Research Articles
From Atoms to Insights: A Comprehensive Guide to the Molecular Dynamics Workflow for Biomolecular Trajectories
This article provides a comprehensive guide to the molecular dynamics (MD) workflow for generating and analyzing atomic trajectories, tailored for researchers, scientists, and drug development professionals.
Force Fields in Molecular Dynamics: Calculating Atomic Forces from Fundamentals to Drug Discovery Applications
This article provides a comprehensive overview of the critical role force fields play in calculating atomic forces for molecular dynamics (MD) simulations.
Atomic Motion Tracking: The Fundamental Principles and Biomedical Applications of Molecular Dynamics
This article provides a comprehensive exploration of Molecular Dynamics (MD) simulations, a computational technique that tracks the physical movements of every atom in a system over time.
Molecular Dynamics Demystified: Simulating Newton's Equations from Theory to Biomedical Application
This article provides a comprehensive guide to the principles and practices of Molecular Dynamics (MD) simulations, a computational technique that solves Newton's equations of motion to model atomic-scale systems.
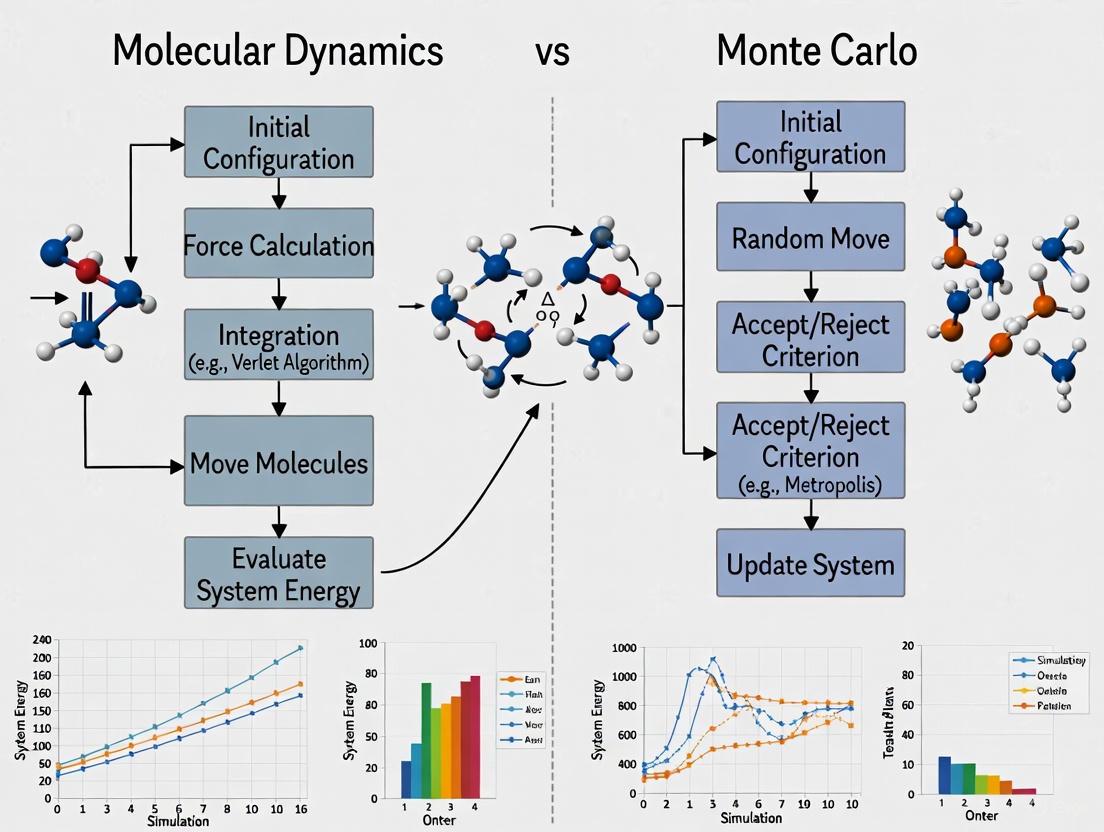
Molecular Dynamics vs Monte Carlo: A Comprehensive Guide for Computational Drug Discovery
This article provides a detailed comparison of Molecular Dynamics (MD) and Monte Carlo (MC) simulation methods for researchers and professionals in computational biology and drug development.
Molecular Dynamics Simulations: A Comprehensive Guide to Best Practices from Setup to AI Integration
This article provides a comprehensive guide to best practices in molecular dynamics (MD) simulations, tailored for researchers, scientists, and drug development professionals.
Beyond Force Metrics: A Practical Guide to Improving Stability and Convergence in Molecular Dynamics Simulations
Molecular dynamics (MD) simulations are indispensable in drug discovery and materials science, yet achieving stable and converged results remains a significant challenge.
Comparative Analysis of Molecular Dynamics Integration Algorithms: From Foundational Principles to Advanced Applications in Drug Discovery
This article provides a comprehensive examination of molecular dynamics (MD) integration algorithms, exploring their foundational principles, methodological applications, optimization strategies, and validation frameworks.

Classical vs. Reactive MD: A Comprehensive Guide to Force Field Selection for Biomedical Research
This article provides a detailed comparison between Classical Molecular Dynamics (MD) and Reactive Force Fields, with a focus on ReaxFF, tailored for researchers and professionals in drug development and biomedical...
Bridging the Gap: A Comprehensive Guide to Validating Molecular Dynamics Diffusion Coefficients with Experimental Measurements
This article provides a comprehensive framework for researchers, scientists, and drug development professionals seeking to validate diffusion coefficients derived from Molecular Dynamics (MD) simulations against experimental data.