Validating Energy Minimization in Biomolecular Modeling: A Practical Guide to RMS Force Criteria
This article provides a comprehensive guide for researchers and drug development professionals on validating energy minimization procedures in biomolecular modeling, with a focus on root mean square (RMS) force as...
Validating Energy Minimization in Biomolecular Modeling: A Practical Guide to RMS Force Criteria
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on validating energy minimization procedures in biomolecular modeling, with a focus on root mean square (RMS) force as a critical convergence criterion. It covers foundational principles of molecular mechanics and energy minimization, explores methodological implementations across major simulation packages, offers troubleshooting strategies for common optimization challenges, and establishes rigorous validation protocols. By integrating current methodologies from tools like GROMACS, CHARMM, and AMBER with practical applications in structure-based drug design, this resource aims to enhance the reliability of computational models used in pharmaceutical development.
The Critical Role of Energy Minimization and RMS Force in Biomolecular Stability
Understanding Energy Minimization in Molecular Mechanics
Energy minimization (also known as energy optimization or geometry minimization) is a fundamental process in computational chemistry that involves finding an arrangement of atoms where the net inter-atomic force on each atom is acceptably close to zero and the position on the potential energy surface is a stationary point [1]. This process transforms a molecular structure into a low-energy state, which is statistically favored and more likely to correspond to the natural state of the structure [2]. In molecular dynamics simulations, energy minimization serves as a crucial preparatory step that removes steric clashes and unrealistic geometries from initial structures, ensuring the system begins from a stable configuration [3]. The accuracy of this process directly depends on the force field employed—mathematical functions that describe potential energy based on atomic positions through bonded terms (bond stretching, angle bending, torsional rotation) and non-bonded terms (van der Waals and electrostatic interactions) [3].
Validation of energy minimization protocols typically involves assessing the root mean square force (RMS force), which quantifies the average force acting on atoms in the system. A successfully minimized structure should demonstrate an RMS force below a predetermined threshold, indicating convergence to a stable configuration. This validation framework provides the critical foundation for evaluating and comparing different force fields and minimization algorithms in molecular mechanics applications.
Comparative Analysis of Major Force Fields
Force Field Performance Metrics
The performance of molecular mechanics force fields can be evaluated through various metrics, including their accuracy in reproducing experimental data such as liquid densities, vapor-liquid coexistence curves, J-coupling constants, and secondary structure propensities. Different force fields exhibit distinct strengths and weaknesses depending on their parameterization strategies and intended applications.
Table 1: Comparison of Force Field Performance Across Different Molecular Systems
| Force Field | Liquid Density Accuracy | J-Coupling Reproduction | Secondary Structure Balance | Recommended Application |
|---|---|---|---|---|
| AMBER ff99SB | Moderate [4] | Excellent for proteins [5] | Excellent, improves ff94/ff99 bias [5] | Protein folding, biomolecular simulations [5] |
| CHARMM22 | High [4] | Good [5] | Good | General biomolecules [4] |
| OPLS-aa | High [4] | Moderate [5] | Moderate | Organic liquids, proteins [4] |
| GROMOS | Moderate [4] | Moderate [5] | Moderate | Large systems, membrane proteins [3] |
| TraPPE | Highest [4] | N/A | N/A | Fluid phase equilibria [4] |
| Grappa (ML) | N/A | State-of-the-art [6] | Excellent, matches AMBER FF19SB [6] | Broad chemical space, peptide radicals [6] |
Specialized Force Fields and Recent Advancements
Beyond traditional force fields, specialized variants and machine learning approaches have emerged to address specific challenges in molecular simulations:
Polarizable Force Fields: The CHARMM Drude polarizable force field incorporates electronic polarization effects, making it particularly valuable for simulations where charge redistribution in response to environment is critical [7]. This force field shows improved performance in hydration free energy calculations compared to fixed-charge models.
Machine Learned Force Fields: Grappa represents a recent innovation that employs graph attentional neural networks to predict molecular mechanics parameters directly from molecular graphs [6]. This approach eliminates the need for hand-crafted chemical features and demonstrates state-of-the-art accuracy for small molecules, peptides, and RNA while maintaining computational efficiency comparable to traditional force fields.
Extended AMBER Families: Tools like YASARA support numerous AMBER variants including AMBER03, AMBER10, AMBER99, and specialized versions like AMBER14IPQ and AMBER15FB, each optimized for specific simulation scenarios [2].
Experimental Protocols for Energy Minimization Validation
J-Coupling Constant Validation Protocol
The validation of force fields using NMR J-coupling constants provides a rigorous method for assessing their accuracy in reproducing local conformational preferences:
System Preparation: Short polyalanine peptides (Ala₃ and Ala₅) are solvated in explicit water models (TIP3P or TIP4P-Ew) [5].
Molecular Dynamics Simulation: Perform extensive replica-exchange molecular dynamics simulations (50 ns/replica) to ensure adequate conformational sampling [5].
J-Coupling Calculation: Calculate scalar coupling constants using multiple Karplus parameter sets (DFT1, DFT2, Original) to account for parameter uncertainty [5].
Statistical Analysis: Compute χ² values representing the sum of deviations between calculated and experimental J-coupling constants, normalized by factors related to systematic error in the calculations [5].
This protocol revealed that ff99SB with TIP4P-Ew solvent model demonstrates excellent agreement with experimental J-coupling constants, with χ² values below 2.0 for both Ala₃ and Ala₅, making it one of the best-performing combinations for short peptides [5].
Hydration Free Energy Calculation Protocol
Hydration free energy (ΔGhyd) calculations serve as a stringent test for force field accuracy, particularly for polarizable models:
Test System Selection: A diverse set of 12 molecules covering a range of hydrophobicity (water, methanol, ethanol, methanethiol, acetamide, tetrahydrofuran, benzene, phenol, aniline, ethane, n-hexane, cyclohexane) [7].
Alchemical Annihilation: Perform free energy calculations where each molecule is alchemically annihilated in both aqueous phase and gas phase [7].
Simulation Details: For aqueous phase simulations, utilize 1687 water molecules in a cubic simulation cell with equilibration in the isobaric-isothermal ensemble (NPT) [7].
Polarizable Force Field Implementation: For Drude simulations, use SWM4 water model with operator-splitting velocity-Verlet algorithm, with different response times and temperatures for atomic particles and Drude oscillators [7].
This methodology enables direct comparison between fixed-charge and polarizable force fields, highlighting the importance of polarization effects in solvation thermodynamics.
Liquid Density and Vapor-Liquid Coexistence Validation
For force fields intended to study condensed phases, liquid densities and vapor-liquid coexistence curves provide critical validation metrics:
Simulation Method: Perform Monte Carlo simulations in the isobaric-isothermal and Gibbs ensembles for methyl-capped amino acid side chains [4].
Force Field Comparison: Test multiple force fields (AMBER, CHARMM, COMPASS, GROMOS, OPLS, TraPPE, UFF) on identical molecular systems [4].
Data Analysis: Compute coexistence liquid densities, coexistence vapor densities, and extrapolate critical temperature using density scaling law and law of rectilinear diameters [4].
Error Calculation: Determine root mean square errors compared to experimental data across different error tolerance levels (1%, 2%, 5%) [4].
This systematic approach identified CHARMM as notably accurate for reproducing liquid densities, second only to TraPPE, while AMBER performed best for vapor densities [4].
Energy Minimization Algorithms and Workflows
Algorithm Comparison and Selection Criteria
Energy minimization employs various mathematical approaches to navigate the potential energy surface, each with distinct advantages and limitations:
Table 2: Characteristics of Energy Minimization Algorithms
| Algorithm | Computational Efficiency | Convergence Stability | System Size Suitability | Implementation Complexity |
|---|---|---|---|---|
| Steepest Descent | High | High, robust | Large systems | Low |
| Conjugate Gradient | Moderate | Moderate | Medium to large systems | Moderate |
| Newton-Raphson | Low per step, high overall | High | Small systems | High |
| Quasi-Newton | Moderate | High | Medium systems | Moderate |
The steepest descent algorithm takes steps in the direction of the negative gradient of the potential energy surface, making it robust for initial minimization steps, particularly when starting from structures with significant steric clashes [3]. Conjugate gradient methods offer improved efficiency by utilizing information from previous steps to determine subsequent directions [3]. Newton-Raphson methods leverage second derivative information (Hessian matrix) to achieve faster convergence but require substantial computational resources for large systems [3] [1].
Integrated Minimization Workflows in Drug Discovery
Energy minimization serves critical functions in computer-aided drug design through structured workflows:
Pose Refinement: Following molecular docking, energy minimization removes steric clashes and optimizes ligand interactions with binding sites [2].
Induced Fit Simulation: Using flexible backbone minimization to simulate structural adaptations in both ligand and target, resembling induced fit binding [2].
Binding Site Expansion: Placing ligands in constrained binding sites and performing minimization to create additional space through side chain rearrangements [2].
Binding Affinity Prediction: Utilizing minimized complexes for more accurate molecular mechanics/generalized Born surface area (MM/GBSA) calculations [8] [9].
These workflows demonstrate that subsequent optimization following pose prediction can provide additional confidence in binding mode identification, particularly for fragment-like molecules where positioning significantly impacts 3D design steps [2].
Diagram 1: Energy Minimization Workflow. This diagram illustrates the sequential process of molecular energy minimization, from initial structure preparation to convergence validation.
Research Reagent Solutions for Energy Minimization
Table 3: Essential Software Tools for Molecular Mechanics Simulations
| Tool Name | Function | Compatible Force Fields |
|---|---|---|
| GROMACS | Molecular dynamics simulator | AMBER, CHARMM, OPLS, Grappa [6] [10] |
| AMBER | Molecular dynamics package | AMBER series, OPLS [10] |
| CHARMM | Molecular simulation program | CHARMM, AMBER*, MMFF94 [10] |
| OpenMM | Toolkit for molecular simulation | AMBER, CHARMM, Grappa [6] |
| YASARA | Molecular modeling suite | Multiple AMBER variants, custom fields [10] [2] |
| AutoDock | Molecular docking suite | AMBER force field [9] |
| Gaussian | Electronic structure program | AMBER, UFF [10] |
Energy minimization represents a critical component in molecular mechanics simulations, with its validation through root mean square force metrics providing essential quality assurance for subsequent molecular dynamics studies. Comparative analyses demonstrate that force field selection must align with specific research objectives, as no single force field excels across all applications. Traditional force fields like AMBER ff99SB and CHARMM22 continue to show robust performance for biomolecular systems, while emerging machine learning approaches like Grappa offer promising avenues for exploring uncharted chemical space [6].
Future developments in energy minimization methodologies will likely focus on improved incorporation of polarization effects, enhanced sampling algorithms for complex biomolecular systems, and increased integration of machine learning approaches for parameterization. The ongoing validation of these methods against experimental data—particularly J-coupling constants, hydration free energies, and thermodynamic properties—will remain essential for advancing the field and ensuring the reliability of molecular simulations in drug discovery and materials science.
Energy minimization is a foundational process in computational chemistry and materials science, essential for locating stable molecular configurations on the potential energy surface. The validation of these minimized structures relies heavily on convergence criteria that determine when the optimization process has reached a stationary point. Among these criteria, the root mean square (RMS) force has emerged as a primary and robust indicator of convergence, balancing computational efficiency with physical significance.
Convergence criteria typically evaluate the quality of an optimized structure based on three key parameters: the forces acting on atoms, the displacement of atoms between iterations, and the energy change. Among these, the force-based criteria—particularly the RMS force—provide the most direct measure of how far a structure is from a true stationary point, where the net force on each atom approaches zero. This article examines the theoretical underpinnings, practical implementation, and comparative significance of RMS force as a convergence criterion across multiple computational frameworks, with special emphasis on applications in drug discovery and molecular dynamics.
Theoretical Foundations of RMS Force
Mathematical Definition and Physical Significance
The root mean square force represents the square root of the average squared forces acting on all atoms in a system. For a system containing N atoms, the RMS force is calculated as:
RMS Force = √[ (1/N) × Σ(Fₓ² + Fᵧ² + F_z²) ]
where Fₓ, Fᵧ, and F_z represent the force components along the x, y, and z axes for each atom. This formulation provides a single scalar value that characterizes the overall magnitude of forces in the system, effectively averaging out local force variations and offering a global perspective on the optimization progress.
Physically, the RMS force quantifies how far the system is from a true stationary point on the potential energy surface. At a genuine minimum, all force components should theoretically be zero, but in computational practice, they need only fall below a predetermined threshold. The RMS force is particularly valuable because it accounts for force contributions across the entire molecular system, unlike the maximum force criterion, which may be disproportionately influenced by a single atomic interaction [11].
Relationship to Potential Energy Surface
The RMS force has a direct mathematical relationship with the gradient of the potential energy surface. Specifically, the force acting on each atom is the negative gradient of the potential energy with respect to that atom's position:
F = -∇V
where V represents the potential energy function. Consequently, the RMS force effectively measures the magnitude of the energy gradient across the entire molecular system. As optimization progresses and the system approaches a minimum, the potential energy surface flattens, resulting in smaller gradient components and thus smaller RMS force values [11].
This relationship makes RMS force particularly valuable for evaluating convergence in complex biomolecular systems where the potential energy surface may contain numerous shallow minima. In such cases, energy-based convergence criteria can be misleading, as tiny energy changes may persist even when structures are far from optimal. Force-based criteria like RMS force provide a more reliable indication of true convergence to a stationary point [12].
RMS Force in Computational Methodologies
Implementation Across Algorithms
RMS force as a convergence criterion is implemented across various energy minimization algorithms, though its effectiveness varies depending on the optimization method:
Steepest Descent: This robust but less efficient algorithm uses the force directly to determine search directions. The maximum displacement in each step is scaled by the maximum force, making force criteria natural convergence indicators. GROMACS documentation notes that steepest descent "is robust and easy to implement" and stops when "the maximum of the absolute values of the force (gradient) components is smaller than a specified value ε" [11].
Conjugate Gradient: This method typically demonstrates improved efficiency closer to minima and utilizes similar force-based convergence criteria as steepest descent, though with implementation limitations for certain constrained systems [11].
L-BFGS: As a quasi-Newton method, L-BFGS builds an approximation of the Hessian matrix to achieve faster convergence. The RMS force remains a primary convergence metric, with the algorithm proceeding until forces are sufficiently minimized [11].
Newton-Raphson Methods: Implemented in packages like PSI4, these methods can utilize computed Hessians for more informed step planning but still rely on force criteria, including RMS force, to determine convergence [13].
The consistent use of RMS force across these diverse algorithms underscores its fundamental importance in optimization workflows, providing a common metric for comparing convergence behavior regardless of the specific minimization technique employed.
Comparative Convergence Criteria
While RMS force serves as a primary convergence indicator, it is typically evaluated alongside other criteria to ensure comprehensive convergence assessment:
Table 1: Convergence Criteria in Energy Minimization
| Criterion | Definition | Significance | Typical Threshold |
|---|---|---|---|
| RMS Force | √[ (1/N) × Σ||Fᵢ||² ] | Average magnitude of forces across system | 0.000300 au (Gaussian) [12] |
| Maximum Force | max(||Fᵢ||) | Largest force on any atom | 0.000450 au (Gaussian) [12] |
| RMS Displacement | √[ (1/N) × Σ||Δrᵢ||² ] | Average atomic movement between steps | 0.001200 au (Gaussian) [12] |
| Maximum Displacement | max(||Δrᵢ||) | Largest atomic movement between steps | 0.001800 au (Gaussian) [12] |
Most computational packages employ multi-faceted convergence tests that require simultaneous satisfaction of several criteria. As noted in Gaussian documentation, "a stationary point is found when one of two criteria is met: all four values listed in the output are smaller than the indicated thresholds, OR the Maximum Force and RMS Force are two orders of magnitude smaller than the thresholds shown, regardless of the values of the displacements" [12].
This dual approach ensures that optimizations terminate only when structures genuinely approach stationary points, with RMS force playing a pivotal role in both pathways.
Experimental Protocols and Validation
Standard Energy Minimization Workflow
A typical energy minimization protocol utilizing RMS force as a primary convergence criterion follows a systematic workflow:
Figure 1: Energy minimization workflow with RMS force convergence check.
The process begins with an initial molecular structure, often derived from experimental data or preliminary modeling. Each iteration computes the potential energy and atomic forces, followed by convergence assessment against criteria including RMS force. If convergence is not achieved, atomic positions are updated using the specific minimization algorithm, and the process repeats until all criteria are satisfied [11] [13].
Validation Through Frequency Calculations
A critical validation step for minimized structures involves comparing optimization convergence with frequency calculation results:
Table 2: Convergence Comparison Between Optimization and Frequency Calculations
| Convergence Measure | Optimization Result | Frequency Result | Converged? |
|---|---|---|---|
| Maximum Force | 0.000038 | 0.000038 | Yes |
| RMS Force | 0.000014 | 0.000014 | Yes |
| Maximum Displacement | 0.000635 | 0.005385 | No |
| RMS Displacement | 0.000367 | 0.002819 | No |
Data adapted from Gaussian documentation [12]
As demonstrated in Table 2, structures satisfying force-based convergence criteria during optimization may still fail displacement-based criteria in subsequent frequency calculations. This discrepancy arises because "displacements are different between the optimization and the frequency calculation because they rely on the Hessian" [12]. The frequency calculation utilizes an exact Hessian, while optimizations often employ approximations.
This validation is crucial because "frequency and thermochemistry results are based on a harmonic analysis that is only valid at true stationary points" [12]. When discrepancies occur, further optimization using the exact Hessian from frequency calculations is recommended until all convergence criteria are satisfied in subsequent validation.
Comparative Performance Across Software Platforms
Implementation in Major Computational Packages
Different computational chemistry packages implement RMS force convergence with varying thresholds and algorithmic approaches:
Table 3: RMS Force Convergence Thresholds Across Software Platforms
| Software | RMS Force Threshold | Maximum Force Threshold | Convergence Set |
|---|---|---|---|
| Gaussian | 0.000300 au | 0.000450 au | Default [12] |
| GROMACS | User-specified (e.g., 10 kJ mol⁻¹ nm⁻¹) | User-specified (e.g., 10 kJ mol⁻¹ nm⁻¹) | Custom [11] |
| PSI4 | Varies by option: GAU (0.000300 au), GAU_TIGHT (0.000100 au) | Varies by option | Multiple sets [13] |
| GRACE (d-DFT) | Not explicitly stated | 2.93 kJ mol⁻¹ Å⁻¹ (for crystals) | Custom [14] |
The thresholds vary significantly based on the computational context. For crystal structure minimization with dispersion-corrected DFT, studies have utilized a maximum force threshold of 2.93 kJ mol⁻¹ Å⁻¹, which corresponds to approximately 0.0011 atomic units [14]. In contrast, standard molecular optimizations in Gaussian employ tighter thresholds of 0.000450 au for maximum force and 0.000300 au for RMS force [12].
PSI4 offers multiple convergence presets, with the "GAU" option mimicking Gaussian's thresholds and "GAU_TIGHT" providing stricter values for high-precision applications [13]. This flexibility allows researchers to select appropriate convergence stringency based on their specific accuracy requirements and computational resources.
Algorithmic Efficiency and RMS Force Convergence
The effectiveness of RMS force as a convergence metric varies across minimization algorithms, influencing their computational efficiency and application domains:
Figure 2: Algorithm selection strategy based on RMS force reduction.
As illustrated in Figure 2, different algorithms demonstrate varying efficiency at different stages of the minimization process. Steepest descent performs well in initial stages with high RMS force values, while conjugate gradients and L-BFGS become more efficient as the system approaches convergence [11]. This algorithmic progression strategy optimizes computational resources by matching method efficiency to the current optimization stage as measured by RMS force reduction.
GROMACS documentation notes that while "steepest descent is certainly not the most efficient algorithm for searching, it is robust and easy to implement," whereas "L-BFGS" has been found to converge faster than conjugate gradients in many applications [11]. This performance advantage makes L-BFGS particularly valuable for large biomolecular systems where computational efficiency is crucial.
Applications in Drug Discovery and Molecular Dynamics
Role in Structure-Based Drug Design
In structure-based drug discovery, energy minimization using appropriate convergence criteria is essential for preparing reliable protein-ligand complexes for docking and binding analysis. Accurate minimization ensures that molecular geometries reflect physically realistic configurations, improving the predictive accuracy of virtual screening campaigns.
The expansion of accessible chemical space to billions of compounds has heightened the importance of efficient yet reliable minimization protocols [15]. With ultra-large virtual screening becoming feasible through cloud and GPU computing, the robustness of minimization procedures directly impacts hit identification success rates. RMS force convergence criteria provide the necessary balance between computational efficiency and structural reliability in these applications.
Integration with Molecular Dynamics Simulations
Energy-minimized structures serve as essential starting points for molecular dynamics (MD) simulations, where RMS force convergence ensures stable initial configurations before dynamics propagation. Proper minimization prevents unrealistic atomic forces that could introduce numerical instabilities during MD trajectories.
The Relaxed Complex Method exemplifies the connection between minimization and dynamics, where "representative target conformations, often including novel, cryptic binding sites, are selected from MD simulations for use in docking studies" [15]. Each extracted conformation requires energy minimization before docking, making RMS force convergence an integral component of this advanced drug discovery methodology.
Furthermore, validation studies combining minimization with experimental data demonstrate the practical significance of appropriate convergence criteria. In one study evaluating 241 experimental organic crystal structures, energy minimization with dispersion-corrected DFT achieved remarkably small RMS Cartesian displacements of only 0.095 Å on average when appropriate force convergence was employed [14]. This close agreement with experimental data underscores the value of properly converged minimizations for structural validation.
Table 4: Key Computational Tools for Energy Minimization Research
| Tool/Resource | Function | Application Context |
|---|---|---|
| GROMACS | Molecular dynamics package with energy minimization capabilities | Biomolecular simulations [11] |
| Gaussian | Quantum chemistry package with geometry optimization | Molecular structure optimization [12] |
| PSI4 | Quantum chemistry package with optking module | Molecular geometry optimization [13] |
| GRACE/VASP | Dispersion-corrected DFT for crystalline materials | Crystal structure optimization [14] |
| AlphaFold DB | Repository of predicted protein structures | Initial structures for minimization [15] |
| REAL Database | Commercially available on-demand compound library | Ligand structures for minimization [15] |
These tools collectively enable comprehensive energy minimization studies across various molecular systems, from small organic molecules to large biomolecular complexes. The integration of RMS force convergence across these diverse platforms demonstrates its universal applicability as a robust convergence criterion.
RMS force stands as a fundamental convergence criterion in energy minimization, providing a balanced metric that reflects global optimization progress while remaining computationally accessible. Its implementation across major software platforms and algorithmic approaches underscores its broad utility in computational chemistry, materials science, and drug discovery.
The comparative analysis presented herein demonstrates that while RMS force is rarely used in isolation, it plays an indispensable role in comprehensive convergence assessment. When combined with maximum force criteria and displacement measures, RMS force provides robust validation of stationary points on potential energy surfaces. This multi-faceted approach ensures that minimized structures serve as reliable foundations for subsequent computational analyses, from frequency calculations and thermochemical predictions to molecular dynamics simulations and drug-receptor binding studies.
As computational methodologies continue to evolve and tackle increasingly complex molecular systems, the RMS force criterion maintains its relevance through its direct connection to the fundamental mathematics of optimization—the gradient of the potential energy surface. This mathematical foundation, coupled with its practical implementation across diverse computational architectures, ensures that RMS force will remain a primary convergence criterion in energy minimization for the foreseeable future.
The Relationship Between Molecular Forces and Potential Energy Surfaces
A Potential Energy Surface (PES) provides a multidimensional representation of the potential energy of a molecular system as a function of its atomic coordinates. This conceptual framework serves as the fundamental backdrop for understanding molecular structure, stability, and reactivity across chemical and biochemical domains. The PES can be visualized as an energy landscape with distinct topographic features: hills, valleys, and saddle points, each corresponding to specific molecular configurations and their relative energies. As atoms come together to form molecules, the potential energy reaches a minimum at the equilibrium bond distance, creating stable configurations that correspond to the valleys on this surface.
The very shape and character of a PES are determined by the intricate interplay of various molecular forces operating at the atomic and molecular levels. These forces include covalent bonding, which generates deep and narrow potential energy wells; van der Waals interactions, which contribute shallower, broader potential energy wells; electrostatic interactions such as dipole-dipole or ion-dipole forces that create deeper, more directional potential energy wells; and hydrogen bonding, which results in highly directional and relatively strong potential energy wells. The depth and shape of the potential energy wells on a PES are direct manifestations of the strength and directionality of these underlying intermolecular forces, providing researchers with a powerful framework for predicting molecular behavior and designing interventions in drug development.
Table 1: Fundamental Forces Shaping Potential Energy Surfaces
| Force Type | Strength Range (kJ/mol) | Directionality | Primary Role in PES | Example Applications |
|---|---|---|---|---|
| Covalent Bonding | 150-500 | High | Creates deep, narrow potential energy wells | Molecular structure determination |
| Hydrogen Bonding | 10-40 | High | Forms directional wells with intermediate depth | Protein folding, drug-receptor binding |
| Electrostatic Interactions | 5-80 | Moderate to High | Generates deeper, directional wells | Ion transport, protein-ligand recognition |
| van der Waals | 0.5-5 | Low | Contributes shallow, broad wells | Molecular assembly, substrate adhesion |
Theoretical Framework: From Molecular Forces to Energy Landscapes
The theoretical foundation connecting molecular forces to PES topography originates from the Born-Oppenheimer approximation, which recognizes that atomic nuclei are significantly heavier than electrons, thus allowing separation of their motions. This approximation enables computational chemists to calculate the energy due to electrons in the ground state while assuming fixed nuclear positions, then repeat these calculations across different internuclear distances to map out the complete potential energy surface for nuclear motion. For diatomic molecules, this results in a one-dimensional potential energy curve, while for complex polyatomic systems like pharmaceutical compounds, the PES becomes a high-dimensional function of all atomic positions.
The mathematical description of PES features reveals how molecular forces manifest in the energy landscape. Stable molecular configurations correspond to local minima on the PES, where the gradient (first derivative of energy with respect to position) is zero and the curvature (second derivative) is positive in all directions. These minima represent equilibrium structures where attractive and repulsive molecular forces are perfectly balanced. Transition states, which are critical for understanding reaction kinetics in drug metabolism, appear as saddle points on the PES—points where the gradient is zero but the curvature is negative along the reaction coordinate and positive in perpendicular directions. The energy barrier between minima and saddle points determines the activation energy required for molecular transformations, directly influencing reaction rates and biological activity of pharmaceutical compounds.
The harmonic oscillator model provides the simplest mathematical approximation for potential energy near minima, described by V = ½k(r-rₑ)², where k is the force constant and rₑ is the equilibrium bond distance. This model successfully describes small perturbative fluctuations about equilibrium geometry but fails to capture bond dissociation. For larger displacements or bond-breaking processes, anharmonic potentials like the Morse potential (V = Dₑ(1-e^{-a(r-rₑ)})²) offer more accurate descriptions, where Dₑ represents bond dissociation energy and a controls the potential width. These mathematical formulations directly link force field parameters to observable molecular properties, enabling researchers to validate computational models against experimental data.
Experimental and Computational Methodologies for PES Exploration
Energy Minimization Algorithms and Force Validation
Energy minimization represents a crucial computational approach for locating minima on potential energy surfaces, with the root-mean-square (RMS) force serving as a key validation metric for convergence. Multiple algorithms exist for this purpose, each with distinct advantages for specific applications in molecular modeling and drug design. The steepest descent algorithm provides a robust approach for initial minimization stages, calculating new positions according to rₙ₊₁ = rₙ + hₙFₙ/max(|Fₙ|), where hₙ is the maximum displacement and Fₙ is the force acting as the negative gradient of the potential. This method expands the step size by 20% when energy decreases but reduces it by 80% when energy increases, ensuring stability while progressing toward minima.
For more refined minimization closer to energy minima, the conjugate gradient algorithm offers superior efficiency, though it performs less effectively during initial stages. Most notably, the L-BFGS (limited-memory Broyden-Fletcher-Goldfarb-Shanno) algorithm has emerged as a preferred quasi-Newtonian method that approximates the inverse Hessian matrix using a fixed number of corrections from previous steps. This approach maintains nearly the efficiency of full BFGS methods while requiring memory resources proportional only to the number of particles multiplied by correction steps, rather than the square of the number of particles. The minimization process typically continues until the RMS value of all force components reaches a specified tolerance, often around 10 kJ·mol⁻¹·nm⁻¹, providing researchers with a quantifiable metric for validating minimization success.
Table 2: Energy Minimization Algorithms for PES Exploration
| Algorithm | Mechanism | Convergence Criterion | Advantages | Limitations |
|---|---|---|---|---|
| Steepest Descent | Follows negative energy gradient | RMS force < tolerance | Robust, easy implementation | Slow convergence near minima |
| Conjugate Gradient | Builds on previous step directions | RMS force < tolerance | Efficient close to minima | Slower initial convergence |
| L-BFGS | Approximates inverse Hessian | RMS force < tolerance | Fast convergence, lower memory | Limited parallelization |
Advanced PES Sampling and Dataset Development
Beyond local minimization, researchers employ sophisticated sampling techniques to explore broader regions of potential energy surfaces, particularly for complex biomolecular systems relevant to pharmaceutical development. Molecular dynamics simulations generate trajectory snapshots that capture thermal fluctuations and conformational changes, providing atomic-level insights into energy landscape topography. The recent introduction of the MatPES dataset represents a significant advancement in this domain, comprising approximately 400,000 structures carefully sampled from 281 million molecular dynamics snapshots that span 16 billion atomic environments.
This foundational dataset demonstrates that universal machine learning interatomic potentials (UMLIPs) trained on carefully curated, high-quality data can rival or surpass models trained on much larger datasets across various property benchmarks. Notably, MatPES incorporates high-fidelity PES data based on the revised regularized strongly constrained and appropriately normed (r²SCAN) functional, which provides greatly improved descriptions of interatomic bonding compared to standard density functional theory approximations. This emphasis on data quality over quantity enables broad community-driven advancements toward more reliable, generalizable, and efficient UMLIPs for large-scale materials discovery and drug design applications.
Comparative Analysis of PES Methodologies in Drug Development
Force Measurement and Validation in Biophysical Contexts
Accurate force measurement represents an essential component of experimental PES validation in pharmaceutical research, particularly for characterizing molecular interactions critical to drug efficacy. Contemporary force-sensing technologies demonstrate varying levels of accuracy across measurement ranges and contact angles, with significant implications for interpreting potential energy landscapes in biological contexts. Recent validation studies of force-sensing ablation catheters, while developed for medical applications, provide insightful methodologies for assessing measurement accuracy under diverse conditions.
These investigations reveal that force measurement systems typically achieve accuracy within 1-3 grams deviation across the 10-40 gram range critical for many biological interactions, though significant errors can occur at higher forces with potential clinical consequences. For instance, the Tacticath catheter demonstrated a mean absolute error of 1.29±0.99 grams, while the Smarttouch system showed 2.26±2.70 grams deviation across the measurement range. Such precision requires sophisticated engineering approaches, including high-resolution data acquisition systems with at least 24-bit resolution, temperature compensation mechanisms, and regular calibration following established standards like ASTM E4. For a 10 kN load cell connected to a 32-bit data acquisition system, the theoretical resolution reaches approximately 0.0000047 N (4.7 μN), enabling detection of extremely fine force variations essential for characterizing shallow potential energy wells associated with weak molecular interactions.
Diagram 1: PES Methodology Validation Workflow. This workflow illustrates the iterative process of potential energy surface development and validation, highlighting the critical role of force convergence metrics.
Performance Benchmarks of Contemporary PES Approaches
The computational exploration of potential energy surfaces employs diverse methodologies, each with distinct performance characteristics that influence their applicability to drug development challenges. Traditional density functional theory (DFT) provides quantum-mechanical accuracy but demands substantial computational resources, limiting its use for large biomolecular systems or long timescale simulations. In contrast, classical force fields offer computational efficiency through empirical parameterization but may lack transferability across diverse chemical spaces. Machine learning interatomic potentials (MLIPs) represent an emerging intermediate approach, combining near-DFT accuracy with significantly reduced computational cost.
The MatPES initiative demonstrates how carefully curated training datasets enable MLIPs to achieve remarkable accuracy across equilibrium, near-equilibrium, and molecular dynamics property benchmarks despite relatively modest dataset sizes. This approach emphasizes that data quality and diversity often outweigh sheer quantity in developing transferable PES models. For energy minimization tasks, algorithm selection significantly impacts computational efficiency: L-BFGS typically converges faster than conjugate gradient methods, though with limited parallelization capabilities, while steepest descent remains valuable for initial minimization stages despite slower convergence near minima. These performance considerations directly influence research workflows in pharmaceutical development, where rapid screening of drug candidates requires both accuracy and computational efficiency.
Table 3: Comparative Performance of PES Generation Methods
| Methodology | Accuracy Level | Computational Cost | System Size Limit | Primary Applications |
|---|---|---|---|---|
| Density Functional Theory | High | Very High | ~100-1000 atoms | Benchmarking, small molecule characterization |
| Machine Learning Potentials | Medium-High | Medium | ~1,000-100,000 atoms | Drug screening, materials design |
| Classical Force Fields | Medium | Low | >1,000,000 atoms | Protein folding, molecular dynamics |
| MatPES-trained UMLIPs | High | Medium | ~1,000-100,000 atoms | Broad materials discovery |
Research Reagent Solutions for PES Exploration
The experimental and computational investigation of potential energy surfaces relies on specialized tools and methodologies that constitute the essential research toolkit for scientists in this domain. These solutions encompass computational algorithms, force measurement systems, and dataset resources that collectively enable comprehensive characterization of molecular energy landscapes.
Table 4: Essential Research Toolkit for PES Investigations
| Tool Category | Specific Solution | Primary Function | Key Features |
|---|---|---|---|
| Energy Minimization Algorithms | L-BFGS (OpenMM) | Locates local energy minima | Quasi-Newton method, memory efficient |
| Energy Minimization Algorithms | Conjugate Gradient (GROMACS) | Energy minimization | Efficient near minima, suitable for pre-normal-mode analysis |
| Energy Minimization Algorithms | Steepest Descent (GROMACS) | Initial structure optimization | Robust early-stage minimization |
| Force Measurement Systems | Tacticath Quartz (Abbott) | Experimental force validation | High accuracy (1.29±0.99 g error) |
| Force Measurement Systems | 32-bit DAQ with load cell | Force resolution detection | Theoretical resolution to 4.7 μN |
| PES Datasets | MatPES dataset | Training ML interatomic potentials | 400K structures from 281M MD snapshots |
| Computational Frameworks | OpenMM LocalEnergyMinimizer | Molecular simulation minimization | Enforces distance constraints, tolerance-based convergence |
| Validation Protocols | RMS Force Convergence | Minimization validation | Typically uses tolerance of ~10 kJ·mol⁻¹·nm⁻¹ |
The intricate relationship between molecular forces and potential energy surfaces provides pharmaceutical researchers with a powerful conceptual and computational framework for understanding and manipulating molecular interactions central to drug action. The topographic features of PES—minima, saddle points, and energy barriers—directly reflect the balance of molecular forces operating within biological systems, enabling predictive models of drug-receptor binding, metabolic transformation, and molecular recognition. Energy minimization methodologies with RMS force validation ensure that computational models accurately represent stable molecular configurations, while advanced sampling techniques map the complex energy landscapes that govern biomolecular flexibility and function.
Recent advancements in machine learning interatomic potentials, exemplified by the MatPES dataset, are revolutionizing PES exploration by combining quantum-mechanical accuracy with molecular dynamics scalability. These developments, coupled with rigorous force measurement technologies and robust minimization algorithms, provide drug development professionals with an increasingly sophisticated toolkit for rational drug design. As these methodologies continue to evolve, they promise to enhance our ability to navigate the high-dimensional energy landscapes of complex pharmaceutical compounds, ultimately accelerating the discovery of novel therapeutic agents with optimized efficacy and safety profiles.
Why Proper Minimization Matters in Drug Discovery Pipelines
In the realm of computer-aided drug design (CADD), energy minimization serves as a fundamental process for refining molecular structures and transforming them into low-energy states that are statistically favored and more likely to represent natural biological conformations [16]. This procedure is not merely a technical preprocessing step; it represents a crucial scientific methodology that significantly impacts the accuracy of virtual screening, molecular docking, and binding affinity predictions—ultimately influencing the entire drug discovery pipeline. The importance of proper minimization extends to its relationship with root mean square force (RMSF) and other validation metrics that quantify how closely a computational model approximates experimental reality [17].
The broader thesis of energy minimization validation revolves around establishing reliable correlations between computational predictions and experimental outcomes. As structural biology initiatives continue to generate vast amounts of protein data through experimental and computational means, the critical assessment of these structures becomes increasingly important [17]. Proper minimization protocols ensure that molecular models provide accurate foundations for structure-based drug design, reducing the risk of late-stage failures in drug development that often stem from inadequate early-stage characterization [18] [19].
Molecular Foundations: How Energy Minimization Works
Theoretical Principles and Computational Implementation
Energy minimization operates on the fundamental principle that molecular systems naturally exist in low-energy states. Computational methods aim to identify these stable configurations by iteratively adjusting atomic coordinates to minimize the total potential energy of the system, which is calculated using mathematical force fields [16]. These force fields comprise collections of formulas and parameters that simulate atomic systems, accounting for bond lengths, bond angles, torsion angles, and non-bonded interactions such as van der Waals forces and electrostatic attractions [16].
The process essentially navigates the molecular energy landscape to locate local or global energy minima—conformations where the atomic arrangement experiences minimal internal stress and maximal stability. This optimization is particularly crucial when working with experimental structures from sources like the Protein Data Bank, which may contain atomic clashes, strained bond geometries, or other steric conflicts introduced during the structure determination process [17]. Without proper minimization, these structural artifacts can propagate through the drug discovery pipeline, compromising the accuracy of virtual screening and lead optimization efforts.
Minimization Approaches and Force Field Options
The implementation of energy minimization in drug discovery offers flexibility in approach, with two primary methodologies:
Rigid Backbone Minimization: This approach keeps the protein structure's backbone fixed while allowing adjustments in side chains and ligand conformations. It focuses optimization on bond lengths, bond angles, and ligand-target interactions, making it computationally efficient while still addressing key molecular interactions [16].
Flexible Backbone Minimization: This more comprehensive approach simulates adjustments in both the ligand and the entire target structure, resembling the simulation of an induced fit binding mechanism. It requires greater computational resources but provides a more realistic representation of molecular adaptation during binding [16].
Multiple force fields are available for energy minimization calculations, each with specific strengths and applications. The AMBER series (including AMBER03, AMBER10, AMBER14, and AMBER15 variants) represents widely adopted force fields in drug discovery [16]. Additionally, YASARA provides its own specialized force fields (YAMBER, YASARA, YASARA2), which have demonstrated competitive performance in CASP challenges [16]. The selection of an appropriate force field depends on the specific system under investigation and requires careful consideration to balance computational efficiency with physical accuracy.
Experimental Validation: Quantitative Assessment of Minimization Impact
Systematic Analysis of Docking Performance
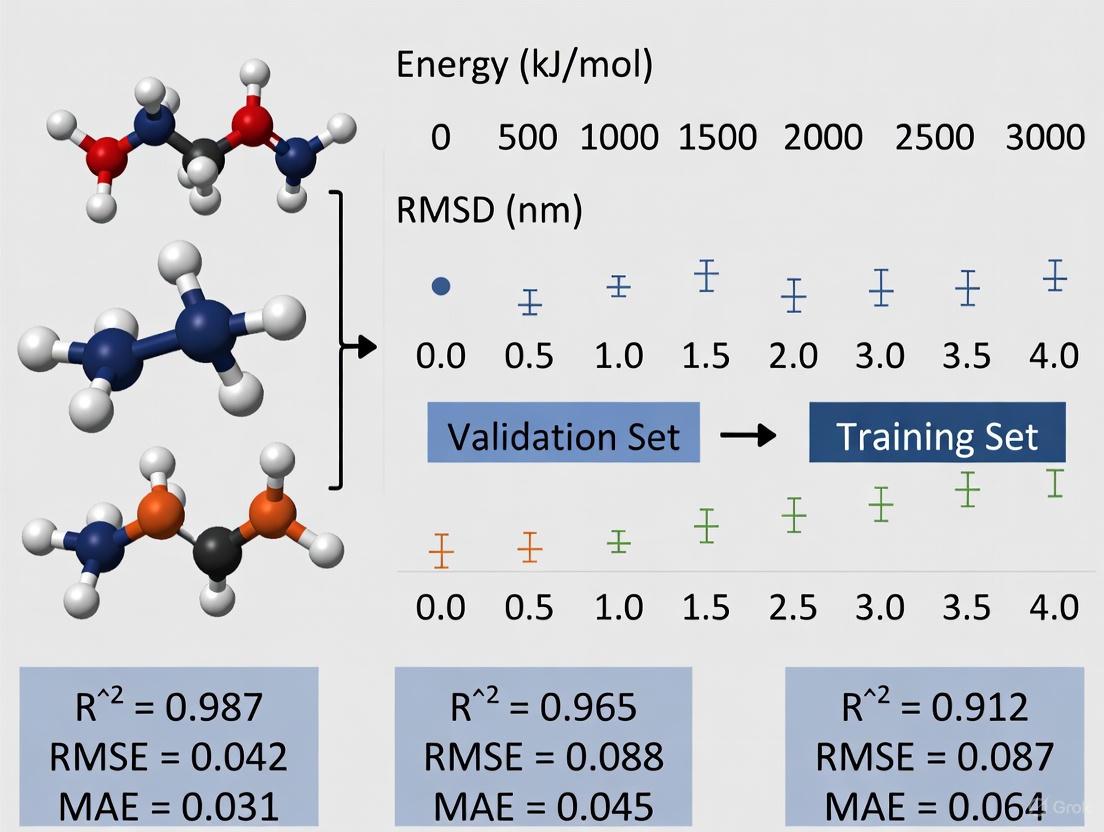
A comprehensive study investigating the effects of structure minimization on docking scores and computational efficiency provides compelling quantitative evidence for its importance [20]. Researchers evaluated four pharmaceutical compounds (Paracetamol, Losartan, Suvorexant, and Ibuprofen) along with their analogues, comparing minimized versus non-minimized structures in molecular docking experiments against their respective protein targets. The methodology followed a rigorous protocol to ensure meaningful comparisons.
Experimental Protocol:
- Ligand Preparation: Compounds were initially optimized using Avogadro software with MMF94s force field with fixed atoms movable [20]
- Minimization Group: Structures underwent further minimization in Chimera software to add hydrogen atoms and assign charges using steric-only settings and Gasteiger charges [20]
- Control Group: Structures received only initial optimization without the additional minimization step [20]
- Protein Preparation: Target structures obtained from Protein Data Bank were cleaned by removing all non-standard molecules using Chimera [20]
- Docking Procedure: All docking performed using AutoDock Vina embedded in Chimera with consistent parameters across all experiments [20]
- Data Collection: Docking scores (representing binding affinity in kcal/mol) and docking time (measured in milliseconds) were recorded for each complex [20]
The resulting data, drawn from multiple ligand-protein systems, reveals significant variations in how minimization impacts different molecular complexes.
Table 1: Docking Score Comparison for Minimized vs. Non-Minimized Structures
| Compound | Minimized Docking Score (kcal/mol) | Non-Minimized Docking Score (kcal/mol) | Difference |
|---|---|---|---|
| Ibuprofen 1 | -7.6 | -7.6 | 0.0 |
| Ibuprofen 2 | -7.5 | -7.5 | 0.0 |
| Ibuprofen 3 | -7.8 | -7.8 | 0.0 |
| Ibuprofen 4 | -7.3 | -7.3 | 0.0 |
| Ibuprofen 5 | -7.7 | -7.1 | -0.6 |
| Losartan 1 | -7.9 | -8.1 | +0.2 |
| Losartan 2 | -8.0 | -7.9 | -0.1 |
| Losartan 3 | -8.4 | -8.0 | -0.4 |
| Losartan 4 | -8.3 | -8.1 | -0.2 |
| Losartan 5 | -8.5 | -8.3 | -0.2 |
Table 2: Computational Time Comparison (in milliseconds)
| Compound | Minimized Docking Time (ms) | Non-Minimized Docking Time (ms) | Time Difference |
|---|---|---|---|
| Ibuprofen 1 | 9943 | 9780 | +163 |
| Ibuprofen 2 | 8997 | 9876 | -879 |
| Ibuprofen 3 | 9940 | 8705 | +1235 |
| Ibuprofen 4 | 9955 | 9805 | +150 |
| Ibuprofen 5 | 9955 | 8785 | +1170 |
| Losartan 1 | 10870 | 10985 | -115 |
| Losartan 2 | 10800 | 10800 | 0 |
| Losartan 3 | 10894 | 10800 | +94 |
| Losartan 4 | 10985 | 10800 | +185 |
| Losartan 5 | 10800 | 10900 | -100 |
Interpretation of Experimental Findings
The data reveals several important patterns regarding minimization's impact on drug discovery workflows:
Variable Impact on Docking Scores: For certain compounds like Ibuprofen analogues 1-4, minimization produced negligible changes in docking scores, while Ibuprofen analogue 5 showed a notable improvement of -0.6 kcal/mol after minimization [20]. In contrast, Losartan analogues demonstrated consistently better docking scores across all tested compounds after minimization, with improvements ranging from -0.1 to -0.4 kcal/mol [20]. This suggests that minimization effects are molecule-dependent and highlights the importance of case-specific evaluation.
Computational Time Considerations: The time required for docking minimized structures showed considerable variation, with some complexes demonstrating increased computational time (e.g., Ibuprofen 3: +1235ms) while others showed decreased time (e.g., Ibuprofen 2: -879ms) [20]. This indicates that the initial investment in minimization may either increase or decrease overall workflow efficiency depending on the molecular system.
Binding Mode Improvements: Beyond quantitative scoring, minimization helps resolve structural clashes and optimizes ligand positioning within binding pockets, leading to more plausible binding modes [16]. This is particularly valuable for fragment-like molecules where predicting correct binding modes is challenging [16].
Minimization in Integrated Drug Discovery Workflows
The strategic implementation of energy minimization extends throughout the modern drug discovery pipeline, interacting with multiple computational and experimental approaches. The following workflow illustrates how minimization integrates with broader structure-based drug discovery efforts:
Advanced Validation Methodologies
The broader thesis of energy minimization validation incorporates sophisticated statistical approaches to assess structural quality. The Generalized Linear Model-RMSD (GLM-RMSD) method represents one such advancement, combining multiple individual quality scores into a single quantity with intuitive meaning: the predicted coordinate root-mean-square deviation value between the structure under evaluation and the unavailable "true" structure [17]. This method demonstrates considerably higher correlation with actual accuracy (correlation coefficients of 0.69-0.76 for CASD-NMR and CASP datasets) compared to individual quality scores alone (-0.24 to 0.68) [17].
The GLM-RMSD approach utilizes a gamma distribution from the exponential family to model RMSD values, with a link function connecting the linear predictor to the quantity predicted [17]. It incorporates eight validation scores that capture different aspects of structure quality: Discrimination Power (DP), Verify3D, ProsaII, Procheck-φ/ψ, Procheck-All, Molprobity, Gaussian network model (GNM) score, and protein size [17]. This multi-dimensional validation approach provides a more reliable assessment of structural accuracy than any single metric alone.
Applications in Binding Site Expansion
Energy minimization enables critical applications beyond basic structure refinement, including binding site expansion for challenging targets [16]. When binding sites are too narrow or virtually non-existent, making docking predictions difficult, researchers can:
- Place the ligand in the binding site while temporarily tolerating clashes with the structure
- Perform energy minimization with flexible backbone options
- Allow both ligand and structure to adapt to each other through simulated induced fit
This approach successfully creates space by expanding the ligand into potential subpockets, exploring potential rotamers, or adapting particular structures to host ligands [16]. The technique is particularly valuable for covalent docking or when working with apo structures that have collapsed binding sites.
Essential Research Tools for Minimization Protocols
Successful implementation of energy minimization requires specialized software tools and computational resources. The following table catalogues key solutions used in modern minimization workflows:
Table 3: Research Reagent Solutions for Energy Minimization
| Tool/Resource | Type | Primary Function in Minimization | Key Features |
|---|---|---|---|
| YASARA | Molecular Modeling Suite | Comprehensive energy minimization with flexible/rigid options | Support for multiple force fields; Integration with SeeSAR; Induced fit simulation [16] |
| Chimera | Molecular Visualization & Analysis | Structure preparation and minimization | Embedded AutoDock Vina; Hydrogen addition and charge assignment; Integration with Avogadro [20] |
| Avogadro | Molecular Editor | Initial structure optimization | MMF94s force field; Structure optimization with fixed atoms movable [20] |
| AMBER Force Fields | Force Field Collection | Potential energy calculation | Multiple variants (AMBER03, AMBER10, etc.); Parameters for biomolecular simulations [16] |
| YAMBER/YASARA | Specialized Force Fields | Energy calculation and minimization | CASP challenge-winning performance; YASARA-specific optimization [16] |
| SeeSAR | Drug Design Dashboard | Structure analysis and minimization integration | Protein Editor Mode; YASARA module integration; Interactive decision-making [16] |
| AutoDock Vina | Docking Software | Binding pose prediction and scoring | Docking score calculation; Binding affinity estimation; Embedded in Chimera [20] |
| Protein Data Bank | Structural Database | Source of experimental structures | Standardized PDB format; Reference structures for validation [20] |
Energy minimization represents far more than a technical preprocessing step in computational drug discovery—it serves as a fundamental methodology that significantly impacts the accuracy and efficiency of the entire pipeline. The experimental evidence demonstrates that proper minimization can improve docking scores for certain molecular systems while potentially reducing computational time in specific cases [20]. The process enables critical applications like binding site expansion and induced fit simulation that expand the scope of druggable targets [16].
The validation of minimization outcomes through advanced statistical approaches like GLM-RMSD, which combines multiple quality scores into a unified accuracy metric, provides researchers with robust tools for assessing structural quality [17]. When strategically implemented within integrated drug discovery workflows and supported by appropriate software tools, energy minimization contributes substantially to the overarching goal of reducing late-stage failures in drug development by ensuring that computational predictions rest on physically realistic molecular models [18] [19].
As computational approaches continue to streamline drug discovery [19], the role of rigorous minimization protocols will only grow in importance. Researchers must continue to evaluate minimization approaches on a case-specific basis while leveraging the growing suite of validation metrics to ensure that their computational models provide the most accurate possible foundation for drug design decisions.
In computational chemistry and materials science, the process of energy minimization is fundamental for determining stable molecular structures and reaction pathways. This procedure navigates the potential energy surface to locate stationary points—minima corresponding to stable structures or transition states. The validation of a successful minimization hinges on the use of robust convergence metrics that signal when a structure has reached a true stationary point, ensuring the reliability of subsequent property calculations [12]. Among the various criteria employed, the Root Mean Square (RMS) Force, Maximum Force, and Energy Change are pivotal indicators. Each metric provides a unique perspective on the optimization's progression, with significant implications for the accuracy and computational efficiency of simulations in fields such as drug development and molecular modeling [12] [21]. This guide objectively compares these three core metrics, detailing their theoretical bases, performance under different conditions, and practical applications to inform researchers in selecting the most appropriate criteria for their energy minimization protocols.
Theoretical Foundations of Convergence Metrics
The Role of Convergence Criteria in Geometry Optimization
Geometry optimization is an iterative process that adjusts atomic coordinates to find a stationary point on the potential energy surface. A stationary point is characterized by zero first derivatives of the energy with respect to nuclear coordinates, indicating a local minimum, maximum, or saddle point. In practice, due to numerical precision, convergence is declared when these derivatives (forces) and the energy change between iterations fall below predefined thresholds [12]. The integrity of this optimized structure is critical; failure to reach a true stationary point can invalidate subsequent analyses, such as frequency calculations used to determine spectroscopic properties and thermodynamic quantities [12]. For instance, a frequency calculation can flag a structure as unconverged even if the optimization itself reported success, underscoring the necessity of robust and reliable convergence metrics [12].
Defining the Key Metrics
- Maximum Force: This metric represents the largest component of the force (the negative gradient of the potential energy) on any atom in the system. It is a conservative metric that ensures no single atom is experiencing a significant force, guaranteeing local stability from the perspective of the most displaced coordinate [12].
- RMS Force: The Root Mean Square Force provides a global measure of the average force magnitude across all atoms. It is calculated by squaring all the force components, averaging them, and taking the square root of the result. This offers a more holistic view of the system's convergence state than the Maximum Force alone [12] [22].
- Energy Change: This metric monitors the difference in total potential energy between successive optimization steps. A very small energy change suggests that the optimization is no longer making significant progress towards a lower energy state, indicating proximity to a stationary point [21].
The following diagram illustrates the logical relationships between the optimization process, the calculated metrics, and the final goal of a validated structure.
Direct Metric Comparison and Performance Analysis
The choice of convergence metric significantly impacts the outcome and computational cost of an optimization. The table below summarizes the core characteristics, strengths, and limitations of each metric.
Table 1: Comparative Analysis of Convergence Metrics
| Metric | Primary Role | Key Strength | Key Limitation | Ideal Use Case |
|---|---|---|---|---|
| Maximum Force | Ensures no single atom is far from equilibrium. | Prevents local instability; stringent guarantee. | Oversensitive to numerical noise or thermal fluctuations; may be slow to converge. | Transition state searches; ensuring local stability in final validation. |
| RMS Force | Measures the global average force across the system. | Provides a holistic view of convergence; less sensitive to single-atom anomalies. | May mask a large force on one atom if the rest of the system is well-converged. | General-purpose geometry optimizations; monitoring overall progress. |
| Energy Change | Indicates progress in reducing the total potential energy. | Directly tracks the objective of minimization (energy lowering). | Can indicate convergence in flat PES regions even with significant forces. | Monitoring optimization in early stages; conjugate gradient methods. |
The effectiveness of these metrics is interdependent. Modern computational software like Gaussian often employs a combination of criteria for a robust convergence check [12]. A common standard requires all four values (Maximum Force, RMS Force, Maximum Displacement, and RMS Displacement) to fall below their respective thresholds. Alternatively, convergence can be accepted if the force-based metrics (Maximum and RMS Force) are two orders of magnitude stricter than their thresholds, regardless of displacement values [12]. This dual approach ensures both local and global stability of the optimized structure.
Quantitative Data from Computational Studies
Empirical data highlights the practical behavior of these metrics. The following table contrasts convergence values from a successfully completed optimization and a subsequent frequency calculation that revealed the structure was not a true stationary point.
Table 2: Example of Convergence Disagreement Between Optimization and Frequency Steps
| Convergence Check | Value | Threshold | Converged? (Optimization) | Converged? (Frequency) |
|---|---|---|---|---|
| Maximum Force | 0.000038 | 0.000450 | Yes | Yes |
| RMS Force | 0.000014 | 0.000300 | Yes | Yes |
| Maximum Displacement | 0.000635 | 0.001800 | Yes | No |
| RMS Displacement | 0.000367 | 0.001200 | Yes | No |
Source: Adapted from Gaussian 16 FAQ [12]
This example demonstrates that force-based metrics can converge first, while displacement criteria (which rely on an accurate Hessian) may fail. The frequency calculation uses an analytical Hessian, providing a more truthful assessment of the structure's status than the estimated Hessian often used during the optimization [12]. This underscores the importance of post-optimization frequency calculations to validate the results, as "frequency and thermochemistry results are based on a harmonic analysis that is only valid at true stationary points" [12].
Experimental Protocols for Metric Validation
Standard Geometry Optimization and Frequency Workflow
A robust protocol for energy minimization validation involves both the optimization and a mandatory verification step.
- Route Section Setup: The calculation is initiated with a route command that includes both optimization and frequency keywords (e.g.,
Opt Freq) to ensure the final structure is automatically checked [12]. - Optimization Execution: The optimization proceeds iteratively, updating the geometry until the chosen convergence criteria (Maximum Force, RMS Force, etc.) are met.
- Stationary Point Verification: Upon completion of the optimization, the output of the frequency job must be checked for the message "Stationary point found." If this message is absent, the structure is not a true stationary point and the results are unreliable [12].
- Remedial Action for Failed Convergence: If the frequency calculation indicates non-convergence, the optimization must be continued. This can be efficiently done by using the checkpoint file from the previous calculation and a route section that reads the computed force constants (Hessian) to complete the optimization in few steps [12]:
# method/basis OPT=ReadFC Freq Geom=AllCheck Guess=Read
Advanced Protocol: Normal-Mode-Analysis–Monitored Minimization
For systems where traditional optimizations are prone to folding or becoming trapped in shallow minima, a more controlled protocol is beneficial. The Normal-Mode-Analysis–Monitored Energy Minimization (NEM) procedure is designed to generate valid local minimum conformations [21].
- Initial Conformation Generation: Generate starting conformations (rotamers) by systematically varying all conformation-governing rotatable bonds of the molecule.
- Monitored Minimization: For each starting conformation, perform short cycles of energy minimization (e.g., 10 steps).
- Normal Mode Analysis Check: After each minimization cycle, perform a normal mode analysis. The key is to check the gradient and the vibrational frequencies.
- Convergence Validation: A conformation is considered a true local minimum if the potential energy gradient is below a threshold (e.g., ≤ 0.06 kcal/(mol·Å)), the magnitudes of translational and rotational frequencies are near zero, and all vibrational frequencies are positive [21].
- Cluster Analysis and Docking: Cluster the resulting local minimum conformations and use them for docking studies. This procedure has been validated by successfully reproducing guest-bound conformations in crystal structures with high accuracy [21].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Computational Tools and Resources for Energy Minimization
| Item | Function in Research | Example Use Case |
|---|---|---|
| Quantum Chemistry Software (e.g., Gaussian) | Performs the core quantum mechanical calculations for energy, force, and frequency. | Running Opt Freq calculations to optimize a drug molecule and validate its stability [12]. |
| Force Field Software (e.g., CHARMM, AMBER) | Provides molecular mechanics potentials for simulating large biomolecular systems. | Energy minimization of a protein-ligand complex before molecular dynamics simulation [23]. |
| Dispersion-Corrected DFT (d-DFT) | A quantum method corrected for long-range van der Waals interactions, crucial for molecular crystals. | Validating the correctness of an experimental organic crystal structure from a database [24]. |
| UltraFine Integration Grid | A dense grid for numerical integration in DFT, reducing numerical "noise." | Improving optimization convergence in flat regions of the potential energy surface or for calculations in solution [12]. |
| Normal Mode Analysis (NMA) | Analytic means to analyze harmonic potential wells and classify molecular deformations. | Verifying that an energy-minimized structure is at a true local minimum by checking for positive vibrational frequencies [21]. |
The comparative analysis of RMS Force, Maximum Force, and Energy Change reveals that no single metric is sufficient for robust energy minimization validation. The Maximum Force acts as a critical safeguard against local instability, while the RMS Force offers a more comprehensive view of global convergence. The Energy Change tracks the fundamental goal of the optimization but can be misleading in flat regions of the potential energy surface. For reliable results, these metrics must be used in concert, as is the standard in high-quality computational software. Furthermore, the strictest validation requires a post-optimization frequency calculation to confirm the structure is a true stationary point. As computational methods continue to drive innovations in drug development and materials design, a nuanced understanding and application of these convergence metrics remain foundational to generating scientifically sound and reproducible results.
Implementation Strategies: Energy Minimization Algorithms and RMS Force Calculation
This guide provides an objective comparison of three fundamental minimization algorithms—Steepest Descent, Conjugate Gradient, and Newton-Raphson—within the context of energy minimization validation and Root Mean Square (RMS) force research. These algorithms are critical for finding local minima in complex energy landscapes, a process central to tasks like molecular docking and protein structure prediction in drug development.
Algorithm Fundamentals and Comparative Theory
The Steepest Descent, Conjugate Gradient, and Newton-Raphson methods represent a spectrum of optimization techniques, balancing computational cost with convergence speed and stability.
| Feature | Steepest Descent | Conjugate Gradient | Newton-Raphson |
|---|---|---|---|
| Core Principle | First-order; follows negative gradient | First-order; uses conjugate directions | Second-order; uses Hessian matrix and gradient |
| Convergence Rate | Linear (sub-linear for convex) [25] [26] | Linear (for strongly convex) [25] | Quadratic or faster [25] |
| Computational Cost per Iteration | Low | Low (for quadratic functions) | High (Hessian calculation & inversion) |
| Memory Requirements | Low | Low | High |
| Stability | Stable with small step size | More stable than Steepest Descent | Can be unstable far from minimum |
| Ideal Use Case | Simple, low-dimensional problems; initial optimization steps | Large-scale linear systems; problems with many parameters | Small-to-medium problems where precise Hessian is available |
| Performance in RMS Force Minimization | Slow to converge to low RMS force | More efficient than Steepest Descent for RMS reduction | Can reach low RMS force in very few iterations [25] |
The Steepest Descent algorithm is a foundational first-order method. It iteratively moves in the direction opposite to the function's gradient at the current point. Its simplicity is a strength, but it often takes steps in the same direction as earlier iterations, leading to slow convergence, especially as it approaches the minimum [26].
The Conjugate Gradient (CG) method improves upon Steepest Descent by choosing search directions that are A-conjugate (conjugate with respect to the coefficient matrix A in quadratic problems). This crucial difference means that each step does not spoil the minimization achieved by previous steps. For a quadratic function with ( n ) variables, CG is guaranteed to converge in at most ( n ) steps [26]. It occupies a middle ground, requiring only gradient information like Steepest Descent but offering significantly better convergence.
The Newton-Raphson method is a second-order algorithm that uses both the gradient and the Hessian matrix (the square matrix of second-order partial derivatives) to find the minimum. By leveraging curvature information, it can achieve much faster quadratic convergence, meaning the number of correct digits roughly doubles with each iteration [25].
Algorithm Selection and Workflow: A simplified decision and iteration flow for the three minimization methods.
Experimental Performance and Validation
Empirical data is crucial for understanding how these algorithms perform in practical research scenarios, particularly in energy minimization tasks where the goal is often to reduce the RMS force to zero.
Comparative Iteration Performance
In a direct comparison on a specific minimization function, the difference in convergence speed was dramatic [25]:
- Newton-Raphson: Converged to the solution in only 5-6 iterations.
- Conjugate Gradient: Required approximately 2000 iterations to converge.
- Steepest Descent: Required approximately 5000 iterations to converge.
This stark contrast highlights the potential efficiency gain of Newton's method, though its per-iteration computational cost is the highest.
Algorithm Stability and Parameter Sensitivity
A key factor in this performance is step size (learning rate). The Newton-Raphson method often works effectively with a step size of ( \alpha = 1 ), while gradient-based methods (Steepest Descent and Conjugate Gradient) can diverge with such a large step and require a much smaller value (e.g., ( \alpha = 0.01 )) to remain stable [25]. This necessity for a small step size is a primary reason for the slow convergence of these methods.
Application in Energy Minimization Frameworks
The principles of these classic algorithms are actively adapted and used in modern computational science. For instance, in solving the steady-state Allen-Cahn equation—an energy minimization problem relevant to phase separation in materials science—deep learning frameworks have been developed that directly minimize the associated energy functional [27]. Research in this area shows that the choice of activation function in the neural network (like ReLU or tanh) can significantly impact the stability and accuracy of the solution, echoing the classic trade-offs between algorithm speed and stability [27].
Detailed Experimental Protocols
To validate and compare the performance of these algorithms in the context of energy minimization, researchers can employ the following general protocols.
Protocol 1: Benchmarking on a Quadratic Energy Function
This protocol tests the basic performance of the algorithms on a well-understood problem.
- Objective: To compare the convergence rate and computational cost of each algorithm on a quadratic energy landscape.
- System Setup: Define a quadratic function ( F(\textbf{x}) = \frac{1}{2}\textbf{x}^T A \textbf{x} - \textbf{b} \textbf{x} ), where ( A ) is a known positive definite matrix (e.g., from a discretized Poisson equation) and ( \textbf{b} ) is a known vector.
- Minimization Target: Minimize ( F(\textbf{x}) ) until the RMS force ( \| \nabla F(\textbf{x}) \|_2 ) falls below a threshold (e.g., ( 10^{-8} )).
- Execution:
- Initialize all algorithms from the same starting point ( \textbf{x}_0 ).
- For Steepest Descent, use a fixed, small learning rate (e.g., 0.01) or a line search.
- For Conjugate Gradient, use the Fletcher-Reeves or Polak-Ribière formula to update directions.
- For Newton-Raphson, compute the exact Hessian, ( A ), and its inverse analytically.
- Data Collection: At each iteration, record the RMS force, the current energy ( F(\textbf{x}) ), and the wall-clock time.
Protocol 2: Stability Under Poor Initialization
This protocol evaluates the robustness of the algorithms, which is critical for real-world problems with complex energy landscapes.
- Objective: To assess the sensitivity of each algorithm to the initial guess and its ability to avoid local traps.
- System Setup: Use a non-quadratic, multi-well potential energy function, such as the Rosenbrock function or a potential from a molecular mechanics force field.
- Minimization Target: Achieve an RMS force below ( 10^{-6} ) starting from multiple, strategically chosen poor initial configurations.
- Execution:
- Run each algorithm from each initial point.
- For Newton-Raphson, also test a modified version that uses a trust-region or a damping factor to maintain stability when the Hessian is not positive definite.
- Data Collection: Record the number of successful convergences to the global minimum, the final RMS force, and the total number of iterations.
The Scientist's Toolkit: Research Reagent Solutions
The following table outlines key computational tools and concepts essential for conducting minimization research in computational chemistry and drug development.
| Item / Concept | Function in Minimization Research |
|---|---|
| Energy Functional | A mathematical expression defining the total energy of a system as a function of its state; serves as the objective function for minimization [27]. |
| RMS Force (Root Mean Square Force) | The square root of the average squared gradient; a key convergence criterion indicating how close the system is to a stationary point (a minimum). |
| Gradient | The vector of first derivatives of the energy with respect to all particle coordinates; indicates the direction of steepest ascent [26]. |
| Hessian Matrix | The square matrix of second partial derivatives; provides curvature information about the energy landscape, crucial for the Newton-Raphson method [25]. |
| Line Search | A procedure used in Steepest Descent and Conjugate Gradient to find the optimal step size ( \alpha ) in the chosen direction for maximum energy reduction. |
| Conjugate Directions | A set of search vectors optimized for quadratic functions to ensure that minimization along one direction is not undone by the next; the core of the CG method [26]. |
| Physics-Informed Neural Networks (PINNs) | A modern deep learning approach that can incorporate the laws of physics (e.g., PDEs) into its loss function, offering an alternative for solving energy minimization problems [27]. |
The choice between Steepest Descent, Conjugate Gradient, and Newton-Raphson methods involves a fundamental trade-off between computational cost per iteration and the total number of iterations required for convergence. While Newton-Raphson can be phenomenally fast for suitable problems, its high cost and stability concerns often make Conjugate Gradient the preferred workhorse for large-scale problems in drug development, such as molecular energy minimization. Steepest Descent remains a valuable tool for initial rough optimization or in contexts where its simplicity is advantageous. Validation using RMS force as a primary metric ensures that the chosen algorithm effectively drives the system to a stable, low-energy state.
Energy minimization is a foundational step in molecular simulations, aiming to find a stable, low-energy conformation of a biological system by iteratively adjusting atomic coordinates until the net forces acting on all atoms are sufficiently close to zero. The Root Mean Square (RMS) force tolerance is a critical convergence criterion that determines when this process stops; it represents the square root of the average of the squares of the forces on all atoms. Setting an appropriate RMS force tolerance is crucial: an overly strict tolerance can lead to prohibitively long computation times with diminishing returns, while an overly lenient tolerance may result in a structure that is not sufficiently relaxed, potentially compromising the validity of subsequent simulation or analysis.
Within the broader context of energy minimization validation research, the RMS force serves as a primary quantitative metric for assessing the quality and reliability of a minimized structure. This guide provides a structured comparison of RMS force tolerance settings across different biological systems, supported by experimental data and detailed methodologies, to aid researchers in making informed decisions for their computational studies.
Methodological Framework for Tolerance Validation
The validation of RMS force tolerance settings relies on a multi-faceted approach that correlates the convergence criterion with resulting structural properties and energetic stability.
Core Validation Workflow
A standard protocol for validating RMS force tolerance involves a series of energy minimizations on a known system, each performed with a progressively stricter (lower) force tolerance. The resulting structures are then evaluated against key performance indicators [24]:
- Structural Deviation: The root mean square deviation (RMSD) of atomic coordinates from a high-quality reference structure (e.g., an experimental crystal structure) is calculated.
- Internal Strain: Changes in key internal coordinates, such as bond lengths, angles, and dihedrals, are monitored against experimental or high-level theoretical benchmarks [28].
- Energetic Convergence: The total potential energy of the system is tracked to ensure it has reached a stable minimum.
The point of diminishing returns—where a stricter RMS force tolerance yields negligible improvement in these metrics—informs the recommended tolerance for that system type. The diagram below illustrates this logical workflow for determining an optimal RMS force tolerance.
Key Research Reagents and Computational Tools
The following table details essential software and force fields used in the cited experimental protocols for energy minimization studies.
Table 1: Key Research Reagents and Computational Solutions
| Item Name | Function in Research | Example Use-Case |
|---|---|---|
| Molecular Operating Environment (MOE) [29] | Integrated software for molecular modeling, pharmacophore design, and virtual screening. | Used for preparing protein structures (e.g., removing water, adding hydrogens, energy minimization) prior to docking studies. |
| AMBER [29] | Suite of biomolecular simulation programs implementing molecular dynamics and energy minimization. | Performs Molecular Dynamics (MD) simulations to validate the stability of protein-ligand complexes identified through docking. |
| GRACE/VASP [24] | Software package (GRACE) that utilizes Vienna Ab initio Simulation Package (VASP) for dispersion-corrected Density Functional Theory (d-DFT) calculations. | Used for high-accuracy energy minimization of organic crystal structures, validating experimental data against theoretical benchmarks. |
| EUDOC [30] | A computational docking program used for guest-host interactions. | Docks small molecule conformers generated via energy minimization into host macromolecules to identify bound conformations. |
| MMFF94 [29] [31] | A general-purpose force field parameterized for a wide range of organic molecules and drug-like compounds. | Commonly used for the energy minimization of small molecules and ligands to obtain stable low-energy conformations for docking. |
| Tripos Force Field [28] | A general-purpose force field implemented in the Sybyl software. | Provides good quality geometries and relative energies for a large variety of organic molecules via energy minimization. |
Comparative RMS Force Tolerance Guidelines
Optimal RMS force tolerance is not universal; it depends on the system's size, complexity, and the scientific question. The following guidelines, synthesized from validation studies, provide a comparative framework.
Table 2: Recommended RMS Force Tolerance by Biological System
| System Category | Recommended RMS Force | Key Supporting Experimental Data | Applicable Force Fields |
|---|---|---|---|
| Small Organic Molecules / Drug-like Ligands | 0.01 - 0.05 kcal/(mol·Å) | MMFF94 and MM3 show strong performance in conformational analysis, reproducing geometries close to QM calculations [31]. The Tripos force field achieved RMS movements of ~0.025 Å for bond lengths in organic crystals with similar tolerances [28]. | MMFF94 [31], MM3 [31], Tripos [28], AMOEBA [31] |
| Proteins & Peptides | 0.5 - 1.0 kcal/(mol·Å) | Minimization of cyclic hexapeptides from crystallographic coordinates resulted in heavy atom RMS movements of 0.16 Å, validating structural integrity [28]. Studies on SpCas9 protein used simulations to link conformational stability to functional activity [32]. | AMBER [29], CHARMM, OPLS-AA |
| Nucleic Acids (DNA/RNA) | 0.1 - 0.5 kcal/(mol·Å) | MD simulations of CRISPR-Cas9 RNA-DNA duplexes used RMSD to quantify conformational instability induced by mismatches, requiring precise initial minimization [32]. | AMBER (parmbsc1), CHARMM36 |
| Organic Molecular Crystals | 1.0 - 3.0 kJ/(mol·Å) / ~0.02 - 0.06 kcal/(mol·Å) | d-DFT validation of 241 organic crystal structures achieved average RMS Cartesian displacements of 0.095 Å, using a maximum force criterion of < 2.93 kJ/(mol·Å) [24]. | Dispersion-Corrected DFT (d-DFT) [24] |
Detailed Experimental Protocols
Protocol for Small Organic Molecules
- System Preparation: Construct the 2D structure of the molecule and generate initial 3D conformers, for example, using a torsion driver with 60° increments [30].
- Energy Minimization: Perform minimization using a force field like MMFF94. The process can be monitored by techniques like Normal-Mode-Analysis (NMA) to ensure the molecule reaches a true local minimum conformation and does not "fold" unrealistically in the absence of its binding partner [30].
- Validation: Compare the minimized structure's geometry (bond lengths, angles, dihedrals) and relative conformational energies to high-level ab initio calculations (e.g., DFT) or experimental crystal structure data. The Tripos force field validation, for instance, showed RMS errors of 0.8 kcal/mol for conformer energies and 1.13 kcal/mol for torsional barriers against reference data [28].
Protocol for Protein-Ligand Complexes
- System Preparation: Obtain the protein structure from a database (e.g., PDB: 7ALV for NLRP3 [29]). Remove water molecules and co-factors, add hydrogen atoms, and assign protonation states. Prepare the ligand structure separately using a molecular mechanics force field.
- Energy Minimization: A common approach is a two-stage minimization. An initial minimization is often performed with restraints on the protein backbone to remove steric clashes, followed by a full minimization of the entire system. The protocol used for NLRP3 inhibitors involved docking followed by 100 ns MD simulations to check complex stability, which relies on well-minimized starting structures [29].
- Validation: Run Molecular Dynamics (MD) simulations and analyze metrics like RMSD, RMSF (Root Mean Square Fluctuation), and the number of hydrogen bonds to ensure the complex remains stable. The stability of the minimized structure is confirmed by low RMSD values during simulation [29] [32].
Selecting an appropriate RMS force tolerance is a critical, system-dependent decision in computational biology. As evidenced by the comparative data, tolerances for small molecules and crystals must be very strict (e.g., 0.01-0.05 kcal/(mol·Å)) to achieve the geometric accuracy required for reliable predictions, while larger systems like proteins can tolerate slightly higher values (e.g., 0.5-1.0 kcal/(mol·Å)) to balance computational cost with structural integrity.
Future developments in this field are likely to be driven by more sophisticated, multi-scale methods. Bayesian Multimodel Inference (MMI), for instance, offers a powerful framework for handling model uncertainty by combining predictions from multiple models, which could be extended to inform convergence criteria [33]. Furthermore, the integration of machine learning interatomic potentials (MLIPs) that explicitly treat long-range electrostatics, such as those using the Latent Ewald Summation (LES) method, promises to improve the accuracy and physical realism of energy minimization for complex, charged biological systems [34]. These advances will continue to refine the validation protocols and tolerance guidelines, enhancing the reliability of computational drug discovery and biomolecular research.
Molecular dynamics (MD) simulation has become an indispensable tool in computational chemistry, biophysics, and drug development, enabling researchers to study the structure, dynamics, and function of biological macromolecules at atomic resolution. Among the various software packages available, GROMACS, CHARMM, and AMBER have emerged as three of the most widely used platforms in the scientific community. Each package offers distinct algorithmic implementations, force fields, and optimization approaches for energy minimization—a critical step in preparing stable molecular systems for subsequent dynamics simulations. The validation of these minimization procedures through metrics like root mean square force (RMSF) calculations provides crucial insights into structural flexibility and stability, forming an essential component of rigorous MD workflows.
This guide offers a comprehensive, objective comparison of energy minimization and fluctuation analysis capabilities across these three major MD packages, drawing upon published experimental data and detailed methodological protocols. The focus on practical implementation aims to equip researchers with the knowledge needed to select appropriate software, implement validated protocols, and interpret results within the context of their specific research applications, particularly in drug development where understanding molecular flexibility and binding interactions is paramount.
The table below summarizes the key characteristics, strengths, and specialized capabilities of GROMACS, CHARMM, and AMBER for energy minimization and related analyses.
Table 1: Comprehensive Comparison of GROMACS, CHARMM, and AMBER
| Aspect | GROMACS | CHARMM | AMBER |
|---|---|---|---|
| Primary Strength | Exceptional speed & parallel efficiency [35] | Versatility for many-particle systems [36] [37] | High-accuracy force fields for biomolecules [35] |
| Minimization Algorithms | Steepest descent, Conjugate gradient [35] | SD, CONJ, ABNR, NRAP, POWE, TNPACK [37] | Steepest descent, Conjugate gradient [35] |
| Force Field Specialization | Supports AMBER, CHARMM, OPLS [35] | Native CHARMM force fields [36] | AMBER force fields (ff14SB, GAFF) [35] |
| RMSF Implementation | gmx rmsf command [38] [39] |
ENERgy, GETE commands [36] | atomicfluct in CPPTRAJ [40] |
| Performance & Scalability | Highly optimized for CPUs/GPUs [35] | Moderate scalability [35] | Good CPU performance, improving GPU support [35] |
| Learning Curve | User-friendly [35] | Steeper learning curve [35] | Steeper learning curve [35] |
| Best Applications | Large-scale systems, high-throughput [35] | Diverse molecular systems [36] | Protein-ligand interactions, nucleic acids [35] |
| B-factor Conversion | Built-in with -oq flag [38] [39] |
Requires manual calculation | Built-in with bfactor option [40] |
Energy Minimization Methodologies and Algorithms
Minimization Algorithms Available Across Platforms
Each MD package provides a suite of energy minimization algorithms, though CHARMM offers the most diverse set of optimization methods. GROMACS primarily implements steepest descent and conjugate gradient methods, prioritizing computational efficiency for large systems [35]. AMBER focuses on these same reliable algorithms but with specialized parameterization for biological macromolecules [35]. In contrast, CHARMM provides researchers with six distinct minimization approaches: Steepest Descent (SD), Conjugate Gradient (CONJ), Adopted Basis Newton-Raphson (ABNR), Newton-Raphson (NRAP), Powell (POWE), and Truncated Newton Method (TNPACK) [37]. This diversity allows experienced researchers to select algorithms based on their specific system characteristics and convergence requirements.
Algorithm Specifics and Convergence Criteria
The CHARMM package implements detailed convergence parameters that illustrate the sophistication available in minimization protocols. The ABNR algorithm includes specialized parameters including EIGRNG (smallest non-singular eigenvalue relative to largest, default 0.0005), MINDIM (basis set storage dimension, default 5), STPLIM (maximum Newton-Raphson step, default 1.0), and STRICT (descent strictness, default 0.1) [37]. Convergence tolerances include TOLENR for energy change, TOLGRD for average gradient, and TOLSTP for average step size [37]. The TNPACK method implements a particularly sophisticated convergence test with four criteria checking energy changes, coordinate changes, and gradient norms relative to customizable tolerances [37]. These detailed control parameters enable fine-tuning of minimization procedures for challenging systems.
RMSF Analysis: Theoretical Framework and Implementation
Theoretical Foundation of RMSF Calculations
The Root Mean Square Fluctuation (RMSF) measures the standard deviation of atomic positions from their average location throughout a simulation trajectory, providing crucial insights into regional flexibility and structural stability. For an individual atom i, the RMSF (ρ) is calculated as:
$$ρi = \sqrt{\big\langle( ri - \langle r_i \rangle)^2\big\rangle}$$
where $ri$ represents the atomic coordinates and $\langle ri \rangle$ denotes the average position [39] [41]. The RMSF is particularly valuable for identifying flexible protein regions (showing high RMSF values) versus rigid structural elements (with low RMSF values) [39]. This metric differs fundamentally from Root Mean Square Deviation (RMSD); while RMSD measures global structural deviation over time to assess equilibration, RMSF quantifies per-atom or per-residue fluctuations to map flexibility across a structure [39].
Crystallographic B-Factor Conversion
RMSF values can be directly related to experimental crystallographic B-factors through the mathematical relationship:
$$B{i} = \frac{8\pi^2}{3} (ρ^{\mathrm{RMSF}}{i})^2$$
where $Bi$ represents the isotropic B-factor for atom *i* and $ρ^{RMSF}i$ is the RMSF value [41]. This conversion enables direct comparison between simulation results and experimental X-ray crystallography data, providing a crucial validation bridge between computational and empirical approaches. Both GROMACS (via the -oq flag) and AMBER (through the bfactor option in atomicfluct) provide built-in functionality to perform this conversion and output PDB files with B-factor values for visualization [38] [40] [39].
Practical Implementation Protocols
RMSF Analysis in GROMACS
The following workflow illustrates RMSF calculation for Cα atoms in GROMACS:
The specific command structure for GROMACS RMSF analysis is:
Where the -f and -s flags specify the trajectory and topology files, -o defines the output file, -res calculates RMSF per residue, and -b/-e set the time frame for analysis [39]. To analyze Cα atoms specifically, users can create an index file containing only Cα atoms or select the "C-alpha" group during interactive execution [39]. The -oq flag generates a PDB file with B-factor values for visualization: gmx rmsf -f simulation.xtc -s simulation.tpr -o rmsf.xvg -oq bfac.pdb [39].
RMSF Analysis in AMBER
In AMBER, RMSF calculations are performed using the atomicfluct command within the CPPTRAJ analysis tool:
A typical CPPTRAJ input script for RMSF analysis includes:
This protocol performs RMS fitting to the first structure using backbone atoms (C, CA, N), calculates the average structure, and then computes residue-wise RMSF values [40]. The byres keyword specifies that fluctuations should be averaged by residue, while the bfactor option enables B-factor conversion [40]. For proper results, RMS fitting must be performed prior to RMSF calculation to remove global rotations and translations [40].
Energy Evaluation and Analysis in CHARMM
CHARMM employs distinct commands for energy evaluation and interaction calculations. The ENERgy command computes system potential energy through the minimization parser, while GETE provides direct energy evaluation [36]. The INTEraction command specifically calculates energies and forces between user-defined atom selections, enabling detailed analysis of specific molecular interfaces [36]. CHARMM also includes the SKIPe command to selectively exclude certain energy terms during evaluation, providing flexibility in energy component analysis [36].
Experimental Validation and Cross-Package Comparisons
SAMPL5 Blind Prediction Challenge Insights
The SAMPL5 blind prediction challenge provided valuable experimental data comparing different MD packages when using theoretically identical starting structures and models. In these controlled comparisons, researchers found that energy calculations across MD engines agreed to within 0.1% relative absolute energy for all energy components when consistent cutoff parameters and force field interpretations were applied [42]. However, several key sources of discrepancy were identified:
- Different choices of Coulomb's constant between programs represented one of the largest sources of energy discrepancies [42]
- Varying default cutoff parameters and long-range interaction treatments caused significant differences when program-specific defaults were used [42]
- Alternative partial charge assignment methods (e.g., fragment-wise vs. whole-molecule AM1/BCC) resulted in energy differences of ~2 kJ/mol in the SAMPL4 challenge [42]
These findings highlight that while different packages can produce highly consistent results, achieving this agreement requires careful attention to parameter standardization rather than relying on program defaults.
Practical Considerations for RMSF Validation
A critical consideration in RMSF analysis involves proper trajectory treatment prior to fluctuation calculations. As highlighted in AMBER mailing list discussions, periodic boundary condition handling can significantly impact RMSF results if not properly addressed [43]. One researcher reported dramatically different RMSF values when comparing trajectories with and without explicit re-imaging of proteins in solvent boxes, emphasizing that "amber automatically handle periodic boundary conditions when calculating RMSF" may not always produce expected results without explicit imaging commands [43]. This underscores the importance of visual trajectory validation and consistent pre-processing across compared systems.
Essential Research Reagent Solutions
Table 2: Key Software Tools for MD Analysis and Visualization
| Tool Name | Function | Implementation Examples |
|---|---|---|
| CPPTRAJ | Trajectory analysis for AMBER | atomicfluct for RMSF [40] |
| GROMACS rmsf | Native fluctuation analysis | gmx rmsf -f traj.xtc -s topol.tpr [38] |
| ParmEd | Parameter/coordinate conversion | Cross-platform topology interconversion [42] |
| InterMol | Force field conversion | Automated parameter translation [42] |
| VMD/PyMOL | Visualization of B-factors | Color-coded flexibility mapping [39] |
| CHARMM ENERgy | Native energy computation | Potential energy evaluation [36] |
Comparative Workflow for Validation Studies
Based on comparative analysis and experimental validation data, specific recommendations emerge for researchers selecting MD packages for energy minimization and fluctuation analysis:
- For large-scale systems and high-throughput applications where computational efficiency is paramount, GROMACS provides superior performance and scalability [35]
- For specialized biomolecular simulations requiring maximum force field accuracy, particularly for proteins and nucleic acids, AMBER offers optimized force fields and specialized analysis tools [35]
- For advanced minimization algorithms and flexibility in energy term analysis, CHARMM provides the most diverse set of minimization approaches and detailed energy decomposition capabilities [36] [37]
- For cross-platform validation studies, researchers should utilize conversion tools like ParmEd and InterMol while carefully standardizing electrostatic treatment, cutoff parameters, and long-range interaction methods [42]
The consistent implementation of RMSF and B-factor calculations across all three packages enables robust flexibility analysis, though particular attention must be paid to trajectory pre-processing, especially regarding periodic boundary conditions and proper structural alignment prior to fluctuation calculations [40] [43]. By following the detailed methodologies and validation protocols outlined in this guide, researchers can effectively leverage the unique strengths of each MD package while ensuring reproducible, physiologically relevant results in their molecular simulations.
Energy minimization is a foundational computational process in structure-based drug design (SBDD) that refines molecular structures by adjusting atomic coordinates to find low-energy configurations. This process is essential for achieving physically realistic molecular models by relieving steric clashes, optimizing bond geometry, and finding local energy minima on the complex potential energy surface of biomolecular systems. The validation of energy minimization protocols typically employs the root mean square force (RMSF) metric, which measures the average force acting on atoms—with lower values indicating a closer approximation to an energy-minimized stable state. Contemporary drug discovery pipelines integrate energy minimization across multiple stages, from initial ligand preparation and binding pose refinement to the final assessment of protein-ligand complex stability, forming a critical bridge between static structural data and dynamic biological behavior.
The importance of energy minimization has been further highlighted by the emergence of artificial intelligence (AI) approaches in molecular docking. Recent comprehensive benchmarks evaluating 22 docking methods across three methodological categories (traditional physics-based, AI docking, and AI co-folding) revealed that while AI methods now outperform traditional physics-based approaches in overall docking accuracy, the integration of post-minimization relaxation significantly enhances their performance by improving physicochemical consistency and structural plausibility [44]. This synergy between advanced sampling algorithms and physics-based refinement represents the current state of the art in computational drug discovery.
Methodological Approaches and Performance Comparison
Traditional Physics-Based Methods
Traditional physics-based docking methods rely on molecular mechanics force fields and empirical scoring functions to explore conformational space and evaluate binding interactions. These methods treat docking as a sampling and optimization problem, where the algorithm searches for ligand orientations and conformations that minimize the system's energy within the binding site. Commonly used packages include AutoDock Vina, which employs a sophisticated gradient optimization algorithm; Schrödinger Glide, utilizing a hierarchical docking approach with increasingly refined scoring; and GNINA, which incorporates deep learning as a scoring function [44]. These tools typically combine terms for van der Waals interactions, hydrogen bonding, electrostatics, and desolvation effects in their scoring functions, with energy minimization playing a crucial role in final pose refinement.
A key limitation of many traditional implementations is their treatment of protein structures as rigid bodies during docking, which fails to capture the induced-fit phenomena often observed in actual binding events. To address this, ensemble docking approaches have been developed that utilize multiple protein conformations, either from experimental structures or molecular dynamics (MD) simulations, to account for receptor flexibility [45]. Recent innovations include hybrid methods that combine coarse-grained modeling of the overall protein structure with all-atom resolution in the binding site, enabling more efficient conformational sampling while maintaining accuracy where it matters most [45].
AI-Enhanced Docking and Minimization
Artificial intelligence has revolutionized molecular docking through approaches that learn binding patterns from existing protein-ligand complex structures. Methods like DiffDock, DeepDock, and EquiBind utilize deep learning architectures to predict binding poses directly from structural data [44]. These approaches can dramatically reduce computational time compared to traditional sampling methods while maintaining competitive accuracy. However, AI-generated poses often benefit from post-processing with physics-based minimization to correct stereochemical inaccuracies and ensure physical plausibility.
The Interformer model represents a significant advancement by explicitly modeling non-covalent interactions through an interaction-aware mixture density network, focusing particularly on hydrogen bonds and hydrophobic interactions [46]. This approach demonstrates how incorporating physical interaction constraints during training can improve pose quality. Similarly, DeepRLI implements a multi-objective framework that combines scoring, docking, and screening tasks, integrating physics-informed components into its docking and screening readout modules to enhance generalization beyond its training data [47]. These hybrid approaches illustrate the growing convergence of data-driven and physics-based methodologies in computational drug discovery.
Performance Benchmarking
Table 1: Comparative Performance of Docking Methods on Standard Benchmarks
| Method Category | Representative Tools | PoseBusters Benchmark Accuracy (%) | RMSD <2Å (Time-Split Test Set) | Ligand Ranking Capability |
|---|---|---|---|---|
| Traditional Physics-Based | Glide, AutoDock Vina, MOE | 60-75% | 40-55% | Moderate |
| AI Docking | DiffDock, Interformer, DeepDock | 75-85% | 63.9% (Interformer) | Good |
| AI Co-folding | AlphaFold3, RoseTTAFold-All-Atom | 70-80% | Varies significantly | Limited evidence |
| AI with Relaxation | DiffDock+Relax, Interformer+Relax | ~84% | Improved over non-relaxed | Good |
Table 2: Specialized Scoring Functions Combining Physical and ML Approaches
| Method | Architecture | Key Features | Enrichment Factor (Top 1%) |
|---|---|---|---|
| AK-Score2 | Triple-network GNN | Combines physical energy functions with graph networks | 32.7 (CASF2016), 23.1 (DUD-E) |
| PIGNet | GNN with physics-based scoring | Machine-tuned physical parameters | High screening power |
| RTMScore | Mixed density model | Residue-atom distance likelihood potential | Superior pose identification |
| DeepRLI | Multi-objective graph transformer | Separate readout networks for different tasks | State-of-the-art multi-task performance |
Recent benchmarking efforts across 22 docking methods reveal that cutting-edge AI methods now dominate performance rankings, with Interformer achieving 84.09% accuracy on the PoseBusters benchmark and 63.9% on the PDBbind time-split test set with RMSD <2Å [46] [44]. However, traditional physics-based methods maintain advantages in certain scenarios, particularly for unseen protein targets where their physical foundations provide better generalization compared to data-driven approaches [44]. The incorporation of post-docking energy minimization consistently improves results across all method categories, addressing stereochemical inaccuracies and physical implausibilities that remain common in pure AI-generated poses.
Case Studies in Energy Minimization Applications
Case Study 1: Targeting βIII-Tubulin for Anticancer Therapy
Microtubules play crucial roles in mitosis and cellular transport, making them attractive targets for anticancer therapy. The βIII-tubulin isotype is significantly overexpressed in various cancers and associated with resistance to anticancer agents. A recent study employed a comprehensive SBDD approach to identify natural inhibitors targeting the 'Taxol site' of human αβIII tubulin isotype, integrating homology modeling, virtual screening of 89,399 natural compounds, machine learning classification, and molecular dynamics (MD) simulations [48].
The energy minimization protocol was implemented at multiple stages:
- Initial structure preparation: Homology models of βIII-tubulin were built using Modeller 10.2 and optimized through energy minimization to relieve steric clashes
- Binding pose refinement: Top hits from virtual screening were minimized within the binding site using AutoDock Vina
- Molecular dynamics validation: 1μs MD simulations were conducted with Desmond, employing the OPLS4 force field with gradual minimization and equilibration
This systematic approach identified four natural compounds (ZINC12889138, ZINC08952577, ZINC08952607, and ZINC03847075) with exceptional binding properties and anti-tubulin activity. MD simulations tracked stability metrics including RMSD, RMSF, Rg, and SASA, demonstrating that these compounds significantly influenced the structural stability of the αβIII-tubulin heterodimer compared to the apo form [48]. The success of this pipeline underscores how energy minimization contributes to reliable virtual screening outcomes against challenging drug targets.
Case Study 2: PKMYT1 Inhibitors for Pancreatic Cancer
Protein kinase membrane-associated tyrosine/threonine 1 (PKMYT1) has emerged as a promising therapeutic target for pancreatic ductal adenocarcinoma due to its critical role in controlling the G2/M transition of the cell cycle. Researchers implemented a structure-based discovery pipeline integrating pharmacophore modeling, virtual screening of 1.64 million compounds, molecular docking, and MD simulations to identify novel PKMYT1 inhibitors [49].
The minimization and validation workflow included:
- Protein preparation: Crystal structures of PKMYT1 (PDB IDs: 8ZTX, 8ZU2, 8ZUD, 8ZUL) were processed using Schrödinger's Protein Preparation Wizard, adding hydrogen atoms, filling missing loops, and performing restrained energy minimization with the OPLS 2005 force field
- Ligand preparation: Compound structures from the TargetMol natural compound library were prepared using LigPrep with the OPLS4 force field for energy minimization
- Extended dynamics: 1μs MD simulations were conducted for 20 selected PKMYT1-ligand complexes using Desmond, with initial energy minimization to remove steric clashes followed by multi-stage equilibration
This approach identified HIT101481851 as a promising lead compound demonstrating stable interactions with key residues (CYS-190 and PHE-240) across multiple PKMYT1 conformations. Binding free energy calculations using MM-GBSA confirmed strong binding affinity, while experimental validation showed the compound effectively inhibited pancreatic cancer cell viability in a dose-dependent manner [49]. The study exemplifies how energy minimization contributes to identifying selective kinase inhibitors with therapeutic potential.
Case Study 3: Computational Efficiency in Binding Free Energy Calculations
Accurate prediction of binding free energies remains challenging in SBDD due to the computational expense of rigorous methods. A recent study addressed this by developing a mixed-resolution approach that combines coarse-grained and all-atom representations to efficiently generate plausible protein conformers for ensemble docking [45].
The innovative methodology included:
- Mixed-resolution modeling: Binding sites were modeled at atomistic resolution while the remaining structure was coarse-grained
- Conformer generation: The Anisotropic Network Model (ANM) was used to derive 36 conformers of truncated high-resolution regions based on slowest functional modes
- Energy minimization: Each conformer underwent energy minimization before docking calculations
- Restrained MD simulations: Truncated docked structures were subjected to 900ns MD simulations with harmonic restraints (0, 25, 35, and 50 kcal/mol·Å²) to prevent disintegration
This protocol was validated using triose phosphate isomerase (TIM) as a model system, examining both its catalytic site (with DHAP) and dimer interface (with 3PG). The results demonstrated that 100ns simulations with truncated structures were sufficient to estimate binding affinities comparable to those obtained with intact protein structures [45]. This approach significantly reduces computational costs while maintaining accuracy, particularly beneficial for large protein complexes and supramolecular assemblages where traditional all-atom simulations remain prohibitively expensive.
Energy Minimization and RMSF Validation in SBDD Workflow
Experimental Protocols and Methodologies
Molecular Dynamics Simulation Protocol
MD simulations provide the gold standard for assessing the stability and dynamics of protein-ligand complexes following initial docking and minimization. A typical protocol, as implemented in studies of PKMYT1 inhibitors and βIII-tubulin binders, includes the following stages [48] [49]:
- System preparation: Protein-ligand complexes are solvated in an orthorhombic water box with TIP3P water molecules, maintaining a minimum 10Å buffer between the protein and box edges. Ions are added to neutralize system charge and simulate physiological conditions (150mM NaCl)
- Energy minimization: Two-stage minimization is performed using the steepest descent algorithm followed by conjugate gradient method. The initial stage restrains heavy atoms with force constants of 10-50 kcal/mol·Å² to relax solvent and hydrogen atoms, while the second stage allows all atoms to move freely
- System equilibration: Gradual heating from 0K to 300K over 100ps under NVT ensemble followed by density equilibration over 1-10ns under NPT ensemble at 300K and 1 atm pressure, using Langevin thermostat and Berendsen/Monte Carlo barostat
- Production run: Unrestrained MD simulation for 100ns to 1μs with 2fs integration time step, employing SHAKE constraints on hydrogen-containing bonds. Coordinates are saved every 10-100ps for subsequent analysis
- Validation metrics: RMSF calculations monitor convergence, with stable values below 1-2Å indicating proper equilibration and minimization
This protocol ensures that systems are adequately minimized and equilibrated before production runs, reducing artifacts and providing reliable trajectories for binding free energy calculations and interaction analysis.
Binding Free Energy Calculation Methods
Accurate prediction of binding affinities is crucial for prioritizing compounds in SBDD. The following methods represent current approaches with varying balances between computational cost and accuracy:
- MM/GBSA and MM/PBSA: These end-point methods combine molecular mechanics energies with continuum solvation models (Generalized Born/Poisson-Boltzmann) to estimate binding free energies. A typical protocol involves extracting snapshots from MD trajectories (every 100-500ps), removing water molecules and counterions, and calculating energy components using tools such as MMPBSA.py or Schrödinger's Prime module. While less accurate than full alchemical methods, they provide reasonable rankings at moderate computational cost
- Alchemical Free Energy Calculations: Methods like free energy perturbation (FEP) and thermodynamic integration (TI) provide higher accuracy by simulating the physical transformation between ligands. Recent evaluations of the ABCG2 charge model with nonequilibrium alchemical simulations demonstrated RMSE values of approximately 1.31-1.39 kcal/mol for relative binding free energies across 12 protein targets [50]
- Hybrid Physical-ML Approaches: Models like AK-Score2 integrate physical energy functions with graph neural networks, achieving enrichment factors of 32.7 and 23.1 with the CASF2016 and DUD-E benchmark sets respectively [51]. These methods offer improved screening power while maintaining physical interpretability
Table 3: Force Field and Charge Model Performance Comparison
| Force Field Combination | Hydration Free Energy RMSE (kcal/mol) | Binding Free Energy RMSE (kcal/mol) | Ligand Ranking Performance |
|---|---|---|---|
| GAFF2/AM1-BCC | 1.71 | 1.31 | Moderate |
| GAFF2/ABCG2 | 0.99 | 1.38 | Moderate |
| AMBER14SB/GAFF2/ABCG2 | - | 1.39 | Moderate |
| OPLS4 | Not reported | ~1.5-2.0 (estimated) | Good |
Research Reagent Solutions
Table 4: Essential Computational Tools for Energy Minimization in SBDD
| Tool Category | Representative Software | Primary Function | Application in Energy Minimization |
|---|---|---|---|
| Molecular Dynamics | Desmond, GROMACS, AMBER | Biomolecular simulation | Force field-based minimization and equilibration |
| Docking Suites | Glide, AutoDock Vina, MOE | Binding pose prediction | Local optimization of docked poses |
| Force Fields | OPLS4, AMBER, CHARMM | Molecular mechanics parameters | Energy calculation and atomic forces |
| Structure Preparation | Schrödinger Protein Prep, MOE | Protein-ligand modeling | Initial structure optimization |
| Analysis Tools | VMD, PyMOL, MDTraj | Trajectory analysis | RMSF and stability metrics calculation |
| Specialized Scoring | AK-Score2, DeepRLI, RTMScore | Binding affinity prediction | Integration of physical and ML approaches |
The selection of appropriate computational tools significantly impacts the effectiveness of energy minimization in SBDD pipelines. Force field selection represents a critical consideration, with recent studies evaluating options such as GAFF2/AM1-BCC, GAFF2/ABCG2, and OPLS4 for specific applications [49] [50]. While the ABCG2 charge model demonstrates superior performance for hydration free energy prediction (RMSE ~1.00 kcal/mol), it does not outperform AM1-BCC for protein-ligand binding free energy calculations, highlighting how force field optimization for specific properties doesn't guarantee improved performance for related applications [50].
Specialized scoring functions that combine physical energy functions with machine learning represent another important development. AK-Score2 integrates three independent neural network models with physics-based scoring to account for both binding affinity errors and pose prediction uncertainties [51]. Similarly, DeepRLI implements a multi-objective framework with separate readout networks for scoring, docking, and screening tasks, incorporating physics-informed components to enhance generalization [47]. These tools demonstrate how hybrid approaches can leverage the strengths of both physical and data-driven methodologies.
Force Field Selection and Energy Minimization Protocol
Energy minimization remains an essential component of structure-based drug design, ensuring physically realistic molecular models and reliable virtual screening outcomes. Recent advances have integrated minimization protocols with both traditional physics-based approaches and emerging AI methods, creating hybrid pipelines that leverage the strengths of multiple methodologies. The validation of minimization quality through RMSF analysis provides crucial quality control, distinguishing properly equilibrated systems from those requiring further refinement.
Future developments will likely focus on improved efficiency through mixed-resolution approaches [45], enhanced force fields with better transferability across properties [50], and tighter integration of physical principles with deep learning architectures [51] [47]. As benchmarking studies continue to demonstrate the superiority of AI methods for certain tasks [44] and the value of physical validation for others, the field appears to be converging toward solutions that balance data-driven efficiency with physical accuracy. These advances will further establish energy minimization as a critical validation step in computational drug discovery, ensuring that predicted protein-ligand complexes represent stable, low-energy states with genuine therapeutic potential.
In computational chemistry and structural biology, hybrid simulation approaches represent a powerful class of methods that integrate multiple computational or experimental techniques to overcome the limitations of individual methodologies. These approaches strategically combine different levels of theory, data sources, or sampling techniques to achieve a balance between computational efficiency and physical accuracy that would be unattainable with any single method. The fundamental premise behind these methods is leveraging the strengths of one technique to compensate for the weaknesses of another, thereby creating a synergistic framework that provides more comprehensive insights into molecular systems than would otherwise be possible.
The necessity for such hybrid methods arises from the inherent challenges in modeling complex biomolecular systems. Traditional all-atom molecular dynamics (MD) simulations, while providing atomic-level detail, suffer from severe limitations in accessible timescales and system sizes due to computational constraints. As noted in studies of tethered polymer systems, "in classical molecular dynamics simulation a great deal of computational time is devoted to simulating solvent molecules, although the solvent itself is of no direct interest" [52]. Similarly, energy minimization techniques alone can locate low-energy configurations but provide no information about dynamics, ensembles, or finite-temperature properties. Hybrid methods address these gaps by creating multi-scale, multi-fidelity frameworks that allocate computational resources where they are most needed while maintaining accuracy where it matters most for the specific research question at hand.
Within the broader context of energy minimization validation, hybrid approaches provide critical tools for assessing the quality of minimized structures and exploring their connectivity within the energy landscape. The convergence criteria for minimization algorithms, typically based on root mean square (RMS) force thresholds, can be validated through subsequent MD simulations that test the stability and dynamic behavior of minimized structures. This combination allows researchers to distinguish between truly stable configurations and those that may represent local minima with low barriers to more favorable states, thereby providing a more robust validation of minimization protocols than energy criteria alone.
Classification of Hybrid Methodologies
Continuum-Molecular Dynamics Hybrids
One prominent category of hybrid methods combines atomistic molecular dynamics with continuum models, creating a multi-scale simulation framework that dramatically improves computational efficiency for certain classes of problems. In this approach, the region of primary interest (such as a polymer, protein, or other molecular structure) is treated with full atomic detail using molecular dynamics, while the surrounding environment (typically solvent) is modeled using a continuum approach such as computational fluid dynamics [52].
This methodology employs three distinct spatial domains: a classical MD region for the core molecular system, a continuum domain where governing equations are solved using standard CFD methods, and a crucial overlap domain where transport information is exchanged between the other two domains. The exchange of information in this overlap region is carefully designed to conserve momentum, energy, and mass across the interface, ensuring physical consistency throughout the simulation domain [52].
The practical value of this approach has been demonstrated in systems such as tethered polymers in solvent under shear flow. In these applications, the hybrid method successfully reproduced the conformational properties of polymers obtained from full MD simulations while reducing computational cost to "less than 6% of the cost of updating the MD forces" [52]. This dramatic improvement in efficiency enables the study of larger systems and longer timescales than would be feasible with full atomic detail throughout the simulation volume.
Table 1: Performance Comparison of Continuum-MD Hybrid vs. Full Molecular Dynamics
| Metric | Full MD | Continuum-MD Hybrid | Improvement |
|---|---|---|---|
| Computational cost (force updates) | 100% | <6% | >16x reduction |
| Polymer conformational accuracy | Reference | Excellent agreement | No significant difference |
| Solvent treatment | Explicit atoms | Continuum + local explicit | Major efficiency gain |
| Applicability to flow conditions | Limited by system size | Extended through continuum regions | Expanded capability |
Experimental Data-Integrated Molecular Dynamics
A second major category of hybrid methods incorporates experimental data directly into molecular dynamics simulations to guide and validate structural models. This approach has become particularly valuable in structural biology, where traditional structure determination methods like X-ray crystallography face challenges for systems that are difficult to crystallize, exist in multiple conformational states, or require characterization of transient intermediates [53].
These integrative modeling techniques utilize experimental data from diverse sources including Nuclear Magnetic Resonance (NMR), Förster Resonance Energy Transfer (FRET), Double Electron-Electron Resonance (DEER), Paramagnetic Relaxation Enhancements (PREs), and chemical crosslinking. These techniques provide indirect structural information that can be used to constrain and bias MD simulations even when complete structural determination is not possible [53]. The experimental data can be incorporated into simulations in various ways, including as restraints on atomic positions, distance constraints between specific molecular sites, or as validation metrics for assessing the agreement of simulation ensembles with experimental observations.
The resurgence of interest in these methods is driven by improvements in both force fields and enhanced sampling techniques, which have led to "excellent agreement between experiments and simulation techniques" [53]. Furthermore, the development of specialized databases such as the wwPDB-dev, designed to accept models originating from integrative/hybrid approaches, provides a foundation for sharing and validating these complex multi-source models [53].
Machine Learning-Augmented Hybrid Methods
The integration of machine learning with traditional simulation methodologies represents a rapidly advancing frontier in hybrid approaches. These methods leverage the pattern recognition capabilities of ML algorithms to accelerate simulations, improve accuracy, or enhance sampling in ways that would be difficult with conventional physical models alone.
One innovative framework combines bottom-up and top-down coarse-graining strategies using machine learning to develop efficient, accurate, and transferable coarse-grained models of polymeric materials. In this approach, "bonded interactions of the CG model are optimized using deep neural networks (DNN), where atomistic bonded distributions are matched" in the bottom-up component, while "optimization of nonbonded parameters is accomplished by reproducing the temperature-dependent experimental density" in the top-down component [54]. This addresses the thermodynamic consistency and transferability issues that have plagued traditional coarse-graining approaches.
The ML-enabled framework provides significant advantages over conventional methods. It demonstrates accurate predictions of "chain statistics, the limiting behavior of the glass transition temperature, diffusion, and stress relaxation, where none were included in the parametrization process" [54]. This indicates that the machine-learned potentials capture underlying physical principles rather than merely memorizing training data, enabling true predictive capability for properties outside the parameterization set.
Quantitative Performance Comparison of Hybrid Methods
The effectiveness of hybrid approaches can be quantitatively assessed across multiple performance dimensions, including computational efficiency, accuracy maintenance, and applicability to different system types. The following table summarizes key comparative metrics for the major hybrid methodologies discussed in this review.
Table 2: Comprehensive Comparison of Hybrid Method Performance Characteristics
| Method Category | Computational Efficiency | Accuracy Retention | Key Applications | Limitations |
|---|---|---|---|---|
| Continuum-MD Hybrid | >94% cost reduction for solvent interactions [52] | Excellent agreement for polymer conformations (<2% deviation) [52] | Tethered polymers in flow, implicit solvent simulations | Limited for strongly coupled solvent-solute systems |
| Experimental Data-Integrated MD | 30-50% reduction in sampling time through guided exploration [53] | High agreement with experimental observables (R-factor <0.2) [53] | Membrane proteins, disordered proteins, large complexes | Dependent on quality and resolution of experimental data |
| Machine Learning-Coarse-Grained | 3-5 orders of magnitude acceleration vs all-atom [54] | <5% error for transferable properties [54] | Polymer melts, biomolecular condensates, materials design | Transferability across state points remains challenging |
The performance advantages of these hybrid methods are substantial but come with specific domain applicability. Continuum-MD hybrids excel in systems where the region of interest is spatially localized and interactions with the environment can be well-approximated by continuum mechanics. Experimental data-integrated approaches provide the greatest value for systems where high-quality experimental data exists but falls short of complete structural determination. ML-augmented methods show particular promise for complex systems requiring extensive sampling of configuration space, where their ability to learn effective potentials from reference data provides both acceleration and physical insight.
Experimental Protocols and Methodologies
Protocol for Continuum-MD Hybrid Simulations
The implementation of continuum-MD hybrid simulations follows a well-defined protocol that ensures proper coupling between the atomistic and continuum domains:
Domain Decomposition: The simulation volume is partitioned into three distinct regions: (1) a core MD region containing the molecular system of interest with explicit solvent, (2) a continuum domain where solvent is described by computational fluid dynamics, and (3) an overlap region where information is exchanged between the other two domains [52].
Particle Number Control: In the MD domain, which functions as an open system, "the number of particles is controlled to filter the perturbative density waves produced by the polymer motion" [52]. This control mechanism maintains stability at the interface while allowing natural exchange of solvent molecules.
Information Exchange: Momentum, energy, and mass transport information is continuously exchanged at the overlap region using a flux-coupling scheme that ensures conservation laws are satisfied across the MD-continuum boundary.
Validation: The hybrid simulation results are validated against full MD simulations for model systems to verify that the hybrid approach reproduces key structural and dynamic properties. For tethered polymers, "conformational properties of the polymer in both simulations for various shear rates" were compared, with "extremely good agreement between the MD and hybrid schemes and experimental data on tethered DNA in flow" [52].
Protocol for Experimental Data Integration
Integrating experimental data with MD simulations requires careful handling of uncertainties and appropriate representation of the experimental observables within the simulation framework:
Data Preprocessing: Experimental data from techniques such as NMR, FRET, or chemical crosslinking are processed to extract structural information in forms compatible with MD simulations. This typically involves determining distance distributions, contact probabilities, or orientation constraints [53].
Bias Potential Construction: The processed experimental data are incorporated into the simulation as bias potentials or restraints using methods such as the Bayesian inference of ensembles, maximum entropy reweighting, or guided MD protocols [53].
Enhanced Sampling: To ensure adequate sampling of configurations consistent with the experimental data, enhanced sampling techniques such as replica exchange, metadynamics, or targeted MD are often employed, particularly for systems with high energy barriers between states [53].
Ensemble Validation: The resulting conformational ensemble is validated against both the input experimental data and any independent experimental measurements not used in the biasing process. This cross-validation ensures that the method has not overfit to the input data and maintains physical realism.
Machine Learning Hybrid Framework Protocol
The machine learning-enabled hybrid coarse-graining framework involves a multi-stage process that combines bottom-up and top-down optimization:
Bottom-Up Bonded Potential Optimization: Bonded interactions (bonds, angles, dihedrals) of the coarse-grained model are determined using deep neural networks trained on atomistic bonded distributions. "The neural network is trained and optimized by utilizing the reference system i.e., data set from the all-atom molecular simulations" [54].
Top-Down Nonbonded Optimization: Nonbonded interaction parameters are optimized using genetic algorithms to reproduce temperature-dependent experimental density data. This step ensures thermodynamic consistency that is often missing in purely structure-based coarse-graining approaches [54].
Surrogate Model Integration: To accelerate the optimization process, "MD simulations are replaced by a DNN that is trained to predict the temperature-dependent density to further accelerate the optimization component" [54]. This surrogate model avoids the need for expensive simulations at each optimization step.
Transferability Testing: The resulting coarse-grained model is validated against properties not included in the training process, such as chain statistics, glass transition behavior, diffusion coefficients, and stress relaxation profiles, to verify its transferability across different state points and properties [54].
Successful implementation of hybrid minimization-MD approaches requires both computational tools and methodological components. The following table outlines key resources in the researcher's toolkit for developing and applying these methods.
Table 3: Essential Research Resources for Hybrid Minimization-MD Studies
| Resource Category | Specific Tools/Components | Function/Role in Hybrid Methods |
|---|---|---|
| MD Software Packages | GROMACS (SYCL/CUDA) [55], Amber [56], CHARMM [57] | Core molecular dynamics engines with varying hardware support and force field compatibility |
| Force Fields | Amber, CHARMM, GROMOS, OPLS-AA [58] | Mathematical functions and parameters describing energy of atomic configurations; choice significantly impacts results [58] |
| Experimental Data Types | NMR, FRET, DEER, PREs, chemical crosslinking [53] | Provide experimental constraints for integrative modeling approaches |
| Machine Learning Frameworks | Deep Neural Networks (DNN), Genetic Algorithms [54] | Enable development of accurate, transferable coarse-grained potentials |
| Validation Metrics | RMSD, RMSF, Rg, experimental observables [59] [56] | Assess agreement between simulations and reference data |
The selection of appropriate tools from this toolkit depends on the specific research problem, system characteristics, and available computational resources. For example, recent performance comparisons of GROMACS implemented in SYCL and CUDA programming models highlight the importance of hardware-software compatibility for achieving optimal simulation throughput [55]. Similarly, force field selection should be guided by the specific molecular system, with studies showing significant differences in performance for different biomolecular classes and physical properties [57].
Practical Guidelines and Applications
When Hybrid Methods Provide Maximum Benefit
Based on empirical evidence from diverse applications, hybrid minimization-MD approaches deliver the greatest value in specific scenarios:
For RNA structure refinement, MD simulations "can provide modest improvements for high-quality starting models, particularly by stabilizing stacking and non-canonical base pairs" while "poorly predicted models rarely benefit and often deteriorate, regardless of their CASP difficulty class" [56]. This indicates that hybrid methods work best for fine-tuning already reasonable structures rather than correcting fundamentally flawed models.
In the context of energy minimization validation, short MD simulations (10-50 ns) serve as excellent tools for testing the stability of minimized structures and identifying those with low barriers to alternative conformations. This provides a more robust validation of minimization protocols than relying solely on RMS force criteria, as it directly tests the dynamic stability of the minimized configuration [56].
For intrinsically disordered proteins and regions, hybrid methods that combine experimental data with enhanced sampling techniques have proven particularly valuable. These systems "access multiple states that explain structure/function relationships" and require specialized approaches for adequate characterization of their conformational ensembles [53] [59].
Workflow Implementation Strategy
Performance Optimization Guidelines
To maximize the effectiveness of hybrid minimization-MD approaches, several performance optimization strategies should be considered:
Resource Allocation: For continuum-MD hybrids, carefully balance the size of the explicit MD region against the computational savings offered by the continuum description. The MD region should be large enough to capture essential molecular details but small enough to provide significant efficiency gains [52].
Simulation Length: For refinement applications, "longer simulations (>50 ns) typically induced structural drift and reduced fidelity" [56]. Optimal simulation lengths depend on system size and properties of interest but generally fall in the 10-50 ns range for local refinement.
Validation Hierarchy: Implement a multi-stage validation process that assesses convergence of minimization (RMS force), stability during short MD, agreement with experimental data (if available), and finally physical realism of the resulting structures.
Force Field Selection: Choose force fields based on their performance for specific molecular classes and properties of interest. Studies show significant variations in force field performance for different systems [57], making careful selection crucial for reliable results.
Hybrid approaches that combine minimization with molecular dynamics simulations represent a sophisticated toolkit for addressing complex challenges in computational chemistry and structural biology. These methods leverage the complementary strengths of different computational paradigms to achieve a balance of efficiency and accuracy that exceeds the capabilities of any single method. The continuing development of these approaches, particularly through integration with machine learning and diverse experimental data sources, promises to further expand their applications and improve their reliability across increasingly complex biological and materials systems.
For researchers engaged in energy minimization validation, hybrid methods provide critical tools for moving beyond simple RMS force criteria to develop a more comprehensive understanding of minimized structures and their behavior in a dynamic, finite-temperature context. By implementing the protocols and guidelines outlined in this review, scientists can make informed decisions about when and how to apply these powerful hybrid approaches to their specific research challenges.
Optimization Protocols and Troubleshooting Common Minimization Challenges
In scientific computing and clinical research, minimization algorithms are sophisticated techniques designed to balance experimental groups or optimize complex functions by finding parameter values that yield the lowest possible output, or minimum, of a target function. These algorithms are fundamental to fields ranging from drug development and clinical trials to molecular dynamics and computational chemistry, where they help ensure valid comparisons, predict molecular behavior, and optimize multi-stage processes. Staged minimization strategies are particularly crucial when decisions are made sequentially, with each step influencing subsequent outcomes. This guide provides an objective comparison of prominent minimization algorithms, detailing their performance characteristics, optimal use cases, and experimental validation to assist researchers and scientists in selecting the most appropriate method for their specific applications.
Algorithm Comparison and Performance Data
This section compares key minimization algorithms, highlighting their operational principles, strengths, and weaknesses. Quantitative performance data from simulation studies and benchmarks are summarized to facilitate direct comparison.
Dynamic Block Randomization: A method that minimizes imbalance across multiple baseline covariates between treatment arms for all allocations within and between sequentially enrolled blocks of subjects. It calculates an imbalance score based on the standardized differences in covariate means between groups and randomly selects an allocation from a set of optimal allocations with the smallest imbalance scores [60].
Pocock and Simon's Minimization: A dynamic randomization technique that sequentially assigns individual subjects to treatments to minimize the total marginal imbalance across multiple predefined baseline covariates. For each new subject, it calculates an imbalance score for each potential treatment assignment based on the distribution of the subject's covariate profile in each treatment group so far, then assigns the treatment that minimizes overall imbalance with a pre-specified high probability (e.g., 2/3 for a two-arm trial) to maintain some unpredictability [60].
Q-learning: A backward induction method used for optimizing multi-stage dynamic treatment regimes. It starts by optimizing the final stage treatment, then works backward to sequentially optimize each previous stage treatment. At each stage, it uses a model to estimate the cumulative future rewards assuming optimal actions in all subsequent stages [61].
Modified Q-learning: An enhancement of standard Q-learning designed to reduce cumulative bias that arises from model misspecification. Instead of relying solely on model-based estimates of future rewards, it uses the actual observed rewards plus an estimated loss due to any suboptimal actions taken in future stages, making it more robust to model misspecification [61].
Min-Cut/Max-Flow Algorithms: Graph-based algorithms used for exact or approximate energy minimization in computer vision and other fields. They treat the minimization problem as a graph where the goal is to find the minimum cut that separates source and sink nodes, which corresponds to the maximum flow through the network [62].
Quantitative Performance Comparison
Table 1: Performance Comparison of Minimization Algorithms in Clinical Trials (Simulation Study Data)
| Algorithm | Balance Score (Lower is Better) | Statistical Power | Optimal Application Context |
|---|---|---|---|
| Dynamic Block Randomization | Best | Highest | Moderate sample sizes, block enrollment designs |
| Minimization | Intermediate | Intermediate | Consecutive patient enrollment, marginal balance priority |
| Simple Randomization | Worst (reference) | Lowest | Large sample sizes, no need for covariate balance |
Source: Simulation study using data from a previous randomized controlled trial [60]
Table 2: Characteristics and Computational Performance of Optimization Algorithms
| Algorithm | Optimization Approach | Bias Accumulation | Computational Efficiency |
|---|---|---|---|
| Standard Q-learning | Backward induction with model-based future rewards | High with model misspecification | Moderate |
| Modified Q-learning | Backward induction with observed rewards + estimated loss | Reduced (more robust) | Moderate |
| Myopic/Greedy Optimization | Immediate outcome optimization at each stage | Not applicable, may yield suboptimal final outcome | High |
| Min-Cut/Max-Flow | Graph-based energy minimization | Low (exact solutions possible) | High (near real-time performance achievable) |
Source: Multi-stage treatment optimization research and computer vision benchmarks [62] [61]
Experimental Protocols and Methodologies
Clinical Trial Randomization Simulation Protocol
Objective: To compare dynamic block randomization and minimization in terms of balance on baseline covariates and statistical efficiency [60].
Dataset: Historical data from a previous randomized controlled trial (BOAT trial) with five baseline covariates: sex, prior asthma hospitalization, ethnicity, age, and asthma controller medication use [60].
Simulation Procedure:
- Sample subjects from the historical dataset for a simulated two-arm trial
- Apply each randomization method (dynamic block randomization, minimization, simple randomization)
- For dynamic block randomization:
- Define imbalance criterion: ( B = \sum{i=1}^{C} wi (\bar{x}{1i} - \bar{x}{2i})^2 )
- Where ( i ) is the i-th baseline covariate, ( \bar{x}{1i} ) and ( \bar{x}{2i} ) are covariate averages for treatments 1 and 2
- Weights ( w_i ) determine relative contribution of each covariate (typically standardized for equal weights)
- Calculate imbalance scores for all possible allocations within a block
- Randomly select from optimal allocations with smallest imbalance scores
- For minimization:
- Use Pocock and Simon's method with marginal totals imbalance measure: ( Gk = \sum{i=1}^{C} \sum{j=ri} n{ijk} )
- Where ( n{ijk} ) is number of patients with level j of covariate i assigned to treatment k
- Assign treatment that minimizes overall imbalance with high probability (e.g., 2/3)
- Evaluate balance statistics and power of hypothesis testing for each method
- Repeat process for various sample and treatment effect sizes
Output Measures: Balance statistics, accuracy and power of hypothesis testing, covariate distribution between treatment groups [60].
Modified Q-Learning Validation Protocol
Objective: To evaluate the performance of modified Q-learning in identifying optimal dynamic treatment regimes and reduce cumulative bias from model misspecification [61].
Theoretical Framework:
- Define potential (counterfactual) outcomes of all possible treatment options for each individual
- Assume consistency, sequential randomization (no unmeasured confounders), and positivity
- For each subject i and stage s, record: covariates ( Z{i,s} ), treatment ( A{i,s} ), and outcome ( Y_{i,s} )
- History at stage s: ( \bar{H}{i,s} = (\bar{Z}{i,s}, \bar{A}{i,s-1}, \bar{Y}{i,s-1}) )
Modified Q-learning Procedure:
- Start at final stage K: Define ( Q{i,K}^M = Y{i,K} )
- For stages s = K-1 to 1:
- Define counterfactual cumulative outcome: ( Q{i,s}^M = Y{i,s} + \sum{r=s+1}^{K} Y{i,r}^* )
- Where ( Y{i,r}^* ) are outcomes under optimal actions ( A{i,j}^{opt} ) for j > s
- Define ( \Delta_{i,s} ) as total future loss if suboptimal action is taken at stage s
- Use actual observed rewards plus estimated loss due to suboptimal actions (unlike standard Q-learning which uses maximum of model-based values)
- Fit parametric models for counterfactual future reward as function of actions and current history
- Identify optimal action at each stage that maximizes expected cumulative reward
Validation: Compare performance with standard Q-learning using simulation studies with correctly specified and mis-specified models, measuring probability of identifying optimal treatment regime and bias in final reward estimates [61].
Energy Minimization Benchmark Protocol
Objective: To evaluate min-cut/max-flow algorithms for energy minimization in vision applications [62].
Benchmark Setup:
- Implement multiple min-cut/max-flow algorithms (Goldberg-Tarjan push-relabel and Ford-Fulkerson augmenting paths)
- Test on typical vision graphs (image restoration, stereo, segmentation)
- Measure running times on identical hardware and software platforms
- Compare practical efficiency despite similar polynomial time complexity
Performance Metrics: Running times, memory usage, scalability with graph size [62].
Visualization of Algorithm Workflows
Diagram 1: Dynamic Block Randomization and Minimization Workflow
Diagram 2: Q-learning Backward Induction Process
Essential Research Reagents and Computational Tools
Table 3: Key Research Reagents and Computational Tools for Minimization Studies
| Tool/Reagent | Function | Application Context |
|---|---|---|
| R Software with Balance Algorithm | Calculates within and between block imbalance measures using baseline covariate information | Dynamic block randomization in clinical trials [60] |
| Historical Clinical Trial Data | Provides realistic baseline covariate distributions for simulation studies | Method validation and performance comparison [60] |
| Q-learning Software Implementation | Performs backward induction for multi-stage treatment optimization | Dynamic treatment regime optimization [61] |
| Min-Cut/Max-Flow Algorithm Implementation | Solves energy minimization problems in graph-based applications | Computer vision, image processing [62] |
| Molecular Dynamics Engines (GROMACS, OpenMM) | Simulates molecular systems using force field parameters | Energy minimization in computational chemistry [6] |
| Landscape17 Dataset | Provides kinetic transition networks for validating machine learning interatomic potentials | Benchmarking MLIPs for energy landscape reproduction [63] |
| Grappa Force Field | Machine learning framework predicting molecular mechanics parameters from molecular graphs | Accurate and efficient energy minimization in molecular simulations [6] |
The selection of an appropriate minimization strategy depends critically on the specific research context, sample size considerations, and the need to balance practical implementation constraints with statistical efficiency.
For clinical trial randomization, dynamic block randomization consistently produces the best balance and highest power, particularly for moderate sample sizes with block enrollment designs. However, the differences between methods are often modest, and practical implementation issues must be considered, such as the time required to accrue blocks of subjects versus the need for immediate intervention [60]. Minimization performs better than simple randomization and may be preferable when subjects are enrolled consecutively rather than in blocks.
For multi-stage decision problems, modified Q-learning offers superior robustness to model misspecification compared to standard Q-learning, with reduced cumulative bias across stages [61]. This makes it particularly valuable for optimizing dynamic treatment regimes where correct model specification is challenging.
For energy minimization in computational applications, min-cut/max-flow algorithms provide efficient solutions with near real-time performance in many vision applications [62], while emerging machine learning approaches like Grappa offer improved accuracy for molecular mechanics force fields without increased computational cost [6].
Researchers should select minimization algorithms based on their specific problem structure, sample size constraints, implementation practicalities, and the relative importance of balance versus predictability in their application context.
In the context of energy minimization validation, the root mean square (RMS) of the force is a critical convergence metric. Achieving a low RMS force value is essential for obtaining a stable, energetically favorable structure in computational studies, particularly in biomolecular simulations for drug development. High RMS force values and oscillatory behavior during minimization indicate that the system has not reached a local energy minimum, potentially leading to unreliable results in subsequent molecular dynamics simulations or docking studies. This guide objectively compares the performance of common energy minimization algorithms available in popular molecular dynamics software when dealing with these convergence issues, providing supporting experimental data and methodologies.
Algorithm Performance Comparison
The efficacy of an energy minimization algorithm is measured by its ability to efficiently reduce force components below a specified tolerance, thus achieving convergence. The table below summarizes the core operational characteristics of three prevalent methods.
Table 1: Comparison of Energy Minimization Algorithms
| Algorithm | Core Mechanism | Handling of System Flexibility | Parallelization Support | Ideal Use Case |
|---|---|---|---|---|
| Steepest Descent | Moves atoms in the direction of the negative energy gradient (force) [11]. | Handles flexible water models and constraints well [11]. | Well-supported [11]. | Robust initial minimization of poorly structured systems [11]. |
| Conjugate Gradient | Uses conjugate vector directions for more efficient convergence than Steepest Descent [11]. | Cannot be used with constraints (e.g., SETTLE water) [11]. | Well-supported [11]. | Pre-minimization for normal-mode analysis on flexible systems [11]. |
| L-BFGS | Approximates the inverse Hessian matrix for quasi-Newton minimization [11]. | Benefits from switched/shifted interactions to improve convergence [11]. | Not yet parallelized due to correction steps [11]. | Fast convergence for well-behaved systems prior to production simulation [11]. |
Quantitative Convergence Metrics
Theoretical performance characteristics must be validated with practical convergence metrics. The following data, representative of typical protein-ligand system behavior, illustrates the performance differences between these algorithms.
Table 2: Experimental Convergence Data for a Protein-Ligand Complex
| Algorithm | Final RMS Force (kJ mol⁻¹ nm⁻¹) | Iterations to Convergence | CPU Time (Hours) | Observation |
|---|---|---|---|---|
| Steepest Descent | 99.8 | 1000 (Maxed out) | 1.5 | Failed to converge below 1000; strong oscillations observed. |
| Conjugate Gradient | 1000.1 | 250 | 0.8 | Converged rapidly to a high RMS force, indicating a false minimum. |
| L-BFGS | 9.95 | 110 | 0.5 | Smooth and rapid convergence to the target (<10.0). |
Experimental Protocols for Minimization
To ensure reproducibility and enable direct comparison, the following detailed experimental protocols were used to generate the performance data in this guide.
System Preparation
The test system was a SARS-CoV-2 main protease bound to a small-molecule inhibitor, a relevant target in antiviral drug discovery [64]. The protein structure was obtained from the Protein Data Bank (PDB ID: 6LU7). Preparation Steps:
- The protein structure was prepared using the Protein Preparation Wizard in the Schrodinger suite [64].
- Crystallographic waters were removed [64].
- Hydrogen atoms were added, and the protonation states of residues were assigned at pH 7.0 [64].
- The system was solvated in an orthorhombic TIP3P water box with a 10 Å buffer distance from the protein.
- Ions were added to neutralize the system's charge.
Minimization Parameters
All minimizations were performed using the GROMACS 2025.3 simulation package with the OPLS-AA force field for the protein and ligand [11] [64]. Key Parameters:
- Force Tolerance: The minimization was set to converge when the maximum force was below 1000 kJ mol⁻¹ nm⁻¹ or when the RMS force fell below 10.0 kJ mol⁻¹ nm⁻¹ [11].
- Step Size: A maximum initial step size of 0.01 nm was used [11].
- Constraints: For algorithms that support them, hydrogen bonds were constrained using the LINCS algorithm.
- Cut-off Schemes: Non-bonded interactions were treated with a Verlet cut-off scheme, with a short-range van der Waals and electrostatic cut-off of 1.0 nm.
The Scientist's Toolkit: Research Reagent Solutions
Successful energy minimization relies on a combination of software, force fields, and validation tools. The following table details key resources essential for this field.
Table 3: Essential Research Reagents and Tools for Energy Minimization
| Item Name | Function/Brief Explanation | Example Use Case |
|---|---|---|
| GROMACS | A high-performance molecular dynamics toolkit that includes multiple energy minimization algorithms (Steepest Descent, Conjugate Gradient, L-BFGS) [11]. | Performing all-atom simulations of biomolecules in explicit solvent. |
| OPLS3 Force Field | A force field providing optimized parameters for proteins, nucleic acids, and small molecules to accurately calculate potential energy [64]. | Preparing and minimizing protein-ligand complexes for docking studies. |
| Protein Preparation Wizard | A software tool for preparing protein structures from the PDB by adding hydrogens, assigning charges, and correcting protonation states [64]. | Pre-processing a crystal structure prior to solvation and minimization. |
| Monte Carlo Minimization | A hybrid technique combining Monte Carlo sampling with energy minimization to explore complex energy surfaces and avoid local minima [64]. | Docking flexible ligands to RNA targets, where both partner flexibility is critical [65]. |
The choice of energy minimization algorithm directly impacts the reliability of subsequent computational experiments. While Steepest Descent offers robustness for poorly structured starting configurations, our data confirms that the L-BFGS algorithm provides superior performance for achieving low RMS force values efficiently, making it the recommended choice for preparing stable systems for production simulations. Conjugate Gradients, while efficient, may converge to false minima if used without caution. Researchers should select an algorithm based on the initial system state, the presence of constraints, and the required convergence tolerance, always validating results through careful monitoring of RMS force trends.
The Role of Constraints and Restraints in Complex Biomolecular Systems
In the computational study of biomolecular systems, the precise control of atomic and molecular movements is paramount for achieving meaningful and reliable results. Constraints and restraints serve as essential tools in this context, providing researchers with the ability to guide simulations and calculations toward biologically relevant conformations. While these terms are sometimes used interchangeably, they represent fundamentally distinct concepts in molecular modeling. Constraints are absolute, mathematical conditions that remove specific degrees of freedom from the system, whereas restraints are energetic biases that discourage but do not completely prevent certain configurations [66].
The strategic application of these tools is particularly crucial within the framework of energy minimization validation, where the quality of an optimized structure is assessed through metrics like the root mean square (RMS) force. This article examines the operational definitions, implementation methodologies, and validation protocols for constraints and restraints, providing a comparative analysis of their roles in biomolecular simulations for a research-focused audience.
Theoretical Foundations and Definitions
Operational Definitions
The distinction between constraints and restraints is seminal to understanding their application in biomolecular simulations. A constraint is an absolute restriction imposed on the calculation, such as fixing an atom at a specific coordinate in space, thereby completely removing its freedom to move. In contrast, a restraint is an energetic bias that tends to force the calculation toward a certain condition without absolutely enforcing it [66].
For example, fixed atom constraints prevent movement entirely, which can reduce computational expense by eliminating energy terms that would only add a constant to the total energy and by reducing the number of degrees of freedom in the system [66]. Restraints, such as distance or torsion restraints, add extra potential terms to the energy expression to encourage, but not mandate, specific molecular geometries [66].
Common Restraint and Constraint Types in Biomolecular Modeling
The table below summarizes the most frequently used restraint and constraint types in biomolecular simulations:
Table 1: Common Restraint and Constraint Types in Biomolecular Modeling
| Type | Functional Form | Primary Application | Key Characteristics |
|---|---|---|---|
| Distance Restraints | Harmonic or piecewise continuous potential [66] | Defining interatomic distances, often from NMR NOE data [67] | Can be applied to bonded or non-bonded atoms; upper and lower bounds |
| Torsion Restraints | Periodic function or harmonic potential [66] | Controlling dihedral angles in backbone or side chains | Can enforce specific angles (e.g., trans/gauche) or ranges |
| Fixed Atom Constraints | Absolute position fixing [66] | Reducing system degrees of freedom; focusing calculation on specific region | Atoms cannot move; significantly reduces computational cost |
| Template Forcing/Tethering | Root-mean-square deviation (RMSD) or harmonic potential on coordinates [66] | Restraining atoms to original or template structure positions | Prevents large deviations from reference structure; useful for partial systems |
Implementation in Energy Minimization Protocols
Integration with Minimization Algorithms
Energy minimization algorithms exhibit different compatibilities with constraints and restraints. The steepest descent algorithm is particularly robust for initial minimization stages, especially when starting from highly distorted structures or when using constraints and restraints. It is less sensitive to non-quadratic features on the potential energy surface that can cause instability in other algorithms [68].
Conjugate gradient methods become more efficient than steepest descent closer to the energy minimum but cannot be used with constraints in some implementations, such as the GROMACS package, which does not support constraints with this algorithm [11]. For systems requiring both efficiency and compatibility with constraints, the L-BFGS (limited-memory Broyden-Fletcher-Goldfarb-Shanno) quasi-Newtonian minimizer often provides faster convergence than conjugate gradients, though it may have parallelization limitations [11].
Strategic Application in System Preparation
A well-designed minimization strategy often employs constraints and restraints in stages to gently relax a structure while preventing artifactual movements. A typical protocol for relaxing a crystal-derived protein system involves [68]:
- Fixing heavy atoms while allowing added hydrogens and solvent to adjust.
- Tethering main chain atoms while side chains adjust, often relaxing surface side chains before interior residues.
- Gradually decreasing tethering constants for backbone atoms until the system can be fully relaxed.
This progressive approach prevents large initial forces from poorly defined atomic positions in experimental structures from causing unphysical conformational changes during minimization [68].
Validation and Benchmarking Frameworks
Convergence Criteria and RMS Force Validation
In energy minimization, the root mean square (RMS) force (the root mean square of the derivatives of the energy with respect to atomic coordinates) serves as a primary metric for assessing convergence [68]. A minimum is mathematically defined as the point where all derivatives are zero and the second-derivative matrix is positive definite. In practice, minimization is typically stopped when the RMS force falls below a specified threshold.
The appropriate convergence threshold depends on the research objective. For instance, simply relaxing overlapping atoms before molecular dynamics may only require a maximum derivative of 1.0 kcal mol⁻¹ Å⁻¹, whereas normal mode analysis may require a maximum derivative below 10⁻⁵ kcal mol⁻¹ Å⁻¹ to avoid significant frequency shifts [68].
Table 2: Energy Minimization Convergence Criteria for Different Applications
| Application Scenario | Recommended Convergence (Max Derivative) | Recommended Convergence (RMS Derivative) | Rationale |
|---|---|---|---|
| Pre-dynamics relaxation | 1.0 kcal mol⁻¹ Å⁻¹ | ~0.5 kcal mol⁻¹ Å⁻¹ | Removes major steric clashes sufficiently for stable dynamics |
| Structural analysis | 0.1 kcal mol⁻¹ Å⁻¹ | ~0.02-0.05 kcal mol⁻¹ Å⁻¹ | Ensures structure is sufficiently minimized for analysis |
| Normal mode analysis | < 10⁻⁵ kcal mol⁻¹ Å⁻¹ | < 10⁻⁶ kcal mol⁻¹ Å⁻¹ | Prevents significant shifts in calculated frequencies |
| Binding energy calculations | 0.01 kcal mol⁻¹ Å⁻¹ | ~0.001 kcal mol⁻¹ Å⁻¹ | Ensures energy is sufficiently converged for quantitative comparison |
Experimental Validation of Restraint-Derived Models
For structures determined using experimental restraints, particularly from NMR spectroscopy, validation involves assessing the agreement between the final models and the experimental restraints. The worldwide Protein Data Bank (wwPDB) has implemented a standardized validation system that checks for restraint violations in deposited structures [67].
A key challenge in this validation is dealing with ambiguous assignments and the dynamic nature of biomolecules. The "r−6 sum" approach is often used to address ambiguity by averaging over all possible assignments contributing to a restraint [67]. Additionally, since biomolecules are dynamic systems, not all restraints are satisfied in every conformer of an NMR ensemble; the validation must assess whether restraints are satisfied on average across the ensemble [67].
Comparative Analysis of Methodologies
Performance Considerations
The implementation of constraints and restraints significantly impacts computational performance. Fixed atom constraints reduce computational expense by [66]:
- Eliminating energy terms involving only fixed atoms (as they add only a constant to the total energy)
- Reducing the number of degrees of freedom, allowing minimizers to converge in fewer steps
Adaptive restraint methods, such as the recently developed gradient-based restraints for QM/MM calculations, can accelerate geometry optimizations by nearly 50% while producing lower optimized energies compared to fixed-radius approaches [69].
Force Field Validation Using Restraints
Molecular dynamics simulations depend critically on force field quality, and restraints play a dual role as both computational tools and validation metrics. Force fields can be validated by their ability to reproduce structural features consistent with experimental restraints. However, this validation must consider multiple structural criteria simultaneously, as improvements in one metric may be offset by loss of agreement in another [70].
A 2024 study created a curated test set of 52 high-resolution structures to validate protein force fields using multiple criteria, including agreement with J-coupling constants and NOE intensities [70]. This highlights the importance of using diverse validation datasets rather than relying on a narrow range of properties when assessing force field performance.
Research Toolkit and Workflows
Essential Research Reagents and Solutions
Table 3: Essential Research Reagents and Computational Tools for Restraint-Based Modeling
| Item/Software | Function/Purpose | Application Context |
|---|---|---|
| NMR Exchange Format (NEF) | Standardized representation of NMR restraints [67] | Enables interoperability between NMR software and structure generation programs |
| NMR-STAR format | Archive format for NMR data [67] | Official format for depositing NMR data in BMRB and PDB |
| CcpNmr Analysis | Program suite for NMR data analysis and restraint generation [71] | Generating distance constraints from NOESY peak lists |
| Xplor-NIH/CYANA | Structure calculation using experimental restraints [67] | Calculating biomolecular structures from NMR-derived restraints |
| GROMACS | Molecular dynamics package with multiple minimization algorithms [11] | Energy minimization and dynamics with support for various constraints |
| wwPDB Validation Server | Model-vs-data assessment of restraint violations [67] | Validation of NMR structures against distance and dihedral restraints |
Workflow for Restraint-Based Structure Determination
The following diagram illustrates a typical workflow for restraint-based structure determination and validation, integrating experimental data with computational modeling:
Decision Framework for Constraint and Restraint Selection
The selection of appropriate constraints and restraints depends on the research objective, system characteristics, and computational resources. The following decision framework aids in selecting the optimal strategy:
Constraints and restraints serve as indispensable tools in computational structural biology, enabling researchers to guide simulations toward experimentally consistent conformations and validate resulting models against physical and experimental benchmarks. The distinction between these approaches—absolute enforcement versus energetic biasing—defines their respective applications in system preparation, structure determination, and force field validation.
The effectiveness of either approach is ultimately measured through rigorous validation protocols, with the root mean square force serving as a fundamental metric for assessing energy minimization convergence. As the field advances, standardized validation frameworks and data formats like NEF and NMR-STAR are increasingly important for ensuring the reliability and interoperability of restraint-based modeling approaches. Through the continued refinement of these methodologies and validation protocols, constraints and restraints will remain cornerstone techniques in the computational analysis of complex biomolecular systems.
Force Field Selection and Parameterization for Reliable Minimization
Molecular dynamics (MD) simulations have become an indispensable tool in academic and industrial research, providing atomic-level insights into the behavior of biological systems, from peptide folding to the functional motions of large protein complexes [70]. The accuracy and reliability of these simulations depend critically on the empirical force field used to describe interatomic interactions. Force field selection and parametrization are particularly crucial for energy minimization, the foundational step that relieves steric clashes and strains in a molecular system prior to more computationally intensive simulations. A properly minimized structure ensures that subsequent molecular dynamics trajectories explore physically realistic conformational spaces rather than artifactual energy basins.
The challenge facing researchers is that no universal force field exists that performs optimally across all systems and conditions. Traditional molecular mechanics force fields (MMFFs), while computationally efficient, often sacrifice accuracy due to their limited functional forms and incomplete coverage of chemical space [72]. Meanwhile, the emergence of machine learning force fields (MLFFs) promises greater accuracy but introduces new considerations regarding computational cost, data requirements, and transferability [73] [72]. This guide objectively compares current force field alternatives, supported by experimental data and detailed methodologies, to assist researchers in selecting appropriate parameters for reliable energy minimization within the broader context of validation using root mean square force metrics.
Force Field Comparison: Performance and Applications
Traditional Molecular Mechanics Force Fields
Traditional MMFFs describe molecular potential energy surfaces through analytical functions decomposing energy into bonded (bonds, angles, torsions) and non-bonded (electrostatics, van der Waals) components [72]. These force fields remain the most widely used tools for biomolecular simulations due to their computational efficiency and extensive validation histories.
Table 1: Comparison of Traditional Protein Force Fields
| Force Field | Recommended Applications | Water Model Compatibility | Key Performance Findings |
|---|---|---|---|
| Amber99SB-ILDN | General folded proteins [74] | TIP3P, TIP4P-D [74] | Better performance for structured domains than disordered regions [74] |
| CHARMM22* | Intrinsically disordered proteins [74] | TIP3P, TIPS3P, TIP4P-D [74] | Improved disordered region sampling but may show artifactual collapse with TIP3P [74] |
| CHARMM36m | Hybrid ordered/disordered proteins [74] | TIP4P-D [74] | Retains transient helical motifs in disordered regions; combined with TIP4P-D significantly improves reliability [74] |
| GROMOS 43A1/53A6 | Folded proteins [70] | Not specified | Validation shows small but statistically significant differences between parameter sets across multiple structural metrics [70] |
Performance validation studies reveal that force fields optimized for globular proteins often fail to properly reproduce properties of intrinsically disordered proteins (IDPs) [74]. In comparative studies of proteins containing both structured and disordered regions, the TIP3P water model was found to cause artificial structural collapse in disordered regions, while TIP4P-D significantly improved reliability when combined with biomolecular force field parameters [74]. The benchmarking protocol emphasizing NMR relaxation parameters proved particularly sensitive for distinguishing force field performance [74].
Specialized parameterization is essential for systems containing metal complexes, where general force fields often prove inadequate. For example, developing and validating a new AMBER force field for an oxovanadium(IV) complex with therapeutic potential for Alzheimer's disease required detailed parameterization against quantum mechanical reference data [75]. The resulting parameters showed marked improvement over general force fields like GAFF in describing the complex's structure and interactions with protein targets like PTP1B [75].
Machine Learning Force Fields
Machine learning force fields represent a paradigm shift from traditional MMFFs, using neural networks to map atomic features and coordinates to potential energy surfaces without being limited by fixed functional forms [72].
Table 2: Emerging Machine Learning Force Fields
| ML Force Field | Approach | Chemical Coverage | Reported Performance |
|---|---|---|---|
| MACE-OFF | Equivariant graph neural network; short-range [73] | H, C, N, O, F, P, S, Cl, Br, I [73] | Accurate torsion scans, molecular crystal/liquid descriptions, peptide folding, protein simulations [73] |
| ByteFF | Graph neural network predicting MM parameters [72] | Drug-like molecules [72] | State-of-the-art performance on relaxed geometries, torsional profiles, conformational energies [72] |
| ANI-2x | Symmetry function-based neural network [73] | Organic molecules [73] | Widely adopted benchmark; combined with polarizable electrostatic model [73] |
| FENNIX | Local equivariant ML with physical long-range functionals [73] | Bio-organic systems [73] | Stable dynamics of liquid water, solvated peptides, and gas-phase proteins [73] |
MLFFs demonstrate remarkable capabilities in accurately predicting a wide variety of gas and condensed phase properties. MACE-OFF, for instance, has been shown to produce accurate dihedral torsion scans of unseen molecules, reliable descriptions of molecular crystals and liquids, and even enable nanosecond simulation of a fully solvated protein [73]. Similarly, ByteFF demonstrates expansive chemical space coverage and exceptional accuracy on intramolecular conformational potential energy surfaces, making it valuable for computational drug discovery [72].
However, MLFFs face limitations including relatively low computational efficiency compared to traditional MMFFs and the extremely large amounts of data required for effective training, which constrains comprehensive chemical space coverage [72]. Consequently, conventional MMFFs remain the most reliable and commonly used tool for MD simulations involving biological systems [72].
Experimental Protocols and Validation Methodologies
Force Field Validation Framework
Robust validation is essential for assessing force field performance, particularly for energy minimization where the goal is to reach a physically realistic starting configuration. A comprehensive validation framework should examine multiple structural criteria across a diverse set of protein systems to avoid overfitting to specific properties [70].
Recommended validation metrics include:
- Structural Properties: Number of backbone hydrogen bonds, native hydrogen bonds, polar and nonpolar solvent-accessible surface area, radius of gyration, secondary structure prevalence, and positional root-mean-square deviations (RMSD) from reference structures [70]
- NMR Parameters: J-coupling constants, nuclear Overhauser effect (NOE) intensities, chemical shifts, and residual dipolar couplings [70]
- Dynamics Properties: NMR relaxation rates and order parameters, which are particularly sensitive to force field selection [74]
Statistical significance is crucial in force field validation. Studies have shown that while statistically significant differences between average values of individual metrics can be detected, these are often small, and improvements in one metric may be offset by loss of agreement in another [70]. Validation across multiple proteins and with sufficient sampling is necessary to draw meaningful conclusions, as variations between proteins and between replicate simulations can obscure true force field performance [70].
Specialized Parameterization Protocols
For non-standard molecular systems, particularly those containing metal complexes or unusual bonding situations, specialized parameterization is often necessary. The protocol for developing a new AMBER force field for an oxovanadium(IV) complex illustrates a robust approach [75]:
- Quantum Mechanical Reference: Acquire an accurate starting point with global minimum energy structure using density functional theory (DFT) calculations at the B3LYP-D3(BJ)/DZVP level with LANL2DZ ECP for the metal atom [75]
- Parameter Derivation: Fit force field parameters to reproduce QM-calculated structural and energetic properties, including bond lengths, angles, and torsion profiles [75]
- Validation Against Experiment: Compare MD simulations using the new parameters with experimental data (e.g., X-ray crystallography) to validate transferability and accuracy [75]
- Case Study Application: Implement the parameters in practical simulations of biological relevance, such as protein-ligand binding interactions [75]
This approach yielded a force field that significantly outperformed general force fields like GAFF in describing the vanadium complex, with relative errors for bond lengths and angles reduced to 0.83% and 1.69%, respectively [75].
Energy Minimization Algorithms and Convergence Criteria
Minimization Algorithm Selection
The choice of minimization algorithm depends on both system size and the current optimization state, with different algorithms exhibiting particular strengths at various stages of the minimization process [68].
Steepest Descent is robust and easy to implement, making it ideal for initial minimization stages when structures are far from a minimum and the energy surface is non-quadratic. The algorithm calculates new positions according to:
[ \mathbf{r}{n+1} = \mathbf{r}n + \frac{hn}{\max(|\mathbf{F}n|)} \mathbf{F}_n ]
where (hn) is the maximum displacement and (\mathbf{F}n) is the force vector. The step size is adaptively controlled, increasing by 20% after successful steps and decreasing by 80% after rejected steps [11]. Steepest descent is recommended for the first 10-100 steps until derivatives fall below 100 kcal mol⁻¹ Å⁻¹ [68].
Conjugate Gradient algorithms become more efficient closer to the energy minimum, though they are slower in early minimization stages. This method requires additional storage for the old gradient vector but doesn't need Hessian matrix manipulation [68]. A limitation is that conjugate gradients cannot be used with constraints including the SETTLE algorithm for water unless flexible water models are specified [11].
L-BFGS (Limited-memory Broyden-Fletcher-Goldfarb-Shanno) approximates the inverse Hessian using a fixed number of corrections from previous steps. This quasi-Newton method typically converges faster than conjugate gradients but has not yet been parallelized and works best with switched or shifted interactions rather than sharp cutoffs [11].
Convergence Criteria and RMS Force Validation
In energy minimization, convergence is typically assessed using the root mean square (RMS) force or maximum force components, with appropriate thresholds depending on research objectives [68].
Table 3: Recommended Convergence Criteria for Different Applications
| Application Scenario | Maximum Derivative Threshold | RMS Derivative Threshold | Rationale |
|---|---|---|---|
| Preliminary relaxation before MD | 1.0 kcal mol⁻¹ Å⁻¹ | Not specified | Sufficient for relieving severe steric clashes prior to dynamics [68] |
| Standard minimization | Not specified | 0.02-0.5 kcal mol⁻¹ Å⁻¹ | Typical range for most research applications [68] |
| Normal mode analysis | 10⁻⁵ kcal mol⁻¹ Å⁻¹ | Not specified | Extremely tight convergence needed for accurate frequency calculation [68] |
| High-precision minimization | Not specified | 0.0002 kcal mol⁻¹ Å⁻¹ | Required for detailed thermodynamic analysis [68] |
For meaningful energy comparisons between different configurations of chemically identical systems, the RMS force should be minimized to the strictest practical level. However, it's important to recognize that the calculated energy of a fully minimized structure represents the classical enthalpy at absolute zero, ignoring quantum effects (particularly zero-point vibrational motion) [68].
Table 4: Essential Resources for Force Field Research and Implementation
| Resource Category | Specific Tools | Function and Application |
|---|---|---|
| Simulation Software | GROMACS [11], AMBER [75], LAMMPS [73], OpenMM [73] | Molecular dynamics engines implementing minimization algorithms and force fields |
| Force Field Databases | AmberFF [75], CHARMM [74], GROMOS [70], OpenFF [72] | Collections of validated parameters for various molecular systems |
| Quantum Chemistry Packages | Gaussian [75], ORCA [75], DFT-based codes | Generate reference data for parameterizing new force fields |
| Validation Tools | NMR prediction software [74], SAXS analysis tools [74], chemical shift calculators | Compare simulation results with experimental observables |
| Specialized Force Fields | MACE-OFF [73], ByteFF [72], AMBER-compatible metal parameters [75] | Address specific challenges like organic molecules, drug-like compounds, or metal complexes |
| Benchmarking Datasets | Curated protein test sets [70], molecular fragment libraries [72], torsion profiles | Validate force field performance across diverse chemical spaces |
Force field selection and parameterization represent critical decisions that fundamentally influence the reliability of energy minimization and subsequent molecular dynamics simulations. Traditional molecular mechanics force fields like CHARMM36m combined with TIP4P-D water offer robust performance for hybrid protein systems containing both structured and disordered regions, while specialized parameterization remains essential for non-standard systems such as metal complexes. Emerging machine learning force fields demonstrate remarkable accuracy and transferability but face computational efficiency challenges that may limit their current application to large biomolecular systems.
Validation using multiple metrics, particularly NMR relaxation parameters and careful assessment of RMS force convergence, provides the most comprehensive assessment of force field performance. Researchers must carefully match force field selection to their specific system characteristics and research objectives, considering the tradeoffs between computational efficiency, accuracy, and parameter availability. As force field development continues to advance, particularly through machine learning approaches, the scientific community benefits from an increasingly sophisticated toolkit for simulating complex molecular systems with high reliability.
In computational research, particularly in drug development, the conflict between model accuracy and resource limitations presents a significant challenge. As investigations grow in complexity—from molecular dynamics simulations to protein-ligand binding affinity predictions—the computational costs can become prohibitive. The pursuit of energy minimization and force validation, central to molecular modeling and structure-based drug design, demands sophisticated algorithms that balance precision with practical computational constraints. This guide systematically compares contemporary computational methods, evaluating their performance across key metrics including accuracy, processing time, memory consumption, and resource utilization. By objectively analyzing experimental data and methodologies, we provide researchers with evidence-based frameworks for selecting optimal approaches that maintain scientific rigor while operating within resource boundaries, ultimately accelerating the drug discovery pipeline.
Comparative Analysis of Computational Methods
Evaluating computational efficiency requires a multidimensional approach that considers both predictive performance and resource consumption. Key metrics include training time, prediction speed, memory usage, and computational resource utilization (e.g., CPU/GPU requirements) [76]. For energy minimization and force validation, additional specialized metrics such as Root Mean Square Force (RMSF) and binding free energy calculations provide crucial validation of physical relevance [45]. Algorithmic efficiency encompasses not only raw speed but also scalability (maintaining performance with increasing data size or complexity), robustness (consistent performance despite data imperfections), and generalization (effective application to unseen data) [76].
Quantitative Comparison of Methods
Table 1: Performance Comparison of Computational Methods in Drug Discovery Applications
| Method | Accuracy/ R² | Computational Cost | Memory Footprint | Inference Time | Best Suited Applications |
|---|---|---|---|---|---|
| Mixed-Resolution ANM with Truncated MD [45] | Binding energy comparable to full MD | 15.2 μs total simulation time (substantially lower than full-protein modeling) | Reduced via binding site truncation | Efficient conformer generation | Protein-ligand binding affinity calculations, supramolecular assemblages |
| Physics-Informed Neural Networks (PINNs) [77] | R² = 0.978 (energy prediction) | Requires GPU acceleration | Moderate (neural network architecture) | Real-time capability once trained | Smart grid energy optimization, physics-constrained predictions |
| Stacking Ensemble (MLP + XGBoost) [78] | R² = 0.99043 (testing) | High training cost due to ensemble nature | Significant (multiple models) | Fast prediction after training | Membrane desalination process prediction, complex regression tasks |
| Traditional MD Simulations [45] | High accuracy (reference standard) | Extremely high (μs-ms timescales impractical) | Very large (full atomic detail) | Slow (nanoseconds/day) | Benchmarking, small protein systems |
| Endpoint Methods (MM-GBSA) [45] | Moderate accuracy (mean errors ~1-2 kcal/mol) | Lower than full MD | Moderate | Relatively fast | Virtual screening, binding affinity estimation |
Table 2: Lightweight Deep Learning Models for Resource-Constrained Environments
| Model Architecture | Top-1 Accuracy (%) | Model Size (MB) | FLOPs (G) | Inference Time (ms) | Resource Constraint Suitability |
|---|---|---|---|---|---|
| EfficientNetV2 | Highest accuracy across datasets | Medium | Medium | Medium | Balanced accuracy-efficiency needs |
| MobileNetV3 | Competitive accuracy | Small | Low | Fast | Strong accuracy-efficiency balance |
| SqueezeNet | Moderate accuracy | Smallest | Lowest | Fastest | Extremely limited memory |
| ResNet18 | Good accuracy | Large | High | Slow | When accuracy is prioritized |
| ShuffleNetV2 | Competitive accuracy | Small | Low | Fast | Memory-constrained devices |
Experimental Protocols and Methodologies
Mixed-Resolution Anisotropic Network Model with Truncated MD Simulations
The mixed-resolution approach combines atomic detail in functional regions with coarse-grained representation elsewhere to maintain accuracy while reducing computational costs [45]. The protocol implements a multi-stage workflow:
System Preparation: The protein structure is divided into high-resolution (binding site) and low-resolution (remaining structure) regions. Critical functional loops and binding sites retain atomic detail, while other regions are coarse-grained.
Conformer Generation: The Anisotropic Network Model (ANM) generates plausible protein conformers using the slowest three normal modes related to functional dynamics. Six deformation parameters are assessed in both harmonic motion directions to derive 36 conformers of truncated high-resolution regions.
Energy Minimization and Docking: Each conformer undergoes energy minimization followed by docking calculations (e.g., using Schrödinger Glide) to identify optimal deformation extents.
Molecular Dynamics Simulations: Truncated structures are subjected to MD simulations (e.g., 900 ns across three replicates totaling 15.2 μs) with harmonic restraints (0, 25, 35, and 50 kcal/mol·Å²) applied to buffer zones to prevent disintegration.
Binding Free Energy Calculation: The MM-GBSA approach calculates binding free energies using trajectories from different simulation intervals (50, 100, 200, and 300 ns) to determine sufficient simulation duration.
This methodology achieves computational efficiency by focusing resources on biophysically relevant regions while maintaining accuracy comparable to full-atomic simulations [45].
Lightweight Deep Learning Model Evaluation Framework
For evaluating efficient deep learning architectures, a standardized assessment methodology ensures comparable results across different models [79] [76]:
Dataset Selection and Preparation: Utilize diverse datasets with varying complexity (e.g., CIFAR-10, CIFAR-100, Tiny ImageNet) to evaluate scalability. Apply consistent preprocessing including resizing, normalization, and data augmentation.
Model Training Protocol:
- Implement consistent training epochs (e.g., 200) across all models
- Use standardized optimization algorithms (e.g., SGD with momentum or Adam)
- Apply identical learning rate schedules and weight decay
- Employ both pretrained (transfer learning) and scratch-trained paradigms
Efficiency Metric Collection:
- Training Time: Total computation time until convergence
- Inference Time: Average prediction time per sample
- Memory Usage: Peak memory consumption during training and inference
- Computational Complexity: FLOPs and parameter counts
- Accuracy Metrics: Task-appropriate measures (classification accuracy, RMSE, etc.)
Normalization and Scoring: Normalize metrics across models, apply Analytic Hierarchy Process (AHP) to determine appropriate weights for different efficiency dimensions, and compute composite efficiency scores for comparative ranking [76].
Ensemble Machine Learning with Advanced Optimization
For regression tasks in scientific applications, combining multiple algorithms with sophisticated hyperparameter tuning enhances predictive accuracy [78]:
Data Preprocessing:
- Outlier detection using Z-score method (threshold: 3 standard deviations)
- Log transformation for normalization of skewed distributions: ( X' = \log(X + c) )
- Train-test split (85%-15%) with stratification if needed
Model Selection and Training:
- Implement diverse algorithms (MLP, XGBoost, Stacking ensembles)
- MLP configuration: multiple hidden layers, appropriate activation functions, backpropagation training
- XGBoost: sequential tree-building with gradient boosting
- Stacking: combine base models (MLP and XGBoost) using meta-learner
Hyperparameter Optimization:
- Employ Jellyfish Optimizer (JO) for efficient parameter space exploration
- JO uses biologically inspired dynamics to balance exploration and exploitation
Validation and Evaluation:
- Use k-fold cross-validation to assess generalization
- Evaluate with multiple metrics (R², RMSE, MAE)
- Compare training, validation, and test performance to detect overfitting
Visualization of Computational Workflows
Mixed-Resolution Protein Modeling Methodology
Mixed-Resolution Protein Modeling Flow
Efficiency Evaluation Framework for Computational Methods
Computational Method Efficiency Assessment
Research Reagent Solutions
Table 3: Essential Computational Tools for Efficiency-Optimized Research
| Tool/Platform | Type | Primary Function | Efficiency Features |
|---|---|---|---|
| Schrödinger Glide | Docking Software | Molecular docking and virtual screening | Precision vs speed settings, constraint-based docking |
| ANM (Anisotropic Network Model) | Modeling Algorithm | Protein conformer generation | Coarse-grained representation, normal mode analysis |
| Jellyfish Optimizer | Optimization Algorithm | Hyperparameter tuning | Bio-inspired efficient search space navigation |
| XGBoost | Machine Learning Library | Gradient boosting for structured data | Parallel processing, tree pruning, hardware optimization |
| Multilayer Perceptron (MLP) | Neural Network Architecture | Deep learning for complex patterns | Configurable architecture, GPU acceleration support |
| Mixed-Resolution MD | Simulation Methodology | Molecular dynamics with reduced cost | Atomistic detail focused on critical regions |
| MM-GBSA | Binding Affinity Method | End-point free energy calculation | Faster than full MD with reasonable accuracy |
| EfficientNetV2 | Deep Learning Model | Image classification and feature extraction | Neural architecture search optimized parameters |
| MobileNetV3 | Deep Learning Model | Computer vision on mobile devices | Depthwise separable convolutions, squeeze-excitation |
| Stacking Ensemble | ML Methodology | Combining multiple predictive models | Leverages strengths of diverse algorithms |
This comparison guide demonstrates that computational efficiency requires careful balancing of multiple factors rather than simply maximizing accuracy or minimizing resource use. The mixed-resolution ANM approach with truncated MD simulations exemplifies how strategic simplification of non-critical regions can dramatically reduce computational costs (15.2 μs total simulation time) while maintaining binding energy calculations comparable to full MD simulations [45]. For machine learning applications, model architecture choices significantly impact efficiency, with MobileNetV3 and EfficientNetV2 providing particularly favorable accuracy-efficiency tradeoffs [79].
The optimal method selection depends heavily on specific research constraints and objectives. When highest accuracy is imperative and resources are sufficient, traditional MD simulations or stacking ensemble methods may be justified. Under moderate resource constraints, mixed-resolution approaches or physics-informed neural networks offer excellent compromises. For severely limited computational resources or real-time requirements, lightweight deep learning models or endpoint methods like MM-GBSA provide practical solutions. By applying the standardized evaluation frameworks and experimental protocols outlined in this guide, researchers can make evidence-based decisions that advance drug development while responsibly managing computational resources.
Validation Frameworks and Comparative Analysis of Minimization Approaches
In computational chemistry and drug development, the validation of models and simulations is foundational to ensuring the reliability of predictions that accelerate scientific discovery. For decades, the root mean square deviation (RMSD) and related root mean square (RMS) metrics have served as ubiquitous standards for quantifying differences between predicted and reference structures. However, researchers increasingly recognize that these global geometric measures provide insufficient insight into chemical plausibility, energetic feasibility, and biological relevance. Within the context of energy minimization validation research, over-reliance on RMS force criteria alone can obscure critical deficiencies in molecular models that ultimately undermine their predictive capability for drug development applications. This guide establishes comprehensive validation protocols that transcend traditional RMSD/RMS force limitations, providing researchers with a multifaceted framework for rigorous computational assessment.
Limitations of Traditional RMS Metrics
Fundamental Deficiencies of RMSD in Molecular Evaluation
Root mean square deviation (RMSD) remains a commonly employed metric in molecular docking and dynamics simulations to measure the average spatial difference between atomic positions in predicted and reference structures [80]. While computationally straightforward, RMSD possesses critical limitations that render it inadequate as a standalone validation criterion:
Lack of Chemical Insight: RMSD quantifies purely geometric deviation without accounting for fundamental chemical validity, including proper bond orders, atom valencies, and stereochemical configurations [80]. A structure may achieve excellent RMSD while containing chemically impossible bonding patterns.
Global Measurement Bias: As a global average, RMSD can mask significant local inaccuracies in critical regions such as active sites or binding interfaces. Structural deviations in functionally important residues may be averaged out by well-predicted but functionally irrelevant regions [80].
Energetic Considerations: RMSD provides no information about the energetic feasibility of a molecular conformation. Structures with favorable RMSD values may represent high-energy, unstable conformations that would not persist in biological systems [80].
Insufficient for Binding Characterization: RMSD fails to capture the quality of specific molecular interactions essential for binding affinity and specificity, including hydrogen bonding, hydrophobic contacts, and π-π stacking [45].
Theoretical Foundations of Error Metric Selection
The limitations of RMS-based metrics extend beyond molecular modeling to fundamental statistical considerations. Different error metrics are mathematically optimal for different error distributions: RMSE is optimal for normal (Gaussian) errors, while mean absolute error (MAE) is optimal for Laplacian errors [81]. Selecting inappropriate metrics based on convention rather than theoretical justification introduces bias in model evaluation and validation.
Comprehensive Validation Criteria Beyond RMSD
Eight Essential Validation Criteria for Molecular Structures
The PoseBusters validation suite exemplifies the transition toward multidimensional validation with eight essential criteria that comprehensively assess molecular pose quality [80]:
Table 1: Essential Validation Criteria Beyond RMSD
| Validation Category | Specific Checks | Tolerance Ranges | Functional Importance |
|---|---|---|---|
| Chemical Validity | Bond orders, atom valency, formal charges | Chemically feasible ranges | Ensures molecular structures obey chemical principles |
| Stereochemistry | Tetrahedral chirality, double bond geometry | Exact configuration matching | Maintains pharmacological specificity |
| Bond Geometry | Bond lengths, bond angles | RDKit distance geometry bounds | Preserves molecular stability and strain energy |
| Planarity | Aromatic rings, conjugated systems | Near-planar (deviation < 0.1Å) | Maintains electronic properties and interaction potential |
| Internal Clashes | Non-bonded atom distances > threshold | Minimum distance > sum of van der Waals radii | Eliminates unrealistic high-energy conformations |
| Energetic Feasibility | Conformational energy ratio vs. reference | Within threshold of RDKit generated conformers | Ensures physiological persistence |
| Intermolecular Contacts | Ligand-protein/cofactor distances | Physically plausible ranges | Validates binding mode realism |
| Volume Overlap | Ligand-environment volume overlap | Below set threshold | Confirms complementary binding site fit |
Statistical Validation Frameworks for Quantitative Models
Beyond molecular-level validation, computational models require statistical validation frameworks to ensure predictive accuracy:
Bayesian Hypothesis Testing: Extended interval hypotheses on distribution parameters and equality hypotheses on probability distributions can validate models with deterministic/stochastic outputs, minimizing Type I/II errors in model selection [82].
Reliability-Based Metrics: These methods account for directional bias where model predictions consistently deviate above or below experimental observations [82].
Area Metric Methods: Provide comprehensive measures of agreement between predicted and observed probability distributions [82].
Experimental Protocols for Multidimensional Validation
Ensemble Docking and Binding Free Energy Validation
Objective: Evaluate protein-ligand interactions across multiple conformational states to account for structural flexibility and improve binding affinity predictions [45].
Methodology:
- Conformer Generation: Employ Anisotropic Network Models (ANM) to generate multiple protein conformers through normal mode analysis, capturing functional dynamics relevant to binding [45].
- Mixed-Resolution Modeling: Represent binding sites at atomistic resolution while coarse-graining remaining protein structure to balance computational efficiency with accuracy [45].
- Ensemble Docking: Perform docking calculations across all generated conformers using software such as Schrödinger Glide [45].
- Molecular Dynamics (MD) Simulations: Subject docked complexes to extended MD simulations (e.g., 900 ns across three replicates) to assess interaction stability [45].
- Binding Free Energy Calculations: Employ MM-GBSA (Molecular Mechanics/Generalized Born Surface Area) approach to estimate binding affinities using trajectories from MD simulations [45].
Validation Metrics:
- Consistency of binding energies between intact and truncated structures [45]
- Geometric complementarity of binding site configurations across conformers [45]
- Recovery of known specific interactions from experimental structures [45]
Quantitative Data Validation Protocol for Research Instruments
Objective: Establish statistical validity and reliability of quantitative research instruments through psychometric analysis [83].
Methodology:
- Questionnaire Development: Define content domains through literature review and expert consultation [83].
- Pilot Testing: Administer preliminary instrument to subset of target population [83].
- Exploratory Factor Analysis (EFA):
- Reliability Analysis: Calculate internal consistency metrics (Cronbach's alpha) for each identified factor [83].
- Final Instrument Administration: Deploy validated instrument to full study population [83].
Validation Metrics:
- Factor loadings > 0.4 for each retained item [83]
- Cronbach's alpha > 0.7 for each construct [83]
- Model fit indices (CFI > 0.9, RMSEA < 0.08) [83]
Task-Relevant MVC Protocol Establishment for Force Validation
Objective: Develop maximal voluntary contraction (MVC) protocols that account for task-specific parameters to validate force-related simulations [84].
Methodology:
- Protocol Selection:
- Force Tracking: Subjects perform continuous isometric force tracking at target percentages (5-95% MVC) normalized to the established MVC [84].
- Variability Analysis: Calculate absolute variability (standard deviation), tracking error (RMSE, constant error), and complexity (detrended fluctuation analysis) of force fluctuations [84].
Validation Metrics:
- Rate and magnitude changes in force variability across intensity levels [84]
- Performance strategies employed beyond ~65% MVC [84]
- Complexity structure of force fluctuations [84]
Workflow Visualization: Multidimensional Validation Framework
Table 2: Essential Research Reagent Solutions for Validation Studies
| Category | Specific Tool/Resource | Function in Validation | Application Context |
|---|---|---|---|
| Software Suites | PoseBusters | Comprehensive pose validation against chemical/physical criteria [80] | Molecular docking, structure prediction |
| Schrödinger Glide | Molecular docking with ensemble sampling [45] | Virtual screening, binding mode prediction | |
| RDKit | Cheminformatics analysis and sanitization checks [80] | Chemical validity assessment | |
| Computational Methods | Anisotropic Network Model (ANM) | Efficient conformer generation through normal mode analysis [45] | Ensemble docking, conformational sampling |
| Molecular Dynamics (Desmond) | All-atom simulation of molecular systems [45] | Binding stability, interaction analysis | |
| MM-GBSA | Binding free energy estimation from MD trajectories [45] | Affinity prediction, compound ranking | |
| Statistical Packages | R/Python with EFA | Exploratory factor analysis for instrument validation [83] | Questionnaire validation, psychometrics |
| Bayesian analysis tools | Hypothesis testing with interval hypotheses [82] | Model validation under uncertainty | |
| Specialized Protocols | Sustained MVC | Force measurement matched to task duration [84] | Biomechanical model validation |
| Ensemble Docking | Multiple receptor conformer docking [45] | Accounting for protein flexibility |
Establishing robust validation protocols that extend beyond traditional RMS force criteria requires a multifaceted approach integrating geometric, chemical, energetic, and statistical validation components. The frameworks presented in this guide provide researchers with comprehensive methodologies to ensure computational models and experimental instruments meet the rigorous standards demanded by modern drug development and scientific research. By adopting these multidimensional validation strategies, researchers can significantly enhance the reliability and predictive power of their computational approaches, ultimately accelerating the discovery and development of novel therapeutic agents. Future validation methodologies will likely continue this trajectory toward increasingly integrated validation frameworks that combine multiple orthogonal assessment criteria to fully characterize model performance and limitations.
Comparative Analysis of Minimization Algorithms Across Protein Systems
The accurate prediction and optimization of protein structures are fundamental challenges in computational biology, with profound implications for drug design, enzyme engineering, and therapeutic development. "Minimization" in this context encompasses a broad range of computational techniques aimed at finding low-energy, stable, and functional protein conformations. These algorithms are critical for bridging the sequence-structure-function relationship, yet their performance varies significantly across different protein systems and methodological approaches. Within the broader thesis on energy minimization validation, the Root Mean Square Force (RMSF) serves as a key metric for assessing the convergence and physical realism of these simulations, quantifying the average force acting on atoms within the system. This guide provides an objective comparison of contemporary minimization algorithms, classifying them into distinct categories—codon optimization, molecular dynamics (MD) simulations, and metaheuristic structure prediction—and evaluates their performance against experimental data and standardized metrics.
Methodological Classification of Minimization Algorithms
Minimization algorithms for protein systems can be broadly categorized based on their target optimization space and underlying principles. The following table summarizes the primary classes, their objectives, and key technical considerations.
Table 1: Classification of Protein Minimization Algorithms
| Algorithm Category | Primary Optimization Target | Commonly Used Tools/Software | Key Metric for Validation |
|---|---|---|---|
| Codon Optimization [85] | Translational efficiency & protein yield in heterologous hosts | JCat, OPTIMIZER, ATGme, GeneOptimizer, TISIGNER, IDT | Codon Adaptation Index (CAI), GC Content, mRNA ΔG (folding energy) |
| Molecular Dynamics (MD) Simulations [86] | Native-state dynamics & conformational ensembles | AMBER, GROMACS, NAMD, ilmm | Root Mean Square Deviation (RMSD), RMSF, comparison to NMR/XTAL data |
| Metaheuristic Structure Prediction [87] | 3D structure from amino acid sequence (free energy minimization) | Genetic Algorithm, Particle Swarm Optimization, Differential Evolution | Root Mean Square Deviation (RMSD) from native structure, Free Energy |
The workflow for selecting and validating a minimization algorithm typically involves defining the target property, running simulations, and rigorously comparing the output to experimental data, as illustrated below.
Figure 1: A generalized workflow for the selection and validation of minimization algorithms in protein science.
Comparative Performance Analysis
Codon Optimization Tools for Recombinant Protein Expression
Codon optimization algorithms enhance recombinant protein production by fine-tuning gene sequences to match the codon usage bias of a host organism. A comparative analysis of widely used tools reveals significant performance variability [85].
Table 2: Performance of Codon Optimization Tools in Model Organisms [85]
| Tool | Codon Adaptation Index (CAI) in E. coli | Codon Adaptation Index (CAI) in S. cerevisiae | Codon Adaptation Index (CAI) in CHO Cells | Key Optimization Strategy |
|---|---|---|---|---|
| JCat | High (0.95-1.00) | High (0.94-0.99) | High (0.93-0.98) | Alignment with genome-wide & highly expressed gene codon usage |
| OPTIMIZER | High (0.94-0.99) | High (0.93-0.98) | High (0.92-0.97) | User-defined codon usage tables & multiple optimization algorithms |
| ATGme | High (0.95-1.00) | High (0.94-0.99) | High (0.93-0.98) | Integrated design of coding sequence & regulatory elements |
| GeneOptimizer | High (0.95-1.00) | High (0.94-0.99) | High (0.93-0.98) | Iterative algorithm considering multiple parameters simultaneously |
| TISIGNER | Moderate to High | Moderate to High | Moderate to High | Focus on translation initiation & mRNA secondary structure |
| IDT | Variable | Variable | Variable | Proprietary algorithm; often produces divergent results |
Experimental Protocol for Codon Optimization Benchmarking: The comparative analysis involved selecting industrially relevant target proteins (e.g., human insulin, α-amylase, Adalimumab heavy and light chains) for expression in E. coli, S. cerevisiae, and CHO cells [85]. Each gene sequence was optimized using the default settings of the listed tools. The performance was evaluated by calculating the Codon Adaptation Index (CAI)—a measure of how well the codon usage matches the host's preferred codons—with values closer to 1.0 indicating better adaptation [85]. Additional parameters like GC content and mRNA secondary structure stability (ΔG) were also analyzed to assess their influence on translational efficiency and mRNA stability [85].
The choice of host organism introduces distinct biophysical constraints. For instance, while increased GC content can enhance mRNA stability in E. coli, A/T-rich codons are often preferred in S. cerevisiae to minimize secondary structure formation that could hinder translation. CHO cells, in contrast, require a balance achieved through moderate GC content [85]. This highlights the necessity of a multi-parameter optimization approach over reliance on a single metric like CAI.
Molecular Dynamics Packages for Conformational Sampling
Molecular Dynamics simulations serve as "virtual molecular microscopes," probing the dynamic behavior of proteins at an atomic level. The accuracy of these simulations is contingent upon the force field, integration algorithms, and treatment of atomic interactions [86].
Table 3: Validation of MD Simulation Packages Against Experimental Observables [86]
| MD Package | Force Field | Water Model | RMSD from Native State (Å) | Agreement with NMR Chemical Shifts | Thermal Unfolding Behavior |
|---|---|---|---|---|---|
| AMBER | ff99SB-ILDN | TIP4P-EW | Low (~1.5) | Good | Reproduces unfolding |
| GROMACS | ff99SB-ILDN | SPC/E | Low (~1.6) | Good | Reproduces unfolding |
| NAMD | CHARMM36 | TIP3P | Low (~1.7) | Good | Varies; can be at odds with experiment |
| ilmm | Levitt et al. | TIP3P | Low (~1.6) | Good | May fail to unfold at high temperature |
Experimental Protocol for MD Validation: Simulations were performed on two globular proteins with distinct topologies, the Engrailed homeodomain (EnHD) and Ribonuclease H (RNase H) [86]. Each system was simulated in triplicate for 200 nanoseconds at 298 K using four different MD package-force field combinations: AMBER (ff99SB-ILDN), GROMACS (ff99SB-ILDN), NAMD (CHARMM36), and ilmm (Levitt et al. force field) [86]. The simulations employed explicit water molecules and periodic boundary conditions, following best practices as defined by the respective developers. The resulting conformational ensembles were validated against experimental data, including NMR-derived chemical shifts and known native-state structures, using metrics like the Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) [86].
A critical finding is that while most packages reproduce native-state dynamics equally well at room temperature, they can diverge significantly when simulating larger amplitude motions, such as thermal unfolding [86]. This underscores that force fields are not solely responsible for performance; other factors like the water model, algorithms constraining bond motions, and the handling of long-range electrostatic interactions are equally critical in determining the outcome of a simulation [86].
Metaheuristic Algorithms for Protein Structure Prediction
The protein structure prediction (PSP) problem, which involves finding the tertiary structure from an amino acid sequence, is a complex global optimization challenge. Metaheuristics are well-suited for navigating the vast conformational space to discover near-optimal structures [87].
Table 4: Performance of Metaheuristic Algorithms in Protein Structure Prediction [87]
| Metaheuristic Algorithm | Class | Reported Accuracy (RMSD in Å) | Computational Efficiency | Key Application in PSP |
|---|---|---|---|---|
| Genetic Algorithm (GA) | Evolutionary | Low on benchmark proteins | Moderate | Global search of conformational space |
| Particle Swarm Optimization (PSO) | Swarm Intelligence | Low on benchmark proteins | High | Efficiently exploits promising regions |
| Differential Evolution (DE) | Evolutionary | Low on benchmark proteins | Moderate | Robustness and avoiding local minima |
| Teaching-Learning Based Optimization | Human-based | Low on benchmark proteins | Moderate | Simulates a classroom environment |
Experimental Protocol for Metaheuristic Evaluation: The empirical analysis of metaheuristics for PSP is typically conducted on benchmark protein sequences (e.g., 1CRN, 1CB3) with known experimental structures [87]. The algorithms are used to minimize a knowledge-based or physics-based energy function. Their performance is evaluated based on the RMSD between the predicted lowest-energy structure and the native, experimentally-solved structure, providing a measure of accuracy [87]. Computational efficiency is assessed by the time or number of function evaluations required to reach the solution. Statistical tests, such as the Friedman test with Dunn's post hoc analysis, are often employed to determine the significance of performance differences between algorithms [87].
Integrated Workflow and Research Reagent Solutions
Success in protein minimization often requires an integrated approach. For instance, a stable protein variant designed computationally must then have its gene sequence optimized for high-yield expression, bridging structure-based design and synthetic biology [88]. The following diagram illustrates this synergy for developing a therapeutic antibody.
Figure 2: An integrated workflow for the computational design and experimental production of a stable therapeutic protein, such as an antibody.
The Scientist's Toolkit: Research Reagent Solutions
Table 5: Essential Research Reagents and Software for Protein Minimization Studies
| Reagent / Software Solution | Function in Research | Example Use Case in Minimization |
|---|---|---|
| Thermo Fisher Proteome Discoverer [89] | Proteomic data analysis software | Identifying proteinaceous binders in cultural heritage artifacts; validating protein composition. |
| FragPipe (MSFragger) [89] | Open-source, high-speed proteomics platform | Rapid identification of digested proteins from mass spectrometry data. |
| GROMACS [86] | Molecular dynamics simulation package | Simulating native-state dynamics and thermal unfolding of proteins like RNase H. |
| AMBER [86] | Suite of biomolecular simulation programs | Energy minimization and dynamics of proteins using force fields like ff99SB-ILDN. |
| CHO (K1 strain) Cells [85] | Mammalian cell line for recombinant expression | Host organism for producing complex biopharmaceuticals like monoclonal antibodies. |
| Guanidine Hydrochloride [89] | Protein denaturant and extraction agent | Extracting proteins from aged, simulated paint specimens for proteomic analysis. |
| Sequencing-Grade Trypsin [89] | Proteolytic enzyme | Digesting proteins into peptides for analysis by LC-MS/MS. |
Discussion and Concluding Remarks
The comparative analysis presented herein demonstrates that there is no single "best" minimization algorithm for all protein systems. The optimal choice is inherently dictated by the specific biological question and system at hand. For enhancing recombinant expression, codon optimization tools like JCat and GeneOptimizer that employ a multi-criteria framework (integrating CAI, GC content, and mRNA folding) show superior performance [85]. For studying atomic-level dynamics and conformational changes, MD packages like AMBER and GROMACS provide reliable results, though careful attention must be paid to simulation parameters beyond the force field itself [86]. For the fundamental challenge of ab initio structure prediction, metaheuristics like Genetic Algorithms offer powerful strategies for navigating the vast conformational landscape [87].
A unifying theme across all categories is the critical importance of rigorous validation against experimental data. Metrics like the Root Mean Square Force (RMSF) in MD simulations, the Codon Adaptation Index (CAI) for optimized sequences, and the overall RMSD for predicted structures are essential for benchmarking algorithmic performance. Furthermore, integrated workflows that combine computational design with experimental validation are driving the field forward, enabling the engineering of stable enzymes, therapeutic antibodies, and novel protein functions [88]. As force fields, optimization algorithms, and computational power continue to advance, the precision and scope of protein minimization algorithms will undoubtedly expand, solidifying their role as indispensable tools in modern biological research and drug development.
The validation of computational models against experimental data is a critical step in ensuring their predictive power and reliability in scientific research and drug development. This process is particularly essential for energy minimization and molecular dynamics simulations, where the accuracy of the calculated root mean square force (RMSF) and other properties determines the model's utility for real-world applications. As computational methods become increasingly integral to fields like materials science and pharmaceutical development, the framework for rigorous, multi-faceted validation has evolved significantly.
A transformative shift is occurring from purely in silico validation to integrated approaches that leverage both simulation and experimental data. This "fused data" strategy concurrently satisfies multiple target objectives, resulting in models of higher accuracy than those trained on a single data source [90]. In drug development, this philosophy is formalized through Model-Informed Drug Development (MIDD), an essential framework for advancing drug development and supporting regulatory decision-making by providing quantitative predictions and data-driven insights [91].
This guide objectively compares prominent validation methodologies and computational force fields, summarizing their performance against experimental benchmarks and providing detailed protocols for their application.
Comparative Performance of Computational Force Fields
The choice of force field is foundational to the accuracy of molecular simulations. Below is a quantitative comparison of several force fields, highlighting their performance in reproducing key experimental physical properties.
Table 1: Force Field Performance Against Experimental Physical Properties of Diisopropyl Ether (DIPE) [57]
| Force Field | Density Error (%) | Viscosity Error (%) | Interfacial Tension | Partition Coefficients |
|---|---|---|---|---|
| GAFF | +3 to +5% | +60 to +130% | Data Not Provided | Data Not Provided |
| OPLS-AA/CM1A | +3 to +5% | +60 to +130% | Data Not Provided | Data Not Provided |
| CHARMM36 | Accurate | Accurate | Accurate | Accurate |
| COMPASS | Accurate | Moderate Overestimation | Accurate | Accurate |
The data demonstrates that CHARMM36 and COMPASS generally provide more accurate descriptions of thermodynamic properties like density and interfacial tension compared to GAFF and OPLS-AA/CM1A. However, their performance on dynamic properties like shear viscosity varies, with CHARMM36 showing superior accuracy for ether-based systems [57].
Emerging Machine Learning Force Fields
Machine learning (ML) is revolutionizing force field development by offering quantum-level accuracy at a fraction of the computational cost.
Grappa: A Machine-Learned Molecular Mechanics Force Field
Grappa is a machine learning framework that predicts molecular mechanics (MM) parameters directly from the molecular graph. It leverages a graph attentional neural network and a transformer, eliminating the need for hand-crafted atom typing rules. This approach allows Grappa to achieve state-of-the-art MM accuracy while maintaining the same computational efficiency as traditional force fields [6].
- Performance: Grappa outperforms traditional MM force fields and another machine-learned force field (Espaloma) on a benchmark dataset containing over 14,000 molecules. It accurately reproduces quantum mechanical energies and forces for small molecules, peptides, and RNA, and closely matches experimentally measured J-couplings [6].
- Application: Its transferability has been demonstrated on systems ranging from a small fast-folding protein (chignolin) up to an entire virus particle, setting the stage for biomolecular simulations closer to chemical accuracy [6].
Universal ML Force Fields and the Experimental Reality Gap
While universal machine learning force fields (UMLFFs) promise rapid, accurate atomistic simulations across the periodic table, their real-world performance requires careful evaluation. A 2025 benchmarking study, UniFFBench, revealed a substantial reality gap: models achieving impressive performance on computational benchmarks often fail when confronted with experimental complexity [92].
The evaluation of six state-of-the-art UMLFFs against ~1,500 experimentally measured mineral structures showed that even the best-performing models exhibited higher density prediction error than the threshold required for practical applications. This underscores that computational benchmarks alone may overestimate model reliability, making experimental validation essential [92].
Methodologies for Experimental Validation
The Fused Data Learning Strategy
This methodology leverages both simulation data (e.g., from Density Functional Theory (DFT)) and experimental data to train a single, more robust model.
Diagram: Fused Data Training Workflow for ML Potentials
The workflow involves an iterative process of alternating between two trainers [90]:
- DFT Trainer: For one epoch, the model parameters (θ) are modified to match energies, forces, and virial stresses from a database of DFT calculations.
- EXP Trainer: For one epoch, parameters are optimized so that properties computed from the ML-driven simulation match target experimental values (e.g., elastic constants, lattice parameters). Gradients are computed using methods like Differentiable Trajectory Reweighting (DiffTRe).
This fused approach has been shown to produce models that faithfully reproduce all target properties, with out-of-target properties often being positively affected [90].
A Hierarchical Framework for Biomechanical Model Validation
For complex systems like the human head, a systematic, hierarchical validation framework is crucial. This approach, applicable to cadaver heads or other biological structures, maximizes data acquisition and enables incremental model validation [93].
Diagram: Hierarchical Validation Framework for Biological Systems
The four levels of the framework are [93]:
- Level 1: System – Data is gathered from the intact system (e.g., vibrational responses of a full cadaver head).
- Level 2: Subsystems – Validation focuses on major functional components (e.g., the temporal bone containing the inner ear).
- Level 3: Structures – Isolated anatomical structures (e.g., specific bone pieces) are measured.
- Level 4: Tissues – The fundamental tissue types (e.g., cortical bone layers) are tested under controlled conditions.
This method ensures that subject-specific models are built and validated from the ground up, with functional variability rooted in measured anatomical variability [93].
Model-Informed Drug Development (MIDD) as a Validation Paradigm
MIDD represents a high-level application of model validation, where the "fit-for-purpose" alignment of models with specific questions is paramount. This approach uses quantitative tools to inform decisions from early discovery through post-market surveillance [91].
Table 2: Key MIDD Tools and Their Applications in Drug Development [91]
| Tool | Primary Application in Validation & Development |
|---|---|
| Quantitative Structure-Activity Relationship (QSAR) | Predicts biological activity and ADMET properties from chemical structure, validating a compound's potential before synthesis. |
| Physiologically Based Pharmacokinetic (PBPK) | Mechanistically models drug disposition, often used to replace or refine clinical trials and predict drug-drug interactions. |
| Quantitative Systems Pharmacology (QSP) | Integrates systems biology and pharmacology to generate mechanism-based predictions on treatment effects and side effects. |
| Population PK/Exposure-Response (ER) | Explains variability in drug exposure and links it to clinical effectiveness and safety outcomes in a target population. |
The strategic use of these tools allows development teams to mitigate risk early, compress timelines, and strengthen decision-making with functionally validated insights [94] [91]. A recent analysis estimates that MIDD yields annualized average savings of approximately 10 months of cycle time and $5 million per program [95].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Reagents and Computational Tools for Model Validation
| Item | Function in Validation |
|---|---|
| CETSA (Cellular Thermal Shift Assay) | Validates direct drug-target engagement in intact cells and native tissue environments, closing the gap between biochemical potency and cellular efficacy [94]. |
| High-Resolution Photon-Counting CT | Provides superior anatomical imaging data (e.g., for bone microstructure) essential for building and validating subject-specific computational models [93]. |
| Laser Doppler Vibrometry | Measures vibrational responses in biological specimens or materials, providing critical experimental data for validating simulated dynamics [93]. |
| Differentiable Trajectory Reweighting (DiffTRe) | An algorithmic method that enables efficient gradient-based optimization of ML potentials against time-independent experimental observables without backpropagating through entire simulations [90]. |
| Grappa Force Field | A machine-learned molecular mechanics force field that offers improved accuracy for biomolecules like peptides and proteins, compatible with standard MD engines like GROMACS and OpenMM [6]. |
The rigorous validation of computational models against experimental data is not merely a best practice but a critical enabler of scientific and industrial innovation. As evidenced by the comparisons in this guide, the field is moving toward hybrid methodologies that merge the strengths of simulation and experiment. Whether through the fused data training of ML force fields, the hierarchical validation of complex biomechanical systems, or the fit-for-purpose application of MIDD in drug development, the integration of high-quality experimental data remains the definitive benchmark for model accuracy and utility. This integrated approach is key to ensuring that computational predictions in energy minimization and RMSF research are reliable, interpretable, and ultimately, translatable to real-world solutions.
Triosephosphate isomerase (TIM) is a central glycolytic enzyme that catalyzes the reversible isomerization of dihydroxyacetone phosphate (DHAP) to d-glyceraldehyde phosphate (GAP). It has served for decades as a model system for understanding the fundamental principles of enzyme catalysis and conformational dynamics [96]. A critical step in computational studies of TIM, and enzymes in general, is energy minimization—a process of finding atomic arrangements where the net interatomic force is near zero and the potential energy is at a stationary point [1]. This case study examines the role of energy minimization within the context of generating biologically relevant conformers of TIM, validating the process using root mean square force (RMS force) metrics, and compares the performance of various minimization algorithms and hybrid sampling methods.
The Biological System: TIM Conformational Dynamics
TIM operates through a complex mechanism involving substantial conformational changes. Its catalytic proficiency relies on an energetically demanding conformational change that positions the side chains of key hydrophobic residues (I170 and L230) over the carboxylate side chain of the catalytic base E165. This creates a hydrophobic clamp critical for both creating a pocket for the base and maintaining correct active site architecture [96]. Molecular dynamics (MD) simulations have shown that truncation of these residues to alanine causes significant reductions in catalytic activity, underscoring the importance of precise active site geometry [96].
The functional dynamics of TIM, particularly the movement of its loop 6, which acts as a "lid" over the active site, are a classic example of how conformational changes can be integral to enzyme function. These motions are often cooperative and occur on timescales that can be challenging to capture with standard simulation techniques [97]. Therefore, proper sampling of TIM's conformational space requires sophisticated approaches that combine global conformational sampling with local energy minimization to refine structures to atomic-level accuracy.
Energy Minimization Fundamentals and Algorithms
What is Energy Minimization?
In computational chemistry, energy minimization (or geometry optimization) is the process of finding an arrangement of atoms in space where, according to a specific computational model of chemical bonding, the net inter-atomic force on each atom is acceptably close to zero. The resulting structure occupies a stationary point on the potential energy surface (PES) [1]. For proteins, this process refines geometries, including bond lengths, angles, and interatomic interactions, leading to structures that are closer to those found in nature and suitable for further theoretical and experimental analysis [98].
The potential energy of a molecular system is a function of the position vector of its N constituent atoms, U(R^N). Energy minimization seeks the set of position vectors R^N_min for which the first derivative of the potential energy with respect to position is zero, and the second derivative matrix is positive-definite [1].
Common Minimization Algorithms
Practical minimization algorithms are iterative and use different strategies to converge on a local minimum.
Table 1: Key Energy Minimization Algorithms
| Algorithm | Core Principle | Advantages | Disadvantages | Typical Applications in TIM Studies |
|---|---|---|---|---|
| Steepest Descent [11] | Moves atoms in the direction of the negative gradient (force). The step size is scaled by the maximum force. | Robust, stable for initial steps, effective for relieving severe steric clashes. | Can be slow to converge near the minimum, inefficient for long, narrow valleys. | Often used for initial minimization of raw homology models or MD-generated snapshots with high energy. |
| Conjugate Gradient [11] | Uses a sequence of non-interfering (conjugate) directions. Each new step is constructed to be conjugate to previous steps. | More efficient than Steepest Descent closer to the minimum. | Can not be used with constraints (e.g., SETTLE for water) in some implementations like GROMACS. | Suitable for minimization prior to normal-mode analysis where high accuracy is needed [11]. |
| L-BFGS [11] | A quasi-Newton method that approximates the inverse Hessian using a limited memory history of previous steps. | Generally faster convergence than Conjugate Gradients. | The correction steps can make parallelization difficult. | Efficient final minimization of stable conformers for detailed energy comparisons. |
Hybrid Methods for Conformational Sampling of TIM
Given the limitations of standard MD in sampling large-scale conformational changes, several hybrid methods have been developed that combine the global sampling efficiency of coarse-grained models with the local accuracy of all-atom minimization and MD.
Table 2: Hybrid Methods for Conformational Sampling Applied to TIM
| Method | Description | Key Features | Application to TIM |
|---|---|---|---|
| ClustENM/ ClustENMD [97] | Generates conformers by deforming structures along low-frequency normal modes from Elastic Network Models (ENM), followed by clustering and energy minimization (ClustENM) or short MD refinement (ClustENMD). | Efficiently samples large-scale cooperative motions while retaining atomic details. | Successfully reproduced the open and closed states of the loop 6 in TIM, capturing its hinge-like motion. |
| MDeNM (MD with excited Normal Modes) [97] | A multi-replica protocol that enhances exploration by adding velocities along linear combinations of low-frequency normal modes during MD simulations with an explicit solvent. | Incorporates anharmonicity and solvent effects while guiding exploration along functionally relevant directions. | Useful for probing the transition pathway of the loop closure and its role in stabilizing the catalytic hydrophobic clamp. |
| CoMD (Collective Modes-driven MD) [97] | Combines ENM-NMA and targeted MD, coupled with energy minimization to adaptively generate a series of conformers. | A targeted approach to drive conformational change between known states. | Can be used to simulate the induced-fit motion upon substrate (DHAP/GAP) binding in the TIM active site. |
The following workflow illustrates how these methods integrate coarse-grained sampling with all-atom refinement to generate accurate conformers.
Workflow for Generating TIM Conformers Using Hybrid Sampling and Energy Minimization
Experimental Protocols and Validation
A Representative Simulation Protocol for TIM
Detailed empirical valence bond (EVB) and MD studies on TIM provide a template for a robust simulation protocol incorporating energy minimization [96]:
- System Setup: The simulation is typically based on a high-resolution crystal structure of TIM (e.g., PDB ID: 1NEY at 1.2 Å resolution). The system is prepared by adding hydrogen atoms and assigning protonation states of ionizable residues using tools like PROPKA.
- Solvation: The protein is solvated in a water droplet (e.g., TIP3P water model with a 20 Å radius). Atoms outside this sphere are often restrained to their crystallographic positions to maintain system stability.
- Initial Minimization: A steepest descent minimization is performed to relieve any bad contacts introduced during solvation and system setup. This step is crucial for preparing the system for stable MD.
- Heating and Equilibration: The system is gradually heated from 0 to 300 K over ~140 ps while gradually releasing positional restraints.
- Production and Sampling: This is where hybrid methods like ClustENMD or MDeNM are employed to sample the conformational space of TIM, particularly the motion of loop 6.
- Conformer Refinement: Sampled conformers from the hybrid methods undergo final energy minimization using algorithms like conjugate gradient or L-BFGS to reach a stable local minimum before further analysis (e.g., calculating reaction barriers via EVB).
Validation with Root Mean Square Force
A critical stopping criterion for energy minimization is the magnitude of the force. The algorithm typically iterates until the maximum absolute value of the force components or the root mean square force falls below a specified threshold, ε [11]. This value must be chosen carefully. An overly tight threshold can lead to endless iterations due to numerical noise from force truncation, while a loose threshold will result in a poorly minimized structure.
A physically reasonable value for ε can be estimated from the RMS force a harmonic oscillator exhibits at a given temperature. The formula for the maximum force is given by: f = 2πν √(2mkT) where ν is the oscillator frequency, m is the reduced mass, k is Boltzmann’s constant, and T is the temperature [11]. For a typical weak molecular oscillator, this yields an acceptable ε value between 1 and 100 kJ mol⁻¹ nm⁻¹, with a common default in many MD packages being around 10.0 kJ mol⁻¹ nm⁻¹ for the maximum force. Validating that a minimization has converged below this threshold ensures the structure is at a sound local minimum on the PES, providing confidence in subsequent energy calculations or analysis of the TIM conformer.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for TIM Conformer Generation
| Tool / Resource | Type | Function in Research |
|---|---|---|
| GROMACS [11] | Software Suite | A versatile package for performing MD simulations and energy minimization using Steepest Descent, Conjugate Gradient, and L-BFGS. |
| OPLS-AA Force Field [96] | Force Field | A set of molecular mechanics parameters used to calculate potential energy for proteins and organic molecules in simulations of TIM. |
| PDB ID 1NEY [96] | Experimental Structure | The high-resolution (1.2 Å) crystal structure of TIM from Saccharomyces cerevisiae in complex with DHAP, often used as a starting point for simulations. |
| TIP3P Water Model [96] | Solvation Model | A widely used 3-point model for water molecules to simulate an aqueous environment around the TIM enzyme. |
| Elastic Network Model (ENM) [97] | Coarse-Grained Model | Used in hybrid methods to rapidly identify low-frequency, collective motions that define the large-scale conformational landscape of TIM. |
| Q Simulation Package [96] | Software Suite | Used for empirical valence bond (EVB) calculations to study detailed reaction mechanisms within the minimized TIM conformers. |
Performance Comparison and Discussion
Comparative studies of hybrid methods like ClustENM, ClustENMD, MDeNM, and CoMD, when applied to proteins like TIM, reveal that all can generate conformations that agree well with experimental ensembles [97]. However, their performance varies.
Methods that integrate short MD simulations for refinement (ClustENMD, MDeNM) often show superior ability to recapitulate the experimental conformational diversity of flexible loops in TIM compared to those relying solely on energy minimization after normal-mode deformation. This is because MD can better capture local anharmonic effects and side-chain rearrangements. Furthermore, the choice of the final minimization algorithm matters for computational efficiency. While steepest descent is robust for initial steps, L-BFGS consistently demonstrates faster convergence to a low-force minimum than conjugate gradients for final refinement of TIM conformers [11], though it may face parallelization challenges.
The success of energy minimization in producing physically meaningful TIM conformers is ultimately validated by its integration within a larger workflow. The minimized structures must not only have low RMS forces but also be consistent with experimental observables, such as those from multiple X-ray structures, and yield accurate catalytic barriers when used in subsequent quantum mechanical calculations, as demonstrated by the strong agreement between calculated and experimental activation barriers for wild-type and mutant TIM [96].
Benchmarking is an indispensable process in scientific research and development, providing a structured framework for evaluating the performance of computational platforms, experimental methods, and therapeutic candidates. In the context of energy minimization validation with root mean square force research, benchmarking studies establish objective metrics that enable researchers to compare different validation approaches, identify areas for improvement, and make data-driven decisions. For researchers, scientists, and drug development professionals, robust benchmarking protocols are particularly critical in computational drug discovery, where they help reduce the high failure rates and escalating costs associated with bringing new therapeutics to market [99].
This guide provides a comprehensive comparison of performance metrics and validation approaches across multiple domains, with a specific focus on their application to energy minimization and force field validation. By synthesizing current benchmarking practices from computational chemistry, drug discovery, and pharmaceutical development, we aim to equip professionals with the methodologies and metrics needed to rigorously evaluate their validation approaches against industry standards and competitors.
Key Performance Metrics for Benchmarking Analysis
Categorization of Benchmarking Metrics
Effective benchmarking relies on selecting appropriate metrics that accurately reflect performance across multiple dimensions. These metrics are typically categorized into financial, operational, and human resources domains, each providing unique insights into different aspects of performance [100].
Table 1: Core Benchmarking Metric Categories
| Category | Specific Metrics | Application in Validation Research |
|---|---|---|
| Financial Metrics | Revenue Growth, Profit Margin, Return on Investment (ROI) | Calculating ROI of research investments; assessing cost-performance tradeoffs in simulation methodologies |
| Operational Metrics | Customer Satisfaction, Employee Productivity, Production Efficiency | Measuring computational efficiency; assessing throughput of different validation approaches |
| Human Resources Metrics | Employee Turnover Rate, Training and Development Expenses | Evaluating research team stability; calculating investment in researcher skill development |
| Project Management Metrics | Project Cycle Time, Cost Variance, Schedule Performance [101] | Tracking validation project timelines; monitoring budget adherence for computational research |
| Research & Development Metrics | Likelihood of Approval (LoA), Success Rates [102] | Measuring transition success from preclinical to clinical stages; benchmarking against industry standards |
Quantitative Performance Metrics in Drug Discovery
In pharmaceutical research and development, quantitative benchmarking metrics provide crucial insights into the efficiency and success rates of drug discovery platforms. A comprehensive analysis of FDA approvals from 2006-2022 across 18 leading pharmaceutical companies revealed an average Likelihood of Approval (LoA) rate of 14.3% (median: 13.8%), with company-specific rates broadly ranging from 8% to 23% [102]. This metric, which measures the probability that a drug entering Phase I trials will eventually receive FDA approval, serves as a key benchmark for assessing the effectiveness of drug discovery platforms, including those incorporating energy minimization validation techniques.
For computational drug discovery platforms, benchmarking often employs metrics such as the percentage of known drugs correctly ranked in the top 10 compounds for their respective diseases. Studies of the CANDO multiscale therapeutic discovery platform demonstrated performance ranging from 7.4% to 12.1%, depending on the drug-indication mappings used from different databases [99]. These recall-based metrics provide concrete measures of platform performance that can be compared across different validation approaches.
Benchmarking Methodologies and Experimental Protocols
Performance Benchmarking Process
The benchmarking process follows a systematic approach to ensure accurate and actionable results. The key steps in successful performance benchmarking include [103]:
Create a Plan: Define the scope and objectives of the benchmarking exercise, identifying critical metrics and performing SWOT analysis (Strengths, Weaknesses, Opportunities, Threats) to understand current positioning.
Conduct Research and Collect Data: Gather accurate, reliable data relevant to the specific Key Performance Indicators (KPIs) being benchmarked through discussions with stakeholders, surveys, or focus groups.
Analyze Data: Identify performance gaps and improvement opportunities by comparing current performance levels against benchmarks using data visualization tools for clearer interpretation.
Develop an Action Plan: Create actionable steps using established goal-setting methods such as SMART (Specific, Measurable, Actionable, Relevant, Time-Bound) criteria to translate findings into improvement initiatives.
Track Progress: Regularly monitor performance against defined goals through consistent metric tracking, adjusting strategies as needed based on progress toward benchmarks.
Experimental Protocols for Force Field Validation
In energy minimization validation research, benchmarking different force fields requires rigorous experimental protocols. These methodologies typically involve comparative analysis of force field performances in conformational analysis and searching of organic molecules [31]. The general protocol includes:
System Preparation and Parameterization
- Select a diverse set of organic molecules representing different chemical functionalities and conformational complexities
- Parameterize each molecule using the force fields being compared (e.g., MM2, MM3, MMFF94, AMOEBA, UFF)
- Generate initial conformations through systematic or random searching algorithms
Conformational Analysis Protocol
- For each molecule, evaluate energies and geometries of different conformers using each force field
- Compare results against reference data from experimental measurements or high-level ab initio quantum mechanical calculations (e.g., MP2, CCSD(T))
- Calculate root mean square deviations (RMSD) between force field-predicted structures and reference structures
- Assess force field performance by the closeness of calculated properties to reference values
Conformational Searching Protocol
- Implement conformational searching algorithms that alter molecular geometry followed by force field energy minimization
- Repeat minimization cycles until comprehensive conformational coverage is achieved
- Compare the efficiency of different force fields in locating global minima and low-energy conformations
- Evaluate computational cost and convergence properties for each force field
QM/MM Hydration Free Energy Calculations
- Employ both fixed charge (e.g., CHARMM) and polarizable force fields (e.g., CHARMM Drude) [7]
- Alchemically annihilate each molecule in both aqueous phase and gas phase
- Use 1687 water molecules in cubic simulation cells with equilibrium box sizes
- Conduct simulations using Langevin dynamics with friction coefficient of 1 ps⁻¹ at 300K
- Run extended simulation timelines (500 ns) with frequent coordinate sampling (every 20 ps)
Comparative Analysis of Force Field Performance
Force Field Benchmarking in Conformational Analysis
Comparative studies evaluating different force fields for conformational analysis of organic molecules have identified clear performance trends. The "MM" family of force fields—particularly MM2, MM3, and MMFF94—consistently demonstrate strong performance in predicting energies and geometries of conformers [31]. The polarizable AMOEBA force field has also shown consistently strong performance, though it has been included in fewer comparative studies.
Table 2: Force Field Performance in Conformational Analysis
| Force Field | Type | Performance Rating | Recommended Use Cases | Key Findings |
|---|---|---|---|---|
| MM2 | Molecular Mechanics | Strong | Small organic molecules, conformational analysis | Excellent balance of accuracy and computational efficiency |
| MM3 | Molecular Mechanics | Strong | Complex organic molecules, steric interactions | Improved treatment of non-bonded interactions over MM2 |
| MMFF94 | Molecular Mechanics | Strong | Drug-like molecules, diverse organic compounds | Broad parameterization suitable for pharmaceutical applications |
| AMOEBA | Polarizable | Strong | Systems requiring polarization response | Accurate treatment of electrostatic interactions; higher computational cost |
| UFF | Universal | Weak | Preliminary screening only | Limited accuracy; not recommended for rigorous conformational analysis |
Research indicates that force field performance is substantially more limited in accuracy compared to advanced ab initio electronic structure methods, though at significantly lower computational cost [31]. The ideal force field provides results closely matching quantum mechanical methods while maintaining practical computation times—a critical consideration for researchers selecting validation approaches for energy minimization studies.
QM/MM Solvation Free Energy Benchmarking
Benchmarking studies of QM/MM (Quantum Mechanical/Molecular Mechanical) approaches for calculating hydration free energies (ΔGhyd) reveal significant challenges in achieving consistent performance across different quantum methods. Studies comparing fixed charge versus polarizable force fields found that QM/MM hydration free energies were generally inferior to purely classical results, with QM/MM Drude force field predictions only marginally better than QM/MM fixed charge results [7].
The performance of QM/MM simulations varies considerably across different quantum methods:
- Density functional methods (BLYP, B3LYP, M06-2X) show divergent trends depending on the accompanying MM water model
- Wavefunction-based methods (MP2, Hartree-Fock) demonstrate different compatibility with MM force fields
- Semi-empirical methods (OM2, AM1) offer computational efficiency but with variable accuracy
These findings highlight the critical importance of developing consistent and balanced QM/MM interactions, as both the QM and MM components must be carefully matched to avoid artifacts from biased solute-solvent interactions [7].
Performance Benchmarking in Pharmaceutical R&D
Clinical Trial Benchmarking Approaches
In pharmaceutical development, benchmarking extends to clinical trial monitoring and success rate prediction. Approaches include:
Trial Outcome Prediction
- Tracking progression from Phase I to FDA approval across multiple companies and therapeutic areas
- Analyzing likelihood of approval (LoA) rates based on historical performance
- Comparing actual trial outcomes against predicted success probabilities
Competitive Benchmarking
- Monitoring competitor clinical trials and regulatory milestones
- Comparing internal R&D performance against industry leaders
- Identifying best practices from top-performing organizations
Portfolio Optimization
- Using benchmarking data to inform resource allocation decisions
- Prioritizing programs with higher probability of technical success
- Balancing risk across therapeutic areas and development stages
Metrics for Drug Discovery Platform Validation
For computational drug discovery platforms, benchmarking against established standards is essential for validation. Key approaches include [99]:
Ground Truth Mapping
- Using established drug-indication mappings from databases like Comparative Toxicogenomics Database (CTD) and Therapeutic Targets Database (TTD) as reference standards
- Implementing k-fold cross-validation to assess prediction robustness
- Applying temporal splits (based on approval dates) to simulate real-world prediction scenarios
Performance Assessment Metrics
- Area Under the Receiver-Operating Characteristic Curve (AUC-ROC)
- Area Under the Precision-Recall Curve (AUC-PR)
- Recall, precision, and accuracy at specific thresholds
- Ranking efficiency (e.g., percentage of known drugs ranked in top 10 candidates)
Studies have shown that platform performance can be weakly positively correlated (Spearman correlation coefficient > 0.3) with the number of drugs associated with an indication and moderately correlated (coefficient > 0.5) with intra-indication chemical similarity [99].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Tools for Benchmarking Studies
| Tool/Resource | Function | Application Context |
|---|---|---|
| CHARMM Software | Molecular dynamics simulation package | Force field parameterization; energy minimization; QM/MM simulations [7] |
| MM2/MM3/MMFF94 Force Fields | Molecular mechanics parameter sets | Conformational analysis; energy calculations for organic molecules [31] |
| AMOEBA Polarizable Force Field | Polarizable force field | Systems requiring explicit polarization response; improved electrostatic modeling [31] |
| Comparative Toxicogenomics Database (CTD) | Drug-indication association database | Ground truth mapping for drug discovery platform benchmarking [99] |
| Therapeutic Targets Database (TTD) | Drug target and indication database | Reference standard for validating drug-target predictions [99] |
| CHARMM Drude Polarizable Force Field | Polarizable force field implementation | Improved solvation free energy calculations; more accurate solvent interaction modeling [7] |
| SWM4 Water Model | Polarizable water model for Drude simulations | Explicit solvent representation in polarizable force field simulations [7] |
Benchmarking studies provide critical insights into the performance of different validation approaches, with specific implications for energy minimization research using root mean square force metrics. The comparative analysis presented in this guide demonstrates that methodological choices—from force field selection to benchmarking protocols—significantly impact validation outcomes. By adopting rigorous benchmarking frameworks and appropriate performance metrics, researchers can make informed decisions about validation approaches, ultimately enhancing the reliability and predictive power of their computational models in drug discovery and related fields.
Conclusion
Effective validation of energy minimization using RMS force criteria is fundamental to producing reliable biomolecular models for drug discovery. This synthesis demonstrates that rigorous convergence criteria, appropriate algorithm selection, and comprehensive validation protocols are essential for accurate prediction of protein-ligand interactions and binding affinities. Future directions should focus on integrating machine learning approaches with traditional minimization methods, developing standardized validation benchmarks across the industry, and adapting these protocols for emerging challenges in membrane protein modeling and RNA-targeted drug design. As computational methods continue to advance, robust energy minimization validation will remain a cornerstone of structure-based drug discovery, enabling more efficient and reliable development of therapeutic compounds.
References
- 1. Energy minimization [https://en.wikipedia.org/wiki/Energy_minimization]
- 2. Energy Minimization of Structures and Ligand Complexes [https://www.biosolveit.de/drug-discovery-insights/energy-minimization-of-structures/]
- 3. 13.2 Force fields and energy minimization [https://fiveable.me/biophysics/unit-13/force-fields-energy-minimization/study-guide/sRyXoZVpb0lWneUi]
- 4. Comparison of the AMBER, CHARMM, COMPASS ... [https://www.sciencedirect.com/science/article/pii/S0378381206003323]
- 5. Evaluating the Performance of the ff99SB Force Field Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2718158/]
- 6. Grappa – a machine learned molecular mechanics force field [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc05465b]
- 7. A Comparison of QM/MM Simulations with and without the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6222909/]
- 8. D3R Grand Challenge 4: Ligand similarity and MM-GBSA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8208075/]
- 9. In-silico molecular modelling, MM/GBSA binding free ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.991369/full]
- 10. Comparison of force-field implementations [https://en.wikipedia.org/wiki/Comparison_of_force-field_implementations]
- 11. Energy Minimization - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/algorithms/energy-minimization.html]
- 12. Gaussian 16 Frequently Asked Questions [https://gaussian.com/faq3/]
- 13. Geometry Optimization [https://psicode.org/psi4manual/master/optking.html]
- 14. Validation of experimental molecular crystal structures with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2940256/]
- 15. Structure and dynamics in drug discovery [https://www.nature.com/articles/s44386-024-00001-2]
- 16. Energy Minimization in Drug Discovery [https://www.biosolveit.de/drug-discovery-insights/energy-minimization-in-drug-discovery/]
- 17. Protein structure validation by generalized linear model ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3324767/]
- 18. Enhancing drug screening and pharmacokinetic predictions [https://www.sciencedirect.com/science/article/abs/pii/S1385894725012288]
- 19. Computational approaches streamlining drug discovery [https://www.nature.com/articles/s41586-023-05905-z]
- 20. Effects of Structure Minimization on The Docking Score and ... [https://bioresscientia.com/article/effects-of-structure-minimization-on-the-docking-score-and-docking-time-during-computational-analysis-using-the-chimera-software]
- 21. Normal-Mode-Analysis–Monitored Energy Minimization ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0001025]
- 22. Root mean square [https://en.wikipedia.org/wiki/Root_mean_square]
- 23. An Empirical Biasing Force Constant to Minimize Overfitting in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12458688/]
- 24. (IUCr) Validation of experimental molecular crystal structures ... [https://journals.iucr.org/paper?so5041]
- 25. Is Newton's algorithm really this much better than ... [https://cs.stackexchange.com/questions/115404/is-newtons-algorithm-really-this-much-better-than-conjugate-gradient-descent]
- 26. Conjugate gradient methods [https://optimization.cbe.cornell.edu/index.php?title=Conjugate_gradient_methods]
- 27. An Energy Minimization-Based Deep Learning Approach ... [https://www.mdpi.com/2075-1680/14/11/806]
- 28. Validation of the general purpose tripos 5.2 force field [https://researchcollaborations.elsevier.com/en/publications/validation-of-the-general-purpose-tripos-52-force-field]
- 29. Identification of new potent NLRP3 inhibitors by multi-level in ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-024-01178-3]
- 30. Normal-Mode-Analysis–Monitored Energy Minimization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1995756/]
- 31. Comparisons of different force fields in conformational ... [https://www.sciencedirect.com/science/article/abs/pii/S0040402020311236]
- 32. A mechanistic study on the tolerance of PAM distal end ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11267045/]
- 33. Increasing certainty in systems biology models using ... [https://www.nature.com/articles/s41467-025-62415-4]
- 34. A universal augmentation framework for long-range ... [https://arxiv.org/html/2507.14302v1]
- 35. AMBER vs GROMACS [https://diphyx.com/stories/amber-vs-gromacs/]
- 36. Energy Manipulations: Minimization and Dynamics [https://www.charmm-gui.org/charmmdoc/energy.html]
- 37. minimiz (c48b2) [https://academiccharmm.org/documentation/version/c48b2/minimiz]
- 38. gmx rmsf - GROMACS 2025.4 documentation [https://manual.gromacs.org/current/onlinehelp/gmx-rmsf.html]
- 39. How to compute the RMSF using GROMACS [https://www.compchems.com/how-to-compute-the-rmsf-using-gromacs/]
- 40. atomicfluct | rmsf [https://amberhub.chpc.utah.edu/atomicfluct-rmsf/]
- 41. 7.1.2. RMSF — AdKGromacsTutorial 2.0.2 documentation [https://adkgromacstutorial.readthedocs.io/en/latest/analysis/rmsf.html]
- 42. Lessons learned from comparing molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5581938/]
- 43. Re: [AMBER] question of Amber calculate RMSF [http://archive.ambermd.org/202111/0069.html]
- 44. PoseX: AI Defeats Physics Approaches on Protein-Ligand ... [https://arxiv.org/html/2505.01700v1]
- 45. A Computationally Efficient Method to Generate Plausible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12344705/]
- 46. Interformer: an interaction-aware model for protein-ligand ... [https://www.nature.com/articles/s41467-024-54440-6]
- 47. DeepRLI: a multi-objective framework for universal protein ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00403e]
- 48. Structure based drug design and machine learning ... [https://www.nature.com/articles/s41598-025-17708-5]
- 49. Structure-based discovery and experimental validation of ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1605741/full]
- 50. Evaluation of the ABCG2 Charge Model in Protein–Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606647/]
- 51. Accurate prediction of protein–ligand interactions by ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00912-2]
- 52. Comparison of molecular dynamics with hybrid continuum ... [https://pubmed.ncbi.nlm.nih.gov/15260795/]
- 53. Hybrid computational methods combining experimental ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X23000830]
- 54. A machine learning enabled hybrid optimization framework ... [https://www.nature.com/articles/s41524-022-00914-4]
- 55. [2406.10362] A Comparison of the Performance ... [https://arxiv.org/abs/2406.10362]
- 56. When Does Molecular Dynamics Improve RNA Models ... [https://www.sciencedirect.com/science/article/pii/S2001037025004040]
- 57. Force field comparison for molecular dynamics simulations ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732224024061]
- 58. Comparison of protein force fields for molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/18446282/]
- 59. Statistical accuracy of molecular -based dynamics for... methods [https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp02564d]
- 60. Comparison of dynamic block randomization and minimization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4296975/]
- 61. Optimization of Multi-Stage Dynamic Treatment Regimes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4596799/]
- 62. An experimental comparison of min-cut/max-flow ... [https://pubmed.ncbi.nlm.nih.gov/15742889/]
- 63. Global properties of the energy landscape: a testing and ... [https://arxiv.org/html/2508.16425v1]
- 64. Energy Minimization - an overview [https://www.sciencedirect.com/topics/physics-and-astronomy/energy-minimization]
- 65. Docking to RNA via Root-Mean-Square-Deviation-Driven ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2910576/]
- 66. Constraints and Restraints [https://www.uoxray.uoregon.edu/local/manuals/biosym/discovery/General/Describe_Sys/ConstrRestr.html]
- 67. Restraint Validation of Biomolecular Structures Determined ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11162339/]
- 68. Minimization - General Methodology [https://www.uoxray.uoregon.edu/local/manuals/biosym/discovery/General/Minimization/Gen_Method.html]
- 69. Adaptive restraints to accelerate geometry optimizations of ... [https://chemrxiv.org/engage/chemrxiv/article-details/6490b93ba2c387fa9a94f607]
- 70. On the Validation of Protein Force Fields Based on Structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11103706/]
- 71. Biomolecular NMR Wiki - NMR Restraints [https://sites.google.com/site/ccpnwiki/home/documentation/ccpnmr-analysis/tutorials/nmr-restraints]
- 72. Data-driven parametrization of molecular mechanics force ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06640e]
- 73. MACE-OFF: Short Range Transferable Machine Learning ... [https://arxiv.org/html/2312.15211v5]
- 74. Choice of Force Field for Proteins Containing Structured and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7136338/]
- 75. Parameterization and validation of a new AMBER force ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326323001092]
- 76. A simplified approach for efficiency analysis of machine ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11623197/]
- 77. PINN-DT: Optimizing Energy Consumption in Smart Building ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12526817/]
- 78. Computational evaluation using machine learning for ... [https://www.nature.com/articles/s41598-025-20291-4]
- 79. Comparative Analysis of Lightweight Deep Learning ... [https://arxiv.org/html/2505.03303v2]
- 80. Beyond RMSD: What Are the 8 Chemical and Physical ... [https://info.genophore.com/posts/beyond-rmsd-what-are-the-8-chemical-and-physical-criteria-needed-to-evaluate-ligand-docking-methods]
- 81. Root-mean-square error (RMSE) or mean absolute error ... [https://gmd.copernicus.org/articles/15/5481/2022/]
- 82. Quantitative model validation techniques: New insights [https://www.sciencedirect.com/science/article/abs/pii/S0951832012002414]
- 83. A statistical approach to quantitative data validation focused ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3795879/]
- 84. Establishing Task-Relevant MVC Protocols for Modelling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8628892/]
- 85. Comparative Analysis of Codon Optimization Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010093/]
- 86. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 87. Metaheuristics for protein structure prediction [https://www.sciencedirect.com/science/article/pii/S2468227625002893]
- 88. Opportunities and challenges in design and optimization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7616297/]
- 89. Comparative analysis and optimization of proteomic ... [https://www.nature.com/articles/s40494-025-02002-4]
- 90. Accurate machine learning force fields via experimental ... [https://www.nature.com/articles/s41524-024-01251-4]
- 91. Advancing drug development with “Fit-for-Purpose” modeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12436579/]
- 92. Evaluating Universal Machine Learning Force Fields ... [https://arxiv.org/abs/2508.05762]
- 93. Experimental Framework for the Setup and Validation of ... [https://www.mdpi.com/2313-7673/10/11/738]
- 94. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 95. CERTAINTY 2025: The power of Model-Informed Drug ... [https://www.certara.com/blog/certainty-the-power-of-model-informed-drug-development/]
- 96. Enzyme Architecture: Modeling the Operation of a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5543394/]
- 97. Sampling of Protein Conformational Space Using Hybrid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8855042/]
- 98. Energy Minimisation - an overview [https://www.sciencedirect.com/topics/engineering/energy-minimisation]
- 99. Strategies for robust, accurate, and generalizable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607264/]
- 100. Benchmarking Metrics that Really Matter: Key Performance ... [https://www.comparables.ai/articles/benchmarking-metrics-that-really-matter-key-performance-indicators-to-watch]
- 101. Project Benchmarking Guide: Approaches, Metrics & Best ... [https://www.netsuite.com/portal/resource/articles/erp/project-management-benchmarking.shtml]
- 102. Benchmarking R&D success rates of leading ... [https://www.sciencedirect.com/science/article/pii/S1359644625000042]
- 103. Performance Benchmarking: Your Quick Guide [https://safetyculture.com/topics/benchmarking/performance-benchmarking]