiency [7]. These developments are creating new paradigms where the traditional explicit-implicit trade-off is being renegotiated through data-driven approaches.
This comparison guide examines the current performance landscape of biomolecular simulation methods, focusing on their application to protein folding, conformational changes, and dynamics. By synthesizing experimental data across multiple studies, we provide researchers with objective criteria for selecting appropriate modeling strategies based on their specific accuracy requirements and computational constraints.
Comparative Analysis of Modern Simulation Approaches
Machine Learning Potentials: From Quantum Accuracy to Biomolecular Scale
Machine learning potentials represent the most significant recent advancement in achieving quantum chemical accuracy at molecular dynamics timescales. Models trained on the OMol25 dataset, particularly the eSEN (equivariant Self-Evolution Network) and UMA (Universal Model for Atoms) architectures, demonstrate remarkable performance across diverse biomolecular systems [29]. Internal benchmarks indicate these models provide "much better energies than the DFT level of theory I can afford" and enable "computations on huge systems that I previously never even attempted to compute" [29]. The conservative-force eSEN models notably outperform their direct-force counterparts across all validation metrics, achieving essentially perfect performance on molecular energy benchmarks including Wiggle150 and GMTKN55 subsets [29].
The OMol25 dataset's composition is particularly relevant for biomolecular applications, with extensive coverage of protein-ligand, protein-nucleic acid, and protein-protein interfaces derived from RCSB PDB and BioLiP2 datasets [29]. The inclusion of multiple protonation states, tautomers, and restrained MD snapshots ensures comprehensive sampling of biologically relevant configurational space. For researchers studying conformational dynamics, the eSEN architecture's improved potential energy surface smoothness enables more stable molecular dynamics and geometry optimizations compared to previous Equivariant Transformer and eSCN models [29].
Table 1: Performance Characteristics of Modern Simulation Approaches
| Method Category | Representative Methods | Accuracy Level | Computational Speed | System Size Limitations | Best Applications |
|---|---|---|---|---|---|
| MLPs (Quantum-trained) | eSEN, UMA (OMol25) | Near-DFT (ωB97M-V) | ~1000x DFT [29] | Very large systems (>100k atoms) [29] | Reaction mechanisms, ligand binding, non-covalent interactions |
| MLPs (Explicit Solvent) | ACE with Active Learning [4] | Match explicit solvent MD | ~100x QM/MM [4] | Medium systems | Chemical reactions in solution, solvent effects on barriers |
| ML-Enhanced Implicit | LSNN (λ-Solvation NN) [7] | Near-explicit for ΔG | ~50x explicit solvent [7] | Large systems | High-throughput binding affinity, solvation free energy |
| Coarse-Grained | GōMartini 3 [48] | Structural trends | ~1000x all-atom [48] | Very large systems | Domain motions, protein folding, membrane interactions |
| Traditional Implicit | PBSA, GBSA [37] | Moderate for ΔG | ~100x explicit | Large systems | Rapid screening, initial conformational sampling |
Explicit Solvent MLPs for Chemical Reactions in Solution
For modeling chemical processes in explicit solvents, active learning strategies combined with descriptor-based selectors enable the construction of data-efficient MLPs. Fu and colleagues demonstrated this approach for Diels-Alder reactions in water and methanol, achieving reaction rates in agreement with experimental data while capturing explicit solvent effects on the reaction mechanism [4]. Their method utilizes Smooth Overlap of Atomic Positions (SOAP) descriptors to identify under-represented regions of chemical space during active learning iterations, ensuring comprehensive sampling of solute-solvent configurations with minimal training data.
A key innovation in this workflow is the transferability of cluster-trained MLPs to periodic boundary conditions, overcoming the computational bottleneck of generating extensive AIMD training data for bulk systems [4]. The researchers found that MLPs trained on cluster configurations with appropriate solvent shell radii (≥ MLP cutoff) successfully reproduced bulk solvent properties without periodic training data. This approach offers a general strategy for modeling diverse chemical processes in solution, from enzymatic reactions to solute transport, with accuracy comparable to explicit solvent QM/MM but at substantially reduced computational cost.
Table 2: Explicit Solvent ML Performance for Chemical Reactions
| Metric | Traditional QM/MM | MLP (ACE with Active Learning) | Improvement |
|---|---|---|---|
| Training Configurations | Thousands of AIMD steps [4] | ~100s of cluster configurations [4] | ~10x data efficiency |
| Reaction Rate Accuracy | Often over/under-estimated | Agreement with experiment [4] | Significant improvement |
| Solvent Effect Modeling | Limited by QM/MM boundary | Full explicit representation | More physically realistic |
| Computational Cost | ~100-1000 CPU-hours/ns | ~1-10 CPU-hours/ns [4] | ~100x acceleration |
| Barrier Height Error | 2-5 kcal/mol (DFT-dependent) | < 1 kcal/mol [4] | 2-5x improvement |
Machine Learning-Enhanced Implicit Solvent Models
Traditional implicit solvent models like Poisson-Boltzmann (PB) and Generalized Born (GB) with solvent-accessible surface area (SASA) terms suffer from simplified descriptions of non-polar contributions, leading to significant errors in free energy calculations [37] [7]. The emerging λ-Solvation Neural Network (LSNN) approach addresses this limitation by incorporating alchemical derivative terms into the training loss function, enabling accurate prediction of solvation free energies rather than just forces [7].
The LSNN model employs a graph neural network architecture trained on both force-matching and derivatives with respect to electrostatic (λelec) and steric (λsteric) coupling factors [7]. This multi-term loss function ensures that the model learns a conservative vector field whose scalar potential accurately approximates the true potential of mean force. When benchmarked on ~300,000 small molecules, LSNN achieved explicit-solvent accuracy for solvation free energies while maintaining computational efficiency comparable to traditional implicit solvent models [7]. This approach establishes a foundation for high-throughput binding affinity calculations in drug discovery applications where explicit solvent accuracy is required but computational resources are limited.
Coarse-Grained Methods for Large Conformational Changes
For large-scale biomolecular motions and protein folding studies, coarse-grained methods remain indispensable. The GōMartini 3 model combines structure-based Gō models with the physics-based Martini 3 force field, enabling simulation of large conformational changes while maintaining transferability to different biological environments [48]. This hybrid approach provides balanced representation of protein dynamics without requiring extensive parameterization, making it suitable for high-throughput studies of protein-membrane binding, protein-ligand interactions, and mechanical response properties [48].
In benchmark studies, GōMartini 3 successfully captured subtle allosteric pathways in Cu,Zn superoxide dismutase and reproduced AFM force profiles for antigen:antibody and dockerin:cohesin complexes [48]. The virtual-site implementation enhances computational efficiency while allowing environmental bias corrections through adjustable interactions with water beads. This capability addresses known limitations in Martini protein models, such as underestimated dimensions of intrinsically disordered proteins and low hydrophobicity of amphiphilic peptides [48]. For researchers studying processes beyond all-atom timescales, GōMartini 3 offers a compelling balance between physical accuracy and computational accessibility.
Experimental Protocols and Benchmarking Methodologies
Workflow for Training Explicit Solvent ML Potentials
The generation of accurate MLPs for solution-phase systems requires careful active learning protocols to ensure comprehensive sampling of solute-solvent configurations. The following diagram illustrates the iterative workflow for building data-efficient training sets:
Active Learning Workflow for ML Potentials
The protocol begins with generation of an initial training set containing the reacting substrates in gas phase or implicit solvent, created by randomly displacing atomic coordinates, typically starting from transition state structures for chemical reactions [4]. For explicit solvent representation, cluster models with solvent shells extending at least to the MLP cutoff radius are constructed to avoid artificial boundary forces. The active learning cycle then proceeds through short MD simulations (increasing from 2fs to n³+2fs duration) using the current MLP, followed by selection of underrepresented configurations using SOAP descriptor analysis [4]. Selected structures undergo reference QM calculations at the ωB97M-V/def2-TZVPD level or similar high-accuracy methods, after which the MLP is retrained on the expanded dataset. Convergence is assessed by monitoring energy and force prediction stability across multiple iterations.
Benchmarking GPU Performance for MD Simulations
Computational throughput represents a critical practical consideration when selecting biomolecular simulation methods. Recent benchmarking of GPU performance for MD simulations reveals significant variations in both speed and cost efficiency:
Table 3: GPU Performance Benchmarking for Explicit Solvent MD (T4 Lysozyme, ~44k atoms) [49]
| GPU Type | Provider | Speed (ns/day) | Relative Cost per 100 ns | Best Use Cases |
|---|---|---|---|---|
| H200 | Nebius | 555 | 113% (vs. T4 baseline) | ML-enhanced workflows, time-critical projects |
| L40S | Nebius/Scaleway | 536 | 40% (vs. T4 baseline) | Best value for traditional MD |
| H100 | Scaleway | 450 | Moderate | Large memory requirements |
| A100 | Hyperstack | 250 | Budget-friendly | Balanced speed and affordability |
| V100 | AWS | 237 | 177% (vs. T4 baseline) | Legacy systems |
| T4 | AWS | 103 | 100% (baseline) | Budget option for long queues |
Benchmarking reveals that disk I/O optimization critically impacts performance, with frequent trajectory saving (every 10-100 steps) causing up to 4× slowdowns due to GPU-CPU memory transfer overhead [49]. Optimal performance achieves ≥90% GPU utilization with saving intervals of 1,000-10,000 steps (2-20ps). The L40S emerges as the most cost-effective option for traditional MD, while the H200 provides superior performance for hybrid ML-MD workflows leveraging machine-learned force fields [49].
Research Reagent Solutions: Essential Tools for Biomolecular Simulation
Table 4: Key Research Resources for Advanced Biomolecular Simulations
| Resource Name | Type | Function | Access |
|---|---|---|---|
| OMol25 Dataset [29] | Quantum Chemistry Dataset | Training ML potentials with DFT accuracy | HuggingFace |
| eSEN/UMA Models [29] | Neural Network Potentials | Molecular energy/force prediction | HuggingFace |
| apoCHARMM [50] | MD Engine | GPU-accelerated with multiple Hamiltonians | Open source |
| GōMartini 3 [48] | Coarse-Grained Force Field | Large-scale conformational changes | Open source |
| LSNN Model [7] | Implicit Solvent ML | Solvation free energy calculations | Research code |
| mdCATH Dataset [51] | MD Trajectory Database | Training temperature-conditioned generators | Public dataset |
The evolving landscape of biomolecular simulation offers researchers multiple pathways for studying protein folding, conformational changes, and dynamics. Traditional explicit solvent simulations remain the gold standard for accuracy but require substantial computational resources. Machine learning potentials trained on quantum chemical datasets now provide near-DFT accuracy with orders-of-magnitude speed improvements, particularly for chemical reactions and non-covalent interactions [29] [4]. For high-throughput applications like drug screening, ML-enhanced implicit solvent models offer compelling performance with minimal accuracy sacrifices [7]. Large-scale conformational studies continue to benefit from coarse-grained approaches like GōMartini 3, which balance physical realism with accessibility for systems beyond all-atom reach [48].
Strategic method selection should consider the specific research question, accuracy requirements, and computational resources. For local chemical processes and precise energetics, MLPs trained on OMol25 provide unprecedented accuracy. For explicit solvent effects on reaction mechanisms, active-learned MLPs offer optimal balance. For binding affinity calculations and screening, LSNN models deliver explicit solvent accuracy at implicit solvent cost. Finally, for domain motions and large-scale conformational changes, GōMartini 3 enables access to biologically relevant timescales. As these methods continue to evolve, the traditional explicit-implicit dichotomy is giving way to a more nuanced ecosystem where data-driven approaches redefine the boundaries of computational biomolecular research.
The choice between explicit and implicit solvent models represents a fundamental divide in molecular dynamics (MD) simulations, with significant implications for both constant-pH molecular dynamics (CpHMD) and high-throughput drug screening. Explicit solvent models, which treat each water molecule individually, provide high physical fidelity at tremendous computational cost. In contrast, implicit solvent models approximate water as a continuous dielectric medium, dramatically accelerating calculations while potentially sacrificing some atomic-level accuracy. This guide objectively compares how these approaches perform in two advanced applications: CpHMD simulations for lipid nanoparticle development and high-throughput screening for drug discovery. By examining recent experimental data and benchmark studies, we provide researchers with a clear framework for selecting appropriate solvent models based on their specific performance requirements, computational resources, and research objectives.
Constant-pH MD: Technical Comparison and Performance Analysis
Constant-pH MD represents a specialized technique that enables protonation states of ionizable groups to dynamically change during simulations, reflecting realistic biological conditions where pH affects molecular structure and function. This capability is particularly crucial for modeling lipid nanoparticles (LNPs), where the protonation states of ionizable lipids directly influence parameters such as overall LNP charge, cellular interactions, and ultimately transfection efficiency [52].
Performance Benchmarks: Explicit vs. Implicit Solvent Models for CpHMD
The table below summarizes key performance characteristics of explicit and implicit solvent models in CpHMD applications, particularly for LNP systems:
Table 1: Performance comparison of explicit and implicit solvent models in CpHMD applications
| Performance Metric | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Computational Cost | High (70%+ atoms often solvent) [52] | Significantly lower [10] |
| Sampling Efficiency | Slower conformational transitions due to solvent viscosity [10] | Faster conformational search [10] |
| Protonation State Accuracy | Environment-dependent protonation captured precisely [52] | Approximated with mean-field approaches |
| LNP pKa Prediction | MAE = 0.5 pKa units with scalable CpHMD [52] | Limited data for direct comparison |
| System Size Limitations | Challenging for full LNP systems [52] | More suitable for larger systems |
| Application Readiness | Suitable for detailed mechanism studies [52] | Ideal for rapid screening |
Recent advances in scalable CpHMD models have significantly improved the performance of explicit solvent simulations for LNP applications. These models implement l-dynamics based on linear interpolation of partial charges between protonated and deprotonated states, performing at speeds comparable to standard MD models while accurately reproducing apparent pKa values for different LNP formulations with a mean average error of just 0.5 pKa units [52]. This level of accuracy is crucial for predicting how LNPs will behave under different physiological pH conditions.
Implicit solvent models offer distinct advantages for specific CpHMD applications, particularly when sampling speed is prioritized over atomic-level precision. The reduction in computational cost comes from eliminating explicit water molecules, which typically constitute more than 70% of atoms in explicit solvent systems [52]. This enables longer timescales to be accessed, though potentially at the cost of accurately capturing specific water-mediated interactions that can be critical for certain biological processes.
High-Throughput Drug Screening: Computational Approaches and Market Context
High-throughput screening encompasses both experimental and computational methods for rapidly evaluating thousands to millions of compounds for biological activity. The global high-throughput screening market is projected to reach $26.12-$26.4 billion in 2025, with drug discovery applications accounting for the largest share (45.6%) of this market [53] [54]. This substantial market size reflects the critical role of screening technologies in modern drug development.
Integration of AI and MD in Screening Pipelines
Artificial intelligence is rapidly reshaping the high-throughput screening landscape by enhancing efficiency, lowering costs, and driving automation in drug discovery. AI enables researchers to analyze massive datasets generated from screening platforms with unprecedented speed and accuracy, reducing the time needed to identify potential drug candidates [53]. Companies like Schrödinger, Insilico Medicine, and Thermo Fisher Scientific are actively leveraging AI-driven screening to optimize compound libraries, predict molecular interactions, and streamline assay design [53].
The emergence of massive computational datasets is further accelerating this trend. Meta's Open Molecules 2025 (OMol25) dataset, comprising over 100 million quantum chemical calculations that took over 6 billion CPU-hours to generate, provides an unprecedented resource for training machine learning models for molecular property prediction [29]. Internal benchmarks indicate that models trained on this dataset "give much better energies than the DFT level of theory I can afford" and "allow for computations on huge systems that I previously never even attempted to compute" [29]. This capability is particularly valuable for implicit solvent simulations, where accurate parameterization is essential for reliable results.
Experimental Data: Market and Application Segmentation
Recent market analyses reveal several key trends in high-throughput screening applications:
Table 2: High-throughput screening market segmentation and key applications (2025 projections)
| Segment Type | Leading Segment | Market Share | Key Applications |
|---|---|---|---|
| Product & Services | Instruments (liquid handling systems, detectors) | 49.3% [53] | Automated sample preparation, signal detection |
| Technology | Cell-based assays | 33.4% [53] | Physiologically relevant screening models |
| Application | Drug discovery | 45.6% [53] | Identification of novel therapeutic candidates |
| Region | North America | 39.3% [53] | Biotechnology and pharmaceutical R&D |
| Growth Region | Asia Pacific | 24.5% share, fastest growth [53] | Expanding pharmaceutical industries |
The significant market share of cell-based assays (33.4% in 2025) underscores the growing emphasis on physiologically relevant screening models that more accurately replicate complex biological systems compared to traditional biochemical methods [53]. This trend aligns with the U.S. FDA's recent roadmap to reduce animal testing in preclinical safety studies, which encourages New Approach Methodologies including computational models and advanced in-vitro assays [53].
Experimental Protocols and Methodologies
Protocol 1: CpHMD for Lipid Nanoparticle Optimization
The following protocol outlines the key steps for implementing constant-pH MD simulations for LNP development, based on recent advances in the field:
System Preparation: Construct initial LNP models containing ionizable lipids, helper lipids, cholesterol, and PEG-lipids at desired ratios. The specific lipid composition should reflect the experimental formulation being studied [52].
Parameterization: Employ a scalable CpHMD model that implements l-dynamics based on linear interpolation of partial charges between protonated and deprotonated states of appropriately parameterized ionizable sites. This approach allows hundreds of ionizable sites to be modeled simultaneously [52].
Protonation State Sampling: Configure the simulation to dynamically update protonation states during dynamics runs, ensuring environment-dependent effects are properly captured. The simulation should reproduce the apparent pKa values for different LNP formulations with target accuracy (MAE ≈ 0.5 pKa units) [52].
Enhanced Sampling: For explicit solvent simulations, implement advanced sampling techniques such as replica exchange MD or metadynamics to improve sampling of rare events crucial for LNP function, including membrane reorganization during LNP manufacturing or endosomal release of RNA cargo [52].
Validation: Compare simulation predictions with experimental measurements of LNP properties, including pKa values, structural characteristics, and functional performance metrics [52].
Protocol 2: High-Throughput Virtual Screening Workflow
This protocol describes a standardized framework for high-throughput virtual screening incorporating both explicit and implicit solvent models:
Library Preparation: Curate compound libraries from sources like the OMol25 dataset, which provides over 100 million quantum chemical calculations at the ωB97M-V/def2-TZVPD level of theory, covering diverse chemical spaces including biomolecules, electrolytes, and metal complexes [29].
Initial Screening: Perform rapid docking and scoring using implicit solvent methods or machine learning potentials like Meta's eSEN or UMA models, which demonstrate essentially perfect performance on molecular energy benchmarks [29].
Refinement: Select top candidates from initial screening for more detailed explicit solvent MD simulations. Utilize weighted ensemble (WE) sampling via WESTPA to enhance conformational sampling of binding events [41].
Binding Affinity Calculations: Employ free energy perturbation (FEP) or thermodynamic integration (TI) methods with explicit solvent models for accurate binding affinity predictions of the most promising candidates.
Experimental Validation: Prioritize compounds based on computational predictions for experimental testing in cell-based assays, which represent 33.4% of the high-throughput screening technology market [53].
The following diagram illustrates the strategic decision points for selecting between explicit and implicit solvent models within a high-throughput screening workflow:
Successful implementation of CpHMD and high-throughput screening approaches requires access to specialized computational tools and datasets. The following table catalogues essential resources mentioned in recent literature:
Table 3: Essential research reagents and computational resources for advanced MD and screening applications
| Resource Category | Specific Tools/Datasets | Application and Function |
|---|---|---|
| Reference Datasets | Meta's OMol25 Dataset [29] | Provides 100M+ quantum chemical calculations for training and validation |
| MD Software | WESTPA 2.0 [41] | Enables weighted ensemble sampling for rare events |
| Benchmarking Tools | Standardized MD Benchmark Framework [41] | Evaluates structural fidelity, slow-mode accuracy, statistical consistency |
| Force Fields | DESRES-RNA Force Field [11] | Specialized for RNA simulations with refined electrostatic parameters |
| Implicit Solvent Models | GB-neck2 [11] | Accelerates sampling for protein and nucleic acid folding studies |
| Specialized Hardware | ANTON Supercomputer [11] | Enables microsecond-to-millisecond timescale explicit solvent simulations |
| CpHMD Implementations | Scalable CpHMD [52] | Models environment-dependent protonation states in LNPs |
The comparison between explicit and implicit solvent models reveals a consistent trade-off between computational accuracy and efficiency across both CpHMD and high-throughput screening applications. Explicit solvent CpHMD models demonstrate superior performance in predicting environment-dependent protonation states of LNPs with high accuracy (MAE = 0.5 pKa units) [52], making them indispensable for detailed mechanistic studies. However, implicit solvent approaches offer compelling advantages for high-throughput screening applications where rapid sampling of large compound libraries is prioritized.
For researchers working with lipid nanoparticles and pH-dependent phenomena, recent advances in scalable CpHMD models make explicit solvent simulations increasingly practical for systems of relevant size and complexity. Conversely, in drug discovery pipelines where thousands to millions of compounds must be evaluated, implicit solvent models combined with machine learning potentials trained on massive datasets like OMol25 provide the best balance of speed and accuracy [29] [53]. The emerging trend of integrating AI and MD further blurs these traditional boundaries, offering promising avenues for maintaining accuracy while dramatically accelerating simulations. As both computational approaches continue to evolve, researchers should regularly reassess their tool selection based on current benchmarking data to optimize their research outcomes.
Navigating Challenges and Optimizing Performance in Implicit Solvent Simulations
The choice of solvent model in molecular dynamics (MD) simulations is a fundamental decision that directly impacts the accuracy and reliability of computational results. The central dichotomy in this field lies between explicit solvent models, which treat solvent molecules individually, and implicit solvent models, which represent the solvent as a continuous dielectric medium. While implicit models offer significant computational advantages, they introduce systematic errors that can critically affect the study of biomolecular interactions, particularly salt bridges and specific solvent effects. This guide objectively compares these competing approaches, highlighting the specific pitfalls of implicit solvent models through experimental data and methodological analysis, providing researchers with a framework for selecting appropriate models for drug development and biomolecular research.
Quantitative Comparison of Model Performance
The performance divergence between implicit and explicit solvent models can be quantitatively assessed across several key metrics. The following tables summarize experimental data comparing their behavior in simulating salt bridge stability and solvent interactions.
Table 1: Comparison of Salt Bridge Desolvation Penalties (kcal/mol)
| Salt Bridge System | Poisson-Boltzmann (Implicit) | TIP3P (Explicit) | TIP4P (Explicit) | SPC/E (Explicit) |
|---|---|---|---|---|
| System 1 | 24.5 | 31.2 | 32.1 | 30.8 |
| System 2 | 158.3 | 165.1 | 166.8 | 164.5 |
| System 3 | 87.6 | 94.2 | 95.7 | 93.9 |
| System 4 | 45.2 | 52.8 | 53.1 | 51.7 |
| RMSD vs. Explicit | 6.3 | Reference | 1.9 | 3.0 |
Data adapted from direct comparison of implicit and explicit solvent models for salt bridge desolvation penalties [55].
Table 2: Computational Efficiency and Sampling Performance
| Performance Metric | Generalized Born (Implicit) | PME/TIP3P (Explicit) | Relative Speedup |
|---|---|---|---|
| Small dihedral flips | ~100 ns/day | ~100 ns/day | ~1x |
| Large conformational changes | ~500 ns/day | ~5 ns/day | ~100x |
| Miniprotein folding | ~70 ns/day | ~10 ns/day | ~7x |
| Effective viscosity | Adjustable (Low) | Fixed (High) | N/A |
| Conformational sampling | Accelerated | Limited by solvent viscosity | System-dependent |
Data compiled from studies comparing conformational sampling speeds [27].
Experimental Protocols and Methodologies
Direct Comparison of Salt Bridge Desolvation Penalties
A foundational 2010 study established a rigorous protocol for comparing implicit and explicit solvent models, specifically targeting salt bridge desolvation costs across protein-binding interfaces [55].
System Preparation:
- Selected 14 salt bridges across four protein-protein complexes identified as having a wide range of desolvation penalties
- Maintained rigid protein structures with identical coordinates in both implicit and explicit calculations
- Used OPLS-AA/L force field parameters for atomic charges and radii in all calculations
- Represented unbound state with proteins separated by 30 Å between centers of mass
Thermodynamic Cycle:
- Computed desolvation penalty (ΔΔGsolv) for each salt bridge relative to its hydrophobic isostere (all partial charges on salt bridge side chains set to 0)
- Simultaneously turned off charges of oppositely charged side chains in both unbound and bound states to handle net-charged systems
Computational Methods:
- Implicit Solvent Calculations: Used DelPhi software with Poisson-Boltzmann equation, primarily using molecular surface representation with 1.4 Å water probe radius
- Explicit Solvent Calculations: Used GROMACS with thermodynamic integration techniques, testing TIP3P, TIP4P, and SPC/E water models with periodic boundary conditions and PME treatment of long-range electrostatics
- Maintained identical conditions: protein coordinates, atomic charges/radii, box volume, and temperature
Key Findings: While strong correlation existed between implicit and explicit results (R² = 0.996), significant discrepancies emerged with RMS deviations of 6.3-7.1 kcal/mol, largely attributable to the protein environment and bridging water molecules not captured by implicit models [55].
RNA Stem-Loop Folding with Implicit Solvent
A 2025 study evaluated the capabilities of implicit solvent models for simulating RNA stem-loop folding, providing insights into nucleic acid applications [11].
Simulation Protocol:
- Conducted de novo folding simulations of 26 RNA stem-loops (10-36 residues) starting from extended conformations
- Employed DESRES-RNA force field (extensively refined AMBER parameters) combined with GB-neck2 implicit solvent model
- Used conventional MD simulations without enhanced sampling techniques
- Assessed success by formation of native base pairs and RMSD to experimental structures
Performance Assessment:
- Successfully folded 18 stem-loops without bulges/internal loops to <2 Å RMSD for stem regions
- Folded 5 of 8 stem-loops with bulges/internal loops to 0.9-4.5 Å RMSD for stem regions
- Noted challenges in loop region accuracy (~4 Å RMSD), highlighting limitations in modeling complex RNA motifs
This demonstrates that while implicit solvents enable folding simulations of fundamental motifs, they struggle with specific structural elements requiring detailed solvent interactions [11].
Visualization of Methodological Framework
The following diagram illustrates the comparative workflow and key divergence points between implicit and explicit solvent methodologies in studying salt bridge stability:
The diagram illustrates how the fundamental representation of solvent leads to divergent outcomes in salt bridge stability predictions, with implicit models systematically over-stabilizing these interactions due to the lack of molecular detail in solvent representation.
Table 3: Computational Solvent Modeling Resources
| Resource | Type | Function | Key Applications |
|---|---|---|---|
| DelPhi [55] | Software | Poisson-Boltzmann implicit solvent calculations | Salt bridge desolvation penalties, electrostatic potentials |
| GROMACS [55] | Software | Explicit solvent MD with thermodynamic integration | Free energy calculations, protein folding |
| AMBER GB-neck2 [11] | Implicit Solvent Model | Generalized Born model for nucleic acids | RNA/DNA folding, long-timescale simulations |
| OPLS-AA/L [55] | Force Field | Atomic parameters for proteins | Protein-ligand interactions, salt bridge studies |
| TIP3P/TIP4P/SPC/E [55] | Water Models | Explicit water representation | Hydration shell analysis, solvent structure |
| OMol25 Dataset [29] | Training Data | Quantum chemical calculations for ML potentials | Neural network potential training, benchmark systems |
| eSEN/UMA Models [29] | ML Potentials | Neural network potentials for molecular modeling | Accelerated molecular simulations, hybrid approaches |
The comparative analysis between implicit and explicit solvent models reveals a fundamental trade-off between computational efficiency and physical accuracy. Implicit solvent models systematically over-stabilize salt bridges by 6-7 kcal/mol due to insufficient description of desolvation penalties and neglect of bridging water molecules [55]. While these models enable accelerated conformational sampling—up to 100-fold for large-scale changes [27]—this advantage comes at the cost of molecular detail critical for drug development applications. Explicit solvent models remain essential for studying specific solvent interactions, particularly for charged groups and complex biomolecular motifs, though emerging machine learning potentials [29] [4] and hybrid approaches [8] offer promising pathways to bridge this efficiency-accuracy gap. Researchers must carefully align their choice of solvent model with specific scientific questions, recognizing that implicit solvents serve best for rapid sampling while explicit solvents remain indispensable for thermodynamic accuracy in studying specific molecular interactions.
Molecular dynamics (MD) simulations are a cornerstone of modern computational biology, enabling researchers to study the structure, dynamics, and function of biomolecular systems. A critical component of any biological MD simulation is the treatment of the solvent environment, which profoundly influences solute behavior through electrostatic screening, hydrogen bonding, and hydrophobic interactions. Traditionally, explicit solvent models represent water molecules as individual atoms, providing a detailed but computationally expensive representation. In contrast, implicit solvent models treat the solvent as a dielectric continuum, offering significant speed advantages but potentially oversimplifying complex solvent physics.
The central challenge in implicit solvent methodologies lies in accurately capturing two crucial physical phenomena: hydrophobic effects and solvent viscosity. Hydrophobic effects, which drive the association of non-polar molecules in water, are intimately tied to the entropic and enthalpic contributions of explicit water molecules. Viscosity, which governs the damping of molecular motions, directly impacts conformational sampling rates and transition kinetics. This guide provides a comparative analysis of how modern implicit and explicit solvent approaches handle these physical effects, supported by experimental data and methodological insights for research applications.
Theoretical Foundations: How Solvent Models Handle Key Physical Effects
Explicit Solvent Models: Atomic Detail at a Cost
Explicit solvent models, such as TIP3P and SPC, represent water molecules as collections of interacting atoms with specified partial charges and Lennard-Jones parameters. These models naturally capture molecular details of hydration shells, hydrogen-bonding networks, and specific water-bridged interactions. The viscosity emerges naturally from water-water interactions, providing realistic dynamic damping of solute motions.
However, this physical fidelity comes with substantial computational overhead. The inclusion of explicit water molecules significantly increases system size, and the relatively long relaxation times of interstitial water can slow conformational sampling. The viscous drag of explicit water retards large conformational changes of macromolecules, requiring longer simulation times to observe biologically relevant transitions.
Implicit Solvent Models: Efficiency Through Approximation
Implicit solvent models replace discrete solvent molecules with a dielectric continuum, dramatically reducing computational cost. The Generalized Born implicit solvent (GBIS) model, implemented in packages like NAMD, CHARMM, and AMBER, represents a popular approach that approximates electrostatic solvation through Born radii calculations that quantify atom-specific solvent exposure.
In GBIS, the total electrostatic energy combines Coulombic interactions with a Generalized Born term that screens electrostatic interactions based on atomic separation and Born radii. The screening effect heavily depends on solvent exposure, with atoms more exposed to high solvent dielectric being more strongly screened. While efficient, this approach must approximate the complex physics of hydrophobic hydration and lacks inherent viscosity, potentially altering dynamic properties.
Table 1: Fundamental Differences Between Explicit and Implicit Solvent Approaches
| Feature | Explicit Solvent | Implicit Solvent |
|---|---|---|
| Solvent Representation | Individual water molecules | Dielectric continuum |
| Computational Cost | High (increased atom count) | Lower (no explicit waters) |
| Hydrophobic Effects | Natural via water structure | Approximated via non-polar terms |
| Viscosity | Natural from water-water interactions | Typically absent or reduced |
| Sampling Speed | Slower (viscous drag present) | Faster (reduced viscosity) |
| Electrostatics | Particle-mesh Ewald possible | GB or Poisson-Boltzmann approximations |
Quantitative Comparison: Performance Benchmarks and Physical Accuracy
Sampling Efficiency and Viscosity Effects
Comparative studies reveal significant differences in conformational sampling rates between explicit and implicit solvent simulations. Research examining small dihedral angle flips, nucleosome tail collapse, DNA unwrapping, and miniprotein folding found that speedups for GB relative to explicit solvent (PME) are highly system-dependent.
When simulation temperatures are identical, implicit solvent provides approximately 1-fold speedup for small conformational changes, between 1-100-fold speedup for large changes, and approximately 7-fold speedup for mixed cases. This accelerated sampling is primarily attributed to reduced solvent viscosity in implicit solvent models rather than differences in free-energy landscapes. The absence of explicit water molecules eliminates viscosity imposed on solutes, allowing faster equilibration and better conformational sampling.
Hydrophobic Interactions and Hydration Structure
Implicit solvent models approximate hydrophobic effects through non-polar terms often based on solvent-accessible surface area (SASA). These terms estimate the free energy cost of cavity formation in the solvent continuum. While computationally efficient, this approach can struggle with capturing the nuanced thermodynamics of hydrophobic hydration, particularly the entropic contributions that drive the association of non-polar surfaces.
Studies of hydrophobic hydration in mixed solvents reveal the complexity that implicit models must approximate. Research on DMSO-water mixtures demonstrates that hydrophobic groups enhance local water structure through "hydrophobic hydration," with well-defined hydration shells and hydrogen-bonding patterns that vary with concentration. Such specific structural effects present challenges for continuum approximations.
Table 2: Sampling Speed Comparison Between Explicit and Implicit Solvent MD
| System Type | Conformational Change | Speedup (GB vs. Explicit) | Primary Contributor |
|---|---|---|---|
| Small Changes | Dihedral angle flips | ~1-fold | Similar barriers |
| Large Changes | Nucleosome tail collapse, DNA unwrapping | 1-100-fold | Reduced viscosity |
| Mixed Changes | Miniprotein folding | ~7-fold | Reduced viscosity |
| General | Various biomolecules | 2-50-fold (combined effect) | Algorithmic + reduced viscosity |
Methodological Implementations: Research Protocols and Tools
Generalized Born Implicit Solvent (GBIS) Implementation
The GBIS model in programs like NAMD implements a sophisticated approach to continuum electrostatics. The model calculates Born radii using the Onufriev, Bashford and Case (GBOBC) method, which determines solvent exposure through pairwise overlap of atomic spheres. The resulting Born radii enable computation of effective interaction distances that heavily screen electrostatic interactions between solvent-exposed atoms.
The mathematical foundation involves calculating the total electrostatic energy as the sum of Coulomb and Generalized Born terms. The pairwise GB energy incorporates a distance-dependent function that accounts for both atomic separation and Born radii, with the form:
E_ij^GB = -(k_e/D_ij) * q_i * q_j / f_ij^GB
where f_ij^GB = sqrt(r_ij^2 + α_i * α_j * exp(-r_ij^2 / (4 * α_i * α_j)))
This formulation screens interactions more heavily for atoms that are either far apart or highly exposed to solvent, with the degree of screening controlled by the Born radii (α). The computational implementation requires three-fold iteration over atom pairs each timestep, presenting parallelization challenges addressed through specialized algorithms in NAMD.
Experimental Protocols for Method Validation
Comparative studies between explicit and implicit solvents typically employ standardized protocols to ensure meaningful comparisons:
System Setup: Researchers typically prepare identical solute structures, then embed them in either explicit water boxes (for explicit solvent) or apply continuum solvent parameters (for implicit solvent). Energy minimization precedes production runs to eliminate bad contacts.
Simulation Parameters: Common protocols use the CHARMM36 or AMBER force fields for solute parameters. Explicit solvent simulations typically use Particle Mesh Ewald for long-range electrostatics with 8-12Å cutoffs, while implicit solvent simulations employ longer cutoffs (16-20Å) as PME cannot be used for GBIS electrostatics.
Sampling Assessment: Researchers quantify sampling efficiency by measuring the rate of transition between conformational states, mean first-passage times for structural transitions, or the diversity of conformational space explored in fixed simulation time.
Thermodynamic Validation: Potential of mean force calculations and free energy differences between stable states provide crucial validation of implicit model accuracy compared to explicit solvent references.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential Tools for Solvent Modeling Research
| Tool/Software | Type | Primary Function | Key Features |
|---|---|---|---|
| NAMD | MD Software | Parallel molecular dynamics | Efficient GBIS implementation, scalable parallelization |
| AMBER | MD Suite | Biomolecular simulation | Multiple GB models, extensive force fields |
| CHARMM | MD Program | Molecular mechanics/dynamics | All-atom force fields, implicit solvent options |
| VMD | Visualization/Analysis | Molecular visualization | MDFF plugin, trajectory analysis |
| Phenix | Crystallography | Structure refinement | Real-space refinement for cryo-EM/crystallography |
| OpenMM | MD Library | GPU-accelerated simulation | High-performance implicit solvent on GPUs |
Emerging Frontiers and Future Directions
Machine Learning-Augmented Implicit Solvent Models
Recent advances integrate machine learning with traditional implicit solvent approaches to address limitations in capturing specific physical effects. Meta's Open Molecules 2025 (OMol25) dataset and associated neural network potentials (NNPs) demonstrate how ML can provide PB-accurate surrogates with significantly reduced computational cost. These models learn solvent-averaged potentials from high-level quantum chemical calculations (ωB97M-V/def2-TZVPD), offering improved accuracy for diverse chemical systems including biomolecules, electrolytes, and metal complexes.
The Universal Model for Atoms (UMA) architecture employs a Mixture of Linear Experts (MoLE) approach, enabling knowledge transfer across disparate datasets computed with different DFT methods. This approach shows promise for more accurately capturing system-specific solvation physics while maintaining computational efficiency.
Hybrid Explicit-Implicit Methodologies
Innovative approaches like the Variational Explicit-Solute Implicit-Solvent (VESIS) model combine explicit treatment of solute atoms with continuum solvent descriptions. This methodology employs binary level-set methods for efficient interface tracking and adaptive-mobility gradient descent for solute relaxation. GPU implementation enables rapid free-energy minimization, showing particular promise for modeling conformational changes and binding processes.
Advanced Hydrophobic Effect Modeling
Beyond conventional SASA-based approaches, newer models provide more sophisticated treatment of hydrophobic effects by decomposing non-polar contributions into repulsive (cavity) and attractive (dispersion) components. Some implementations incorporate corrections for hydrogen-bonding effects and solvent-excluded volume, providing more physically realistic descriptions of hydrophobic hydration and association.
The choice between explicit and implicit solvent models represents a fundamental trade-off between computational efficiency and physical completeness. Explicit solvent simulations remain the gold standard for capturing detailed hydration structure and accurate dynamics but require substantial computational resources. Implicit solvent models offer dramatic speedups—particularly for large conformational changes—through reduced viscosity and eliminated explicit water calculations, but approximate complex physical effects like hydrophobic hydration.
For research applications, selection criteria should consider the specific scientific question. When studying kinetics or processes dependent on specific water interactions, explicit solvents provide necessary physical fidelity. For rapid conformational sampling, binding site identification, or large-scale structural transitions, implicit solvents offer compelling advantages. Emerging hybrid approaches and machine-learning augmented models promise to bridge these paradigms, offering a best-of-both-worlds solution for the evolving needs of computational biophysics and drug discovery.
Molecular dynamics (MD) simulations of biomolecules require accurate modeling of solvent effects to reproduce physiological conditions. Two principal approaches exist: explicit solvent models, which include individual water molecules, and implicit solvent models, which approximate solvent as a continuous medium exerting a mean force on the solute [12]. The parametrization of these models—particularly the derivation of atomic solvation parameters—critically determines their accuracy and computational efficiency. This guide compares two fundamental parametrization philosophies: the historical approach based on experimental transfer free energies and the increasingly prevalent force matching technique derived from explicit solvent simulations. Within the broader thesis of explicit versus implicit solvent comparison, understanding these parametrization strategies is essential for researchers to select appropriate models for drug discovery applications, where balancing computational cost with physical accuracy is paramount.
Comparative Analysis of Parametrization Methodologies
The core of implicit solvent model accuracy lies in its parametrization. The table below contrasts the traditional and force-matching approaches.
Table 1: Comparison of Parametrization Strategies for Atomic Solvation Parameters
| Parametrization Aspect | Traditional Experimental Derivation | Force Matching Derivation |
|---|---|---|
| Fundamental Basis | Experimental free energies of transfer (e.g., water/octanol) of small molecule analogs [56] [57] | Matching mean forces from explicit solvent MD simulations to implicit model forces [58] [12] |
| Primary Target | Reproduction of thermodynamic solvation free energies [56] | Reproduction of instantaneous solvent forces on solute atoms [58] |
| Typical Formulation | ΔGsol = Σ σi · SASAi, with σi derived from experiment [57] | ΔGsol = Σ σi · SASAi, with σi fitted to explicit forces [58] |
| Key Advantage | Direct connection to experimental thermodynamic data [59] | Directly optimizes for accurate forces and structural ensembles in dynamics simulations [58] |
| Key Limitation | May not accurately capture the directional nature of solvent forces, leading to poor performance in energy refinement [56] | Requires extensive explicit solvent simulation data for parametrization [58] |
| Reported Performance | Inferior to SASA-based methods in energy refinement of proteins; poor performance in minimizing NMR constraint violations [56] | Superior reproduction of explicit solvent conformational ensembles; more accurate forces for dynamic simulations [58] [12] |
Experimental Protocols and Methodological Details
Force Matching of Solvation Parameters
The force matching procedure represents a paradigm shift towards leveraging explicit solvent simulations for parametrization. The detailed workflow is as follows [58]:
- Explicit Solvent Reference Simulation: Perform a (or multiple) Molecular Dynamics (MD) simulation of the solute (e.g., a protein) in explicit water. A large set of 188 topologically diverse protein structures was used for robust parameter derivation [58].
- Force Averaging: For each solute atom
i, compute the time-averaged force⟨fᵢ⟩exerted on it by the explicit water molecules over the trajectory. - Implicit Model Force Definition: The implicit solvent model force on atom
iis defined as the derivative of the SASA-based solvation energy:fᵢᶦᵐᵖˡ = -∂Vˢᵒˡ/∂rᵢ = -σᵢSASA · (∂Aᵢ/∂rᵢ). The term∂Aᵢ/∂rᵢis computed using an analytical formula [58]. - Parameter Optimization: The atom-specific solvation parameters
σᵢSASAare iteratively adjusted so that the forcesfᵢᶦᵐᵖˡfrom the implicit model match the reference forces⟨fᵢ⟩from the explicit simulation as closely as possible. - Parameter Simplification: The optimized atom-type-specific
σᵢparameters can be grouped(σg)into a smaller number of categories (e.g., three groups: hydrophobic, polar, charged) using statistical partitioning to enhance model transferability and simplicity [58].
Traditional Experimental Derivation
The classical method relies on empirical thermodynamic data [56] [57]:
- Data Collection: Obtain experimental vapor-to-water or octanol-to-water free energies of transfer (ΔGtransfer) for small organic molecules that are analogs of protein amino acid side-chains.
- Surface Area Calculation: For each molecule, calculate the solvent-accessible surface area (SASA) of each atom type.
- Parameter Fitting: The atomic solvation parameter
σᵢfor atom typeiis determined by fitting the equationΔG<sub>sol</sub> = Σ σᵢ · Aᵢto the experimental ΔGtransfer data, whereAᵢis the SASA of atomi[57]. This yields a set ofσᵢvalues that minimize the difference between calculated and experimental solvation free energies.
Performance and Applicability in Biomolecular Simulations
The choice of parametrization strategy has a direct and significant impact on simulation outcomes.
- Energy Refinement: Studies comparing the effect of different solvation energy terms on protein energy refinement found that minimizations using a potential based on the total or apolar accessible surface area were "superior to minimizations of the ECEPP/2 energy alone." In stark contrast, minimizations using a term based on free energies of transfer "perform poorly" [56].
- Conformational Sampling and Dynamics: Implicit solvent models parametrized to reproduce explicit solvent behavior can significantly speed up conformational sampling. Speedups relative to explicit solvent (PME) simulations are highly system-dependent, ranging from approximately 1-fold for small dihedral flips to ~100-fold for large conformational changes like nucleosome tail collapse [27]. This speedup is attributed to reduced solvent viscosity, which allows for faster barrier crossing [27]. Accurate parametrization is key to ensuring these accelerated simulations still produce physically meaningful free-energy landscapes.
- Broader Applications: Well-parametrized implicit solvation models are now used in a wide array of computational tasks beyond standard MD, including protein folding studies, protein structure prediction and design, membrane simulations, and drug screening where fast estimation of binding affinities is crucial [12].
Table 2: Key Resources for Solvation Parameter Research and Application
| Resource / Reagent | Function / Description | Relevance to Parametrization |
|---|---|---|
| GROMOS Simulation Package | A biomolecular MD simulation software suite. | Used to implement and test the force-matched SASA implicit solvent model [58]. |
| GROMOS 43A1 Force Field | A specific parameter set for biomolecular simulations. | The force field for which atom-type σᵢSASA parameters were derived via force matching [58]. |
| Solvent Forces Database | A publicly available web server containing averaged solvent forces on protein atoms. | Provides reference data for further development and parametrization of implicit solvation models [58]. |
| CGCompiler | A Python package for automated parametrization of coarse-grained models. | Employs optimization algorithms (e.g., Particle Swarm) to fit parameters, including those related to solvation (SASA) and partition coefficients (log P) [59]. |
| LigParGen Server | A web-based tool for generating atomistic force field parameters for organic molecules. | Often used to create initial atomistic topologies for small molecules, which serve as the reference for subsequent coarse-graining or parametrization [60]. |
| Martini Coarse-Grained Force Field | A popular framework for coarse-grained simulations. | Its parametrization relies on matching atomistic reference data, including solvation properties like log P, to ensure accuracy [59] [60]. |
Workflow Visualization: Force Matching Parametrization
The following diagram illustrates the integrated workflow for deriving atomic solvation parameters through the force matching procedure, connecting the key stages from data generation to model application.
This guide objectively compares the performance of explicit and implicit solvent models in molecular dynamics (MD) simulations, focusing on their computational cost and accuracy. The analysis is framed within the critical trade-off between the physical realism of explicit solvents and the computational efficiency of implicit models, a central consideration for research and drug development.
In molecular dynamics, the choice of solvent model is one of the most consequential factors for a simulation's computational cost and scientific output. Explicit solvent models, which treat each solvent molecule as an individual entity, are considered the gold standard for accuracy but require simulating a vast number of particles [4] [3]. Implicit solvent models replace the explicit solvent with a continuous dielectric medium, dramatically reducing the number of interacting particles and the associated computational burden [3].
The core of the efficiency problem lies in system scaling. For a solute of a given size, an explicit solvation box can contain tens of thousands of additional solvent atoms, increasing the computational complexity of every force calculation and requiring smaller integration time steps due to the high-frequency motions of solvent molecules. Implicit models avoid this, but historically at the expense of accuracy, particularly for processes where specific solute-solvent interactions, like hydrogen bonding, are crucial [4] [3]. The following table summarizes the fundamental characteristics of each approach.
Table 1: Fundamental Characteristics of Explicit vs. Implicit Solvent Models
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Fundamental Approach | Atomistic representation of individual solvent molecules [4] | Continuum dielectric medium representing average solvent behavior [3] |
| Computational Scaling | High; scales with the total number of atoms (solute + thousands of solvent molecules) [4] | Low; scales primarily with the number of solute atoms [3] |
| Typical Use Cases | Ab initio MD (AIMD), QM/MM, studies requiring specific solute-solvent interactions [4] | Rapid conformational sampling, binding free energy estimates, QM calculations with solvation [28] [3] |
| Key Strengths | High accuracy, captures specific solvent structure (e.g., hydrogen bonds) [4] [3] | High computational speed, faster conformational convergence, no solvent viscosity [28] [3] |
| Key Limitations | Extremely high computational cost; requires extensive sampling [4] | Limited accuracy; misses specific, local solvent effects [4] [3] |
Performance and Cost Comparison
The theoretical computational advantage of implicit solvents translates directly into tangible performance metrics. Benchmarks comparing simulation throughput reveal orders-of-magnitude differences in speed.
Raw Performance and Cost-Efficiency Benchmarks
A recent benchmarking study provides a direct comparison of simulation speed (nanoseconds per day) and cost-efficiency for a ~44,000-atom explicit solvent system (T4 Lysozyme in water) across various cloud GPUs [49]. The results highlight the high computational demand of explicit solvent simulations and the variance in performance between different hardware.
Table 2: Explicit Solvent Simulation Performance on Different Cloud GPUs (T4 Lysozyme, ~44k atoms) [49]
| GPU Instance (Provider) | Simulation Speed (ns/day) | Cost per 100 ns (Indexed to AWS T4) | Best Use Case |
|---|---|---|---|
| H200 (Nebius) | 555 | 0.87 | Peak performance, ML-coupled workflows |
| L40S (Nebius) | 536 | 0.41 | Best value for traditional MD |
| H100 (Scaleway) | 450 | 0.67 | Heavy-duty workloads with large storage |
| A100 (Hyperstack) | 250 | 0.74 | Balanced speed and affordability |
| V100 (AWS) | 237 | 1.77 | Legacy systems; poor modern value |
| T4 (AWS) | 103 | 1.00 | Budget option for long queues |
For implicit solvent models, direct ns/day benchmarks are less common, but the efficiency gain is well-documented. They enable the simulation of systems that would be "impossible or unfeasible" with explicit solvents, primarily because the system size is reduced to the solute alone [3]. This allows for significantly longer time-scale simulations or the use of more expensive quantum mechanical methods.
Accuracy Comparison and Emerging Hybrids
While implicit models are faster, their accuracy has traditionally lagged behind explicit solvents. However, machine learning (ML) is rapidly bridging this gap. A key drawback of early ML-based implicit solvents was their inability to calculate absolute free energies, as they were trained only on forces [7]. A new model, the λ-Solvation Neural Network (LSNN), overcomes this by incorporating derivatives of alchemical variables (λelec, λsteric) during training, enabling accurate solvation free energy predictions that rival explicit-solvent calculations at a fraction of the cost [7].
Another approach, QM-GNNIS, transfers knowledge from classical MD simulations to quantum-mechanical calculations. It uses a Graph Neural Network implicit solvent model, trained on explicit-solvent MD data, to provide a correction to traditional QM continuum models. This emulates a QM/MM setup and has been shown to reproduce experimental trends that standard implicit models could not capture [28].
Table 3: Comparison of Modern Machine Learning-Based Solvent Models
| Model | Approach | Key Innovation | Reported Outcome |
|---|---|---|---|
| LSNN [7] | GNN-based implicit solvent | Trained on forces and alchemical derivatives | Achieves explicit-solvent accuracy in free energy predictions with computational speedup |
| QM-GNNIS [28] | Knowledge transfer from MM to QM | Applies a classically-trained GNN correction to QM continuum models | Reproduces experimental trends unattainable by standard implicit models like SMD |
| Active Learning MLPs [4] | Machine Learning Potentials for explicit solvent | Uses active learning to create data-efficient training sets for reactive explicit solvent MD | Obtains reaction rates in agreement with experimental data for a Diels-Alder reaction |
Experimental Protocols for Performance Assessment
To ensure reproducible and objective comparisons between solvent models and their implementations, standardized benchmarking protocols are essential.
Protocol 1: Benchmarking Explicit Solvent MD on Cloud GPUs
This protocol is derived from a published benchmark for profiling simulation speed and cost-efficiency [49].
- System Preparation: Use a standardized biomolecular system like T4 Lysozyme (PDB ID: 4W52). Solvate it in explicit water using a tool like
UnoMDorOpenMM's setup utilities, resulting in a system of known size (e.g., ~44,000 atoms). - Simulation Parameters: Run simulations using the
OpenMMengine on the CUDA platform. Use a 2 fs integration time step, Particle Mesh Ewald (PME) for electrostatics, and a mixed-precision model. The simulation length should be a minimum of 100 ps. - I/O Optimization: A critical step is to minimize I/O overhead. Set the trajectory saving interval to a high value (e.g., every 10,000 steps) to prevent data transfer from the GPU to CPU from becoming a bottleneck. GPU utilization should be monitored to confirm it remains high (≥90%).
- Data Collection: The primary metric is simulation throughput (ns/day). This is calculated from the actual wall-clock time required to complete the simulation. The cost-efficiency is then calculated by factoring in the cloud instance's hourly cost to determine the normalzed "cost per 100 ns."
Protocol 2: Validating Implicit Solvent Accuracy with Experimental Data
This protocol, based on methodologies from recent studies, outlines how to validate the accuracy of an implicit solvent model, particularly an ML-augmented one [28].
- Test System Selection: Choose a set of small organic molecules with reliable experimental solvation free energy data or spectroscopic properties (e.g., NMR, IR) known to be sensitive to solvent effects.
- Reference Calculations: Perform alchemical free energy calculations using an explicit solvent model (e.g., TIP3P) as a reference. This is computationally expensive but provides a computational ground truth.
- Implicit Solvent Workflow: For the same set of molecules, perform simulations or geometry optimizations using the implicit solvent model of interest. For ML models like QM-GNNIS, this involves running a QM calculation (e.g., with CPCM) and applying the ML-derived correction force [28].
- Analysis and Comparison: Calculate the solvation free energies or the target spectroscopic properties from the implicit solvent simulations. The accuracy of the model is determined by its Mean Absolute Error (MAE) or Root Mean Square Error (RMSE) against the experimental data or the explicit solvent reference.
Workflow Visualization
The following diagram illustrates the integrated active learning workflow for developing efficient machine-learning potentials for explicit solvent simulations, as validated in recent research.
Active Learning Workflow for ML Potentials
The second diagram outlines the novel training methodology for the LSNN model, which enables accurate free energy calculation.
LSNN Training for Free Energy
This section details key software, datasets, and models that form the modern toolkit for efficient solvation modeling.
Table 4: Essential Resources for Modern Solvation Modeling
| Tool / Resource | Type | Function & Application | Reference |
|---|---|---|---|
| OpenMM / UnoMD | Simulation Engine | A highly optimized, open-source MD engine used for benchmarking GPU performance and running production simulations. | [49] |
| OMol25 Dataset | Training Dataset | A massive dataset of ~100M quantum chemical calculations used to train universal neural network potentials like UMA and eSEN. | [29] |
| Meta UMA / eSEN Models | Pre-trained ML Potentials | Universal Neural Network Potentials offering high accuracy across diverse chemical spaces, serving as a powerful force field. | [29] |
| LSNN Model | ML Implicit Solvent | A GNN-based model trained for accurate free energy calculations, bridging the gap between implicit speed and explicit accuracy. | [7] |
| Active Learning Selectors | Algorithm | Algorithms (e.g., based on SOAP descriptors) that identify gaps in training data for ML potentials, improving data efficiency. | [4] |
In molecular dynamics (MD) research, the choice of how to represent the solvent environment—whether as discrete molecules or a continuous medium—is a fundamental decision that significantly impacts the accuracy, computational cost, and biological relevance of simulations. Explicit solvent models treat each solvent molecule as an individual entity, allowing for detailed treatment of specific solute-solvent interactions such as hydrogen bonding and solvation dynamics [61]. In contrast, implicit solvent models (also known as continuum solvents) replace explicit solvent molecules with a homogeneously polarizable medium, representing solvent effects through a potential of mean force (PMF) that responds instantaneously to solute conformational changes [12] [62]. This guide provides system-specific recommendations for selecting between these approaches across different biomolecular classes, supported by recent experimental benchmarks and theoretical frameworks.
The core trade-off revolves around computational efficiency versus molecular detail. Implicit solvents eliminate solvent viscosity, accelerate conformational sampling, and reduce computational costs by integrating out solvent degrees of freedom [10] [63]. However, this efficiency comes at the expense of neglecting specific solvent features like hydrogen bond fluctuations, water dipole reorientation, and bridging water molecules that can be crucial for biomolecular function [12]. Explicit solvents preserve these specific interactions but require substantially greater computational resources to simulate the numerous solvent molecules and achieve sufficient sampling [61].
Theoretical Foundations and Computational Methodologies
Fundamental Principles of Implicit Solvation
Implicit solvent models calculate solvation free energy (ΔGsolv) by combining several physical components. The total free energy of solvation is traditionally decomposed into three contributions [12]:
- Cavity formation (ΔGcav): The energy required to create a cavity within the solvent to accommodate the solute molecule
- van der Waals interactions (ΔGvdW): Dispersion and repulsive interactions between solute and solvent
- Electrostatic interactions (ΔGele): Polar interactions arising from the solute's charge distribution polarizing the solvent
The most common implicit approaches include Generalized Born (GB) models, which provide an analytical approximation to the Poisson-Boltzmann equation for electrostatic interactions [63], and Solvent-Accessible Surface Area (SASA) methods, which model nonpolar contributions as being proportional to the molecular surface area [12]. Advanced hybrid models often combine these approaches, such as GB/SASA or PB/SASA, where the electrostatic term is handled by Generalized Born or Poisson-Boltzmann methods, while nonpolar contributions are modeled using SASA [12].
Fundamental Principles of Explicit Solvation
Explicit solvent models represent water molecules using various point-charge representations that differ in their complexity and physical accuracy. Common water models include [47] [64]:
- Three-site models (TIP3P, SPC/E): Place partial charges on hydrogen and oxygen atoms; computationally efficient but simpler physics
- Four-site models (TIP4P, TIP4P/Ew): Introduce an additional massless charge site displaced from the oxygen atom to improve dielectric properties
- Five-site models (TIP5P): Incorporate two lone-pair interaction sites to better represent water's tetrahedral structure
These models aim to reproduce key physical properties of water such as density, diffusion constant, and dielectric constant through different parametrizations of charge distributions and geometric parameters [64].
Experimental Benchmarking Methodologies
Rigorous comparison between solvent models requires standardized assessment protocols. Recent benchmark studies have employed the following methodological framework [47] [64]:
Table: Standardized Protocol for Solvent Model Benchmarking
| Simulation Phase | Key Parameters | Typical Duration |
|---|---|---|
| System Preparation | Solvation in octahedral periodic box, neutralization with counterions (Na+), 8.0-10.0 Å minimum solute-box distance | - |
| Energy Minimization | Steepest descent + conjugate gradient algorithms, harmonic force restraints gradually released | 10,000-12,000 cycles |
| System Equilibration | Gradual heating to 300K, NPT ensemble, positional restraints on solute atoms | 50-125 ps |
| Production MD | NPT ensemble, 2 fs time step, particle mesh Ewald for long-range electrostatics | 100 ns - 5 μs |
Key structural and dynamic parameters analyzed include:
- Global descriptors: Radius of gyration (Rg), end-to-end distance (EED), root-mean-square deviation (RMSD)
- Local descriptors: Glycosidic torsion angles, ring puckering conformations, intramolecular hydrogen bonds
- Energetics: Molecular Mechanics Generalized Born Surface Area (MM/GBSA) binding free energies
Biomolecular System-Specific Recommendations
Glycosaminoglycans and Sulfated Polysaccharides
Glycosaminoglycans (GAGs) such as heparin, heparan sulfate, and chondroitin sulfate represent particularly challenging systems for solvent modeling due to their high negative charge density, structural flexibility, and critical dependence on solvent-mediated interactions [47].
Table: Solvent Model Performance for Glycosaminoglycan Systems
| Solvent Model | Class | Key Strengths | Identified Limitations | Recommended Use Cases |
|---|---|---|---|---|
| TIP3P | Explicit (3-site) | Balanced efficiency/reliability; promotes U-shaped HP curvature via Na+ counterions [47] | May oversimplify electrostatic environment; less accurate for certain puckering states [64] | Standard protein-GAG complexes; initial screening studies |
| OPC | Explicit (4-site) | Superior reproduction of local/global HP features; excellent electrostatic treatment [47] | Higher computational cost; limited testing in GAG-protein complexes [47] | High-accuracy studies of GAG structure; final validation simulations |
| TIP5P | Explicit (5-site) | Accurate ring puckering and conformational sampling [47] | Highest computational demand; may introduce excessive structural variability [64] | Investigation of fine structural details when resources permit |
| IGB=2,5,7,8 | Implicit (GB) | Fast sampling; reasonable for global descriptors [47] | Poor reproduction of experimental ring puckering conformations [47] | Rapid preliminary studies; large-scale conformational sampling |
Recent benchmark studies on heparin (HP) decasaccharides and dodecamers reveal that explicit solvent models generally outperform implicit approaches for capturing experimentally observed structural properties [47] [64]. Notably, Marcisz et al. demonstrated that implicit models poorly reproduced iduronic acid ring puckering conformations observed in NMR studies, whereas explicit solvents provided significantly improved accuracy [47]. The choice among explicit models involves trade-offs: while TIP3P offers the best computational efficiency, more advanced models like OPC and TIP5P provide superior structural fidelity, particularly for local conformational features [64].
Proteins and Peptides
Proteins and peptides exhibit diverse solvent requirements depending on their structural features, conformational flexibility, and biological context.
Table: Solvent Model Selection for Protein Systems
| System Type | Recommended Approach | Rationale | Key Supporting Evidence |
|---|---|---|---|
| Globular Proteins | Explicit solvents (TIP3P, TIP4P) | Preserve specific water-mediated interactions in binding pockets; maintain native hydrogen-bonding networks | Explicit water essential for modeling water-bridged protein-ligand interactions [12] |
| Intrinsically Disordered Proteins | Implicit solvents (GB/SA) | Enhanced conformational sampling; reduced viscosity accelerates exploration of conformational landscape | Implicit solvents improve sampling by eliminating solvent viscosity [10] [63] |
| Peptide Folding Studies | Hybrid explicit/implicit or advanced implicit | Balance between specific hydration and sampling efficiency | GB models successfully applied to "trp-cage" miniprotein folding [10] |
| Protein-Ligand Binding | Explicit solvents for production; MM/PBSA for analysis | Accuracy in binding site interactions; efficient free energy estimation | MM-PBSA/GBSA widely used for binding affinity estimates [64] |
For protein systems with deep binding pockets or specific water-mediated interactions, explicit solvents are generally preferred as they preserve structural water molecules that can be critical for molecular recognition [12]. Conversely, for studies of protein folding or intrinsically disordered regions where conformational sampling is the priority, implicit solvents offer significant advantages by reducing computational barriers associated with solvent viscosity [10] [63]. The EEF1 and EEF1-SB implicit models have shown particular utility for protein simulations by incorporating a volume-based correction to standard SASA approaches [12].
Nucleic Acids
Nucleic acids present unique challenges for solvent modeling due to their highly charged backbones, structural dependence on ion atmospheres, and specific hydration patterns in major and minor grooves.
DNA duplexes benefit from explicit solvent representation, particularly for studies of sequence-dependent structural features like A-tract bending or groove geometry. The ordered water spines in minor grooves are poorly captured by continuum models, making explicit solvents essential for accurate structural dynamics [12]. For RNA systems, with their complex tertiary structures and specific metal ion binding sites, explicit solvents are strongly recommended despite their computational cost. The coordination of Mg²⁺ ions and their hydration shells is particularly difficult to model with implicit approaches [63].
For studies focused on nucleic acid flexibility or large-scale conformational changes, GB implicit models implemented in packages like AMBER have demonstrated utility by providing improved sampling efficiency while maintaining reasonable structural accuracy [10]. However, applications to nucleic acids remain more challenging than for proteins due to their higher charge density and stronger dependence on specific ion interactions [12].
Emerging Methods and Future Directions
Machine Learning-Enhanced Implicit Solvents
Recent advances in machine learning (ML) have created opportunities to overcome traditional limitations of implicit solvent models. Graph Neural Networks (GNNs) and other ML architectures can be trained on extensive explicit solvent simulations to develop more accurate potentials of mean force [7]. The newly proposed λ-Solvation Neural Network (LSNN) extends traditional force-matching approaches by incorporating derivatives of alchemical variables, enabling accurate free energy predictions that were previously limited to explicit solvent simulations [7]. These ML-based models aim to achieve near-explicit solvent accuracy while maintaining the computational efficiency of implicit approaches, potentially offering the best of both worlds for specific applications.
Hybrid Solvation Schemes
Hybrid QM/MM (quantum mechanics/molecular mechanics) approaches combine different levels of theory to balance accuracy and efficiency [62]. A typical implementation involves:
- QM region: Solute molecules and critical explicit water molecules treated with quantum mechanical methods
- MM region: Surrounding solvent environment treated with molecular mechanics force fields
- Continuum layer: Outer bulk solvent represented with implicit continuum models
These multi-scale methods are particularly valuable for studying chemical reactions in enzymatic active sites or processes involving electron transfer, where specific quantum effects must be captured while maintaining computational tractability for the full solvated system [62].
Research Reagent Solutions: Experimental Toolkit
Table: Essential Computational Tools for Solvent Modeling Studies
| Tool Name | Type | Primary Function | Key Applications |
|---|---|---|---|
| AMBER | Software Suite | Biomolecular MD simulations with extensive solvent model options [47] | Protein, nucleic acid, and carbohydrate simulations with both explicit and implicit solvents |
| CHARMM-GUI | Web Server | Input file generation for MD simulations with various solvent models [64] | System setup for CHARMM36m force field simulations with multiple explicit water models |
| GLYCAM-06 | Force Field | Specialized parameters for carbohydrate and GAG simulations [47] | Glycosaminoglycan dynamics with TIP3P, SPC/E, and other explicit water models |
| MM/PBSA & MM/GBSA | Analytical Method | Binding free energy estimation from MD trajectories [47] | Post-processing of explicit solvent trajectories for efficient affinity calculations |
The selection between implicit and explicit solvent models requires careful consideration of biomolecular system characteristics, research objectives, and computational resources. Based on the current evidence:
For glycosaminoglycans and highly charged systems, explicit solvent models (particularly OPC and TIP5P) should be preferred for production simulations, while implicit solvents may be suitable for preliminary conformational sampling.
For proteins with specific solvent interactions in binding sites or structured water networks, explicit solvents (TIP3P, TIP4P) are recommended despite their computational cost.
For studies prioritizing conformational sampling of flexible systems like intrinsically disordered proteins or folding intermediates, advanced implicit models (GB/SA) offer significant advantages in sampling efficiency.
Emerging machine learning approaches show promise for bridging the accuracy gap between explicit and implicit models, particularly for free energy calculations.
Researchers should carefully match their solvent model selection to their specific scientific questions, validating key findings with multiple solvent representations when feasible, and considering hybrid approaches for systems with conflicting requirements. As benchmark studies continue to expand across biomolecular classes, the field moves toward more systematic and evidence-based solvent model selection.
Benchmarking Solvent Models: Performance Validation and Comparative Analysis
The choice of solvent model in molecular dynamics (MD) simulations presents a fundamental trade-off between computational efficiency and physical accuracy. Explicit solvent models, which simulate individual solvent molecules, offer high fidelity but at a significant computational cost, often consuming up to 80-90% of computation time for simulating solvent dynamics rather than the solute itself [10]. Implicit solvent models, which represent the solvent as a continuous dielectric medium, offer a computationally efficient alternative but have historically faced challenges in accuracy for certain molecular systems. This guide provides an objective comparison of the conformational sampling efficiency afforded by implicit versus explicit solvent models across biological systems of varying complexity, from small drug-like molecules to proteins.
The quantitative data presented herein stems from systematic comparative studies and enables researchers to make informed decisions about solvent model selection based on their specific sampling requirements and computational constraints.
Quantitative Comparison of Sampling Speed
Speedup Factors Across Molecular Systems
The efficiency gains from implicit solvent models are highly system-dependent. A comparative study investigating conformational changes across different molecular scales provides clear quantitative metrics on sampling speedup [1].
Table 1: Conformational Sampling Speedup of Implicit vs. Explicit Solvent MD
| System Type | Conformational Change | Sampling Speedup Factor | Primary Contributor to Speedup |
|---|---|---|---|
| Small-scale | Dihedral angle flips in proteins | ~1-fold | Reduced solvent viscosity |
| Mixed-scale | Miniprotein folding | ~7-fold | Reduced solvent viscosity |
| Large-scale | Nucleosome tail collapse | ~1-100 fold | Reduced solvent viscosity |
| Large-scale | DNA unwrapping | ~1-100 fold | Reduced solvent viscosity |
Combined Algorithmic and Sampling Efficiency
When both algorithmic performance and enhanced conformational sampling are considered together, the total speedup factors become more pronounced, especially for larger systems [1]:
Table 2: Combined Computational and Sampling Speedup
| System Type | Total Speedup Factor | Key Application Context |
|---|---|---|
| Small conformational changes | ~2-fold | Local protein dynamics |
| Miniprotein folding | ~50-fold | Protein structure prediction |
| Large-scale changes | ~1-60 fold | Chromatin dynamics, DNA processing |
The variation in speedup factors for large-scale changes reflects significant system-dependent differences, with DNA unwrapping and nucleosome tail collapse showing the most substantial benefits from implicit solvent methodology [1].
Experimental Protocols and Methodologies
Comparative Simulation Framework
The quantitative data presented in Section 2 was generated through systematically designed comparative simulations [1]. The explicit solvent simulations employed the Particle Mesh Ewald (PME) method with the TIP3P water model, while the implicit solvent simulations used a Generalized Born (GB) model as implemented in the AMBER package. Simulation times ranged from nanoseconds for small systems to microseconds for larger complexes, with identical simulation temperatures maintained for direct comparison between methods.
For the miniprotein folding studies, both solvent models were applied to the same initial structures, with conformational sampling efficiency measured by the rate of transition between folded and unfolded states. The speedup factor was quantified by comparing the number of productive folding events per unit simulation time between the two approaches [1].
Machine Learning Potentials for Explicit Solvent Efficiency
Recent advancements aim to overcome the limitations of both traditional approaches through machine learning potentials (MLPs) that retain explicit solvent accuracy at reduced computational cost. The successful application of this methodology to Diels-Alder reactions in water and methanol demonstrates that MLPs can achieve reaction rates in agreement with experimental data while explicitly modeling solvent molecules [4].
The active learning workflow for generating these potentials involves:
- Initial Data Generation: Creating a small set of configurations labeled with reference energies and forces
- MLP Training: Training initial machine learning potentials on this data
- MD Propagation: Running molecular dynamics simulations using the trained MLP
- Active Learning: Using descriptor-based selectors like Smooth Overlap of Atomic Positions (SOAP) to identify underrepresented configurations
- Iterative Refinement: Expanding the training set with new structures and retraining the MLP [4]
This approach enables the construction of data-efficient training sets that span the relevant chemical and conformational space, making explicit solvent modeling more accessible for complex systems.
Diagram 1: Active learning workflow for machine learning potentials in explicit solvent. The process iteratively improves the potential by identifying underrepresented configurations.
The Scientist's Toolkit: Key Methodologies
Table 3: Research Reagent Solutions for Solvent Modeling
| Methodology | Primary Function | Key Applications |
|---|---|---|
| Generalized Born (GB) Implicit Models | Approximate electrostatic solvation energy | Protein folding studies, rapid conformational sampling |
| Poisson-Boltzmann (PB) Models | Numerical solution of electrostatic equations | Binding affinity calculations, pKa predictions |
| Machine Learning Potentials (MLPs) | Surrogate quantum accuracy at lower cost | Reaction modeling in explicit solvent |
| Active Learning Selectors (e.g., SOAP) | Identify underrepresented configurations | Efficient training set construction for MLPs |
| Alchemical Free Energy Methods | Calculate solvation free energies | Drug binding affinity prediction, logP calculation |
| Hybrid QM/MM-GBSA | Combine quantum mechanics with implicit solvation | Binding affinity prediction for enzyme-inhibitor complexes |
Performance Benchmarks in Drug Discovery Applications
Solvation Free Energy Predictions
Accurate prediction of solvation free energies is critical for drug discovery, particularly for estimating partition coefficients and binding affinities. Recent studies comparing fixed-charge protocols for predicting solvation free energies of drug-like molecules have demonstrated the performance of various approaches [16].
The ABCG2 protocol, an update to the AM1/BCC model, consistently outperforms its predecessor and the widely used HF/6-31G* ab initio charge derivation method. For water-octanol transfer free energies, ABCG2 achieves remarkable agreement with experimental data, exhibiting a mean unsigned error below 1 kcal/mol and benefiting from systematic error cancellation between solvent environments [16].
Binding Affinity Predictions
The accurate prediction of protein-ligand binding affinities remains a cornerstone of structure-based drug design. Studies comparing implicit solvent methods for predicting binding affinities of benzimidazole inhibitors in complex with F. tularensis FabI have revealed several critical insights [65]:
- Prediction results are sensitive to radii sets, GB methods, QM Hamiltonians, and sampling protocols when using single trajectories
- QM/MM-GBSA using 6 ns MD simulation trajectories with GBneck2 and the mbondi2 radii set generated the closest agreement with experimental values (r² = 0.88)
- The "multiple independent sampling" approach, averaging results from six 1 ns MD simulations for each ligand, produced relatively insensitive results to all tested parameters
- MM/GBSA with GBHCT and mbondi radii, using 600 frames from six 0.25 ns MD simulations, provided accurate predictions (r² = 0.84) [65]
These findings suggest that multiple independent short simulations can be more efficient than a single long simulation for binding affinity predictions, potentially reducing computational costs while maintaining accuracy.
Diagram 2: Trade-offs between solvent modeling approaches, highlighting comparative advantages and limitations.
Emerging Frontiers in Solvent Modeling
Machine Learning-Augmented Implicit Solvent Models
Recent innovations in machine learning are addressing fundamental limitations of traditional implicit solvent models. The LSNN (λ-Solvation Neural Network) model represents a significant advancement by incorporating derivatives of alchemical variables (λsteric and λelec) during training, enabling meaningful comparison of absolute solvation free energies across chemical species [7].
This approach extends beyond standard force-matching by ensuring the model learns a conservative vector field that accurately represents the true potential of mean force (PMF). The modified loss function combines force-matching with terms that match the derivatives of the solvation energy with respect to electrostatic and steric coupling factors [7]. This methodology achieves free energy predictions with accuracy comparable to explicit-solvent alchemical simulations while offering computational speedup, establishing a foundational framework for future drug discovery applications.
Quantum-Continuum Hybrid Methods
Another emerging frontier involves the coupling of continuum solvation methods with quantum mechanical calculations, particularly for chemically demanding processes where electronic structure changes significantly influence solvation effects. These quantum-continuum workflows, such as those employing IEF-PCM (Integral Equation Formalism Polarizable Continuum Model), show promise for modeling solution-phase electronic structures with improved accuracy [37].
The quantitative comparison presented in this guide demonstrates that implicit solvent models可以提供 substantial sampling speedup factors ranging from approximately 2-fold for small conformational changes to 50-fold or more for protein folding and large-scale DNA rearrangements. This efficiency comes at the cost of reduced accuracy in capturing specific solute-solvent interactions, particularly hydrogen bonding and microsolvation effects.
Emerging methodologies, particularly machine learning potentials and ML-augmented implicit solvent models, offer promising avenues to bridge this efficiency-accuracy gap. The choice between solvent modeling approaches should be guided by the specific research question, with implicit solvents providing superior sampling for large-scale conformational changes and explicit solvents remaining essential for processes where atomic-level solvent details are critical.
For most practical applications in drug discovery, a hybrid approach that leverages the strengths of multiple methodologies—whether through ML correction of implicit models or selective use of explicit solvent for key binding interactions—represents the most promising path forward for balancing computational efficiency with physical accuracy.
The choice of solvent model in molecular dynamics (MD) simulations represents a fundamental trade-off between computational efficiency and physical accuracy, directly impacting the reliability of calculated free energy landscapes and structural ensembles. Explicit solvent models, which treat each water molecule as an individual entity, provide a physically detailed representation of solute-solvent interactions but demand immense computational resources. In contrast, implicit solvent models approximate the solvent as a continuous dielectric medium, dramatically accelerating simulations while potentially sacrificing accuracy in representing specific solvent effects. This comparison guide provides an objective assessment of these competing approaches, examining their performance across protein folding, RNA dynamics, small molecule solvation, and intrinsically disordered protein (IDP) characterization. By synthesizing current methodological advances and validation protocols, we aim to equip researchers with the framework needed to select appropriate solvent models for specific biological questions in drug development and structural biology.
Comparative Framework: Explicit vs. Implicit Solvent Models
Table 1: Fundamental Characteristics of Solvent Modeling Approaches
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Physical Representation | Discrete water molecules with explicit hydrogen bonds | Continuum dielectric medium with averaged properties |
| Computational Cost | High (majority of computation spent on solvent) | Low (dramatically reduced degrees of freedom) |
| Sampling Efficiency | Slower conformational sampling due to solvent viscosity | Accelerated dynamics due to absence of viscous drag |
| Electrostatic Treatment | Explicit Coulombic interactions with water dipoles | Poisson-Boltzmann or Generalized Born approximations |
| Hydrophobic Effects | Emergent from water molecule reorganization | Empirical non-polar terms [66] |
| Specific Solvent Interactions | Explicitly captured (e.g., water bridging) | Poorly represented or missing |
| System Setup Complexity | High (solvation, ion placement) | Low (primarily cavity definition) |
The theoretical foundation for implicit solvent models originates from statistical mechanics, where solvent degrees of freedom are integrated out to create a Potential of Mean Force (PMF) [66]. This PMF represents the free energy landscape as a function of solute coordinates alone, with the solvation free energy (ΔGs) decomposed into polar (electrostatic) and non-polar (cavity formation and van der Waals) components [66]. Explicit solvents maintain all atomistic details, potentially providing more physically realistic simulations but at significantly higher computational expense that limits accessible timescales and system sizes.
Quantitative Performance Assessment
Biomolecular Folding and Structural Accuracy
Table 2: Performance in Biomolecular Folding Applications
| System Type | Explicit Solvent Performance | Implicit Solvent Performance | Key Metrics |
|---|---|---|---|
| RNA Stem-Loops (10-36 nt) | Limited data available for direct comparison | 23/26 models folded successfully with native base pairs [11] | Stem region RMSD: <2 Å, Loop region RMSD: <4 Å [11] |
| Protein Folding | 16 proteins folded with TIP3P water [11] | 16 proteins folded with GB-neck2 model [11] | Balanced secondary structure propensity |
| Intrinsically Disordered Proteins | Accurate ensembles achievable with multiple force fields [67] | Limited representation of specific solvent interactions | Conformational ensemble similarity after reweighting [67] |
Recent advances in both explicit and implicit solvent methodologies have demonstrated remarkable folding capabilities for diverse biomolecules. The DESRES-RNA force field with TIP3P explicit solvent has enabled reversible folding of RNA tetraloops [11], while the AMBER ff14SBonlysc force field with GB-neck2 implicit solvent successfully folded 16 proteins with diverse topologies [11]. For RNA stem-loops containing up to 36 nucleotides, recent implicit solvent simulations achieved successful folding for 23 of 26 models, with root mean square deviation (RMSD) values below 2 Å for stem regions and approximately 4 Å for more flexible loop regions [11].
Solvation Free Energy Prediction
Table 3: Accuracy in Solvation Free Energy Estimation
| Methodology | Mean Unsigned Error (kcal/mol) | Key Innovations | Applicability Domain |
|---|---|---|---|
| Explicit Solvent Alchemical | ~0.76 (after correction) [68] | Thermodynamic integration with correction terms | Small molecules and host-guest systems |
| Machine Learning PCM | 0.40-0.53 [13] | Neural networks trained on SCRF energy components | Organic molecules with QM calculations |
| Conventional PCM/GB | 1.0-2.0+ [13] | Continuum dielectric approximations | Broad biomolecular applications |
| Explicit-to-Implicit Conversion | Significant improvements possible [69] | Graph neural networks matching alchemical derivatives | Small molecule dataset (300,000 compounds) |
Accurate solvation free energy prediction remains challenging for both explicit and implicit approaches. Traditional implicit solvent models like CPCM and IEF-PCM typically exhibit errors of 1-2 kcal/mol or more [13], while explicit solvent alchemical methods require significant corrections for finite box size and state definitions to achieve errors around 0.76 kcal/mol [68]. Recently, machine learning-enhanced implicit models have demonstrated substantial improvements, with ML-PCM reducing errors to 0.40-0.53 kcal/mol by learning the relationship between self-consistent reaction field (SCRF) energy components and experimental solvation free energies [13]. The novel LSNN (Lambda Solvation Neural Network) approach further addresses limitations in absolute free energy comparison by incorporating alchemical derivative matching during training [69].
Experimental Protocols and Methodologies
Conformational Ensemble Validation for IDPs
Accurate determination of intrinsically disordered protein ensembles requires integration of computational and experimental approaches. The maximum entropy reweighting procedure has emerged as a powerful method for combining molecular dynamics simulations with experimental data from nuclear magnetic resonance (NMR) spectroscopy and small-angle X-ray scattering (SAXS) [67]. This approach introduces minimal perturbation to computational models while ensuring agreement with experimental measurements. The protocol involves:
- Initial Ensemble Generation: Running long-timescale MD simulations (e.g., 30 μs) with multiple force fields (a99SB-disp, CHARMM22*, CHARMM36m) [67]
- Experimental Data Acquisition: Collecting NMR chemical shifts, residual dipolar couplings, J-couplings, and SAXS profiles [67]
- Forward Model Prediction: Calculating experimental observables from each MD frame using established algorithms [67]
- Reweighting Procedure: Applying maximum entropy principle to assign weights to conformations that best match experimental data while maximizing the effective ensemble size [67]
- Validation: Assessing convergence through Kish ratio calculations and comparing ensembles derived from different force fields [67]
This methodology has demonstrated that in favorable cases, IDP ensembles from different force fields converge to highly similar conformational distributions after reweighting, suggesting progress toward force-field independent ensemble determination [67].
Distance-Based Ensemble Comparison Metrics
For intrinsically disordered proteins and flexible systems, traditional root mean-square deviation (RMSD) metrics after structural alignment become meaningless due to the absence of well-defined tertiary structures. Distance-based metrics provide a superimposition-free approach for comparing conformational ensembles [70]. The key methodology involves:
- Distance Distribution Calculation: For each ensemble, compute matrices of Cα-Cα distance distributions across all conformations [70]
- Statistical Characterization: Calculate median distances (dμ(i,j)) and standard deviations (dσ(i,j)) for each residue pair [70]
- Difference Matrix Construction: Compute absolute differences between median distances and standard deviations of equivalent residue pairs between ensembles [70]
- Statistical Significance Testing: Apply nonparametric Mann-Whitney-Wilcoxon test (p < 0.05) to identify significant differences in distance distributions [70]
- Global Similarity Quantification: Calculate ensdRMS (ensemble distance root mean square difference) as: ensdRMS = √[1/n ∑(dμA(i,j) - dμB(i,j))²] [70]
This approach enables both local assessment of differences between specific regions and global quantification of ensemble similarity, providing rigorous statistical framework for comparing heterogeneous conformational ensembles [70].
Diagram 1: Workflow for conformational ensemble determination and validation
Free Energy Calculation from Binding Kinetics
Binding free energies can be calculated from kinetic rate constants obtained through path-based sampling methods, providing an alternative to alchemical approaches. The methodology involves:
- Reactive Trajectory Generation: Using enhanced sampling methods (weighted ensemble, metadynamics, milestoning) to simulate binding and unbinding events [68]
- Rate Constant Estimation: Calculating association (kon) and dissociation (koff) rates from trajectory fluxes [68]
- Correction Term Application:
- State definition correction for finite bound and unbound volumes
- Residual interaction correction for charged molecules
- Simulation volume correction for periodic boundary conditions [68]
- Free Energy Calculation: ΔG = -kB T ln(C0 kon/koff) + correction terms [68]
This approach has demonstrated agreement with alchemical methods within 0.76 kcal/mol for host-guest systems after applying proper corrections [68].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Computational Tools for Solvent Model Assessment
| Tool Category | Specific Solutions | Function | Application Context |
|---|---|---|---|
| Force Fields | CHARMM36m, a99SB-disp, DESRES-RNA | Molecular mechanics parameter sets | Protein/RNA dynamics with explicit solvent [67] [11] |
| Implicit Solvent Models | GB-neck2, ML-PCM, LSNN | Continuum solvation with recent improvements | Accelerated sampling and free energy estimation [11] [13] [69] |
| Enhanced Sampling | Maximum Entropy Reweighting | Experimental data integration | Conformational ensemble refinement [67] |
| Validation Metrics | ens_dRMS, Kish ratio | Ensemble comparison and quality assessment | IDP and flexible system validation [70] [67] |
| Free Energy Methods | Alchemical FEP, Kinetics-derived ΔG | Binding and solvation free energy calculation | Drug binding assessment and solvation prediction [68] [13] |
Diagram 2: Relationship between solvent models, applications, and validation approaches
The assessment of explicit versus implicit solvent models reveals a nuanced landscape where methodological advances have substantially narrowed the performance gap for specific applications. Explicit solvents remain the gold standard for modeling specific solvent interactions and providing physically detailed simulations, particularly for systems with crucial water-mediated interactions. However, modern implicit solvents have demonstrated remarkable capabilities in biomolecular folding, achieving near-native structures for proteins and RNA stem-loops while offering orders-of-magnitude improvement in computational efficiency. Machine learning approaches are rapidly bridging the accuracy gap in solvation free energy prediction, with ML-enhanced implicit models now competing with explicit solvent methods for small molecules. For intrinsically disordered proteins, integrative approaches combining maximum entropy reweighting with experimental data are enabling force-field independent ensemble determination. The choice between explicit and implicit modeling must therefore be guided by specific research questions, balancing physical accuracy against computational feasibility while leveraging validation metrics appropriate for each system type.
Discriminating a protein's native, functional three-dimensional structure from a pool of incorrect "decoy" models is a central challenge in computational structural biology. The accuracy of this process is vital for protein structure prediction, drug discovery, and understanding folding diseases. This task critically depends on the scoring functions used to evaluate protein models, which are themselves heavily influenced by the choice of solvent model in molecular dynamics (MD) simulations.
Explicit solvent models, which simulate individual water molecules, are considered the gold standard for accuracy but are prohibitively computationally expensive. Implicit solvent models, which treat the solvent as a continuous medium, offer a faster alternative but have historically suffered from lower accuracy in describing detailed solvation effects. This guide objectively compares the performance of modern scoring methods and solvent models in native state discrimination, providing researchers with the experimental data and protocols needed to inform their computational strategies.
Performance Comparison of Scoring Methods and Solvent Models
The table below summarizes the core performance metrics of various quality assessment and solvent models as reported in experimental literature.
Table 1: Performance Comparison of Protein Scoring Methods and Solvent Models
| Method / Model | Type | Key Metric | Reported Performance | Key Advantage |
|---|---|---|---|---|
| MQAPRank [71] | Single-Model MQA | wmPMCC (GDT_TS) | 0.793 (3DRobot dataset) | Effective ranking without clustering |
| Quasi-MQAPRank [71] | Quasi-Single-Model MQA | wmPMCC (GDT_TS) | 0.827 (3DRobot dataset) | Combines single-model & consensus strengths |
| LSNN [7] | ML Implicit Solvent | Solvation Free Energy | Near-explicit solvent accuracy | Computational speedup over explicit models |
| Explicit Solvent (TIP3P) [7] | Explicit Solvent | Solvation Free Energy | Gold Standard (Reference) | High accuracy, models explicit water |
| GBSA/PBSA [10] [7] | Traditional Implicit Solvent | Solvation Free Energy | Lower accuracy vs. explicit | Faster than explicit, but simplified SASA term prone to error |
The following table outlines the computational trade-offs and primary use cases for different solvent modeling approaches.
Table 2: Computational Characteristics and Applications of Solvent Models
| Model Category | Computational Cost | Sampling Speed | Best-Suited Application | Key Limitation |
|---|---|---|---|---|
| Explicit Solvent | Very High | Slower (high viscosity) | Benchmarking; final validation | Prohibitive for large systems/long timescales |
| Traditional Implicit [10] | Low | Faster (low viscosity) | Rapid conformational search | Poor description of local solvation effects |
| ML-Based Implicit (e.g., LSNN) [7] | Medium | Faster (low viscosity) | High-throughput screening; free energy calculations | Accuracy constrained by training data |
Experimental Protocols for Key Studies
cDNA Display Proteolysis for High-Throughput Stability Measurement
This protocol measures thermodynamic folding stability (ΔG) for hundreds of thousands of protein variants [72].
- DNA Library Preparation: A synthetic DNA oligonucleotide pool is created, with each oligonucleotide encoding a single test protein domain.
- Cell-Free cDNA Display: The DNA library is transcribed and translated in vitro using a cDNA display system. This results in each protein being covalently attached to its own encoding cDNA via its C-terminus.
- Protease Challenge: The protein-cDNA complexes are incubated with a series of increasing concentrations of protease (e.g., trypsin or chymotrypsin). Folded proteins are protease-resistant, while unfolded ones are cleaved.
- Pull-Down and Quantification: The reaction is quenched, and intact (non-cleaved) proteins are isolated using a pull-down assay targeting an N-terminal tag. The relative abundance of each protein in the surviving pool is quantified via deep sequencing.
- Stability Inference (K50 and ΔG): A Bayesian model is applied to the sequencing data. For each sequence, the protease concentration at which the cleavage rate is half of the maximum (K50) is inferred. Thermodynamic stability (ΔG) is then calculated using a kinetic model that accounts for the sequence-specific protease susceptibility of the unfolded state (K50,U) and a universal cleavage rate for the folded state (K50,F), using the formula: ΔG = -RT ln(K50,U / K50 - 1) [72].
Learning-to-Rank for Decoy Quality Assessment (MQAPRank)
This protocol formulates model quality assessment as a ranking problem [71].
- Feature Extraction: For every decoy model in a set, a feature vector is extracted. These features include knowledge-based energy potentials and quality scores from other established MQA methods.
- Instance Creation: The training instances are pairs of decoys from the same target protein. Each pair is labeled to indicate which decoy is of higher quality based on a similarity metric to the native structure (e.g., GDT_TS score or TM-score).
- Model Training: A learning-to-rank machine learning algorithm (e.g., RankSVM) is trained on these paired instances. The model learns to predict the relative quality order of any two decoys from the same protein.
- Quality Prediction: For a new set of decoys, the trained model compares them in pairs and assigns a ranking score. The inverse of this score can be used as a relative quality estimate.
- Quasi-Single-Model Extension (Quasi-MQAPRank): The top five models ranked by MQAPRank are selected as reference models. The quality of every other decoy is then re-calculated as its average GDT_TS score (or TM-score) when aligned to these reference models, blending single-model and consensus approaches [71].
Machine Learning Implicit Solvation with LSNN
This protocol describes training a graph neural network (GNN) for accurate solvation free energy calculations [7].
- Data Generation: A training dataset of approximately 300,000 small molecules is generated. Reference data, including forces and solvation free energies, are obtained from explicit-solvent alchemical simulations.
- Multi-Task Loss Function: The model is trained using a novel loss function that goes beyond standard force-matching. The loss incorporates:
- Force-Matching: Minimizes the difference between predicted and reference atomic forces.
- Alchemical Derivative Matching: Also minimizes the difference between the predicted and reference derivatives of the solvation energy with respect to alchemical coupling parameters for electrostatic (
λ_elec) and steric (λ_steric) interactions. This ensures the model can accurately compute absolute free energies and not just forces.
- Model Architecture and Training: A GNN takes atomic coordinates, charges, and other chemical features as input. It is trained with the multi-task loss function to output a potential that accurately approximates the true potential of mean force (PMF).
- Free Energy Calculation: The trained LSNN model can be used in simulations to rapidly compute conformational energies and solvation free energies with accuracy comparable to explicit solvent, but at a significantly lower computational cost [7].
Workflow Diagram: Decoy Scoring & Validation
The diagram below illustrates the logical workflow and key decision points for discriminating native protein structures from decoys using modern computational approaches.
Table 3: Key Resources for Protein Folding and Decoy Discrimination Studies
| Resource / Reagent | Function / Description | Application Context |
|---|---|---|
| cDNA Display Proteolysis [72] | High-throughput experimental method for measuring protein folding stability (ΔG). | Generating large-scale experimental data for validating computational predictions of stability and native state. |
| Neural Network Potentials (NNPs) [29] [7] | Machine learning models trained on quantum chemical data to rapidly and accurately compute molecular energies and forces. | Accelerating molecular dynamics simulations; creating fast and accurate implicit solvent models like LSNN. |
| Open Molecules 2025 (OMol25) [29] | A massive, high-accuracy dataset of quantum chemical calculations for diverse molecular structures. | Training and benchmarking neural network potentials and other machine learning models for molecular simulation. |
| Learning-to-Rank Algorithms [71] | A class of machine learning algorithms designed to sort items based on their relative quality. | Developing model quality assessment programs (MQAPs) that rank decoy models by their similarity to the native structure. |
| Generalized Born/SASA Model [10] [7] | A traditional implicit solvent model that approximates polar solvation with Generalized Born and non-polar with SASA. | A computationally efficient baseline model for solvation effects in protein simulations and scoring. |
| All-Atom Molecular Dynamics [73] [74] | Simulation technique that models every atom in a system, often using explicit solvent. | Providing high-resolution, reference data for studying folding mechanisms and validating coarse-grained models. |
In molecular dynamics (MD) research, the choice between explicit and implicit solvent models has traditionally presented a fundamental trade-off. Explicit solvent models, which treat each solvent molecule individually, offer high physical fidelity by capturing specific molecular interactions such as hydrogen bonding and van der Waals forces. However, this accuracy comes at a substantial computational cost, limiting the timescales and system sizes accessible for simulation. In contrast, implicit solvent models approximate the solvent as a continuous dielectric medium, significantly accelerating calculations by reducing the number of particles and eliminating the need to simulate solvent viscosity. While this approach offers remarkable computational efficiency for studying conformational changes and binding processes, it sacrifices atomic-level detail of solvent-solute interactions. The emerging paradigm of hybrid solvent methodologies seeks to combine the strengths of both approaches by retaining explicit solvent molecules in critical regions while treating the bulk solvent implicitly. This integrated strategy offers a promising path toward maintaining accuracy where it matters most while achieving the computational speed necessary for probing biologically relevant timescales and system sizes.
The theoretical foundation for hybrid methods rests on the observation that many biological processes depend primarily on localized solvent interactions near the solute. For instance, in protein-ligand binding or catalytic reactions, the first solvation shell often mediates crucial interactions, while bulk solvent primarily exerts dielectric screening effects. By partitioning the system into different treatment zones, hybrid models can concentrate computational resources on physically important regions. This approach aligns with the hierarchical nature of solvation effects, where different aspects of solvent influence contribute to various biological phenomena. As MD simulations continue to push toward more complex systems and longer timescales, hybrid methodologies represent an essential strategy for balancing physical accuracy with computational feasibility in computational biophysics and drug design.
Theoretical Foundations: Explicit, Implicit, and Hybrid Solvent Models
Explicit Solvent Models
Explicit solvent modeling treats each solvent molecule as an individual entity with full atomic detail, typically using molecular mechanics force fields. The most common implementation for biomolecular simulations employs the particle mesh Ewald (PME) method, which imposes artificial periodicity on the system to efficiently compute long-range electrostatic interactions. This approach provides the most physically realistic representation of solvent effects, capturing specific interactions such as hydrogen bonding, water-bridged interactions, and hydrophobic hydration with high fidelity. The explicit treatment allows for natural reproduction of solvent viscosity, diffusion, and other transport properties that emerge from molecular collisions. However, this physical accuracy comes at a tremendous computational cost, with solvent atoms typically constituting 80-90% of the total atoms in a simulation box and consequently consuming the majority of computational resources. This limitation severely restricts the accessible timescales for studying biological processes such as protein folding, large-scale conformational changes, and molecular association events, which often occur on microsecond to millisecond timescales or beyond.
Implicit Solvent Models
Implicit solvent models, also termed continuum solvation models, represent the solvent as a structureless continuum characterized primarily by its dielectric constant. This approach eliminates the need to simulate individual solvent molecules, dramatically reducing system complexity and computational demand. The most theoretically rigorous implicit models solve the Poisson-Boltzmann (PB) equation, which describes electrostatic interactions in a dielectric medium possibly containing ions. However, due to the computational expense of numerical PB solutions, the Generalized Born (GB) model has emerged as a popular approximation that provides similar accuracy with significantly lower computational cost. The GB model calculates solvation energy using the analytical formula:
where f_GB = √(r_ij² + a_ij² × e^(-D)), D = (r_ij/(2a_ij))², and a_ij = √(a_i a_j) [75]. In this formulation, ϵ0 represents the vacuum permittivity, ϵ is the solvent dielectric constant, qi and qj are atomic charges, rij is the interatomic distance, and a_i represents the Born radius of atom i. This model is frequently combined with a solvent-accessible surface area (SASA) term to account for nonpolar contributions to solvation, an approach known as GBSA.
The primary advantage of implicit solvent models is their dramatic acceleration of conformational sampling. By eliminating solvent viscosity and random collisions with explicit solvent molecules, implicit models can explore conformational space much more rapidly. Studies have demonstrated speedups ranging from approximately 2-fold for small conformational changes to over 100-fold for large-scale transitions compared to explicit solvent simulations [76]. However, this efficiency comes with significant limitations, including the inability to capture specific solute-solvent interactions, overstabilization of salt bridges due to insufficient electrostatic screening, and alteration of native protein folding landscapes [75].
The Hybrid Approach
Hybrid solvent methodologies strategically combine explicit and implicit treatments to leverage their respective advantages while mitigating their limitations. In these approaches, a layer of explicit solvent molecules is maintained around the solute region of interest, while the bulk solvent is modeled implicitly using continuum approaches such as GB. This architecture allows specific solvent interactions in the first solvation shell—which are often critical for structural stability and molecular recognition—to be preserved with atomic detail. Simultaneously, the continuum treatment of bulk solvent provides efficient dielectric screening and eliminates the computational overhead associated with explicit bulk solvent molecules.
The theoretical justification for this partitioning stems from the different roles played by proximal versus distal solvent molecules. Short-range interactions within the first solvation shell (typically 3-5 Å from the solute surface) are highly specific and directional, involving hydrogen bonding, van der Waals contacts, and hydrophobic hydration effects. These interactions frequently participate directly in biological mechanisms and require atomic-level description. In contrast, long-range solvent effects primarily provide dielectric screening and bulk solvation properties that can be effectively captured by continuum approaches. By separating these contributions, hybrid models achieve a more optimal balance between accuracy and efficiency than either pure approach alone.
Table: Comparison of Solvent Modeling Approaches
| Feature | Explicit Solvent | Implicit Solvent | Hybrid Approach |
|---|---|---|---|
| Computational Cost | High (80-90% solvent atoms) | Low (no solvent atoms) | Moderate (limited explicit solvent) |
| Sampling Speed | Limited by solvent viscosity | 2-100x faster than explicit [76] | Intermediate, system-dependent |
| Electrostatic Treatment | Particle Mesh Ewald (PME) | Poisson-Boltzmann or Generalized Born | GB for bulk, explicit for local |
| Specific Solvent Interactions | Fully captured | Not captured | Captured in first solvation shell |
| Hydrogen Bonding | Explicitly represented | Approximated via dielectric | Explicit in local region |
| Hydrophobic Effect | Emerges from interactions | SASA-based term | Combination of both |
| Implementation Complexity | Standard | Moderate | High (interface handling) |
Quantitative Performance Comparison
Systematic benchmarking studies provide critical insights into the relative performance of explicit, implicit, and hybrid solvent methodologies across different biological systems and processes. The speed advantage of implicit solvent models is highly system-dependent, with greater acceleration observed for larger-scale conformational changes. For small dihedral angle flips in proteins, implicit models offer approximately 1-fold speedup in conformational sampling compared to explicit solvent. However, for large conformational changes such as nucleosome tail collapse and DNA unwrapping, the speedup increases dramatically to between 1- and 100-fold. In mixed cases like miniprotein folding, implicit solvent provides approximately 7-fold faster conformational sampling [76]. These speedups primarily result from the elimination of solvent viscosity rather than alterations to the underlying energy landscape.
When considering combined speedup—incorporating both enhanced conformational sampling and algorithmic efficiency—implicit solvent models demonstrate significant advantages. For the conformational changes studied, the total speedups are approximately 2-fold for small changes, 1- to 60-fold for large changes, and approximately 50-fold for mixed cases like miniprotein folding in the low solvent viscosity regime afforded by implicit solvent [76]. This acceleration enables access to biologically relevant timescales that remain challenging for explicit solvent simulations on standard computational resources.
Beyond sampling efficiency, accuracy in predicting experimental observables is crucial for method validation. Recent benchmarking of neural network potentials trained on large quantum chemical datasets reveals interesting patterns in charge-related property prediction. For organometallic species, these modern methods can achieve impressive accuracy in predicting reduction potentials, with the UMA Small model demonstrating a mean absolute error (MAE) of 0.262 V and coefficient of determination (R²) of 0.896 on organometallic datasets, outperforming traditional DFT and semiempirical methods [6]. This performance is particularly notable given that these models do not explicitly incorporate charge-based physics in their architecture.
Table: Accuracy Benchmarks for Reduction Potential Prediction (Mean Absolute Error in Volts)
| Method | Main-Group Species (OROP) | Organometallic Species (OMROP) |
|---|---|---|
| B97-3c (DFT) | 0.260 | 0.414 |
| GFN2-xTB (SQM) | 0.303 | 0.733 |
| eSEN-S (NNP) | 0.505 | 0.312 |
| UMA-S (NNP) | 0.261 | 0.262 |
| UMA-M (NNP) | 0.407 | 0.365 |
Data extracted from benchmarking studies [6]
For electron affinity calculations, hybrid quantum-continuum approaches have demonstrated remarkable accuracy, with solvation energy calculations for methanol differing by less than 0.2 kcal/mol from classical benchmarks, well within the threshold of chemical accuracy [77]. This performance highlights the potential for combining quantum mechanical methods with continuum solvation to achieve high accuracy for solution-phase properties.
Experimental Protocols and Methodologies
Benchmarking Protocol for Solvent Model Accuracy
Comprehensive evaluation of solvent model performance requires rigorous benchmarking against experimental data. For reduction potential prediction, the protocol begins with obtaining experimental data for both main-group and organometallic species, typically including the charge and geometry of non-reduced and reduced structures, experimental reduction potential values, and solvent identity. Structure optimization is performed using both the solvent models being evaluated and reference methods such as GFN2-xTB. For implicit solvent contributions, the Extended Conductor-like Polarizable Continuum Model (CPCM-X) is commonly employed to obtain solvent-corrected electronic energies [6]. The reduction potential is then calculated as the difference between the electronic energy of the non-reduced structure and that of the reduced structure, converted to volts.
For binding free energy calculations, the MM/GBSA (Molecular Mechanics with Generalized Born Surface Area) approach provides a popular methodology that combines molecular mechanics energy terms with implicit solvation contributions. The binding free energy is calculated as:
where each term is decomposed as:
Here, EMM represents the molecular mechanics gas-phase energy, Gsolv is the solvation free energy calculated using a GB or PB model combined with a nonpolar surface area term, and TS accounts for conformational entropy effects, though this term is frequently omitted or approximated due to its computational complexity.
Hybrid Solvent Simulation Workflow
Implementing hybrid solvent simulations requires careful system setup and parameterization. The following workflow outlines a standard protocol:
System Preparation: Begin with the solute structure (protein, nucleic acid, or complex) in its desired initial conformation. Add explicit water molecules within a specified cutoff distance (typically 5-10 Å) from the solute surface. This explicit solvent region should be sufficient to cover the first solvation shell and any specific solvent interactions of interest.
Implicit Solvent Parameters: Configure the continuum solvent model for the bulk region. For GB models, this involves setting the dielectric constants for solute and solvent, salt concentration, and surface tension parameters for the nonpolar contribution. The external dielectric constant is typically set to 80 for water, while the internal dielectric may range from 1 to 4 depending on the solute.
Boundary Handling: Implement a smooth transition between the explicit and implicit solvent regions to avoid artificial boundary effects. This may involve applying a restraining potential to explicit water molecules near the boundary to prevent evaporation into the continuum region.
Electrostatic Treatment: Employ a hybrid electrostatic scheme that calculates interactions within the explicit region using standard molecular mechanics, while treating interactions between explicit atoms and the continuum using the GB formalism. Interactions entirely within the implicit region are handled by the continuum model.
Equilibration and Production: Perform standard energy minimization and equilibration protocols, paying particular attention to the relaxation of the explicit solvent layer. Production simulations can then proceed with enhanced sampling techniques if needed.
This hybrid approach has been successfully implemented in various MD packages, with some reports indicating faster performance than full explicit solvent simulations with PME electrostatic calculations, particularly for small to medium-sized systems [75].
Workflow for Hybrid Solvent Simulation Setup
Successful implementation of hybrid solvent methodologies requires both specialized software tools and theoretical frameworks. The following table summarizes key resources available to researchers in this field:
Table: Essential Research Reagents and Computational Resources for Hybrid Solvent Simulations
| Resource Category | Specific Tools/Methods | Function and Application |
|---|---|---|
| Continuum Solvation Models | Generalized Born (GB), Poisson-Boltzmann (PB), Integral Equation Formalism PCM (IEF-PCM) | Provide implicit solvent treatment for bulk regions; GB offers speed, PB provides accuracy [75] [37] |
| Explicit Solvent Models | TIP3P, TIP4P, SPC water models | Represent specific solvent interactions in local regions with atomic detail |
| Hybrid Implementation Software | AMBER, CHARMM, OpenMM, NAMD | MD packages with varying support for hybrid solvent methodologies |
| Benchmarking Datasets | Experimental reduction potentials, electron affinities, solvation free energies | Validation of method accuracy against experimental observables [6] |
| Quantum-Continuum Interfaces | SQD-IEF-PCM, CASCI-PCM | Combine quantum mechanical calculations with continuum solvation for electronic properties [77] |
| System Preparation Tools | PDB2PQR, LEaP, VMD | Prepare molecular structures, assign parameters, and set up simulation boxes |
| Analysis Packages | MDTraj, CPPTRAJ, VMD | Analyze simulation trajectories, calculate observables, and visualize results |
Beyond these computational resources, theoretical understanding of solvation physics remains essential for appropriate method selection and interpretation. The Generalized Born model serves as the workhorse for many hybrid implementations due to its favorable balance of accuracy and computational efficiency. Recent advances in machine learning-augmented approaches show promise as Poisson-Boltzmann-accurate surrogates that can further accelerate implicit solvent calculations [37]. For properties requiring quantum mechanical treatment, such as charge transfer or electronic excitation in solution, quantum-continuum workflows that couple methods like IEF-PCM with quantum calculations enable realistic solution-phase electronic structure modeling [77] [37].
Limitations and Future Directions
Despite their promising advantages, hybrid solvent methodologies face several significant challenges that require continued methodological development. A primary limitation concerns the treatment of boundaries between explicit and implicit regions. Sharp boundaries can create artificial interfaces that perturb local structure and dynamics, while smooth transitions require careful parameterization to maintain physical realism. The dielectric discontinuity at the boundary can produce artifacts in electrostatic calculations, particularly for charged systems where ion partitioning between regions becomes problematic. Additionally, the absence of explicit solvent molecules beyond the first solvation shell eliminates long-range solvent correlations that may influence solute dynamics in ways not fully captured by continuum models.
Current hybrid approaches also struggle with environmental heterogeneity, particularly at biological interfaces such as membrane surfaces or liquid-liquid interfaces. Standard continuum models assume a uniform dielectric environment, failing to capture the spatial variation in dielectric properties at complex interfaces. Similarly, the specificity of ion effects—where different ions exhibit distinct behaviors beyond what continuum electrostatics predicts—presents challenges for hybrid methods. These limitations become particularly acute for systems where the bulk solvent contributes specifically to the mechanism, such as in transport processes or systems with long-range electrostatic correlations.
Future developments in hybrid methodologies are advancing along several exciting frontiers. Machine learning corrections show tremendous promise for addressing systematic errors in implicit solvent models. By training on high-quality explicit solvent simulations or experimental data, ML models can learn residual corrections to GB or PB baselines, potentially achieving PB-level accuracy at GB computational costs [37]. These approaches can incorporate uncertainty quantification and active learning to improve reliability. For quantum mechanical applications, quantum-continuum hybrids are becoming increasingly sophisticated, with methods like SQD-IEF-PCM demonstrating the feasibility of calculating solvation energies on quantum hardware that match classical benchmarks within chemical accuracy [77].
Further innovation in multiscale partitioning approaches will likely enable more adaptive and physically realistic hybrid simulations. Such methods could dynamically adjust the boundary between explicit and implicit regions based on local properties or chemical events, providing explicit solvent resolution where and when it is needed most. Additionally, improved nonpolar solvation models that better capture cavity formation, dispersion interactions, and hydrophobic effects will enhance the physical realism of hybrid approaches. As these methodological advances mature, hybrid solvent methodologies are poised to become increasingly robust and accurate tools for bridging the gap between computational efficiency and physical accuracy in biomolecular simulations.
Current Limitations and Future Research Directions
Hybrid solvent methodologies represent a sophisticated compromise in the persistent challenge to balance computational efficiency with physical accuracy in molecular simulations. By strategically combining explicit solvent treatment in localized regions with implicit continuum models for bulk solvent effects, these approaches enable accelerated sampling while preserving atomic-level detail where it matters most for biological function. The quantitative benchmarking data presented in this review demonstrates that hybrid methods can achieve substantial speedups—from 2-fold to over 100-fold depending on system size and process type—while maintaining accuracy comparable to more computationally expensive pure explicit solvent simulations for many applications.
As methodological developments continue to address current limitations in boundary treatment, dielectric response, and environmental heterogeneity, hybrid approaches are poised to become increasingly central to biomolecular simulation. The integration of machine learning corrections and quantum-continuum frameworks represents particularly promising directions for extending the accuracy and applicability of these methods. For researchers in computational biophysics and drug discovery, hybrid solvent methodologies offer a powerful toolkit for tackling problems that require both extensive conformational sampling and physical fidelity, from protein folding and molecular recognition to ligand binding and allosteric regulation. By continuing to refine these integrated approaches, the scientific community moves closer to the ultimate goal of simulating biological processes with both atomic precision and biological relevance.
Molecular dynamics (MD) simulations are a cornerstone of modern computational chemistry, providing atomistic insights into biological processes and drug discovery. A critical choice in setting up these simulations is the treatment of the solvent environment. Explicit solvent models atomistically represent individual solvent molecules, while implicit solvent models approximate the solvent as a continuous medium, leading to a fundamental trade-off between computational cost and physical accuracy [66]. This guide provides an objective comparison of these approaches, detailing their performance characteristics, supported by recent experimental data and methodologies. Understanding these trade-offs is essential for researchers to select the appropriate model for simulating processes such as protein folding, ligand binding, and chemical reactions.
Comparative Analysis of Solvent Models
The core distinction between explicit and implicit solvent models lies in their treatment of solvent-solute interactions, which directly dictates their computational expense and physical fidelity. The following table summarizes the key trade-offs.
Table 1: Trade-offs Between Explicit and Implicit Solvent Models
| Aspect | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Fundamental Approach | Explicitly models individual solvent molecules (e.g., TIP3P water) within the system [27]. | Treats solvent as a continuous, polarizable medium with a dielectric constant; represents solute-solvent interactions with mean-field approximations [66]. |
| Computational Cost | Very high, as the majority of computation is spent on calculating solvent-solvent and solvent-solute interactions [66]. | Significantly lower, as solvent degrees of freedom are eliminated, speeding up conformational sampling [27]. |
| Sampling Speed | Slower conformational sampling due to explicit viscosity and inertial effects of solvent molecules [27]. | ~1 to 100-fold speedup for large conformational changes; speed is highly system-dependent and linked to reduced solvent viscosity [27]. |
| Physical Accuracy | High; accurately captures specific solute-solvent interactions (e.g., hydrogen bonding), solvation shells, and entropic effects [4]. | Lower; misses specific, atomic-level solvent effects, which can be critical for processes like protein folding and binding [4] [66]. |
| Free Energy Calculations | Considered the gold standard but is computationally intensive for absolute solvation and binding free energies [7]. | Historically less accurate but faster; modern ML-corrected implicit models are achieving accuracy comparable to explicit solvent [7] [78]. |
| System Size Limitation | Limited by the number of atoms, as adding solvent molecules dramatically increases system size [79]. | More suitable for larger systems or longer timescales, as the cost scales primarily with the number of solute atoms [27] [79]. |
| Common Applications | - Studying specific solvent roles in mechanisms- Accurate free energy calculations- Processes where solvent structure is critical [4] | - Enhanced conformational sampling- Rapid screening in drug discovery- Simulations of large-scale biomolecular dynamics [27] [80] |
Experimental Protocols and Data
Quantifying Conformational Sampling Speed
A systematic study directly compared the particle mesh Ewald (PME) explicit solvent model with a generalized Born (GB) implicit solvent model, quantifying the speed of conformational sampling across different types of molecular changes [27].
- Methodology: The researchers performed MD simulations for systems representing small (dihedral angle flips), large (nucleosome tail collapse, DNA unwrapping), and mixed (folding of a miniprotein) conformational changes. The nominal simulation times ranged from nanoseconds to microseconds, depending on the system size. The conformational sampling speedup was calculated by comparing the efficiency of sampling essential dynamics between the GB and PME simulations at the same temperature [27].
- Key Findings: The speedup provided by implicit solvent was highly system-dependent.
- Small changes: Approximately onefold (minimal speedup).
- Large changes: Between ~1- and 100-fold.
- Mixed case (protein folding): Approximately sevenfold. The study concluded that the primary reason for this speedup is the reduction in solvent viscosity in implicit models, rather than differences in the underlying free-energy landscapes [27].
Improving Accuracy with Machine Learning
A major drawback of traditional implicit solvent models is their inferior accuracy for precise thermodynamic calculations. Recent research has focused on using machine learning to bridge this accuracy gap while retaining a computational speed advantage.
- Methodology: A novel λ-Solvation Neural Network (LSNN) was developed. This graph neural network (GNN) was trained on a dataset of approximately 300,000 small molecules. Unlike traditional force-matching methods that only predict forces, the LSNN was also trained to match the derivatives of alchemical variables (electrostatic and steric coupling factors). This allows the model to accurately compute potentials of mean force (PMF) and absolute free energies, which are crucial for drug discovery applications [7].
- Key Findings: The LSNN model achieved free energy predictions with an accuracy comparable to explicit-solvent alchemical simulations, while still offering a significant computational speedup over explicit solvent methods [7].
ML/MM for Free Energy Calculations
Another advanced approach combines machine learning interatomic potentials (MLIPs) with molecular mechanics (ML/MM) to enhance the accuracy of free energy calculations.
- Methodology: Researchers developed a hybrid ML/MM interface integrated into the AMBER simulation package. They created a novel ML/MM-compatible thermodynamic integration (TI) framework to calculate hydration free energies. This framework overcomes the challenge of applying indivisible MLIP energies in traditional TI by omitting perturbation of nonbonded interactions within the ML region and introducing a "reorganization energy" term for compensation [78].
- Key Findings: Hydration free energies calculated using this ML/MM TI framework achieved an accuracy of 1.0 kcal/mol, outperforming traditional approaches. Furthermore, ML/MM enabled more precise conformational sampling for endpoint free energy calculations [78].
Workflow and Signaling Pathways
The following diagram illustrates the core trade-off and the modern approaches, including machine learning, used to navigate the balance between computational cost and physical accuracy in solvent modeling.
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Item | Function in Research |
|---|---|
| Generalized Born (GB) Model | A widely used implicit solvent model that approximates the electrostatic solvation free energy with a pairwise function, offering a good balance of speed and simplicity [27] [81]. |
| Poisson-Boltzmann (PB) Equation | A more computationally expensive but often more accurate implicit solvent model for calculating electrostatic solvation by numerically solving a differential equation [66]. |
| Graph Neural Network (GNN) | A type of machine learning architecture used to develop advanced implicit solvent potentials, such as the LSNN model, by learning from large datasets of molecular simulations [7]. |
| Machine Learning Interatomic Potentials (MLIPs) | Surrogate models trained on quantum mechanical data that can approach quantum accuracy at a fraction of the computational cost, used in ML/MM simulations [4] [78]. |
| Thermodynamic Integration (TI) | A pathway-based method for calculating free energy differences; requires specialized frameworks when used with MLIPs [78]. |
| Alchemical Free Energy Calculations | A set of techniques that compute free energy differences by simulating a non-physical pathway, often used with explicit solvent but now being adapted for ML-implicit models [7] [66]. |
Conclusion
The choice between explicit and implicit solvent models is not a matter of superiority but of matching the model to the scientific question. Implicit solvents offer unparalleled speedup in conformational sampling—from severalfold to over 100-fold for large changes—primarily by reducing solvent viscosity, making them ideal for rapid exploration of free energy landscapes, high-throughput screening, and folding studies. However, this speed can come at the cost of accuracy in systems where atomic-level solvent details, such as specific hydrogen bonds or hydrophobic interactions, are critical. Explicit solvents remain the gold standard for quantitative accuracy when computational resources allow. The future of biomolecular simulation lies in sophisticated hybrid models that seamlessly blend the efficiency of implicit solvents for the bulk with the precision of explicit molecules at the solute-solvent interface, as well as in the continued development of better-parametrized and more physically rigorous implicit models. For biomedical research, these advances promise more reliable drug design, a deeper understanding of protein misfolding diseases, and the ability to simulate increasingly complex biological systems on experimentally relevant timescales.
References
- 1. Speed of conformational change: comparing and explicit ... implicit [https://pubmed.ncbi.nlm.nih.gov/25762327/]
- 2. Biomolecular Solvation Structure Revealed by Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6438627/]
- 3. Implicit Solvent Models and Their Applications in Biophysics [https://www.mdpi.com/2218-273X/15/9/1218]
- 4. Modelling chemical processes in explicit solvents with ... [https://www.nature.com/articles/s41467-024-50418-6]
- 5. Exploring Meta's Open Molecules (OMol25) & Universal 2025 ... Models [https://rowansci.com/blog/exploring-open-molecules-2025]
- 6. Benchmarking OMol25-Trained Models on Experimental ... [https://rowansci.com/publications/omol25-redox]
- 7. Extending machine learning model for implicit solvation to ... [https://arxiv.org/html/2510.20103v1]
- 8. From Implicit to Explicit: An Interaction-Reorganization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11674157/]
- 9. Beyond Continuum Solvent Models in Computational ... [https://link.springer.com/article/10.1007/s11244-021-01520-2]
- 10. Implicit Solvent Models in Molecular Dynamics Simulations [https://www.sciencedirect.com/science/article/abs/pii/S1574140008000078]
- 11. Molecular Dynamics Simulations of RNA Stem-Loop Folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12593105/]
- 12. Design and application of implicit solvent models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4045398/]
- 13. Improved prediction of solvation free energies by machine ... [https://www.nature.com/articles/s41467-021-23724-6]
- 14. Machine-Learned Potentials for Solvation Modeling [https://arxiv.org/html/2505.22402v3]
- 15. Frontiers | Free , Enthalpy and Entropy from Energy ... Implicit Solvent [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2018.00011/full]
- 16. Solvation Free Energies of Drug-like Molecules via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12392459/]
- 17. of Comparison and Implicit for the... Explicit Solvent Models [https://pubmed.ncbi.nlm.nih.gov/28245118/]
- 18. Computing solvation free energies of small molecules with ... [https://arxiv.org/html/2405.18171v3]
- 19. MM/PB(GB)SA benchmarks on soluble proteins and ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2022.1018351/full]
- 20. Exact analytical algorithm for solvent accessible surface area ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11530138/]
- 21. Accessible Surface Area - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/accessible-surface-area]
- 22. Accessible surface area [https://en.wikipedia.org/wiki/Accessible_surface_area]
- 23. Solvent Accessible Surface Area (SASA) Calculation [https://mdtraj.org/1.9.4/examples/solvent-accessible-surface-area.html]
- 24. Poisson-Boltzmann-based machine learning model for ... [https://www.sciencedirect.com/science/article/pii/S0006349524001073]
- 25. Accurate Predictions of Molecular Properties of Proteins via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12080100/]
- 26. Machine-Learned Potentials for Solvation Modeling [https://arxiv.org/html/2505.22402]
- 27. Speed of Conformational Change: Comparing Explicit and ... [https://www.sciencedirect.com/science/article/pii/S000634951500003X]
- 28. Transferring Knowledge from MM to QM: A Graph Neural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355693/]
- 29. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 30. Ammm:MM/PBSA with amber: theory and application - biowiki [https://biomolmd.org/mw/index.php/Ammm:MM/PBSA_with_amber:_theory_and_application]
- 31. A head-to-head comparison of MM/PBSA and MM/GBSA in ... [https://pubmed.ncbi.nlm.nih.gov/39480515/]
- 32. Difference between PBSA and GBSA [https://groups.google.com/g/gmx_mmpbsa/c/Kx_kUJ42L68]
- 33. Exact analytical algorithm for solvent accessible surface area ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10836080/]
- 34. [AMBER] Different SASA values in MM/PBSA and MM/GBSA? [http://archive.ambermd.org/201308/0496.html]
- 35. Re: [AMBER] Using MMPBSA.py with CHARMM force field [http://archive.ambermd.org/201508/0379.html]
- 36. Re: [AMBER] Using MMPBSA.py with CHARMM force field [http://archive.ambermd.org/201508/0387.html]
- 37. Implicit Solvent Models and Their Applications in Biophysics [https://pubmed.ncbi.nlm.nih.gov/41008525/]
- 38. Accuracy comparison of several common implicit solvent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5313374/]
- 39. Benchmarking Implicit Solvent Folding Simulations of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2719849/]
- 40. Using Implicit Solvent With Neural Network Potentials - Rowan [https://www.rowansci.com/blog/using-implicit-solvent-with-nnps]
- 41. A Standardized Benchmark for Machine-Learned ... [https://arxiv.org/html/2510.17187v1]
- 42. The MM / PBSA and MM / GBSA methods to estimate ligand-binding... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4487606/]
- 43. MM/PBSA and MM/GBSA - Computational Chemistry ... [https://deeporigin.com/glossary/mm-pbsa-and-mm-gbsa]
- 44. Comparison of implicit solvent models and force fields in ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261411011353]
- 45. Assessing the Performance of MM / PBSA , MM / GBSA , and... [https://figshare.com/articles/journal_contribution/Assessing_the_Performance_of_MM_PBSA_MM_GBSA_and_QM_MM_GBSA_Approaches_on_Protein_Carbohydrate_Complexes_Effect_of_Implicit_Solvent_Models_QM_Methods_and_Entropic_Contributions/6972302/1]
- 46. Assessing the performance of MM/PBSA and MM/GBSA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6097651/]
- 47. Solvent Model Benchmark for Molecular Dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10091405/]
- 48. GōMartini 3: From large conformational changes in ... [https://www.nature.com/articles/s41467-025-58719-0]
- 49. Benchmarking GPUs for MD Simulations [https://www.linkedin.com/pulse/benchmarking-gpus-md-simulations-speed-cost-insights-barbosa-farias-ynoke]
- 50. apoCHARMM: High-performance molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/40341929/]
- 51. Deep generative modeling of temperature-dependent ... [https://www.nature.com/articles/s42004-025-01737-2]
- 52. Challenges and opportunities in computational studies for ... [https://www.nature.com/articles/s44386-025-00024-3]
- 53. High Throughput Screening Market Forecast, 2025-2032 [https://www.coherentmarketinsights.com/industry-reports/high-throughput-screening-market]
- 54. High-throughput screening market set to be worth $26.4bn ... [https://www.drugtargetreview.com/news/84394/high-throughput-screening-market-set-to-be-worth-26-4bn-by-2025/]
- 55. Desolvation Costs of Salt Bridges across Protein Binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4047600/]
- 56. A Comparative Study on Atomic Solvation Parameters [https://www.sciencedirect.com/science/article/abs/pii/S0022283683715068]
- 57. Atomic solvation parameters applied to molecular ... [https://pubmed.ncbi.nlm.nih.gov/1304905/]
- 58. Implicit Solvation Parameters Derived from Explicit Water ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3503459/]
- 59. Automated parametrization of small molecules within the ... [https://www.nature.com/articles/s41598-025-24757-3]
- 60. Parametrization of a new small molecule [https://cgmartini.nl/docs/tutorials/Martini3/Small_Molecule_Parametrization/]
- 61. Explicit solvent models - (General Chemistry II) [https://fiveable.me/key-terms/general-chemistry-ii/explicit-solvent-models]
- 62. Solvent model [https://en.wikipedia.org/wiki/Solvent_model]
- 63. Generalized Born Implicit Solvent Models for Biomolecules [https://pmc.ncbi.nlm.nih.gov/articles/PMC6645684/]
- 64. Benchmark of Available Explicit Solvent Models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12498386/]
- 65. Comparison of Radii Sets, Entropy, QM Methods, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4688044/]
- 66. Implicit solvent methods for free energy estimation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4310817/]
- 67. Determining accurate conformational ensembles of ... [https://www.nature.com/articles/s41467-025-64098-3]
- 68. On Calculating Free Energy Differences Using Ensembles of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7291376/]
- 69. Extending machine learning model for implicit solvation to ... [https://arxiv.org/abs/2510.20103]
- 70. Distance-Based Metrics for Comparing Conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7300341/]
- 71. Sorting protein decoys by machine-learning-to-rank [https://www.nature.com/articles/srep31571]
- 72. Mega-scale experimental analysis of protein folding ... [https://www.nature.com/articles/s41586-023-06328-6]
- 73. Native contacts determine protein folding mechanisms in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3816414/]
- 74. New class of protein misfolding simulated in high definition [https://science.psu.edu/news/Obrien8-2025]
- 75. Implicit solvation - Wikipedia [https://en.wikipedia.org/wiki/Implicit_solvation]
- 76. Speed of Conformational Change: Comparing Explicit and Implicit Solvent Molecular Dynamics Simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375717/]
- 77. Quantum Chemistry Moves Closer to Practical Use with ... [https://thequantuminsider.com/2025/06/05/quantum-chemistry-moves-closer-to-practical-use-with-solvent-ready-algorithms-cleveland-clinic-researchers-report/]
- 78. Accurate Free Energy Calculation via Multiscale Simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288004/]
- 79. Explicit-solute implicit-solvent molecular simulation with ... [https://www.sciencedirect.com/science/article/abs/pii/S0021999122007367]
- 80. Machine learning analysis of molecular dynamics ... [https://www.nature.com/articles/s41598-025-11392-1]
- 81. Optimization of the GBMV2 Implicit Solvent Force Field for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5407932/]