Research Articles
Molecular Mechanics Energy Calculations: Principles, Force Fields, and Applications in Drug Discovery
This article provides a comprehensive overview of molecular mechanics (MM) energy calculations, a foundational computational method for modeling molecular systems in structural biology and drug development.
Electromagnetic Potential Energy and Maximum Force: Computational Strategies for Drug Discovery
This article provides a comprehensive exploration of electrostatic potential energy and force calculations, detailing their critical role in structure-based drug discovery.
Energy Minimization in MD Simulation: A Foundational Guide for Robust Biomolecular Research
This article provides a comprehensive guide to the critical role of energy minimization within the molecular dynamics (MD) simulation workflow for researchers and drug development professionals.
Steepest Descent vs Conjugate Gradient: Algorithmic Breakdown and Applications in Drug Discovery
This article provides a comprehensive explanation of the Steepest Descent and Conjugate Gradient algorithms, tailored for researchers and professionals in drug development.
Force Field Parameters for Energy Minimization: A Modern Guide for Biomedical Researchers
This article provides a comprehensive guide to force field parameters for energy minimization in molecular dynamics simulations, tailored for researchers and professionals in drug development.
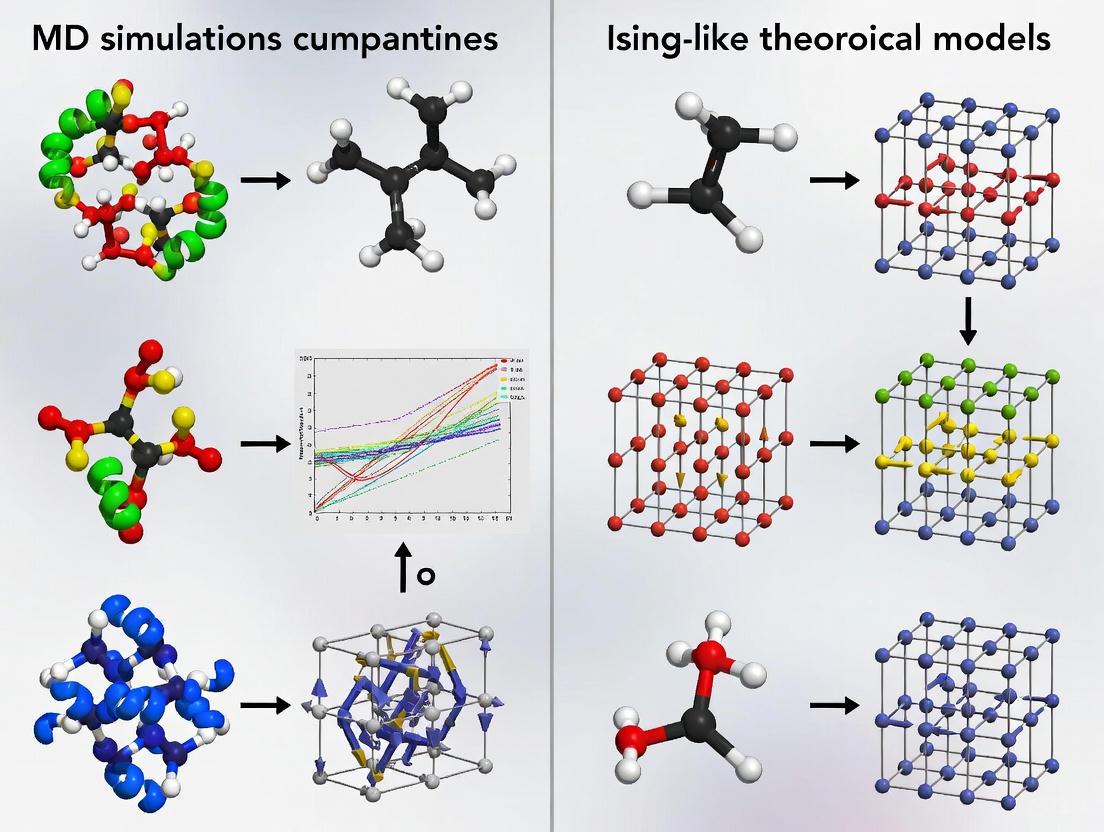
Bridging Scales in Drug Discovery: Molecular Dynamics Simulations and Ising Models in Computational Biology
This article provides a comprehensive comparison between Molecular Dynamics (MD) simulations and Ising-like theoretical models, two powerful computational approaches in modern drug discovery and biomedical research.
Evaluating Secondary Structure Reproduction in MD Simulations: A Guide for Biomolecular Researchers
Molecular dynamics (MD) simulation is a powerful computational technique for studying protein structure and dynamics at the atomistic level.
Validating Force Fields for Small Protein Folding: A Guide to Accuracy, Methods, and Best Practices
Accurately validating molecular mechanics force fields is paramount for reliable simulations of small protein folding, a process critical for understanding biological function and guiding drug discovery.
Explicit vs. Implicit Solvent Models in Protein Folding: A Comprehensive Guide to Accuracy, Speed, and Application
This article provides a detailed comparison of explicit and implicit solvent models for protein folding simulations, tailored for researchers and drug development professionals.
A Practical Guide to RMSD Analysis: Validating Molecular Dynamics Simulations in Biomedical Research
This comprehensive guide details Root Mean Square Deviation (RMSD) analysis for validating Molecular Dynamics (MD) simulations, a critical technique in computational biology and drug development.