Surrogate Model-Assisted Molecular Dynamics (SMA-MD): A Revolutionary Approach for Accelerated Biomolecular Sampling and Drug Discovery
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) represents a paradigm shift in computational biochemistry, integrating deep generative models with traditional molecular simulations to overcome the critical challenge of sampling molecular equilibrium ensembles.
Surrogate Model-Assisted Molecular Dynamics (SMA-MD): A Revolutionary Approach for Accelerated Biomolecular Sampling and Drug Discovery
Abstract
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) represents a paradigm shift in computational biochemistry, integrating deep generative models with traditional molecular simulations to overcome the critical challenge of sampling molecular equilibrium ensembles. This article provides a comprehensive exploration of SMA-MD, beginning with its foundational principles that address the limitations of conventional Molecular Dynamics. We detail the methodological workflow, from leveraging generative models for enhanced sampling of slow degrees of freedom to statistical reweighting and short simulations for thermodynamic property prediction. Practical guidance on troubleshooting common challenges, such as handling explicit solvent models, is provided alongside empirical validation demonstrating SMA-MD's superior performance in generating more diverse, lower-energy conformational ensembles. For researchers and drug development professionals, this synthesis highlights SMA-MD's transformative potential in accurately predicting solvation free energies and other crucial properties, ultimately accelerating therapeutic design.
The Foundations of SMA-MD: Overcoming the Boltzmann Sampling Challenge in Molecular Dynamics
In fields ranging from drug discovery to materials science, the accurate prediction of thermodynamic properties depends on effectively sampling the Boltzmann distribution. This distribution, defined as μ(𝐱) = Z⁻¹exp(-βu(𝐱)), where u(𝐱) is the potential energy of the system configuration 𝐱, β is the inverse temperature, and Z is the partition function, represents the equilibrium state of a molecular system [1]. Conventional Molecular Dynamics (MD) simulations attempt to generate samples from this distribution by numerically integrating Newton's equations of motion over time. However, this approach faces a fundamental limitation: the timescales accessible to simulation are often insufficient to adequately explore the complex, high-dimensional energy landscape of biologically relevant systems. This sampling bottleneck becomes particularly severe for molecules with slow, torsional degrees of freedom or systems featuring multiple metastable states separated by high energy barriers, where MD simulations tend to become trapped in local energy minima, failing to provide statistically representative conformational ensembles within practical computational timeframes [2].
The consequences of this sampling challenge extend directly to industrial applications, particularly in pharmaceutical development. For example, in researching treatments for conditions like spinal muscular atrophy (SMA), understanding molecular mechanisms and binding affinities relies on accurate thermodynamic predictions [1]. When conventional MD fails to adequately sample the Boltzmann distribution, computed observables such as free energy differences, binding affinities, and conformational populations remain unreliable, potentially leading to suboptimal therapeutic candidates advancing in development pipelines. This introduction examines the technical foundations of this critical bottleneck and sets the stage for understanding how surrogate model-assisted approaches offer a transformative solution.
The Computational Anatomy of the Sampling Bottleneck
Energy Landscapes and Timescale Disparities
The core challenge in conventional MD stems from the complex topography of molecular energy landscapes. Biomolecular systems typically exhibit a rough energy surface with numerous local minima separated by energy barriers of varying heights. The probability of transitioning between these minima decreases exponentially with the barrier height, following Arrhenius kinetics. This results in metastable states where systems remain trapped for timescales that can far exceed those practical for simulation [1]. For instance, in the context of protein-ligand interactions relevant to drug discovery, key conformational changes often occur on microsecond to second timescales, while state-of-the-art MD simulations typically reach only microsecond durations even with specialized hardware. This orders-of-magnitude disparity means that conventional MD cannot reliably generate the statistically independent samples needed for converged thermodynamic averages.
The severity of this sampling problem scales dramatically with system size and complexity. For a system of N atoms, the configuration space Ω ⊆ ℝ³N has an exponentially large volume that must be explored. Conventional MD navigates this space through local steps guided by the energy gradient, making it exceptionally difficult to traverse between distant regions of configuration space that might correspond to important functional states. Enhanced sampling methods like replica exchange molecular dynamics or metadynamics attempt to address this through sophisticated biasing strategies, but these require careful selection of collective variables and still face limitations in high-dimensional systems [1].
Quantitative Scaling Limitations
Table 1: Computational Scaling of Conventional MD Versus Theoretical Requirements
| System Size (N atoms) | MD Steps to Convergence | Practical MD Time Window | Theoretical Requirement |
|---|---|---|---|
| Small molecule (<50 atoms) | 10⁷-10⁹ steps | Nanoseconds-microseconds | Microseconds-milliseconds |
| Protein domain (~1000 atoms) | 10⁹-10¹¹ steps | Microseconds | Milliseconds-seconds |
| Protein-ligand complex (~10,000 atoms) | >10¹² steps | << Microsecond | Seconds-minutes |
| Macromolecular assembly (>100,000 atoms) | >10¹⁴ steps | << Nanosecond | Hours-days |
The computational burden of conventional MD manifests not only in simulation time but also in memory requirements and analysis overhead. Each simulated nanosecond for a typical protein-ligand system requires approximately 24 hours of wall-clock time on standard computing resources, making the collection of statistically independent samples for reliable ensemble averages practically prohibitive [2]. This fundamental limitation has motivated the development of novel approaches that can more efficiently explore configuration space without being constrained by the timescale barriers of conventional dynamics.
Surrogate Model-Assisted Molecular Dynamics (SMA-MD): A Paradigm Shift
Theoretical Foundation and Mechanistic Workflow
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) represents a fundamental reimagining of the conformational sampling problem. Rather than relying solely on physical dynamics to explore configuration space, SMA-MD leverages deep generative models to directly sample slow molecular degrees of freedom, followed by statistical reweighting and short MD simulations to refine the ensemble and ensure proper Boltzmann statistics [2]. This approach effectively decouples the exploration of configuration space from the limitations of physical timescales, allowing the system to jump between metastable states that would be inaccessible to conventional MD within practical simulation windows.
The theoretical foundation of SMA-MD rests on constructing a surrogate model ρ₁(𝐱) that approximates the true Boltzmann distribution μ(𝐱). This model is trained on available simulation data and is designed to enable efficient sampling and likelihood evaluation. The critical innovation lies in using this surrogate to generate initial conformational ensembles that already approximate the equilibrium distribution, then employing importance weighting and short MD simulations to correct any discrepancies and recover unbiased Boltzmann statistics [2]. This hybrid strategy maintains physical accuracy while overcoming the timescale limitations that plague conventional approaches.
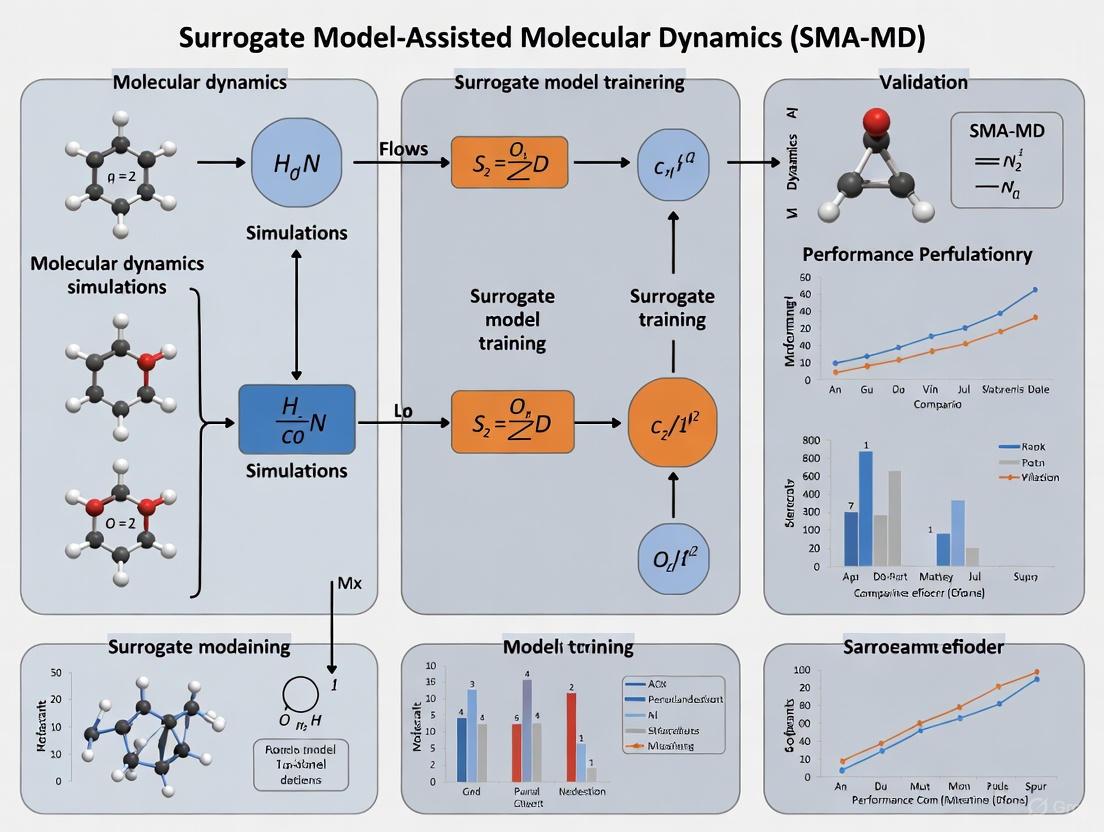
The SMA-MD Protocol: A Step-by-Step Methodology
The SMA-MD procedure implements a structured pipeline for generating conformational ensembles that effectively sample the Boltzmann distribution. The protocol consists of three integrated phases that combine machine learning generation with physical validation:
Phase 1: Surrogate Model Training and Configuration
- Step 1.1: System preparation and initialization using preprocessing.py to format input structures and define the relevant configuration space [2].
- Step 1.2: Training of the deep generative model (torsional diffusion) using train.py within the e3nn-env environment, specifying dataset paths and hyperparameters in parameters.py [2].
- Step 1.3: Model validation against held-out simulation data to ensure proper learning of the energy landscape.
Phase 2: Enhanced Conformational Sampling
- Step 2.1: Generation of initial conformational ensembles using sample.py within the e3nn-env environment, leveraging the trained surrogate model to explore slow degrees of freedom [2].
- Step 2.2: Energy evaluation of generated structures using energy_evaluation.py within the openmm-env to compute potential energies [2].
Phase 3: Statistical Reweighting and Refinement
- Step 3.1: Importance reweighting of the generated ensemble using weights wᵢ ∝ μ(𝐱ᵢ)/ρ₁(𝐱ᵢ) to recover unbiased Boltzmann statistics [1].
- Step 3.2: Molecular dynamics fine-tuning using md_finetuning.py within the openmm-env to relax structures and ensure physical realism [2].
- Step 3.3: Ensemble validation through comparison with experimental observables and convergence assessment.
This protocol has demonstrated empirical success in generating more diverse and lower-energy ensembles than conventional MD simulations, while maintaining the physical accuracy required for reliable thermodynamic calculations [2].
Research Reagent Solutions: Computational Tools for SMA-MD
Table 2: Essential Research Tools and Environments for SMA-MD Implementation
| Tool/Environment | Function | Implementation in SMA-MD |
|---|---|---|
| SMA-MD v1.b | Core procedure for conformational sampling | Primary framework combining generative modeling with MD [2] |
| e3nn-env | Specialized Python environment | Training and sampling of generative models [2] |
| openmm-env | Molecular dynamics environment | Energy evaluation and MD fine-tuning [2] |
| Torsional Diffusion | Conformer generation algorithm | Surrogate model for enhanced sampling [2] |
| Boltzmann Generators | Deep learning sampling approach | Alternative framework for equilibrium sampling [1] |
| HollowFlow | Efficient likelihood evaluation | Addresses computational bottlenecks in large systems [1] |
| CUDA-enabled GPU | Hardware acceleration | Essential for practical training and inference times [2] |
The SMA-MD methodology depends on specialized computational tools and environments that enable the integration of deep generative modeling with molecular dynamics. The e3nn-env provides the necessary infrastructure for training and sampling from equivariant neural network-based surrogate models, which are particularly suited for molecular systems due to their natural incorporation of rotational and translational symmetries [2]. The complementary openmm-env offers a validated ecosystem for running physics-based simulations with the AMBER, CHARMM, and other force fields, ensuring that the refinement phase maintains physical accuracy.
A critical innovation in scaling these approaches to biologically relevant systems is HollowFlow, which addresses the prohibitive computational cost of likelihood evaluation in large systems. By enforcing a block-diagonal Jacobian structure through non-backtracking graph neural networks, HollowFlow reduces the number of backward passes required for likelihood computation from scaling as 𝒪(N) to 𝒪(1) in system size N, achieving speed-ups of up to 10²× for systems of 55 particles [1]. This breakthrough enables the application of SMA-MD principles to increasingly complex molecular systems that would otherwise be computationally intractable.
Application to Therapeutic Development: The SMA Case Study
The practical implications of enhanced sampling methods extend directly to pharmaceutical development, particularly for complex genetic disorders like spinal muscular atrophy (SMA). SMA is caused by mutations in the SMN1 gene leading to deficient levels of survival motor neuron (SMN) protein, ultimately resulting in progressive motor neuron degeneration [3] [4]. Understanding the molecular mechanisms of SMN protein function and its interactions with potential therapeutic compounds represents an ideal application domain for SMA-MD approaches.
Recent advances in SMA treatment have produced multiple targeted therapies, including nusinersen (Spinraza), onasemnogene abeparvovec (Zolgensma), and risdiplam (Evrysdi), all aimed at increasing SMN protein levels [4] [5] [6]. These therapies operate through distinct mechanisms—nusinersen is an antisense oligonucleotide that modifies SMN2 splicing, onasemnogene abeparvovec is a gene replacement therapy delivering SMN1 via AAV9, and risdiplam is a small molecule SMN2 splicing modifier [4]. The development of next-generation SMA therapeutics requires detailed understanding of molecular interactions and binding thermodynamics that can be dramatically accelerated through enhanced sampling approaches.
Clinical trials continue to optimize SMA treatment paradigms, with recent studies including the DEVOTE trial (testing higher Spinraza doses), STEER trial (evaluating intrathecal Zolgensma), RAINBOWFISH trial (assessing Evrysdi in presymptomatic infants), and SAPHIRRE trial (testing apitegromab combination therapies) [6]. The complexity of these therapeutic mechanisms and their potential interactions creates a pressing need for efficient molecular sampling methods to understand structure-function relationships at unprecedented resolution.
Quantitative Performance Benchmarks and Validation
Computational Efficiency Metrics
The performance advantages of SMA-MD over conventional sampling approaches manifest in multiple dimensions, from sampling diversity to computational efficiency. Empirical evaluations demonstrate that SMA-MD generates more diverse conformational ensembles with lower potential energies compared to conventional MD simulations of equivalent computational cost [2]. This improved efficiency stems directly from the ability of the surrogate model to make large, informed jumps through configuration space rather than being constrained by local energy barriers.
Table 3: Performance Comparison of Sampling Methods for Molecular Systems
| Performance Metric | Conventional MD | Enhanced Sampling MD | SMA-MD |
|---|---|---|---|
| Sampling Diversity | Low (local traps) | Moderate | High (informed jumps) |
| Time to Convergence | Exponential | Polynomial | Near-linear |
| Likelihood Evaluation | Not required | Not required | 𝒪(1) with HollowFlow |
| System Size Scaling | 𝒪(N²) | 𝒪(N²) | 𝒪(1) with innovations |
| Energy Landscape Coverage | Incomplete | Improved | Comprehensive |
For the specific challenge of likelihood evaluation—a critical component for reweighting generated ensembles—the HollowFlow innovation provides dramatic improvements. In tests on a 55-particle Lennard-Jones system (LJ55), HollowFlow achieved a 102× speed-up compared to conventional approaches, reducing the scaling of backward passes from 𝒪(N) to 𝒪(1) with system size [1]. This breakthrough demonstrates how specialized architectures can overcome fundamental bottlenecks that have previously limited the application of advanced sampling methods to biologically relevant systems.
Thermodynamic Property Prediction
Beyond raw sampling efficiency, the ultimate validation of any enhanced sampling method lies in its ability to accurately predict experimental observables. SMA-MD has demonstrated particular promise in estimating implicit solvation free energies, a critical property in drug discovery and binding affinity prediction [2]. By combining broad configuration space exploration through generative modeling with physical refinement through short MD simulations, SMA-MD achieves an optimal balance between exploration and physical accuracy that exceeds what either approach can accomplish independently.
The reweighting procedure central to SMA-MD ensures that, despite being generated through a learned surrogate model, the final ensemble properly represents the true Boltzmann distribution. This is accomplished through importance weights wᵢ ∝ μ(𝐱ᵢ)/ρ₁(𝐱ᵢ), which correct any discrepancies between the surrogate model distribution ρ₁(𝐱) and the target Boltzmann distribution μ(𝐱) [1]. The result is unbiased estimation of thermodynamic observables with statistical confidence that would require orders-of-magnitude more computation using conventional approaches.
The critical bottleneck in conventional Molecular Dynamics—its inability to adequately sample the Boltzmann distribution for complex molecular systems within practical timeframes—represents a fundamental challenge across computational chemistry and biology. Surrogate Model-Assisted Molecular Dynamics addresses this limitation through a principled integration of deep generative modeling with physical simulation, enabling comprehensive exploration of configuration space while maintaining physical fidelity. The SMA-MD protocol demonstrates quantitatively superior performance in generating diverse, low-energy conformational ensembles and accurately predicting thermodynamic properties like solvation free energies.
Looking forward, several emerging trends promise to further expand the impact of SMA-MD approaches. The development of increasingly efficient architectures like HollowFlow will continue to push the size limits of addressable systems, while integration with experimental data will enhance model validation and refinement. Additionally, the application of these methods to specific therapeutic challenges—such as understanding the molecular mechanisms of SMA treatments and designing next-generation therapeutics—will provide tangible benefits to drug development pipelines. As these computational innovations mature, they will increasingly transform how researchers sample molecular complexity, ultimately accelerating the discovery of novel therapeutics for challenging conditions like spinal muscular atrophy and beyond.
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) represents a paradigm shift in computational molecular simulation. Traditional Molecular Dynamics (MD) is a powerful technique for studying microscopic phenomena by numerically integrating Newton's equations of motion for each particle in a molecular system [7]. However, its application to biologically relevant timescales and system sizes remains computationally prohibitive. SMA-MD addresses this fundamental limitation by integrating deep generative models as surrogate systems that learn the underlying distribution of molecular trajectories, enabling rapid exploration of configuration space and diverse downstream tasks that are not straightforward to address with MD itself [7] [8]. This approach moves beyond merely accelerating simulations toward creating flexible multi-task models that can be conditioned on specific structural or dynamic constraints for specialized applications.
The core innovation of SMA-MD lies in reformulating the surrogate modeling problem from learning single-point equilibrium distributions or transition densities to generative modeling of full trajectories viewed as time-series of 3D molecular structures [7]. This "molecular video" perspective incorporates temporal dynamics explicitly into the learning framework, enabling the model to capture both structural and dynamical properties of molecular systems. By appropriately conditioning these generative models on specific frames or parts of the system, SMA-MD can be adapted to diverse tasks including forward simulation, transition path sampling, trajectory upsampling, and dynamics-conditioned molecular design [7].
Core Computational Framework
Foundational MD Theory
Molecular dynamics simulation is based on integrating the equations of motion for each particle i in a molecular configuration, typically described by: [ Mi\ddot{\mathbf{x}}i = -\nabla{\mathbf{x}i}U(\mathbf{x}1\ldots\mathbf{x}N) ] where (Mi) is the mass, (\mathbf{x}i) is the position, and (U) is the potential energy function [7]. In practice, these equations are often modified with thermostats to model contact with surroundings, such as in the Langevin thermostat: [ d\mathbf{x}i = \mathbf{p}i/Mi\,dt,\quad d\mathbf{p}i = -\nabla{\mathbf{x}i}U\,dt - \gamma\mathbf{p}i\,dt + \sqrt{2Mi\gamma kT}\,d\mathbf{w} ] where (\mathbf{p}i) are the momenta, (\gamma) is the friction coefficient, and (d\mathbf{w}) represents Wiener noise [7]. This formulation converges to the Boltzmann distribution (p(\mathbf{x}1\ldots\mathbf{x}_N) \propto e^{-U/kT}), connecting dynamics to thermodynamic equilibrium.
Generative Modeling of Trajectories
The SMA-MD framework introduces a novel approach where generative models learn the joint probability distribution of entire molecular trajectories: [ p(\mathbf{X}^{(1:T)})=p(\mathbf{X}^{(1)},\mathbf{X}^{(2)},\ldots,\mathbf{X}^{(T)}) ] where (\mathbf{X}^{(t)}) represents the molecular configuration at time t [7]. This differs fundamentally from previous approaches that learned either the autoregressive transition density (p(\mathbf{X}^{(t+1)}|\mathbf{X}^{(t)})) or the equilibrium distribution (p(\mathbf{X})). Table: Comparison of MD Surrogate Modeling Approaches
| Approach | Target Distribution | Capabilities | Limitations |
|---|---|---|---|
| Boltzmann Generators | Equilibrium distribution (p(\mathbf{X})) | Efficient equilibrium sampling | No dynamical information |
| Transition Density Models | Single-step (p(\mathbf{X}^{(t+1)}|\mathbf{X}^{(t)})) | Forward simulation | Error accumulation in long trajectories |
| SMA-MD (Full Trajectory) | Joint (p(\mathbf{X}^{(1:T)})) | Forward/backward simulation, path sampling, upsampling, inpainting | Higher computational cost for training |
The generative model is typically parameterized using all-atom molecular trajectories in terms of residue offsets and sidechain torsions with respect to conditioning key frames, obtaining a generative modeling task over a 2D array of SE(3)-invariant tokens rather than residue frames or point clouds [7]. This representation ensures rotational and translational invariance while capturing essential molecular degrees of freedom.
Multi-Fidelity Optimization Framework
SMA-MD incorporates a multi-fidelity optimization strategy that uses Gaussian process surrogate modeling to build inexpensive models of physical properties as a function of force field parameters [8]. This approach enables rapid evaluation of approximate objective functions, greatly accelerating searches over parameter space and enabling the use of optimization algorithms capable of searching more globally [8].
The iterative framework performs global optimization with differential evolution at the surrogate level, followed by validation at the simulation level and surrogate refinement [8]. This addresses the fundamental limitation of traditional force field optimization where the computational expense of physical property simulations restricts the size of training datasets and number of optimization steps possible.
SMA-MD Workflow Architecture
The following diagram illustrates the integrated architecture of the SMA-MD framework, showing how traditional molecular dynamics components interact with deep generative models:
Key Technical Capabilities
Multi-Task Application Framework
SMA-MD enables diverse scientific applications through appropriate conditioning of the generative model:
Forward Simulation: Given the initial frame of a trajectory, the model samples a potential time evolution of the molecular system, serving as a familiar surrogate forward simulator of the reference dynamics [7].
Interpolation (Transition Path Sampling): Given the frames at two endpoints of a trajectory, the model samples a plausible path connecting them, which is important for studying reactions and conformational transitions [7].
Upsampling: Given a trajectory with timestep Δt between frames, the model upsamples the "framerate" by a factor of M to obtain a trajectory with timestep Δt/M, inferring fast motions from trajectories saved at less frequent intervals [7].
Inpainting: Given part of a molecule and its trajectory, the model generates the rest of the molecule and its time evolution to be consistent with the known part, enabling dynamics-scaffolded molecular design [7]. Table: Quantitative Performance of SMA-MD on Tetrapeptide Systems
| Task | Evaluation Metric | SMA-MD Performance | Baseline Method |
|---|---|---|---|
| Forward Simulation | Free Energy Surface Accuracy | High correlation with reference MD | Limited by simulation time |
| Transition Path Sampling | Path Likelihood | Realistic paths between metastable states | Not directly addressable |
| Trajectory Upsampling | Fast dynamics recovery | Accurate inference of sub-sampled motions | Information loss |
| Molecular Inpainting | Sequence Recovery | Higher than inverse folding methods | Limited by static frames |
Novel Inverse Problem Solving
A distinctive capability of SMA-MD is addressing inverse problems not straightforward to solve even with MD itself [7]. While forward simulation aligns with the typical modeling paradigm of approximating the data-generating process, tasks like transition path sampling, upsampling, and inpainting represent novel capabilities on scientifically important inverse problems.
For molecular inpainting, preliminary results show that SMA-MD obtains much higher sequence recovery than inverse folding methods based on one or two static frames [7]. This suggests that dynamical information provides additional constraints for biomolecular design that go beyond static structural information.
Experimental Protocols
SMA-MD Model Training Protocol
Objective: Train a generative model on molecular trajectory data for multi-task applications.
Materials and Reagents: Table: Research Reagent Solutions for SMA-MD Implementation
| Reagent/Software | Function | Specifications |
|---|---|---|
| MD Simulation Dataset | Training data for generative model | All-atom trajectories with sufficient sampling of relevant states |
| Scalable Interpolant Transformer (SiT) | Generative backbone architecture | Flow-based model for trajectory generation |
| Hyena Architecture | Long-context processing | Replacement for time-wise attention in long trajectories |
| Gaussian Process Models | Surrogate for physical properties | Accelerates parameter optimization |
| OpenFF Evaluator | Simulation workflow driver | Automated physical property simulations |
Procedure:
Trajectory Data Preparation:
- Collect MD trajectories representing the system of interest
- Parameterize all-atom molecular trajectories in terms of residue offsets and sidechain torsions with respect to conditioning key frames [7]
- Format data as a 2D array of SE(3)-invariant tokens
Model Architecture Selection:
- Implement Scalable Interpolant Transformer (SiT) as the flow-based generative backbone [7]
- For long trajectories (>100k frames), replace time-wise attention with Hyena architecture for long-context processing [7]
- Design appropriate conditioning mechanisms for different tasks (initial frame, endpoints, partial structures)
Training Protocol:
- Train model to maximize likelihood of trajectories in the dataset
- Use appropriate regularization to prevent overfitting
- Validate on held-out trajectory segments
Multi-Task Adaptation:
- For forward simulation: condition on initial frame only
- For transition path sampling: condition on endpoint frames
- For upsampling: condition on sparse trajectory frames
- For inpainting: condition on known part of molecular system [7]
Multi-Fidelity Force Field Optimization Protocol
Objective: Optimize force field parameters using Gaussian process surrogates to accelerate physical property matching.
Procedure:
Surrogate Model Construction:
- Select training set of physical properties (densities, enthalpies of vaporization, solvation free energies, etc.) [8]
- Perform initial simulations across parameter space to build training data for surrogates
- Construct Gaussian process models for each physical property as a function of force field parameters [8]
Iterative Optimization:
- Perform global optimization using differential evolution at the surrogate level [8]
- Validate promising parameter sets with full molecular dynamics simulations
- Refine surrogates with new simulation data
- Iterate until convergence of objective function
Validation and Testing:
- Assess optimized parameters on hold-out test sets
- Evaluate transferability to similar molecules not in training set [8]
- Compare performance against previous parameter sets
Implementation Considerations
Computational Requirements
SMA-MD implementation requires significant computational resources for both the initial MD simulations to generate training data and for training the generative models. The use of multi-fidelity optimization with Gaussian process surrogates reduces the overall computational cost by minimizing the number of expensive MD simulations required for parameter optimization [8].
Limitations and Future Directions
Current limitations include the need for substantial training data, potential distribution shift issues when applying models to novel chemical space, and challenges in modeling extremely long-timescale processes. Future work should focus on developing more sample-efficient training methods, incorporating physical constraints directly into the model architecture, and extending the approach to more complex biomolecular systems.
Molecular dynamics (MD) simulations are an indispensable tool for understanding the function of biomolecules at an atomistic level [9]. However, a critical limitation of conventional MD simulations is their restriction to relatively short timescales, which are often insufficient to sample slow biological processes, such as large-scale conformational changes in proteins or complex ligand-binding events [10]. This timescale problem results in inadequate sampling of the underlying free energy landscape, limiting the accuracy and predictive power of the simulations [10]. Enhanced sampling techniques have been developed to overcome the energetic barriers that trap conventional MD simulations in local minima, thereby enabling a more thorough exploration of conformational space [9] [10].
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) is a novel procedure designed to address this fundamental challenge [2]. It integrates deep generative models with enhanced sampling and statistical reweighting to efficiently generate broad, thermodynamically representative conformational ensembles. This application note details the specific protocols for implementing SMA-MD, framing its key advantages within the broader context of accelerating and improving molecular simulations for drug discovery and biomolecular research.
Key Advantages of SMA-MD
The SMA-MD procedure delivers two primary, interconnected advantages over conventional simulation approaches, leading to more accurate and computationally efficient characterization of molecular thermodynamics.
Table 1: Core Advantages of SMA-MD over Conventional MD
| Feature | Conventional MD | SMA-MD | Impact on Research |
|---|---|---|---|
| Sampling of Slow Degrees of Freedom | Relies on thermal fluctuations, often resulting in incomplete sampling of slow motions [10]. | Uses a deep generative model (Torsional Diffusion) to proactively sample slow torsional modes [2]. | Enables the study of large-scale conformational changes and rare events that are otherwise inaccessible. |
| Diversity of Conformational Ensemble | Can be trapped in local energy minima, producing a narrow, non-representative set of structures. | Generates more diverse and lower-energy ensembles than conventional MD [2]. | Provides a more complete picture of the accessible states of a molecule, crucial for understanding function and binding. |
| Thermodynamic Accuracy | Directly samples the force field's energy landscape, which can be inefficient. | Employs statistical reweighting followed by short MD simulations to refine the ensemble toward the Boltzmann distribution [2]. | Yields ensembles suitable for computing equilibrium properties, such as solvation free energies [2]. |
| Computational Efficiency | May require prohibitively long simulation times to achieve sufficient sampling. | Leverages a surrogate model to guide sampling, reducing the need for ultra-long simulations [2]. | Lowers the computational cost of obtaining well-sampled ensembles, accelerating research timelines. |
SMA-MD Experimental Protocol
The following section provides a detailed, step-by-step protocol for executing the complete SMA-MD procedure as described in the original work [2].
Prerequisites and Environment Setup
- System Requirements: Ensure access to a high-performance computing cluster with at least one CUDA-enabled GPU [2].
- Software Installation: Clone the SMA-MD repository from GitHub (
olsson-group/sma-md) and install the required dependencies [2]. - Conda Environments: Create and activate the two required Conda environments:
e3nn-env: Used for training and sampling from the generative model.openmm-env: Used for molecular dynamics and energy evaluation tasks [2].
Step-by-Step Workflow
The SMA-MD workflow consists of four major phases, which are also visualized in the diagram below.
Workflow Diagram Title: SMA-MD Protocol Workflow
Phase 1: Pre-processing
- Objective: Prepare the input data for the generative model.
- Protocol:
- Run the
preprocessing.pyscript. - Specify the paths to your molecular dataset and all necessary (hyper-)parameters in the
./parameters.pyfile [2]. This includes defining the molecular system and any specific sampling requirements.
- Run the
Phase 2: Generative Sampling with Torsional Diffusion
- Objective: Enhance the sampling of slow torsional degrees of freedom.
- Protocol:
- Training (If required): To train the torsional diffusion surrogate model on your dataset, run the
train.pyscript using thee3nn-envenvironment [2]. - Sampling: To generate a diverse initial ensemble of conformers, run the
sample.pyscript using thee3nn-envenvironment [2]. This step leverages the deep generative model to overcome energy barriers and explore conformational space more broadly than conventional MD.
- Training (If required): To train the torsional diffusion surrogate model on your dataset, run the
Phase 3: Statistical Reweighting
- Objective: Refine the generated ensemble towards the correct Boltzmann distribution.
- Protocol:
Phase 4: Molecular Dynamics Finetuning
- Objective: Further relax and validate the reweighted ensemble.
- Protocol:
Successful implementation of SMA-MD relies on a suite of software tools and computational resources. The table below catalogs the key components.
Table 2: Essential Research Reagents and Computational Resources for SMA-MD
| Item Name | Function / Role in the Workflow | Key Details |
|---|---|---|
| Torsional Diffusion | Deep generative model for sampling molecular conformers. | Based on the work by Jing et al. (2023); used in Phase 2 to generate initial conformational diversity [2]. |
| OpenMM | High-performance MD simulation toolkit. | Used for energy evaluation (Phase 3) and short MD finetuning (Phase 4) within the openmm-env [2]. |
| REform | Python library for statistical reweighting of ensembles. | Required dependency; installed via pip; crucial for the statistical reweighting in Phase 3 [2]. |
| e3nn | Euclidean neural networks library. | Provides the underlying framework for the generative model in e3nn-env (Phase 2) [2]. |
| CUDA | Parallel computing platform. | Mandatory for GPU acceleration, which is required for training and sampling with the generative model [2]. |
| Anaconda/Miniconda | Python package and environment manager. | Essential for managing the two complex and separate software environments (e3nn-env and openmm-env) [2]. |
Technical Application: Calculating Implicit Solvation Free Energies
A primary application of SMA-MD is the computation of thermodynamic properties, such as implicit solvation free energies [2]. The enhanced sampling and diverse ensembles generated by SMA-MD lead to more accurate and converged estimates of these properties compared to conventional MD. The logical flow of this application is outlined below.
Diagram Title: Solvation Free Energy Calculation
- Execute SMA-MD: Follow the protocol in Section 3 to generate a comprehensive, reweighted conformational ensemble for the molecule of interest.
- Compute Solvation Energy: For each conformer in the final ensemble, calculate its solvation free energy using an implicit solvent model (e.g., Generalized Born or Poisson-Boltzmann).
- Calculate Weighted Average: The final, thermodynamic solvation free energy is the statistically reweighted average of the solvation energies of all conformers in the ensemble. The diversity and thermodynamic accuracy of the SMA-MD ensemble ensure this estimate is more reliable than one derived from a less complete sampling method.
Surrogate Model-Assisted Molecular Dynamics represents a significant advancement in computational molecular science. By integrating deep generative models to enhance the sampling of slow degrees of freedom, followed by rigorous statistical reweighting, SMA-MD generates more diverse and thermodynamically accurate conformational ensembles than conventional simulation approaches. The detailed protocols and tools outlined in this application note provide researchers with a clear pathway to apply SMA-MD to challenging problems in drug discovery and biomolecular mechanism, ultimately enabling more reliable prediction of thermodynamic properties and a deeper understanding of molecular function.
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) represents a paradigm shift in computational molecular science, integrating deep generative models with physics-based simulations to sample the equilibrium ensembles of molecules. The accurate prediction of thermodynamic properties, crucial for drug discovery and materials design, hinges on effective sampling from the underlying Boltzmann distribution. Conventional approaches, notably Molecular Dynamics (MD), face significant challenges due to the vast separation of timescales between femtosecond-level integration steps and the millisecond-level transitions often required for full exploration of a molecule's conformational landscape. Enhanced sampling techniques have only partially bridged this gap, remaining sensitive to hyperparameters and difficult to apply generally. SMA-MD emerges as a novel procedure that strategically leverages deep generative models to enhance the sampling of slow degrees of freedom, subsequently applying statistical reweighting and short simulations to recover the equilibrium distribution. This framework directly addresses the sampling bottlenecks of conventional MD, offering a pathway to more diverse, lower-energy ensembles and enabling the computation of previously inaccessible thermodynamic properties [2] [11].
Core Theoretical Framework
The SMA-MD framework is architecturally founded upon a sequential integration of three methodological pillars: generative modeling for conformational exploration, statistical reweighting for ensemble correction, and molecular dynamics for local refinement and validation.
Generative Models for Conformational Exploration
Generative models are deep learning frameworks that parameterize and enable the sampling of high-dimensional, multimodal distributions. Within SMA-MD, they are specifically trained to sample molecular configurations conditioned on the identity of a molecular system, providing an end-to-end paradigm for sampling equilibrium distributions that circumvents the sequential bottlenecks of physical simulation. A defining property of these models is their capacity to draw statistically independent samples with fixed computational cost, thereby overcoming the curse of correlated samples that severely limits the efficiency of molecular dynamics. These models, termed ensemble emulators, often utilize architectural elements from state-of-the-art protein structure prediction networks, such as AlphaFold2, to achieve transferability across diverse protein sequences. By conditioning a diffusion model on features extracted from Multiple Sequence Alignments, these emulators can produce a distribution of structures that recall experimentally observed conformational states with significantly improved diversity compared to single-point predictions [11].
Statistical Reweighting and the Boltzmann Distribution
The ensembles generated by deep generative models do not, a priori, represent the equilibrium Boltzmann distribution. The generated ensemble is therefore subjected to a statistical reweighting procedure. This critical step assigns a statistical weight to each generated conformation, ensuring the final ensemble accurately reflects the true thermodynamic probabilities as defined by the system's potential energy. The process often involves evaluating the energy of generated conformations using a classical force field or a coarse-grained potential, then applying reweighting techniques such as Boltzmann weighting or more sophisticated methods like Multistate Bennet Acceptance Ratio to compute equilibrium properties from the non-equilibrium samples. This step effectively grounds the data-driven generative process in the physical energy landscape [2].
Molecular Dynamics for Fine-Tuning and Validation
The final component of the SMA-MD workflow involves short molecular dynamics simulations initiated from the reweighted ensemble. These simulations serve a dual purpose: they act as a local sampler to refine the generated structures and relax any high-energy atomic clashes, and they provide a means to validate the thermodynamic quality of the reweighted ensemble. By running multiple, short, and independent simulations from different starting points in the reweighted ensemble, SMA-MD can confirm the stability of the predicted conformations and compute dynamic properties not accessible from the static generative model alone. This synergy between global generative sampling and local physical simulation is the cornerstone of the SMA-MD approach [2].
The logical and procedural relationships between these core theoretical components are visualized in the following workflow:
Quantitative Performance Comparison
The empirical performance of SMA-MD and related AI-based ensemble methods can be evaluated across several key dimensions, including system size, transferability, and the nature of training data. The table below synthesizes data from various state-of-the-art methods, providing a comparative overview of their capabilities and scope.
Table 1: Performance and Scope of AI-Based Methods for Sampling Protein Ensembles
| Method | Category | Largest System Demonstrated | Transferability | Training Data |
|---|---|---|---|---|
| SMA-MD [2] | Generative Model & MD | Small Molecules | Specific Molecules | Molecular Datasets |
| DiG [11] | Generative Model | 306 AA | Monomers | PDB + 100 µs MD + Force Field |
| AlphaFlow [11] | Generative Model | PDB-based (up to 768 AA) | Monomers | PDB + 380 µs MD |
| UFConf [11] | Generative Model | PDB-based | Monomers | PDB |
| Charron et al. [11] | Coarse-grained ML Potential | 189 AA | Monomers & Protein-Protein Interactions | 100 µs MD |
| Boltzmann Generators [11] | Generative Model (Exact Likelihood) | 58 AA | No (Per-System Training) | 1 ms MD + Force Field |
The data reveals a trade-off between system size, transferability, and methodological complexity. Generative models pre-trained on large structural databases (e.g., DiG, AlphaFlow) demonstrate strong transferability to monomeric proteins of substantial size. In contrast, methods like SMA-MD and Boltzmann Generators, which more tightly integrate with physical potentials and simulations, have thus far been applied to smaller systems but offer a direct link to the underlying energy landscape, which is crucial for computing thermodynamic properties like free energies [11].
Application Notes & Protocols
This section provides a detailed, actionable protocol for implementing the SMA-MD procedure, from environment setup to the computation of thermodynamic properties.
Protocol: SMA-MD for Conformational Ensemble Generation
Objective: To generate a Boltzmann-weighted conformational ensemble for a target molecule and compute its implicit solvation free energy.
I. Prerequisites and Environment Setup
- Software & Hardware:
- Install Anaconda or Miniconda with Python 3.9.
- Ensure access to a CUDA-enabled GPU.
- Clone the SMA-MD repository:
git clone https://github.com/olsson-group/sma-md
- Environment Configuration:
- SMA-MD utilizes two separate Conda environments.
- Create and activate the
e3nn-envfor training and sampling from the generative model. - Create and activate the
openmm-envfor all molecular dynamics and energy evaluation tasks. The dependencies are complex; follow the installation steps in the repository precisely to avoid conflicts [2].
II. Data Preprocessing
- Input Preparation: Prepare the input data file for your target molecule, specifying its atomic structure and any relevant chemical information.
- Run Preprocessing: Execute the preprocessing script to format the data for the generative model.
- Critical Parameter: All dataset paths and molecular indexes must be correctly specified in
./parameters.py[2].
- Critical Parameter: All dataset paths and molecular indexes must be correctly specified in
III. Generative Model Sampling
- Activate Environment: Activate the
e3nn-env. - Sampling: Run the sampling script to generate a diverse set of candidate conformations. This step uses a trained surrogate model (e.g., Torsional Diffusion) to explore the molecule's torsional space.
- Output: A file (
raw_ensemble.pkl) containing the generated, non-equilibrium ensemble of structures [2].
- Output: A file (
IV. Statistical Reweighting
- Activate Environment: Switch to the
openmm-envfor energy calculations. - Energy Evaluation: Calculate the potential energy of every generated conformation in the desired state (e.g., in vacuum for solvation free energy calculations) using a classical force field.
- Reweighting Algorithm: Implement a reweighting scheme. For the Boltzmann distribution, the weight for a conformation i with energy E_i is proportional to exp(-E_i / k_B T), where k_B is Boltzmann's constant and T is the temperature. Normalize the weights so that they sum to 1.
- Note: This step effectively filters out high-energy, non-physical conformations that may have been generated and assigns correct thermodynamic probabilities [2].
V. Molecular Dynamics Fine-Tuning
- Structure Relaxation: Use the reweighted ensemble as a starting point for short, unbiased MD simulations to locally relax the structures and alleviate any residual steric strains.
- Rationale: These short simulations ensure atomic-level realism and stability of the final ensemble members [2].
VI. Computation of Thermodynamic Properties
- Free Energy Calculation: To compute an implicit solvation free energy, repeat the Energy Evaluation step (IV.2) for the final ensemble in the solvated state. With the energies of each conformation in both vacuum and solvated states, the free energy difference can be computed using methods such as the Zwanzig equation or Bennet Acceptance Ratio, applied over the reweighted ensemble [2].
- Ensemble Analysis: Analyze the
final_ensemble.pklto compute other desired properties, such as root-mean-square fluctuations (RMSF), radius of gyration, or dihedral angle distributions.
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details key computational "reagents" and resources essential for conducting SMA-MD research.
Table 2: Essential Research Reagents and Solutions for SMA-MD
| Item | Function/Description | Example/Note |
|---|---|---|
| Generative Model | Surrogate model for exploring conformational space; learns to generate plausible molecular structures. | Torsional Diffusion [2]. |
| Force Field | Physical potential used for energy evaluation during reweighting and MD fine-tuning. | Classical all-atom force fields (e.g., AMBER, CHARMM). |
| Molecular Dynamics Engine | Software to perform short, fine-tuning simulations for local relaxation. | OpenMM [2]. |
| Reweighting Algorithm | Statistical method to correct the generated ensemble to the equilibrium Boltzmann distribution. | Boltzmann reweighting; Multistate Bennet Acceptance Ratio (MBAR). |
| Training Datasets | Large-scale structural and dynamic data used to pre-train or inform generative models. | Protein Data Bank (PDB); molecular dynamics trajectories (e.g., ATLAS [13], mdCATH [17]) [11]. |
Visualization of Logical Pathways
The conceptual journey from a molecular system to a thermodynamically valid ensemble involves a clear, hierarchical decision process. The following diagram outlines this high-level logical pathway, connecting the core theoretical components and their outcomes.
Implementing SMA-MD: A Step-by-Step Workflow from Generative Sampling to Free Energy Calculations
Application Note: Surrogate Model-Assisted Molecular Dynamics for Spinal Muscular Atrophy Drug Discovery
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder and a leading genetic cause of infant mortality, with an estimated incidence of approximately 1 in 10,000 live births [12] [13]. This devastating disease results from biallelic mutations in the survival motor neuron 1 (SMN1) gene, leading to insufficient levels of SMN protein and subsequent degeneration of alpha motor neurons in the spinal cord [14] [12]. The severity of SMA is partially modulated by the copy number of the SMN2 gene, a paralog that produces only a small fraction of functional SMN protein due to alternative splicing that predominantly excludes exon 7 [12] [13].
The recent development of disease-modifying therapies (DMTs) for SMA has transformed the therapeutic landscape. Three primary pharmacological approaches have received regulatory approval: nusinersen (an antisense oligonucleotide), onasemnogene abeparvovec (a gene therapy), and risdiplam (a small-molecule SMN2 splicing modifier) [12] [13] [15]. These therapies share the common objective of increasing functional SMN protein levels, albeit through distinct molecular mechanisms. However, treatment response varies considerably based on factors including SMA type, age at treatment initiation, SMN2 copy number, and disease duration [16] [13]. This heterogeneity underscores the critical need for advanced computational approaches to optimize therapeutic strategies and identify novel drug candidates.
The Three-Stage SMA-MD Procedure integrates molecular dynamics (MD) simulations with machine learning-based surrogate models to accelerate the discovery and optimization of SMA therapeutics. This workflow architecture addresses the profound computational challenges associated with simulating large biomolecular systems over biologically relevant timescales, enabling rapid screening of compound libraries and detailed investigation of molecular interactions governing SMN2 splicing modulation.
Clinical and Molecular Context of SMA
SMA Classification and Natural History
SMA demonstrates a broad spectrum of clinical severity, historically classified into types based on age of onset and maximum motor function achieved [17] [13]. The traditional classification system and natural history are summarized in Table 1.
Table 1: Clinical Classification of Spinal Muscular Atrophy
| SMA Type | Age of Onset | Maximum Motor Function | SMN2 Copy Number | Natural History |
|---|---|---|---|---|
| Type I (most severe) | <6 months | Never sits independently | 2 copies (80%) | Progressive weakness, respiratory failure, early mortality |
| Type II (intermediate) | 6-18 months | Sits independently, never walks independently | 3 copies (82%) | Slowly progressive, scoliosis, respiratory complications |
| Type III (milder) | >18 months | Walks independently | 3-4 copies | Gradual loss of ambulation, normal lifespan |
| Type IV (adult-onset) | Adulthood | Walks independently | 4-8 copies | Mild proximal weakness, slow progression |
Approved SMA Therapies and Mechanisms
The three currently approved disease-modifying therapies for SMA target the fundamental molecular pathology through distinct approaches, as detailed in Table 2.
Table 2: Approved Disease-Modifying Therapies for SMA
| Therapy | Mechanism of Action | Administration Route | Key Clinical Trials | Efficacy Findings |
|---|---|---|---|---|
| Nusinersen | ASO that binds ISS-N1 in SMN2 intron 7, promoting exon 7 inclusion | Intrathecal injection | ENDEAR (Type I), CHERISH (Type II/III) | 51% motor milestone response in Type I vs. 0% control; significant HFMSE improvement in later-onset |
| Risdiplam | Small molecule that modulates SMN2 splicing to include exon 7 | Oral daily | FIREFISH (Type I), SUNFISH (Type II/III) | 41% of infants sat without support for ≥5 seconds; significant motor function improvements |
| Onasemnogene abeparvovec | AAV9-mediated SMN1 gene replacement | Single intravenous infusion | SPR1NT (presymptomatic) | 100% of presymptomatic infants sat independently, 92% walked with assistance |
The Three-Stage SMA-MD Procedure: Experimental Protocol
Stage 1: System Preparation and Surrogate Model Training
Molecular System Construction
Objective: Prepare accurate structural models of the SMN2 pre-mRNA splicing complex for molecular dynamics simulations.
Materials and Reagents:
- Source PDB Files: Obtain crystal structures of splicing factors (hnRNPA1, U2AF65, U1 snRNP) from Protein Data Bank
- SMN2 RNA Sequence: Construct SMN2 pre-mRNA containing exon 7, intron 7, and flanking exonic regions
- Small Molecule Libraries: Compound collections for screening (e.g., ZINC20, Enamine, in-house repositories)
- Molecular Visualization Software: PyMOL 3.0 or ChimeraX for structural analysis
- Force Field Parameters: RNA-specific force fields (OL3, DES-AMBER) and small molecule parameterization (GAFF2)
Protocol:
- Retrieve and Prepare Spliceosome Components:
- Download PDB structures 5X9M (U1 snRNP), 6PWQ (hnRNPA1-RNA complex), and 4PJO (U2AF65)
- Remove crystallographic water molecules and additives using PyMOL
- Add missing hydrogen atoms and side chains using MODELLER
- Generate protonation states appropriate for physiological pH (7.4)
Construct SMN2 Pre-mRNA Model:
- Build SMN2 RNA sequence (exon 6 - intron 7 - exon 7 - intron 8 - exon 8) using UCSF ChimeraX
- Incorporate known structural motifs including the ISS-N1 target sequence (5'-UAGUUUUA-3') in intron 7
- Fold RNA structure using RNAComposer with constraints from chemical mapping data
Dock Small Molecule Binders:
- Prepare risdiplam and analogs using LigPrep (Schrödinger) with OPLS4 force field
- Perform molecular docking to ISS-N1 region using AutoDock-GPU
- Select top 50 poses based on binding energy for MD simulation
Surrogate Model Development
Objective: Train machine learning models to predict binding free energies from simplified molecular descriptors.
Procedure:
- Feature Engineering:
- Calculate molecular descriptors (Morgan fingerprints, RDKit 2D descriptors)
- Extract interaction fingerprints from short MD trajectories (50 ps)
- Generate quantum chemical features (partial charges, HOMO-LUMO gap) for representative compounds
- Model Training:
- Implement gradient boosting regression (XGBoost) with 5-fold cross-validation
- Train on dataset of 500 compounds with known binding affinities to RNA targets
- Validate model performance using root mean square error (RMSE) and R² metrics
- Target prediction accuracy of RMSE < 1.5 kcal/mol for binding free energies
Stage 2: High-Throughput Binding Affinity Screening
Workflow Architecture and Execution
Objective: Rapidly screen large compound libraries (10,000-100,000 molecules) for SMN2 ISS-N1 binding.
Computational Resources:
- High-Performance Computing Cluster: Minimum 100 nodes, 20 cores per node
- GPU Acceleration: NVIDIA A100 or V100 GPUs for MD simulations
- Parallel Processing Framework: Apache Spark for distributed scoring
Protocol:
- Library Preparation:
- Filter compound libraries for drug-like properties (Lipinski's Rule of Five)
- Generate 3D conformations using OMEGA (OpenEye)
- Standardize tautomer and protonation states at pH 7.4
Surrogate Model Screening:
- Calculate molecular descriptors for entire library
- Apply trained surrogate models to predict binding affinities
- Select top 1,000 compounds for short MD simulations
Short MD Validation:
- Solvate ligand-RNA complexes in TIP3P water box with 10 Å buffer
- Add neutralizing ions (Na⁺, Cl⁻) to physiological concentration (150 mM)
- Energy minimize using steepest descent algorithm (5,000 steps)
- Perform 10 ns MD simulations at 310 K with 2 fs timestep
- Calculate MM/PBSA binding energies for final validation
Stage 3: Free Energy Perturbation and Mechanism of Action
Alchemical Free Energy Calculations
Objective: Precisely quantify relative binding affinities for top candidate compounds.
Materials:
- Software: Schrödinger Desmond, OpenMM, GROMACS
- Force Fields: RNA.OL3 for RNA, GAFF2 for small molecules, TIP3P water
- Analysis Tools: MDAnalysis, PyEMMA, VMD
Protocol:
- System Preparation:
- Build simulation systems for top 20 compounds identified in Stage 2
- Solvate in octahedral water boxes with 10 Å buffer distance
- Neutralize with ions and add 150 mM NaCl
Equilibration Protocol:
- Energy minimization: 5,000 steps steepest descent
- NVT equilibration: 100 ps with positional restraints on heavy atoms
- NPT equilibration: 1 ns with Berendsen barostat (1 atm) and Langevin thermostat (310 K)
Production FEP Simulations:
- Run 20 ns alchemical transitions between compound pairs
- Use 24 lambda windows with soft-core potentials
- Calculate ΔΔG values using Multistate Bennett Acceptance Ratio (MBAR)
- Perform error analysis with block averaging and bootstrap methods
Splicing Modulation Analysis
Objective: Characterize molecular mechanisms of SMN2 splicing modulation.
Procedure:
- Trajectory Analysis:
- Extract 1 µs aggregate simulation data per compound
- Calculate hydrogen bonding occupancy with key ISS-N1 nucleotides
- Monitor conformational changes in exon 7 5' splice site
- Quantify protein-RNA interaction dynamics for hnRNPA1 displacement
- Experimental Validation Prioritization:
- Rank compounds by binding affinity and synthetic accessibility
- Select top 5-10 candidates for in vitro splicing assays
- Design analogs to optimize favorable interactions identified in simulations
Workflow Visualization and Implementation
Three-Stage SMA-MD Workflow Architecture
SMN2 Splicing Regulation and Therapeutic Targeting
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for SMA-MD
| Reagent/Tool | Category | Source/Provider | Function in SMA-MD Workflow |
|---|---|---|---|
| hnRNPA1 Protein | Recombinant Protein | Thermo Fisher Scientific | Key splicing repressor protein for binding studies and complex construction |
| SMN2 RNA Constructs | Nucleic Acids | Integrated DNA Technologies | Target sequence for docking and MD simulations of splicing modulation |
| Risdiplam Analogs | Small Molecule Library | MedChemExpress | Reference compounds for validation and analog design |
| AMBER20 | Molecular Dynamics Software | University of California, San Diego | Production MD simulations and free energy calculations |
| Desmond | MD Simulation | Schrödinger | GPU-accelerated MD for high-throughput screening |
| OpenMM | MD Engine | Stanford University | Customizable platform for alchemical free energy calculations |
| AutoDock-GPU | Docking Software | Scripps Research | High-throughput molecular docking of compound libraries |
| XGBoost | Machine Learning | Open Source | Surrogate model implementation for binding affinity prediction |
| MDAnalysis | Analysis Tool | Open Source | Trajectory analysis and feature extraction from MD simulations |
| ChimeraX | Visualization | UCSF | Molecular visualization and model building |
The Three-Stage SMA-MD Procedure represents a robust computational framework that synergistically combines molecular dynamics simulations with machine learning approaches to accelerate the discovery of novel SMA therapeutics. By integrating detailed structural models of the SMN2 splicing apparatus with efficient screening methodologies, this workflow addresses critical bottlenecks in traditional drug discovery pipelines.
The clinical urgency for improved SMA treatments is underscored by the limitations of current therapies, including variable treatment responses, administration challenges, and incomplete efficacy in older patients with established disease [16] [13]. The SMA-MD workflow directly addresses these challenges by enabling rapid identification of novel splicing modulators that may offer improved efficacy, blood-brain barrier penetration, and administration profiles.
Future developments will focus on incorporating enhanced sampling techniques to capture rare conformational transitions in the spliceosome, integrating quantum mechanical/molecular mechanical (QM/MM) methods for investigating chemical modifications, and expanding the framework to include multi-target approaches addressing both SMN-dependent and SMN-independent pathways [14] [12] [13]. The continued validation of this computational framework against experimental splicing assays and clinical outcomes will further refine its predictive accuracy and utility in the ongoing effort to develop optimized therapies for spinal muscular atrophy.
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) is an advanced computational procedure designed to sample the equilibrium ensemble of small molecules more effectively than conventional Molecular Dynamics (MD) simulations [18] [19]. The accurate prediction of thermodynamic properties, such as implicit solvation free energies, is crucial in drug discovery and materials design. This task relies on proper sampling from the underlying Boltzmann distribution, which can be challenging with standard simulation approaches [18]. The entire SMA-MD protocol consists of three primary stages: 1) leveraging deep generative models for initial conformational exploration, 2) statistical reweighting of the generated ensemble, and 3) running short simulations for refinement [2] [19]. This application note provides a detailed experimental protocol for the first and foundational stage: using deep generative models to enhance the sampling of a molecule's slow degrees of freedom, thereby generating a diverse and low-energy initial conformational ensemble [2].
Experimental Principles and Workflow
The core principle of this initial stage is to employ a deep generative model, specifically a torsional diffusion model, to explore the conformational landscape of a small molecule more broadly and efficiently than traditional MD [2]. Conventional MD simulations can be computationally expensive and may become trapped in local energy minima, failing to adequately sample the full conformational space within practical timeframes. The torsional diffusion model acts as a surrogate, learning the underlying distribution of molecular conformations and generating a diverse set of candidate structures that cover a wider range of the molecule's potential energy surface [2]. This procedurally generated ensemble serves as a high-quality starting point for the subsequent stages of statistical reweighting and short MD simulations, which collectively refine the ensemble to produce a Boltzmann-ranked set of conformations [19].
Workflow Visualization
The following diagram illustrates the logical sequence and data flow for Stage 1 of the SMA-MD procedure:
Materials and Reagents
Research Reagent Solutions
Table 1: Essential computational tools and environments for implementing Stage 1 of SMA-MD.
| Item Name | Function/Application in the Protocol |
|---|---|
| SMA-MD Codebase (v1.b) | The primary software package containing all necessary scripts for preprocessing, training, sampling, and energy evaluation [2]. |
| Anaconda/Miniconda | A package and environment management system used to create isolated Python environments with specific dependency versions [2]. |
| e3nn-env | A specific Conda environment required for running the training and sampling scripts of the torsional diffusion generative model [2]. |
| openmm-env | A specific Conda environment required for running molecular dynamics-related tasks, including energy evaluation and fine-tuning [2]. |
| Terason 2000 System (5-MHz probe) | An ultrasound system used for quantitative muscle echogenicity in validation studies [20]. |
| Adobe Photoshop | Used for image analysis to quantify tissue luminosity from ultrasound data [20]. |
Detailed Experimental Protocol
Step 1: Software and Environment Setup
- Obtain the SMA-MD code: Clone the official repository from GitHub using the command:
git clone https://github.com/olsson-group/sma-md[2]. - Navigate to the directory: Change into the cloned repository's directory:
cd sma-md[2]. - Install
reformdependency: Navigate to thereformdirectory and install the package using pip:pip install .[2]. - Create and activate Conda environments: The procedure requires two separate Conda environments. Follow the repository's instructions to create
e3nn-env(for generative model tasks) andopenmm-env(for MD tasks) [2].
Step 2: Data Preprocessing
- Configure parameters: Before execution, specify all necessary parameters, including dataset paths and hyperparameters, in the
./parameters.pyfile [2]. - Run preprocessing script: Execute the
preprocessing.pyscript using thee3nn-envenvironment. This step prepares the input data for the generative model [2].
Step 3: Training the Generative Model
- Activate the correct environment: Ensure the
e3nn-envConda environment is active [2]. - Execute training: Run the
train.pyscript. This will train the torsional diffusion model on the prepared dataset to learn the distribution of molecular conformations [2].
Step 4: Conformational Sampling
- Remain in the
e3nn-envenvironment. - Generate the initial ensemble: Execute the
sample.pyscript. This uses the trained torsional diffusion model to generate a diverse set of molecular conformers, constituting the output of Stage 1 [2].
Step 5: Energy Evaluation and MD Fine-Tuning (Preparation for Stage 2)
- Switch environments: Activate the
openmm-envConda environment [2]. - Perform energy evaluation: Run the
energy_evaluation.pyscript to analyze the generated ensemble [2]. - Initiate short simulations: Execute
md_finetuning.pyto begin the refinement of the generated ensemble with short MD simulations, bridging to the full SMA-MD procedure [2].
Data Analysis and Interpretation
Key Performance Metrics
Table 2: Quantitative metrics for evaluating the performance of the initial conformational exploration stage.
| Metric | Description | Interpretation |
|---|---|---|
| Ensemble Diversity | The structural variety of conformers generated, often measured by the root-mean-square deviation (RMSD) between members. | Higher diversity indicates better exploration of the conformational landscape, helping to avoid getting trapped in local minima [18]. |
| Average Conformer Energy | The mean potential energy of the generated conformers, calculated using a molecular mechanics forcefield. | A lower average energy suggests the model is preferentially generating more stable, physically realistic structures [18]. |
| Luminosity Ratio (LR) | In validation studies, the ratio of muscle luminosity to subcutaneous fat luminosity in quantitative ultrasound. | Increased LR correlates with greater disease severity in Spinal Muscular Atrophy (SMA), e.g., Type 2: 3.85 ± 1.3 vs. Normal: 1.27 ± 0.26 [20]. |
Troubleshooting and Optimization
Table 3: Common issues encountered during Stage 1 implementation and recommended solutions.
| Problem | Potential Cause | Suggested Solution |
|---|---|---|
| Installation failures | Complex dependencies or conflicting package versions. | Strictly use the Conda environments (e3nn-env, openmm-env) as specified in the prerequisites [2]. |
| Poor quality generated conformers | Insufficient training data or suboptimal hyperparameters in parameters.py. |
Review and adjust the hyperparameters in ./parameters.py. Ensure the training dataset is representative and of high quality [2]. |
| Low correlation between LR and strength | Heterogeneous patient population or measurement error. | Ensure a homogeneous subject group and standardized measurement protocols, as correlation can be moderate (e.g., r = -0.588 in SMA) [20]. |
Statistical reweighting is a cornerstone technique in computational chemistry for reconciling theoretical simulations with experimental data. Within the framework of Surrogate Model-Assisted Molecular Dynamics (SMA-MD), reweighting serves as the critical step that refines a diverse set of conformations generated by a deep generative model, biasing them toward the experimentally observed Boltzmann equilibrium distribution [18]. This process transforms a broadly sampled conformational ensemble into a physically accurate one, enabling the precise computation of thermodynamic properties. Ensemble refinement addresses the inverse problem of determining the statistical weights of ensemble members by integrating experimental measurements, thereby providing faithful descriptions of dynamic biomolecules, such as intrinsically disordered proteins, which are crucial in drug development [21].
Theoretical Foundation of Bayesian Ensemble Refinement (BioEn)
The Bayesian Ensemble Refinement (BioEn) method provides a robust mathematical framework for statistical reweighting [21]. It is a generalization of the earlier Ensemble Refinement of SAXS (EROS) method. The core principle involves optimizing the statistical weights ( w_\alpha ) of N ensemble members (with ( \alpha = 1, ..., N )) to maximize the posterior probability given the experimental data.
The fundamental objective is to minimize the negative log-posterior function: [ L = \frac{1}{2} \chi^2 - \theta S_{KL} ] where:
- ( \chi^2 = \sum{i=1}^{M} \frac{ \left( Yi - \langle yi \rangle \right)^2 }{\sigmai^2} ) quantifies the agreement between experimental observations ( Yi ) and ensemble-averaged calculated values ( \langle yi \rangle = \sum{\alpha=1}^{N} w\alpha y_{i\alpha} ).
- ( S{KL} = - \sum{\alpha=1}^{N} w\alpha \ln \left( \frac{w\alpha}{w\alpha^0} \right) ) is the Kullback-Leibler divergence, which acts as a regularization term that penalizes significant deviations from the reference weights ( w\alpha^0 ).
- The parameter ( \theta ) expresses confidence in the reference ensemble, often derived from a molecular dynamics force field or, in the context of SMA-MD, a surrogate model [21] [18].
The solution is found by optimizing the weights ( w\alpha ) under the constraints ( \sum{\alpha=1}^{N} w\alpha = 1 ) and ( w\alpha > 0 ). The uniqueness of the optimal solution is guaranteed by the convexity of the negative log-posterior ( L ) [21].
Table 1: Key Components of the BioEn Negative Log-Posterior
| Component | Mathematical Expression | Description |
|---|---|---|
| χ² (Goodness-of-fit) | ( \chi^2 = \sum{i=1}^{M} \frac{ \left( Yi - \langle yi \rangle \right)^2 }{\sigmai^2} ) | Measures discrepancy between experimental data and ensemble-averaged predictions. |
| SKL (Regularization) | ( S{KL} = - \sum{\alpha=1}^{N} w\alpha \ln \left( \frac{w\alpha}{w_\alpha^0} \right) ) | Kullback-Leibler divergence; penalizes large deviations from the reference ensemble. |
| θ (Confidence Parameter) | Scalar parameter | Balances the influence of the experimental data against the prior information from the reference ensemble. |
Efficient Numerical Optimization Methods
To solve this constrained optimization problem efficiently for large N (number of structures) and M (number of data points), two complementary unconstrained formulations are employed [21]:
Log-Weights Optimization: The problem is recast in terms of the variables ( g\alpha = \ln w\alpha ). This transformation implicitly handles the positivity and normalization constraints, allowing the use of efficient gradient-based algorithms like L-BFGS. The gradient of ( L ) with respect to ( g\mu ) is given by: [ \frac{\partial L}{\partial g\mu} = w\mu \left[ \sum{i=1}^{M} \frac{(Yi - \langle yi \rangle)y{i\mu}}{\sigmai^2} + \theta \left( \ln w\mu - \ln w\mu^0 + 1 \right) \right] - \delta\mu ] where ( \delta\mu ) is a constant ensuring normalization [21].
Generalized Forces Optimization: This lower-dimensional approach solves for the M Lagrange multipliers ( \lambdai ) (generalized forces) associated with the M experimental constraints. The optimal weights can be expressed analytically as: [ w\alpha = \frac{w\alpha^0 \exp \left[ \sum{i=1}^{M} \lambdai y{i\alpha} \right]}{Z(\lambda)} ] where ( Z(\lambda) ) is the normalization partition function. The optimization then minimizes a convex function of the ( \lambda_i ) [21].
The choice between methods depends on the specific problem dimensions; the log-weights method is typically efficient for moderate N, while the generalized forces method is superior for very large N and moderate M [21].
Application Notes for SMA-MD Integration
The SMA-MD procedure explicitly incorporates statistical reweighting as a final refinement step [18]. The surrogate model, a deep generative model, accelerates the sampling of slow degrees of freedom and generates a diverse initial conformational ensemble. Subsequently, this ensemble is statistically reweighted against experimental or high-fidelity theoretical data. Finally, short, conventional molecular dynamics simulations are performed to validate and relax the reweighted structures [18].
A key consideration, especially when deriving reference weights from a surrogate model, is the accurate estimation of the confidence parameter ( \theta ). This parameter can be determined through cross-validation against held-out experimental data or based on the estimated uncertainty of the surrogate model's predictions. The reweighting step ensures that the final ensemble reflects the true Boltzmann distribution, which is critical for accurate computation of properties like implicit solvation free energies [18].
Diagram 1: SMA-MD workflow with statistical reweighting. The reweighting stage is crucial for converting the broadly sampled ensemble from the generative model into a physically accurate equilibrium ensemble.
Experimental Protocol: Reweighting an MD Ensemble of a Disordered Peptide
This protocol details the application of the BioEn method to refine an all-atom molecular dynamics ensemble of the disordered penta-alanine peptide (Ala-5) using NMR J-couplings as experimental data [21].
Materials and Setup
Table 2: Research Reagent Solutions for Ala-5 Ensemble Refinement
| Reagent / Resource | Description | Function in the Protocol |
|---|---|---|
| Molecular System | Ala-5 peptide in explicit solvent. | The intrinsically disordered model system for refinement. |
| Simulation Software | Software package with MD capabilities (e.g., GROMACS, AMBER). | Generates the initial unbiased conformational ensemble. |
| Force Field | AMBER99SB*-ILDNP-Q. | Provides the reference potential energy function and initial weights ( w_\alpha^0 ). |
| Experimental Data | NMR J-couplings for Ala-5. | The experimental observables ( Y_i ) used for refinement. |
| Back-Calculation Tool | Software to compute J-couplings from atomic coordinates. | Calculates the observable value ( y_{i\alpha} ) for each structure α. |
| Reweighting Software | Implementation of the BioEn method (e.g., custom code). | Performs the numerical optimization to find the optimal weights ( w_\alpha ). |
Step-by-Step Procedure
Generate Reference Ensemble:
- Perform a long, unbiased molecular dynamics simulation of Ala-5 using the AMBER99SB*-ILDNP-Q force field in explicit solvent.
- Save a large number of snapshots (e.g., N > 100,000) from the equilibrated portion of the trajectory. This set of structures, along with their uniform reference weights ( w_\alpha^0 = 1/N ), constitutes the reference ensemble [21].
Process Experimental Data:
- Collect experimental NMR J-coupling values ( Y_i ) for the Ala-5 peptide.
- Estimate the total uncertainty ( \sigmai ) for each datum, combining experimental error and the uncertainty in calculating ( yi ) from a structure [21].
Compute Theoretical Observables:
- For every saved snapshot ( \alpha ) in the reference ensemble, calculate the corresponding J-coupling values ( y_{i\alpha} ).
Perform Bayesian Ensemble Reweighting:
- Choose an optimization method (log-weights or generalized forces) based on the ensemble size N and number of data points M.
- Select a value for the confidence parameter ( \theta ). This can be determined via cross-validation: systematically varying ( \theta ), refining the ensemble for each value, and predicting held-out experimental data to find the optimum.
- Execute the BioEn optimization algorithm to minimize ( L ), obtaining the refined weights ( w\alpha ). Monitor convergence of the ( \chi^2 ), ( S{KL} ), and the gradient norms [21].
Validate and Analyze the Refined Ensemble:
- Verify that the ensemble-averaged observables ( \langle yi \rangle ) calculated from the refined weights agree with the experimental ( Yi ).
- Analyze the conformational changes in the refined ensemble. For Ala-5, reweighting with J-couplings consistently increased the population of polyproline-II (PPII) conformations and decreased α-helical-like populations, providing a more accurate structural model [21].
Table 3: Example Results from Ala-5 Reweighting [21]
| Conformational State | Population in Reference Ensemble | Population after BioEn Reweighting |
|---|---|---|
| Polyproline-II (PPII) | Baseline | Increased |
| α-helical-like | Baseline | Decreased |
Advanced Applications and Recent Methodologies
Statistical reweighting principles are being applied and extended in various advanced computational contexts.
Binding Site Identification in RNA with SHAMAN
The SHAMAN method identifies small-molecule binding sites in dynamic RNA ensembles by combining molecular dynamics simulations with enhanced sampling using molecular probes [22]. Its parallel architecture consists of a "mother" simulation that explores the RNA's conformational landscape and multiple "shadow" replicas, each containing a different probe molecule. The probes sample the binding landscape on the RNA conformations provided by the mother simulation using metadynamics. The resulting probe densities are analyzed to identify binding sites (SHAMAPs), which are ranked by the probe's binding free energy ( \Delta G ) [22]. In a benchmark on riboswitches and viral RNAs, SHAMAN successfully identified all experimentally known binding sites, ranking them among the most favorable sites [22].
Thermodynamic Interpolation with Generative Models
Emerging deep generative models like Boltzmann Generators and the related Thermodynamic Interpolation (TI) offer a powerful, simulation-free approach to sampling equilibrium distributions [23]. These models use normalizing flows—invertible neural networks—to learn a direct mapping from a simple latent distribution (e.g., a Gaussian) to the complex Boltzmann distribution of a molecular system. The TI framework enables the generation of ensembles across multiple thermodynamic states (e.g., temperatures) from a single trained model. Furthermore, these models allow for the direct calculation of free energy differences between states, providing a versatile and efficient alternative to traditional reweighting of simulation data [23].
Diagram 2: Logical relationship of the ensemble reweighting problem, its Bayesian solution, and inputs/outputs.
Within the framework of Surrogate Model-Assisted Molecular Dynamics (SMA-MD), Stage 3 represents the critical phase where computational efficiency is translated into validated, quantitative predictions. SMA-MD is a procedure designed to sample the equilibrium ensemble of molecules more effectively than conventional molecular dynamics (MD) by leveraging deep generative models to enhance the sampling of slow degrees of freedom [19]. This initial enhanced sampling is followed by statistical reweighting and, crucially, short simulations for final validation and property prediction [19]. This stage ensures that the ensembles generated are not only diverse and low in energy but also thermodynamically meaningful and suitable for the accurate computation of properties such as implicit solvation free energies, which are vital in drug discovery [19].
The primary objective of Stage 3 is to anchor the statistically reweighted ensembles in physically accurate, albeit short, MD simulations. While generative models excel at exploring conformational space, final short simulations serve to validate the thermodynamic quality of these structures and perform the ultimate property prediction. This protocol details the application of short simulations for these purposes, providing a robust methodology for researchers and drug development professionals.
Experimental Workflow and Signaling Pathways
The following diagram illustrates the logical workflow and data flow for Stage 3 of the SMA-MD protocol, from the initial input to the final property prediction.
Workflow for Final Validation and Property Prediction
Application Notes and Protocols
Protocol: Configuration of Short MD Simulations
Objective
To define the parameters for short, explicit-solvent MD simulations that will validate the reweighted conformational ensemble and serve as the basis for thermodynamic property prediction.
Detailed Methodology
This protocol initiates the final validation phase by setting up the short MD simulations.
System Preparation:
- Input: Take the molecular structures from the reweighted ensemble generated in the previous stage of SMA-MD [19].
- Solvation: Solvate each molecule in an appropriate explicit solvent box (e.g., TIP3P water). The box size should ensure a minimum distance of 1.2 nm between the solute and the box edges.
- Neutralization: Add a physiological concentration of ions (e.g., 0.15 M NaCl) to mimic a biological environment. Neutralize the system's total charge by adding counter-ions.
Force Field Selection:
- Employ a modern, class II force field such as CHARMM36, OPLS-AA/M, or GAFF2. The choice should be consistent with the class of molecules being studied (e.g., proteins, small organic molecules).
- Obtain small molecule parameters from the force field's companion parameterization suite (e.g., CGenFF for CHARMM, the General Force Field for OPLS).
Simulation Parameterization:
- Energy Minimization: Perform a two-step minimization: first, with positional restraints on the solute (force constant of 1000 kJ/mol·nm²) to relax the solvent and ions; second, an unrestrained minimization of the entire system until the maximum force is below 1000 kJ/mol·nm.
- Equilibration:
- Conduct a 100 ps simulation in the NVT ensemble, using a thermostat like the Nosé-Hoover with a coupling constant of 1.0 ps. Maintain restraints on solute heavy atoms.
- Follow with a 100 ps simulation in the NPT ensemble, using a barostat such as Parrinello-Rahman with a coupling constant of 2.0 ps. Maintain isotropic pressure coupling. Continue restraints on solute heavy atoms.
- Production MD: Launch an ensemble of short production simulations. The length can vary from 1 to 10 ns per replica, depending on the property of interest. These are "short" relative to the timescales required for full conformational sampling but are sufficiently long to validate the ensemble and compute equilibrium properties. Save coordinates every 10 ps for analysis.
Protocol: Execution of Ensemble Simulations
Objective
To run the configured simulations efficiently across high-performance computing (HPC) resources, generating the necessary trajectory data for analysis.
Detailed Methodology
- Software Selection: Use a high-performance MD package such as GROMACS, OpenMM, or NAMD. The choice may depend on hardware compatibility and force field support.
- Parallelization: Configure the simulation to leverage GPU acceleration. A single simulation replica should be assigned to one GPU for optimal performance.
- Replica Strategy: For each molecule in the test set, run a minimum of 5 simulation replicas. This is achieved by initiating simulations from different random velocity seeds to decorrelate the trajectories and provide error estimates for computed properties.
- Monitoring: Implement automated checks for simulation stability, including potential energy, density, and root-mean-square deviation (RMSD) trends. Failed simulations should be restarted from the equilibration phase.
Protocol: Trajectory Analysis and Ensemble Validation
Objective
To analyze the simulation trajectories, validate the quality and convergence of the sampled ensemble, and compute structural properties.
Detailed Methodology
Stability Assessment:
- Calculate the backbone (or heavy-atom) RMSD relative to the initial energy-minimized structure to confirm the simulation has stabilized.
- Plot the radius of gyration (Rg) to monitor compactness and identify potential unfolding or collapse.
Conformational Cluster Validation:
- Cluster Analysis: Perform cluster analysis (e.g., using the GROMACS
clustertool with the GROMOS algorithm) on the combined trajectory from all replicas for a given molecule. Use the backbone atoms for proteins or all heavy atoms for small molecules with a cutoff of 0.15-0.25 nm. - Comparison: Compare the population of the top clusters from the short MD simulations to the clusters predicted by the SMA-MD generative model. A strong correlation validates the model's predictive accuracy for the equilibrium distribution.
- Cluster Analysis: Perform cluster analysis (e.g., using the GROMACS
Property Calculation (Structural):
- Compute properties such as:
- Root-mean-square fluctuation (RMSF) of residue side chains or molecular fragments.
- Solvent Accessible Surface Area (SASA) for the entire molecule or specific hydrophobic patches.
- Intramolecular Hydrogen Bonds.
- Compute properties such as:
Protocol: Free Energy Calculation for Property Prediction
Objective
To utilize the validated simulation ensembles for the prediction of key thermodynamic properties, such as solvation free energy, a critical parameter in drug discovery [19] [24].
Detailed Methodology
Implicit Solvation Free Energy (ΔG_solv):
- This is a primary application where SMA-MD has shown utility [19].
- Method: Use the Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) or the Generalized Born Surface Area (MM/GBSA) method.
- Procedure: Extract a statistically independent set of snapshots (e.g., 500) from the production phase of the combined simulation trajectories. For each snapshot, calculate the energy in vacuum and in the implicit solvent model, and compute the surface area term. The free energy is given by:
ΔG_solv = <E_MM>_solv - <E_MM>_gas + <G_solv> - T<S_MM>- Where
E_MMis the molecular mechanics energy,G_solvis the solvation free energy from PB/GB, andS_MMis the conformational entropy (often omitted due to high computational cost and error). The angled brackets represent the ensemble average.
- Interpretation: The calculated ΔG_solv provides a quantitative measure of solubility, a fundamental property in drug design.
Binding Free Energy (Optional, for ligand-receptor complexes):
- If the system involves a binding event, more advanced techniques like alchemical free energy perturbation (FEP) can be applied, using the short simulations to define the relevant bound and unbound states.
Data Presentation and Analysis
Table 1: Quantitative Properties Predictable from Stage 3 Short Simulations
| Property Category | Specific Property | Calculation Method | Typical Values / Range | Relevance in Drug Discovery |
|---|---|---|---|---|
| Thermodynamic | Implicit Solvation Free Energy (ΔG_solv) | MM/PBSA or MM/GBSA [19] | -5 to +50 kJ/mol | Predicts solubility, permeability, and ADMET profiles [24] |
| Structural | Radius of Gyration (Rg) | Trajectory Analysis | Molecule-dependent (Å to nm) | Indicates molecular compactness and folding state |
| Root-Mean-Square Fluctuation (RMSF) | Trajectory Analysis | 0.1 - 5.0 Å | Identifies flexible regions and potential binding sites | |
| Dynamic | Intramolecular H-Bond Count | Trajectory Analysis | Integer count | Impacts stability and solvent exposure |
Performance Comparison: SMA-MD vs. Conventional MD
Table 2: Empirical Comparison of SMA-MD and Conventional MD Workflows
| Metric | SMA-MD with Short Simulations | Conventional MD | Implication |
|---|---|---|---|
| Ensemble Diversity | More diverse [19] | Limited by simulation time | Better coverage of conformational space |
| Sampled Conformer Energy | Lower energy [19] | Higher energy local minima | More thermodynamically relevant structures |
| Time to Sample Slow DOF | Accelerated via generative model | Limited by molecular vibration timescales | Faster convergence for property prediction |
| Solvation Free Energy Accuracy | High (validated by short simulations) | Variable, depends on convergence | More reliable prediction for novel molecules |
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Solutions
| Item Name | Function / Purpose | Example Software/Package |
|---|---|---|
| High-Performance MD Engine | Executes the short, explicit-solvent MD simulations with high efficiency and GPU acceleration. | GROMACS, OpenMM, NAMD |
| Force Field Parameterization Suite | Provides necessary atomic parameters and topologies for small organic molecules within a chosen force field. | CGenFF (for CHARMM), ACPYPE (for GAFF), LigParGen |
| Trajectory Analysis Toolkit | A suite of tools for calculating RMSD, Rg, RMSF, SASA, hydrogen bonds, and performing cluster analysis. | MDAnalysis, GROMACS built-in tools, cpptraj |
| Contrast-Rich Visualization Suite | Generates publication-quality diagrams of molecular structures, trajectories, and results, ensuring accessibility. | VMD, PyMOL, Matplotlib (with accessible color palettes) [25] [26] |
| Free Energy Calculator | Computes implicit solvation free energies from the simulation ensemble using MM/PBSA or MM/GBSA. | g_mmpbsa, AmberTools |
Implicit solvation models provide a computationally efficient framework for estimating solvation free energies, a critical parameter in drug discovery. By representing the solvent as a continuous medium rather than individual explicit molecules, these methods enable rapid assessment of ligand binding affinities and stability of molecular complexes. This document outlines the practical application of these models within the context of Surrogate Model-Assisted Molecular Dynamics (SMA-MD) research, providing detailed protocols and quantitative comparisons for research professionals.
The foundational principle of implicit solvation is the Potential of Mean Force (PMF), which represents the solvent-averaged effect on solute molecules [27]. The solvation free energy (ΔGs) quantifies the energy required to transfer a solute from vacuum to solvent and is typically decomposed into polar (electrostatic) and non-polar (cavitation and van der Waals) components [27]. For binding energy calculations, this is expressed as ΔGbind = ΔGspl - ΔGsl - ΔGsp, where the subscripts denote the protein-ligand complex, ligand, and protein, respectively [27].
Key Implicit Solvation Models and Quantitative Comparison
- Poisson-Boltzmann (PB) Model: Solves the PB equation numerically to describe electrostatic interactions in a solvent containing ions [28]. It is computationally expensive but considered highly accurate.
- Generalized Born (GB) Model: An approximation to the PB equation that models the solute as a set of spheres with different dielectric constants [27] [28]. It offers a favorable balance of speed and accuracy.
- COSMO (COnductor-like Screening Model): Treats the solvent as a perfect conductor, providing another efficient approximation for electrostatic screening [29].
- Polarized Continuum Model (PCM): A variant of continuum models that determines the solvent reaction field via an apparent surface charge [29].
- Accessible Surface Area (ASA) Models: Calculate non-polar solvation contributions based on linear relationships between solvent-accessible surface area and transfer free energies [28].
Accuracy Comparison Across Model Types
The following table summarizes the performance of various implicit solvent models in predicting solvation and desolvation energies for different molecular types, as compared to explicit solvent calculations and experimental data [29].
Table 1: Accuracy comparison of implicit solvent models for different molecular systems
| Molecular System | Property Calculated | Implicit Models Tested | Correlation with Explicit Solvent/Experiment | Typical Discrepancy |
|---|---|---|---|---|
| Small Molecules | Hydration Free Energy | PCM, GB, COSMO, PB | R = 0.87-0.93 (vs. experiment) [29] | Varies by model and parameterization |
| Small Molecules | Solvation Energy | PCM, GB, COSMO, PB | R = 0.82-0.97 (vs. explicit solvent) [29] | Varies by model and parameterization |
| Proteins | Polar Solvation Energy | PCM, GB, COSMO, PB | R = 0.65-0.99 (vs. explicit solvent) [29] | Up to 10 kcal/mol [29] |
| Protein-Ligand Complexes | Desolvation Penalty | PCM, GB, COSMO, PB | R = 0.76-0.96 (vs. explicit solvent) [29] | Up to 10 kcal/mol [29] |
Emerging Machine Learning Approaches
Traditional implicit solvent models face limitations in accuracy for precise thermodynamic calculations. Recent advances in machine learning (ML) have led to novel approaches that overcome these limitations:
- Lambda Solvation Neural Network (LSNN): A graph neural network (GNN)-based model trained not only on forces but also on the derivatives of alchemical variables. This ensures that solvation free energies can be meaningfully compared across different chemical species, a drawback of force-matching alone [30]. Trained on ~300,000 small molecules, LSNN achieves accuracy comparable to explicit-solvent alchemical simulations while offering computational speedups [30].
- Gaussian Process Surrogate Modeling: A multi-fidelity optimization technique that builds inexpensive models of physical properties as a function of force field parameters (e.g., Lennard-Jones parameters) [8]. This approach accelerates parameter searches by enabling fast evaluation of approximate objective functions, allowing for more global optimization against large training sets of experimental data [8].
Experimental Protocols
Workflow for Implicit Solvation Free Energy Calculation
The following diagram illustrates the standard protocol for calculating solvation free energies using an implicit solvent model, from structure preparation to final analysis.
Protocol for Solvation Free Energy Calculation
Objective: To compute the solvation free energy (ΔGs) of a small molecule ligand using a Generalized Born (GB) implicit solvent model.
Materials:
- Ligand Structure File (e.g., .mol2, .pdb)
- Molecular Dynamics Software with implicit solvent capabilities (e.g., AMBER, GROMACS, OpenMM)
- Force Field Parameters (e.g., GAFF, OpenFF)
- Partial Charge Assignment Tool (e.g., ANTECHAMBER, RESP)
Procedure:
- Ligand Preparation:
- Obtain the 3D structure of the ligand. If needed, generate reasonable 3D coordinates using a tool like Open Babel or CORINA.
- Add hydrogens and perform a preliminary geometry optimization using molecular mechanics or semi-empirical quantum mechanics (e.g., with the PM7 method [29]).
Parameterization:
- Assign force field atom types to the ligand.
- Calculate partial atomic charges. A common method is the AM1-BCC approach, which is efficient and provides good results for solvation free energies [8].
- Generate the topology file for the ligand in the required format for your simulation software.
Vacuum Energy Calculation:
- Place the ligand in a large, empty simulation box (vacuum conditions).
- Perform a full energy minimization to remove any steric clashes.
- Record the potential energy of the ligand in vacuum (U_ss). This is the intra-solute energy [27].
Implicit Solvent Calculation:
- Place the minimized ligand structure into a new simulation box.
- Configure the implicit solvent model (e.g., select the GB model, set the solvent dielectric constant to 78.5 for water, and the solute dielectric to 1.0).
- Perform another energy minimization under the implicit solvent conditions.
- Record the total potential energy, which now includes the solute-solvent interaction energy.
Free Energy Calculation:
- The solvation free energy is calculated as the difference between the total potential energy in implicit solvent and the potential energy in vacuum: ΔGs = Gimplicit - Uss [27].
- For greater accuracy, especially for binding free energies, consider performing alchemical free energy perturbation (FEP) or thermodynamic integration (TI) calculations, where the ligand is decoupled from the solvent in a series of steps [27] [30].
SMA-MD Enhanced Workflow
The following diagram illustrates how surrogate models can be integrated into the parameter optimization process to accelerate and improve implicit solvation calculations.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential software and resources for implicit solvation calculations in drug discovery
| Tool Name | Type | Primary Function | Application Note |
|---|---|---|---|
| APBS | Software Package | Numerical solver for the Poisson-Boltzmann equation [29]. | Considered a gold standard for electrostatic calculations but computationally demanding for high-throughput screening. |
| DISOLV | Software Package | Implements PCM, COSMO, and S-GB models on a smooth solvent boundary [29]. | Used successfully in post-processing and gridless docking procedures with the MMFF94 force field. |
| GBNSR6 | Software Library | A Generalized Born model implementation known for accuracy with small molecules [29]. | Often cited as one of the most accurate GB models for hydration free energy predictions. |
| OpenFF Evaluator | Workflow Driver | Automates physical property simulations for force field training and validation [8]. | Essential for standardizing and automating the calculation of training sets for surrogate model development. |
| LSNN | Machine Learning Model | Graph Neural Network for implicit solvation and free energy calculations [30]. | Represents a next-generation approach, offering explicit-solvent accuracy with implicit-solvent speed. |
Implicit solvent models provide an essential tool for efficient solvation free energy calculations in drug discovery. While traditional models like PB and GB offer a good balance of speed and accuracy, emerging machine learning approaches, such as LSNN and Gaussian process surrogates, integrated within an SMA-MD framework, are pushing the boundaries of both accuracy and computational efficiency. The protocols and comparisons provided here serve as a practical guide for researchers applying these methods to real-world drug development challenges.
Optimizing SMA-MD Performance: Addressing Computational Challenges and Implementation Pitfalls
In the context of Surrogate Model-Assisted Molecular Dynamics (SMA-MD), a primary challenge is the effective integration of explicit solvent models with the potential energy-based reweighting procedures central to the methodology. SMA-MD leverages deep generative models to enhance the sampling of molecular conformational spaces, after which the generated ensemble is reweighted using statistical mechanics principles and refined with short molecular dynamics (MD) simulations [2] [18]. While explicit solvent models provide a more physically realistic representation of solvent effects by modeling individual solvent molecules, their use introduces significant complexities for reweighting. The core of the challenge lies in the fact that potential energy-based reweighting schemes can be overwhelmed by the immense number of solvent-solvent interactions, whose energy contributions can "drown out" the signal from the solute and solute-solvent interactions of interest [18]. This application note details this specific challenge and provides protocols for navigating it within a SMA-MD research framework.
The Explicit Solvent Challenge in SMA-MD
Core Principles of SMA-MD and Reweighting
The SMA-MD approach is designed to overcome the sampling limitations of conventional molecular dynamics. It consists of three key stages:
- Surrogate Sampling: A deep generative model (e.g., Torsional Diffusion) is used to generate a diverse set of molecular conformations, efficiently exploring slow degrees of freedom [2].
- Statistical Reweighting: The generated ensemble is reweighted to recover the underlying Boltzmann distribution. This often involves calculating the potential energy of each conformation to determine its correct statistical weight [2] [18].
- MD Finetuning: Short, conventional MD simulations are run to locally relax and validate the reweighted ensemble [2].
The integrity of the entire workflow depends on the accuracy of the reweighting step. For a conformation, the probability in the Boltzmann distribution is proportional to ( e^{-\beta E} ), where ( E ) is the potential energy of the system. In implicit solvent models, where the solvent is represented as a continuous dielectric medium, the energy calculation is computationally efficient and dominated by the solute's energy [31]. This makes reweighting straightforward. In contrast, explicit solvent models include the coordinates and interactions of thousands of individual solvent molecules, causing the total potential energy, ( E{total} ), to be dominated by solvent-solvent terms (( E{solvent-solvent} )) that are largely irrelevant to the solute's conformational distribution.
As one researcher directly questioned regarding the SMA-MD method, "how does this method propose to address Boltzmann ensembles with explicit solvent, whose water-water potential energies may drown out the easy potential energy-based reweighting?" [18]. This highlights that the energy of the solute and its immediate environment becomes a small fluctuation on top of a very large, conformationally insensitive background solvent energy, making precise reweighting computationally problematic.
Comparative Analysis of Solvent Models
The table below summarizes the key characteristics of implicit and explicit solvent models relevant to the SMA-MD reweighting challenge.
Table 1: Comparison of Solvent Models for Use in SMA-MD Reweighting
| Feature | Implicit Solvent Models | Explicit Solvent Models |
|---|---|---|
| Solvent Representation | Continuum dielectric medium [31] | Discrete molecules (e.g., TIP3P water) [32] |
| Computational Cost | Lower | Significantly higher [31] |
| Physical Realism | Limited; lacks specific molecular interactions [32] [31] | High; captures explicit hydration shells and water dynamics [32] |
| Suitability for Potential Energy-Based Reweighting | High. Energy is a direct function of solute conformation. | Low. Total energy is dominated by solvent-solvent terms, drowning out solute conformational energy [18]. |
| Key Limitations for SMA-MD | May inaccurately represent specific solvation effects (e.g., hydrogen bonds) [31] | Direct reweighting of the full potential energy is statistically inefficient and computationally prohibitive [18]. |
Protocols for Managing Explicit Solvent in SMA-MD
To circumvent the reweighting challenge, researchers can employ alternative strategies that leverage the strengths of explicit solvent without directly relying on the total potential energy. The following workflow and protocols outline a practical approach.
Figure 1: A hybrid explicit-implicit solvent workflow for free energy calculation. This protocol uses explicit solvent for simulation but an endpoint method for efficient energy computation.
Protocol: Endpoint Free Energy Calculation Using Interaction Energy
This protocol is adapted from the Interaction-Reorganization Solvation (IRS) method [32] and is designed to compute solvation free energies using explicit solvent MD simulations without requiring total potential energy reweighting of the entire ensemble.
Principle: The solvation free energy (( \Delta G{solv} )) is decomposed into an interaction energy component (( \Delta G{int} )) and a reorganization component (( \Delta G{reo} )). The key insight is that ( \Delta G{int} ) can be computed directly from an explicit solvent MD simulation as an ensemble average, avoiding the need for reweighting the entire system's energy [32].
Procedure:
- System Setup:
- Prepare the solute molecule (e.g., a small drug-like molecule) in a simulation box with explicit solvent molecules (e.g., TIP3P water) and necessary ions to neutralize the system.
- Molecular Dynamics Simulation:
- Run an MD simulation (e.g., using OpenMM, GROMACS, or AMBER) in the NPT ensemble to generate a Boltzmann-distributed ensemble of the solute in explicit solvent.
- Simulation Parameters:
- Ensure the simulation is long enough for the solute conformational space to be well-sampled (e.g., tens to hundreds of nanoseconds).
- Trajectory Analysis and Energy Calculation:
- From the saved trajectory, extract a set of uncorrelated solute-solvent snapshots.
- For each snapshot, calculate the solute-solvent interaction energy (( E{int} )). In a typical molecular mechanics force field, this is the sum of the electrostatic (( E{ele} )) and van der Waals (( E{vdw} )) interactions between every atom of the solute and every atom of the solvent [32].
- Compute the ensemble average of the interaction energy, ( \langle E{int} \rangle ).
- Free Energy Component Calculation:
- The interaction free energy is calculated as ( \Delta G{int} = \langle E{int} \rangle - T \Delta S{int} ). In many practical applications, the entropy term (( T \Delta S{int} )) is approximated or combined with the reorganization term [32].
- The reorganization energy (( \Delta G{reo} )), which accounts for cavity formation and solvent polarization, is often modeled as a linear function of the solvent-accessible surface area (SASA) and the interaction energy itself: ( \Delta G{reo} \approx \gamma \cdot \text{SASA} + f(\Delta G{int}) ) [32].
- The final solvation free energy is given by: ( \Delta G{solv} \approx \Delta G{int} + \Delta G{reo} ). The parameters (( \gamma ), etc.) are typically determined by fitting to a training set of molecules with known experimental solvation free energies [32].
Protocol: Integration of SMA-MD with a Hybrid Solvent Approach
This protocol modifies the standard SMA-MD workflow to make it compatible with explicit solvent simulations by strategically using implicit solvent.
Procedure:
- Surrogate Sampling (Implicit Solvent): Perform the initial generative sampling and reweighting stages using an implicit solvent model. This leverages the computational efficiency and direct reweighting capability of the continuum model to generate a diverse, Boltzmann-weighted ensemble of solute conformations [2] [31].
- Cluster Analysis: Cluster the reweighted ensemble from Step 1 to identify representative, low-energy conformers for further analysis.
- Explicit Solvent Validation and Refinement:
- For each representative conformer, set up an explicit solvent MD simulation as described in Protocol 3.1.
- Run short MD simulations (e.g., 1-10 ns) to locally relax the structure in a physically realistic solvent environment and to assess the stability of the conformer.
- Targeted Free Energy Calculation: Use the snapshots from the explicit solvent simulations as input for the endpoint method described in Protocol 3.1 to calculate relevant thermodynamic properties, such as solvation free energy or binding affinity. This step replaces the direct potential-energy reweighting of the full SMA-MD ensemble in explicit solvent.
The Scientist's Toolkit: Research Reagents & Computational Solutions
The following table details key software and methodological "reagents" essential for implementing the protocols described above.
Table 2: Essential Research Reagents and Computational Solutions
| Tool/Solution | Type | Primary Function in Protocol | Key Notes |
|---|---|---|---|
| SMA-MD Codebase [2] | Software Package | Core framework for surrogate model sampling and reweighting. | Requires two Conda environments (e3nn-env, openmm-env). Integrates Torsional Diffusion for conformer generation. |
| OpenMM [2] | MD Simulation Engine | Performing implicit and explicit solvent MD simulations for refinement and energy calculation. | GPU acceleration is highly recommended. Compatible with the SMA-MD workflow. |
| Interaction-Reorganization Solvation (IRS) [32] | Computational Method | Calculating solvation free energies from explicit solvent MD trajectories. | Avoids total energy reweighting by using interaction energy and SASA. |
| Generalized Born (GB) Model [31] | Implicit Solvent Model | Efficient conformational sampling and reweighting in the initial SMA-MD phase. | Faster but less accurate than explicit solvent. Models solvent as a continuum. |
| Hydrogen Mass Repartitioning (HMR) [33] | Simulation Technique | Enabling longer integration time steps (4 fs) in explicit solvent MD, speeding up calculations. | Increases the mass of hydrogen atoms, allowing a larger timestep while maintaining stability. |
| Gaussian Process Surrogates [34] | Surrogate Model | Accelerating force field parameter optimization by approximating physical properties. | Can be used to pre-screen parameters or conditions before running expensive explicit solvent simulations. |
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) addresses a critical bottleneck in computational biophysics: the prohibitive cost of simulating complex molecular processes at biologically relevant timescales. Adaptive sampling and surrogate model refinement techniques form a synergistic framework that intelligently allocates computational resources to regions of the molecular configuration space that yield the highest information gain. These methodologies shift the paradigm from exhaustive sampling to targeted data acquisition, enabling researchers to explore complex energy landscapes and rare events with unprecedented efficiency. For drug development professionals, these techniques enable more rapid and accurate prediction of ligand binding affinities, protein folding pathways, and allosteric mechanisms—fundamental processes in rational drug design.
This article presents application notes and experimental protocols for implementing adaptive sampling and model refinement within SMA-MD research, with specific consideration for challenges in pharmaceutical development.
Theoretical Foundation and Key Concepts
The Role of Surrogate Models in Molecular Dynamics
In SMA-MD, surrogate models (also called metamodels or emulators) approximate the expensive molecular mechanics force field or the mapping from molecular configurations to quantities of interest. These statistical or machine learning models run at a fraction of the computational cost of full MD simulations, allowing for rapid exploration of conformational space.
The core challenge lies in constructing accurate surrogates with minimal full MD data. Adaptive sampling addresses this by treating surrogate modeling as an active learning process where training points are selected sequentially based on the current model's uncertainty and the research objective. The refinement process ensures the surrogate evolves to become increasingly accurate in regions critical for predicting molecular mechanisms and thermodynamic properties.
Mathematical Principles of Adaptive Sampling
Effective adaptive sampling strategies balance exploration (sampling regions of high uncertainty) with exploitation (sampling regions likely to improve model accuracy for specific predictions). Recent methodologies achieve this balance through formal acquisition functions. For instance, some approaches frame the residual loss of the surrogate model as an unnormalized probability density function, using deep generative models to sample from this distribution and refine the training set [35].
Other techniques incorporate mechanisms from meta-heuristic algorithms, such as using random walks with an acceptance criterion inspired by simulated annealing to maintain the exploration-exploitation balance [36]. In the context of molecular dynamics, these principles translate to sampling strategies that preferentially initiate new simulations from configurations where the surrogate model is uncertain about the potential energy or where the predicted probability of transitioning to a new metastable state is high.
Adaptive Sampling Techniques: Protocols and Applications
Deep Adaptive Sampling for Surrogate Modeling (DAS²)
The DAS² methodology generalizes deep adaptive sampling to parametric settings, making it suitable for problems where molecular behavior depends on external parameters such as pH, ionic concentration, or temperature [35].
Protocol 1: Deep Adaptive Sampling Workflow
Objective: Construct an accurate surrogate model for a parametric molecular system with minimal full MD simulations.
Materials and Software:
- Molecular Dynamics Engine: (e.g., GROMACS, NAMD, OpenMM)
- Differentiable Programming Framework: (e.g., PyTorch, TensorFlow, JAX)
- Deep Generative Model: Normalizing Flow or Variational Autoencoder implementation
- High-Performance Computing (HPC) cluster with GPU acceleration
Procedure:
- Initial Surrogate Training:
- Define the parametric space (e.g., temperature range 300-310K, pH 7.0-7.4).
- Execute a small set of initial full MD simulations (e.g., 10-50 runs) using a space-filling design (e.g., Latin Hypercube) across the parametric and conformational space.
- Train an initial Physics-Informed Neural Network (PINN) as the surrogate model on this dataset. The loss function includes the molecular mechanics residual.
Residual-Based Probability Mapping:
- Treat the PINN residual as an unnormalized probability density function over the combined spatial and parametric variables.
- Train a deep generative model (e.g., Normalizing Flow) to approximate this residual-induced distribution. This model learns to generate samples where the current surrogate is inaccurate.
Generative Sampling and Dataset Augmentation:
- Generate new sample configurations and parameters from the trained generative model.
- Execute targeted full MD simulations at these newly sampled points.
Model Refinement:
- Augment the training dataset with results from the new MD simulations.
- Retrain the surrogate model on the expanded dataset.
Convergence Checking:
- Iterate steps 2-4 until the surrogate model's predictive accuracy, measured by cross-validation error or uncertainty estimation, falls below a pre-defined threshold. Monitor key thermodynamic observables (e.g., free energy difference) for stability.
Application Note:
This protocol is particularly effective for mapping multi-dimensional free energy landscapes as functions of environmental parameters. In drug design, this enables rapid prediction of how a lead compound's binding affinity varies with physiological conditions.
Meta-Heuristic Informed Sampling
This technique combines adaptive sampling with global optimization algorithms, ideal for identifying rare events and global minima on complex energy landscapes [36].
Protocol 2: Balanced Sampling via Simulated Annealing Acceptance
Objective: Efficiently locate globally stable conformational states and transition pathways.
Materials and Software:
- Surrogate Model: Cubic Radial Basis Function (RBF) or Gaussian Process model.
- Genetic Algorithm Library: (e.g., DEAP)
- Molecular Dynamics Engine
Procedure:
- Initialization:
- Build a preliminary surrogate model from an initial set of MD simulations.
- Define a random walk procedure in molecular conformational space (e.g., using collective variables).
Candidate Generation:
- At each iteration, use a genetic algorithm to locate the putative global optimum (e.g., minimum energy structure) on the current surrogate model.
- Simultaneously, perform a random walk step from the previous sample to propose a new candidate configuration.
Controlled Acceptance:
- Accept or reject the new candidate configuration based on a probabilistic simulated annealing-style criterion that considers both the surrogate-predicted energy (exploitation) and the model's uncertainty at that point (exploration).
- This acceptance function should become progressively more selective over computational time.
Targeted Simulation and Update:
- Execute a full MD simulation from the accepted configuration(s).
- Use the new data to update and refine the surrogate model.
- Repeat steps 2-4 until the global minimum conformation is satisfactorily identified and verified.
Application Note:
This protocol is highly effective for studying protein folding and ligand docking, where the goal is to find the most stable conformation among many local minima. The balance of exploration and exploitation prevents the sampling from becoming trapped in metastable states.
Quantitative Comparison of Sampling Strategies
Table 1: Comparative Analysis of Adaptive Sampling Techniques for SMA-MD
| Technique | Core Mechanism | Best-Suited MD Applications | Computational Overhead | Key Metric for Refinement |
|---|---|---|---|---|
| Deep Adaptive Sampling (DAS²) [35] | Deep generative model sampling from residual distribution | Parametric studies (e.g., pH, temperature), free energy surface mapping | High (requires training generative model) | Residual loss of the Physics-Informed Neural Network |
| Meta-Heuristic Balanced Sampling [36] | Random walk with simulated annealing acceptance + genetic algorithm | Global optimization (e.g., protein folding, binding site discovery) | Medium (depends on optimization algorithm complexity) | Balance between surrogate-predicted energy and model uncertainty |
The Scientist's Toolkit: Essential Research Reagents and Computational Materials
Successful implementation of the protocols requires careful selection of computational tools and theoretical constructs.
Table 2: Research Reagent Solutions for SMA-MD
| Item / Reagent | Function / Purpose | Implementation Example |
|---|---|---|
| Physics-Informed Neural Network (PINN) | Serves as the foundational surrogate model; approximates the force field or energy function while obeying physical laws. | A deep network where the loss function includes terms for molecular dynamics residuals (e.g., forces from energy gradients). |
| Normalizing Flow Model | A deep generative model used in DAS² to sample from the residual-induced distribution for adaptive data acquisition. | RealNVP or Glow architecture trained on the PINN's residual loss. |
| Cubic Radial Basis Function (RBF) | A simple, interpretable surrogate model used in meta-heuristic approaches for rapid global optimization. | scipy.interpolate.Rbf with function='cubic' for building an interpolant from MD data. |
| Genetic Algorithm (GA) | A global optimization meta-heuristic used to locate the optimal configuration on the current surrogate surface. | DEAP library configured to minimize the surrogate-predicted energy. |
| Simulated Annealing Scheduler | Provides the temperature parameter for the acceptance criterion, controlling the exploration-exploitation balance over time. | A logarithmic cooling schedule: ( T(k) = T_0 / \ln(1+k) ) for iteration ( k ). |
Workflow Visualization and Logical Relationships
The following diagrams illustrate the core workflows and logical structures of the primary techniques discussed.
Deep Adaptive Sampling (DAS²) Workflow
Meta-Heuristic Sampling Logic
Adaptive sampling and surrogate model refinement represent a paradigm shift in molecular dynamics, transforming it from a purely simulation-based discipline to a data-driven, learning-augmented science. The protocols outlined for Deep Adaptive Sampling and Meta-Heuristic Informed Sampling provide concrete roadmaps for researchers to implement these advanced strategies. By strategically guiding computational resources to where they are most informative, these techniques drastically reduce the cost of probing long-timescale biological phenomena and multi-parameter pharmaceutical problems. As these methodologies mature, they promise to accelerate drug discovery by enabling more exhaustive in silico screening and more accurate predictions of in vivo molecular behavior, ultimately compressing the timeline from target identification to viable therapeutic candidate.
In computational research fields such as Molecular Dynamics (MD), practitioners are frequently confronted with a fundamental trade-off: the high computational cost of accurate, high-fidelity simulations against the need for extensive sampling to achieve statistical reliability. Surrogate models, also known as metamodels, present a powerful solution to this challenge by acting as fast-to-evaluate approximations of more complex, computationally expensive models [37] [38]. The core objective is to accelerate exploration and analysis while maintaining acceptable accuracy guarantees.
Within this domain, multi-fidelity methods have emerged as a sophisticated strategy. These methods leverage low-cost surrogate models to speed up computations and make occasional recourse to expensive high-fidelity models to establish accuracy guarantees [39]. A critical insight of modern surrogate modeling is that the surrogate and high-fidelity models are used in concert; poor predictions by surrogate models can be compensated for with more frequent access to the high-fidelity model. This introduces a central design trade-off: should one invest computational resources to improve the accuracy of the surrogate model, or simply make more frequent recourse to the expensive high-fidelity model? [39] This balancing act is the central focus of these application notes.
Theoretical Foundation: The Efficiency-Accuracy Trade-off
Formalizing the Trade-off
The design of a surrogate modeling framework involves optimizing a cost function that accounts for two primary expenses:
- Approximation Cost (
C_approx): The computational cost required to construct and improve the surrogate model's fidelity. - Sampling Cost (
C_samp): The computational cost incurred from querying the high-fidelity model to compensate for the surrogate's inaccuracies and to validate results.
The total computational cost (C_total) can be expressed as:
C_total = C_approx + C_samp
The relationship between these costs is often inverse; a higher investment in C_approx typically yields a more accurate surrogate, which in turn reduces the number of necessary high-fidelity samples, lowering C_samp. The optimal balance is achieved when C_total is minimized for a required level of output accuracy [39].
Context-Aware Surrogate Modeling
Traditional model reduction methods aim to create surrogates that are so accurate they can replace the high-fidelity model entirely. However, a context-aware approach recognizes that within a multi-fidelity framework, the surrogate is not a standalone replacement. Its purpose is to work in tandem with the high-fidelity model, and its optimal fidelity is therefore often lower than what would be required for a traditional replacement model [39]. This principle is key to achieving significant runtime speedups, which have been demonstrated to reach up to an order of magnitude in practical examples [39].
Application in Molecular Dynamics (SMA-MD Research)
MD-Enabled Surrogate Modeling Framework
In the specific context of Surrogate Model-Assisted Molecular Dynamics (SMA-MD) for materials science, an MD-enabled surrogate modeling framework can be developed to capture complex constitutive and damage behavior at the atomic scale [37]. This approach is particularly valuable for modeling composite materials, where the multiscale composition and difficulty of experimental characterization at small scales present significant challenges.
Table 1: Key Phases of an MD-Enabled Surrogate Modeling Framework
| Phase | Key Activities | Output |
|---|---|---|
| Problem Setup & Data Generation | Run selected MD simulations; model constituents and interfaces separately; ensure distinct behaviors are reflected. | A foundational dataset of high-fidelity MD simulations. |
| Surrogate Model Selection & Training | Select appropriate force fields; embed time step data; train multi-task GRU-based neural networks on MD data. | A trained surrogate model capable of inferring rate-dependent response. |
| Validation & Deployment | Test the model against MD simulations with unseen loading patterns; demonstrate robustness and generalization. | A validated, fast-to-evaluate surrogate for constitutive response and failure prediction. |
Research Reagent Solutions: The Computational Toolkit
The following table details essential "research reagents" – in this context, computational tools and datasets – required for implementing an SMA-MD framework.
Table 2: Key Research Reagent Solutions for SMA-MD
| Item | Function / Explanation | Relevance to SMA-MD |
|---|---|---|
| High-Fidelity MD Simulator | Software (e.g., LAMMPS, GROMACS) that performs atomic-scale simulations based on Newton's equations of motion and interatomic potentials. | Generates the ground-truth data used for training and validating the surrogate model. Its high computational cost motivates the use of surrogates. |
| Force Fields | Mathematical representations of the potential energy surface governing atomic interactions (e.g., CHARMM, AMBER). | Critical for producing comparable and physically meaningful results among different material constituents in MD simulations [37]. |
| MD Simulation Dataset | A curated collection of input strain paths and corresponding output mechanical responses (stress, damage) from MD simulations. | Serves as the training data for the surrogate model. It should encompass a wide range of expected loading conditions. |
| Multi-task GRU-based Neural Network | A recurrent neural network architecture designed to handle sequential data. "Multi-task" indicates simultaneous prediction of multiple outputs (e.g., stress and failure). | Captures the path-dependent and rate-dependent constitutive response directly from the strain path input, leveraging embedded time-step information [37]. |
| Polynomial Regression (PR) Model | A surrogate model that fits a polynomial function to the input-output data. | A simple, efficient model for establishing baseline performance; efficient for model generation and determining influential design variables [38]. |
| Kriging-based Model (Gaussian Process) | A probabilistic surrogate model that provides not just a prediction but also an estimate of uncertainty at any point in the input space. | Often provides higher accuracy and is better for global optimization and max-min searches due to its ability to predict a broader range of objective values [38]. |
Experimental Protocols for Key Analyses
Protocol 1: Context-Aware Surrogate Model Training for Multi-Fidelity Importance Sampling
This protocol outlines the procedure for training a context-aware surrogate model to be used in a multi-fidelity importance sampling scheme, directly addressing the core trade-off.
Objective: To construct a surrogate model for a biasing density that minimizes the total computational cost (C_total) in a Bayesian inverse problem or importance sampling context.
Materials/Software:
- High-fidelity model (e.g., a full MD simulation).
- Low-fidelity model family (e.g., a coarse-grained MD model or a machine learning model of reduced complexity).
- Computational environment for model training and testing.
Procedure:
- Define Cost Metrics: Quantify the computational cost of a single high-fidelity model evaluation (
C_HF) and the cost of a single low-fidelity model evaluation (C_LF). - Theoretical Trade-off Analysis: For a given computational budget, theoretically model the trade-off between the number of high-fidelity samples and the fidelity of the surrogate. The goal is to find the point where the combined cost of building the surrogate and compensating with high-fidelity samples is minimized [39].
- Construct Surrogate: Build the surrogate model (the biasing density) using the selected low-fidelity approach. The key is to stop at a lower fidelity than traditional methods would, as dictated by the trade-off analysis.
- Perform Multi-Fidelity Sampling: Execute the importance sampling, using the surrogate to propose samples and the high-fidelity model to compute accurate weights for a subset of these samples.
- Validate and Iterate: Assess the statistical accuracy of the result (e.g., in estimating an integral or posterior distribution). If necessary, adjust the fidelity of the surrogate model and repeat.
Protocol 2: GRU-Based Surrogate Model for MD Constitutive Response
This protocol provides a detailed methodology for creating a machine learning-based surrogate for MD simulations, as referenced in [37].
Objective: To train a surrogate model that can predict the rate-dependent and path-dependent constitutive response and failure of a composite material, bypassing the need for full MD simulations after training.
Materials/Software:
- MD simulation software with appropriate force fields.
- A dataset of MD simulations under various strain paths.
- Machine learning library (e.g., TensorFlow, PyTorch) with support for GRUs.
Procedure:
- Data Generation: Run MD simulations for each material constituent (e.g., fiber, matrix, interface) separately. Apply a diverse set of strain paths to capture a wide range of material behaviors.
- Data Preprocessing: Embed the time step information directly into the input data for the model. This allows the model to infer the rate-dependent response based solely on the given strain path [37].
- Model Architecture: Design a multi-task GRU-based neural network. The GRU layers will capture the temporal (path-) dependencies in the strain data. The multi-task output heads will simultaneously predict the constitutive response (e.g., stress tensor) and a failure indicator.
- Model Training: Train the network on the generated MD data. Use a loss function that combines the errors from the constitutive response prediction and the failure prediction tasks.
- Generalization Testing: Validate the model's robustness by testing its predictions against the results of new, unseen MD simulations with loading patterns not present in the training data [37].
Visualization of Workflows and Relationships
The following diagrams, generated with Graphviz DOT language, illustrate the core logical relationships and workflows described in these notes.
Diagram 1: Multi-Fidelity SMA-MD Workflow. This chart illustrates the iterative process of balancing surrogate model construction cost (C_approx) against high-fidelity sampling cost (C_samp) to achieve an optimal computational strategy.
Diagram 2: GRU Surrogate Model Architecture. This depicts a multi-task neural network that takes a strain path as input and uses Gated Recurrent Units (GRUs) to simultaneously predict constitutive response and material failure.
The strategic balance between computational efficiency and sampling accuracy is not merely a technical consideration but a fundamental aspect of modern computational science, particularly in demanding fields like molecular dynamics. The adoption of a context-aware, multi-fidelity perspective allows researchers to escape the rigid constraints of traditional model reduction. By consciously accepting a lower-fidelity surrogate that is optimized to work in concert with—rather than replace—high-fidelity models, significant speedups of an order of magnitude become achievable [39]. As demonstrated in SMA-MD research for composites, machine learning models like multi-task GRUs provide a powerful and flexible means to implement these surrogates, effectively capturing complex, path-dependent physical phenomena [37]. The continued development and systematic application of these principles will be crucial for tackling increasingly complex multiscale and multiphysics problems in drug development and materials science.
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) research leverages machine learning to overcome the fundamental time-scale limitations of conventional molecular dynamics simulations. This approach employs generative artificial intelligence to explore molecular configuration spaces more efficiently and uses reweighting protocols to recover accurate thermodynamic statistics from enhanced sampling methods. The integration of these technologies has created a powerful paradigm for accelerating molecular discovery in drug development and materials science.
Generative models learn the underlying probability distribution of molecular configurations, enabling researchers to sample relevant regions of conformational space without performing computationally expensive simulations for every candidate structure. Meanwhile, reweighting techniques allow for the recovery of canonical Boltzmann distributions from biased or enhanced sampling simulations, ensuring that thermodynamic properties can be accurately calculated. When combined, these approaches form a cohesive framework that significantly accelerates molecular design and optimization workflows for scientific and industrial applications.
Generative Architectures for Molecular Systems
Architectural Taxonomy and Performance Characteristics
Several probabilistic generative architectures have emerged as particularly effective for molecular simulation tasks. Based on comprehensive benchmarking studies, three frameworks demonstrate distinct performance advantages across different molecular data characteristics [40]:
- Neural Spline Flows (NSF) utilize a series of invertible transformations to map simple distributions to complex ones. They excel at capturing probability density differences and mode asymmetry in low-dimensional data but experience decreased accuracy in high-dimensional spaces [40].
- Conditional Flow Matching (CFM) models demonstrate superior performance for high-dimensional data with low complexity but show diminished capability when modeling complex, multimodal distributions [40].
- Denoising Diffusion Probabilistic Models (DDPM) progressively add noise to data through a forward process, then learn to reverse this process to generate new samples. They most accurately model complex, multimodal distributions (as in peptide dihedral angle distributions) but are less accurate than other methods at high data dimensionality [40].
Table 1: Performance Characteristics of Probabilistic Generative Models for Molecular Data
| Model | Dimensionality Strength | Multimodal Complexity Handling | Computational Efficiency | Primary Molecular Application |
|---|---|---|---|---|
| Neural Spline Flows (NSF) | Low-dimensional data | Excellent for asymmetric modes | Moderate | Free energy estimation for collective variables |
| Conditional Flow Matching (CFM) | High-dimensional data | Limited for complex multimodality | High | High-dimensional molecular descriptor generation |
| Denoising Diffusion Probabilistic Models (DDPM) | Low-to-mid dimensional data | Superior for complex multimodality | Lower (iterative denoising) | Peptide conformation sampling, small molecule design |
Architecture Selection Protocol
Based on the empirical findings, the following structured protocol is recommended for selecting appropriate generative architectures in SMA-MD research:
Dataset Dimensionality Assessment: Quantify the intrinsic dimensionality of your molecular system using principal component analysis or other dimensionality reduction techniques [41]. For systems with low intrinsic dimensionality (≤50 dimensions), NSF or DDPM are preferred. For high-dimensional systems (>50 dimensions), CFM typically provides superior performance [40].
Modal Complexity Evaluation: Analyze the potential energy surface or free energy landscape for multimodal characteristics. For systems with complex, asymmetric probability distributions (such as peptide dihedral angles), DDPM demonstrates the strongest performance. For simpler, unimodal distributions, CFM is typically sufficient [40].
Training Data Volume Consideration: Evaluate the amount of available simulation data. In low-data regimes, NSF and CFM generally outperform DDPM, which typically requires larger training datasets to achieve optimal performance [40].
Sampling Speed Requirements: For applications requiring rapid generation of molecular configurations, CFM provides the fastest sampling, followed by NSF. DDPM's iterative denoising process makes it computationally more intensive for generation tasks [40].
Reweighting Protocols for Enhanced Sampling
Population-Based Reweighting Methodology
Reweighting protocols are essential for recovering canonical Boltzmann distributions from enhanced sampling simulations. While traditional methods often use potential energy in reweighting, these approaches can suffer from inaccuracies due to large energy fluctuations in complex biomolecules [42]. Population-based reweighting offers a robust alternative that mitigates these issues.
The fundamental principle of population-based reweighting involves modifying the biomolecular potential energy surface by applying a scaling factor λ (ranging from 0 to 1) to create a flattened landscape: ( U^*(x) = λU(x) ) [42]. This modification enhances conformational sampling while maintaining the ability to recover the canonical distribution through population statistics rather than energetic terms.
The mathematical foundation for population-based reweighting derives from statistical mechanics. For a scaled potential energy surface, the modified probability distribution becomes: [ p^(x) = \frac{e^{-βλU(x)}}{Z^} ] where ( Z^* ) is the partition function for the scaled system [42]. The canonical distribution is recovered using: [ p(x) = \frac{p^(x)}{∑_i p^(xi)e^{β(λ-1)U(xi)}} ] This approach effectively groups similar configurations together during reweighting, reducing the impact of energetic noise that plagues traditional reweighting methods [42].
Enhanced Reweighting Implementation Protocol
The following detailed protocol enables effective implementation of population-based reweighting for SMA-MD applications:
System Preparation and Enhanced Sampling
- Configure the molecular system using standard simulation packages (GROMACS, AMBER, or OpenMM).
- Implement potential energy scaling by applying a scaling factor λ (typically 0.5-0.8 for balanced enhancement) to the biomolecular Hamiltonian.
- Execute the enhanced sampling simulation, ensuring sufficient conformational exploration.
Dimensionality Reduction and Microstate Definition
- Extract molecular configurations from the enhanced trajectory at regular intervals.
- Perform dimensionality reduction using principal component analysis (PCA) or other nonlinear techniques (t-SNE, UMAP) to identify essential degrees of freedom [41].
- Define microstates by binning the reduced dimensionality space, with bin sizes typically corresponding to 1-12° for dihedral angles [42].
Population-Based Reweighting Calculation
- Calculate the population of each microstate in the enhanced sampling simulation: ( p^*(xi) = Ni/N{total} ), where ( Ni ) is the count in microstate i.
- Apply the population-based reweighting formula to recover canonical probabilities for each microstate.
- Calculate free energy differences using: ( ΔG = -kBT \ln(pi/pj) ), where ( pi ) and ( p_j ) represent probabilities of different states.
Validation and Convergence Assessment
- Compare results with conventional MD where feasible.
- Perform convergence testing by examining free energy differences across different trajectory segments.
- Validate molecular properties against experimental data when available.
Integrated SMA-MD Framework: TrustMol Case Study
The TrustMol framework represents an advanced implementation of trustworthy inverse molecular design that integrates generative modeling with uncertainty-aware optimization [43]. This approach addresses two critical challenges in SMA-MD: accurate forward modeling of molecular properties and reliable inversion for molecular design.
TrustMol Architecture and Implementation
TrustMol employs a novel SELFIES-Graph-Property Variational Autoencoder (SGP-VAE) that incorporates three information sources to create a well-behaved latent space [43]:
Molecular String Representation: Uses SELFIES (SELF-referencing Embedded Strings) to ensure all decoded molecules are chemically valid [43].
3D Structural Information: Reconstructs 3D molecular graphs to embed structural similarity within the latent space [43].
Property Prediction: Directly predicts molecular properties from latent vectors, organizing the latent space according to property values [43].
This multi-task learning approach creates a latent space where proximity corresponds to both structural and property similarity, enabling more accurate surrogate modeling of the molecular property landscape.
TrustMol Experimental Protocol
The following protocol details the implementation of the TrustMol framework for inverse molecular design:
Latent Space Construction
- Train the SGP-VAE using molecular structures, their 3D geometries, and associated properties.
- Implement the triplet loss function that simultaneously optimizes for SELFIES reconstruction, graph structure reconstruction, and property prediction.
- Validate latent space smoothness by interpolating between known molecular structures and verifying chemical validity and property continuity.
Uncertainty-Aware Surrogate Model Training
- Sample representative latent-property pairs using the novel reacquisition method to ensure comprehensive coverage of the latent space [43].
- Train an ensemble of property predictors to model the mapping from latent space to property space.
- Quantify epistemic uncertainty using the variance of predictions across the ensemble.
Uncertainty-Guided Molecular Optimization
- Initialize a random molecular latent vector ( z_0 ).
- Implement iterative optimization using the objective function: [ z^* = \arg \minz |p{target} - Φ(z)| + α \cdot \mathcal{U}(z) ] where ( Φ(z) ) is the surrogate model prediction, ( \mathcal{U}(z) ) is the epistemic uncertainty, and α is a regularization parameter [43].
- Decode the optimized latent vector ( z^* ) to obtain the final molecular structure.
Table 2: TrustMol Framework Components and Functions
| Component | Implementation | Function in Inverse Molecular Design |
|---|---|---|
| SGP-VAE | Multi-decoder variational autoencoder | Creates property-aware latent space with smooth transitions between valid molecular structures |
| Latent-Property Reacquisition | Active learning-based sampling | Ensures training data representativeness for surrogate model |
| Ensemble Surrogate | Multiple neural networks with varied initialization | Provides both property prediction and uncertainty quantification |
| Uncertainty-Guided Optimization | Regularized objective function | Balances property optimization with exploration of reliable regions |
Essential Research Reagents and Computational Tools
Table 3: Research Reagent Solutions for SMA-MD Implementation
| Research Reagent | Type/Format | Function in SMA-MD Workflow |
|---|---|---|
| Molecular Dynamics Packages (GROMACS, AMBER, OpenMM) | Software suite | Provides foundation for conventional and enhanced sampling simulations |
| Enhanced Sampling Plugins (PLUMED, Colvars) | Library/plugin | Implements advanced sampling algorithms and collective variable analysis |
| Probabilistic Generative Libraries (PyTorch, TensorFlow Probability) | Deep learning frameworks | Enables implementation of NSF, CFM, and DDPM architectures |
| Molecular Representation Tools (SELFIES, SMILES, Graph representations) | Data format/parser | Standardizes molecular structure encoding for machine learning applications |
| Dimensionality Reduction Tools (scikit-learn, UMAP) | Software library | Reduces molecular trajectory data to essential degrees of freedom |
| Free Energy Estimation Tools (MBAR, WHAM) | Analysis algorithm | Calculates thermodynamic properties from simulation data |
The integration of carefully selected generative architectures with robust reweighting protocols creates a powerful foundation for Surrogate Model-Assisted Molecular Dynamics research. By matching architectural strengths to specific molecular data characteristics and implementing population-based reweighting to mitigate energetic noise, researchers can significantly accelerate molecular discovery while maintaining thermodynamic accuracy. The TrustMol framework demonstrates how uncertainty quantification and multi-task learning can further enhance the trustworthiness of inverse molecular design, providing a comprehensive approach to addressing the complex challenges in computational drug development and materials science. As these methodologies continue to evolve, they promise to expand the accessible timescales and complexity of molecular systems amenable to computational design and optimization.
Empirical Validation: Benchmarking SMA-MD Against Conventional Molecular Dynamics
Within the framework of Surrogate Model-Assisted Molecular Dynamics (SMA-MD) research, the accurate assessment of computational sampling is paramount. SMA-MD itself is a procedure designed to sample the equilibrium ensemble of molecules by leveraging deep generative models to enhance the sampling of slow degrees of freedom, followed by statistical reweighting and short simulations [18] [2]. The primary goal is to generate a conformational ensemble that is both diverse and thermodynamically representative, thereby enabling accurate prediction of thermodynamic properties crucial for drug discovery and materials design [18]. This application note details the performance metrics and protocols essential for evaluating the quality of generated ensembles, focusing on two cornerstone concepts: ensemble diversity and energy landscape exploration.
Core Performance Metrics
The quality of a conformational ensemble generated by SMA-MD or related methods can be quantified using a suite of metrics that assess its structural diversity and its coverage of the underlying free energy landscape (FEL). The following table summarizes the key performance metrics.
Table 1: Key Performance Metrics for Conformational Ensemble Assessment
| Metric Category | Specific Metric | Definition and Purpose | Interpretation |
|---|---|---|---|
| Structural Diversity | Root Mean Square Deviation (RMSD) | Measures the average distance between atoms of superimposed structures. Assesses structural variation within the ensemble [44]. | Lower RMSD values indicate higher structural similarity; a diverse ensemble will sample a wide range of RMSD values. |
| Radius of Gyration (Rg) | Measures the compactness of a molecular structure [44] [45]. | Tracking Rg helps identify extended vs. compact conformations, contributing to diversity assessment. | |
| Energy & Stability | Potential Energy | The sum of the bonded and non-bonded interaction energies computed by the force field [18]. | A broader sampling of low-energy states indicates better exploration of stable conformations. |
| Free Energy (ΔG) | The energy associated with a basin in the FEL, derived from the probability of observing a conformational state [45]. | Lower free energy minima correspond to more stable, highly populated states. | |
| Free Energy Landscape Topology | RG-RMSD-based Free Energy Landscape (FEL) | A 2D projection of the FEL using Rg and RMSD as collective variables to visualize stable states and transition paths [44]. | Reveals the number, depth, and barrier heights between metastable conformers. |
| Conformational Markov Network (CMN) | A graph representation where nodes are conformational states and edges are transition probabilities, unveiling the FEL's mesoscopic structure [46]. | Identifies basins of attraction, dwell times, and rate constants between conformational states. |
Experimental Protocols for Metric Evaluation
Protocol: Constructing a Free Energy Landscape from an Ensemble
This protocol details the generation of a 2D free energy landscape, a standard method for visualizing the thermodynamic and kinetic stability of sampled conformations [44] [45].
- Trajectory Preparation: Perform molecular dynamics (MD) simulations using a production-level engine (e.g., AMBER, GROMACS) or an SMA-MD pipeline [44] [2]. Save a sufficient number of frames (e.g., ≥10,000) to ensure adequate sampling of conformational states [45].
- Collective Variable (CV) Calculation: For every saved frame, compute two collective variables that describe the system's essential dynamics. Common choices include:
- Boltzmann Inversion: Create a 2D histogram of the two chosen CVs. The free energy change (ΔG) for the conformations in bin i is calculated using: ΔGi = -kBT ln(ni / nmax) where kB is Boltzmann's constant, T is the simulation temperature, ni is the population of the i-th bin, and nmax is the population of the most populated bin [45]. This yields a landscape where the deepest well (global minimum) is set to zero.
- Visualization: Plot the resulting ΔG values as a contour or surface plot. The x and y axes are the chosen CVs (e.g., RMSD and Rg), and the z-axis (often represented by color contours) is the relative free energy [45].
Protocol: Assessing Ensemble Diversity via Structural Clustering
This protocol uses structural clustering to quantify the diversity of conformations in the generated ensemble.
- Frame Alignment: Superimpose all trajectory frames onto a common reference structure (e.g., the first frame or an average structure) to remove global rotation and translation.
- RMSD Matrix Calculation: Compute an all-to-all pairwise RMSD matrix for the aligned conformational snapshots.
- Clustering Algorithm: Apply a clustering algorithm (e.g., hierarchical clustering, k-means, or DBSCAN) to the RMSD matrix to group structurally similar conformations. Each resulting cluster represents a distinct metastable state.
- Diversity Quantification: Calculate the following to assess diversity:
- Number of Clusters: A larger number of significant clusters indicates higher structural diversity.
- Population Distribution: The relative population of each cluster, which can be related to the free energy of that state (ΔG ~ -ln(Population)) [46].
- Inter-Cluster RMSD: The average RMSD between the centroids of different clusters; a higher value suggests the ensemble covers broader conformational space.
Workflow Diagram
The following diagram illustrates the logical workflow for evaluating conformational ensembles using the protocols described above, integrating both SMA-MD and traditional simulation approaches.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
This section lists key software tools and computational methods that form the essential "research reagents" for conducting SMA-MD research and analyzing conformational ensembles.
Table 2: Key Research Reagents and Computational Solutions
| Tool/Solution | Type | Function in Ensemble Analysis |
|---|---|---|
| SMA-MD Codebase [2] | Software Pipeline | Implements the core Surrogate Model-Assisted Molecular Dynamics procedure, integrating generative modeling with molecular dynamics. |
| MD Simulation Engines (e.g., AMBER) [44] | Software | Performs the molecular dynamics simulations that generate the conformational trajectories for analysis. |
| Generative Models (e.g., Torsional Diffusion) [2] | Algorithm/Model | Acts as the surrogate model in SMA-MD to enhance sampling of slow torsional degrees of freedom. |
| Conformational Markov Network (CMN) [46] | Analysis Framework | Provides a mesoscopic description of the Free Energy Landscape, revealing basins, pathways, and kinetics. |
| MD DaVis [45] | Analysis Software | Specialized tool for constructing, visualizing, and comparing free energy landscapes from simulation trajectories. |
| MMPBSA.py (AMBER) [44] | Analysis Tool | Calculates binding free energies from MD trajectories using Molecular Mechanics/Generalized Born Surface Area methods. |
| Landscape17 Benchmark [47] | Dataset & Test Suite | Provides kinetic transition networks for validating machine learning interatomic potentials on kinetic properties. |
Accurate sampling of molecular conformational ensembles is fundamental to advancements in structural biology and rational drug design. The thermodynamic properties and biological functions of molecules are dictated by their dynamic energy landscapes rather than single, static structures. Conventional Molecular Dynamics (cMD) simulations have long been the cornerstone for exploring these landscapes, providing atomic-level resolution and insights into molecular motion. However, their computational cost and limited capacity to sample rare events or slow degrees of freedom present significant bottlenecks. Surrogate Model-Assisted Molecular Dynamics (SMA-MD) emerges as a transformative approach that integrates deep generative models with physics-based simulations to overcome these limitations, offering a more efficient path to sampling equilibrium ensembles. This application note provides a detailed, comparative analysis of these two methodologies, equipping researchers with the data and protocols needed to select and implement the appropriate sampling strategy for their projects.
Conventional MD (cMD) relies on numerically solving Newton's equations of motion for a system of atoms, using empirical force fields to calculate energies and forces. While highly accurate, its sampling efficiency is constrained by the simulation timestep (typically 1-2 femtoseconds) and the need to simulate over micro- to milliseconds to observe biologically relevant transitions. This often makes it prohibitively expensive for adequate sampling of conformational space, particularly for flexible biomolecules like intrinsically disordered proteins (IDPs) or RNA [48].
SMA-MD is a multi-stage procedure designed to enhance the sampling of slow degrees of freedom [2]. It first leverages a deep generative model, trained on a dataset of molecular conformations, to propose a diverse set of candidate structures. This ensemble is then statistically reweighted, followed by short, conventional MD simulations for local relaxation and energy evaluation. This hybrid approach aims to generate more diverse and lower-energy ensembles than cMD alone [2].
The table below summarizes the core methodological differences and performance outcomes of the two approaches.
Table 1: Head-to-Head Comparison of SMA-MD and Conventional MD
| Feature | SMA-MD (Surrogate Model-Assisted MD) | Conventional MD (cMD) |
|---|---|---|
| Core Principle | Hybrid approach combining deep generative models with short MD simulations for refinement and validation [2]. | Physics-based simulation using numerical integration of equations of motion with empirical force fields [49]. |
| Sampling Mechanism | Generative model proposes structures; MD refines and validates [2]. | Time-dependent exploration of the energy landscape from an initial structure [49]. |
| Computational Efficiency | Higher efficiency for sampling slow degrees of freedom and rare events; reduces need for long simulation times [2]. | Computationally expensive; requires long simulation times (µs-ms) for adequate sampling, limited by timestep [48]. |
| Diversity of Ensembles | Generates more diverse conformational ensembles by learning from data and exploring broader space [2]. | Risk of being trapped in local energy minima near the starting conformation; lower diversity without enhanced sampling [48]. |
| Energy of Ensembles | Empirical results show generation of ensembles with lower energy states compared to cMD [2]. | Aims to sample the Boltzmann distribution but may miss low-energy states due to insufficient sampling [48]. |
| Applicability to IDPs/RNA | Potentially highly effective for highly flexible systems like IDPs and RNA by learning complex conformational distributions [48]. | Struggles with the vast conformational space of IDPs and RNA; often insufficient sampling of transient states [49] [48]. |
| Key Limitations | Dependence on quality and size of training data; model interpretability; integration of physical laws is complex [48]. | Extremely high computational cost; force field inaccuracies; poor sampling efficiency for rare events [49] [48]. |
A practical demonstration of cMD's limitations is evident in RNA refinement. A 2025 benchmark study found that short cMD simulations (10–50 ns) could provide modest improvements for high-quality starting RNA models, but poorly predicted models rarely benefit and often deteriorate. Furthermore, longer simulations (>50 ns) typically induced structural drift and reduced fidelity, challenging the assumption that longer cMD runs are inherently better for refinement [49].
Experimental and Computational Protocols
Protocol for SMA-MD Implementation
The following protocol is adapted from the official SMA-MD repository [2].
Objective: To generate a thermodynamically representative conformational ensemble of a small molecule using SMA-MD.
Prerequisites:
- Anaconda/Miniconda with Python 3.9
- CUDA-enabled GPU
- Installation of the
sma-mdpackage from GitHub [2]
Procedure:
Environment Setup
Data Preprocessing
- Prepare your input molecular structure file (e.g., in .pdb or .sdf format).
- Run the preprocessing script to featurize the data for the generative model.
Surrogate Model Training
- Train a torsional diffusion model on a relevant dataset of molecular conformers.
- Hyperparameters and dataset paths are specified in
./parameters.py.
Conformational Sampling
- Sample a large ensemble of candidate conformers from the trained generative model.
Energy Evaluation and Reweighting
- Switch to the OpenMM environment and evaluate the energy of each sampled conformation.
- Statistically reweight the ensemble to approximate the Boltzmann distribution.
MD Fine-Tuning
- Perform short MD simulations from the top-weighted conformations to relax local strains and validate stability.
Protocol for Conventional MD Refinement of RNA Structures
This protocol is based on a 2025 benchmark study that established best practices for using cMD in RNA model refinement [49].
Objective: To refine a predicted RNA 3D model using short, restrained MD simulations.
Prerequisites:
- AMBER 22 package with PMEMD.CUDA [49]
- AMBER ff99bsc0χOL3 (χOL3) force field for RNA [49]
- System with GPU acceleration
Procedure:
System Preparation
- Use the
tleapmodule from AMBER to prepare the initial RNA structure. - Solvate the RNA in a truncated octahedral TIP3P water box with a 10 Å buffer.
- Neutralize the system with Na+ ions.
- Use the
Energy Minimization
- Perform a two-stage minimization to remove steric clashes.
- Stage 1: 10,000 cycles with positional restraints on RNA backbone phosphorus and oxygen atoms (restraint_wt = 20.0 kcal mol⁻¹ Å⁻²).
- Stage 2: 10,000 cycles with no restraints.
System Heating
- Gradually heat the system from 100 K to 300 K over 500 ps (250,000 steps) under an NVT ensemble.
- Maintain positional restraints on RNA backbone atoms (restraint_wt = 20.0 kcal mol⁻¹ Å⁻²).
- Use a Langevin thermostat with a collision frequency of 5.0 ps⁻¹.
System Equilibration
- Equilibrate in four phases under NVT conditions:
- Phase 1: 200 ps with backbone restraints at 10.0 kcal mol⁻¹ Å⁻².
- Phase 2: 200 ps with backbone restraints at 5.0 kcal mol⁻¹ Å⁻².
- Phase 3: 200 ps with backbone restraints at 1.0 kcal mol⁻¹ Å⁻².
- Phase 4: 2 ns without any restraints.
Production Simulation
- Run a production simulation under constant pressure (NPT ensemble) for a recommended duration of 10-50 ns [49].
- Use a Monte Carlo barostat for pressure control and a Langevin thermostat.
- Critical Note: The benchmark study strongly advises against extended simulations (>50 ns) for refinement purposes, as they often induce structural drift [49].
Analysis
- Analyze the trajectory using tools like
cpptrajto calculate Root Mean Square Deviation (RMSD) and identify the most stable, low-energy conformation from the production run.
- Analyze the trajectory using tools like
Workflow Visualization
The following diagram illustrates the logical sequence and key differences between the SMA-MD and conventional MD workflows.
SMA-MD vs cMD Workflow
The Scientist's Toolkit: Essential Research Reagents & Software
Table 2: Key Resources for Conformational Sampling Studies
| Item / Resource | Function / Description | Example / Source |
|---|---|---|
| AMBER | A suite of biomolecular simulation programs used for conventional MD simulations, including energy minimization, dynamics, and analysis [49]. | https://ambermd.org/ |
| ff99bsc0χOL3 | A well-validated, RNA-specific force field for AMBER, crucial for accurate simulation of RNA structures and dynamics [49]. | Included with AMBER |
| OpenMM | A high-performance, open-source toolkit for molecular simulation, used in the SMA-MD pipeline for energy evaluation and MD fine-tuning [2]. | https://openmm.org/ |
| SMA-MD Code | The core software package implementing the Surrogate Model-Assisted Molecular Dynamics protocol [2]. | https://github.com/olsson-group/sma-md |
| Torsional Diffusion | A deep learning model for molecular conformer generation, which can serve as the generative surrogate model in SMA-MD [2]. | Jing et al., 2023 |
| SHAMAN | An advanced computational technique that uses probes and metadynamics to identify small-molecule binding sites in dynamic RNA ensembles [22]. | Nature Communications (2024) |
| CUDA-enabled GPU | Essential hardware for accelerating both MD simulations (e.g., via PMEMD.CUDA) and the training/inference of deep generative models. | NVIDIA GPUs |
The choice between SMA-MD and conventional MD is not a matter of simple replacement but strategic selection. Conventional MD remains the method of choice for probing local dynamics, validating specific structural models, and simulating processes where physical accuracy at short timescales is paramount. However, for the daunting task of efficiently exploring vast conformational spaces, generating diverse structural ensembles, and accessing rare events, SMA-MD presents a transformative, data-driven alternative. By integrating the power of deep generative models with the physical rigor of MD, SMA-MD addresses the critical sampling bottleneck, offering a more efficient and comprehensive path to understanding molecular thermodynamics. This is particularly impactful for drug discovery efforts targeting highly dynamic and therapeutically relevant biomolecules like RNA. Integrating these advanced computational methods paves the way for a new era in rational drug design.
Surrogate Model-Assisted Molecular Dynamics (SMA-MD) represents a transformative approach in computational chemistry and drug discovery, designed to overcome the inherent sampling limitations of conventional molecular dynamics (MD) simulations. Accurate prediction of thermodynamic properties is crucial in various fields such as drug discovery and materials design. This task relies on sampling from the underlying Boltzmann distribution, which is challenging using conventional approaches such as simulations [18]. The SMA-MD procedure enhances the sampling of slow degrees of freedom and generates more diverse and lower-energy conformational ensembles, enabling more accurate computation of thermodynamic properties like implicit solvation free energies [18]. This Application Note provides a detailed quantitative and methodological framework for implementing SMA-MD, with specific emphasis on its validation through lower energy ensembles and improved thermodynamic property prediction.
The superior performance of SMA-MD over conventional MD is demonstrated through key metrics that highlight its enhanced sampling efficiency and thermodynamic accuracy. The following table summarizes the core quantitative findings from empirical evaluations of the SMA-MD methodology.
Table 1: Quantitative Performance Metrics of SMA-MD vs. Conventional MD
| Metric | SMA-MD Performance | Conventional MD Performance | Significance |
|---|---|---|---|
| Ensemble Diversity | Higher | Lower | SMA-MD accesses a broader region of conformational space [18] |
| Energy Levels | Lower energy ensembles | Higher energy ensembles | SMA-MD identifies more stable molecular configurations [18] |
| Sampling Efficiency | Enhanced sampling of slow degrees of freedom | Limited by simulation timescales | Deep Generative Models overcome energy barriers [18] |
| Application Potential | Accurate implicit solvation free energies | Challenging for complex molecules | Improved thermodynamic property prediction [18] |
SMA-MD Experimental Protocol
This section provides a detailed, step-by-step protocol for executing the core SMA-MD workflow to generate and validate conformational ensembles.
Protocol: SMA-MD for Ensemble Generation and Validation
Objective: To generate a diverse, low-energy conformational ensemble of a small molecule and compute its thermodynamic properties.
Materials & Prerequisites:
- Initial Structure: A 3D molecular structure file (e.g., PDB, MOL2).
- Software: Access to a deep generative model framework (e.g., PyTorch, TensorFlow) and an MD simulation package (e.g., GROMACS, OpenMM, AMBER).
- Force Field: A suitable molecular mechanics force field and solvent model.
Procedure:
Deep Generative Model Sampling
- Input: The initial 3D molecular structure.
- Process: Train or utilize a pre-trained deep generative model to sample molecular conformations. This model is designed to explore the molecule's torsional space and other slow degrees of freedom that are poorly sampled by conventional MD.
- Output: A preliminary, broad ensemble of molecular conformations.
Statistical Reweighting
- Input: The preliminary ensemble from Step 1.
- Process: Apply statistical reweighting techniques (e.g., based on the Boltzmann distribution) to assign appropriate statistical weights to each generated conformation. This step ensures the ensemble reflects the true thermodynamic equilibrium.
- Output: A reweighted conformational ensemble.
Targeted Molecular Dynamics Simulations
- Input: Selected structures from the reweighted ensemble.
- Process: Perform short, conventional MD simulations initiated from the reweighted conformations. These simulations locally explore the conformational space around each starting point and validate the stability of the predicted structures.
- Output: A refined and validated conformational ensemble.
Validation and Analysis
- Cluster Analysis: Perform clustering (e.g., using RMSD) on the final ensemble to identify dominant conformational states.
- Energy Analysis: Compare the potential energy distribution of the SMA-MD ensemble against an ensemble generated from a long, conventional MD simulation.
- Property Calculation: Compute the target thermodynamic property, such as the implicit solvation free energy, from the finalized ensemble.
Workflow Visualization
The following diagram illustrates the logical flow and key components of the SMA-MD protocol.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of the SMA-MD protocol relies on a combination of computational tools and theoretical frameworks. The following table catalogues the essential "research reagents" for this methodology.
Table 2: Essential Research Reagents for SMA-MD Implementation
| Item | Function / Description | Application in Protocol |
|---|---|---|
| Deep Generative Model | A machine learning model that learns the underlying data distribution of molecular conformations to generate novel, plausible structures. | Samples initial conformational ensemble, overcoming slow degrees of freedom [18]. |
| Reweighting Algorithm | A statistical method (e.g., Binless WHAM/MBAR) to correct the weights of sampled structures to match the Boltzmann distribution. | Corrects biases in the generated ensemble to recover true thermodynamics [18]. |
| Molecular Dynamics Engine | Software that performs numerical simulation of molecular motion based on classical mechanics. | Executes short, targeted simulations to refine and validate the ensemble [18]. |
| Implicit Solvent Model | A computational method that represents solvent as a continuous medium rather than explicit molecules, reducing computational cost. | Enables efficient calculation of solvation free energies from the final ensemble [18]. |
| Boltzmann Distribution | The fundamental probability distribution for states in a system at thermodynamic equilibrium. | Serves as the theoretical target for the reweighted conformational ensemble [18]. |
The accurate prediction of solvation free energy is a cornerstone of computational chemistry, with profound implications for drug discovery and materials science. In biological systems, which are primarily aqueous, predicting the hydration free energy of small molecules deepens our understanding of dissolution mechanisms and provides critical theoretical support for rational drug design [50]. Traditional implicit solvent models, which treat the solvent as a continuous medium rather than explicit molecules, offer computational efficiency but often sacrifice accuracy, particularly for complex molecular systems [29].
The integration of machine learning (ML) techniques with physical models presents a promising pathway to overcome these limitations. Recent advancements have demonstrated that ML can enhance the accuracy of solvation free energy predictions by almost an order of magnitude without substantial additional computational costs [51]. Furthermore, within the specific context of Surrogate Model-Assisted Molecular Dynamics (SMA-MD), these approaches enable more efficient sampling of molecular conformational ensembles, leading to improved estimation of thermodynamic properties like implicit solvation free energies [2] [18].
This case study examines the key methodologies, performance benchmarks, and experimental protocols that are driving these accuracy enhancements, providing researchers with practical insights for implementing these approaches in computational drug development.
Performance Comparison of Computational Approaches
The table below summarizes the performance of various modern computational approaches for predicting solvation free energy, highlighting their methodologies and reported accuracy.
Table 1: Performance Comparison of Solvation Free Energy Prediction Methods
| Method Name | Core Methodology | Test Dataset | Key Features | Reported Error (MUE) |
|---|---|---|---|---|
| Improved ML Scheme [50] | Ensemble ML with KNN imputation | FreeSolv (642 molecules) | 2D features only; KNN for missing data | 0.53 kcal/mol |
| LSNN Model [52] | Graph Neural Network (GNN) | ~300,000 small molecules | Derivatives matching for alchemical variables; combines polar & non-polar terms | Comparable to explicit solvent |
| ML-PCM [51] | Machine-Learning Polarizable Continuum Model | Benchmark experimental data | Uses SCRF energy components; neural network mapping | 0.40 - 0.53 kcal/mol |
| SMA-MD [2] [18] | Surrogate Model-Assisted Molecular Dynamics | Molecular conformers | Deep generative models for sampling; reweighting & short simulations | Improved ensemble diversity & energy |
Detailed Experimental Protocols
Lightweight Machine Learning Scheme
This protocol outlines a high-accuracy, resource-efficient method for predicting hydration free energy using classical ML models on the FreeSolv database [50].
Data Set Acquisition and Splitting
- Source the FreeSolv database (v0.52), containing 642 small neutral organic molecules.
- Assign 47 molecules from the SAMPL4 blind challenge as a dedicated test set.
- Use the remaining 595 molecules for model training and validation to ensure no data leakage.
Feature Preprocessing and Engineering
- Descriptor Generation: Convert molecular structures into numerical descriptors using multiple fingerprint methods (APFP, ECFP6) and topological/physical-chemical descriptors (TOPOL, MolProps). Create concatenated descriptors (e.g., MolPropsECFP6).
- Handling Missing Data: Remove non-numeric features initially. Impute remaining missing values using the K-Nearest Neighbors (KNN) algorithm.
- Data Normalization: Scale all feature data to a consistent range to ensure model stability.
Model Training and Evaluation
- Algorithm Selection: Train and compare multiple ML models, including Support Vector Machine (SVM), Random Forest (RF), and Extreme Gradient Boosting (XGB).
- Ensemble Modeling: Employ ensemble learning techniques to combine predictions from multiple base models for improved accuracy and robustness.
- Performance Validation: Evaluate the final model on the held-out SAMPL4 test set, reporting the Mean Unsigned Error (MUE).
Graph Neural Network for Implicit Solvation (LSNN)
This protocol describes training a GNN-based implicit solvent model that is suitable for free energy calculations by going beyond simple force-matching [52].
Model Architecture Setup
- Implement a Graph Neural Network (GNN) where atoms are represented as nodes and bonds as edges.
- Design the network to output both solvation forces and the derivatives of the solvation energy with respect to alchemical coupling parameters (
λ_elecandλ_steric).
Loss Function Definition
- Use a multi-component loss function that goes beyond traditional force-matching:
ℒ = w_F * (Force_Error) + w_elec * (dG/dλ_elec_Error) + w_steric * (dG/dλ_steric_Error) - Empirically tune the weights (
w_F,w_elec,w_steric) to balance the contribution of each term.
- Use a multi-component loss function that goes beyond traditional force-matching:
Model Training
- Train the model on a large dataset (e.g., ~300,000 small molecules) using reference data from explicit-solvent simulations.
- Optimize model parameters to minimize the combined loss function, ensuring the model learns a consistent potential energy landscape.
Free Energy Calculation
- Use the trained LSNN model in alchemical free energy simulation protocols (e.g., Thermodynamic Integration). The model's accurate derivatives with respect to
λvariables enable precise calculation of solvation free energies.
- Use the trained LSNN model in alchemical free energy simulation protocols (e.g., Thermodynamic Integration). The model's accurate derivatives with respect to
Surrogate Model-Assisted Molecular Dynamics (SMA-MD)
This protocol uses deep generative models to enhance conformational sampling for calculating implicit solvation free energies [2] [18].
Generative Sampling
- Train a deep generative model (e.g., a diffusion model or variational autoencoder) on molecular structures to learn the underlying degrees of freedom.
- Sample a diverse set of molecular conformers from the generative model, effectively exploring low-energy states and slow conformational transitions that are difficult to access with standard MD.
Statistical Reweighting
- Apply statistical mechanics reweighting techniques (e.g., Boltzmann reweighting) to correct for any biases introduced by the generative model. This step ensures the generated ensemble represents the true equilibrium distribution.
Molecular Dynamics Finetuning
- Perform short, conventional MD simulations starting from the reweighted ensemble of conformers. This step refines local geometry and validates the stability of the sampled structures.
Solvation Free Energy Estimation
- Compute the solvation free energy for each conformer in the final ensemble using an implicit solvation model (e.g., GB, PB, or an ML-enhanced model like ML-PCM).
- Calculate the overall thermodynamic average by weighting the solvation free energy of each conformer according to its Boltzmann probability.
Workflow Visualization
Figure 1: A unified workflow for enhanced solvation free energy calculations, integrating both direct ML prediction and SMA-MD sampling paths.
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Tool/Resource | Type | Primary Function | Relevance to Protocol |
|---|---|---|---|
| FreeSolv Database [50] | Database | Benchmark dataset of experimental and calculated solvation free energies for small molecules. | Provides standardized data for training and testing ML models (Protocol 3.1). |
| K-Nearest Neighbors (KNN) [50] | Algorithm | Handles missing values in feature data by imputation from similar molecules. | Critical pre-processing step in lightweight ML scheme to maintain dataset integrity (Protocol 3.1). |
| Graph Neural Network (GNN) [52] | Model Architecture | Learns representation of molecular structure and properties directly from graph data (atoms/bonds). | Core of the LSNN model that captures complex atomic interactions for implicit solvation (Protocol 3.2). |
| Alchemical Coupling Parameters (λ) [52] | Mathematical Parameter | Scales intermolecular interactions in alchemical free energy calculations. | Enables accurate free energy comparisons across molecules by extending the loss function (Protocol 3.2). |
| Generative Model (e.g., Diffusion) [2] | Model Architecture | Samples diverse molecular conformations beyond local minima found by MD. | Enhances conformational sampling in SMA-MD by exploring slow degrees of freedom (Protocol 3.3). |
| Boltzmann Reweighting [2] [18] | Statistical Method | Corrects biases in a generated ensemble to recover the true equilibrium distribution. | Essential step in SMA-MD to ensure sampled conformers represent the correct thermodynamics (Protocol 3.3). |
Conclusion
Surrogate Model-Assisted Molecular Dynamics represents a significant advancement in computational molecular science, effectively addressing the long-standing challenge of Boltzmann distribution sampling that has limited conventional MD approaches. By strategically integrating deep generative models with statistical reweighting and targeted simulations, SMA-MD generates more diverse and thermodynamically favorable conformational ensembles, enabling more accurate prediction of crucial properties like solvation free energies. The empirical validation demonstrating superior performance over traditional methods, coupled with practical strategies for overcoming implementation challenges such as explicit solvent environments, establishes SMA-MD as a powerful tool for researchers and drug development professionals. Future directions should focus on expanding applications to complex biological systems, integrating with multi-omics data in biomedical research, and further optimizing computational efficiency for high-throughput drug screening. As this methodology matures, it holds immense promise for accelerating rational drug design and materials development by providing unprecedented access to biomolecular conformational landscapes.
References
- 1. HollowFlow: Efficient Sample Likelihood Evaluation using ... [https://arxiv.org/html/2510.21542]
- 2. olsson-group/sma-md [https://github.com/olsson-group/sma-md]
- 3. Investigating New Treatments for Spinal Muscular Atrophy [https://news.feinberg.northwestern.edu/2025/09/16/investigating-new-treatments-for-spinal-muscular-atrophy/]
- 4. Advancing personalized spinal muscular atrophy care [https://pmc.ncbi.nlm.nih.gov/articles/PMC12405325/]
- 5. Drugs for Spinal Muscular Atrophy (SMA) 2025 to Grow at ... [https://www.datainsightsmarket.com/reports/drugs-for-spinal-muscular-atrophy-sma-301494]
- 6. SMA clinical trial results: what's new? [https://www.institut-myologie.org/en/2025/02/19/sma-clinical-trial-results-whats-new/]
- 7. Generative Modeling of Molecular Dynamics Trajectories [https://arxiv.org/html/2409.17808v1]
- 8. Using physical property surrogate models to perform ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10259372/]
- 9. Conformational landscapes of membrane proteins ... [https://www.sciencedirect.com/science/article/pii/S0005273617303516]
- 10. Editorial: Advanced Sampling and Modeling in Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8633950/]
- 11. AI-based Methods for Simulating, Sampling, and Predicting Protein... [https://arxiv.org/html/2509.17224v1]
- 12. Pharmacological Therapies of Spinal Muscular Atrophy [https://pmc.ncbi.nlm.nih.gov/articles/PMC10605203/]
- 13. Transforming Spinal Muscular Atrophy: From Pivotal Trials ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383623/]
- 14. A qualitative, mixed-method approach to reaching ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535128/]
- 15. Twice-Weekly Outpatient Rehabilitation Intervention for ... [https://www.researchprotocols.org/2023/1/e46363]
- 16. Therapeutic Decision-Making Under Uncertainty in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9338192/]
- 17. An overview of the Cure SMA membership database [https://pmc.ncbi.nlm.nih.gov/articles/PMC6004903/]
- 18. Generation of conformational ensembles of small ... [https://chemrxiv.org/engage/chemrxiv/article-details/65604ef5cf8b3c3cd7009e1f]
- 19. Generation of conformational ensembles of small ... [https://research.chalmers.se/en/publication/540902]
- 20. Assessing spinal muscular atrophy with quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2918474/]
- 21. Efficient Ensemble Refinement by Reweighting - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6727217/]
- 22. Identifying small-molecules binding sites in RNA ... [https://www.nature.com/articles/s41467-024-49638-7]
- 23. Thermodynamic Interpolation: A Generative Approach to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11912209/]
- 24. Extrapolative prediction of small-data molecular property ... [https://www.nature.com/articles/s41524-023-01194-2]
- 25. Text has enhanced contrast | ACT Rule | WAI [https://www.w3.org/WAI/standards-guidelines/act/rules/09o5cg/]
- 26. Color contrast - Accessibility - MDN Web Docs - Mozilla [https://developer.mozilla.org/en-US/docs/Web/Accessibility/Guides/Understanding_WCAG/Perceivable/Color_contrast]
- 27. Implicit solvent methods for free energy estimation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4310817/]
- 28. Implicit solvation [https://en.wikipedia.org/wiki/Implicit_solvation]
- 29. Accuracy comparison of several common implicit solvent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5313374/]
- 30. Extending machine learning model for implicit solvation to ... [https://arxiv.org/abs/2510.20103]
- 31. Implicit Solvent Models and Their Applications in Biophysics [https://www.mdpi.com/2218-273X/15/9/1218]
- 32. From Implicit to Explicit: An Interaction-Reorganization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11674157/]
- 33. Speeding up MD simulations in explicit . – Dr Anthony Nash... solvent [https://distributedscience.wordpress.com/2017/06/19/speeding-up-md-simulations-in-explicit-solvent/]
- 34. Using physical property surrogate models to perform ... [https://pubs.rsc.org/en/content/articlehtml/2023/dd/d2dd00138a]
- 35. Deep adaptive sampling for surrogate modeling without ... [https://arxiv.org/abs/2402.11283]
- 36. Adaptive sampling techniques of surrogate optimization for ... [https://ui.adsabs.harvard.edu/abs/2025IJMMD.tmp...44E/abstract]
- 37. Molecular dynamics enabled data-driven modeling of ... [https://www.sciencedirect.com/science/article/pii/S004578252500502X]
- 38. A machine learning-based comparative analysis of surrogate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10405017/]
- 39. [2010.11708] Context-aware surrogate modeling for ... [https://arxiv.org/abs/2010.11708]
- 40. A survey of probabilistic generative frameworks for ... [https://arxiv.org/html/2411.09388v1]
- 41. Unsupervised Learning Methods for Molecular Simulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8391792/]
- 42. Population Based Reweighting of Scaled Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3808002/]
- 43. Trustworthy Inverse Molecular Design via Alignment with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12279213/]
- 44. Structural and free energy landscape analysis for the ... [https://www.nature.com/articles/s41598-024-69816-3]
- 45. Free Energy Landscapes — MD DaVis 0.4.1 documentation [https://md-davis.readthedocs.io/en/latest/guides/free_energy_landscapes.html]
- 46. Exploring the Free Energy Landscape: From Dynamics to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2694367/]
- 47. a testing and training arena for machine learned potentials [https://arxiv.org/abs/2508.16425]
- 48. Use of AI-methods over MD simulations in the sampling of ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1542267/full]
- 49. When does molecular dynamics improve RNA models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12513224/]
- 50. Enhancing Accuracy and Feature Insights in Hydration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12268727/]
- 51. Improved prediction of solvation free energies by machine ... [https://www.nature.com/articles/s41467-021-23724-6]
- 52. Extending machine learning model for implicit solvation to ... [https://arxiv.org/html/2510.20103v1]