Research Articles
Bridging the Chemical Space Gap: Next-Generation Force Fields for Accelerated Drug Discovery
The rapid expansion of synthetically accessible chemical space presents a critical challenge for traditional molecular mechanics force fields, which often lack parameters for novel drug-like molecules.
Overcoming Sampling Limitations in Force Field Validation: Strategies for Reliable Biomolecular Simulations
Accurate molecular dynamics simulations are paramount for modern drug discovery and structural biology, yet their predictive power is critically limited by insufficient sampling and force field inaccuracies.
Correcting Bonded Parameter Imbalance in Biomolecular Force Fields: From Foundations to Next-Generation Solutions
Accurate biomolecular force fields are fundamental to reliable molecular dynamics simulations in drug discovery and structural biology.
Systematic Error Reduction in Force Field Parameter Optimization: Advanced Strategies for Biomolecular Simulation and Drug Discovery
This comprehensive review addresses the critical challenge of systematic errors in force field parameterization, which significantly impacts the reliability of molecular simulations in drug discovery and biomolecular research.
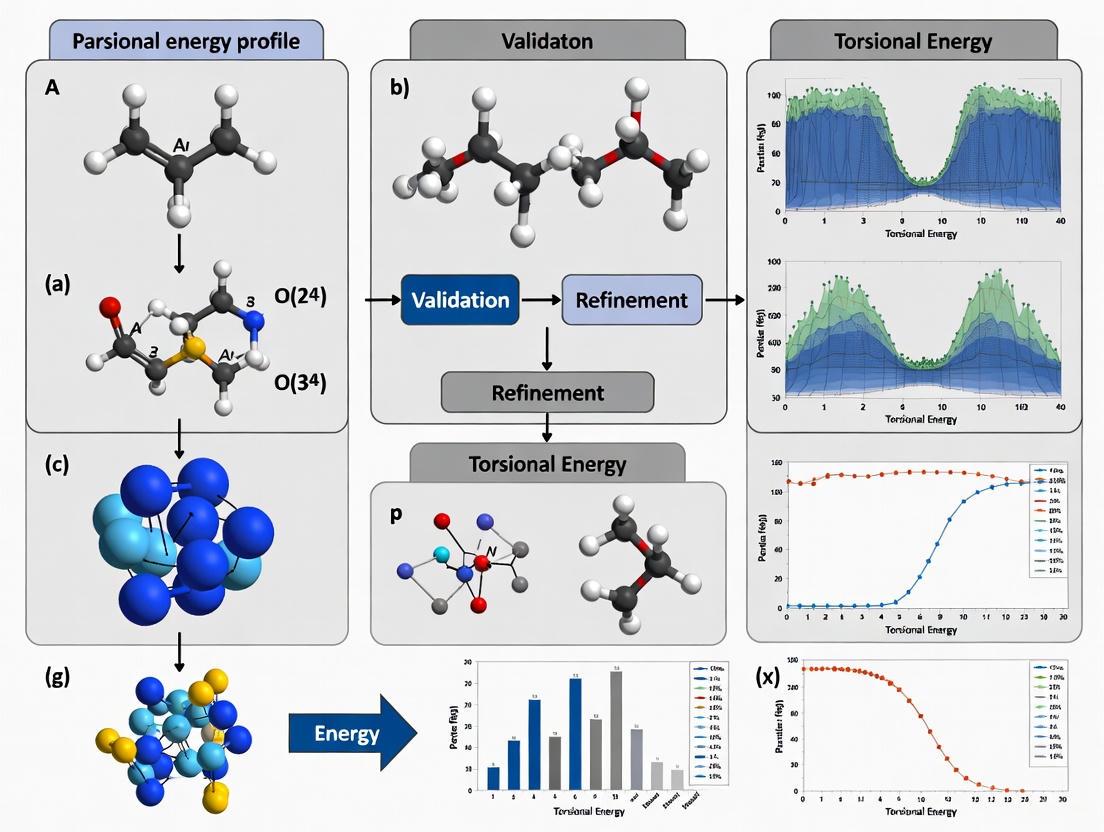
Advances in Torsional Energy Profiles: Improving Accuracy in Small Molecule Force Fields for Drug Discovery
Accurate torsional energy profiles are critical for reliable molecular dynamics simulations in drug discovery, directly impacting the prediction of binding affinities and molecular conformations.
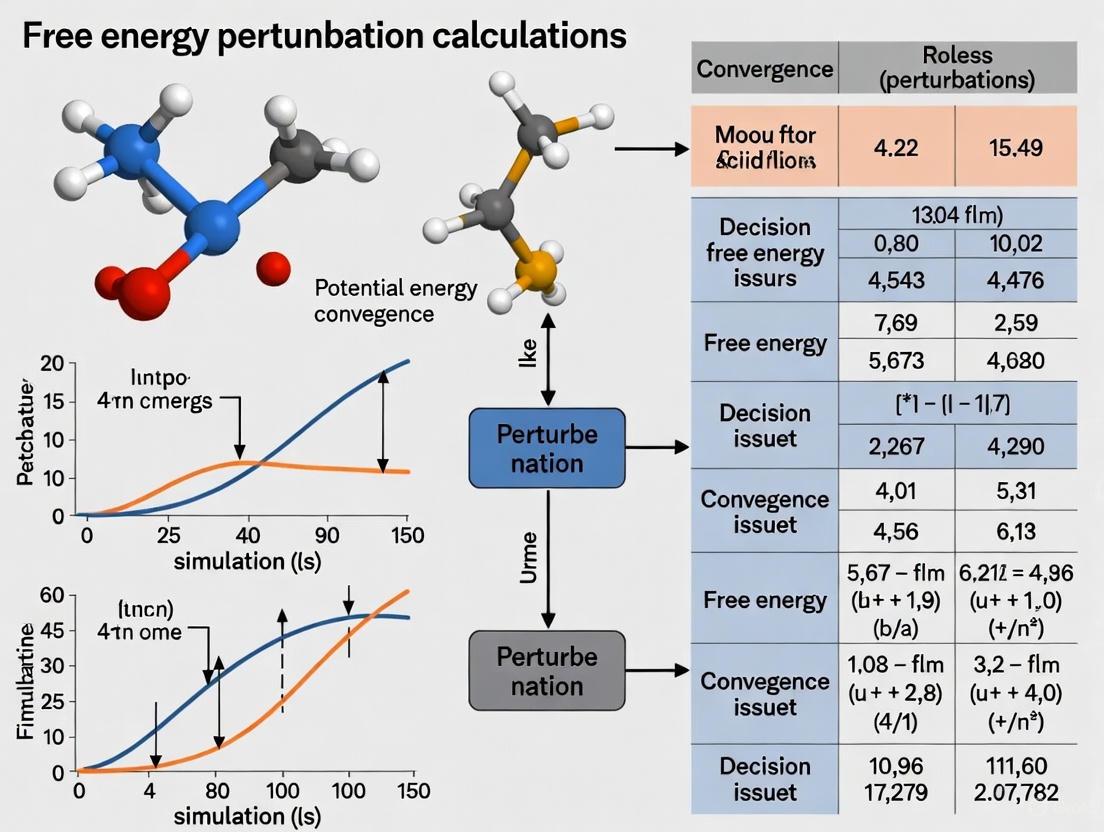
Converging on Accuracy: A Practical Guide to Fixing FEP Sampling and Convergence Issues
Free Energy Perturbation (FEP) calculations have become an indispensable tool in computational drug discovery for predicting binding affinities.
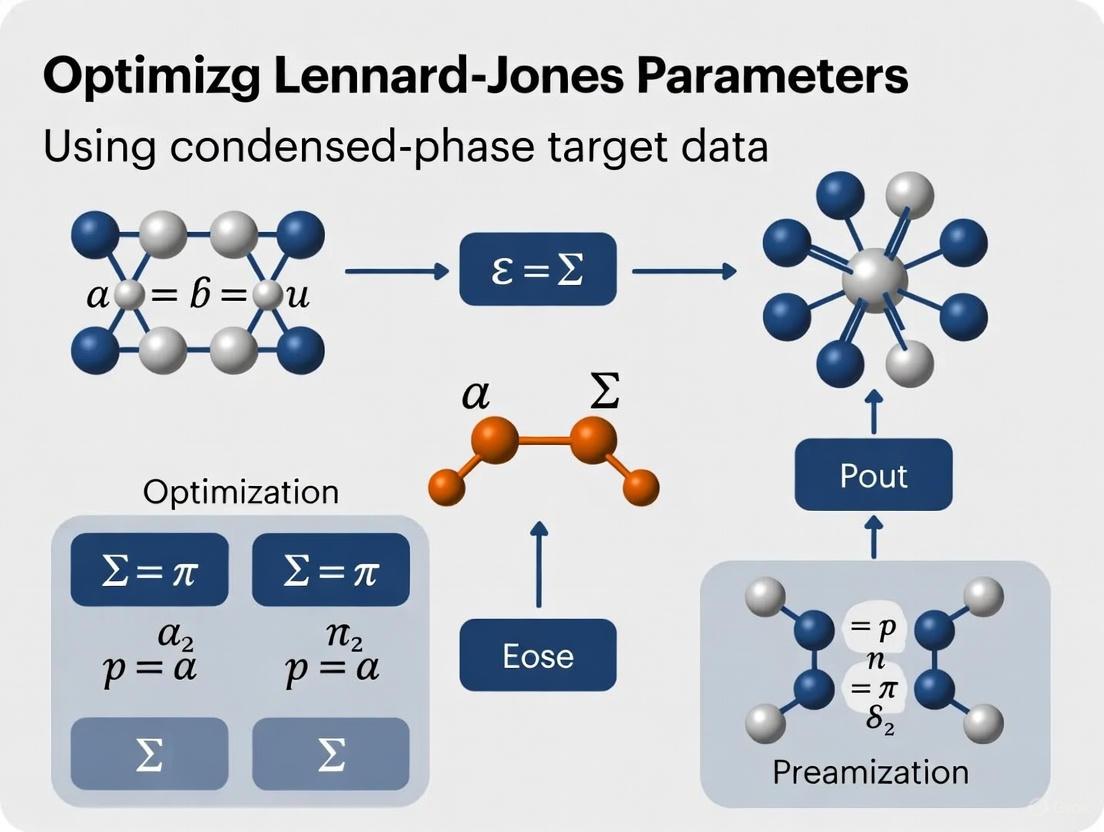
Optimizing Lennard-Jones Parameters with Condensed-Phase Data: A Guide for Robust Force Field Development
This article provides a comprehensive guide for researchers and scientists in drug development on the advanced optimization of Lennard-Jones (LJ) parameters using condensed-phase target data.
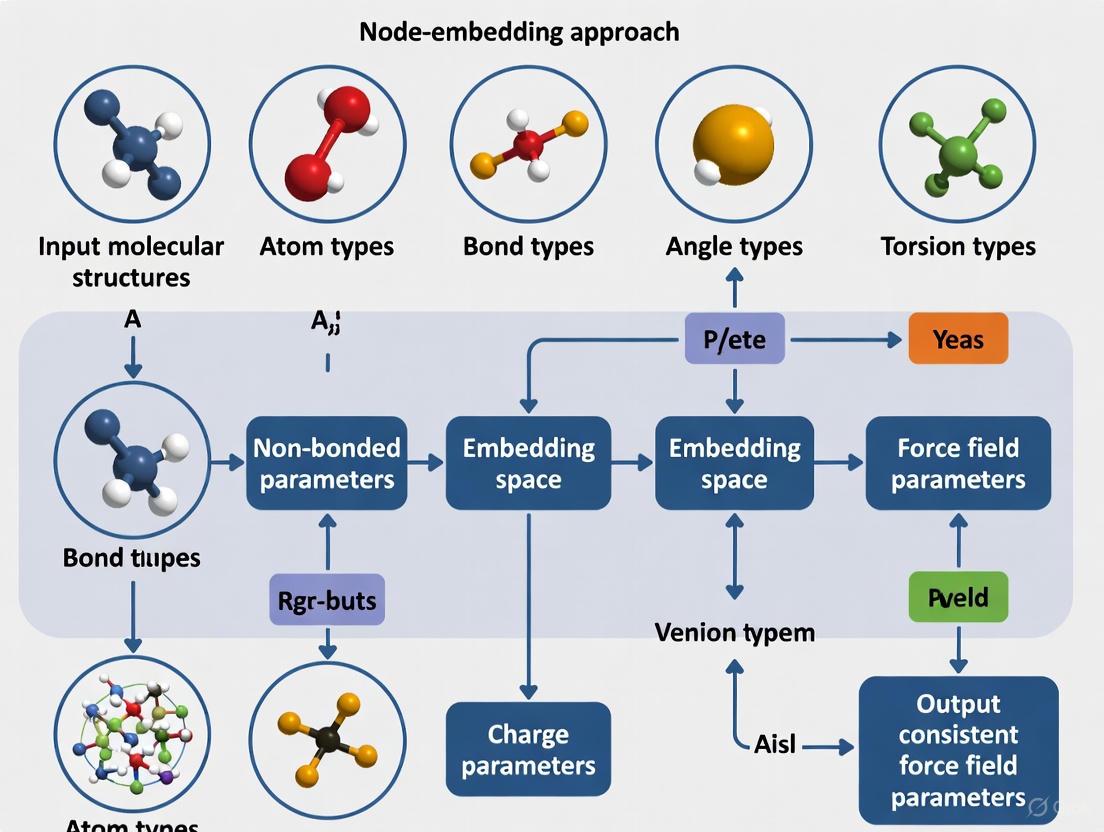
Node-Embedding Approaches for Consistent Force Field Parameter Assignment: From Molecular Graphs to Accurate Simulations
This article explores the transformative shift in molecular mechanics force field development from traditional, discrete atom-typing to modern, data-driven node-embedding approaches.
Iterative Optimization for Force Field Development: A Modern Guide to Enhanced Accuracy and Efficiency
This article provides a comprehensive guide to iterative optimization procedures for developing molecular mechanics force fields, a critical tool for computational drug discovery and materials science.
Bayesian Inference of Conformational Populations (BICePs): A Guide to Advanced Parameter Refinement in Structural Biology and Drug Development
This article explores Bayesian Inference of Conformational Populations (BICePs), a powerful algorithm that refines computational models against sparse and noisy experimental data.