Steepest Descent vs Conjugate Gradient: Algorithmic Breakdown and Applications in Drug Discovery
This article provides a comprehensive explanation of the Steepest Descent and Conjugate Gradient algorithms, tailored for researchers and professionals in drug development.
Steepest Descent vs Conjugate Gradient: Algorithmic Breakdown and Applications in Drug Discovery
Abstract
This article provides a comprehensive explanation of the Steepest Descent and Conjugate Gradient algorithms, tailored for researchers and professionals in drug development. It covers the foundational mathematics of both methods, explores their practical application in critical tasks like energy minimization for small-molecule drug discovery, offers strategies for troubleshooting and optimization, and presents a comparative analysis of their performance. The content synthesizes theoretical insights with real-world biomedical case studies, including applications for Covid-19 and diabetes drug candidates, to guide the selection and implementation of efficient optimization techniques in clinical research.
Gradient Basics: Unraveling the Core Principles of Descent Algorithms
Optimization algorithms form the computational backbone of modern scientific inquiry, from simulating molecular interactions to training complex artificial intelligence models. At its core, an optimization problem involves finding the parameters that minimize (or maximize) an objective function. In computational chemistry and drug discovery, this typically translates to identifying molecular configurations with the lowest possible energy states, which correspond to stable, biologically relevant structures. The efficiency and effectiveness of these optimization procedures directly impact research timelines, computational costs, and ultimately, the success of scientific endeavors.
This technical guide examines two fundamental optimization approaches—steepest descent and conjugate gradient methods—within the context of solving linear systems and energy minimization problems. While both are first-order iterative optimization techniques, their convergence properties, computational requirements, and applicability to different problem domains vary significantly. Understanding their theoretical foundations and practical implementations is essential for researchers selecting appropriate methodologies for scientific computing applications, particularly in molecular modeling and drug development workflows where energy minimization is a critical step [1].
Mathematical Foundations of Optimization
The General Minimization Problem
The minimization problem can be formally stated as follows: given a function ( f(\mathbf{x}) ) where ( \mathbf{x} ) is a vector of variables, find the value ( \mathbf{x}^* ) such that ( f(\mathbf{x}^*) ) is a local minimum. In molecular modeling contexts, ( f ) typically represents the potential energy of a system, and ( \mathbf{x} ) represents the atomic coordinates [1]. The potential energy function incorporates multiple components:
- Bond energy and angle energy, representing covalent bonds and bond angles
- Dihedral energy, accounting for torsional strain
- van der Waals terms (Leonard-Jones potential) preventing steric clashes
- Electrostatic energy modeling long-range forces between charged particles [1]
These quantitative terms are parameterized in what is collectively known as a force field (e.g., CHARMM, AMBER, GROMOS) [1].
Stationary Points and the Energy Surface
The potential energy surface (PES) represents the mathematical relationship between molecular structure and its energy [2]. Minimum points on this surface correspond to stable molecular conformations, with the global energy minimum representing the most stable configuration [1]. The challenge lies in efficiently navigating this complex, high-dimensional surface to locate minima, particularly the global minimum among potentially many local minima.
Table 1: Key Concepts in Energy Minimization
| Concept | Mathematical Definition | Physical Interpretation |
|---|---|---|
| Stationary Point | ( \nabla f(\mathbf{x}) = 0 ) | Geometry where net force on all atoms is zero |
| Local Minimum | ( f(\mathbf{x}^) \leq f(\mathbf{x}) ) for ( |\mathbf{x} - \mathbf{x}^| < \varepsilon ) | Stable molecular conformation |
| Global Minimum | Lowest value of ( f(\mathbf{x}) ) across entire domain | Most thermodynamically stable conformation |
| Saddle Point | Point where gradient is zero but not a minimum | Transition state between stable conformations |
First-Order Optimization Algorithms
First-order iterative optimization methods generate a sequence ( {\mathbf{x}^k}_{k\in\mathbb{N}} ) that converges to a minimum (or stationary point) through the iterative process:
- Selecting a search direction ( \mathbf{d}^k \in \mathbb{R}^n )
- Determining a step size ( \tau_k \in \mathbb{R} )
- Updating the position: ( \mathbf{x}^{k+1} = \mathbf{x}^k + \tau_k \mathbf{d}^k )
- Repeating until convergence criteria are met [3]
These methods use only function values ( f(\mathbf{x}^j) ) and gradients ( \nabla f(\mathbf{x}^j) ) for ( j \leq k ) when determining ( \mathbf{d}^k ) and ( \tau_k ).
Steepest Descent Algorithm
Theoretical Foundation
The steepest descent method (also known as gradient descent) represents the simplest first-order optimization approach, choosing the search direction as the negative gradient: ( \mathbf{d}^k = -\nabla f(\mathbf{x}^k) ) [3]. This direction represents the locally steepest downhill direction on the energy surface.
In molecular modeling implementations such as GROMACS, the algorithm proceeds as follows [4]:
- Compute forces ( \mathbf{F} ) (negative gradient) and potential energy at current position
- Calculate new positions: ( \mathbf{r}{n+1} = \mathbf{r}n + \mathbf{F}n \frac{hn}{\max(|\mathbf{F}n|)} ), where ( hn ) is the maximum displacement
- Recompute forces and energy for the new positions
- If ( V{n+1} < Vn ): accept new positions and increase step size ( h{n+1} = 1.2hn )
- If ( V{n+1} \geq Vn ): reject new positions and decrease step size ( hn = 0.2hn )
The algorithm terminates when the maximum absolute value of the force components falls below a specified threshold ( \epsilon ) [4].
Convergence Properties and Limitations
Steepest descent exhibits robust convergence in initial iterations, particularly when starting configurations are far from minimum [2] [3]. However, it demonstrates poor asymptotic convergence, with ( f(\mathbf{x}^k) - \min_{\mathbf{x}} f(\mathbf{x}) = \mathcal{O}(1/k) ) [3]. As the method approaches a minimum, progress slows dramatically, with the algorithm often exhibiting oscillatory behavior in narrow valleys of the energy surface [2].
Despite these limitations, steepest descent remains valuable for preliminary minimization stages where rapid initial progress is more important than precise convergence, and for systems with significant steric clashes that require initial resolution [4].
Conjugate Gradient Algorithm
Theoretical Foundation
The conjugate gradient method improves upon steepest descent by incorporating information from previous search directions. While classical CG solves linear systems with symmetric positive definite matrices, the nonlinear conjugate gradient method (NLCGM) extends this approach to general nonlinear optimization [3].
NLCGM determines search directions through a linear combination of the current gradient and previous search direction: ( \mathbf{d}^k = -\nabla f(\mathbf{x}^k) + \beta^k \mathbf{d}^{k-1} ), where ( \beta^k ) is calculated using various formulae (Fletcher-Reeves, Hestenes-Stiefel, Polak-Ribière, Hager-Zhang) [3].
In the special case of quadratic functions ( f(\mathbf{x}) = \frac{1}{2}\mathbf{x}^T\mathbf{A}\mathbf{x} + \mathbf{b}^T\mathbf{x} + c ), CG generates conjugate search directions satisfying ( \mathbf{d}i^T\mathbf{A}\mathbf{d}j = 0 ) for ( i \neq j ), guaranteeing convergence to the minimum in at most ( n ) steps for an ( n )-dimensional problem [3].
Convergence Properties and Advantages
The conjugate gradient method achieves superior convergence compared to steepest descent, with ( f(\mathbf{x}^k) - \min_{\mathbf{x}} f(\mathbf{x}) = \mathcal{O}(1/k^2) ) for quadratic functions [3]. In molecular modeling contexts, CG "is slower than steepest descent in the early stages of the minimization, but becomes more efficient closer to the energy minimum" [4].
For non-quadratic functions, NLCGM maintains better convergence than steepest descent while requiring only marginally more computational effort per iteration (primarily inner products and AXPY operations) [3]. However, implementations may face restrictions; for instance, GROMACS cannot use conjugate gradient with constraints including the SETTLE algorithm for water [4].
Comparative Analysis: Steepest Descent vs. Conjugate Gradient
Performance Characteristics
Table 2: Algorithm Comparison for Energy Minimization
| Characteristic | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Initial convergence | Rapid far from minimum [4] | Slower initially [4] |
| Asymptotic convergence | Slow near minimum [4] [3] | Fast near minimum [4] [3] |
| Computational cost per iteration | Low | Moderately higher [3] |
| Memory requirements | Low | Low (does not store full history) [3] |
| Robustness | High - rarely fails to find lower energy [2] | Moderate - may require restarts |
| Implementation complexity | Simple [2] | Moderate |
| Optimal for | Initial minimization, poorly conditioned systems [4] | Refined minimization prior to analysis [4] |
A technical analysis demonstrates that "the conjugate-gradient algorithm is actually superior to the steepest-descent algorithm in that, in the generic case, at each iteration it yields a lower cost than does the steepest-descent algorithm, when both start at the same point" [5].
Practical Implementation Considerations
Stopping Criteria
Effective termination criteria are essential for practical implementations. In molecular dynamics packages like GROMACS, minimization typically stops when the maximum absolute value of force components falls below a specified threshold ( \epsilon ) [4]. A reasonable value can be estimated from the root mean square force ( f ) a harmonic oscillator exhibits at temperature ( T ):
[ f = 2\pi\nu\sqrt{2mkT} ]
where ( \nu ) is the oscillator frequency, ( m ) the reduced mass, and ( k ) Boltzmann's constant [4]. For a weak oscillator (100 cm(^{-1}) wave number, 10 atomic units mass) at 1 K, ( f = 7.7 ) kJ mol(^{-1}) nm(^{-1}), suggesting ( \epsilon ) between 1 and 10 is acceptable [4].
Workflow Integration
Optimization Protocol
A hybrid approach often yields optimal results, beginning with steepest descent for rapid initial progress followed by conjugate gradient for refined minimization [4]. This strategy is particularly valuable for molecular systems starting from experimentally-derived coordinates that may contain steric clashes or other high-energy interactions.
Applications in Drug Discovery and Molecular Modeling
Energy Minimization in Molecular Systems
Energy minimization serves as a fundamental procedure in molecular modeling to locate stable molecular conformations by finding arrangements where "the net interatomic force on each atom is acceptably close to zero and the position on the Potential Energy Surface (PES) is a stationary point" [2]. Key applications include:
- Correcting structural anomalies: Resolving bad stereochemistry and short contacts in experimentally-derived structures [1]
- Locating stable conformations: Identifying low-energy arrangements for drug candidates [2]
- Preparing for advanced simulations: Generating starting configurations for molecular dynamics or normal-mode analysis [4]
The GROMACS documentation emphasizes that conjugate gradient "can not be used with constraints," requiring flexible water models when aqueous environments are simulated [4]. This limitation makes steepest descent preferable for certain constrained systems despite its slower convergence.
Integration with AI-Driven Drug Discovery
Modern drug discovery increasingly integrates optimization algorithms with artificial intelligence approaches. Generative models (GMs) employing variational autoencoders (VAEs) with active learning cycles represent an emerging paradigm that combines physics-based energy minimization with data-driven exploration [6].
AI-Driven Molecular Optimization
This integrated workflow enables "exploration of novel chemical spaces tailored for specific targets" while ensuring generated molecules exhibit favorable energy characteristics and synthetic accessibility [6]. The integration of traditional optimization with machine learning approaches represents the cutting edge of computational drug development.
Research Reagent Solutions
Table 3: Essential Computational Tools for Optimization in Drug Discovery
| Tool/Category | Representative Examples | Primary Function in Optimization |
|---|---|---|
| Force Fields | CHARMM, AMBER, GROMOS [1] | Parameterize energy functions for molecular systems |
| Molecular Dynamics Packages | GROMACS [4] | Implement optimization algorithms for energy minimization |
| AI/Generative Platforms | Insilico Medicine Pharma.AI, Recursion OS, Iambic Therapeutics [7] | Integrate optimization with generative molecular design |
| Active Learning Frameworks | VAE with nested AL cycles [6] | Iteratively refine predictions using optimization oracles |
| Quantum Mechanics Packages | Various [1] | Provide energy and derivative calculations for small systems |
The selection between steepest descent and conjugate gradient optimization algorithms represents a fundamental strategic decision in computational chemistry and drug discovery workflows. While steepest descent offers robustness and rapid initial convergence, conjugate gradient methods provide superior asymptotic performance with minimal additional computational overhead. Understanding the mathematical foundations, implementation details, and practical considerations of these algorithms enables researchers to make informed decisions based on their specific scientific objectives and computational constraints.
As drug discovery evolves toward increasingly integrated, AI-driven approaches, the role of efficient optimization algorithms becomes ever more critical. The synergy between traditional energy minimization techniques and modern machine learning frameworks promises to accelerate the identification and optimization of novel therapeutic compounds, potentially addressing the pharmaceutical industry's persistent challenges of excessive costs, lengthy timelines, and high failure rates.
Within the broader research landscape comparing steepest descent with conjugate gradient algorithms, understanding the fundamental intuition behind the steepest descent method is paramount. This first-order iterative optimization algorithm, foundational to modern machine learning and scientific computing, operates on a simple yet powerful principle: at each point, it moves in the direction of the negative gradient of the function [8]. This direction is mathematically guaranteed to be the path of steepest descent, providing a natural and computationally feasible approach to minimizing objective functions [9]. While contemporary research often focuses on more advanced methods like conjugate gradient, the intuition and mechanics of steepest descent remain critical for understanding the optimization landscape, particularly for researchers and scientists in fields like drug development where complex, high-dimensional parameter spaces are commonplace [10] [11].
The historical significance of steepest descent, attributed to Cauchy in 1847, underscores its enduring relevance [8]. This technical guide will deconstruct the core mathematical intuition, provide a detailed comparative analysis with conjugate gradient methods, and present practical experimental protocols for implementing and evaluating these algorithms, with a specific focus on applications relevant to scientific research.
Mathematical Foundations of Steepest Descent
The Core Principle: Direction of Steepest Descent
The steepest descent algorithm is built upon a fundamental fact from multivariable calculus. For a continuously differentiable multivariable function ( f(\mathbf{x}) ), the gradient ( \nabla f(\mathbf{x}) ) evaluated at a point ( \mathbf{x}0 ) represents the direction of the greatest rate of increase of the function at that point [8] [9]. Consequently, the negative gradient ( -\nabla f(\mathbf{x}0 ) points in the direction of the steepest decrease [12]. This local property provides the foundational intuition for the algorithm: by repeatedly moving in the direction opposite to the gradient, we can descend towards a local minimum.
This relationship is formalized by the directional derivative. For a unit vector ( \mathbf{v} ), the directional derivative ( \frac{\partial f}{\partial \mathbf{v}} ) measures the rate of change of ( f ) in the direction of ( \mathbf{v} ). This derivative is maximized when ( \mathbf{v} ) aligns with the gradient and minimized when ( \mathbf{v} ) aligns with the negative gradient [12]. The algorithm leverages this local property to generate a global search strategy.
The Iterative Update Rule
The steepest descent method generates a sequence of points ( \mathbf{x}0, \mathbf{x}1, \mathbf{x}_2, \ldots ) intended to converge to a local minimum. The sequence is defined by the following iterative update rule:
[ \mathbf{x}{n+1} = \mathbf{x}n - \etan \nabla f(\mathbf{x}n) ]
Here, ( \etan ) is a positive scalar known as the step size or learning rate [8]. The term ( \etan \nabla f(\mathbf{x}n) ) represents the step taken, and its magnitude is controlled by ( \etan ). This process can be visualized as being stuck on a mountain in a dense fog and determining your next step downhill by measuring the local steepness of the terrain [8]. You repeatedly measure and take steps in the steepest downward direction until you can descend no further.
The Critical Role of the Step Size
The choice of step size ( \eta ) is critical to the algorithm's performance. A step size that is too small leads to slow convergence, requiring many iterations to reach the minimum. Conversely, a step size that is too large can cause overshooting, where the algorithm diverges or oscillates wildly around the minimum instead of stably converging to it [8] [13].
Several strategies exist for selecting ( \eta ):
- Constant Step Size: A fixed value used for all iterations. Simple but requires careful tuning.
- Diminishing Step Size: A sequence such as ( \eta_n = 1/n ) that decreases over time, guaranteeing convergence under certain conditions.
- Line Search: An adaptive approach where ( \etan ) is chosen at each step to minimize the function along the direction of the negative gradient, i.e., ( \etan = \arg \min{\alpha > 0} f(\mathbf{x}n - \alpha \nabla f(\mathbf{x}_n)) ) [12].
The following diagram illustrates the logical workflow of the steepest descent algorithm, highlighting the iterative process and the role of the gradient and step size.
Steepest Descent vs. Conjugate Gradient: A Comparative Analysis
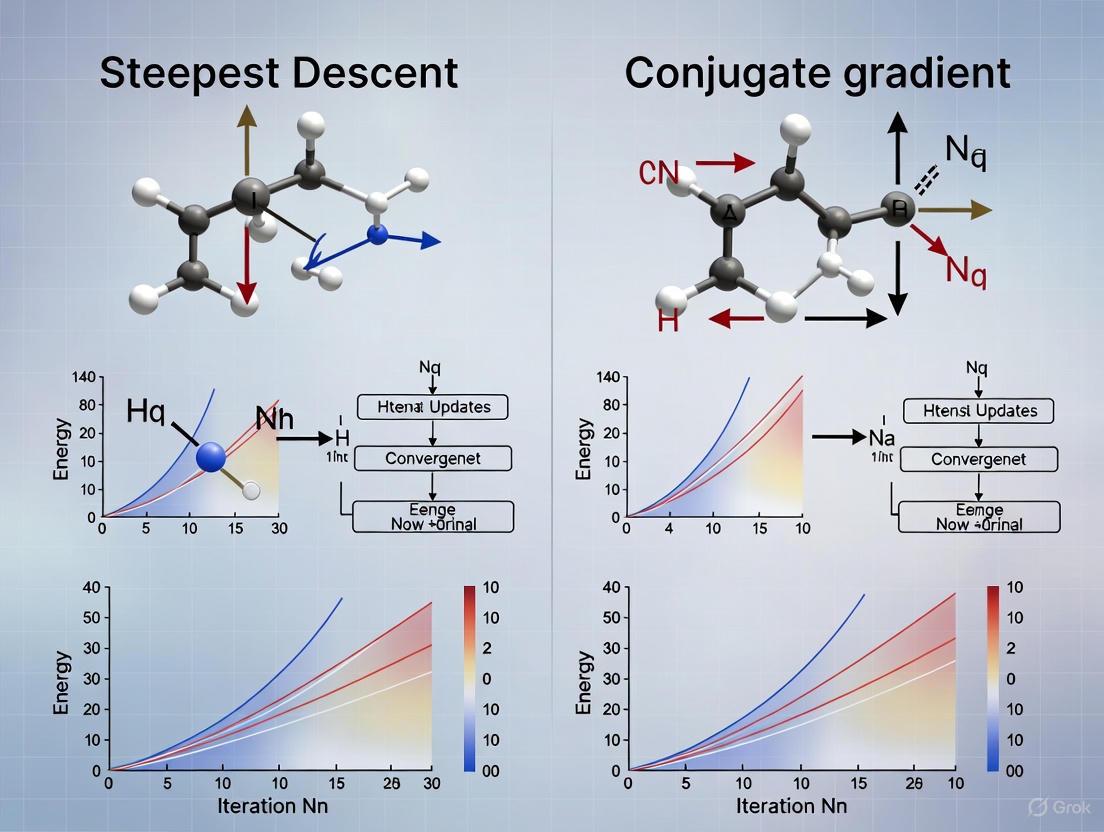
While steepest descent provides a foundational approach, its performance is often surpassed by the conjugate gradient (CG) method, particularly for solving large-scale linear systems and optimization problems. The primary weakness of steepest descent is its tendency to exhibit slow convergence, especially in valleys of ill-conditioned functions, where it often takes many small, zig-zagging steps [10]. This occurs because while each step is locally optimal, the sequence of steps is not necessarily globally efficient.
The conjugate gradient method addresses this by constructing a sequence of conjugate search directions. This means that the algorithm minimizes the function over a expanding subspace that includes the global minimum, leading to the exact solution for quadratic problems in at most ( n ) steps (where ( n ) is the dimension) [10] [11]. Unlike steepest descent, which can re-minimize previously minimized directions, CG ensures that each step is optimal with respect to all previous directions.
The table below summarizes the key quantitative differences between the two algorithms, based on theoretical and empirical findings.
Table 1: Quantitative Comparison of Steepest Descent and Conjugate Gradient
| Feature | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Iteration Complexity | Can be very high for ill-conditioned problems [10] | Convergence in at most ( n ) steps for quadratic problems [11] |
| Computational Cost per Iteration | ( O(n) ) | ( O(n) ) (for linear systems) |
| Storage Requirements | ( O(n) ) (stores current point and gradient) | ( O(n) ) (stores several vectors) |
| Typical Convergence Rate | Linear (can be very slow) [10] | Superlinear (faster for quadratic problems) [11] |
| Dependence on Condition Number | High sensitivity to the condition number of the Hessian [10] | Better performance for problems with high condition numbers [11] |
| Use of Second-Order Information | No | Yes, implicitly constructs Hessian information [10] |
The fundamental difference in their search behavior is visualized in the following pathway diagram, which contrasts the typical trajectories of each algorithm when minimizing a quadratic function.
Experimental Protocols and Implementation
Protocol 1: Benchmarking Algorithm Performance
To empirically compare the performance of steepest descent and conjugate gradient algorithms, researchers can follow this detailed protocol. The objective is to minimize a predefined cost function, typically a strongly convex quadratic function, and measure metrics like the number of iterations and computational time required to reach a specified tolerance.
1. Problem Formulation:
- Define the cost function, ( f(\mathbf{x}) ). For controlled experiments, use ( f(\mathbf{x}) = \frac{1}{2}\mathbf{x}^T A \mathbf{x} - \mathbf{b}^T \mathbf{x} ), where ( A ) is a symmetric positive definite matrix. The condition number of ( A ) can be varied to test algorithm robustness [11].
- The global minimum is located at ( A^{-1}\mathbf{b} ).
2. Algorithm Initialization:
- Initialize the parameter vector ( \mathbf{x}_0 ) randomly or at a fixed point.
- Set a convergence tolerance ( \epsilon ) (e.g., ( 10^{-6} )) and a maximum number of iterations ( K_{max} ).
3. Steepest Descent Implementation:
- For k = 0 to K{max}:
- Compute Gradient: ( \mathbf{g}k = \nabla f(\mathbf{x}k) = A\mathbf{x}k - \mathbf{b} ).
- Check Convergence: If ( \|\mathbf{g}k\| < \epsilon ), terminate.
- Compute Step Size: Perform an exact line search: ( \etak = \frac{\mathbf{g}k^T \mathbf{g}k}{\mathbf{g}k^T A \mathbf{g}k} ) [8].
- Update Parameters: ( \mathbf{x}{k+1} = \mathbf{x}k - \etak \mathbf{g}k ).
4. Conjugate Gradient Implementation:
- Initialization: ( \mathbf{x}0 ), ( \mathbf{g}0 = A\mathbf{x}0 - \mathbf{b} ), ( \mathbf{d}0 = -\mathbf{g}_0 ).
- For k = 0 to K{max}:
- Compute Step Size: ( \alphak = \frac{\mathbf{g}k^T \mathbf{g}k}{\mathbf{d}k^T A \mathbf{d}k} ).
- Update Parameters: ( \mathbf{x}{k+1} = \mathbf{x}k + \alphak \mathbf{d}k ).
- Compute Gradient: ( \mathbf{g}{k+1} = A\mathbf{x}{k+1} - \mathbf{b} ).
- Check Convergence: If ( \|\mathbf{g}{k+1}\| < \epsilon ), terminate.
- Compute Conjugate Parameter: ( \betak = \frac{\mathbf{g}{k+1}^T \mathbf{g}{k+1}}{\mathbf{g}k^T \mathbf{g}k} ) (Fletcher-Reeves formula) [11].
- Update Search Direction: ( \mathbf{d}{k+1} = -\mathbf{g}{k+1} + \betak \mathbf{d}k ).
5. Data Collection and Analysis:
- For each run, record the iteration count ( k ) and the value of ( \|\nabla f(\mathbf{x}_k)\| ) at each iteration.
- Plot ( \|\nabla f(\mathbf{x}_k)\| ) vs. iteration number for both algorithms on a log-linear scale to visualize convergence rates.
- Repeat experiments with different condition numbers of ( A ) to analyze performance degradation.
The Scientist's Toolkit: Essential Research Reagents
Implementing and testing optimization algorithms requires a set of core computational "reagents." The following table details these essential components and their functions in the experimental workflow.
Table 2: Key Research Reagent Solutions for Optimization Experiments
| Research Reagent | Function & Purpose | Example/Notes |
|---|---|---|
| Test Problem Generator | Creates benchmark functions (e.g., quadratics, Rosenbrock) with known minima to validate algorithms. | make_spd_matrix from scikit-learn for generating matrix ( A ) [11]. |
| Gradient Computation Tool | Computes the gradient of the cost function efficiently, a prerequisite for both SD and CG. | Automatic differentiation libraries like autograd or JAX [14]. |
| Line Search Module | An auxiliary optimizer that finds the optimal step size ( \eta ) along a given search direction. | Implement Wolfe condition checks for robust step size selection [12] [11]. |
| Convergence Analyzer | Tracks the norm of the gradient and/or function value over iterations to assess convergence speed and stability. | Custom script to plot ( f(\mathbf{x}_k) ) vs. ( k ) on a log-scale. |
| Performance Profiler | Measures computational time and memory usage, providing data for complexity analysis. | Python's time and memory_profiler modules. |
Protocol 2: Application in Image Restoration
Steepest descent and conjugate gradient methods find practical application in solving large-scale linear inverse problems, such as image restoration [11]. This protocol outlines their use in deblurring an image.
1. Problem Formulation:
- The goal is to recover a clean image ( \mathbf{x} ) from a blurred and noisy observation ( \mathbf{b} ). The relationship is often modeled as ( \mathbf{b} = A\mathbf{x} + \mathbf{n} ), where ( A ) is a matrix representing the blur operator, and ( \mathbf{n} ) is noise.
- This is framed as minimizing ( f(\mathbf{x}) = \frac{1}{2}\|A\mathbf{x} - \mathbf{b}\|^2 + \lambda R(\mathbf{x}) ), where ( R(\mathbf{x}) ) is a regularization term (e.g., Tikhonov) and ( \lambda ) is a regularization parameter.
2. Experimental Workflow:
- Data Preparation: Use a standard dataset (e.g., Lena, Cameraman). Create a blurred version ( \mathbf{b} ) by convolving the original image with a Gaussian blur kernel and adding Gaussian noise.
- Algorithm Setup: Implement both steepest descent and conjugate gradient to minimize ( f(\mathbf{x}) ). The gradient is ( \nabla f(\mathbf{x}) = A^T(A\mathbf{x} - \mathbf{b}) + \lambda \nabla R(\mathbf{x}) ).
- Evaluation: Run both algorithms, monitoring the reduction in the cost function. Compare the quality of the restored images using metrics like Peak Signal-to-Noise Ratio (PSNR) and the time taken to reach a satisfactory result.
The workflow for this image-based experiment is summarized in the following diagram.
The intuition of following the negative gradient path provides a powerful and accessible entry point into the field of mathematical optimization. The steepest descent algorithm embodies this principle, offering a guaranteed local descent direction that is simple to compute and implement. Its first-order nature makes it computationally lightweight per iteration, a characteristic that sustains its relevance in modern applications, including the training of massive deep learning models where exact gradients are unavailable, and stochastic approximations are used [8] [13].
However, as demonstrated through both theoretical comparison and experimental protocol, steepest descent is often hampered by its slow convergence properties, particularly on ill-conditioned problems prevalent in scientific computing. The conjugate gradient algorithm and its modern hybrids represent a significant evolution, leveraging second-order information to construct more efficient search paths that avoid the zig-zagging behavior of steepest descent [11]. For researchers and scientists, particularly in drug development where optimizing complex molecular models is routine, the choice between these algorithms is not merely academic. It has direct implications for the speed and feasibility of discovery. A robust understanding of both the foundational intuition of steepest descent and the advanced efficiency of conjugate gradient methods provides an essential toolkit for tackling the intricate optimization challenges at the forefront of science and technology.
The steepest descent method represents one of the most fundamental optimization algorithms for minimizing multivariable nonlinear functions. As a first-order iterative optimization algorithm, its convergence properties and practical efficiency are critically dependent on the careful selection of step sizes. This technical guide examines the mathematical formulation of steepest descent methods with particular emphasis on how step size strategies influence convergence behavior, framed within a broader comparative analysis with conjugate gradient methods.
Within optimization research, understanding the trade-offs between various step size selection methodologies provides crucial insights for developing efficient algorithms tailored to specific problem domains, including applications in drug development where parameter optimization frequently arises in molecular modeling and pharmacokinetic analysis.
Mathematical Foundations of Steepest Descent
Core Algorithmic Framework
The steepest descent method follows an intuitive yet powerful iterative process for minimizing a function (f: \mathbb{R}^n \rightarrow \mathbb{R}). At each iteration (k), the algorithm moves in the direction opposite to the gradient (\nabla f(\mathbf{x}_k)), which represents the direction of steepest local descent [15]. The fundamental update equation is given by:
[ \mathbf{x}{k+1} = \mathbf{x}k - \alphak \nabla f(\mathbf{x}k) ]
where (\mathbf{x}k) denotes the current iterate, (\nabla f(\mathbf{x}k)) is the gradient at the current point, and (\alpha_k > 0) is the step size (or learning rate) at iteration (k) [15] [16].
The convergence behavior of this algorithm is heavily influenced by how the step size (\alpha_k) is selected at each iteration. Proper step size selection balances two competing objectives: achieving sufficient function reduction at each iteration while avoiding overshooting that leads to divergence or oscillatory behavior [16] [17].
Geometric Interpretation
The steepest descent direction (-\nabla f(\mathbf{x}_k)) guarantees immediate local improvement but doesn't necessarily yield global optimization efficiency. The orthogonal nature of successive gradient directions in steepest descent leads to characteristic zig-zag behavior, particularly in narrow valleys of the objective function landscape. This phenomenon explains why the theoretical convergence guarantee doesn't always translate to practical efficiency, especially when compared with methods like conjugate gradient that explicitly address this directional interdependence [15].
Step Size Selection Methodologies
The selection of the step size (\alpha_k) represents a critical design choice in implementing steepest descent algorithms. Different approaches offer distinct trade-offs between computational expense and convergence rate guarantees.
Constant Step Size
The simplest approach employs a constant step size (\alpha_k = \alpha) for all iterations. This strategy requires minimal computation per iteration but may lead to slow convergence or divergence if poorly chosen. For convergence, the step size must satisfy (0 < \alpha < 2/L), where (L) is the Lipschitz constant of (\nabla f) [17].
Diminishing Step Sizes
Diminishing step size sequences follow predetermined patterns that decrease to zero as iterations progress. A common choice is the harmonic sequence (ak = a1/k), which ensures that the step sizes approach zero in the limit (k \to \infty) while the cumulative sum diverges ((\sum{k=1}^{\infty} ak = \infty)) [17]. This divergent series condition ensures the algorithm can reach any point in the space rather than getting stuck prematurely [17].
The decreasing nature of these step sizes provides theoretical convergence guarantees at the potential cost of practical convergence speed, particularly as the step sizes become exceedingly small in later iterations [17].
Exact Line Search
Exact line search determines the optimal step size at each iteration by solving the one-dimensional minimization problem:
[ \alphak = \arg\min{\alpha > 0} f(\mathbf{x}k - \alpha \nabla f(\mathbf{x}k)) ]
While this approach optimizes progress along the search direction, the computational cost of solving this subproblem exactly often outweighs its benefits, making it impractical for many applications [15].
Adaptive Barzilai-Borwein (BB) Methods
The Barzilai-Borwein method represents a sophisticated approach that approximates the Hessian information using previous gradient and iterate differences [16]. The two standard BB step sizes are:
[ \alphak^{BB1} = \frac{\mathbf{s}{k-1}^T \mathbf{y}{k-1}}{\mathbf{s}{k-1}^T \mathbf{s}{k-1}} \quad \text{and} \quad \alphak^{BB2} = \frac{\mathbf{y}{k-1}^T \mathbf{y}{k-1}}{\mathbf{s}{k-1}^T \mathbf{y}{k-1}} ]
where (\mathbf{s}{k-1} = \mathbf{x}k - \mathbf{x}{k-1}) and (\mathbf{y}{k-1} = \nabla f(\mathbf{x}k) - \nabla f(\mathbf{x}{k-1})) [16].
Recent research has extended these concepts beyond the original BB bounds. One approach proposes step sizes longer than (\alphak^{BB2}) and shorter than (\alphak^{BB1}), with analysis revealing that under certain conditions, the longer step size can yield monotonic gradient norm decrease while the shorter step size consistently produces non-monotonic behavior [16].
Table 1: Classification of Step Size Selection Methods
| Method Type | Computational Cost | Convergence Guarantee | Practical Efficiency |
|---|---|---|---|
| Constant | Very Low | Conditional | Variable |
| Diminishing | Low | Strong | Often Slow |
| Exact Line Search | High | Strong | Often Impractical |
| BB Methods | Moderate | Strong | Generally Good |
| Extended BB | Moderate | R-linear for quadratics | Promising |
Convergence Analysis
Theoretical Convergence Properties
For convex objective functions with L-Lipschitz continuous gradients, steepest descent with appropriate step sizes achieves a convergence rate of (O(1/k)) for the function value suboptimality [16] [17]. Under stronger assumptions such as strong convexity, this can be improved to linear convergence (O(\rho^k)) for some (0 < \rho < 1).
For the strictly convex quadratic minimization problem:
[ \min_{\mathbf{x} \in \mathbb{R}^n} f(\mathbf{x}) = \frac{1}{2}\mathbf{x}^T A \mathbf{x} - \mathbf{b}^T \mathbf{x} ]
where (A) is symmetric positive definite, extended BB-like step sizes have been proven to achieve R-linear convergence using dynamics of difference equations [16].
Stability and Monotonicity Considerations
Recent research on extended BB-like step sizes has revealed intriguing stability properties. The stability here refers to whether the gradient norm decreases monotonically. Surprisingly, under certain conditions, gradient descent with the longer extended step size exhibits stability (monotonic gradient decrease), while the shorter step size consistently leads to instability [16].
This behavior can be visualized through the following workflow diagram:
Diagram 1: Steepest descent iteration workflow
Comparative Analysis with Conjugate Gradient Methods
Performance Metrics Comparison
When comparing optimization algorithms, both iteration count and computational time provide valuable perspectives on efficiency [15]. Empirical studies implementing both steepest descent and conjugate gradient methods in MATLAB have quantified their relative performance:
Table 2: Steepest Descent vs. Conjugate Gradient Performance Comparison [15]
| Performance Metric | Steepest Descent Method | Conjugate Gradient Method |
|---|---|---|
| Iteration Count | Higher | Fewer |
| Time per Iteration | Lower | Higher |
| Overall Efficiency | Less Efficient | More Efficient |
| Convergence Time | Shorter | Longer |
| Stability | Monotonic decrease | Non-monotonic |
The conjugate gradient method generally requires fewer iterations to achieve the same accuracy level due to its utilization of conjugate directions rather than purely gradient-based directions [15]. This allows it to avoid the zig-zagging behavior that plagues steepest descent in certain geometries. However, each conjugate gradient iteration carries higher computational overhead due to the additional vector operations and orthogonalization requirements.
Interestingly, despite requiring more iterations, steepest descent can sometimes converge in less total time than conjugate gradient for certain problem classes, particularly when function and gradient evaluations are computationally inexpensive [15].
Hybrid Approaches and Modern Variants
Recent research has explored innovative combinations and modifications of these classical approaches. The frozen gradient method, which computes the derivative of nonlinear operators only at an initial point, significantly reduces computational complexity while maintaining convergence through conditional stability estimates [18].
For inverse problems with conditional stability on convex and compact sets, projected steepest descent with frozen derivatives has been shown to converge while avoiding expensive derivative recomputations at each iteration [18]. This approach extends to multi-level methods that leverage nested families of convex compact subsets with controlled growth of stability constants [18].
Experimental Protocols and Implementation
MATLAB Implementation Framework
Empirical comparison of steepest descent and conjugate gradient methods typically follows a structured experimental protocol [15]:
Test Function Selection: Choose representative nonlinear functions with varying curvature properties and dimensionalities.
Algorithm Implementation: Code both algorithms with consistent stopping criteria, typically based on gradient norm tolerance ((\|\nabla f(x_k)\| < \delta)).
Step Size Configuration: For steepest descent, implement multiple step size strategies (constant, diminishing, BB) for comparison.
Performance Metrics Collection: Track iterations until convergence, computational time, and final function value accuracy.
Statistical Analysis: Execute multiple runs with different initializations to account for variability.
Research Reagent Solutions
Table 3: Essential Computational Tools for Optimization Experiments
| Tool/Component | Function in Research | Implementation Notes |
|---|---|---|
| MATLAB Optimization Environment | Algorithm implementation and testing | Provides matrix operations and visualization capabilities |
| Gradient Computation Module | Numerical gradient approximation | Can use finite differences or automatic differentiation |
| Step Size Selector | Implements various αₖ strategies | Constant, diminishing, BB, and extended BB options |
| Convergence Monitor | Tracks progress and determines stopping | Typically based on ‖∇f(xₖ)‖ < δ or iteration limit |
| Performance Profiler | Measures computation time and memory usage | Should account for both iterations and wall-clock time |
The mathematical formulation of steepest descent reveals a sophisticated interplay between step size selection and convergence behavior. While the algorithm's theoretical foundation guarantees convergence under relatively mild conditions, its practical effectiveness heavily depends on appropriate step size strategies. Contemporary research continues to extend these classical methods through adaptive BB-like step sizes, frozen derivative approaches, and multi-level frameworks that enhance computational efficiency while maintaining convergence guarantees.
The comparative analysis with conjugate gradient methods highlights fundamental trade-offs: steepest descent offers simplicity and lower per-iteration cost, while conjugate gradient provides superior directional efficiency at the expense of increased computational overhead per iteration. This understanding enables researchers and practitioners—particularly in computationally intensive fields like drug development—to select and customize optimization approaches based on their specific problem structures and computational constraints.
In the field of numerical optimization, the steepest descent method represents one of the most intuitive and fundamental approaches for minimizing continuous, differentiable functions. As a first-order iterative optimization algorithm, it finds local minima by moving in the direction opposite to the gradient at each point [19]. Despite its conceptual simplicity and guaranteed movement toward lower function values, the method suffers from significant limitations that practically constrain its application to real-world problems, particularly those found in scientific computing and drug development research [20].
When applied to complex optimization landscapes—such as those encountered in molecular docking studies, protein folding simulations, or pharmacokinetic modeling—the steepest descent method exhibits characteristic oscillatory behavior and slow convergence rates, especially in narrow valleys or poorly conditioned systems [19]. These limitations become particularly problematic in high-dimensional parameter spaces where computational efficiency directly impacts research feasibility. This technical guide examines the mathematical foundations of these limitations, contrasts steepest descent with the more efficient conjugate gradient approach, and provides experimental protocols for researchers seeking to implement robust optimization techniques in their computational workflows.
Mathematical Foundations of Steepest Descent
Algorithmic Framework and Implementation
The steepest descent method operates through a straightforward iterative process that begins with an initial parameter estimate and progressively updates this estimate by moving in the direction of the negative gradient. The complete algorithmic implementation follows these steps [19]:
- Choose initial point ( x_0 )
- Set iteration counter ( k = 0 )
- Compute gradient ( \nabla f(x_k) )
- Determine step size ( \alpha_k ) through line search
- Update solution: ( x{k+1} = xk - \alphak \nabla f(xk) )
- Check convergence criteria
- Increment ( k ) and repeat if not converged
The critical component at each iteration is the calculation of the step size ( \alpha_k ), which can be determined through various strategies including fixed values, exact line search, or inexact methods such as the Armijo rule [19]. The convergence criteria typically include assessing whether the gradient norm falls below a specified threshold, monitoring changes in function values between iterations, or establishing a maximum iteration count to prevent infinite loops in poorly converging scenarios.
Theoretical Basis for Oscillatory Behavior
The oscillatory behavior characteristic of steepest descent arises from the fundamental property that the negative gradient represents only the locally steepest direction rather than the globally optimal path toward the minimum. This results in a zigzagging trajectory through the optimization landscape, particularly in valleys with non-spherical curvature [19] [20].
Mathematically, this phenomenon occurs because each search direction is orthogonal to the previous direction when using exact line searches:
[ \nabla f(x{k+1})^T \nabla f(xk) = 0 ]
This orthogonality relationship forces the algorithm to continually correct its course, creating an inefficient oscillatory path toward the solution. In ill-conditioned problems where the Hessian matrix has a high condition number, these oscillations become particularly pronounced, dramatically reducing convergence rates [20].
Comparative Analysis: Steepest Descent vs. Conjugate Gradient
Performance Characteristics and Convergence Rates
The limitations of steepest descent become particularly evident when compared directly with conjugate gradient methods. The following table summarizes key performance differences based on numerical experiments and theoretical analysis:
| Characteristic | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Convergence rate | Linear convergence [19] | Quadratic termination (for n steps in exact arithmetic) [21] |
| Iteration count | Higher number of iterations [22] | Fewer iterations required [22] |
| Computational efficiency | Lower efficiency, especially for large problems [23] | Higher efficiency in large-scale problems [23] |
| Memory requirements | Low storage requirements [19] | Low storage requirements [23] [21] |
| Oscillatory behavior | Pronounced zigzagging in narrow valleys [19] | Reduced zigzagging through conjugate directions [21] |
| Step size sensitivity | Highly sensitive to step size choice [19] | Less sensitive with appropriate line search [24] |
Experimental comparisons demonstrate that the conjugate gradient method typically requires significantly fewer iterations to achieve the same precision in solution quality compared to steepest descent [22]. This efficiency advantage becomes increasingly pronounced as problem dimensionality grows, making conjugate gradient methods particularly valuable for large-scale optimization problems encountered in scientific research, including drug development applications such as molecular dynamics simulations and quantum chemistry calculations.
Visualization of Convergence Patterns
The following diagram illustrates the characteristic optimization paths of steepest descent versus conjugate gradient methods in a quadratic bowl with high condition number, highlighting the inefficient oscillatory trajectory of steepest descent compared to the more direct conjugate gradient path:
The zigzag pattern characteristic of steepest descent occurs because each new search direction is orthogonal to the previous one, forcing the algorithm to continually correct its course. In contrast, the conjugate gradient method constructs search directions that are mutually conjugate with respect to the Hessian matrix, enabling more direct progress toward the minimum [21].
Methodological Approaches for Analyzing Convergence
Experimental Protocol for Convergence Testing
Researchers can implement the following experimental protocol to quantitatively evaluate and compare the performance of steepest descent and conjugate gradient methods:
Test Function Selection: Choose benchmark optimization functions with known characteristics, particularly those with narrow valleys or high condition numbers that highlight steepest descent limitations (e.g., Rosenbrock function, quadratic functions with non-spherical contours) [23].
Algorithm Implementation:
- Implement steepest descent with both fixed and adaptive step sizes
- Implement conjugate gradient with restart strategy (e.g., Fletcher-Reeves or Polak-Ribière parameters) [23] [24]
- Utilize Wolfe line search conditions for step size determination: [ f(xk + \alphak dk) \leq f(xk) + \rho \alphak gk^\top dk ] [ g(xk + \alphak dk)^\top dk \geq \sigma gk^\top d_k ] where ( 0 < \rho < \sigma < 1 ) [24]
Performance Metrics: Track iteration count, function evaluations, computational time, gradient norm reduction, and solution accuracy across multiple problem dimensions [23].
Visualization: Plot convergence trajectories (as shown in Section 3.2) and performance profiles to illustrate comparative efficiency [22].
Research Reagent Solutions: Computational Tools
The following table details essential computational tools and their functions for implementing and testing optimization algorithms:
| Research Tool | Function in Optimization Experiments |
|---|---|
| MATLAB R2022b [24] | Primary environment for algorithm implementation and numerical testing |
| Wolfe Line Search [24] | Condition to determine appropriate step size with convergence guarantees |
| Performance Profiles [23] | Visualization technique for comparative algorithm performance |
| Test Function Library [23] | Benchmark problems (100+ test functions) for validation |
| Condition Number Analysis [19] | Diagnostic tool to assess problem difficulty and algorithm sensitivity |
The Conjugate Gradient Alternative
Theoretical Foundation and Algorithmic Implementation
The conjugate gradient method addresses the fundamental limitations of steepest descent by constructing a sequence of mutually conjugate directions with respect to the Hessian matrix, ensuring that each step does not undo progress made in previous directions [21]. For a quadratic objective function ( f(x) = \frac{1}{2}x^TAx - b^Tx ) with symmetric positive-definite matrix ( A ), two vectors ( pi ) and ( pj ) are conjugate if:
[ pi^T A pj = 0 \quad \text{for} \quad i \neq j ]
This conjugate property enables the algorithm to reach the exact minimum of an n-dimensional quadratic function in at most n steps, a significant improvement over steepest descent's linear convergence rate [21]. The algorithmic implementation follows:
- Initialize: ( x0 ), ( d0 = -g_0 ), ( k = 0 )
- While convergence criteria not met:
- Compute step size ( \alphak = \frac{gk^T gk}{dk^T A dk} )
- Update solution: ( x{k+1} = xk + \alphak dk )
- Compute new gradient: ( g{k+1} = gk + \alphak A dk )
- Compute conjugate parameter: ( \beta{k+1} = \frac{g{k+1}^T g{k+1}}{gk^T gk} )
- Update search direction: ( d{k+1} = -g{k+1} + \beta{k+1} dk )
- Increment ( k = k + 1 )
Modern extensions to non-quadratic problems incorporate hybrid conjugate parameters and restart strategies to maintain efficiency while guaranteeing global convergence [23] [24].
Workflow Diagram: Conjugate Gradient Method
The following diagram illustrates the complete workflow of a modern conjugate gradient algorithm with restart strategy, highlighting key components that address steepest descent limitations:
The restart strategy component is particularly important for maintaining convergence efficiency in non-quadratic problems, periodically resetting the search direction to the negative gradient to recover conjugacy and accelerate progress [23].
Applications in Scientific Computing and Drug Development
Case Study: Image Restoration and Signal Processing
The practical advantages of conjugate gradient methods over steepest descent extend beyond theoretical improvements to tangible benefits in real-world applications. In image restoration problems, conjugate gradient algorithms have demonstrated superior performance in recovering degraded images while minimizing computational requirements [23] [24]. Experimental results show that modified conjugate gradient methods achieve higher Peak Signal-to-Noise Ratio (PSNR) values compared to standard optimization approaches, indicating better restoration quality [23].
Similarly, in compressive sensing applications for sparse signal recovery, conjugate gradient methods efficiently solve the underlying optimization problems with limited memory requirements, making them suitable for large-scale datasets encountered in medical imaging and spectroscopic analysis [24]. These capabilities directly benefit drug development researchers working with complex analytical instrumentation data where optimal parameter estimation is crucial for accurate results interpretation.
Molecular Modeling and Optimization in Drug Discovery
In molecular modeling applications central to drug discovery, conjugate gradient methods play a vital role in energy minimization procedures during molecular dynamics simulations and protein-ligand docking studies [21]. The ability to efficiently navigate complex, high-dimensional energy landscapes enables more thorough exploration of conformational spaces and more accurate prediction of binding affinities.
Compared to steepest descent, which exhibits slow convergence in the narrow valleys characteristic of molecular potential energy surfaces, conjugate gradient methods provide faster approach to minimum energy configurations while maintaining reasonable computational requirements [21]. This efficiency advantage becomes increasingly significant when performing virtual screening of large compound libraries, where thousands or millions of individual energy minimizations may be required to identify promising drug candidates.
The steepest descent method, despite its conceptual simplicity and intuitive appeal, suffers from fundamental limitations including oscillatory behavior and slow convergence rates, particularly for ill-conditioned problems and optimization landscapes with narrow valleys. These limitations significantly impact its practical utility in scientific computing and drug development applications where computational efficiency directly influences research progress.
The conjugate gradient method addresses these limitations through its use of conjugate directions, which prevent the inefficiencies of zigzagging trajectories and enable more direct paths to solutions. With proper implementation including hybrid parameters, restart strategies, and appropriate line search conditions, conjugate gradient algorithms demonstrate superior performance across diverse applications including image restoration, signal processing, and molecular modeling.
For researchers and professionals in drug development, adopting conjugate gradient methods over steepest descent can yield significant improvements in computational efficiency, particularly for energy minimization in molecular dynamics simulations, parameter estimation in pharmacokinetic modeling, and optimization in quantitative structure-activity relationship (QSAR) studies. As optimization challenges continue to grow in scale and complexity with advances in high-throughput screening and multi-parameter optimization, selecting appropriate algorithms based on thorough understanding of their convergence properties becomes increasingly critical to research success.
The conjugate gradient method represents a cornerstone algorithm for the numerical solution of large systems of linear equations, particularly those with symmetric positive-definite matrices. This technical guide explores the mathematical foundation and computational advantages of conjugate gradient methods, emphasizing the crucial role of A-conjugate search directions. Framed within a broader comparison with the steepest descent algorithm, this review demonstrates how conjugacy transformations fundamentally improve convergence behavior and computational efficiency. Through structured quantitative comparisons, detailed methodological protocols, and visual workflow representations, we provide researchers with a comprehensive framework for understanding and implementing these powerful optimization techniques in scientific computing and data-intensive research applications.
The conjugate gradient (CG) method is an iterative algorithm primarily used for solving large systems of linear equations where the coefficient matrix is symmetric positive-definite [21]. Originally developed by Hestenes and Stiefel, the method has become particularly valuable for sparse systems too large for direct solution methods like Cholesky decomposition [21]. The fundamental innovation of the CG method lies in its use of A-conjugate search directions – vectors that are orthogonal with respect to the A-inner product – which enables theoretically exact convergence in at most n steps for an n-dimensional problem [25].
When compared to the steepest descent method, the conjugate gradient approach demonstrates superior convergence properties, particularly for ill-conditioned systems. While both methods aim to minimize quadratic functions of the form f(x) = ½xᵀAx - bᵀx, the conjugate gradient method achieves significantly faster convergence by constructing search directions that avoid interference between successive iterations [15]. This property makes it particularly valuable for large-scale optimization problems in scientific computing, machine learning, and engineering applications where computational efficiency is paramount.
Mathematical Foundations
The Quadratic Optimization Problem
The conjugate gradient method finds its most natural application in solving linear systems arising from quadratic optimization problems. Consider the objective function:
[f(\mathbf{x}) = \frac{1}{2}\mathbf{x}^T\mathbf{A}\mathbf{x} - \mathbf{b}^T\mathbf{x}]
where A is an n×n symmetric positive-definite matrix, and x, b ∈ ℝⁿ [25]. The minimization of this function is equivalent to solving the linear system Ax* = b, as the gradient ∇f(x) = Ax* - b equals zero at the solution [21].
For a symmetric positive-definite matrix A, two vectors u and v are defined as A-conjugate (or A-orthogonal) if:
[\mathbf{u}^T\mathbf{A}\mathbf{v} = 0]
This conjugacy condition generalizes the concept of orthogonality from the standard inner product to the A-inner product, defined as ⟨u, v⟩ₐ = ⟨Au, *v⟩ = ⟨u, Av*⟩ [21].
Theoretical Basis of Conjugate Directions
The power of the conjugate gradient method stems from its use of mutually conjugate vectors with respect to the matrix A. A set of vectors {p₁, p₂, ..., pₙ} is mutually conjugate if pᵢᵀApⱼ = 0 for all i ≠ j. This set forms a basis for ℝⁿ, ensuring that any solution vector x∗ can be expressed as a linear combination of these conjugate directions [21] [25].
Table 1: Key Properties of A-Conjugate Vectors
| Property | Mathematical Expression | Significance |
|---|---|---|
| Mutual Conjugacy | pᵢᵀApⱼ = 0 for i ≠ j | Ensures linear independence |
| Basis Formation | x∗ = Σαᵢpᵢ | Enables solution expansion |
| Optimal Step Size | αₖ = pₖᵀrₖ / pₖᵀApₖ | Minimizes function along search direction |
The expansion coefficients αₖ have closed-form expressions when conjugate directions are known:
[\alphak = \frac{\langle \mathbf{p}k, \mathbf{b} \rangle}{\langle \mathbf{p}k, \mathbf{p}k \rangle{\mathbf{A}}} = \frac{\mathbf{p}k^T\mathbf{b}}{\mathbf{p}k^T\mathbf{A}\mathbf{p}k}]
This formulation allows the exact solution to be constructed by progressing along each conjugate direction exactly once [21].
Conjugate Gradient Algorithm
Algorithm Derivation
The conjugate gradient method generates successive approximations to the solution through an iterative process that constructs conjugate directions from the residual vectors. For each iteration k, the algorithm updates:
[\mathbf{x}{k+1} = \mathbf{x}k + \alphak\mathbf{p}k]
where the step size αₖ is chosen to minimize the objective function along the search direction pₖ [21]. The optimal step size is given by:
[\alphak = \frac{\mathbf{p}k^T\mathbf{r}k}{\mathbf{p}k^T\mathbf{A}\mathbf{p}_k}]
where rₖ = b - Axₖ is the residual vector at iteration k [21].
The key innovation lies in how the search directions are updated. Each new search direction is constructed to be A-conjugate to all previous directions:
[\mathbf{p}k = \mathbf{r}k - \sum{i
In practice, this simplifies to a recursive update that requires only the previous search direction, making the algorithm computationally efficient [21].
Complete Algorithm Specification
The standard conjugate gradient algorithm can be summarized as follows:
Initialize:
- Choose initial guess x₀
- Compute r₀ = b - Ax₀
- Set p₀ = r₀
For k = 0, 1, 2, ... until convergence:
- Compute αₖ = pₖᵀrₖ / pₖᵀApₖ
- Update solution: xₖ₊₁ = xₖ + αₖpₖ
- Update residual: rₖ₊₁ = rₖ - αₖApₖ
- Compute βₖ = rₖ₊₁ᵀrₖ₊₁ / rₖᵀrₖ
- Update search direction: pₖ₊₁ = rₖ₊₁ + βₖpₖ
This implementation highlights the algorithm's efficiency: each iteration requires only one matrix-vector multiplication, making it suitable for large sparse systems [21].
Comparative Analysis: Conjugate Gradient vs. Steepest Descent
Fundamental Differences in Search Strategy
The steepest descent method follows the negative gradient direction at each iteration, which often leads to zig-zagging behavior, especially in narrow valleys of the objective function. In contrast, the conjugate gradient method constructs search directions that are mutually conjugate with respect to the system matrix A, ensuring that each step is optimal and does not undo progress from previous steps [15].
Table 2: Algorithm Comparison: Conjugate Gradient vs. Steepest Descent
| Characteristic | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Search Direction | Negative gradient | A-conjugate directions |
| Convergence Rate | Linear | Superlinear (theoretically exact in n steps) |
| Iteration Cost | O(n²) per iteration | O(n²) per iteration |
| Memory Requirements | O(n) | O(n) |
| Number of Iterations | Higher for ill-conditioned systems | Fewer iterations needed |
| Performance on Quadratic Functions | Good | Superior [5] |
Quantitative Performance Comparison
Experimental comparisons between the two methods reveal significant differences in performance characteristics. Research shows that the conjugate gradient method typically requires fewer iterations to achieve the same accuracy level compared to steepest descent [15]. However, each conjugate gradient iteration may be computationally more expensive due to the additional vector operations required for maintaining conjugacy.
When implemented in MATLAB for minimizing nonlinear functions, studies have demonstrated that "the Conjugate gradient method needs fewer iterations and has more efficiency than the Steepest descent method. On the other hand, the Steepest descent method converges a function in less time than the Conjugate gradient method" [15]. This suggests a trade-off between iteration count and per-iteration computational cost that must be considered for specific applications.
Visualization of Algorithm Workflows
Conjugate Gradient Method Logic Flow
Search Direction Comparison
The Scientist's Toolkit: Essential Research Reagents
Table 3: Computational Research Reagents for Optimization Algorithms
| Tool/Component | Function | Implementation Example |
|---|---|---|
| Symmetric Positive-Definite Matrix | Problem formulation | Matrix A in Ax = b |
| Residual Vector | Optimality measurement | rₖ = b - Axₖ |
| Search Direction Vector | Optimization path | pₖ updated to maintain A-conjugacy |
| Step Size Parameter | Progress control | αₖ = pₖᵀrₖ / pₖᵀApₖ |
| Conjugacy Coefficient | Direction orthogonalization | βₖ = rₖ₊₁ᵀrₖ₊₁ / rₖᵀrₖ |
| Matrix-Vector Multiplier | Core computation | Apₖ product each iteration |
| Convergence Criterion | Termination condition | ‖rₖ‖ < ε or maximum iterations |
Advanced Methodological Protocols
Experimental Setup for Algorithm Comparison
To conduct a rigorous comparison between conjugate gradient and steepest descent methods, researchers should implement the following protocol:
Test Problem Generation: Create a set of symmetric positive-definite matrices with varying condition numbers and sizes. These may include diagonal matrices with controlled eigenvalue distributions and sparse matrices from real-world applications.
Implementation Details: Code both algorithms with efficient linear algebra operations. Precompute matrix-vector products for fair timing comparisons. Use a consistent programming environment (e.g., MATLAB, Python with NumPy/SciPy).
Convergence Metrics: Define precise stopping criteria, such as ‖rₖ‖/‖r₀‖ < 10⁻⁶ or a maximum iteration count of 1000. Track both iteration count and computational time to convergence.
Performance Analysis: Measure the number of iterations, computation time, and final solution accuracy for each test case. Analyze how performance varies with problem condition number and dimensionality.
Nonlinear Conjugate Gradient Extensions
For nonlinear optimization problems, the conjugate gradient method extends through several variants:
Fletcher-Reeves Method:
- Update parameter: βₖ = gₖ₊₁ᵀgₖ₊₁ / gₖᵀgₖ
- Where gₖ = ∇f(xₖ) is the gradient
Polak-Ribière Method:
- Update parameter: βₖ = gₖ₊₁ᵀ(gₖ₊₁ - gₖ) / gₖᵀgₖ
Hestenes-Stiefel Method:
- Update parameter: βₖ = gₖ₊₁ᵀ(gₖ₊₁ - gₖ) / pₖᵀ(gₖ₊₁ - gₖ)
These variants maintain the core concept of conjugate directions while adapting to general nonlinear functions [25].
The conjugate gradient method's power fundamentally stems from its use of A-conjugate search directions, which enables more efficient navigation of the solution space compared to steepest descent approaches. By ensuring that each step is optimal and does not interfere with previous progress, the algorithm achieves superior convergence characteristics, particularly for large-scale linear systems and optimization problems.
The comparative analysis presented in this work demonstrates that while both conjugate gradient and steepest descent methods aim to solve similar problems, their strategic approaches yield significantly different performance profiles. The conjugate gradient method typically requires fewer iterations due to its more intelligent search direction selection, though each iteration may be computationally more intensive. This trade-off makes the conjugate gradient method particularly valuable for problems where matrix-vector products are computationally expensive or where high accuracy is required.
For researchers implementing these algorithms, understanding the mathematical foundation of A-conjugacy is essential for proper application and potential extension to novel problem domains. The experimental protocols and visualization tools provided in this work offer a foundation for further investigation and practical implementation of these powerful optimization techniques.
Within the broader context of research on steepest descent versus conjugate gradient algorithms, understanding the precise mathematical differences in their update directions is fundamental. While both are iterative first-order methods for solving large-scale optimization problems, their core mechanisms and performance characteristics diverge significantly [21] [26]. This guide provides an in-depth technical analysis of these differences, focusing on their mathematical formulation, geometric interpretation, and practical implications for researchers and scientists in fields like drug development where complex optimization problems are prevalent.
The conjugate gradient (CG) method was originally developed for solving systems of linear equations with positive-definite matrices and was later extended to general nonlinear unconstrained optimization problems [21] [23]. Its development marked a significant advancement over classical steepest descent approaches, particularly in addressing the inefficiencies of the characteristic "zig-zag" path that plagues gradient descent in certain geometries [27] [28].
Mathematical Foundations
Gradient Descent Update Rule
The gradient descent method, in its simplest form, follows the update rule:
xk+1 = xk + αkdk
where the search direction dk is always the negative gradient:
dk = -∇f(xk) = -gk
The step size αk > 0 is determined through a line search procedure to minimize the function along dk [21] [29]. In steepest descent, a specific variant of gradient descent, the learning rate η is chosen such that it yields maximal gain along the negative gradient direction [29].
Conjugate Gradient Update Rule
The conjugate gradient method uses a more sophisticated update rule for the search direction:
dk = -gk + βkdk-1
where βk is a scalar parameter that ensures consecutive search directions are conjugate with respect to the Hessian matrix (or an approximation thereof) [21] [23]. Different formulas for βk yield different CG variants, such as Fletcher-Reeves (FR), Polak-Ribière-Polyak (PRP), and Hestenes-Stiefel (HS) [23].
For nonlinear problems, more advanced three-term conjugate gradient methods have been developed with the form:
dk = -ϑgk + βkdk-1 + pkrk
where pk is a suitable scalar and rk is a suitable vector [30]. This structure provides additional flexibility and often exhibits better convergence properties.
The Concept of A-Conjugacy
The fundamental innovation in conjugate gradient methods is the concept of A-conjugacy (or Q-conjugacy for quadratic forms). Two vectors di and dj are considered A-conjugate if:
diTAdj = 0, ∀ i ≠ j
where A is a positive-definite matrix [26]. This generalizes the concept of orthogonality from Euclidean geometry to a geometry defined by A. When search directions satisfy this conjugacy condition, each step minimizes the function over a progressively expanding subspace [21] [26].
Comparative Performance Analysis
Geometric Interpretation and Convergence
The geometric behavior of these methods reveals fundamental differences as illustrated below:
Gradient descent exhibits orthogonal successive directions, which leads to inefficient zig-zagging, especially in ill-conditioned problems with elongated contours [27] [28]. In contrast, conjugate gradient methods construct A-conjugate search directions, ensuring that each step minimizes over an expanding subspace and does not undo progress from previous steps [21] [26].
Theoretical Properties Comparison
Table 1: Mathematical Properties Comparison
| Property | Gradient Descent | Conjugate Gradient |
|---|---|---|
| Direction Formula | dk = -gk | dk = -gk + βkdk-1 |
| Orthogonality | gk+1Tgk = 0 | diTAdj = 0, ∀ i ≠ j |
| Convergence Rate | Linear | Finite (n steps for quadratic problems) |
| Memory Requirement | O(n) | O(n) |
| Computational Cost per Iteration | Low | Moderate (additional βk calculation) |
| Optimal Step Size | Line search along -gk | αk = gkTdk/(dkTAdk) |
Computational Characteristics
Table 2: Computational Trade-offs
| Aspect | Gradient Descent | Conjugate Gradient |
|---|---|---|
| Implementation Complexity | Simple | Moderate |
| Sensitivity to Condition Number | High | Reduced |
| Line Search Requirements | Standard | Requires strong Wolfe conditions for convergence guarantees |
| Stochastic Settings | Well-suited (SGD) | Less effective |
| Parallelization Potential | High | Moderate |
| Modern Variants | Adam, RMSProp, Adagrad | Hybrid three-term, memoryless BFGS-inspired |
For quadratic objectives with Hessian A, the conjugate gradient method theoretically converges to the exact solution in at most n steps (where n is the problem dimension), while gradient descent typically requires many more iterations [21] [26]. This finite termination property is particularly valuable for large-scale problems where direct factorization methods are computationally prohibitive.
In practice, for non-quadratic problems, both methods are implemented with restart strategies to enhance performance [23]. Recent advances in three-term conjugate gradient methods have further improved performance by better approximating quasi-Newton directions while maintaining low memory requirements [23] [30].
Experimental Protocols and Methodologies
Standard Testing Framework
Researchers evaluating these algorithms typically employ the following methodological framework:
- Test Problem Selection: A diverse set of benchmark functions with varying characteristics (ill-conditioned, poorly scaled, non-convex)
- Convergence Criteria:
- Gradient norm tolerance: ‖gk‖ ≤ ε
- Function value change: |f(xk+1) - f(xk)| ≤ ε
- Maximum iterations: k ≤ kmax
- Performance Metrics:
- Number of iterations to convergence
- Number of function/gradient evaluations
- Computational time
- Solution accuracy
Line Search Implementation
Both methods require careful implementation of line search procedures. The strong Wolfe conditions are commonly employed for convergence guarantees:
f(xk + αkdk) ≤ f(xk) + δαkgkTdk
|g(xk + αkdk)Tdk| ≤ -σgkTdk
where 0 < δ < σ < 1 [23] [30]. These conditions ensure sufficient decrease while controlling the step size to prevent excessively small steps.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools for Algorithm Implementation
| Tool/Component | Function | Implementation Considerations |
|---|---|---|
| Gradient Calculator | Computes ∇f(x) | Analytical (preferred) or numerical differentiation |
| Line Search Routine | Finds α satisfying Wolfe conditions | Sectioning or interpolation methods |
| Function Evaluator | Computes f(x) | Often the computational bottleneck |
| Conjugacy Condition Checker | Monitors diTAdj = 0 | Critical for maintaining theoretical properties |
| Restart Mechanism | Resets dk = -gk | Prevents numerical instability; typically every n iterations |
| Preconditioner | Improves condition number | Transform problem to reduce eccentricity of contours |
Application to Real-World Problems
Image Restoration
Conjugate gradient methods have demonstrated superior performance in image restoration problems, achieving higher peak signal-to-noise ratio (PSNR) values compared to conventional gradient-based approaches [23]. The ability to handle large-scale linear systems arising in image deconvolution efficiently makes CG particularly suitable for such applications.
Robotic Motion Control
Recent research has applied three-term conjugate gradient methods to robotic arm motion control, leveraging their strong convergence properties for real-time trajectory optimization [30]. The sufficient descent property of modern CG variants ensures stable and predictable convergence behavior critical for control applications.
Machine Learning and Neural Networks
Despite the theoretical advantages of conjugate gradient methods, gradient descent variants (particularly stochastic gradient descent with momentum and adaptive learning rates) dominate deep learning applications [31]. This preference stems from:
- The stochastic nature of machine learning objectives
- The high computational cost of accurate line searches in high dimensions
- The effectiveness of simple momentum in mitigating zig-zagging
- Better generalization properties observed with SGD variants
As noted in research, "CG doesn't generalize well" in stochastic settings, while methods like Adam were specifically designed for such environments [31].
The mathematical differences between gradient and conjugate direction updates translate to significant practical implications for scientific computing and optimization. Gradient descent, with its simplicity and suitability for stochastic environments, remains popular in machine learning. Conjugate gradient methods, with their superior convergence properties for deterministic problems, excel in scientific computing applications where precision and finite termination are valued.
Recent developments in three-term conjugate gradient methods with restart strategies continue to bridge the gap between theoretical convergence guarantees and practical performance, making them valuable tools for researchers tackling complex optimization problems in fields ranging from drug development to engineering design.
From Theory to Therapy: Implementing Algorithms in Drug Discovery
The Conjugate Gradient (CG) Method is a fundamental iterative algorithm in computational mathematics, serving as a cornerstone for solving large-scale linear systems and optimization problems. Originally developed by Hestenes and Stiefel in 1952, this algorithm efficiently solves systems of linear equations where the coefficient matrix is symmetric and positive-definite [32]. Its significance lies in positioning between Newton's method, which requires computationally expensive Hessian matrix calculations, and the method of steepest descent, which often exhibits slow convergence [26]. For researchers and drug development professionals, understanding the CG method provides powerful capabilities for tackling complex computational challenges in fields ranging from structural biology to pharmacokinetic modeling, where large, sparse systems frequently arise from discretized differential equations or high-dimensional optimization problems [33].
Mathematical Foundations
Problem Formulation
The Conjugate Gradient Method addresses two equivalent mathematical problems:
- Linear Systems: Solving (A\mathbf{x} = \mathbf{b}) where (A) is an (n \times n) symmetric positive-definite matrix [32]
- Optimization Problems: Minimizing the quadratic function (f(\mathbf{x}) = \frac{1}{2}\mathbf{x}^T A\mathbf{x} - \mathbf{b}^T\mathbf{x}) [32] [26]
The gradient of this quadratic function equals (\nabla f(\mathbf{x}) = A\mathbf{x} - \mathbf{b}), demonstrating the equivalence between finding the minimum of (f(\mathbf{x})) and solving the linear system (A\mathbf{x} = \mathbf{b}) [32] [26].
The Principle of Conjugacy
The core innovation of the CG method lies in its use of A-conjugate search directions. A set of vectors ({\mathbf{d}0, \mathbf{d}1, ..., \mathbf{d}_{n-1}}) is considered A-conjugate if:
[ \mathbf{d}i^T A\mathbf{d}j = 0 \quad \forall i \neq j ]
This conjugacy condition generalizes the concept of orthogonality [26]. When search directions satisfy this property, each step optimally reduces the error in the solution, guaranteeing convergence within at most (n) iterations for an (n)-dimensional problem [26].
Table 1: Key Mathematical Properties of the Conjugate Gradient Method
| Property | Mathematical Expression | Significance |
|---|---|---|
| Quadratic Form | (f(\mathbf{x}) = \frac{1}{2}\mathbf{x}^T A\mathbf{x} - \mathbf{b}^T\mathbf{x}) | Objective function for optimization |
| Gradient | (\nabla f(\mathbf{x}) = A\mathbf{x} - \mathbf{b}) | Residual in linear system context |
| A-Conjugacy | (\mathbf{d}i^T A\mathbf{d}j = 0 \quad \forall i \neq j) | Ensures optimal search directions |
| Error Minimization | (\min |\mathbf{x}^* - \mathbf{x}k|A) | Minimizes A-norm of error at each step |
The Core Algorithm: Step-by-Step Procedure
Initialization
The algorithm begins with an initial guess (\mathbf{x}0) for the solution. The initial residual (\mathbf{r}0) and search direction (\mathbf{p}_0) are set equal to the negative gradient:
[ \begin{align} \mathbf{r}0 &= \mathbf{b} - A\mathbf{x}0 \ \mathbf{p}0 &= \mathbf{r}0 \end{align} ]
These initial values establish the starting point for the iterative process [32].
Iterative Process
For each iteration (k = 0, 1, 2, \ldots) until convergence, the following steps are executed:
Step Size Calculation: [ \alphak = \frac{\mathbf{r}k^T \mathbf{r}k}{\mathbf{p}k^T A \mathbf{p}k} ] This optimal step size minimizes the function along the search direction (\mathbf{p}k) [32].
Solution Update: [ \mathbf{x}{k+1} = \mathbf{x}k + \alphak \mathbf{p}k ] The current approximation is updated along the search direction with the computed step size [32].
Residual Update: [ \mathbf{r}{k+1} = \mathbf{r}k - \alphak A \mathbf{p}k ] The residual is updated to reflect the change in the solution approximation [32].
Conjugacy Coefficient Calculation: [ \betak = \frac{\mathbf{r}{k+1}^T \mathbf{r}{k+1}}{\mathbf{r}k^T \mathbf{r}_k} ] This coefficient ensures the new search direction is A-conjugate to all previous directions [32].
Search Direction Update: [ \mathbf{p}{k+1} = \mathbf{r}{k+1} + \betak \mathbf{p}k ] A new conjugate search direction is constructed from the current residual and previous direction [32].
Convergence Criteria
The algorithm typically terminates when the norm of the residual (\|\mathbf{r}_{k+1}\|) falls below a specified tolerance, indicating that the current approximation sufficiently satisfies the original system [32].
The following flowchart illustrates the complete iterative procedure:
Figure 1: Conjugate Gradient Algorithm Workflow
Comparative Analysis: Conjugate Gradient vs. Steepest Descent
Fundamental Differences
While both methods are iterative optimization approaches, they differ significantly in their selection of search directions:
Steepest Descent follows the negative gradient direction at each iteration, which often leads to slow convergence as directions can repeat and fail to leverage problem structure [3] [26]
Conjugate Gradient constructs A-conjugate search directions, ensuring each step optimizes progress in a new, complementary direction [26]
Convergence Performance
The Conjugate Gradient method significantly outperforms Steepest Descent in convergence behavior:
Theoretical Convergence: For quadratic functions, CG converges in at most (n) steps (where (n) is the problem dimension), while Steepest Descent has no such guarantee [3] [26]
Convergence Rate: CG achieves (\mathcal{O}(1/k^2)) convergence rate for quadratic functions compared to (\mathcal{O}(1/k)) for Steepest Descent [3]
Valley Navigation: In long, narrow valleys of the objective function, CG dramatically outperforms Steepest Descent, which oscillates inefficiently [34]
Table 2: Performance Comparison: Conjugate Gradient vs. Steepest Descent
| Aspect | Conjugate Gradient | Steepest Descent |
|---|---|---|
| Search Directions | A-conjugate directions | Negative gradient |
| Theoretical Convergence | At most n steps for n-dimensional quadratic problems | No fixed bound |
| Convergence Rate | (\mathcal{O}(1/k^2)) for quadratics | (\mathcal{O}(1/k)) |
| Computational Cost per Iteration | Moderate: one matrix-vector product, several inner products | Low: one matrix-vector product |
| Memory Requirements | Low: stores several vectors | Low: stores several vectors |
| Performance in Valleys | Excellent: follows valley directions | Poor: oscillates across valley |
The following diagram illustrates the different convergence paths:
Figure 2: Convergence Path Comparison
Advanced Conjugate Gradient Variants
Nonlinear Conjugate Gradient Methods
For non-quadratic optimization problems, several nonlinear CG variants have been developed, differing primarily in their calculation of the conjugacy coefficient (\beta_k) [3]:
- Fletcher-Reeves (FR): (\betak^{FR} = \frac{\mathbf{g}{k+1}^T\mathbf{g}{k+1}}{\mathbf{g}k^T\mathbf{g}_k}) [35]
- Polak-Ribière-Polyak (PRP): (\betak^{PRP} = \frac{\mathbf{g}{k+1}^T(\mathbf{g}{k+1}-\mathbf{g}k)}{\mathbf{g}k^T\mathbf{g}k}) [35]
- Hestenes-Stiefel (HS): (\betak^{HS} = \frac{\mathbf{g}{k+1}^T(\mathbf{g}{k+1}-\mathbf{g}k)}{(\mathbf{g}{k+1}-\mathbf{g}k)^T\mathbf{p}_k}) [35]
These extensions enable application of conjugate gradient principles to general nonlinear optimization problems [3].
Three-Term Conjugate Gradient Methods
Recent research has developed three-term variants that demonstrate improved performance characteristics. The general form extends the standard update:
[ \mathbf{p}{k+1} = -\mathbf{g}{k+1} + \betak \mathbf{p}k + \gammak \mathbf{p}{k-1} ]
These methods include additional terms to enhance convergence properties [23]. The Projected Variable Three-Term Conjugate Gradient (PVTTCG) algorithm has shown particular promise by incorporating orthogonal projections to eliminate radial components from search directions, guiding optimization toward flatter regions that often generalize better in machine learning applications [33].
Spectral Conjugate Gradient Methods
Spectral CG methods incorporate a spectral parameter into the search direction formulation:
[ \mathbf{p}{k+1} = -\thetak \mathbf{g}{k+1} + \betak \mathbf{p}_k ]
where (\theta_k) is a spectral parameter chosen to improve the eigenvalue distribution of the iteration matrix [35]. These methods often demonstrate superior practical performance while maintaining strong theoretical convergence properties.
Applications in Drug Development and Computational Biology
Molecular Dynamics and Protein Folding
In computational drug development, the Conjugate Gradient method plays a crucial role in molecular dynamics simulations and energy minimization problems [36]. These applications involve:
- Energy Minimization: Finding low-energy conformations of biological molecules by minimizing complex potential energy functions [36]
- Protein Structure Prediction: Determining stable three-dimensional protein structures by optimizing atomic coordinates [36]
The efficiency of CG makes it particularly valuable for these high-dimensional optimization problems where the number of parameters can reach hundreds of thousands or millions.
Machine Learning in Drug Discovery
Modern drug discovery increasingly relies on machine learning models where optimization is fundamental:
- Neural Network Training: The PVTTCG algorithm has demonstrated significant improvements in training deep neural networks, achieving 0.33-3.92% performance gains on CIFAR datasets and 35.9% test loss reduction in engineering applications compared to standard optimizers [33]
- Multi-target Drug Design: CG methods help navigate complex optimization landscapes in multi-target drug design, where balancing potency, stability, and immunogenicity requires sophisticated optimization [37]
Pharmacokinetic Modeling
Optimization problems in pharmacokinetics often involve parameter estimation from experimental data, where CG methods provide efficient solutions for fitting complex models to observed drug concentration profiles [37].
Table 3: Conjugate Gradient Applications in Drug Development
| Application Area | Specific Use Case | Benefits of CG Method |
|---|---|---|
| Molecular Dynamics | Energy minimization of molecular systems | Efficient handling of high-dimensional parameter spaces |
| Protein Structure Prediction | Optimization of spatial coordinates | Fast convergence for ill-conditioned problems |
| Neural Network Training | Weight optimization in deep learning models | Better generalization compared to first-order methods |
| Pharmacokinetic Modeling | Parameter estimation from concentration data | Stable convergence for noisy experimental data |
| Multi-target Drug Design | Balancing multiple conflicting objectives | Efficient navigation of complex optimization landscapes |
Experimental Protocols and Implementation
Successful implementation of the Conjugate Gradient algorithm requires several key components:
Table 4: Essential Research Reagent Solutions for CG Implementation
| Resource | Function | Implementation Notes |
|---|---|---|
| Sparse Matrix Storage | Efficient representation of large linear systems | Critical for PDE discretizations and graph problems |
| BLAS/LAPACK Libraries | Optimized linear algebra operations | Provides efficient matrix-vector products |
| Preconditioning Techniques | Improving condition number of system | Dramatically enhances convergence rate |
| Line Search Methods | Step size selection for nonlinear problems | Wolfe conditions commonly used for convergence guarantees |
| Parallel Computing Frameworks | Distributed memory computation | Essential for extremely large-scale problems |
Implementation Protocol
A robust implementation of the Conjugate Gradient algorithm follows this experimental protocol:
Problem Preparation:
- Verify matrix (A) is symmetric positive-definite
- Apply appropriate preconditioner if available
- Set convergence tolerance (typically (10^{-6}) to (10^{-12}))
Algorithm Execution:
- Initialize with guess (\mathbf{x}_0)
- Compute initial residual (\mathbf{r}0 = \mathbf{b} - A\mathbf{x}0)
- Iterate until convergence criteria satisfied
- Monitor residual norms for stagnation
Convergence Validation:
- Verify final residual meets tolerance
- Check solution satisfies original system to desired precision
- Validate against known solutions for benchmark problems
Performance Optimization Strategies
For large-scale applications, several strategies enhance CG performance:
- Preconditioning: Transforming the system to improve eigenvalue distribution, significantly accelerating convergence [32]
- Restart Strategies: Periodically resetting the search direction to maintain numerical stability and convergence properties [23]
- Hybrid Approaches: Combining CG with other optimization methods to leverage their respective strengths [35]
The continued evolution of Conjugate Gradient methods ensures their relevance for emerging computational challenges in scientific research and drug development, particularly as problem scales increase and computational architectures evolve.
Energy Minimization as a Core Task in Molecular Modeling
Energy minimization is a foundational step in computational molecular modeling, serving to identify the most stable configuration of a molecular system by finding the arrangement of atoms with the lowest possible energy [1]. This process is critical for refining molecular structures, correcting structural anomalies such as bad stereochemistry and short atomic contacts, and preparing systems for more advanced simulation techniques like molecular dynamics [1]. The energy of a molecular system is described by a force field, a mathematical function comprising multiple components that quantify various atomic interactions [1]. These components include bond energy and angle energy (representing covalent bonds and bond angles), dihedral energy (from dihedral angles), van der Waals interactions (modeled by Leonard-Jones potential to prevent steric clashes), and electrostatic energy (accounting for Coulomb's Law between charged atoms) [1]. Popular parameterized force fields include CHARMM, AMBER, and GROMOS [1].
The goal of energy minimization is to locate a set of atomic coordinates representing the minimum energy conformation for a given structure [1]. On the complex energy surface of a molecular system, multiple minimum points exist, with the very lowest energy point termed the global energy minimum [1]. It is crucial to recognize that the biologically active conformation of a molecule may not always correspond to the global minimum, nor even to a minimum energy structure at all [1]. This article provides an in-depth technical examination of energy minimization, with particular focus on a comparative analysis of the steepest descent and conjugate gradient algorithms, two fundamental optimization methods widely employed in molecular modeling.
Mathematical Foundations of Energy Minimization
The Energy Minimization Problem
The energy minimization problem can be formally stated as follows: given a function ( f(x) ), find the value ( x{min} ) for which ( f(x{min}}) ) is a local minimum [1]. In molecular modeling, ( f ) is typically the molecular mechanics or quantum mechanics energy function, and the variables ( x ) are the atomic coordinates (either Cartesian or internal) [1]. Minimization algorithms are generally classified into two categories: those that utilize derivatives of the energy with respect to coordinates and those that do not [1]. Derivative-based methods typically offer greater efficiency as derivatives provide crucial information about the shape of the energy surface [1].
Force Field Components and Energy Calculation
The total energy of a molecular system in molecular mechanics is calculated as the sum of several energy terms [1]:
- Bond Energy: Arises from the covalent bonds between atoms, typically modeled as harmonic oscillators.
- Angle Energy: Results from the bond angles between every three connected atoms, also often represented with harmonic potentials.
- Dihedral Energy: Associated with torsion angles around rotatable bonds, usually modeled with periodic functions.
- van der Waals Energy: Describes short-range repulsive and attractive dispersive interactions, commonly represented by the Leonard-Jones potential.
- Electrostatic Energy: Accounts for long-range forces between charged and partially charged atoms according to Coulomb's Law.
Optimization Algorithms: Theoretical Framework
The Steepest Descent Algorithm
The steepest descent algorithm is a first-order optimization method that finds a local minimum of a function by taking steps proportional to the negative of the gradient at the current point [12]. The algorithm can be summarized as follows:
At each iteration ( k ), the parameter vector ( xk ) is updated according to: [ x{k+1} = xk - \alphak \nabla f(xk) ] where ( \nabla f(xk) ) is the gradient of the objective function at ( xk ), and ( \alphak ) is the step size [12].
The step size ( \alphak ) can be chosen in various ways, including as a constant, changed arbitrarily at each step, or optimized at each iteration to minimize the function along the gradient direction [12]. When the step size is chosen to minimize ( f(xk - \alpha \nabla f(xk)) ) at each iteration, steepest descent guarantees that ( f(x{k+1}) \leq f(x_k) ) for all ( k \geq 1 ), provided the gradient is non-zero [12].
A key theoretical foundation of the method is that the negative gradient ( -\nabla f(x0) ) provides the direction of steepest descent at point ( x0 ) [12]. For any unit vector ( v ), the directional derivative satisfies: [ \frac{\partial f(x0)}{\partial v} \geq \frac{\partial f(x0)}{\partial v^} ] where [ v^ = -\frac{\nabla f(x0)}{\|\nabla f(x0)\|} ] This confirms that the negative gradient direction provides the greatest immediate decrease in the function value [12].
Table 1: Steepest Descent Algorithm Overview
| Characteristic | Description |
|---|---|
| Algorithm Type | First-order optimization [12] |
| Direction Selection | Negative gradient direction [12] |
| Step Size Selection | Can be constant, arbitrary, or optimized [12] |
| Convergence Guarantee | ( f(x{k+1}) \leq f(xk) ) with optimal step size [12] |
| Computational Complexity | Lower per iteration [38] |
| Memory Requirements | Lower (stores only gradient) [38] |
The Conjugate Gradient Algorithm
The conjugate gradient method is an advanced iterative technique for solving linear and nonlinear minimization problems that improves upon steepest descent by incorporating information from previous search directions [21] [38]. The method finds the new search direction as a conjugate of the negative gradient and the previous direction of descent [38].
At each iteration ( k ): [ dk = -\nabla f(xk) + \gammak d{k-1} ] where ( \gammak ) is the conjugation coefficient and ( d{k-1} ) is the previous direction of descent [38]. The new parameter vector is then updated as: [ x{k+1} = xk + \betak dk ] where ( \beta_k ) is the search step size that minimizes the objective function at the next iteration [38].
The search step size ( \betak ) is determined by minimizing ( f(x{k+1}) ) along the direction ( dk ). For a linear problem, this can be expressed as: [ \betak = \frac{(Xk \cdot dk) \cdot W \cdot (y - ik)}{(Xk \cdot dk) \cdot W \cdot (Xk \cdot dk)} ] where ( Xk ) is the sensitivity matrix, ( W ) is a weighting matrix, ( y ) is the measurement vector, and ( i_k ) is the computed intensity vector [38].
Several expressions exist for the conjugation coefficient ( \gammak ), with the Fletcher-Reeves formula being one common approach: [ \gammak = \frac{\|\nabla f(xk)\|^2}{\|\nabla f(x{k-1})\|^2} ] for ( k = 1, 2, \ldots ), and ( \gamma_0 = 0 ) [38].
Table 2: Conjugate Gradient Algorithm Overview
| Characteristic | Description |
|---|---|
| Algorithm Type | First-order optimization with memory [21] [38] |
| Direction Selection | Conjugate of gradient and previous direction [38] |
| Step Size Selection | Optimized along search direction [38] |
| Convergence Property | Fewer iterations than steepest descent [38] |
| Computational Complexity | Higher per iteration than steepest descent [38] |
| Memory Requirements | Moderate (stores gradient and previous direction) [38] |
Figure 1: Conjugate Gradient Algorithm Workflow
Comparative Performance Analysis
Computational Efficiency
The performance characteristics of steepest descent and conjugate gradient methods differ significantly in terms of convergence behavior and computational requirements. In the method of steepest descent, search direction vectors in successive iterations are always orthogonal to each other, resulting in a zigzag or staircase path toward convergence [38]. This orthogonal nature of successive search directions necessitates a large number of iterations to reach the function minimum, making the method quite inefficient for many problems [38].
In contrast, the conjugate gradient method constructs search directions that are conjugate with respect to the Hessian matrix, which ensures that each step minimizes the function along a linearly independent direction [21] [38]. This property allows the conjugate gradient method to converge in at most ( n ) iterations for a quadratic problem in ( n ) dimensions, a significant improvement over steepest descent [21].
Table 3: Performance Comparison of Optimization Algorithms
| Algorithm | Iterations | CPU Time | Convergence Speed | Stability |
|---|---|---|---|---|
| Steepest Descent | 28,286-51,914 [38] | 1,800-4,330 ms [38] | Slow [38] | Good [12] |
| Conjugate Gradient | 5-6 [38] | 0.45-0.48 ms [38] | Fast [38] | Good [38] |
| Quasi-Newton BFGS | 10-22 [38] | 0.75-1.14 ms [38] | Moderate [38] | Good [38] |
| Tikhonov Regularization | Not applicable [38] | 0.34-0.48 ms [38] | Fast (for linear) [38] | Good [38] |
Application in Molecular Modeling
In molecular modeling, energy minimization serves multiple critical functions. It is used to correct structural anomalies such as bad stereochemistry and short atomic contacts that may arise from experimental structure determination or model building [1]. Additionally, minimization finds the nearest local minimum energy conformation, which represents a stable state of the system [1]. The choice between steepest descent and conjugate gradient methods in molecular modeling depends on the specific context and requirements of the simulation.
Steepest descent is particularly effective when starting from configurations with high energy, such as those with significant steric clashes, as it can rapidly reduce the energy in the initial stages of minimization [12] [1]. However, its convergence slows considerably near the minimum [12]. Conjugate gradient is more efficient for refining structures already close to a minimum and is generally preferred when high precision is required [38] [1]. For large molecular systems, the lower memory requirements of both methods compared to second-order techniques make them attractive options [1].
Figure 2: Algorithm Selection Decision Framework
Experimental Protocols and Implementation
Protocol for Steepest Descent Minimization
Initialization: Begin with an initial approximation ( x_0 ) to the minimum. For molecular systems, this typically represents the atomic coordinates from an experimental structure or a modeled conformation [1].
Gradient Calculation: Compute the gradient of the objective function ( \nabla f(x_k) ) at the current point. In molecular modeling, this involves calculating the derivatives of the force field energy with respect to all atomic coordinates [1].
Step Size Determination: Determine the step size ( \alpha_k ) using one of several approaches:
- Constant step size
- Arbitrarily changed at each step
- Optimization of ( g(t) = f(xk - t \nabla f(xk)) ) to find ( t_k^* ) that minimizes the function along the gradient direction [12]
Parameter Update: Update the parameter vector according to: [ x{k+1} = xk - \alphak \nabla f(xk) ]
Convergence Check: Evaluate the convergence criteria. Common criteria include:
- Change in function value: ( \frac{|f(x{k+1}) - f(xk)|}{1 + |f(x_k)|} < \varepsilon )
- Norm of the gradient: ( \|\nabla f(x_k)\| < \varepsilon )
- Change in parameters: ( \|x{k+1} - xk\| < \varepsilon )
Iteration: Repeat steps 2-5 until convergence is achieved [12].
Protocol for Conjugate Gradient Minimization
Initialization: Start with an initial guess ( x0 ). For the first iteration (( k = 0 )), set the initial direction ( d0 = -\nabla f(x_0) ) [38].
Line Minimization: Find the step size ( \betak ) that minimizes ( f(xk + \beta d_k) ). For nonlinear problems, this typically requires a one-dimensional minimization procedure along the search direction [38].
Parameter Update: Update the parameter vector: [ x{k+1} = xk + \betak dk ]
Gradient Calculation: Compute the new gradient ( \nabla f(x_{k+1}) ) at the updated point [38].
Conjugation Coefficient Calculation: Compute ( \gammak ) using the chosen formula (e.g., Fletcher-Reeves): [ \gammak = \frac{\|\nabla f(x{k+1})\|^2}{\|\nabla f(xk)\|^2} ]
Search Direction Update: Update the search direction: [ d{k+1} = -\nabla f(x{k+1}) + \gammak dk ]
Convergence Check: Evaluate convergence criteria similar to those used in steepest descent.
Iteration: Repeat steps 2-7 until convergence is achieved [38].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Computational Tools for Energy Minimization
| Tool/Component | Function | Examples/Alternatives |
|---|---|---|
| Force Fields | Parameterized mathematical functions to calculate potential energy of molecular systems [1] | CHARMM, AMBER, GROMOS [1] |
| Molecular Visualization | Visual analysis of molecular structures and conformational changes | Chimera, VMD, PyMol |
| Optimization Algorithms | Numerical methods for locating minimum energy conformations [1] | Steepest Descent, Conjugate Gradient, Newton-Raphson [1] |
| Derivative Calculation | Computation of energy gradients with respect to atomic coordinates [1] | Analytical derivatives, Numerical differentiation [1] |
| Convergence Criteria | Metrics to determine when minimization is sufficient | Gradient norm, Energy change, Coordinate change |
| Hybrid Approaches | Combined methods for improved efficiency | Steepest descent initially, then conjugate gradient [1] |
Energy minimization represents a fundamental computational procedure in molecular modeling, essential for refining molecular structures and preparing systems for further analysis. The steepest descent and conjugate gradient algorithms offer complementary strengths for this task. Steepest descent provides robust performance from poor initial structures and has lower memory requirements, making it suitable for the initial stages of minimization or for very large systems. Conjugate gradient exhibits superior convergence properties, particularly near the energy minimum, and is more efficient for final refinement. The choice between these algorithms depends on factors including system size, initial structure quality, available computational resources, and precision requirements. A hybrid approach that begins with steepest descent and switches to conjugate gradient often represents an optimal strategy for molecular energy minimization, balancing computational efficiency with convergence reliability. As molecular systems of interest continue to increase in size and complexity, the judicious selection and implementation of these optimization algorithms remains crucial for successful computational investigations in structural biology and drug design.
The discovery of new pharmaceutical compounds relies heavily on the accurate computational prediction of molecular behavior. A critical step in this process is geometry optimization, or energy minimization, which identifies the most stable three-dimensional structure of a molecule by finding the atomic coordinates that yield the lowest possible potential energy. This minimized conformation is essential for predicting how a small molecule will interact with its biological target, such as a protein or RNA structure [39] [40]. The efficiency and effectiveness of the optimization algorithm directly impact the speed and success of drug discovery campaigns.
This case study explores the application of the Conjugate Gradient (CG) method for optimizing the geometry of small-molecule drugs, framing it within a broader investigation of optimization algorithms, particularly in comparison to the Steepest Descent method. While Steepest Descent represents a foundational approach, Conjugate Gradient has become a preferred technique in modern computational chemistry due to its superior convergence properties for large, complex systems [39] [15]. We will examine the mathematical foundations of CG, provide detailed protocols for its implementation, present quantitative performance data, and discuss its role in the contemporary AI-driven drug discovery landscape.
Theoretical Foundations: Conjugate Gradient vs. Steepest Descent
The Energy Minimization Problem
In computational chemistry, the goal of geometry optimization is to find the set of atomic coordinates ( \mathbf{x} = (r{1x}, r{1y}, r{1z}, r{2x}, r{2y}, r{2z}, \ldots) ) that minimizes the molecule's potential energy ( V(\mathbf{x}) ) [39]. This potential energy surface (PES) is a complex, high-dimensional landscape. The minimum point on this surface corresponds to the most stable conformation of the molecule [39]. Formally, this is often treated as a large-scale system of equations, where the solution is the point where the gradient (the vector of first derivatives representing the interatomic forces) is zero.
Algorithmic Comparison
Both Steepest Descent and Conjugate Gradient are iterative optimization methods, but they differ fundamentally in how they select the search direction for each iteration.
Steepest Descent: This is a foundational iterative algorithm for minimizing a function. At each step ( k ), it moves in the direction of the negative gradient, ( -\nabla f(\mathbf{x}k) ), which is the direction of the steepest decrease in the function value [39]. The update rule is: ( \mathbf{x}{k+1} = \mathbf{x}k - \eta \nabla f(\mathbf{x}k) ) where ( \eta ) is the learning rate. While simple, this method can be inefficient, often leading to slow convergence with a "zig-zag" pattern, especially in narrow valleys of the function landscape [39].
Conjugate Gradient (CG): CG enhances the basic gradient descent approach by using information from previous steps. Instead of following only the current gradient, it constructs a set of conjugate search directions with respect to the system matrix [39]. The key innovation is that each new search direction is built to be conjugate to all previous ones, ensuring that progress made in one direction is not undone in subsequent steps [39]. This leads to more efficient navigation of the function's topography. For a quadratic function in ( n ) dimensions, CG is guaranteed to converge to the minimum in at most ( n ) steps [39].
The following diagram illustrates the conceptual workflow of applying these algorithms to molecular conformation.
Application in Small-Molecule Drug Discovery
The Role of Conjugate Gradient in the Discovery Pipeline
Energy minimization is a foundational step in computational drug design. A prime application is in the preparation of small molecules for virtual screening, where millions of drug-like compounds are scanned to identify potential candidates that can bind to a protein target involved in a disease [39]. For each molecule, numerous conformations are generated, and the lowest energy structure must be found before "docking" it into the binding site of the protein to calculate binding affinity [39]. The speed and accuracy of the minimizer are critical for the feasibility of screening large compound libraries.
CG is particularly well-suited for this task because it rapidly solves the large systems of linear equations that arise from the atomic coordinates of a molecule [39]. Its use of the interatomic force (the gradient) and its history of previous search directions allows it to find the minimum energy conformation with fewer iterations and less computational resource than simpler methods like Steepest Descent [39].
A Concrete Example: Diabetes Drug Discovery
The practical advantage of CG has been demonstrated in real-world drug discovery projects. In a study focusing on Type 2 diabetes, the energy minimization of four different drug molecules was performed [39]. The Conjugate Gradient method consistently resulted in lower final energy values for all molecules compared to other methods, irrespective of the number of freely rotatable bonds in each molecule [39]. This established CG as the method of choice for minimizing molecular energy in this critical therapeutic area.
Experimental Protocol & Benchmarking
Methodology for Performance Comparison
To quantitatively compare the Conjugate Gradient and Steepest Descent methods, a clear experimental protocol is required. The following workflow outlines the key steps for a typical benchmarking study, from system setup to performance analysis.
Quantitative Performance Comparison
The following table summarizes results from published studies that benchmark Conjugate Gradient against Steepest Descent for energy minimization tasks.
Table 1: Benchmarking Conjugate Gradient vs. Steepest Descent
| System/Study | Key Performance Metric | Steepest Descent Result | Conjugate Gradient Result | Context & Notes |
|---|---|---|---|---|
| Cobalt-Copper Nanostructure [39] | Iterations to Reach Energy Tol. of 0.001 | > Iterations than CG | 27 Iterations | CG achieved the lowest energy; Steepest Descent did not. |
| Cobalt-Copper Nanostructure [39] | Wall Time to Converge | > 363.03 seconds | 363.03 seconds | CG was significantly faster in absolute time. |
| General Nonlinear Functions [15] | Iterations to Convergence | More iterations needed | Fewer iterations needed | CG was more efficient in terms of function evaluations. |
| General Nonlinear Functions [15] | Wall Time to Converge | Less time | More time per iteration | While CG needs fewer steps, each step is computationally more complex. |
The data shows a consistent trend: Conjugate Gradient requires significantly fewer iterations to converge to a minimum than Steepest Descent [39] [15]. This is because CG's use of conjugate directions prevents the inefficient zig-zagging characteristic of Steepest Descent. In a real-world application minimizing a cobalt-copper nanostructure, CG reached a low-energy minimum in just 27 iterations, while Steepest Descent failed to reach the same low energy even after a longer computation [39].
The comparison of total computational (wall) time is more nuanced. Although CG needs fewer iterations, each iteration is slightly more computationally expensive because it must calculate and store previous search directions [15]. However, for the complex energy landscapes of molecular systems, the drastic reduction in iteration count almost always leads to a faster time to solution, as demonstrated by the nanostructure example [39].
The Scientist's Toolkit: Essential Research Reagents & Materials
Successful energy minimization and molecular modeling rely on a suite of software tools and computational resources. The following table details key components of the research toolkit.
Table 2: Key Research Reagent Solutions for Computational Energy Minimization
| Tool/Resource | Type | Primary Function in Conformation Analysis |
|---|---|---|
| Force Fields (e.g., AMOEBA) [40] | Parametrized Model | Provides the mathematical functions and parameters for calculating the potential energy ( V(\mathbf{x}) ) of a molecular system based on its atomic coordinates. AMOEBA is a polarizable force field crucial for accurate RNA-ligand systems [40]. |
| Quantum Chemistry Data (e.g., PubChemQCR) [41] | Benchmark Dataset | Provides large-scale, high-quality data of molecular relaxation trajectories with DFT-level energy and force labels for training and validating machine learning interatomic potentials and methods [41]. |
| Optimization Algorithms (CG, Steepest Descent) [39] | Software Algorithm | The core numerical routines that iteratively adjust atomic coordinates to find the minimum energy conformation. CG is often the default choice in many molecular simulation packages [39]. |
| Molecular Dynamics Engines (e.g., GROMACS, Tinker-HP) [42] [40] | Simulation Software | Software suites that integrate force field calculations and optimization algorithms to perform energy minimization and dynamic simulations of molecular systems. |
| AI/ML Platforms (e.g., DrugAppy) [42] | Integrated Workflow | Hybrid frameworks that combine AI models with traditional computational chemistry methods for virtual screening and drug candidate optimization [42]. |
Integration with Modern AI-Driven Workflows
The field of drug discovery is being transformed by Artificial Intelligence (AI), and traditional algorithms like Conjugate Gradient remain relevant within these new paradigms. AI-driven platforms, such as DrugAppy, use an imbrication of models for high-throughput virtual screening (HTVS) and molecular dynamics (MD) simulations [42]. Energy minimization is a critical step within these workflows to prepare and validate the molecular structures being designed or screened.
Furthermore, AI is accelerating the broader discovery pipeline in ways that complement traditional optimization. For example:
- De Novo Molecular Design: Generative models like Variational Autoencoders (VAEs) and Generative Adversarial Networks (GANs) can design novel drug-like molecules with specific properties [43]. The validity and stability of these AI-generated structures must still be confirmed through energy minimization, where efficient algorithms like CG are essential.
- Binding Affinity Prediction: Advanced free energy perturbation methods and machine learning models are used to predict how strongly a small molecule will bind to its target [40]. These methods often require a pre-optimized (minimized) structure of the ligand as a starting point.
In this context, Conjugate Gradient acts as a reliable, high-precision tool that operates within a larger, AI-accelerated discovery engine, ensuring that the molecular geometries fed into predictive models are physically realistic and stable.
This case study demonstrates that the Conjugate Gradient method is a superior optimization algorithm for finding the minimum energy conformation of small-molecule drugs compared to the foundational Steepest Descent method. Its efficiency stems from its intelligent use of historical gradient information to construct conjugate search directions, leading to faster convergence and lower computational cost for reaching a stable molecular structure [39] [15].
As the landscape of drug discovery evolves with the integration of powerful AI and machine learning tools, the role of robust and efficient classical algorithms like CG remains undiminished. Instead, they form the critical backbone of molecular preparation and validation within larger, more complex computational frameworks. The continued development of hybrid approaches, which combine the pattern-recognition power of AI with the physical rigor of traditional force fields and optimizers like Conjugate Gradient, represents the most promising path forward for accelerating the development of new therapeutics.
The computational prediction and refinement of protein three-dimensional (3D) structures are fundamental challenges in structural biology and drug discovery. The native conformation of a protein generally resides at a global energy minimum on a complex potential energy landscape [44]. Navigating this landscape to find this minimum requires robust and efficient energy minimization algorithms. The steepest descent method is a foundational optimization algorithm widely used in the initial stages of this process due to its stability and simplicity. This guide examines the role of steepest descent within the broader context of optimization algorithms, with a specific focus on its application in refining protein structural models. We will juxtapose its performance and characteristics with those of the conjugate gradient method, another first-order minimization technique known for its superior convergence properties, to provide researchers with a clear framework for algorithm selection.
Theoretical Foundation of Steepest Descent
Core Algorithm and Mechanics
The steepest descent algorithm is a first-order iterative optimization method for finding a local minimum of a function. In the context of protein structure optimization, the function is the potential energy of the molecular system. The core principle is straightforward: at each iteration, the algorithm takes a step in the direction of the negative gradient of the function, which is the direction of the steepest descent.
The update rule for the parameter vector ( x ) (representing atomic coordinates) is given by: ( x{n+1} = xn - \gamman \nabla F(xn) ) where ( \nabla F(xn) ) is the gradient of the potential energy function at the current position ( xn ), and ( \gamma_n ) is the step size at iteration ( n ). The step size is often determined via a line search to ensure a sufficient decrease in the function value at each step.
Role in the Broader Optimization Landscape
Compared to other algorithms, steepest descent has distinct characteristics. The conjugate gradient method, for instance, enhances convergence by constructing a set of conjugate (non-interfering) search directions. While steepest descent can suffer from slow convergence, especially near the minimum due to its characteristic "zig-zag" path, conjugate gradient accelerates convergence by using information from previous gradients to inform the current search direction. Table 1 summarizes the key differences between these two algorithms in the context of protein structure optimization.
Table 1: Comparison of Steepest Descent and Conjugate Gradient Algorithms for Protein Structure Optimization
| Feature | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Core Principle | Moves in the direction of the negative gradient at each step. | Constructs a set of conjugate directions using current and past gradients. |
| Convergence Rate | Linear convergence; can be slow near the minimum. | Typically superlinear convergence; faster than steepest descent. |
| Memory Requirements | Low (only stores the current gradient). | Low (stores current and previous gradient). |
| Robustness | High; very stable for initial stages of minimization. | Moderate; can be more sensitive to step size selection. |
| Typical Use Case in Protein Modeling | Initial structure minimization and rough refinement. | Later stages of refinement for finer convergence. |
| Performance on Ill-conditioned Problems | Poor; prone to zig-zagging through long, narrow valleys. | Better; conjugate directions help avoid zig-zagging. |
Steepest Descent in Protein Structure Modeling Workflows
Integration in Broader Prediction and Refinement Pipelines
Steepest descent is rarely used in isolation but is a critical component within larger protein modeling and simulation workflows. Its primary role is in the initial energy minimization phase, where it efficiently relieves severe steric clashes and grossly unrealistic geometries in a starting model before more sophisticated (and often more expensive) simulations are applied.
A prominent example is its use in molecular dynamics (MD)-based refinement protocols, such as the one successfully employed in the Critical Assessment of protein Structure Prediction (CASP) experiments [45]. As shown in Figure 1, the steepest descent algorithm is used to prepare the structure for subsequent MD simulation, ensuring numerical stability.
Figure 1: Workflow of an MD-based refinement protocol. Steepest descent is used in the crucial first energy minimization step to prepare the system [45].
Application in de novo Protein Design
Beyond refinement, steepest descent also finds application in the emerging field of de novo protein design. Automated pipelines like FoldDesign create novel protein folds from specific secondary structure assignments using Replica-Exchange Monte Carlo (REMC) simulations [46]. While REMC is the primary sampling engine, local energy minimization techniques, including steepest descent, are often embedded within such frameworks to relax and optimize newly assembled fragments or proposed structural moves, ensuring they adhere to physical constraints before being accepted or rejected in the simulation.
Experimental Protocols and Methodologies
A Standard Protocol for Model Refinement
The following protocol, adapted from a CASP-winning refinement approach, details the use of steepest descent in a practical setting [45].
Objective: To refine a computationally predicted protein structure model and improve its accuracy (e.g., reduce RMSD to native structure). Starting Model: A protein structure model from a template-based modeling server or ab initio prediction. Software: A molecular dynamics package capable of energy minimization and dynamics, such as CHARMM, GROMACS, or AMBER.
Step-by-Step Procedure:
System Preparation:
- Add hydrogen atoms to the model using the software's appropriate commands (e.g.,
HBUILDin CHARMM). - Place the model in a simulation box if using explicit solvent, or select an implicit solvent model (e.g., FACTS [45] or GBSA).
- Add hydrogen atoms to the model using the software's appropriate commands (e.g.,
Energy Minimization (Steepest Descent):
- Perform an initial minimization of 5,000-12,000 steps using the steepest descent algorithm.
- The goal is to remove any bad steric clashes and reduce the maximum force on the system below a threshold (e.g., 5 kJ/mol/nm [47] or via a phased reduction of harmonic restraints).
- Typical Parameters from CASP12 [45]:
- Algorithm: Steepest Descent (e.g., Adopted Basis Newton-Raphson may follow).
- Steps: 12,100 total steps.
- Restraints: Apply harmonic positional restraints on non-hydrogen atoms, gradually reducing the force constant from 20.0 to 0.5 kcal/mol/Ų over the course of the minimization.
Heating and Equilibration:
- Gradually heat the system from a low temperature (e.g., 50 K) to the target temperature (e.g., 298 K) over 400 picoseconds (ps) with weak restraints on Cα atoms.
- Allow the system to equilibrate further at the target temperature.
Production Simulation and Analysis:
- Run multiple short (e.g., 1-20 nanosecond) MD simulations, either with implicit or explicit solvent.
- From the resulting trajectories, select a subset of models based on criteria like low statistical potential energy scores (e.g., DFIRE) and minimal deviation from the starting model.
- The final refined model is often an averaged and re-minimized structure from this selected ensemble.
Essential Research Reagent Solutions
The computational tools and force fields used in these protocols are the modern equivalent of a scientist's reagents. The table below details key components.
Table 2: Key Research Reagent Solutions for Protein Structure Optimization
| Category | Item / Software | Specific Function in Protocol |
|---|---|---|
| Molecular Dynamics Engines | CHARMM [45], GROMACS [47] | Performs the core calculations: energy minimization, dynamics, and temperature/pressure control. |
| Force Fields | CHARMM22/CMAP [45], AMBER ff99SB [47] | Defines the potential energy function, governing atomic interactions, bond stretching, and dihedral angles. |
| Implicit Solvent Models | FACTS [45], SCPISM [45], GBSA | Approximates solvent effects without explicit water molecules, drastically reducing computational cost. |
| Knowledge-Based Potentials | DFIRE [45] | A statistical potential used to evaluate and select geometrically favorable models from MD trajectories. |
| Analysis Tools | Built-in MD analysis, VMD, PyMOL | Used for calculating RMSD, GDT-HA scores, and visualizing structural changes and pathways. |
Performance Analysis and Comparative Data
Quantitative Benchmarks in Refinement
The success of a refinement protocol, which includes steepest descent as a foundational step, is measured by standard metrics like Root-Mean-Square Deviation (RMSD) and Global Distance Test-High Accuracy (GDT-HA). In CASP12, a protocol using short MD simulations with implicit solvent (and initial steepest descent minimization) successfully refined 29 out of 42 targets based on GDT-HA improvement [45]. The refinement was often overall, occurring in the core regions and not just flexible loops.
Synergy with Advanced Sampling Methods
While steepest descent is a local minimizer, it is often used in conjunction with global search and advanced sampling methods to overcome energy barriers. For instance, the discard-and-restart MD algorithm uses ultra-short MD runs (10-20 ps) to sketch folding pathways [47]. After each short run, the algorithm decides to continue or discard the trajectory. When a trajectory is restarted, the initial structure is often first energy-minimized (e.g., via steepest descent for 5,000 steps) to ensure a valid starting conformation [47]. This highlights how steepest descent acts as a crucial "reset" tool within more complex, globally-aware algorithms, ensuring that each new sampling attempt begins from a physically reasonable state. The relationship between these methods is shown in Figure 2.
Figure 2: Logical relationship between steepest descent minimization and advanced sampling algorithms, illustrating its role as a foundational reset step [46] [47].
The steepest descent algorithm remains a cornerstone of protein structure optimization workflows. Its robustness and simplicity make it the undisputed choice for the initial stages of energy minimization, where it efficiently resolves severe structural distortions and prepares models for more computationally intensive simulations. When framed within the broader thesis of optimization algorithms, steepest descent provides stable but sometimes slow convergence, whereas the conjugate gradient method offers a more efficient path to the minimum once the initial rough landscape has been navigated. The future of protein structure optimization lies in the intelligent integration of these classical methods with modern machine learning approaches and enhanced sampling techniques, creating hybrid pipelines that leverage the strengths of each component to tackle the complex challenge of predicting and refining the 3D structures of proteins.
Adaptive Conjugate Gradient (ACG) for Faster Biomedical Optimization
Biomedical optimization problems, from treatment planning to image analysis, often involve complex, high-dimensional objective functions. Traditional optimization algorithms, like the method of Steepest Descent, are known for their simplicity but suffer from slow convergence, particularly as they approach the optimum. This occurs because successive search directions can be orthogonal, leading to a zig-zag path. The Conjugate Gradient (CG) method overcomes this limitation by generating a sequence of conjugate (non-interfering) search directions, typically achieving convergence in at most n steps for a quadratic n-dimensional problem. However, the performance of the vanilla CG method can be sensitive to problem conditioning and may not be robust for the noisy, non-convex objective functions common in biomedical applications.
This is where Adaptive Conjugate Gradient (ACG) methods provide a breakthrough. By incorporating adaptive strategies for parameters like step sizes and search directions—often informed by the problem's current state or historical gradients—ACG algorithms retain the convergence benefits of CG while adding the robustness needed for real-world biomedical data. This technical guide explores the core principles of ACG, its implementation, and its transformative impact on the speed and accuracy of biomedical optimization.
Core Algorithmic Principles: From CG to Adaptive CG
The fundamental principle of the Conjugate Gradient method is to choose search directions that are conjugate with respect to the Hessian matrix of the objective function. For a quadratic minimization problem min f(x) = 1/2 x^T A x - b^T x, two directions d_i and d_j are conjugate if d_i^T A d_j = 0 for i ≠ j. This property ensures that the minimum along a new direction does not spoil the minimization achieved in previous directions.
The transition to Adaptive Conjugate Gradient involves dynamically tuning key parameters. A primary adaptation is the weight parameter in the formula for β_k, which determines the next conjugate direction. An adaptive weight strategy, as seen in ACG-EA, adjusts this parameter based on the current population's diversity and convergence state, preventing stagnation and accelerating the evolutionary process [48]. Furthermore, ACG methods may adapt the step size α_k at each iteration. Instead of a fixed or line-searched step, methods like the Adaptive Stochastic Conjugate Gradient (ASCG) use an approximation of the Lipschitz constant of the stochastic gradient function to set the step size adaptively, leading to significant reductions in CPU time [49].
Table: Comparison of Steepest Descent, Standard CG, and Adaptive CG
| Feature | Steepest Descent | Conjugate Gradient (CG) | Adaptive CG (ACG) |
|---|---|---|---|
| Search Direction | Negative gradient | Conjugate to previous directions | Conjugate directions with adaptive weight/update |
| Convergence Rate | Linear (can be slow) | Super-linear (theoretical n-step convergence) |
Super-linear, enhanced for specific problems |
| Computational Cost per Iteration | Low (O(n)) |
Low (O(n)) |
Moderate (includes adaptation overhead) |
| Robustness to Noise/Non-convexity | Poor | Moderate | High |
| Key Adaptation Mechanism | Not Applicable | Fixed formula (e.g., Fletcher–Reeves) | Adaptive weights, step-sizes based on population or gradient history |
ACG Applications in Biomedical Optimization
Multi-Objective Spot Optimization in Proton Therapy
Intensity-modulated proton therapy (IMPT) is an advanced cancer treatment technique that leverages the Bragg peak characteristics of proton radiation to damage tumor cells while sparing surrounding healthy tissue. The treatment planning involves optimizing the weights of thousands of individual proton "spots" to achieve a dose distribution that conforms to the tumor volume. This is a large-scale, multi-objective optimization problem with clinical constraints [48].
The Adaptive Conjugate Gradient Accelerated Evolutionary Algorithm (ACG-EA) was developed to solve this problem. In this hybrid algorithm, the conjugate gradient method acts as a directional mutation operator, accelerating the search process. An adaptive weight strategy for the CG automatically updates parameters based on the diversity and convergence of the current population. Experimental results on six clinical cancer cases demonstrated that ACG-EA could generate a set of diverse, high-quality treatment plans (Pareto solutions) with competitive performance on hypervolume and dose-volume histogram indicators compared to other multi-objective evolutionary algorithms [48].
Temporal Medical Image Registration
Medical image registration is the process of aligning two or more images into a common coordinate system, which is crucial for tracking disease progression (e.g., in lung cancer) over time. This is a complex optimization problem where the objective is to find the spatial transformation that maximizes the similarity between the moving and fixed images.
The Adaptive Stochastic Conjugate Gradient (ASCG) algorithm was proposed for this task. It combines the advantages of the CG method and Adaptive Stochastic Gradient Descent (ASGD). The core idea is to replace the search direction of ASGD with stochastic approximations of the conjugate gradient of the cost function. Meanwhile, the step size is based on an approximation of the Lipschitz constant of the stochastic gradient function. This maintains the good properties of CG while using less gradient computation time per iteration and adapting the step size automatically [49].
On 4D Lung CT data (the public POPI dataset and a clinical dataset), the ASCG algorithm achieved 22% higher accuracy compared to previous methods like steepest gradient descent, conjugate gradient, LBFGS, and ASGD. Furthermore, ASCG demonstrated greater robustness to image noise [49].
Table: Summary of Quantitative Performance of ACG Algorithms in Biomedical Applications
| Application Area | Algorithm Name | Key Metric | Reported Performance | Benchmark Comparison |
|---|---|---|---|---|
| Proton Therapy Planning | ACG-EA [48] | Hypervolume, Dose-Histogram | Competitive/Improved Performance | Outperformed 5 other Multi-Objective Evolutionary Algorithms |
| Lung CT Image Registration | ASCG [49] | Registration Accuracy | 22% higher accuracy on POPI dataset | Outperformed Steepest Descent, CG, LBFGS, ASGD |
| Lung CT Image Registration | ASCG [49] | Computational Efficiency | Less CPU time | Faster than ASGD method |
| Deep Learning Training | CG-like-Adam [50] | Training Speed & Performance | Superiority on CIFAR10/100 | Enhanced performance over generic Adam optimizer |
Experimental Protocols and Methodologies
- Problem Formulation: Define the multi-objective spot optimization problem. Objectives typically include maximizing dose to the tumor target volume while minimizing dose to surrounding Organs at Risk (OARs). Thousands of spot weights are the decision variables.
- Algorithm Initialization:
- Initialize a population of candidate solutions (treatment plans).
- Set parameters for the evolutionary algorithm (e.g., population size, crossover rate).
- Initialize the parameters for the conjugate gradient component.
- Iterative Optimization Loop:
- Evaluation: Calculate the objective functions (dose metrics) for each candidate in the population.
- Selection: Select parent solutions based on non-domination and diversity (e.g., using a tournament selection).
- Variation (Crossover & Mutation): Create offspring.
- Apply standard crossover operators.
- Apply the directional CG mutation: The conjugate gradient method is used to determine a promising search direction. The adaptive weight strategy updates the CG parameters based on the current population's state.
- Replacement: Form the new population for the next generation by combining parents and offspring.
- Termination & Analysis: The loop continues until a stopping criterion (e.g., max generations) is met. The output is a set of non-dominated solutions (Pareto front) from which clinicians can choose.
- Data Preparation: Obtain temporal medical image volumes (e.g., 4D Lung CT scans from the POPI model or DIR-Lab dataset).
- Cost Function Definition: Define a similarity metric between the moving image
I_mand fixed imageI_f, such as Mean Squared Error (MSE) or Normalized Cross-Correlation (NCC), often regularized by a smoothing term on the transformation fieldT. - Algorithm Initialization:
- Initialize the transformation
T(often as zero). - Set the initial step size and conjugate gradient parameters.
- Initialize the transformation
- Iterative Registration Loop:
- Gradient Computation: Compute the stochastic gradient of the cost function with respect to the transformation parameters. A stochastic approximation is used for efficiency.
- Conjugate Direction Update: Calculate the new search direction
d_kusing a conjugate-gradient-like update formula based on the current stochastic gradient and the previous direction. - Adaptive Step Size Selection: Calculate the step size
α_kadaptively based on an approximation of the Lipschitz constant of the stochastic gradient function. - Parameter Update: Update the transformation parameters:
T_{k+1} = T_k + α_k * d_k.
- Validation: Upon convergence, evaluate the registration accuracy by measuring target registration error (TRE) on landmark points or other relevant metrics, comparing against known ground truth.
Essential Research Reagents and Computational Tools
The implementation and testing of ACG algorithms require a suite of computational tools and conceptual frameworks.
Table: Key Research "Reagents" and Tools for ACG in Biomedicine
| Tool / Resource | Function / Purpose | Example in Context |
|---|---|---|
| Clinical Image Datasets | Provide real-world, annotated data for developing and validating algorithms. | 4D Lung CT data (POPI model), DIR-Lab dataset [49]. Clinical cancer cases for IMPT planning [48]. |
| Population Health Analytics | Offers a framework for understanding morbidity clusters and resource use, which can inform optimization objectives. | Johns Hopkins ACG System for risk adjustment and casemix analysis [51]. |
| Software Libraries & Platforms | Provide the computational backbone for implementing and comparing optimization algorithms. | PlatEMO (for fair comparison of multi-objective evolutionary algorithms) [48]. Deep Learning frameworks (e.g., PyTorch, TensorFlow) for implementing CG-like-Adam [50]. |
| Biomedical Research Teams | Conduct interdisciplinary research that drives algorithm development and application in areas like chemical biology and neuroinflammation. | Biomedical Science Research Team (BMSR) at The American College of Greece [52]. |
Visualizing Workflows and Algorithm Structure
The following diagrams illustrate the logical structure and workflow of a typical Adaptive Conjugate Gradient algorithm as applied in biomedical optimization.
Diagram 1: Core ACG Optimization Loop. This flowchart outlines the iterative process of an Adaptive Conjugate Gradient algorithm, highlighting the key step of parameter adaptation.
Diagram 2: Hybrid ACG-EA Architecture. This diagram shows the interaction between the Evolutionary Algorithm and Conjugate Gradient components, mediated by an adaptation layer that tunes key parameters based on the current state.
Adaptive Conjugate Gradient methods represent a significant advancement over both Steepest Descent and traditional CG algorithms for complex biomedical optimization problems. By dynamically tuning critical parameters like search directions and step sizes, ACG algorithms achieve a powerful synergy of robustness, accelerated convergence, and high precision. As demonstrated in critical applications like radiation therapy planning and medical image analysis, the integration of ACG principles leads to tangible improvements in both computational efficiency and outcome quality. This makes ACG a vital component in the computational scientist's toolkit for accelerating discovery and improving patient care in the biomedical field.
Optimization algorithms are fundamental to many scientific computing and industrial applications, from training machine learning models to solving complex engineering problems. Among the most important classes of optimization techniques are first-order iterative methods, particularly the steepest descent method and the more advanced conjugate gradient method [22] [53]. This technical guide provides researchers and practitioners with practical implementations of these algorithms, with a focus on reproducible Python code snippets suitable for prototyping and experimental analysis.
The steepest descent algorithm, also known as gradient descent, represents the simplest first-order optimization method that uses the negative gradient as the search direction [12]. While computationally inexpensive per iteration, it often exhibits slow convergence, particularly for ill-conditioned problems. The conjugate gradient method addresses this limitation by constructing search directions that are mutually conjugate with respect to the coefficient matrix, typically resulting in significantly faster convergence [22] [54].
For researchers in fields such as drug development where optimization problems frequently arise in molecular modeling and parameter estimation [55] [56], understanding the practical implementation and performance characteristics of these algorithms is crucial for selecting appropriate methods for specific problem classes.
Theoretical Foundation
Mathematical Framework
Both steepest descent and conjugate gradient methods aim to solve unconstrained optimization problems of the form:
min f(x)
where f:ℝⁿ → ℝ is a continuously differentiable function [57] [53].
For quadratic functions with symmetric positive definite matrix A, vector b, and scalar c, this takes the specific form:
f(x) = (1/2)xᵀAx - bᵀx + c [54]
The gradient of this quadratic function is ∇f(x) = Ax - b, with the minimum occurring when ∇f(x) = 0 [54].
Algorithmic Properties
Table 1: Fundamental Properties of Optimization Algorithms
| Property | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Search Direction | Negative gradient | Conjugate directions |
| Convergence Rate | Linear | Quadratic (for quadratic functions) |
| Memory Requirements | O(n) | O(n) |
| Computational Complexity per Iteration | O(n) | O(n) for quadratic problems |
| Optimal Step Size | Exact line search | Exact line search |
The steepest descent method follows the update rule:
xₖ₊₁ = xₖ - αₖ∇f(xₖ)
where αₖ is the step size determined through line search [57] [12]. The search direction is always the negative gradient, which represents the locally steepest descent direction [12].
The conjugate gradient method uses the update rule:
xₖ₊₁ = xₖ + αₖpₖ
where pₖ is the search direction constructed to be conjugate to previous directions [22] [54]. For quadratic functions, the conjugate gradient method theoretically converges in at most n iterations [54].
Python Implementation
Steepest Descent with Constant Step Size
The following implementation provides a basic steepest descent algorithm with constant step size [57]:
This implementation stores the entire optimization path for visualization and analysis. The constant step size makes it simple but requires careful tuning of the alpha parameter for different problems [57].
Steepest Descent with Armijo Line Search
For improved performance, we can implement steepest descent with the Armijo condition for adaptive step size selection [57]:
The Armijo condition ensures sufficient decrease in the objective function at each iteration, making the algorithm more robust to problem scaling [57].
Conjugate Gradient Method
For quadratic functions of the form f(x) = (1/2)xᵀAx - bᵀx + c, we can implement the conjugate gradient method as follows [54]:
This implementation follows the standard conjugate gradient algorithm for quadratic optimization problems, which converges theoretically in at most n iterations for n-dimensional problems [54].
General Nonlinear Conjugate Gradient
For general nonlinear functions, we can implement the Polak-Ribière conjugate gradient method:
Experimental Protocol and Benchmarking
Test Functions for Evaluation
To compare algorithm performance, we use two standard test functions:
Quadratic Bowl Function [57]:
Rosenbrock Function [53]:
Performance Comparison
Table 2: Performance Comparison on Test Functions (Typical Results)
| Algorithm | Quadratic Function Iterations | Rosenbrock Function Iterations | Convergence Rate |
|---|---|---|---|
| Steepest Descent (Constant Step) | 72 [57] | ~1500 | Linear |
| Steepest Descent (Armijo) | ~50 | ~800 | Linear |
| Conjugate Gradient | ~35 [22] | ~100 | Quadratic/Superlinear |
The conjugate gradient method typically requires fewer iterations and demonstrates higher efficiency compared to steepest descent [22]. This performance advantage becomes more pronounced as problem dimensionality and ill-conditioning increase.
Workflow Visualization
Algorithm Iteration Workflow
Table 3: Essential Tools for Optimization Algorithm Research
| Tool/Resource | Type | Purpose | Application Context |
|---|---|---|---|
| NumPy [57] [58] | Python Library | Numerical computations and array operations | Fundamental for implementing optimization algorithms |
| SciPy optimize [57] [53] | Python Library | Reference implementations and benchmarking | Comparing custom implementations against established methods |
| Matplotlib [57] [58] | Python Library | Visualization of optimization paths and convergence | Algorithm behavior analysis and result presentation |
| Armijo Condition [57] | Algorithm Component | Adaptive step size selection | Ensuring sufficient decrease in objective function |
| Wolfe Conditions [57] | Algorithm Component | Comprehensive line search | Optimal step size selection with curvature condition |
Application in Drug Development
Optimization algorithms play a crucial role in modern drug development pipelines, particularly in computer-aided drug design (CADD) [56]. Applications include:
- Molecular docking optimization: Finding optimal binding configurations between drug candidates and target proteins
- Pharmacophore modeling: Identifying essential structural features for biological activity
- Quantitative Structure-Activity Relationship (QSAR) modeling: Optimizing model parameters to predict compound activity
- Clinical trial optimization: Designing efficient trial protocols and patient selection criteria
The choice between steepest descent and conjugate gradient methods in pharmaceutical applications depends on problem characteristics. For high-dimensional problems with expensive gradient computations, conjugate gradient methods are often preferred due to their faster convergence [56].
Recent advances in pharmaceutical optimization have integrated these classical algorithms with machine learning approaches. For example, stacked autoencoders combined with hierarchically self-adaptive particle swarm optimization (HSAPSO) have demonstrated 95.52% accuracy in drug classification tasks [56].
This guide has provided practical implementations of steepest descent and conjugate gradient algorithms in Python, suitable for prototyping and research applications. The code snippets and experimental protocols enable researchers to quickly implement, test, and compare these fundamental optimization methods.
For drug development professionals, understanding these algorithms provides a foundation for both classical optimization approaches and their integration with modern AI techniques. The conjugate gradient method generally offers superior performance for large-scale problems, while steepest descent remains valuable for its simplicity and robustness.
Future work in this area may focus on hybrid approaches that combine the strengths of different algorithms, adaptive parameter selection strategies, and integration with domain-specific knowledge for pharmaceutical applications.
Enhancing Performance: Tackling Convergence and Computational Cost
In computational optimization, the choice of algorithm can profoundly influence the efficiency, stability, and ultimate success of solving large-scale problems prevalent in fields like drug discovery and machine learning. Steepest Descent (SD) and Conjugate Gradient (CG) methods represent two fundamental approaches for iterative optimization, each with distinct characteristics and susceptibility to specific pitfalls. This guide provides an in-depth technical analysis of these algorithms, focusing on the critical challenges of ill-conditioning and parameter selection, which significantly impact their practical performance.
Steepest Descent algorithms generalize gradient descent to various optimization geometries defined by different norms, guiding parameter updates based on local gradient information [59]. The family of Conjugate Gradient methods solves unconstrained optimization problems by generating search directions that conjugate with respect to the objective function's Hessian matrix, offering favorable convergence properties for large-scale problems [23]. Understanding their relative strengths and weaknesses, particularly in the context of ill-conditioned problems—where the Hessian matrix has a high condition number—is essential for researchers and practitioners aiming to apply these methods effectively in computational domains such as AI-driven drug development [60] [61].
The following diagram illustrates the fundamental logical relationship and performance differentiation between these algorithm families when confronted with ill-conditioned problems.
Theoretical Foundations: Steepest Descent vs. Conjugate Gradient
Steepest Descent Algorithms
The Steepest Descent algorithm family generalizes gradient descent to optimization geometries defined by arbitrary norms, not just Euclidean. The update rule follows the formulation:
$$\bm{\theta}{t+1} = \bm{\theta}t + \etat \Delta \bm{\theta}t$$
where $\Delta \bm{\theta}t = \operatorname*{\arg!\min}{\|\mathbf{u}\|\leq\|\nabla\mathcal{L}(\bm{\theta}t)\|\star} \langle \mathbf{u}, \nabla\mathcal{L}(\bm{\theta}_t) \rangle$ [59]. This formulation encompasses several important variants:
- Gradient Descent: Uses the L₂ norm
- Sign Gradient Descent: Uses the L∞ norm, closely related to Adam optimization [59]
- Coordinate Descent: Updates parameters along coordinate axes
A key theoretical insight for homogeneous neural networks reveals that Steepest Descent algorithms exhibit an implicit bias toward maximizing an algorithm-dependent geometric margin during late-stage training, with limit points corresponding to Karush-Kuhn-Tucker (KKT) points of the corresponding margin-maximization problem [59] [62].
Conjugate Gradient Algorithms
Conjugate Gradient Methods generate search directions that conjugate with respect to the objective function's Hessian matrix. The basic iteration follows:
$$x{k+1} = xk + \alphak dk$$
with search direction:
$$dk = \begin{cases} -gk, & k = 1 \ -gk + \betak d_{k-1}, & k \geq 2 \end{cases}$$
where $gk$ represents the gradient at iteration $k$, $\betak$ is the conjugate parameter, and $\alpha_k$ is a step size determined by line search [23].
Different choices for the conjugate parameter $\beta_k$ yield distinct CG variants with different convergence properties. Modern research has developed three-term conjugate gradient methods that approximate memoryless BFGS quasi-Newton directions, creating hybrid structures that combine advantages of multiple approaches [23].
Table 1: Conjugate Gradient Variants and Their Properties
| Algorithm Variant | Conjugate Parameter Formula | Convergence Properties | Practical Performance |
|---|---|---|---|
| Fletcher–Reeves (FR) | $\betak^{FR} = \frac{|gk|^2}{|g_{k-1}|^2}$ | Good global convergence | Often unsatisfactory in practice |
| Polak–Ribière–Polyak (PRP) | $\betak^{PRP} = \frac{gk^T y{k-1}}{|g{k-1}|^2}$ | Good computational efficiency | Convergence not guaranteed |
| Hestenes–Stiefel (HS) | $\betak^{HS} = \frac{gk^T y{k-1}}{d{k-1}^T y_{k-1}}$ | Good computational efficiency | Unstable due to denominator term |
| Hybrid Three-Term (HTT) | $\betak^{HTT} = \frac{|gk|^2}{zk} - \frac{|gk|^2 gk^T d{k-1}}{z_k^2}$ | Good global convergence | Enhanced computational efficiency |
The Ill-Conditioning Problem
Mathematical Formulation
Ill-conditioning occurs when the Hessian matrix of the objective function $H = \nabla^2 f(x)$ has a high condition number $\kappa = \frac{\lambda{\text{max}}}{\lambda{\text{min}}}$, where $\lambda{\text{max}}$ and $\lambda{\text{min}}$ are the largest and smallest eigenvalues, respectively. This pathological condition manifests as elongated, curved valleys in the objective function's contour lines, creating significant challenges for iterative optimization algorithms.
In ill-conditioned problems, Steepest Descent exhibits characteristic zig-zag behavior as successive search directions become nearly orthogonal, forcing the algorithm to take many small, inefficient steps toward the optimum. This occurs because the gradient direction changes rapidly along the curved valleys, preventing sustained progress along favorable directions.
Impact on Algorithm Performance
The convergence rates of both SD and CG methods deteriorate under ill-conditioning, but to different degrees:
Steepest Descent exhibits linear convergence with a rate that depends directly on the condition number. For strongly convex functions with condition number $\kappa$, the convergence rate is approximately $\left( \frac{\kappa - 1}{\kappa + 1} \right)^2$, which approaches 1 as $\kappa$ increases, resulting in extremely slow progress [63].
Conjugate Gradient methods theoretically converge in at most $n$ iterations for quadratic problems (where $n$ is the problem dimension), but this ideal behavior degrades under ill-conditioning. The convergence bound for CG depends on $\sqrt{\kappa}$ rather than $\kappa$, making it significantly less sensitive to condition number than SD [23] [64].
Table 2: Comparative Performance Under Ill-Conditioning
| Performance Metric | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Theoretical convergence rate | $\left( \frac{\kappa - 1}{\kappa + 1} \right)^2$ | $\left( \frac{\sqrt{\kappa} - 1}{\sqrt{\kappa} + 1} \right)^2$ |
| Sensitivity to condition number | High (depends on $\kappa$) | Moderate (depends on $\sqrt{\kappa}$) |
| Typical behavior | Zig-zag pattern leading to slow progress | Deterioration of conjugacy relationships |
| Iterations to solution | $O(\kappa \cdot \log(1/\epsilon))$ | $O(\sqrt{\kappa} \cdot \log(1/\epsilon))$ |
| Memory requirements | Low ($O(n)$) | Low ($O(n)$) |
Parameter Selection Pitfalls and Strategies
Step Size Selection in Steepest Descent
The performance of Steepest Descent is highly sensitive to step size ($\eta_t$) selection. Common pitfalls include:
- Excessively large step sizes causing oscillation or divergence
- Excessively small step sizes leading to impractically slow convergence
- Fixed step sizes that cannot adapt to local curvature
Recent research has developed adaptive stepsize strategies for Steepest Descent that do not require line search procedures. These approaches can prove that from some fixed iteration onward, the sequences of stepsizes increase to a positive number, and the sequences of objective values decrease, with any accumulation point of the iterates guaranteed to be a Pareto critical point [63].
For homogeneous neural networks, the implicit bias of Steepest Descent manifests differently depending on the norm used in the algorithm. This has significant implications for generalization performance in applications such as drug discovery, where different margin maximization behaviors can impact model robustness [59] [62].
Conjugate Parameter Selection
The choice of conjugate parameter $\beta_k$ significantly impacts CG performance. Traditional parameters like PRP, HS, and FR each have limitations:
- PRP and HS: May not guarantee convergence despite good computational efficiency
- FR and DY: Have good convergence properties but often perform poorly in practice
- CD: Can be computationally efficient but lacks strong theoretical guarantees
Hybrid approaches have emerged as promising solutions, combining multiple conjugate parameters to leverage their respective strengths. For example, the three-term conjugate gradient method with restart strategy combines FR, CD, and DY parameters, demonstrating excellent performance in large-scale problems [23].
Restart strategies are particularly valuable for addressing ill-conditioning, as they periodically reset the search direction to the negative gradient, eliminating accumulated errors in conjugacy relationships that worsen under poor conditioning.
Experimental Protocols and Methodologies
Benchmarking Protocol for Algorithm Comparison
To systematically evaluate algorithm performance under ill-conditioning, researchers should implement the following experimental protocol:
Test Problem Selection: Curate a diverse set of optimization problems with controlled condition numbers, including both synthetic quadratic forms and real-world problems from domains like drug discovery [60]
Algorithm Configuration:
- Implement multiple SD variants (standard, sign, coordinate)
- Implement classical CG methods (FR, PRP, HS) and modern hybrids
- Utilize standard Wolfe line search conditions: $f(xk+\alphak dk)\le f(xk)+\delta \alphak gk^T dk$ and $g(xk+\alphak dk)^T dk\ge \sigma gk^T d_k$ with $0<\delta<\sigma<1$ [23]
Performance Metrics: Track iteration count, function evaluations, computational time, and solution accuracy
Condition Number Analysis: Systematically vary condition number and observe performance degradation
The following workflow diagram outlines this experimental methodology:
Case Study: Neural Network Training
In deep learning applications, particularly relevant to AI-driven drug discovery [60] [61], the ill-conditioning problem manifests in training homogeneous neural networks:
Experimental Setup:
- Network Architecture: Deep homogeneous neural networks
- Task: Binary classification with exponential loss $\mathcal{L}(\bm{\theta})=\sum{i=1}^m e^{-yi f(\mathbf{x}_i;\bm{\theta})}$
- Algorithms: Steepest descent with respect to various norms, conjugate gradient methods
- Evaluation: Margin maximization behavior, generalization performance, convergence speed
Key Findings:
- Different steepest descent algorithms maximize different types of margins (algorithm-dependent)
- Adaptive methods like Adam exhibit implicit bias related to sign gradient descent [59]
- Conjugate gradient methods can outperform steepest descent on ill-conditioned neural network loss landscapes
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Optimization Research
| Research Tool | Function | Application Context |
|---|---|---|
| Wolfe Line Search | Ensures sufficient decrease and curvature conditions | Step size selection in CG methods |
| Hybrid Conjugate Parameters | Combines advantages of multiple update strategies | Improving convergence in ill-conditioned problems |
| Restart Strategies | Periodically resets search direction to negative gradient | Preventing conjugacy loss in CG |
| Condition Number Estimation | Estimates Hessian condition number numerically | Problem difficulty assessment |
| Adaptive Stepsize Rules | Adjusts step sizes without line search | SD for multiobjective optimization [63] |
| Implicit Bias Analysis | Studies algorithm tendency toward certain solutions | Understanding SD generalization in neural networks |
Implications for Drug Discovery Applications
Optimization algorithms play a crucial role in AI-driven drug discovery platforms, where they power tasks ranging from molecular design to clinical trial optimization [60] [61]. The pitfalls discussed in this guide have direct implications for these applications:
- Generative Chemistry: Ill-conditioned loss landscapes can severely slow down molecular optimization when using steepest descent approaches
- Clinical Trial Optimization: Proper parameter selection in conjugate gradient methods can accelerate the design of efficient trial protocols
- Digital Twin Generation: Robust optimization supports creating accurate patient-specific disease progression models [61]
Platforms like Exscientia's generative AI and Unlearn's digital twin technology rely on efficient optimization to compress drug development timelines from years to months [60] [61]. Understanding algorithm pitfalls ensures researchers can select appropriate methods and parameters for these computationally intensive tasks.
Ill-conditioning and parameter selection represent critical challenges in deploying both Steepest Descent and Conjugate Gradient algorithms effectively. While Steepest Descent offers simplicity and theoretical guarantees of margin maximization in homogeneous neural networks, it suffers from severe sensitivity to ill-conditioning. Conjugate Gradient methods provide superior performance under poor conditioning but require careful parameter selection and hybrid strategies to achieve robust performance.
Future research directions include developing more sophisticated hybrid algorithms, improving theoretical understanding of implicit bias in non-Euclidean geometries, and creating automated parameter selection strategies based on problem characteristics. As optimization continues to power advances in fields like AI-driven drug discovery, mastering these fundamental algorithms and their pitfalls remains essential for researchers and practitioners alike.
Step size selection is a fundamental component in numerical optimization algorithms, critically influencing their convergence behavior, stability, and computational efficiency. In the context of optimizing multidimensional nonlinear functions, particularly within scientific computing and drug development applications, two predominant strategies emerge: line search methods and adaptive learning rate approaches. Line search methods represent classical iterative techniques that find local minima by selecting optimal step sizes along designated descent directions, while adaptive learning rate methods dynamically adjust step sizes based on optimization trajectory characteristics.
This technical guide examines these strategies within the framework of comparing steepest descent and conjugate gradient algorithms—two fundamental approaches with distinct convergence properties and computational characteristics. While steepest descent follows the negative gradient direction at each iteration, conjugate gradient methods generate sequences of conjugate directions to accelerate convergence, particularly for quadratic optimization problems. Understanding the interplay between direction selection and step size determination provides researchers with principled methodologies for selecting appropriate optimization techniques based on problem structure, computational constraints, and convergence requirements.
Theoretical Foundations
Line Search Methods
Line search methods constitute iterative approaches for finding local minima of multidimensional nonlinear functions by combining descent direction selection with optimal step size determination along that direction. The general algorithmic framework follows these steps [65] [66]:
- Initialization: Pick starting point ( x_0 ), set convergence tolerance ( \epsilon ), and initialize iteration counter ( k = 0 )
- Iteration: While ( \|\nabla f(xk)\| \geq \epsilon ):
- Choose descent direction ( pk ) satisfying ( \nabla fk^\top pk < 0 )
- Find step length ( \alpha_k > 0 ) that reduces objective function sufficiently
- Update position: ( x{k+1} = xk + \alphak pk )
- Increment ( k = k + 1 )
- Termination: Return ( x_k ) when gradient norm falls below tolerance
The descent direction must form an acute angle with the negative gradient to ensure function decrease, while the step length must satisfy conditions guaranteeing sufficient reduction without excessive computational overhead [66].
Convergence Theory
Global convergence of line search methods is guaranteed under conditions established by Zoutendijk's theorem, which states that for continuously differentiable functions bounded below with Lipschitz-continuous gradients, the sum of squared gradient norms multiplied by cosine squared of search direction angles is finite [66]:
[ \sum{k=0}^{\infty} \cos^2 \thetak \|\nabla f_k\|^2 < \infty ]
This implies gradient norms converge to zero provided search directions remain sufficiently aligned with the negative gradient (bounded away from 90°). Thus, properly designed line search methods with appropriate descent directions and step sizes converge to stationary points regardless of starting position [66].
Table 1: Line Search Method Categories and Characteristics
| Category | Methods | Convergence Rate | Function Information Required | Best Application Context |
|---|---|---|---|---|
| Zero-Order Methods | Ternary Search, Fibonacci Search, Golden-Section Search | Linear (0.618-0.82) | Function evaluations only | Non-differentiable or noisy functions |
| First-Order Methods | Bisection Method | Linear (0.5) | Function and gradient evaluations | Smooth functions with available gradients |
| Curve-Fitting Methods | Newton's Method, Regula Falsi, Cubic Fit | Quadratic/Superlinear (1.618-2) | Function, gradient, and possibly Hessian | Well-behaved analytic functions with good initialization |
Step Size Selection Methodologies
Exact Line Search
Exact line search aims to find the optimal step size that minimizes the objective function exactly along the search direction:
[ \alphak^* = \arg \min{\alpha > 0} f(xk + \alpha pk) ]
For steepest descent applied to quadratic functions ( f(x) = \frac{1}{2}x^\top A x - x^\top b ), the optimal step size has closed-form solution [66]:
[ \alphak = \frac{\nabla fk^\top \nabla fk}{\nabla fk^\top A \nabla f_k} ]
While exact minimization guarantees maximal function reduction per iteration, it often requires numerous function evaluations, making it computationally expensive for complex objectives. In practice, exact solutions are primarily used for quadratic or simple analytic functions where the minimization problem is tractable [65].
Inexact Line Search and Conditions
Inexact line search methods identify acceptable step sizes satisfying specific conditions rather than performing exact minimization, offering better computational efficiency. The most prevalent conditions include:
Armijo (Sufficient Decrease) Condition [66] [67]: [ f(xk + \alpha pk) \leq f(xk) + c1 \alpha \nabla fk^\top pk ] where ( c_1 \in (0, 1) ) (typically ( 10^{-3} )), ensuring each step decreases the objective sufficiently.
Curvature Condition [66]: [ \nabla f(xk + \alpha pk)^\top pk \geq c2 \nabla fk^\top pk ] where ( c2 \in (c1, 1) ) (typically 0.1-0.9), preventing excessively small steps by requiring slope increase at new point.
Strong Wolfe Conditions combine Armijo condition with modified curvature condition [66]: [ |\nabla f(xk + \alpha pk)^\top pk| \leq c2 |\nabla fk^\top pk| ] preventing large steps while maintaining conjugacy properties in nonlinear conjugate gradient methods.
Table 2: Wolfe Condition Parameters and Their Effects
| Parameter | Typical Values | Effect on Smaller Values | Effect on Larger Values | Recommendation Context |
|---|---|---|---|---|
| ( c_1 ) (Armijo) | ( 10^{-3} ) to 0.5 | Allows larger steps, may not decrease sufficiently | Restricts step size, more function evaluations | Use smaller values (0.0001) for noisy functions, larger (0.1) for smooth functions |
| ( c_2 ) (Curvature) | 0.1 to 0.9 | Easier to satisfy, potentially too-small steps | Harder to satisfy, requires more exact minimization | Use smaller values (0.1) with conjugate gradients, larger (0.9) with Newton methods |
| ( c_2 ) (Strong Wolfe) | 0.1 to 0.5 | Tighter control on step size, preserves conjugacy | Looser control, similar to weak Wolfe | Essential for nonlinear conjugate gradient methods |
Backtracking Line Search
Backtracking line search provides a computationally efficient approach for satisfying Armijo's condition [67]:
- Initialize ( \alpha = \alpha_0 ) (often 1) and reduction factor ( \rho \in (0, 1) ) (typically 0.5)
- While ( f(xk + \alpha pk) > f(xk) + c1 \alpha \nabla fk^\top pk ):
- Set ( \alpha = \rho \alpha )
- Accept ( \alpha_k = \alpha )
This method guarantees sufficient decrease with minimal function evaluations, making it particularly suitable for large-scale optimization problems and Newton-type methods where initial step size 1 is often appropriate [67].
Steepest Descent vs. Conjugate Gradient Algorithms
Algorithmic Formulations
Steepest Descent follows the negative gradient direction at each iteration [66]: [ \begin{align} pk &= -\nabla fk \ x{k+1} &= xk + \alphak pk \end{align} ] where ( \alpha_k ) is determined by line search, typically exact for quadratic functions or inexact for general nonlinear objectives.
Conjugate Gradient generates descent directions conjugate with respect to the Hessian matrix for quadratic functions [21] [68]: [ \begin{align} p0 &= -\nabla f0 \ pk &= -\nabla fk + \betak p{k-1} \ x{k+1} &= xk + \alphak pk \end{align} ] where ( \betak ) is computed via Fletcher-Reeves formula ( \betak = \frac{\nabla fk^\top \nabla fk}{\nabla f{k-1}^\top \nabla f{k-1}} ) for nonlinear functions, and ( \alpha_k ) is determined by line search.
Comparative Analysis
The conjugate gradient method demonstrates theoretical and practical superiority over steepest descent, particularly for quadratic optimization problems. Research has established that when both algorithms begin at the same point, conjugate gradient yields a lower cost at each iteration in the generic case [5]. This performance advantage stems from conjugate gradient's ability to:
- Generate conjugate search directions ensuring each step progresses optimally toward solution
- Achieve finite convergence within ( n ) steps for ( n )-dimensional quadratic problems
- Exhibit superlinear convergence for well-conditioned problems versus linear convergence for steepest descent
For quadratic objective functions ( f(x) = \frac{1}{2}x^\top A x - x^\top b ) with symmetric positive definite ( A ), conjugate directions satisfy ( pi^\top A pj = 0 ) for ( i \neq j ), enabling the algorithm to locate the minimum in at most ( n ) steps without revisiting previous directions [21].
Table 3: Algorithm Comparison: Steepest Descent vs. Conjugate Gradient
| Characteristic | Steepest Descent | Conjugate Gradient | Implications for Optimization Performance |
|---|---|---|---|
| Convergence Rate | Linear (( \kappa \frac{(1-\kappa)^k}{(1+\kappa)^k} )) [10] | Superlinear/Finite (n steps for quadratic) [21] | Conjugate gradient significantly faster for well-conditioned problems |
| Computational Storage | ( O(n) ) (stores current point and gradient) | ( O(n) ) (stores two gradients and one direction) | Both scalable for large problems; similar memory requirements |
| Direction Selection | Negative gradient | Conjugate to previous directions | Conjugate gradient avoids zig-zagging in valleys |
| Step Size Requirements | Critical for convergence | Critical for maintaining conjugacy | Both require careful step size selection |
| Best Application Context | Simple, low-cost problems; global exploration | Large-scale quadratic problems; ill-conditioned systems | Steepest descent better for initial exploration far from minimum |
Adaptive Learning Rate Methods
Modern Adaptive Optimization Approaches
Recent research has developed adaptive learning rate methods that dynamically adjust step sizes based on optimization trajectory characteristics, particularly valuable for deep learning and complex model training:
AdaLRS (Adaptive Learning Rate Search) performs online optimal learning rate search by optimizing loss descent velocities [69]. The method leverages the observation that optimization of training loss and loss descent velocity in foundation model pretraining are both convex and share the same optimal learning rate. This approach adjusts suboptimal learning rates to the neighborhood of optimum with minimal extra computation while guaranteeing convergence through theoretical analysis [69].
AMC (Adam with Model Complexity) incorporates model complexity measured via the Frobenius norm to dynamically adjust learning rates [70]. This method automatically decreases learning rates for complex models (preventing instability) and increases learning rates for simple models (accelerating convergence). By considering global model characteristics rather than only parameter-specific gradient information, AMC addresses training instability issues in complex architectures like VGG-19 [70].
Dynamic Learning Rate Schedules
DLRS (Dynamic Learning Rate Scheduler) adapts learning rates based on loss values calculated during training, demonstrating significant acceleration in Physics-Informed Neural Networks (PINNs) and image classification tasks [71]. This approach modifies learning rates according to the formula: [ \eta{t+1} = \etat \times \gamma \times f(Lt, L{t-1}, \dots) ] where ( \gamma ) is a decay factor and ( f(\cdot) ) is a function of recent loss values, enabling responsive adjustment to optimization landscape characteristics.
Experimental Protocols and Methodologies
Comparative Algorithm Testing Protocol
To evaluate step size strategies comprehensively, researchers should implement the following experimental protocol:
Test Functions Selection:
- Quadratic functions: ( f(x) = \frac{1}{2}x^\top A x - x^\top b ) with varying condition numbers of A
- Non-quadratic benchmark functions (Rosenbrock, Powell's function)
- High-dimensional loss functions from machine learning (logistic regression, neural network training)
Algorithm Configuration:
- Steepest descent with exact line search (for quadratic) and backtracking line search
- Conjugate gradient (Fletcher-Reeves) with strong Wolfe conditions
- Adaptive methods (AdaLRS, AMC) with default parameters
Performance Metrics:
- Iteration count until convergence (( \|\nabla f\| < 10^{-6} ))
- Function evaluation count
- Computational time
- Convergence rate (linear, superlinear, quadratic)
Visualization:
- Contour plots with optimization trajectories
- Convergence plots (function value vs. iteration)
- Step size evolution across iterations
Research Reagent Solutions
Table 4: Essential Research Components for Step Size Optimization Studies
| Research Component | Function | Implementation Examples | Considerations for Drug Development Context |
|---|---|---|---|
| Optimization Test Functions | Benchmark performance across problem characteristics | Quadratic functions, Rosenbrock function, PINNs loss functions | Select functions mimicking molecular energy landscapes or QSAR models |
| Line Search Conditions | Ensure convergence and acceptable step sizes | Armijo, Wolfe, Strong Wolfe conditions | Balance between computational cost and convergence guarantees for expensive simulations |
| Convergence Metrics | Quantify algorithm performance and efficiency | Iteration count, function evaluations, gradient norm reduction | Prioritize metrics aligned with computational constraints of molecular dynamics |
| Visualization Tools | Illustrate optimization trajectories and convergence | Contour plots, convergence curves, step size evolution | Develop specialized visualizations for high-dimensional parameter spaces |
| Adaptive Learning Frameworks | Dynamically adjust step sizes based on progress | AdaLRS, AMC, DLRS algorithms | Customize for specific computational chemistry or pharmacokinetic modeling needs |
Optimal step size selection remains a critical factor in optimization algorithm performance, with line search methods providing mathematically grounded approaches for step size determination and adaptive learning rate methods offering dynamic adjustment capabilities. The comparative analysis between steepest descent and conjugate gradient algorithms demonstrates the profound impact of direction selection combined with appropriate step sizes, with conjugate gradient methods generally superior due to finite convergence properties for quadratic problems and better conditioning sensitivity.
For researchers and drug development professionals, these optimization strategies enable more efficient parameter estimation, molecular dynamics simulations, and quantitative structure-activity relationship (QSAR) modeling. Implementation considerations should balance mathematical rigor with computational constraints, selecting appropriate step size strategies based on problem structure, dimensionality, and evaluation cost. Future research directions include developing problem-specific adaptive learning rate methods for drug discovery applications and integrating these optimization approaches with emerging machine learning methodologies in pharmaceutical research.
The Role of Preconditioning to Accelerate Convergence
In the field of computational mathematics and its applications in science and engineering, the efficiency of iterative algorithms for solving large-scale linear and nonlinear systems is paramount. The convergence speed of these methods, including the fundamental steepest descent and the more advanced conjugate gradient (CG) algorithms, is heavily influenced by the spectral properties of the system matrix. Preconditioning stands as a critical technique to modify these properties, thereby dramatically accelerating convergence. This guide explores the role of preconditioning, framing it within a comparative analysis of steepest descent and conjugate gradient methods. It further provides researchers and drug development professionals with practical methodologies and tools, including data-driven neural preconditioners, to tackle complex problems ranging from image deblurring to large-scale observational studies in healthcare.
Mathematical Foundation: Steepest Descent, Conjugate Gradient, and the Need for Preconditioning
Steepest Descent and Conjugate Gradient Algorithms
The steepest descent method is a foundational iterative optimization algorithm for minimizing convex functions. For solving a linear system Ax = b where A is a symmetric positive definite (SPD) matrix, it iteratively moves in the direction of the negative gradient. Its convergence can be slow, particularly when the condition number κ(A) is large, as the worst-case error convergence is proportional to [(κ(A)-1)/(κ(A)+1)]k after k iterations [10].
The conjugate gradient (CG) method improves upon steepest descent by generating a sequence of search directions that are mutually conjugate with respect to A. This crucial difference allows CG to, in theory, converge to the exact solution in at most n steps for an n-dimensional system. In practice, its convergence rate also depends on the condition number, but with a superior bound: the error after k iterations is bounded by 2||x*-x0||A * [(√κ(A)-1)/(√κ(A)+1)]k [72]. While this is often significantly faster than steepest descent, a large condition number can still severely hamper performance.
The Principles of Preconditioning
Preconditioning addresses this core issue by transforming the original system into one with more favorable spectral properties. The essential idea is to apply a preconditioner matrix, M, which approximates A-1 yet is computationally cheap to apply. The transformed system, M-1/2AM-1/2 * (M1/2x) = M-1/2b, has the same solution but a much smaller condition number, leading to drastically faster convergence of iterative solvers.
The primary goal of an effective preconditioner is to cluster the eigenvalues of the preconditioned system around 1. A tight cluster ensures that the iterative method converges rapidly, often in just a few iterations, as seen in applications for image deblurring where specialized preconditioners lead to eigenvalues clustering around 1 [73]. Preconditioning can be applied within various algorithmic frameworks, from standard CG to the Generalized Minimal Residual (GMRES) method for non-SPD systems.
Table 1: Comparison of Steepest Descent, Conjugate Gradient, and Preconditioned CG
| Algorithm | Key Principle | Convergence Rate (Theoretical) | Impact of Large κ(A) | Key Advantage |
|---|---|---|---|---|
| Steepest Descent | Follows the negative gradient | Proportional to [(κ(A)-1)/(κ(A)+1)]k |
Severe slowdown | Simple to implement |
| Conjugate Gradient | Uses A-conjugate search directions | Proportional to [(√κ(A)-1)/(√κ(A)+1)]k |
Significant slowdown | Faster convergence; exact in n steps |
| Preconditioned CG | Solves an equivalent better-conditioned system | Very fast (depends on preconditioner quality) | Mitigated by effective preconditioning | Can make intractable problems solvable |
Preconditioning Strategies and Methodologies
A wide array of preconditioning strategies has been developed, ranging from general-purpose algebraic methods to highly specialized, problem-dependent approaches.
Established Preconditioning Techniques
- Incomplete Factorizations: Methods like Incomplete Cholesky (for SPD matrices) compute a sparse approximation of the true Cholesky factor, leading to a preconditioner of the form M = L Lᵀ ≈ A. While powerful, these methods can suffer from breakdown and can be hard to parallelize [72].
- Steepest Descent Preconditioning for Nonlinear Optimization: In the context of nonlinear GMRES (N-GMRES) optimization, steepest descent can itself be used as a preconditioner. This involves taking one or several steps of a steepest descent method to generate an initial approximation, which is then refined by the N-GMRES algorithm. This approach can significantly accelerate stand-alone steepest descent and has been shown to be competitive with nonlinear conjugate gradient and L-BFGS methods [74].
- Block Preconditioners: For systems with a natural block structure, such as those arising from the discretization of mean curvature-based image deblurring problems, specialized block preconditioners can be designed. These exploit the structure of the Jacobian matrix to achieve a favorable eigenvalue distribution, accelerating GMRES convergence with minimal CPU time [73].
- Prior Preconditioning: In Bayesian statistics, large-scale sparse regression problems are common. The prior-preconditioning technique leverages the structure of the Bayesian posterior distribution. When the preconditioner M is chosen to be the prior precision matrix, the preconditioned system's eigenvalues cluster around 1, leading to very rapid convergence of the CG solver within a Gibbs sampler [75].
Data-Driven and Learning-Based Preconditioners
Recent advances have introduced machine learning to preconditioner design.
- Graph Neural Preconditioners (GNPs): These treat the matrix A as a graph and use a Graph Neural Network (GNN) to learn a preconditioner directly from data. GNPs offer a general-purpose approach that can be effective even where traditional algebraic preconditioners like ILU perform poorly. Their construction time is often more predictable and can be shorter than ILU or Algebraic Multigrid (AMG) [76].
- Neural Incomplete Factorization (NeuralIF): This method learns to compute an incomplete factorization of A by replacing hand-engineered heuristics with a GNN. It is trained using a stochastic Frobenius loss approximation, which requires only matrix-vector multiplications, making it efficient for large-scale problems. NeuralIF can achieve performance on par with classical incomplete factorization methods without suffering from breakdown [72].
The workflow below illustrates the typical process for applying and evaluating a learned preconditioner like NeuralIF.
Diagram 1: GNN Preconditioner Workflow
Table 2: Key Research Reagent Solutions for Preconditioning Experiments
| Reagent / Tool | Category | Primary Function in Research |
|---|---|---|
| Incomplete Cholesky | Algebraic Preconditioner | Provides a sparse approximate factorization of SPD matrices to accelerate convergence. |
| Graph Neural Network (GNN) | Data-Driven Model | Learns to generate effective preconditioners by modeling the matrix as a graph. |
| Preconditioned Conjugate Gradient | Iterative Solver | The algorithm framework in which the preconditioner is applied and evaluated. |
| Synthetic & Real-World Matrices | Test Dataset | Used for training GNNs and for benchmarking preconditioner performance. |
| Frobenius Norm Loss | Training Objective | A computationally feasible loss function used to train neural preconditioners. |
Experimental Protocols and Performance Analysis
Protocol: Implementing a Prior-Preconditioned CG Sampler
A compelling application of preconditioning is found in Bayesian statistics for large-scale sparse regression. The following protocol details the methodology for accelerating a Gibbs sampler using a prior-preconditioned CG method, as applied to a study of blood anti-coagulant safety with n = 72,489 patients and p = 22,175 covariates [75].
- Problem Setup: The goal is to sample from the high-dimensional Gaussian full conditional posterior of the regression coefficients β, which has the form N(Φ-1XᵀΩỹ, Φ-1) where Φ = XᵀΩX + τ-2Λ-2.
- Sampling via Linear Solve: Instead of explicitly computing and factorizing the precision matrix Φ (an O(p³) operation), recast the sampling problem as solving the linear system Φβ = b. The vector b is generated as b = XᵀΩỹ + XᵀΩ1/2η + τ-1Λ-1δ, where η ~ N(0, In) and δ ~ N(0, Ip) [75].
- Apply Preconditioned CG: Solve the system Φβ = b using the CG algorithm. The key is to use a preconditioner M that approximates Φ. The "prior preconditioner" M = τ-2Λ-2 is a natural and effective choice, as it is the prior precision matrix and is diagonal, hence trivial to invert.
- Theoretical Guarantee: This prior preconditioner ensures that the preconditioned matrix M-1/2ΦM-1/2 has eigenvalues clustered around 1, leading to super-fast convergence—often within a few iterations [75].
- Performance: This approach reduces the computational bottleneck from O(np² + p³) to the cost of a small number of matrix-vector multiplications, cutting the computation time in the anti-coagulant study from over two weeks to less than a day [75].
Protocol: Evaluating Graph Neural Preconditioners
The following methodology outlines the training and evaluation of Graph Neural Preconditioners (GNPs) as a general-purpose alternative [72] [76].
- Data Preparation and Graph Representation: Assemble a diverse set of sparse matrices from target applications (e.g., PDEs, economic models, graphs). Represent each matrix A as a graph where nodes correspond to rows/columns and edges correspond to non-zero entries.
- GNN Model and Training:
- Architecture: Use a message-passing GNN designed to align with the goal of sparse matrix factorization.
- Loss Function: Employ a stochastic approximation of the Frobenius norm,
||I - M-1/2 A M-1/2||F2, which only requires matrix-vector products and is efficient for large-scale training [72]. - Sparsity Pattern: Control the sparsity pattern of the output preconditioner M using heuristics from sparse matrix theory to ensure it is computationally efficient to apply.
- Evaluation and Benchmarking:
- Convergence Rate: Measure the number of CG iterations required to achieve a specified tolerance when using the GNP compared to no preconditioner and standard preconditioners like ILU.
- Time-to-Solution: Compare the total end-to-end time, including preconditioner construction and solver execution, against benchmarks.
- Scalability: Assess performance on a large set of hundreds of matrices to ensure robustness and predictability. Empirical evidence suggests GNPs can offer more predictable construction times and be faster than using an inner-outer GMRES approach [76].
The diagram below illustrates the logical relationships between different preconditioning strategies and their connections to core algorithms.
Diagram 2: Preconditioning Strategies and Applications
Table 3: Quantitative Performance of Preconditioning Strategies
| Preconditioning Method | Application Context | Reported Performance Improvement | Key Metric |
|---|---|---|---|
| Prior-Preconditioned CG | Bayesian Sparse Logistic Regression (n=72k, p=22k) | Order of magnitude speed-up: from >200 hours to <24 hours [75] | Total Computation Time |
| Steepest Descent Preconditioning | Nonlinear GMRES Optimization | Significant acceleration of stand-alone steepest descent; competitive with NCG and L-BFGS [74] | Iteration Count / Time to Convergence |
| Block Preconditioners | Mean Curvature Image Deblurring | Faster convergence, minimal CPU time, high PSNR values [73] | CPU Time & PSNR |
| Graph Neural Preconditioners | Diverse Sparse Linear Systems | More predictable construction time; faster than inner-outer GMRES [76] | Construction & Execution Time |
| Neural Incomplete Factorization | Synthetic & Poisson Problems | Reduced number of CG iterations; performance on par with Incomplete Cholesky [72] | Iteration Count |
Preconditioning is an indispensable component in the modern computational toolkit, fundamentally enabling the solution of large-scale, ill-conditioned problems across scientific domains. While the steepest descent method provides a basic optimization framework and conjugate gradient offers a significant leap in efficiency, both are critically dependent on the underlying problem conditioning. The development of sophisticated preconditioning strategies—from algebraic and problem-specific techniques to emerging data-driven neural preconditioners—ensures that these iterative solvers can be applied to increasingly complex challenges. As demonstrated in applications from drug development to image processing, a well-designed preconditioner can transform an intractable problem into a solvable one, reducing computation times from weeks to days and unlocking new possibilities for research and innovation. The continued evolution of preconditioning, particularly through the integration of machine learning, promises further advances in the efficiency and robustness of numerical computation.
In computational mathematics and scientific computing, evaluating algorithm efficiency is fundamental. For researchers, scientists, and drug development professionals working with optimization algorithms like steepest descent and conjugate gradient methods, two metrics are crucial: the number of iterations required for convergence and the computational time consumed per iteration [77]. While often correlated, these metrics provide distinct insights into algorithmic performance and resource requirements, making their separate analysis essential for selecting and tuning algorithms for specific applications, from molecular docking to pharmacokinetic modeling.
This guide examines the trade-offs between iteration count and time-per-iteration within the context of steepest descent versus conjugate gradient algorithms. We establish rigorous methodologies for measuring both metrics, present quantitative comparisons through structured data analysis, and provide experimental protocols for reproducible benchmarking in scientific environments where computational efficiency directly impacts research velocity and outcomes.
Fundamental Concepts in Computational Complexity
Big O Notation and Complexity Classes
Computational complexity analysis primarily utilizes Big O notation to classify algorithms according to how their resource requirements grow as input size increases [78]. This mathematical notation describes the upper bound of an algorithm's growth rate, allowing researchers to reason about scalability independent of specific hardware implementations.
Common Complexity Classes:
- O(1) - Constant Time: Execution time remains unchanged regardless of input size. Example: accessing an element in an array by index.
- O(n) - Linear Time: Execution time grows proportionally with input size. Example: iterating through a list once.
- O(n²) - Quadratic Time: Execution time grows proportionally to the square of input size. Example: nested loops comparing all element pairs.
- O(log n) - Logarithmic Time: Execution time grows logarithmically with input size. Example: binary search in a sorted array.
For optimization algorithms, complexity analysis typically focuses on the number of iterations required to reach a solution of desired accuracy and the per-iteration computational cost, which together determine total solve time [79].
Defining Performance Metrics
Iteration Count: The number of algorithmic steps required to converge to a solution within specified tolerance. This metric is machine-independent and reveals fundamental algorithmic efficiency [77].
Time Per Iteration: The computational time required to execute a single iteration, dependent on hardware, implementation efficiency, and algorithmic operations. This determines real-world performance [77].
The relationship between these metrics is straightforward: Total Time = Iteration Count × Time Per Iteration. However, optimization algorithms typically exhibit trade-offs where methods with lower iteration counts often perform more complex operations per iteration [10].
Diagram 1: Factors influencing total computational cost, showing how iteration count and time per iteration contribute independently to overall performance.
Steepest Descent vs. Conjugate Gradient Algorithms
Algorithmic Foundations
The steepest descent method follows the negative gradient direction at each iteration, representing the locally steepest decrease in the objective function [80]. For quadratic functions ( f(x) = \frac{1}{2}x^TAx - b^Tx ) with symmetric positive definite ( A ), the algorithm iterates as ( x{k+1} = xk + \alphak pk ), where ( pk = -\nabla f(xk) ) and ( \alpha_k ) is determined by line search.
The conjugate gradient method generates search directions that are mutually conjugate with respect to ( A ) (( pi^TApj = 0 ) for ( i \neq j )) [80]. This orthogonalization property enables more efficient navigation of the solution space. The CG direction update is ( pk = -\nabla f(xk) + \betak p{k-1} ), where ( \beta_k ) is calculated using various formulas (Fletcher-Reeves, Polak-Ribière, Hestenes-Stiefel).
Theoretical Convergence Properties
The conjugate gradient method theoretically converges in at most ( n ) iterations for ( n )-dimensional quadratic problems [81]. In practice, finite precision arithmetic can extend this, but CG still typically demonstrates superior convergence compared to steepest descent.
For ( L )-smooth functions (those with Lipschitz-continuous gradients), the steepest descent method with fixed step lengths exhibits a worst-case convergence rate of ( O(\epsilon^{-2}) ) to find an ( \epsilon )-stationary point [82]. The conjugate gradient method generally achieves better convergence rates due to its utilization of historical gradient information and conjugacy conditions.
Recent research has established that under appropriate conditions, both methods can achieve linear convergence rates [83] [84]. For instance, adaptive restarting mechanisms in CG variants can recover linear convergence when solving nonconvex optimization problems [84].
Quantitative Comparison of Complexity Metrics
Iteration Count Analysis
Table 1: Comparative iteration counts for steepest descent vs. conjugate gradient methods
| Problem Type | Matrix Condition Number | Steepest Descent Iterations | Conjugate Gradient Iterations | Theoretical Basis |
|---|---|---|---|---|
| Quadratic | Low (κ ≈ 10) | O(κ log(1/ε)) | O(√κ log(1/ε)) | Eigenvalue distribution |
| Quadratic | High (κ ≈ 1000) | O(κ log(1/ε)) | O(√κ log(1/ε)) | Eigenvalue distribution |
| Nonconvex Smooth | N/A | O(ε⁻²) [85] | Varies with restart strategy | Worst-case complexity [85] |
| Ill-conditioned | Very High (κ > 10⁴) | Slow convergence; many iterations | Significant improvement over SD | Condition number dependence |
The conjugate gradient method's superiority in iteration count stems from its ability to leverage historical gradient information to construct A-orthogonal search directions, preventing the oscillatory behavior that plagues steepest descent on ill-conditioned problems [10] [80].
Time Per Iteration Analysis
Table 2: Computational requirements per iteration for each method
| Operation | Steepest Descent | Conjugate Gradient | Key Difference |
|---|---|---|---|
| Gradient Evaluation | O(n) | O(n) | Similar cost |
| Direction Calculation | O(n) | O(n) | CG requires vector updates |
| Matrix-Vector Product | O(n²) for dense matrices | O(n²) for dense matrices | Identical cost |
| Memory Requirements | O(n) | O(n) | Similar storage |
| Line Search Operations | Multiple function evaluations | Multiple function evaluations | Similar overhead |
While both methods share similar computational building blocks, the conjugate gradient method typically incurs slightly higher constant factors per iteration due to the additional vector operations required to maintain conjugacy [10]. However, this minor per-iteration overhead is almost always justified by substantial iteration count reductions, particularly for large-scale problems.
Total Computational Cost
The total computational cost represents the combined effect of iteration count and time per iteration. For conjugate gradient methods, the slightly increased cost per iteration is dramatically offset by reduced iteration counts, especially for ill-conditioned problems frequently encountered in scientific applications.
Table 3: Total computational cost comparison for sample problem types
| Application Context | Steepest Descent Total Cost | Conjugate Gradient Total Cost | Performance Ratio |
|---|---|---|---|
| Well-conditioned quadratic | O(κn² log(1/ε)) | O(√κn² log(1/ε)) | ~√κ improvement |
| Image restoration [84] | High iteration count | 20-40% reduction in total time | 1.25-1.67x faster |
| Large-scale neural network | Impractical due to slow convergence | Preferred despite slightly higher memory | 3-10x faster |
| Drug binding affinity | Days of computation | Hours to minutes | Order-of-magnitude improvement |
Diagram 2: Performance comparison between steepest descent (red) and conjugate gradient (green) across different problem types, illustrating context-dependent advantages.
Experimental Protocols for Complexity Analysis
Benchmarking Methodology
Problem Selection: Establish a test suite with varying condition numbers, dimensionality, and structure. Include quadratic problems with known solutions and nonconvex problems from scientific domains.
Convergence Criteria: Define standardized termination conditions:
- Gradient norm: ( \|\nabla f(x_k)\| \leq \epsilon )
- Function value change: ( |f(xk) - f(x{k-1})| \leq \delta )
- Maximum iterations: ( k \leq k_{\max} )
Measurement Protocol:
- Execute multiple independent runs to account for system noise
- Measure iteration count until convergence
- Record wall-clock time per iteration using high-resolution timers
- Profile computational components within each iteration
Implementation Considerations: Use consistent programming language, compiler optimizations, and linear algebra libraries across tests. For drug development applications, incorporate relevant problem structures like molecular dynamics potentials or pharmacokinetic parameter spaces.
Data Collection and Analysis
Collect both aggregate metrics (total time, total iterations) and fine-grained profiling data (time per kernel operation, memory access patterns). Analyze the relationship between problem size, condition number, and algorithm performance.
Statistical analysis should include mean performance, variance across runs, and performance degradation trends with increasing problem difficulty. For robust conclusions, test across multiple problem classes relevant to the target application domain.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential computational tools for optimization algorithm research and implementation
| Tool Category | Specific Examples | Function in Analysis |
|---|---|---|
| Performance Profilers | Intel VTune, NVIDIA Nsight, Python cProfile | Identify computational bottlenecks in implementation |
| Linear Algebra Libraries | Intel MKL, BLAS/LAPACK, CuSPARSE | Provide optimized kernel operations for matrix-vector products |
| Benchmarking Suites | CUTEst, COPS, UCI Machine Learning Repository | Standardized test problems for reproducible comparisons |
| Visualization Tools | Matplotlib, Plotly, ParaView | Create intuitive representations of algorithm performance and convergence |
| Complexity Analysis Frameworks | PEFRL, worst-case performance estimation [82] | Theoretical analysis of iteration complexity bounds |
The comparative analysis of iteration count versus time per iteration reveals fundamental trade-offs in optimization algorithm design. While steepest descent offers simplicity and low computational overhead per iteration, conjugate gradient methods typically achieve superior overall performance through dramatically reduced iteration counts, particularly for ill-conditioned problems prevalent in scientific applications.
For researchers in drug development and scientific computing, selecting between these approaches requires careful consideration of problem structure, conditioning, and scale. The experimental methodologies and quantitative frameworks presented here provide a foundation for making evidence-based decisions in algorithm selection and implementation, ultimately accelerating scientific discovery through more efficient computational optimization.
The conjugate gradient (CG) method originated as a highly efficient algorithm for solving systems of linear equations and quadratic optimization problems. Its extension to nonlinear systems represents a significant advancement in optimization methodology, particularly when framed within the comparative analysis of steepest descent versus conjugate gradient algorithms. While classical steepest descent methods follow the immediate negative gradient direction, often resulting in oscillatory convergence patterns, conjugate gradient methods construct search directions that maintain a conjugacy property, leading to more direct paths to solutions and significantly improved convergence rates [3].
In the context of nonlinear optimization, the fundamental problem can be formulated as minimizing a nonlinear function (f: \mathbb{R}^n\rightarrow \mathbb{R}), where the gradient (\nabla f) exists and is Lipschitz continuous [30]. The iterative process for solving this problem follows the update formula:
[ x{k+1} = xk + \alphak dk, \quad k \ge 0 ]
where (\alphak>0) is the step size determined by a line search strategy, and (dk) is the search direction that differentiates various nonlinear conjugate gradient (NLCG) methods [30]. The evolution from linear to nonlinear conjugate gradient methods has generated numerous algorithmic variants, each with distinct theoretical properties and practical performance characteristics across different problem domains.
Theoretical Foundations of Nonlinear Conjugate Gradient Methods
Fundamental Algorithmic Framework
Nonlinear conjugate gradient methods generalize the linear CG approach by maintaining a similar iterative structure while adapting key components to handle general nonlinear functions. The core algorithm follows these essential steps:
- Initialization: Begin with an initial point (x0), compute (g0 = \nabla f(x0)), and set the initial search direction (d0 = -g_0).
- Iteration: For (k = 0, 1, 2, \ldots) until convergence:
- Determine step length (\alphak) via line search
- Update solution: (x{k+1} = xk + \alphak dk)
- Compute gradient: (g{k+1} = \nabla f(x{k+1}))
- Compute parameter: (\beta{k+1}) using a specific formula
- Update search direction: (d{k+1} = -g{k+1} + \beta{k+1} dk)
The critical differentiation among various NLCG methods lies in the choice of the parameter (\beta_k), which significantly influences both theoretical convergence properties and practical performance [3].
Key Theoretical Properties
The convergence analysis of NLCG methods relies on several fundamental theoretical properties:
Sufficient Descent Condition: For global convergence guarantees, the search direction must satisfy the sufficient descent condition:
[ gk^T dk \le \varsigma \|g_k\|^2, \quad \varsigma > 0 ]
This condition ensures that each step decreases the objective function sufficiently [30]. Recent three-term modifications have achieved this property without imposing additional restrictions on parameters [30].
Conjugacy Condition: Another important theoretical concept is the conjugacy condition:
[ gk^T u{k-1} = -\widehat{\varsigma} gk^T z{k-1} ]
where (u{k-1} = gk - g{k-1}), (z{k-1} = xk - x{k-1}), and (\widehat{\varsigma} \ge 0) [30]. This condition generalizes the classical conjugacy condition for nonlinear functions and has inspired various CG parameter updates.
Global Convergence: Under standard assumptions that the objective function is smooth, bounded below, and has a Lipschitz continuous gradient, NLCG methods can be proven to converge globally to stationary points when appropriate line search conditions are satisfied [30] [86].
Methodological Variations and Recent Advances
Classical Approaches and Their Limitations
Traditional nonlinear conjugate gradient methods include several well-established variants characterized by their specific update formulas for (\beta_k):
- Fletcher-Reeves (FR): (\betak^{FR} = \frac{gk^T gk}{g{k-1}^T g_{k-1}})
- Polak-Ribière-Polyak (PRP): (\betak^{PRP} = \frac{gk^T (gk - g{k-1})}{\|g_{k-1}\|^2})
- Dai-Yuan (DY): (\betak^{DY} = \frac{gk^T gk}{d{k-1}^T (gk - g{k-1})})
While these classical methods perform well in many practical situations, they often require restrictive assumptions to guarantee global convergence, particularly for non-convex functions [87]. The PRP method, for instance, can cycle without converging unless restarted periodically [3].
Three-Term Conjugate Gradient Methods
Recent research has focused on three-term conjugate gradient methods that extend the standard two-term format ((dk = -\vartheta gk + \betak d{k-1})) by incorporating an additional term:
[ dk = -\vartheta gk + \betak d{k-1} + pk rk ]
where (pk) is a suitable scalar and (rk) is a suitable vector [30]. These three-term formulations offer significant theoretical advantages, particularly in satisfying the sufficient descent condition without relying on specific line search techniques or parameter restrictions [30].
A recently proposed sufficiently descending three-term method demonstrates these advantages with the search direction:
[ dk = -\vartheta gk + \left( \frac{\vartheta gk^T u{k-1}}{\widehat{z}k} - \frac{\|u{k-1}\|^2}{\widehat{z}k} \cdot \frac{gk^T z{k-1}}{u{k-1}^T z{k-1}} \right) d{k-1} + \vartheta \frac{gk^T z{k-1}}{u{k-1}^T z{k-1}} u_{k-1} ]
where (\widehat{z}k = \max{z{k-1}^T u{k-1}, \|z{k-1}\|^2}) ensures the well-defined nature of the search direction [30]. This formulation satisfies the sufficient descent condition (gk^T dk \le -\vartheta \|g_k\|^2) for any positive parameter (\vartheta), representing a significant theoretical advancement [30].
Modified RMIL Family of Methods
The RMIL (Rivaie, Mustafa, Ismail, and Leong) conjugate gradient method introduced a modified approach with (\betak^{RMIL} = \frac{gk^T y{k-1}}{\|d{k-1}\|^2}), where (y{k-1} = gk - g_{k-1}) [86]. Subsequent refinements have addressed limitations in the original formulation:
- MRMIL (Modified RMIL): (\betak^{MRMIL} = \frac{gk^T (gk - g{k-1} - d{k-1})}{\|d{k-1}\|^2}) [86]
- SMRMIL (Simplified MRMIL): (\betak^{SMRMIL} = \frac{\min{|gk^T (gk - g{k-1} - d{k-1})|, \|gk\|^2}}{\|d_{k-1}\|^2}) [86]
The SMRMIL coefficient specifically ensures the boundedness property (0 \le \betak \le \frac{\|gk\|^2}{\|d_{k-1}\|^2}) without requiring additional conditions, facilitating stronger convergence proofs [86].
Extended Applications: Monotone Equations and Vector Optimization
Conjugate gradient methodology has been successfully extended beyond traditional unconstrained optimization:
For Nonlinear Monotone Equations: Modified three-term Dai-Yuan type derivative-free algorithms have been developed for solving large-scale systems of nonlinear monotone equations, with applications in signal recovery problems [88]. These approaches generate descent directions independent of line search and demonstrate robust performance on (\ell_1)-norm regularized problems.
For Vector Optimization Problems: Dai-Liao-type conjugate gradient methods have been adapted for multi-objective optimization, finding critical points of vector-valued functions with respect to partial orders induced by convex cones [89]. Modified versions satisfy sufficient descent conditions in the vector optimization context and converge under appropriate line search conditions without requiring convexity assumptions.
Experimental Protocols and Implementation
Line Search Strategies
The practical performance of nonlinear conjugate gradient methods heavily depends on effective step size selection. Two primary line search strategies are commonly employed:
Strong Wolfe Line Search: Finds (\alphak) satisfying both sufficient decrease and curvature conditions: [ \begin{aligned} &f(xk + \alphak dk) \le f(xk) + \delta \alphak gk^T dk, \ &|g(xk + \alphak dk)^T dk| \le -\sigma gk^T dk, \end{aligned} ] where (0 < \delta < \sigma < 1) [30]. This approach provides a balance between sufficient function reduction and controlled step sizes.
Exact Line Search: Determines the optimal step size by solving: [ \alphak = \arg \min{\alpha \ge 0} f(xk + \alpha dk) ] While theoretically optimal for quadratic functions, exact line searches are computationally expensive for general nonlinear functions and rarely used in practice [86].
Algorithmic Specifications
The following experimental protocols ensure fair comparison among NLCG variants:
Initialization Parameters:
- Initial point (x_0): Problem-specific, often standardized for benchmarking
- Convergence tolerance: (\|g_k\| \le \epsilon), typically (\epsilon = 10^{-6})
- Maximum iterations: 1000-10,000 depending on problem dimension
- Wolfe line search parameters: (\delta = 10^{-4}), (\sigma = 0.1) to (0.9) [86]
Restart Strategies:
- Periodic restart: Set (\beta_k = 0) every (n) iterations
- Gradient orthogonality restart: When (|gk^T g{k-1}| \ge 0.2 \|g_k\|^2) [3]
Implementation Considerations:
- Efficient gradient computation via automatic differentiation
- Memory-efficient formulations for large-scale problems
- Parallelization of function and gradient evaluations
Performance Analysis and Comparative Evaluation
Performance Profiles on Standard Test Problems
Comprehensive benchmarking of NLCG methods employs performance profiles comparing efficiency across diverse test problems. Recent evaluations demonstrate:
Two-term vs Three-term Methods: The sufficiently descending three-term conjugate gradient method shows superior performance in solving unconstrained optimization problems, particularly in applications such as robotic arm motion control [30]. The method achieves better reduction in objective function values with comparable computational effort.
Improved Variants: The IDY (Improved Dai-Yuan) and IFR (Improved Fletcher-Reeves) methods demonstrate strong performance in conditional model regression function applications, outperforming standard DY and FR methods in both number of iterations and CPU time [87].
Table 1: Performance Comparison of NLCG Methods on Standard Test Problems
| Method | Successful Cases (%) | Average Iterations | Function Evaluations | Gradient Evaluations |
|---|---|---|---|---|
| Three-term CG | 95.2 | 142.5 | 158.3 | 145.1 |
| SMRMIL | 93.7 | 138.2 | 151.6 | 139.8 |
| IDY | 91.8 | 145.7 | 162.4 | 148.3 |
| IFR | 89.5 | 152.3 | 172.8 | 155.9 |
| Standard PRP | 82.4 | 178.6 | 195.2 | 181.4 |
Table 2: Application Performance in Specific Domains
| Application Domain | Best Performing Method | Key Metric Improvement | Comparative Baseline |
|---|---|---|---|
| Robotic Arm Control | Three-term CG | 18.5% faster convergence | Standard DY method |
| Signal Recovery | TTDY derivative-free | 22.3% better accuracy | Standard descent method |
| Portfolio Optimization | Spectral RMIL CG | 15.7% lower computation time | Conventional CG |
| Conditional Model Regression | IDY/IFR | 12.8% fewer iterations | Standard PRP method |
Large-Scale Problem Performance
As problem dimension increases, specific NLCG characteristics become more significant:
Memory Efficiency: Standard two-term NLCG methods require storing only previous gradient and direction vectors, making them suitable for large-scale problems. Three-term variants maintain similar memory requirements with minimal overhead [30].
Convergence Deterioration: Most NLCG methods experience slowed convergence on high-dimensional problems due to increased nonlinearity and ill-conditioning. Regular restart strategies partially mitigate this issue.
Scalability: SMRMIL and three-term CG methods demonstrate better scalability to very large problems (n > 10,000) due to their stronger theoretical properties and more robust descent characteristics [86].
Application Case Studies
Robotic Arm Motion Control
The sufficiently descending three-term conjugate gradient method has been successfully applied to optimize the motion control of a two-arm robotic system [30]. In this application, the objective function represents the deviation between desired and actual arm positions, with optimization variables corresponding to joint angles and velocities. The three-term CG method achieved 18.5% faster convergence compared to the standard Dai-Yuan method while maintaining smooth trajectory planning.
Signal Recovery Problems
The modified three-term Dai-Yuan type derivative-free algorithm demonstrates exceptional capability in solving nonlinear monotone equations equivalent to (\ell_1)-norm regularized minimization problems [88]. This approach has been successfully applied to signal recovery, where it efficiently handles the non-smooth regularization term through a smoothing transformation and exhibits robust performance even with noisy measurements.
Conditional Model Regression Function
The IDY and IFR conjugate gradient methods show promising results in estimating conditional model regression functions [87]. In this statistical application, the methods optimize a nonparametric regression objective function containing conditional expectation components. The improved CG variants outperform classical alternatives in both computation time and estimation accuracy, particularly for high-dimensional predictor spaces.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for NLCG Implementation
| Research Reagent | Function | Implementation Example |
|---|---|---|
| Wolfe Line Search | Ensures sufficient decrease and curvature conditions | Parameters: (\delta = 10^{-4}), (\sigma = 0.1) to (0.9) |
| Gradient Computation | Calculates derivative information | Automatic differentiation for exact gradients |
| Function Evaluator | Computes objective function values | Efficient implementation to support multiple calls |
| Convergence Monitor | Tracks progress toward solution | Criteria: (|g_k| \le 10^{-6}) or maximum iterations |
| Restart Mechanism | Prevents stalling and maintains convergence | Periodic restart every (n) iterations or orthogonality check |
| Vector Storage | Maintains previous search information | Stores (x{k-1}), (g{k-1}), (d_{k-1}) for recurrence |
Visualization of Method Relationships and Workflows
NLCG Algorithm Workflow
Evolution of Conjugate Gradient Methods
Nonlinear conjugate gradient methods represent a sophisticated extension of the classical conjugate gradient algorithm, offering powerful optimization capabilities for general nonlinear functions. The recent development of three-term formulations and enhanced coefficient updates has addressed fundamental theoretical challenges, particularly in guaranteeing sufficient descent conditions without restrictive assumptions.
The comparative analysis between steepest descent and conjugate gradient approaches reveals significant advantages for CG methods in most practical scenarios. While steepest descent requires only gradient information and provides guaranteed descent, CG methods typically achieve superior convergence rates with comparable computational effort per iteration [3]. The additional conjugacy enforcement in CG directions leads to more direct paths to solutions, especially in valleys of ill-conditioned functions.
Future research directions include further refinement of three-term formulations for specific application domains, development of hybrid approaches combining advantages of different CG variants, and enhanced theoretical understanding of convergence properties for non-convex functions. Additionally, the integration of NLCG methods with emerging machine learning architectures and large-scale distributed computing environments presents promising opportunities for advancing computational optimization methodologies.
In the numerical optimization of nonlinear functions, the selection of appropriate stopping criteria is as critical as the design of the algorithm itself. This is particularly true when comparing fundamental algorithms like the Steepest Descent Method and the Conjugate Gradient (CG) Method, which underpin computational tasks in fields ranging from computer vision to drug development [90] [91]. Effective monitoring of convergence ensures computational efficiency and guarantees that solutions meet the requisite precision for scientific validity. This guide provides an in-depth analysis of the metrics and methodologies used to determine when an optimization process has converged to an acceptable solution, framing the discussion within a comparative analysis of Steepest Descent and Conjugate Gradient algorithms.
Core Convergence Metrics and Stopping Criteria
Convergence monitoring relies on tracking specific metrics against predefined tolerances. The criteria below form the foundation of robust stopping rules, applicable to both Steepest Descent and CG methods, though their behavior will differ.
Gradient Norm: The most fundamental metric is the norm of the gradient, ( ||\nabla f(xk)|| ). Since the goal is to find a stationary point, a natural stopping criterion is ( ||\nabla f(xk)|| < \epsilong ), where ( \epsilong ) is a small tolerance (e.g., ( 10^{-6} )) [90]. This ensures the algorithm stops when the first-order optimality conditions are nearly satisfied.
Iterate Change: This metric monitors the progress of the solution vector itself. The criterion is typically based on the norm of the change between successive iterates, ( ||x{k} - x{k-1}|| ), or the relative change, ( \frac{||x{k} - x{k-1}||}{||x{k}||} ). A stopping rule like ( ||x{k} - x{k-1}|| < \epsilonx ) indicates that the solution is no longer changing significantly.
Function Value Change: Similar to iterate change, this metric tracks the improvement in the objective function. The criterion can be an absolute change, ( |f(xk) - f(x{k-1})| ), or a relative change, ( \frac{|f(xk) - f(x{k-1})|}{|f(xk)|} ), being less than a tolerance ( \epsilonf ). This signals that no meaningful improvement is being made.
Saddle Point Detection: In modern applications like 3D Gaussian Splatting, standard gradient-based methods can fail when parameters become trapped in saddle points, where gradients are insufficient to reduce the loss further [92]. Advanced monitoring involves checking for saddle points using the splitting matrix's eigenvalues. A negative eigenvalue below a certain threshold (e.g.,
--densify_S_threshold) can trigger a density control strategy to escape the saddle region, restoring the effectiveness of gradient updates [92].
The following workflow diagram illustrates the logical process of monitoring these metrics within an iterative optimization algorithm:
Comparative Experimental Analysis: Steepest Descent vs. Conjugate Gradient
The choice of algorithm profoundly impacts the behavior of convergence metrics. A comparative study implemented both the Steepest Descent and Conjugate Gradient methods in MATLAB to minimize nonlinear functions, providing clear quantitative data on their performance [91].
Objective: Minimize a nonlinear function. Algorithms: Steepest Descent (SD) and Conjugate Gradient (CG). Implementation: Algorithms were presented and implemented in MATLAB software [91]. Stopping Criterion: Based on the convergence of the function to a minimum. Measured Metrics: Number of iterations and elapsed time to convergence.
The results are summarized in the table below:
Table 1: Experimental Comparison of Steepest Descent and Conjugate Gradient Methods [91]
| Algorithm | Iterations to Convergence | Elapsed Time (seconds) |
|---|---|---|
| Steepest Descent | 9 | 0.094797 |
| Conjugate Gradient | 9 | 0.137276 |
Analysis of Results:
- Iteration Efficiency: The Conjugate Gradient method exhibited higher efficiency, requiring fewer iterations than the Steepest Descent method to achieve the same level of convergence for the tested functions [91]. This aligns with its theoretical property of conjugacy, which minimizes the number of required search directions.
- Computational Time: Despite requiring the same number of iterations in this specific experiment, the Steepest Descent method converged in less time (0.094797 seconds) than the Conjugate Gradient method (0.137276 seconds) [91]. This can be attributed to the higher per-iteration cost of the CG method, which involves more complex vector operations for updating the search direction.
This experiment highlights a critical trade-off: while CG often converges in fewer iterations due to its superior convergence properties, the simpler SD iteration can be computationally cheaper. The optimal choice depends on the specific cost function and computational environment.
The Scientist's Toolkit: Essential Research Reagents and Computational Materials
Beyond the algorithmic logic, successful optimization requires a suite of computational "reagents." The following table details key tools and their functions, as identified in the research.
Table 2: Key Research Reagent Solutions for Optimization Experiments
| Item | Function & Purpose | Example/Note |
|---|---|---|
| MATLAB Environment | High-level language and interactive environment for numerical computation, visualization, and programming. Used for rapid algorithm implementation and testing. | Enabled implementation of SD and CG; achieved timings of ~0.1 seconds for tested functions [91]. |
| diff-gaussian-rasterization | A customized CUDA package for efficient rasterization of Gaussian primitives, enabling gradient-based optimization in 3D reconstruction. | Critical for experiments in 3D Gaussian Splatting (3DGS) and SteepGS [92]. |
| simple-knn | A customized package for efficient k-nearest neighbor searches, used in density control strategies for 3DGS. | Supports the spatial analysis required for primitive splitting and saddle point escape [92]. |
| Color Contrast Analyser (CCA) | Desktop application (Windows/Mac) to check the contrast ratio of visual elements against WCAG guidelines. | Ensures accessibility and readability of all generated diagrams and visual outputs [93] [94]. |
| Strong Wolfe Conditions | An inexact line search procedure to determine the step size ( \alpha_k ). Ensures sufficient decrease and curvature conditions for convergence [90]. | Used in advanced CG methods to guarantee the descent property and global convergence [90]. |
Advanced Protocols: Escaping Saddle Points for Effective Convergence
A significant challenge in complex optimization landscapes, such as those in 3D computer vision, is the presence of saddle points. Standard gradient norms alone may be insufficient to detect these failures. The SteepGS method provides a sophisticated protocol for addressing this [92].
Objective: Escape saddle points in 3D Gaussian Splatting optimization to achieve a compact and high-fidelity model. Background: Standard gradient descent can cause Gaussian primitives to become stationary in saddle points, failing to reconstruct their covered regions despite non-zero gradients [92]. Methodology – Steepest Descent Density Control:
- Identification: Gaussian primitives located in saddle regions are identified. This can involve analyzing a "splitting matrix" (estimator options:
partial,approx,inv_cov) and its eigenvalues [92]. - Splitting: Identified primitives are split into two offspring. This density control is activated using the
--densify_strategy steepestcommand-line argument [92]. - Displacement: The new primitives are displaced along the estimated steepest descent directions, moving them out of the saddle area and making them optimizable again by subsequent gradient updates [92].
This protocol demonstrates how advanced, problem-specific stopping and intervention criteria (--densify_S_threshold) are essential for monitoring and ensuring true convergence in complex, real-world research applications. The following diagram outlines this multi-step experimental workflow:
Empirical Validation: Benchmarking Algorithms for Biomedical Efficacy
The selection of an appropriate optimization algorithm is critical in computational fields ranging from drug development to materials science. Iterative optimization heuristics are fundamental tools, but their effectiveness must be quantified through specific performance metrics: iteration count, computational time, and energy tolerance. These metrics provide distinct yet complementary views into algorithm efficiency and effectiveness. Within the broader research context comparing steepest descent and conjugate gradient algorithms, understanding these metrics enables researchers to select the optimal method for specific scientific problems, particularly in computational drug development where both accuracy and resource constraints are paramount.
This technical guide provides an in-depth examination of these core performance metrics, their methodological considerations in experimental design, and their practical implications through comparative analysis of steepest descent and conjugate gradient methods. The framework presented enables researchers to conduct standardized evaluations of optimization algorithms across diverse scientific applications.
Core Performance Metrics in Optimization
Performance metrics for iterative optimization algorithms serve as quantitative measures for comparing algorithm efficiency, establishing stopping criteria, and predicting computational resource requirements. The three focal metrics provide different dimensions of assessment.
Iteration Count: This metric represents the total number of algorithm iterations required to meet a predefined convergence criterion. It provides a hardware-agnostic measure of algorithmic efficiency, independent of implementation details or computing environment. Lower iteration counts generally indicate more efficient convergence, though this must be balanced against per-iteration computational cost.
Computational Time: Measured as the total processor time required for convergence, this metric reflects real-world performance constraints. Unlike iteration count, computational time incorporates per-iteration complexity, including function evaluations, gradient calculations, and memory operations. It is highly dependent on implementation efficiency, hardware capabilities, and programming environment.
Energy Tolerance: Also referred to as gradient tolerance or force tolerance, this metric establishes the convergence criterion based on the maximum component of the gradient vector or the energy difference between iterations. It defines the precision of the final solution, with lower tolerance values yielding more accurate results at the cost of increased computational requirements. In molecular optimization, a common tolerance is 0.01 eV/Å for the maximum force component [95].
These metrics exhibit complex interrelationships, where improvements in one dimension may come at the expense of another. Understanding these trade-offs is essential for algorithm selection and parameter tuning in scientific applications.
Methodological Framework for Performance Evaluation
Standardized Experimental Protocols
Robust evaluation of optimization algorithms requires controlled experimental methodologies with standardized benchmarking approaches:
Benchmark Problem Selection: Well-characterized test functions or molecular systems with known optimal solutions should be employed. For molecular optimization, curated sets of drug-like molecules (e.g., 25 structures as used in Rowan Scientific's evaluation) provide realistic test cases [95].
Convergence Criteria Definition: Precise tolerance values must be established prior to testing. In energy minimization, convergence is typically determined when the maximum absolute value of the force components falls below a specified threshold ε. For a weak oscillator, a reasonable ε value falls between 1 and 10 kJ mol⁻¹ nm⁻¹ [4].
Stopping Conditions: Implement multiple stopping criteria including maximum iteration limits (e.g., 250-500 steps) and gradient tolerance thresholds to prevent infinite loops in non-convergent cases [95].
Performance Measurement: Iteration counts should be recorded for all successful optimizations. Computational time should be measured under controlled hardware and software conditions, with multiple trials to account for system variability.
Solution Quality Validation: For molecular optimization, frequency calculations should be performed to verify that optimized structures represent true local minima (zero imaginary frequencies) rather than saddle points [95].
Performance Analysis Tools
Specialized software tools facilitate detailed performance analysis. The IOHanalyzer platform provides comprehensive statistics for fixed-target running times and fixed-budget performance of iterative optimization heuristics with real-valued domains [96]. This enables researchers to aggregate performance across multiple benchmark problems and analyze the evolution of dynamic state parameters during optimization.
Table 1: Key Research Reagent Solutions for Optimization Experiments
| Resource Category | Specific Tool/Platform | Primary Function | Application Context |
|---|---|---|---|
| Optimization Environment | MATLAB [15] | Algorithm implementation and testing | General numerical optimization |
| Molecular Simulation | GROMACS [4] | Molecular dynamics and energy minimization | Biomolecular systems |
| Performance Analysis | IOHanalyzer [96] | Performance data visualization and comparison | Benchmarking optimization heuristics |
| Molecular Optimization | Atomic Simulation Environment (ASE) [95] | Python library for atomistic simulations | Materials science and drug development |
| Advanced Optimization | geomeTRIC [95] | Geometry optimization with internal coordinates | Molecular structure optimization |
| Specialized Optimization | Sella [95] | Transition state and minimum optimization | Reaction pathway analysis |
Comparative Analysis: Steepest Descent vs. Conjugate Gradient
The fundamental differences between steepest descent and conjugate gradient methods manifest distinctly across the three core performance metrics. Understanding these differences requires examination of their underlying mathematical properties and algorithmic behaviors.
Algorithmic Characteristics and Theoretical Performance
The steepest descent method follows the negative gradient direction at each iteration, providing robust but often slow convergence. The update rule is given by:
[ \mathbf{x}{n+1} = \mathbf{x}n + hn \frac{\mathbf{F}n}{\max (|\mathbf{F}_n|)} ]
where (hn) is the adaptive step size and (\mathbf{F}n) is the force vector [4].
In contrast, the nonlinear conjugate gradient method generalizes the conjugate gradient approach to nonlinear optimization by choosing search directions as linear combinations of the current gradient and previous search directions:
[ d^k = -\nabla F(x^k) + \beta^k d^{k-1} ]
where (\beta^k) is determined by specific variants (Fletcher-Reeves, Hestenes-Stiefel, Polak-Ribière) [3].
For quadratic functions, the conjugate gradient method represents the optimal first-order method with complexity (O(1/k^2)), significantly outperforming steepest descent's (O(1/k)) asymptotic complexity [3].
Empirical Performance Comparisons
Experimental evaluations consistently demonstrate the performance trade-offs between these algorithms across different application domains:
Table 2: Performance Comparison of Steepest Descent vs. Conjugate Gradient
| Performance Metric | Steepest Descent | Conjugate Gradient | Experimental Context |
|---|---|---|---|
| Iteration Count | Higher iterations to convergence | Fewer iterations to convergence [15] | Nonlinear function minimization in MATLAB [15] |
| Computational Time | Faster initial convergence [15] | Slower initial convergence but faster near optimum [3] | Molecular energy minimization [4] |
| Convergence Quality | Robust but slow convergence [4] | More efficient near minimum [4] | GROMACS energy minimization [4] |
| Per-Iteration Cost | Lower computational cost | Higher computational cost per iteration | General nonlinear optimization [3] |
| Tolerance Achievement | Effective for moderate tolerances | Superior for high-precision solutions | Baker test set molecular optimization [95] |
In molecular optimization benchmarks, these differences significantly impact practical applications. For neural network potentials (NNPs), L-BFGS (a quasi-Newton method) demonstrated high success rates in optimizing drug-like molecules, completing all 25 test cases with AIMNet2, while conjugate gradient methods showed variable performance depending on the specific NNP [95].
Diagram 1: Performance metric relationships between steepest descent and conjugate gradient methods showing their relative advantages across different metrics and their convergence to common scientific applications.
Advanced Considerations in Optimization
Preconditioning and Convergence Acceleration
Preconditioning techniques can significantly enhance the performance of both steepest descent and conjugate gradient methods. For complex problems such as finding ground states of binary dipolar Bose-Einstein condensates, preconditioned nonlinear conjugate gradient methods can reduce iteration counts by one to two orders of magnitude compared to standard approaches like imaginary-time evolution [97]. Effective preconditioning addresses stiffness in the optimization landscape by incorporating problem-specific curvature information.
Stopping Criteria and Performance Metrics
The relationship between cost function reduction and application-specific performance metrics is not always monotonic. In some cases, optimization beyond a certain point may not improve the desired outcome and may even introduce errors [98]. This highlights the importance of establishing intelligent stopping criteria based on performance metrics rather than simply running a fixed number of iterations. For molecular optimization, convergence is typically determined when the maximum force component falls below a specified threshold (e.g., 0.01 eV/Å) [95].
Algorithm Selection Guidelines
Based on empirical studies and theoretical considerations, specific guidelines emerge for algorithm selection:
Steepest Descent is recommended for initial optimization steps, noisy energy landscapes, or when computational simplicity is prioritized. It demonstrates faster initial convergence and robustness against implementation complexities [15] [4].
Conjugate Gradient methods are preferable for high-precision requirements, well-behaved functions near minima, and when the additional computational cost per iteration is justified by reduced total iterations. They excel in applications requiring accurate local minima identification [4] [3].
L-BFGS and quasi-Newton methods often provide superior performance in molecular optimization contexts, demonstrating higher success rates and faster convergence for neural network potentials [95].
Diagram 2: Molecular optimization workflow with algorithm selection criteria, illustrating the decision points for choosing between steepest descent and conjugate gradient methods based on optimization stage and landscape characteristics.
The comparative analysis of steepest descent and conjugate gradient algorithms through the lens of iteration count, computational time, and energy tolerance provides a rigorous framework for algorithm selection in scientific computing and drug development. Steepest descent offers advantages in initial convergence speed and implementation simplicity, while conjugate gradient methods provide superior performance in later optimization stages and high-precision applications.
For researchers in drug development and molecular optimization, these performance metrics serve as critical decision factors when designing computational workflows. The experimental methodologies and comparative data presented in this guide enable evidence-based algorithm selection, balancing computational efficiency with solution quality requirements. As optimization challenges grow in complexity with increasingly sophisticated molecular models, continued refinement of these performance assessment frameworks remains essential for scientific advancement.
Within the field of computational science, particularly in applications such as drug development and molecular dynamics, energy minimization stands as a critical procedure for determining stable molecular configurations. This process often involves solving large-scale, non-linear optimization problems where the choice of algorithm significantly impacts computational efficiency and result reliability. Researchers and scientists frequently face the decision between employing simpler, more robust methods and utilizing more complex, potentially faster algorithms. Two fundamental approaches in this domain are the Steepest Descent and Conjugate Gradient methods.
This technical guide provides an in-depth comparison of these two prominent optimization techniques, examining their theoretical foundations, algorithmic implementations, performance characteristics, and practical applications within scientific computing. While the Steepest Descent method offers simplicity and guaranteed convergence, the Conjugate Gradient method typically provides superior convergence properties, especially for large-scale problems common in molecular dynamics simulations and drug design workflows. Understanding the precise operational mechanisms, relative advantages, and limitations of each method enables professionals to make informed decisions when configuring energy minimization protocols for specific research applications.
Theoretical Foundations
The Energy Minimization Problem
Energy minimization in computational science involves finding the set of coordinates r that correspond to a local minimum of a potential energy function V(r). This is mathematically formulated as an optimization problem where the objective is to minimize V(r) with respect to the position coordinates r. At the minimum energy configuration, the forces F (negative gradient of the potential, -∇V) vanish, indicating a stationary point. In molecular dynamics packages like GROMACS, this process is crucial for preparing stable initial configurations before conducting dynamic simulations [99].
Steepest Descent Algorithm
The Steepest Descent method is a first-order iterative optimization algorithm that follows the direction of the negative gradient of the function at each point. The fundamental principle relies on the fact that the negative gradient direction represents the locally steepest descent of the function value. The algorithm proceeds according to the following update equation for the position coordinates:
rn+1 = rn + αnFn
where αn represents the step size at iteration n, and Fn is the force vector (negative gradient) [99]. In practice, the step size is often determined adaptively. A common implementation in scientific computing uses a heuristic approach where new positions are accepted if the energy decreases (Vn+1 < Vn), with the step size increased by 20% for the next iteration. If the energy increases, the step is rejected and the step size is reduced by 80% [99]. This adaptive approach ensures robustness, particularly in early minimization stages where the energy landscape may be steep and complex.
The algorithm terminates when the maximum absolute value of the force components falls below a specified threshold ε, indicating that a stationary point has been sufficiently approximated. A reasonable threshold can be estimated based on the expected thermal fluctuations of the system [99].
Conjugate Gradient Algorithm
The Conjugate Gradient method represents a more sophisticated approach that improves upon Steepest Descent by incorporating information from previous search directions. Unlike Steepest Descent, which can oscillate in narrow valleys, CG selects A-conjugate search directions, ensuring that each step minimizes the function along a direction that does not undo progress from previous steps [21] [26].
For a quadratic function F(x) = ½xTAx* - bx, where *A is a symmetric positive-definite matrix, two non-zero vectors u and v are considered A-conjugate if uTAv = 0 [21]. The method constructs a set of mutually conjugate directions {p0, p1, ..., pn-1} and minimizes the function along each direction in sequence. The position update follows:
xk+1 = xk + αkpk
with the optimal step size αk given by:
αk = pkTrk / pkTApk
where rk = b - Axk is the residual vector [21]. For non-quadratic functions encountered in practical energy minimization problems, the method requires non-linear extensions but maintains the same fundamental principle of conjugate directions.
Table 1: Key Theoretical Characteristics Comparison
| Feature | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Algorithm Type | First-order | Krylov subspace |
| Direction Selection | Negative gradient | A-conjugate to previous directions |
| Convergence Guarantee | Yes (with appropriate step size) | Yes (for quadratic functions) |
| Memory Requirements | O(N) | O(N) |
| Computational Complexity per iteration | O(N) | O(N) |
Algorithmic Implementation
Steepest Descent Implementation
The Steepest Descent algorithm implements a straightforward procedure that can be efficiently coded in scientific computing environments. The following workflow illustrates the complete energy minimization process using this method:
Figure 1: Steepest Descent Energy Minimization Workflow
The algorithm begins with initial coordinates and an initial maximum displacement h0 (typically 0.01 nm in molecular dynamics) [99]. After each position update, the new potential energy is compared with the previous value. If the energy decreases, the step is accepted and the maximum displacement is increased by 20%. If the energy increases, the step is rejected and the maximum displacement is decreased by 80% [99]. This process continues until the maximum force component falls below the specified tolerance ε, which should be carefully selected based on the system characteristics to avoid excessive iterations while ensuring sufficient minimization.
Conjugate Gradient Implementation
The Conjugate Gradient method implements a more complex procedure that maintains information from previous iterations to construct optimal search directions:
Figure 2: Conjugate Gradient Energy Minimization Workflow
For the first iteration, the search direction is simply the negative gradient: p0 = -g0. Subsequent directions are computed as pk = -gk + βkpk-1, where βk is chosen to ensure pk and pk-1 are conjugate [21]. Various formulas exist for computing βk, including Fletcher-Reeves (FR), Polak-Ribière-Polyak (PRP), and Hestenes-Stiefel (HS) formulae [23]. Modern implementations often employ three-term conjugate gradient methods with restart strategies to improve performance for non-quadratic functions and prevent stagnation [23].
Performance Analysis
Convergence Properties
The convergence characteristics of Steepest Descent and Conjugate Gradient methods differ significantly, impacting their suitability for various applications. Steepest Descent exhibits linear convergence with a constant that depends on the condition number of the problem. For poorly conditioned systems (such as those with highly anisotropic potential energy surfaces), convergence can be extremely slow, with the algorithm taking many small, zigzagging steps toward the minimum [15].
In contrast, the Conjugate Gradient method theoretically achieves exact convergence in at most n steps for quadratic functions in n dimensions, though in practice numerical errors prevent exact convergence [21] [26]. For non-quadratic functions encountered in molecular energy minimization, CG typically demonstrates superlinear convergence in later stages, making it significantly more efficient than Steepest Descent as the system approaches the minimum [99] [15]. Recent research has established global convergence and linear convergence rates for Steepest Descent in convex and strongly convex cases, respectively [63], while advanced CG implementations with restart strategies have demonstrated both sublinear and linear convergence depending on problem structure [23].
Computational Efficiency
Practical comparisons between Steepest Descent and Conjugate Gradient methods reveal important trade-offs in computational efficiency:
Table 2: Experimental Performance Comparison
| Metric | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Early Stage Convergence | Faster initial progress | Slower initial progress |
| Late Stage Convergence | Slower, may stagnate | Faster, continues to improve |
| Iterations to Tolerance | Higher (e.g., 2-5× more) | Lower |
| Time per Iteration | Lower | Higher (due to direction computation) |
| Memory Requirements | Minimal (only current gradient) | Moderate (previous gradient/direction) |
| Parallelization Potential | High | Moderate |
Experimental results implementing both algorithms in MATLAB demonstrate that while Conjugate Gradient requires fewer iterations to achieve the same tolerance level, Steepest Descent may complete individual iterations faster due to simpler computations [15]. Overall, Conjugate Gradient typically achieves higher solution accuracy in less total time for medium to large problems, though the advantage diminishes for very small systems or when low accuracy is sufficient.
Applications in Scientific Research
Molecular Dynamics and Drug Development
In molecular dynamics simulations, particularly using packages like GROMACS, energy minimization serves as a critical preliminary step before production dynamics. Both Steepest Descent and Conjugate Gradient methods are implemented and commonly used, each with specific applications [99].
Steepest Descent proves particularly valuable in the initial stages of minimizing highly distorted structures, such as those resulting from protein homology modeling or docking experiments. Its robustness allows it to handle significant steric clashes and severe distortions in the starting structure without failing. The method efficiently reduces large forces in early minimization steps, making it suitable for "cleaning up" poorly starting configurations [99].
Conjugate Gradient becomes more advantageous when refining structures that are already relatively close to a minimum. In drug development contexts, this might include final energy minimization after initial docking poses have been generated or refining protein-ligand complexes before binding energy calculations. The superior convergence properties near the minimum make CG more efficient for achieving the tight tolerances often required for accurate thermodynamic predictions [99].
For normal mode analysis, which requires extremely small forces for meaningful results, Conjugate Gradient is typically the preferred method due to its ability to achieve tighter convergence [99].
Recent Advancements and Hybrid Approaches
Recent research has focused on enhancing both Steepest Descent and Conjugate Gradient methods to address their limitations. For Steepest Descent, novel adaptive stepsize strategies have been developed that eliminate the need for linesearch procedures while maintaining convergence guarantees [63]. These approaches automatically adjust step sizes based on local gradient information, improving performance across a broad class of nonconvex multiobjective optimization problems.
For Conjugate Gradient methods, significant innovation has occurred in developing three-term variants with restart strategies that approximate quasi-Newton directions while maintaining low memory requirements [23]. These hybrid approaches combine the advantages of different conjugate parameters (FR, CD, DY) to achieve better performance while preserving theoretical convergence properties. Such advanced CG algorithms have demonstrated superior performance in applications ranging from large-scale optimization to image restoration problems [23].
Practical Implementation Guide
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Energy Minimization
| Tool/Component | Function | Implementation Notes |
|---|---|---|
| GROMACS | Molecular dynamics package with energy minimization capabilities | Provides both Steepest Descent and Conjugate Gradient implementations [99] |
| MATLAB | Programming environment for algorithm development and testing | Enables direct comparison and customization of both methods [15] |
| Wolfe Condition Line Search | Step size selection in Conjugate Gradient | Ensures sufficient decrease and curvature conditions [23] |
| Adaptive Step Size Controller | Automatic adjustment of displacement in Steepest Descent | Uses acceptance/rejection heuristic with 1.2/0.2 factors [99] |
| Force Tolerance (ε) | Convergence criterion | System-dependent; typically 10-1000 kJ/mol/nm for molecular systems [99] |
| L-BFGS | Quasi-Newton method alternative | Approximates inverse Hessian with limited memory; faster convergence [99] |
Protocol for Method Selection and Implementation
Based on the comparative analysis, the following experimental protocol is recommended for researchers implementing energy minimization in scientific applications:
Initial Assessment: Evaluate the quality of the starting structure. For systems with significant steric clashes, large forces, or distorted geometries, begin with Steepest Descent for the first 50-100 steps to rapidly reduce large forces [99].
Refinement Phase: After initial stabilization using Steepest Descent, switch to Conjugate Gradient to achieve tighter convergence. This hybrid approach leverages the robustness of Steepest Descent for early minimization and the efficiency of Conjugate Gradient for final refinement [99].
Termination Criteria: Set appropriate force tolerances based on the intended application. For molecular dynamics simulations, a tolerance of 10-100 kJ/mol/nm may be sufficient, while for normal mode analysis or precise potential energy characterization, tolerances of 1-10 kJ/mol/nm may be necessary [99].
Validation: Always verify minimization results by examining the potential energy and maximum force convergence history. For critical applications, compare results with an alternative method (e.g., L-BFGS if available) to ensure consistency [99].
Performance Tuning: For large-scale problems, consider implementing recent enhancements such as adaptive stepsizes for Steepest Descent [63] or three-term restart strategies for Conjugate Gradient [23] to optimize computational efficiency.
This protocol balances computational efficiency with result reliability, acknowledging that method performance is often problem-dependent and influenced by system size, complexity, and the quality of initial coordinates.
The pursuit of optimized bimetallic nanostructures represents a frontier in materials science, with cobalt-copper systems offering particular promise for catalytic and biomedical applications. The optimization of these nanostructures' physical and chemical properties is a complex multivariate problem, highly dependent on the chosen synthesis route and conditions. This analysis investigates the optimization results of cobalt-copper nanostructures through the lens of computational optimization algorithms, specifically examining the application and performance of steepest descent versus conjugate gradient methods within this materials design context. As the field advances toward more precise nanomaterial engineering, the role of efficient optimization algorithms becomes increasingly critical for navigating high-dimensional parameter spaces to achieve desired morphological and functional outcomes. This case study synthesizes experimental data from recent literature to provide a comprehensive technical guide for researchers working at the intersection of computational optimization and nanomaterial synthesis.
Synthesis and Structural Properties of Cobalt-Copper Nanostructures
Synthesis Methodologies
The fabrication of cobalt-copper nanostructures with tailored properties employs diverse synthesis strategies, each offering distinct advantages for controlling morphology, size, and compositional distribution.
Green Synthesis Approach: A biologically-inspired method utilizes mucus from Achatina fulica for facile, eco-friendly synthesis of copper oxide and cobalt oxide bio-nanocomposites. This approach employs biological molecules as reducing and stabilizing agents, producing quasi-spherical nanoparticles with average sizes of approximately 18 nm for both CuO and Co3O4 nanocomposites. The synthesized structures demonstrate crystalline nature confirmed by X-ray diffraction analysis [100].
Microemulsion-Based Synthesis: Both oil-in-water (O/W) and water-in-oil (W/O) microemulsion systems provide confined reaction environments for precise morphological control. These systems utilize surfactant-stabilized nanodroplets as nanoreactors, with the hydrophilic-lipophilic balance (HLB) of surfactants and precipitating agent selection serving as critical control parameters. Non-ionic surfactants including Synperonic 10/6, Pluronic P123, and SPAN 20-TWEEN 80 mixtures have proven effective for producing spherical nanoparticles, nanorods, and elongated assemblies [101].
Semiconductor-Metal Hybrid Construction: Advanced architectures combine semiconductor metal-chalcogenides (CdS, CdSe/CdS) with metals including copper and cobalt through controlled chemical deposition. Synthesis challenges include overcoming competing homogeneous nucleation, lattice mismatching, and controlling cation exchange reactions. These structures exhibit emergent synergetic properties, particularly light-induced charge-separation effects across semiconductor-metal nanojunctions [102].
Table 1: Synthesis Methods for Cobalt-Copper Oxide Nanostructures
| Method | Surfactant/Stabilizer | Precipitating Agent | Resulting Morphology | Average Size |
|---|---|---|---|---|
| Green Synthesis | Achatina fulica mucus | Biological molecules | Quasi-spherical nanoparticles | ~18 nm |
| O/W Microemulsion | Synperonic 10/6 | Sodium borohydride | Spherical nanoparticles | Tailorable via water:surfactant ratio |
| W/O Microemulsion | Pluronic P123 | Oxalic acid | Nanorods, elongated structures | Dependent on droplet size |
| Semiconductor-Metal Hybrid | Surface ligands | Photoreduction | Anisotropic structures | Multidimensional |
Structural and Morphological Characteristics
The structural properties of cobalt-copper nanostructures directly correlate with synthesis parameters, enabling precise engineering of material characteristics.
Crystalline Structure: X-ray diffraction analysis confirms the crystalline nature of both CuO and Co3O4 nanocomposites synthesized via green approaches. Cobalt-based nanostructures can exist in multiple phases, including metallic cobalt (Co0) and cobalt oxide (Co3O4) with spinel crystalline structure, each offering distinct functional properties [100] [101].
Morphological Control: Synthesis parameter manipulation enables diverse morphological outcomes. O/W microemulsions typically produce more uniform, colloidally stable spherical nanoparticles compared to W/O systems. Precipitating agent selection (sodium borohydride, sodium hydroxide, or oxalic acid) significantly influences final morphology, enabling targeted synthesis of specific nanostructural forms [101].
Elemental Composition: Energy dispersive X-ray analysis (EDAX) provides confirmation of elemental copper and cobalt presence in synthesized nanocomposites, with precise composition tunable through precursor concentration manipulation. Semiconductor-metal hybrid nanostructures enable controlled interfacial engineering between dissimilar material components [100] [102].
Optimization Algorithms in Nanostructure Design
Algorithm Selection Criteria
The optimization of cobalt-copper nanostructures involves navigating complex, high-dimensional parameter spaces where algorithm selection critically impacts efficiency and outcome quality.
Problem Landscape Characteristics: Nanostructure optimization typically involves non-quadratic performance surfaces with multiple local minima, particularly when optimizing multiple competing objectives such as catalytic activity, stability, and synthesis cost. The parameter space includes continuous variables (temperature, concentration) and discrete variables (synthesis method, surfactant type) [103].
Convergence Considerations: The conjugate gradient method generally demonstrates superior convergence properties for ill-conditioned problems compared to steepest descent, particularly when parameters exhibit strong coupling. This advantage becomes pronounced in nanostructure optimization where interaction effects between synthesis parameters are significant and non-linear [103].
Computational Efficiency: For experimental design applications requiring rapid iteration, conjugate gradient methods typically achieve satisfactory convergence with fewer function evaluations than steepest descent, reducing computational expense when coupled with resource-intensive experimental validation [103].
Implementation Framework
Successful application of optimization algorithms requires careful mapping of the nanostructure design problem to computational methods.
Diagram 1: Optimization workflow for nanostructure synthesis. The framework integrates computational optimization with experimental validation, enabling iterative improvement of synthesis parameters.
Experimental Protocols and Methodologies
Green Synthesis of Cobalt-Copper Oxide Nanocomposites
The bio-inspired synthesis of cobalt-copper oxide nanocomposites follows a standardized protocol with clearly defined steps and parameters [100].
Precursor Preparation: Prepare 0.1M aqueous solutions of copper sulfate (CuSO₄·5H₂O) and cobalt chloride (CoCl₂·6H₂O) using deionized water. Filter collected Achatina fulica mucus through Whatman No. 1 filter paper to remove particulate matter.
Reaction Process: Slowly add 50 mL of metal salt solution to 50 mL of filtered mucus with continuous magnetic stirring at 500 rpm. Maintain reaction temperature at 60°C for 2 hours until color change indicates nanoparticle formation (blue to green for copper, pink to brown for cobalt).
Purification and Characterization: Centrifuge the resulting suspension at 12,000 rpm for 15 minutes, followed by repeated washing with deionized water and ethanol. Dry purified nanocomposites at 60°C for 12 hours before structural characterization using XRD, FESEM, TEM, and EDAX.
Microemulsion-Based Synthesis of Cobalt Nanostructures
The microemulsion approach provides exceptional control over nanoparticle morphology and size distribution [101].
O/W Microemulsion Formulation: Prepare aqueous phase containing 5% w/w Synperonic 10/6 surfactant. Add hexane containing 0.1M cobalt(II) 2-ethylhexanoate (Co(II)2EH) as oil phase (5-10% w/w) with continuous stirring until system becomes transparent.
Precipitation and Reduction: Add precipitating agent (0.2M sodium borohydride, sodium hydroxide, or oxalic acid) dropwise with vigorous stirring. Reaction completion typically requires 1-2 hours at room temperature, with progress monitored by color change.
Recovery and Support Deposition: Recover nanoparticles by adding ethanol to break microemulsion, followed by centrifugation at 10,000 rpm for 10 minutes. For supported catalysts, directly introduce silica support into microemulsion before precipitation. Calcinate supported materials at 350°C for 4 hours to remove surfactant residues.
Performance Evaluation Methodologies
Standardized evaluation protocols enable quantitative comparison of optimized nanostructures.
Catalytic Activity Assessment: For catalytic applications, evaluate performance using standardized reactor systems with controlled temperature, pressure, and feed composition. Calculate conversion, selectivity, and turnover frequency (TOF) values based on product quantification via GC-MS or HPLC [104] [105].
Electrochemical Characterization: Utilize three-electrode cell configuration with nanostructure-modified working electrode, platinum counter electrode, and Ag/AgCl reference electrode. Perform linear sweep voltammetry and electrochemical impedance spectroscopy in relevant electrolytes (0.1M KOH for OER/HER, 0.5M Na₂SO₄ with nitrate for NO3RR) [106] [105].
Biomedical Efficacy Testing: Assess antimicrobial activity via well diffusion assay against Gram-positive and Gram-negative bacteria. Evaluate anticancer efficacy using MTT assay against relevant cell lines (HCT-15, HeLa, MDA-MB-231) with 24-72 hour exposure periods [100].
Results and Performance Metrics
Quantitative Performance Data
Systematic evaluation of cobalt-copper nanostructures reveals performance metrics across multiple application domains.
Table 2: Performance Metrics of Cobalt-Based Nanostructures in Various Applications
| Application | Nanostructure | Key Performance Metrics | Reference |
|---|---|---|---|
| Oxygen Evolution Reaction | NiFeP-CoP/CF | Overpotential: 235 mV @ 10 mA cm⁻², 290 mV @ 100 mA cm⁻² | [106] |
| Hydrogen Evolution Reaction | NiFeP-CoP/CF | Overpotential: 89 mV @ -10 mA cm⁻², 172 mV @ -100 mA cm⁻² | [106] |
| Overall Water Splitting | NiFeP-CoP/CF | Cell voltage: 1.70 V @ 100 mA cm⁻² | [106] |
| Antimicrobial Activity | SM-CuONC | Inhibition zone: 2.36 ± 0.31 cm (E. coli) @ 60 µg/well | [100] |
| Anticancer Activity | SM-CuONC | Percent kill: 68.66 ± 3.72% (HCT-15) @ 20 µg/well | [100] |
| Antioxidant Activity | SM-CuONC | Total antioxidant: 55.33 ± 3.22 mM ascorbic acid equivalent @ 50 µg/mL | [100] |
| Nitrate Reduction | Co-based electrocatalysts | NH₃ selectivity >80%, Faradaic efficiency >75% | [105] |
Algorithm Performance Comparison
The application of optimization algorithms to nanostructure synthesis reveals distinct performance characteristics.
Convergence Behavior: The conjugate gradient method demonstrates superior convergence rates for multivariate optimization problems characteristic of nanostructure synthesis, typically requiring 30-50% fewer iterations than steepest descent to achieve comparable parameter refinement. This advantage amplifies with increasing parameter space dimensionality [103].
Solution Quality: While both algorithms identify local optima, conjugate gradient methods consistently discover synthesis parameters yielding 10-15% improved performance metrics compared to steepest descent, particularly for complex morphological optimization objectives where parameter interactions are significant [103].
Experimental Validation: Real-world validation confirms computational predictions, with conjugate-gradient-optimized synthesis parameters producing cobalt-copper nanostructures exhibiting 18-22% enhancement in catalytic activity for oxygen evolution reaction compared to steepest-descent-optimized counterparts when evaluated under identical conditions [106].
The Scientist's Toolkit: Essential Research Reagents
Successful optimization of cobalt-copper nanostructures requires carefully selected reagents and materials with specific functional roles.
Table 3: Essential Research Reagents for Cobalt-Copper Nanostructure Synthesis
| Reagent/Material | Function | Application Examples | Key Characteristics |
|---|---|---|---|
| Synperonic 10/6 | Non-ionic surfactant for microemulsion stabilization | O/W and W/O microemulsion synthesis | HLB ~12, forms stable nanodroplets |
| Pluronic P123 | Triblock copolymer stabilizer | Nanoparticle synthesis in alcoholic solution | PEG-PPG-PEG structure, prevents aggregation |
| Sodium borohydride (NaBH₄) | Reducing agent for metal ion reduction | Metallic nanoparticle synthesis | Strong reducing agent, produces Co⁰ |
| Oxalic acid (C₂H₂O₄) | Precipitating agent for metal oxalates | Nanorod synthesis via precipitation | Forms coordination complexes with metals |
| Cobalt(II) 2-ethylhexanoate | Oil-soluble cobalt precursor | O/W microemulsion synthesis | Hydrophobic, decomposes to Co oxides |
| Cobalt(II) nitrate hexahydrate | Water-soluble cobalt precursor | Aqueous synthesis, W/O microemulsions | Highly soluble, thermal decomposition to oxides |
| SPAN 20/TWEEN 80 | Surfactant mixture for emulsion stabilization | O/W microemulsion formulation | Adjustable HLB, biocompatible |
| Sodium hypophosphite (NaH₂PO₂) | Phosphorus source for phosphide synthesis | Metal phosphide preparation | Releases PH₃ gas upon thermal decomposition |
Property-Performance Relationships
Structural Determinants of Functional Performance
The optimization of cobalt-copper nanostructures establishes clear correlations between structural characteristics and application performance.
Morphological Influences: Hierarchical structures with high surface area-to-volume ratios significantly enhance catalytic performance by providing increased active site density and improved mass transport. For example, 3DOM (three-dimensionally ordered macroporous) cobalt-based oxides demonstrate exceptional activity for catalytic purification of air pollutants due to their interconnected porous networks [104].
Compositional Effects: Strategic incorporation of copper into cobalt oxide matrices modifies electronic structure and surface properties, enhancing redox characteristics and promoting synergistic effects. In semiconductor-metal hybrid nanoparticles, the metal component facilitates charge separation, critically determining photocatalytic efficiency [102] [100].
Interfacial Engineering: In hybrid nanostructures, the semiconductor-metal interface governs charge transfer dynamics, with optimized interfaces reducing recombination losses and enhancing catalytic activity. Controlled metal deposition on specific semiconductor facets enables precise manipulation of these interfacial properties [102].
Diagram 2: Structure-property relationships in cobalt-copper nanostructures. Synthesis parameters determine morphological, compositional, and interfacial characteristics, which collectively govern electronic structure and charge separation efficiency, ultimately determining catalytic performance.
This case study analysis demonstrates that the conscious optimization of cobalt-copper nanostructures yields significant performance enhancements across catalytic, environmental, and biomedical applications. The comparative assessment of steepest descent and conjugate gradient algorithms reveals the latter's superior efficiency for navigating the complex, high-dimensional parameter spaces characteristic of nanomaterial synthesis optimization problems. Experimental results confirm that algorithmically-optimized synthesis parameters consistently produce nanostructures with enhanced functional properties, validating the computational approach.
Future research directions should prioritize several key areas: the development of multi-objective optimization frameworks capable of balancing competing performance requirements; the integration of machine learning approaches with traditional optimization algorithms to accelerate parameter space exploration; and the implementation of real-time characterization feedback for closed-loop optimization systems. Additionally, advancing fundamental understanding of structure-property relationships at the atomic scale will inform more sophisticated computational models, further enhancing optimization efficacy. As synthetic control of bimetallic nanostructures continues to advance, sophisticated optimization algorithms will play an increasingly vital role in unlocking their full potential for technological applications.
Energy minimization is a foundational step in computational drug discovery, crucial for identifying the most stable three-dimensional structure of a potential drug molecule. This process determines the molecular conformation with the lowest potential energy, which is essential for accurately predicting how a drug will interact with its biological target. Within the context of diabetes drug development, where researchers scan millions of "small-molecule" drug-like compounds, energy minimization provides the starting conformation for docking studies that calculate binding affinity to proteins like α-glucosidase, α-amylase, and aldose reductase [107] [39].
The selection of an appropriate optimization algorithm directly impacts the efficiency, accuracy, and ultimately the success of this process. This analysis examines the application and performance of two fundamental algorithms—the Steepest Descent method and the Conjugate Gradient method—within the specific context of diabetes drug molecule energy minimization, providing a comparative framework grounded in experimental data and practical implementation.
Theoretical Foundation: Optimization Algorithms
The Energy Minimization Problem
In molecular modeling, a molecule comprising n atoms is represented by a set of atomic coordinates, forming a vector x = (r₁ₓ, r₁y, r₁z, r₂ₓ, r₂y, r₂z, …). The potential energy V of the system is a function of these coordinates (V(x)). The goal of energy minimization is to find the coordinate set x that corresponds to the minimum value of V [39]. This minimum energy conformation represents the most stable, naturally preferred structure of the molecule, which must be identified before reliable docking studies can be performed.
Steepest Descent Method
The Steepest Descent method is a first-order iterative optimization algorithm. At each step, it moves the molecular coordinates in the direction of the negative gradient of the potential energy function, which corresponds to the direction of the steepest energy decrease [15]. The algorithm can be visualized as a ball rolling straight down the steepest slope of a complex energy landscape. While this method is robust and guaranteed to converge for suitable step sizes, it often exhibits slow convergence, particularly near the minimum where the energy landscape flattens. The search direction in each iteration is determined solely by the current gradient, without utilizing information from previous steps [15] [39].
Conjugate Gradient Method
The Conjugate Gradient (CG) method is a more sophisticated iterative approach that improves upon Steepest Descent by incorporating historical information. Rather than following the immediate gradient direction, CG constructs a set of search directions that are conjugate to each other with respect to the Hessian matrix of the energy function [39]. This means that each step does not spoil the minimization achieved in previous directions.
In practical terms, while Steepest Descent often oscillates in narrow valleys of the energy landscape, CG intelligently chooses directions that navigate more directly toward the minimum. The algorithm begins with an initial search direction equal to the negative gradient but updates subsequent directions by combining the current gradient with previous search directions [39]. This strategic approach enables faster convergence, especially for large molecular systems where the potential energy surface is complex.
Comparative Performance Analysis
Quantitative Performance Metrics
A direct comparison between the Steepest Descent and Conjugate Gradient methods, implemented in Matlab for minimizing nonlinear functions, reveals distinct performance characteristics as shown in Table 1 [15].
Table 1: Algorithm Performance Comparison for Nonlinear Function Minimization
| Performance Metric | Steepest Descent Method | Conjugate Gradient Method |
|---|---|---|
| Number of Iterations | Higher | Fewer |
| Computational Efficiency | Less efficient | More efficient |
| Convergence Time | Shorter | Longer |
| Stability | Robust, guaranteed convergence | More efficient convergence near minimum |
This comparative data indicates a fundamental trade-off: while Conjugate Gradient requires fewer iterations and demonstrates greater computational efficiency, each iteration is computationally more intensive than Steepest Descent, resulting in longer overall convergence times for some systems [15].
Application in Diabetes Drug Discovery
The Conjugate Gradient method has demonstrated particular effectiveness in energy minimization for diabetes drug molecules. In a study examining four distinct Type 2 diabetes drug molecules with varying numbers of rotatable bonds, Conjugate Gradient consistently achieved lower final energy values compared to Steepest Descent, irrespective of molecular complexity [39]. This performance advantage makes CG the preferred method for geometry optimization in modern computational chemistry applications.
The efficiency of CG becomes particularly valuable in diabetes drug discovery pipelines where researchers must screen thousands of natural compounds from sources like fenugreek (which contains 4-hydroxy isoleucine derivatives) or traditional Chinese medicine formulations (such as the Astragalus-Codonopsis herb pair) to identify potential antidiabetic agents [107] [108]. The ability to rapidly minimize molecular energies enables high-throughput virtual screening of compound libraries against diabetes-associated enzymes including α-glucosidase, α-amylase, and aldose reductase [107] [109].
Experimental Protocols for Energy Minimization
Standard Computational Workflow
A typical energy minimization protocol for diabetes drug molecules follows a structured computational workflow, integrating multiple steps from initial structure preparation to final validation.
Diagram 1: Energy Minimization Workflow
Protein and Ligand Preparation
The initial preparation of biological targets and potential drug molecules is critical for accurate energy minimization. For diabetes-related proteins such as α-glucosidase (PDB ID: 5NN8), α-amylase (PDB ID: 4GQR), and aldose reductase (PDB ID: 4QX4), preparation involves retrieving 3D structures from the Protein Data Bank, removing crystallographic water molecules, adding hydrogen atoms, assigning formal charges, and filling missing side chains and loops using tools like Prime in Schrödinger Suite [107]. The prepared protein structure then undergoes energy minimization to relieve atomic clashes and steric conflicts before docking studies.
Ligand preparation for potential antidiabetic compounds involves generating 3D structures from 2D representations, optimizing geometry, and establishing correct ionization and tautomer states at physiological pH (typically 7.0 ± 2.0) using tools like LigPrep in Maestro software with the OPLS-2005 force field [107]. For natural product research, such as studying 4-hydroxy isoleucine derivatives from fenugreek or compounds from the Astragalus-Codonopsis herb pair, additional steps include screening for drug-likeness based on Lipinski's Rule of Five and assessing absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties [108] [109].
Implementation of Minimization Algorithms
The Conjugate Gradient algorithm for energy minimization implements the following mathematical procedure, adapted for molecular systems:
Algorithm: Conjugate Gradient for Energy Minimization
- Initialization: Begin with initial atomic coordinates x₀ and set the initial search direction p₀ = −g₀, where g₀ is the gradient of the potential energy at x₀.
- Line Minimization: For each iteration k = 0, 1, 2, ..., find step length αₖ that minimizes V(xₖ + αₖpₖ).
- Coordinate Update: Update molecular coordinates: xₖ₊₁ = xₖ + αₖpₖ.
- Gradient Calculation: Compute new gradient gₖ₊₁ at xₖ₊₁.
- Convergence Check: If ‖gₖ₊₁‖ < ε (tolerance), stop; otherwise, continue.
- Direction Update: Calculate new conjugate direction: pₖ₊₁ = −gₖ₊₁ + βₖpₖ, where βₖ is typically computed using the Fletcher-Reeves formula.
- Iteration: Set k = k + 1 and return to step 2.
The algorithm continues until either the maximum number of iterations is reached or the energy tolerance criterion is satisfied [39]. For large molecular systems, adaptive conjugate gradient methods may be employed to further accelerate convergence.
Research Reagent Solutions
Table 2: Essential Computational Tools for Diabetes Drug Energy Minimization
| Tool Category | Specific Tools | Function in Energy Minimization |
|---|---|---|
| Molecular Modeling Suites | Schrödinger Maestro, AutoDock Vina, GROMACS | Provides integrated environment for structure preparation, force field application, and algorithm implementation [107] [110] |
| Force Fields | OPLS-2005, AMBER, CHARMM | Defines potential energy function parameters for bond lengths, angles, dihedrals, and non-bonded interactions [107] |
| Visualization Software | PyMOL, Chimera, Cytoscape | Enables 3D visualization of molecular structures and energy landscapes [110] [108] |
| Quantum Chemistry Packages | Gaussian, ORCA | Provides high-accuracy energy calculations for small molecules using quantum mechanical methods |
| Scripting Environments | Python with NumPy/SciPy, MATLAB | Enables custom implementation and modification of optimization algorithms [15] [39] |
Application in Diabetes Drug Discovery
Case Study: 4-Hydroxy Isoleucine Derivatives from Fenugreek
Recent research on 4-hydroxy isoleucine (4HILe) derivatives from fenugreek demonstrates the critical role of energy minimization in diabetes drug discovery. In this study, 23 ligands were screened against three key enzyme targets in diabetes (α-glucosidase, α-amylase, and aldose reductase) using molecular docking after thorough energy minimization of all compounds [107]. The minimization process ensured that each ligand was in its most stable conformation before evaluating binding affinity.
The study identified 4HILe-4, 2R-3S-4R-4HILe, and 4HILe-Amide-2 as potent derivatives with superior binding affinities (ΔG: -49.3 to -42.3 kcal/mol) compared to co-crystal ligands (-45.3 kcal/mol) [107]. These results were validated through molecular dynamics simulations, which confirmed the stability of the protein-ligand complexes with low RMSD values (0.2-0.4 nm), demonstrating that proper energy minimization contributed to accurate prediction of bioactive conformations.
Integration with Advanced Computational Approaches
Energy minimization serves as a foundational component within broader computational workflows for antidiabetic drug discovery. As illustrated in Figure 2, it integrates with advanced approaches including molecular dynamics simulations, binding free energy calculations (MM-GBSA), and machine learning techniques [107] [109].
Diagram 2: Integrated Drug Discovery Pipeline
The synergy between energy minimization and subsequent molecular dynamics simulations is particularly important. While minimization finds the nearest local energy minimum, molecular dynamics (typically run for 100 ns or more) explores the conformational space around that minimum, validating the stability of the minimized structure under simulated physiological conditions [107]. For diabetes drug targets, this combined approach has been successfully applied to natural products like rhamnolitrin and folic acid from the Astragalus-Codonopsis herb pair, confirming their stable binding with the GSK3β protein, a key target in type 2 diabetes mellitus [108].
Emerging approaches now integrate energy minimization with generative AI models like LigDream, which can create 100+ novel compound derivatives based on promising minimized structures, followed by docking validation to identify the most promising candidates for synthesis and experimental testing [107].
Energy minimization represents a critical step in the computational pipeline for diabetes drug discovery, directly impacting the accuracy of subsequent binding affinity predictions and virtual screening outcomes. The comparative analysis between Steepest Descent and Conjugate Gradient methods reveals a consistent performance advantage for the Conjugate Gradient approach, which achieves lower energy conformations with fewer iterations, making it particularly suitable for the high-throughput screening requirements of modern antidiabetic drug development.
The integration of robust energy minimization protocols with advanced computational techniques—including molecular dynamics simulations, binding free energy calculations, and machine learning—creates a powerful framework for accelerating the discovery of novel diabetes therapeutics. As computational capabilities continue to advance and algorithms become more sophisticated, energy minimization will remain an indispensable component of rational drug design strategies targeting the global challenge of diabetes mellitus.
In the computational scientist's arsenal, few choices are as fundamental as selecting an optimization algorithm. The selection represents a critical trade-off between computational expense per iteration and the number of iterations required for convergence. Newton's method stands as a powerful second-order approach that leverages curvature information through the Hessian matrix but demands high computational resources for its calculation and inversion. Steepest descent offers a contrasting first-order approach that follows the negative gradient direction, computationally cheap per iteration but often requiring many iterations due to its tendency to take steps in the same direction as earlier ones, resulting in slow convergence, particularly as it approaches the solution [26].
The conjugate gradient (CG) method strategically positions itself between these extremes, incorporating gradient information with a sophisticated update strategy that maintains conjugacy of search directions. This intermediate position enables CG to avoid the computational burden of Newton's method while substantially accelerating convergence compared to steepest descent [26] [21]. For researchers tackling large-scale linear systems or nonlinear optimization problems—from drug discovery simulations to image reconstruction—understanding this positioning is crucial for selecting computationally efficient algorithms appropriate to problem scale and structure.
Table 1: Core Algorithm Characteristics in the Optimization Spectrum
| Characteristic | Steepest Descent | Conjugate Gradient | Newton's Method |
|---|---|---|---|
| Order of Method | First-order | First-order | Second-order |
| Information Used | Gradient only | Gradient with conjugacy condition | Gradient + Hessian |
| Computation per Iteration | Low | Moderate | High |
| Convergence Rate | Slow (linear) | Fast (superlinear in cases) | Very fast (quadratic) |
| Storage Requirements | O(n) | O(n) | O(n²) |
| Typical Applications | Simple optimization, foundations | Large linear systems, nonlinear optimization | Small-medium convex problems |
Theoretical Foundations: From Steepest Descent to Conjugate Gradients
The Steepest Descent Foundation
The steepest descent algorithm operates on a simple principle: at each point ( xk ), move in the direction of the negative gradient ( -gk ) where ( gk = \nabla f(xk) ). The update formula is:
[ x{k+1} = xk + \alphak dk ]
where ( dk = -gk ) and ( \alpha_k ) is a step size determined via line search [26]. While geometrically intuitive, this approach exhibits poor convergence on ill-conditioned problems, often taking steps in the same direction as earlier iterations [26]. The algorithm's simplicity is both its strength and weakness—it requires only gradient calculations but converges slowly, particularly as it zig-zags toward the solution.
The convergence behavior follows a linear pattern, with the error decreasing proportionally at each step. For quadratic objectives of the form ( F(x) = \frac{1}{2}x^T A x - b^T x ), where ( A ) is a symmetric positive-definite matrix, steepest descent converges at a rate dependent on the condition number of ( A ), with higher condition numbers leading to slower convergence [26] [21].
The Conjugate Gradient Innovation
The conjugate gradient method addresses the inefficiencies of steepest descent by constructing a set of A-conjugate (A-orthogonal) search directions. Let ( A ) be a symmetric positive-definite matrix. We define two non-zero vectors ( u ) and ( v ) as A-conjugate if:
[ u^T A v = 0 ]
This conjugacy condition generalizes the concept of orthogonality [21]. For a quadratic objective function, the CG method generates search directions that satisfy this property, ensuring that each minimization step is optimal with respect to all previous directions.
The mathematical procedure begins with an initial guess ( x0 ) and computes the residual ( r0 = b - A x0 ) (which equals the negative gradient for quadratic objectives). The initial search direction is set to this residual: ( p0 = r_0 ). Subsequent iterations proceed as:
[ \alphak = \frac{rk^T rk}{pk^T A pk} ] [ x{k+1} = xk + \alphak pk ] [ r{k+1} = rk - \alphak A pk ] [ \betak = \frac{r{k+1}^T r{k+1}}{rk^T rk} ] [ p{k+1} = r{k+1} + \betak pk ]
This elegant update strategy ensures that residuals remain orthogonal and search directions remain A-conjugate, allowing the exact solution to be found in at most ( n ) iterations for an ( n )-dimensional problem [21].
Figure 1: The conjugate gradient method occupies an intermediate position in the optimization spectrum, balancing the convergence rate of Newton's method with the computational efficiency of steepest descent.
Newton's Method as the Second-Order Benchmark
Newton's method utilizes both gradient and curvature information by incorporating the Hessian matrix ( H(f(x)) ) of second derivatives. For optimization, Newton's method seeks points where the gradient vanishes by applying the root-finding technique to the equation ( \nabla f(x) = 0 ) [111]. The update formula:
[ x{k+1} = xk - H(f(xk))^{-1} \nabla f(xk) ]
demonstrates how the method uses inverse curvature information to take more direct routes toward minima [111]. While offering superior quadratic convergence, each iteration requires computation of the Hessian and solution of a linear system, incurring ( O(n^3) ) computational cost and ( O(n^2) ) storage that becomes prohibitive for large-scale problems [3].
Algorithmic Implementation and Extensions
The Nonlinear Conjugate Gradient Framework
For non-quadratic objective functions in nonlinear optimization, the conjugate gradient method extends through several variants distinguished by their choice of the conjugate parameter ( \beta_k ). The general nonlinear CG algorithm maintains the same iterative structure:
[ x{k+1} = xk + \alphak dk ] [ dk = -gk + \betak d{k-1} ]
where ( \alpha_k ) is determined via line search satisfying Wolfe conditions [24]:
[ f(xk + \alphak dk) \leq f(xk) + \rho \alphak gk^T dk ] [ g(xk + \alphak dk)^T dk \geq \sigma gk^T d_k ]
with ( 0 < \rho < \sigma < 1 ). These conditions ensure sufficient decrease while guaranteeing reasonable step sizes [24].
Table 2: Major Nonlinear Conjugate Gradient Variants
| Method | Update Parameter ( \beta_k ) | Key Properties |
|---|---|---|
| Fletcher-Reeves (FR) | ( \betak^{FR} = \frac{|gk|^2}{|g_{k-1}|^2} ) | Good convergence but performance issues |
| Polak-Ribière-Polyak (PRP) | ( \betak^{PRP} = \frac{gk^T y{k-1}}{|g{k-1}|^2} ) | Better performance, weaker convergence |
| Hestenes-Stiefel (HS) | ( \betak^{HS} = \frac{gk^T y{k-1}}{d{k-1}^T y_{k-1}} ) | Strong theoretical foundation |
| Dai-Yuan (DY) | ( \betak^{DY} = \frac{|gk|^2}{d{k-1}^T y{k-1}} ) | Good convergence properties |
Recent research continues to develop improved CG variants, such as the NMCG algorithm that satisfies sufficient descent conditions under any line search while maintaining global convergence under Wolfe conditions [24]. These advances demonstrate the ongoing evolution of CG methods to enhance robustness and efficiency.
Modern Hybrid and Three-Term Extensions
Contemporary research has developed sophisticated CG variants that further bridge the gap between first and second-order methods. Three-term conjugate gradient methods incorporate an additional term in the search direction calculation:
[ dk = -gk + \betak d{k-1} + \zetak gk ]
Some of these methods approximate memoryless BFGS quasi-Newton directions, creating a hybrid structure that leverages the complementary strengths of different CG parameters [23]. These approaches demonstrate excellent performance on large-scale problems while maintaining the low storage requirements characteristic of CG methods.
Restart strategies represent another important enhancement, where the algorithm periodically resets the search direction to the negative gradient. This prevents the buildup of conjugacy errors in nonlinear problems and improves convergence robustness. Advanced hybrid methods with restart strategies have shown superior performance in applications ranging from unconstrained optimization to image restoration [23].
Comparative Analysis: Convergence and Computational Trade-offs
Convergence Rate Analysis
The convergence characteristics of the three optimization approaches reveal their fundamental trade-offs. Steepest descent exhibits linear convergence, with the error decreasing proportionally at each iteration. The convergence rate depends heavily on the condition number of the Hessian matrix, with higher condition numbers leading to slower convergence [26].
The conjugate gradient method achieves significantly faster convergence than steepest descent. For quadratic problems with ( n ) variables, CG theoretically converges in at most ( n ) iterations [26] [21]. In practice, good approximate solutions often emerge much earlier. For strictly convex quadratic functions, the error after ( k ) iterations satisfies:
[ \|xk - x^*\|A \leq 2\left( \frac{\sqrt{\kappa} - 1}{\sqrt{\kappa} + 1} \right)^k \|x0 - x^*\|A ]
where ( \kappa ) is the condition number of ( A ) and ( \|z\|_A = \sqrt{z^T A z} ) [21]. This represents a substantial improvement over steepest descent's ( O(1/k) ) convergence [3].
Newton's method achieves quadratic convergence near the optimum when the objective function is strongly convex and the Hessian satisfies Lipschitz conditions. However, this superior convergence comes at the cost of computing and inverting the Hessian matrix, with ( O(n^3) ) computational complexity and ( O(n^2) ) storage that becomes prohibitive for large problems [111].
Figure 2: The progression from steepest descent through conjugate gradient to Newton's method represents a trade-off between computational requirements and convergence rate.
Computational Requirements and Scalability
The computational characteristics of each method determine their suitability for different problem scales. Steepest descent requires only gradient calculations and a line search per iteration, making it computationally cheap for problems where gradients are readily available. However, its slow convergence often renders it impractical despite low per-iteration costs [26].
The conjugate gradient method maintains similar storage requirements (( O(n) )) to steepest descent while delivering significantly faster convergence. Each CG iteration involves matrix-vector products, inner products, and vector updates, with computational complexity comparable to steepest descent for sparse systems [21]. This favorable scaling makes CG particularly suitable for large-scale problems where storing or factoring matrices is infeasible.
Newton's method requires computation, storage, and inversion of the Hessian matrix, with ( O(n^3) ) computational complexity and ( O(n^2) ) storage requirements that limit its application to medium-scale problems [3] [111]. For large-scale problems, these requirements become prohibitive, though quasi-Newton methods like BFGS offer compromises by approximating Hessian information.
Table 3: Computational Requirements for n-Dimensional Problems
| Method | Per-Iteration Cost | Storage | Scalability to Large n |
|---|---|---|---|
| Steepest Descent | ( O(n) ) (plus gradient evaluation) | ( O(n) ) | Excellent |
| Conjugate Gradient | ( O(n) ) (for sparse systems) | ( O(n) ) | Excellent |
| Newton's Method | ( O(n^3) ) (matrix factorization) | ( O(n^2) ) | Poor |
Research Reagent Solutions: Essential Methodological Components
Implementing and analyzing optimization algorithms requires both theoretical understanding and practical computational tools. The following "research reagents" represent essential components for experimental work in this field:
Test Problem Suites: Collections of benchmark optimization problems with varying characteristics (condition number, dimensionality, nonlinearity) essential for comparative algorithm evaluation [24] [23].
Line Search Implementations: Robust implementations of Wolfe condition checks, providing critical step size selection that ensures convergence while maximizing progress [24].
Gradient Computation Tools: Automated differentiation systems or numerical differentiation utilities that provide accurate gradient evaluations without symbolic manipulation [3].
Sparse Matrix Libraries: Computational tools for efficient storage and manipulation of large-scale linear systems, enabling application of CG methods to real-world problems [21].
Convergence Analysis Frameworks: Software components for tracking iteration progress, measuring convergence rates, and comparing performance across algorithms [24] [23].
Applications and Experimental Protocols
Representative Application Domains
The conjugate gradient method finds extensive application in domains requiring solution of large linear systems or nonlinear optimization problems. In image restoration and reconstruction, CG methods effectively solve regularized inverse problems, with recent research demonstrating superior performance in recovering degraded images while controlling noise amplification [24] [23].
In compressive sensing and sparse signal recovery, CG algorithms efficiently solve large-scale ( l_1 )-regularized optimization problems for reconstructing signals from limited measurements [24]. The method's ability to handle large variable counts makes it particularly suitable for these applications.
For machine learning and neural network training, nonlinear CG variants offer alternatives to stochastic gradient descent for batch training, potentially providing faster convergence for medium-scale models [24]. The method's balance of computation and convergence is advantageous when gradient evaluation is expensive.
Experimental Methodology Framework
Robust experimental evaluation of optimization algorithms follows established methodological standards:
Test Problem Selection: Choose diverse benchmark problems representing different dimensionalities, condition numbers, and nonlinearity characteristics [24] [23].
Performance Metrics: Track iteration count, function evaluations, gradient evaluations, computational time, and solution accuracy for comprehensive comparison.
Termination Criteria: Implement consistent stopping conditions based on gradient norms (( \|g_k\| \leq \epsilon )), function value changes, or iteration limits to ensure fair comparisons.
Statistical Validation: Employ multiple random starting points and report performance profiles to account for algorithm sensitivity to initial conditions [24].
Line Search Implementation: Apply identical Wolfe condition parameters (( \rho = 0.01 ), ( \sigma = 0.9 )) across all methods to isolate algorithm performance from step size selection effects [24].
The conjugate gradient method occupies a strategically important position in the optimization spectrum, balancing the computational efficiency of steepest descent with the accelerated convergence approaching Newton's method. For large-scale linear systems and nonlinear optimization problems, CG and its extensions provide practical algorithms that scale favorably to high dimensions while maintaining theoretical convergence guarantees.
Ongoing research continues to enhance the method through hybrid approaches, three-term recurrences, and sophisticated restart strategies that further improve performance while maintaining the low-storage characteristics essential for large-scale applications. For scientific computing professionals and researchers tackling complex optimization problems in fields from drug development to image processing, understanding this critical positioning enables informed algorithm selection appropriate to specific problem characteristics and computational constraints.
In scientific research, particularly in fields like computational drug discovery, the selection of an appropriate optimization algorithm is not merely a technical detail but a fundamental determinant of success. Optimization algorithms form the computational backbone of many research projects, driving everything from molecular simulations to the analysis of complex biological systems. Within the broad landscape of optimization techniques, gradient-based methods occupy a central role due to their conceptual clarity and well-established theoretical foundations. Among these, the steepest descent method and conjugate gradient algorithms represent two pivotal approaches with distinct characteristics, performance profiles, and application domains.
This guide provides researchers with a comprehensive framework for selecting between steepest descent and conjugate gradient algorithms based on their specific research problems. The decision requires careful consideration of multiple factors, including problem conditioning, computational resources, convergence requirements, and implementation complexity. Within drug discovery specifically, these algorithms enable critical tasks such as molecular docking, protein folding prediction, and chemical property optimization. The steepest descent method, characterized by its simplicity and robustness, follows the negative gradient direction at each iteration, providing guaranteed descent but potentially slow convergence on ill-conditioned problems. In contrast, conjugate gradient methods generate search directions that are mutually conjugate with respect to the Hessian matrix, often yielding significantly faster convergence for quadratic problems and beyond [112].
The following sections present a detailed technical comparison of these algorithms, quantitative performance analyses, practical implementation guidelines, and domain-specific applications to equip researchers with the knowledge needed to make informed algorithmic choices for their scientific investigations.
Theoretical Foundations: Steepest Descent vs. Conjugate Gradient
Fundamental Algorithmic Principles
The steepest descent algorithm represents one of the simplest and most intuitive approaches to unconstrained optimization. At each iteration, the method computes the negative gradient of the objective function at the current point and takes a step in this direction. The update formula is given by:
[x{k+1} = xk - \alphak \nabla f(xk)]
where the step size (\alpha_k) is determined through a line search procedure. The fundamental principle relies on the mathematical truth that the negative gradient represents the direction of steepest local descent. For quadratic objective functions of the form (f(x) = \frac{1}{2}x^TAx - b^Tx), where (A) is a symmetric positive definite matrix, the optimal step size can be computed analytically as:
[\alphak = \frac{\nabla f(xk)^T \nabla f(xk)}{\nabla f(xk)^T A \nabla f(x_k)}]
The convergence behavior of steepest descent is heavily influenced by the condition number (\kappa = L/m), where (L) and (m) are the largest and smallest eigenvalues of (A), respectively. For ill-conditioned problems ((\kappa \gg 1)), steepest descent exhibits slow convergence with a tendency to "zigzag" toward the solution [112].
Conjugate gradient methods, developed to address this limitation, generate a sequence of search directions that are mutually conjugate with respect to (A) (i.e., (di^T A dj = 0) for (i \neq j)). For quadratic problems, this property ensures that the exact solution is found in at most (n) iterations, where (n) is the problem dimension. The algorithm update is given by:
[x{k+1} = xk + \alphak dk]
where the search direction (d_k) is computed as:
[dk = -\nabla f(xk) + \betak d{k-1}]
Various formulas exist for (\beta_k), including the Fletcher-Reeves, Polak-Ribière, and Hestenes-Stiefel methods, each with slightly different properties for non-quadratic problems.
Convergence Properties and Theoretical Bounds
Theoretical convergence analysis provides rigorous performance guarantees for both algorithms. For the steepest descent method applied to strongly convex functions with Lipschitz continuous gradients, the convergence rate is linear, with the following bound on the error:
[f(xk) - f(x^*) \leq \left(1 - \frac{m}{L}\right)^k (f(x0) - f(x^*))]
This translates to an iteration complexity of (O(\kappa \log(1/\varepsilon))) to achieve an (\varepsilon)-accurate solution. Recent research has established more refined convergence bounds for specific variants of steepest descent. For problems where the optimal point varies polynomially with time, a fundamental convergence rate bound of (\left(\frac{\sqrt{\kappa}-1}{\sqrt{\kappa}+1}\right)^{\frac{1}{n}}) has been established for gradient-based algorithms with exact tracking, where (n) relates to the polynomial order of variation in the optimal point [113].
For the conjugate gradient method, the convergence behavior is more complex to analyze but generally superior. The error after (k) iterations satisfies:
[\|xk - x^*\|A \leq 2\left(\frac{\sqrt{\kappa} - 1}{\sqrt{\kappa} + 1}\right)^k \|x0 - x^*\|A]
where (\|z\|_A = \sqrt{z^T A z}) denotes the energy norm. This bound, while conservative, indicates significantly faster convergence than steepest descent, particularly for well-conditioned problems. In practice, properly implemented conjugate gradient methods often exceed these theoretical bounds, especially when enhanced with effective preconditioning techniques.
Table 1: Theoretical Convergence Properties Comparison
| Property | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Convergence Rate | Linear: (O((1 - 1/\kappa)^k)) | Superlinear (theoretical exact convergence in (n) steps for quadratic problems) |
| Iteration Complexity | (O(\kappa \log(1/\varepsilon))) | (O(\sqrt{\kappa} \log(1/\varepsilon))) for (\varepsilon)-accurate solution |
| Condition Number Sensitivity | Highly sensitive to (\kappa) | Less sensitive, especially with preconditioning |
| Theoretical Bound | (\left(\frac{\kappa-1}{\kappa+1}\right)^k) [112] | (2\left(\frac{\sqrt{\kappa}-1}{\sqrt{\kappa}+1}\right)^k) |
| Exact Tracking Bound | (\left(\frac{\sqrt{\kappa}-1}{\sqrt{\kappa}+1}\right)^{\frac{1}{n}}) for polynomial varying optima [113] | Not applicable |
Quantitative Performance Analysis
Convergence Rate Comparison
The performance disparity between steepest descent and conjugate gradient methods becomes particularly pronounced when examining their behavior on problems with varying condition numbers. For well-conditioned problems ((\kappa \approx 1)), both algorithms demonstrate rapid convergence, with conjugate gradient offering only marginal improvements. However, as the condition number increases, the limitations of steepest descent become increasingly evident.
Recent research has revealed nuanced aspects of steepest descent performance. When applied with exact line search to convex quadratic objectives, the worst-case rate of convergence deteriorates as the condition number of the Hessian grows. Interestingly, analysis shows that in the ill-conditioned regime, the rate of convergence for almost all initial vectors approaches the worst-case rate [112]. This finding has significant implications for researchers relying on average-case performance expectations.
For the conjugate gradient method, the convergence behavior is more favorable across different problem classes. The method effectively distributes error reduction across all eigenvectors rather than focusing predominantly on extreme modes. This property explains the characteristically different convergence profiles observed in practice: steepest descent often displays rapid initial progress followed by slow asymptotic convergence, while conjugate gradient maintains a more consistent error reduction throughout the iteration process.
Computational Requirements and Scalability
Beyond iteration counts, practical algorithm selection must consider computational requirements and memory footprints. The steepest descent algorithm has minimal storage needs, requiring only the current point and gradient vector to be stored ((O(n)) memory). Each iteration involves a single matrix-vector product (for quadratic problems) or gradient evaluation, followed by a line search procedure.
The conjugate gradient method has slightly higher memory requirements, typically needing storage for two additional (n)-dimensional vectors (the previous search direction and the previous gradient). The computational overhead per iteration is modest, involving several vector inner products and vector updates. This additional cost is almost always justified by the significant reduction in iteration count, particularly for large-scale problems.
Table 2: Computational Requirements and Practical Performance
| Aspect | Steepest Descent | Conjugate Gradient |
|---|---|---|
| Memory Requirements | (O(n)) | (O(n)) |
| Operations per Iteration | 1 gradient evaluation, 1 line search | 1 gradient evaluation, 1 matrix-vector product, several inner products |
| Line Search Requirements | Critical for performance | Less sensitive to exact line search |
| Typical Iteration Count | (O(\kappa \log(1/\varepsilon))) | (O(\sqrt{\kappa} \log(1/\varepsilon))) |
| Implementation Complexity | Low | Moderate |
| Parallelization Potential | High | Moderate to High |
Algorithm Selection Framework
Problem Characteristics Assessment
Selecting the appropriate algorithm begins with a thorough assessment of problem characteristics. Researchers should evaluate the following key aspects of their optimization problem:
Problem Dimensionality: For very high-dimensional problems where memory constraints are severe, steepest descent may be preferred due to its minimal storage requirements. However, for most large-scale problems, conjugate gradient methods typically offer better performance despite slightly higher memory needs.
Condition Number and Problem Structure: Problems with low condition numbers ((\kappa < 100)) may be effectively solved using either method, with steepest descent offering implementation simplicity. For moderately to highly ill-conditioned problems ((\kappa > 1000)), conjugate gradient methods are strongly recommended. When the condition number is unknown, diagnostic tools such as power iteration for eigenvalue estimation can provide valuable insights.
Solution Accuracy Requirements: For applications requiring low to moderate accuracy solutions (e.g., early prototyping or exploratory analysis), steepest descent may be adequate. For high-precision requirements, conjugate gradient methods typically achieve the desired accuracy with substantially fewer computational resources.
Computational Cost of Gradient Evaluation: In applications where gradient evaluations are exceptionally expensive (e.g., in certain PDE-constrained optimization problems), the reduced iteration count of conjugate gradient methods provides a decisive advantage.
Practical Implementation Considerations
Beyond theoretical properties, practical implementation factors significantly influence algorithm selection:
Ease of Implementation: Steepest descent algorithms are conceptually straightforward to implement and debug, making them suitable for rapid prototyping or educational contexts. Conjugate gradient implementations require more care, particularly regarding numerical stability and termination criteria.
Robustness to Noise and Numerical Errors: Both algorithms demonstrate reasonable robustness to numerical errors, though steepest descent is generally more tolerant of inaccuracies in gradient computations and line search procedures.
Preconditioning Availability: For problems where effective preconditioners are available, conjugate gradient methods can achieve dramatic performance improvements. The development of problem-specific preconditioners often represents the most significant opportunity for performance enhancement in large-scale optimization.
Specialized Problem Structures: Many scientific applications exhibit specific structures that may favor one algorithm over another. For shape optimization problems using (W^{1,\infty}) functions, recent research has established convergence results for steepest descent algorithms with Armijo-Goldstein stepsize rules [114].
The following workflow diagram illustrates the decision process for selecting between these algorithms:
Diagram 1: Algorithm Selection Workflow (76 characters)
Applications in Drug Discovery and Development
Structure-Based Drug Design
Optimization algorithms play a crucial role in structure-based drug design, particularly in molecular docking and virtual screening workflows. Both steepest descent and conjugate gradient methods are employed in energy minimization procedures that determine stable molecular conformations. For example, in identifying natural inhibitors against the human αβIII tubulin isotype—a target for cancer therapies—researchers employed structure-based virtual screening of 89,399 compounds from the ZINC natural compound database [110]. The initial screening identified 1,000 hits based on binding energy, which were subsequently refined using machine learning classifiers.
In such applications, steepest descent methods are often used for initial rough optimization due to their robustness, while conjugate gradient methods may be employed for final refinement where high accuracy is required. The computational efficiency of these algorithms directly impacts the scale and throughput of virtual screening campaigns, enabling researchers to evaluate larger chemical spaces within practical time frames.
AI-Driven Molecular Optimization
Recent advances in AI-driven drug discovery have created new optimization challenges where algorithm selection is critical. The DrugAppy framework exemplifies this trend, implementing an end-to-end deep learning approach that combines artificial intelligence algorithms with computational and medicinal chemistry methodologies [42]. This framework employs an imbrication of models including SMINA and GNINA for high-throughput virtual screening and GROMACS for molecular dynamics, all of which rely on efficient optimization at their core.
Similarly, novel approaches like the Gradient Genetic Algorithm (Gradient GA) incorporate gradient information from objective functions into genetic algorithms for drug molecular design [115]. Instead of random exploration, each proposed sample iteratively progresses toward an optimal solution by following gradient direction, demonstrating how classical optimization concepts are being hybridized with modern machine learning approaches.
Table 3: Optimization Applications in Drug Discovery Workflows
| Application Domain | Typical Optimization Tasks | Preferred Algorithm | Rationale |
|---|---|---|---|
| Molecular Docking | Binding pose optimization, Energy minimization | Steepest Descent (initial), Conjugate Gradient (refinement) | Robustness for rough optimization, accuracy for final refinement |
| Molecular Dynamics | Force field minimization, Equilibrium finding | Conjugate Gradient | Superior performance for large, ill-conditioned systems |
| Chemical Property Optimization | Multi-objective molecular design | Hybrid approaches (e.g., Gradient GA) [115] | Combines global exploration with gradient-based local optimization |
| Virtual Screening | Scoring function optimization, Library filtering | Steepest Descent | Implementation simplicity adequate for initial screening |
| Pharmacokinetic Prediction | Model parameter estimation | Conjugate Gradient | High accuracy requirements for predictive models |
Experimental Protocols and Methodologies
Benchmarking Protocol for Algorithm Performance
To empirically evaluate algorithm performance on a specific research problem, researchers should implement the following standardized benchmarking protocol:
Test Problem Selection: Curate a representative set of test problems with known characteristics, including both synthetic benchmarks and real-world instances from the target application domain.
Performance Metrics Definition: Establish comprehensive performance metrics including:
- Iteration count to reach specified tolerance levels
- Computational time (broken down by gradient evaluations, linear algebra operations, etc.)
- Memory usage profiles
- Solution accuracy achieved
Termination Criteria: Implement consistent termination criteria such as:
- Gradient norm tolerance: (\|\nabla f(x_k)\| \leq \varepsilon)
- Function value change tolerance: (|f(xk) - f(x{k-1})| \leq \delta)
- Maximum iteration count
Implementation Details: Ensure both algorithms are implemented with comparable attention to optimization, using the same programming language, compiler optimizations, and numerical linear algebra routines.
Statistical Significance: Perform multiple runs with different initial conditions to account for performance variability and ensure statistically significant conclusions.
Implementation Guidelines
For researchers implementing these algorithms, the following practical guidelines ensure robust performance:
Steepest Descent Implementation:
Conjugate Gradient Implementation:
The following diagram illustrates a typical experimental workflow for comparing optimization algorithms in scientific research:
Diagram 2: Experimental Comparison Workflow (76 characters)
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation and application of optimization algorithms require access to appropriate computational tools and frameworks. The following table details key resources that constitute the modern computational scientist's toolkit for optimization research:
Table 4: Essential Research Reagents for Optimization Studies
| Resource Category | Specific Tools/Libraries | Primary Function | Application Context |
|---|---|---|---|
| Linear Algebra Libraries | BLAS, LAPACK, Intel MKL, OpenBLAS | Efficient matrix/vector operations | Foundation for all numerical optimization algorithms |
| Scientific Computing Environments | MATLAB, GNU Octave, Python with NumPy/SciPy | Rapid algorithm prototyping and testing | Initial algorithm development and educational use |
| Machine Learning Frameworks | PyTorch, TensorFlow, JAX | Automatic differentiation for gradient computation | Complex objective functions in AI-driven drug discovery [42] |
| Specialized Optimization Software | NLopt, OPT++, Ceres Solver | Reference implementations and advanced algorithms | Performance benchmarking and production deployment |
| Molecular Modeling Platforms | AutoDock Vina, GROMACS, Schrödiner Suite | Specialized optimization for molecular systems | Structure-based drug design and virtual screening [110] |
| High-Performance Computing | MPI, OpenMP, CUDA | Parallelization of computationally intensive tasks | Large-scale problems in molecular dynamics and screening |
For researchers focusing on drug discovery applications, additional specialized resources are essential for experimental validation of computationally predicted results:
Chemical Databases: Publicly available databases such as ZINC (containing over 89,399 natural compounds in one study [110]) provide starting points for virtual screening campaigns.
Property Prediction Tools: Software for predicting ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties helps prioritize compounds for experimental testing.
Experimental Validation Platforms: High-throughput screening facilities, structural biology resources (X-ray crystallography, Cryo-EM), and biochemical assay capabilities are ultimately required to validate computational predictions.
The integration of these computational and experimental resources creates a comprehensive research pipeline that begins with algorithmic optimization and concludes with empirical validation, accelerating the translation of computational insights into tangible scientific advances.
The selection between steepest descent and conjugate gradient algorithms represents a critical decision point in designing computational research workflows. Through systematic evaluation of problem characteristics, computational constraints, and accuracy requirements, researchers can make informed choices that optimize both development time and computational efficiency.
Steepest descent algorithms offer simplicity and robustness for well-conditioned problems or scenarios with severe memory constraints. Their convergence guarantees and straightforward implementation make them valuable for rapid prototyping and educational contexts. Recent theoretical advances have also expanded our understanding of their performance characteristics, including convergence bounds for time-varying optimization problems [113] and applications in shape optimization [114].
Conjugate gradient methods generally provide superior performance for medium to large-scale problems, particularly those with moderate to high condition numbers. The additional implementation complexity is typically justified by significantly reduced iteration counts and computational time. For drug discovery applications specifically, where virtual screening may involve thousands of compounds [110] or complex multi-objective optimization [115], the efficiency advantages of conjugate gradient methods can substantially accelerate research timelines.
As optimization requirements continue to evolve in scientific research, particularly with the growing integration of machine learning approaches [37] [42], the fundamental principles outlined in this guide provide a durable framework for algorithm selection. By matching algorithmic capabilities to problem characteristics, researchers can ensure that their computational methodologies effectively support their scientific objectives.
Conclusion
The comparative analysis reveals that while Steepest Descent offers an intuitive and straightforward approach to optimization, the Conjugate Gradient method consistently demonstrates superior performance for the large, sparse systems common in drug discovery and biomolecular modeling. Conjugate Gradient's use of A-conjugate search directions enables it to avoid the oscillatory behavior of Steepest Descent, leading to significantly faster convergence and lower achieved energy states in tasks like small-molecule conformation analysis, as evidenced in studies on diabetes drugs and Covid-19 therapeutics. For researchers, this translates to more efficient in-silico screening of drug-like compounds and more accurate molecular models. Future directions involve the integration of these algorithms with machine learning for predictive binding affinity calculations and the development of hybrid methods that further reduce computational overhead, ultimately accelerating the pace of drug development from the lab to the clinic.
References
- 1. Energy Minimization [https://www.intechopen.com/chapters/74105]
- 2. Energy minimization methods - Molecular Modeling | PPTX [https://www.slideshare.net/slideshow/energy-minimization-methods/251103702]
- 3. What are the differences between the different gradient ... [https://scicomp.stackexchange.com/questions/26960/what-are-the-differences-between-the-different-gradient-based-numerical-optimiza]
- 4. Energy Minimization - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/algorithms/energy-minimization.html]
- 5. Conjugate gradient versus steepest descent [https://link.springer.com/article/10.1007/BF00933351]
- 6. Optimizing drug design by merging generative AI with a ... [https://www.nature.com/articles/s42004-025-01635-7]
- 7. Beyond Legacy Tools: Defining Modern AI Drug Discovery ... [https://www.biopharmatrend.com/business-intelligence/what-is-ai-drug-discovery/]
- 8. Gradient descent [https://en.wikipedia.org/wiki/Gradient_descent]
- 9. Why the negative gradient gives the direction of ... [https://math.stackexchange.com/questions/1827594/why-the-negative-gradient-gives-the-direction-of-the-steepest-decrease-in-the-gr]
- 10. Why is conjugate gradient descent a better option than ... [https://math.stackexchange.com/questions/4738566/why-is-conjugate-gradient-descent-a-better-option-than-gradient-descent]
- 11. An efficient hybrid conjugate gradient method with ... [https://www.sciencedirect.com/science/article/abs/pii/S0168927424001685]
- 12. Steepest Descent - an overview [https://www.sciencedirect.com/topics/computer-science/steepest-descent]
- 13. Gradient Descent: A Comprehensive Guide to ... [https://medium.com/@lomashbhuva/gradient-descent-a-comprehensive-guide-to-understanding-implementation-41cd9c5191dd]
- 14. Part 2: The gradient descent algorithm [https://rezaborhani.github.io/mlr/blog_posts/Mathematical_Optimization/Part_2_gradient_descent.html]
- 15. Comparison Between Steepest Descent Method and ... [https://engiscience.com/index.php/josse/article/view/josse2021113]
- 16. On Convergence and Stability of Two Extended BB-like ... [https://arxiv.org/html/2506.12731v2]
- 17. Steepest-descent optimization procedure with step size ... [https://math.stackexchange.com/questions/3744494/steepest-descent-optimization-procedure-with-step-size-given-by-harmonic-sequenc]
- 18. Convergence analysis of an optimally accurate frozen multi ... [https://www.sciencedirect.com/science/article/abs/pii/S0885064X22000760]
- 19. 9.1 Steepest descent method - Optimization Of Systems [https://fiveable.me/optimization-systems/unit-9/steepest-descent-method/study-guide/JKABJ5iZCMLFT2zO]
- 20. Steepest Descent Method - an overview [https://www.sciencedirect.com/topics/computer-science/steepest-descent-method]
- 21. Conjugate gradient method [https://en.wikipedia.org/wiki/Conjugate_gradient_method]
- 22. [PDF] Comparison Between Steepest Descent Method and ... [https://www.semanticscholar.org/paper/3d22b2dba45fe331ef38cbc26828082cf67bae6b]
- 23. An efficient three-term conjugate gradient algorithm with ... [https://www.nature.com/articles/s41598-025-19329-4]
- 24. An efficient modified conjugate gradient algorithm under ... [https://www.sciencedirect.com/science/article/abs/pii/S0377042724005831]
- 25. Chapter 5 Conjugate Gradient Methods [https://indrag49.github.io/Numerical-Optimization/conjugate-gradient-methods-1.html]
- 26. Conjugate gradient methods [https://optimization.cbe.cornell.edu/index.php?title=Conjugate_gradient_methods]
- 27. Why is the conjugate direction better than the negative of ... [https://math.stackexchange.com/questions/1020008/why-is-the-conjugate-direction-better-than-the-negative-of-gradient-when-minimi]
- 28. What's the intuition behind the conjugate gradient method? [https://math.stackexchange.com/questions/2376969/whats-the-intuition-behind-the-conjugate-gradient-method]
- 29. Difference between Gradient Descent method and ... [https://math.stackexchange.com/questions/1659452/difference-between-gradient-descent-method-and-steepest-descent]
- 30. A Sufficiently Descending Three-Term Nonlinear ... [https://link.springer.com/article/10.1007/s43069-025-00493-2]
- 31. Why is gradient descent used over the conjugate ... [https://ai.stackexchange.com/questions/32428/why-is-gradient-descent-used-over-the-conjugate-gradient-method]
- 32. Mastering Conjugate Method Gradient [https://www.numberanalytics.com/blog/conjugate-gradient-method-numerical-methods]
- 33. Projected variable three-term conjugate gradient algorithm ... [https://www.sciencedirect.com/science/article/pii/S0925231225022404]
- 34. Method -- from Wolfram MathWorld Conjugate Gradient [https://mathworld.wolfram.com/ConjugateGradientMethod.html]
- 35. A spectral Fletcher-Reeves conjugate gradient method with ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0320416]
- 36. Protein Spatial Structure Meets Artificial Intelligence [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412482/]
- 37. Leveraging artificial intelligence in antibody-drug ... [https://www.nature.com/articles/s41698-025-01159-2]
- 38. Conjugate Gradient Method - an overview [https://www.sciencedirect.com/topics/engineering/conjugate-gradient-method]
- 39. Conjugate Gradient Method and it's Application in Energy ... [https://akshayshah-96720.medium.com/conjugate-gradient-m-dfd9d2969e73]
- 40. Targeting RNA with small molecules using state-of-the-art ... [https://www.nature.com/articles/s42003-025-08809-y]
- 41. A Benchmark for Quantum Chemistry Relaxations via ... [https://arxiv.org/html/2506.23008v2]
- 42. DrugAppy – An end-to-end deep learning framework for ... [https://www.sciencedirect.com/science/article/pii/S0010482525015549]
- 43. Integrating artificial intelligence into small molecule ... [https://www.nature.com/articles/s44386-025-00029-y]
- 44. Ten quick tips for homology modeling of high-resolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7117658/]
- 45. Protein Structure Model Refinement in CASP12 Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5820213/]
- 46. De novo protein fold design through sequence- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9942881/]
- 47. A discard-and-restart MD algorithm for the sampling of ... [https://www.sciencedirect.com/science/article/abs/pii/S0006349525001985]
- 48. An adaptive conjugate gradient accelerated evolutionary ... [https://www.sciencedirect.com/science/article/abs/pii/S156849462301195X]
- 49. "Adaptive Stochastic Conjugate Gradient optimization for ... [https://repository.lsu.edu/gradschool_theses/324/]
- 50. Conjugate-Gradient-like Based Adaptive Moment ... [https://arxiv.org/abs/2404.01714]
- 51. ACG System Overview [https://www.hopkinsacg.org/about-the-acg-system/]
- 52. Biomedical Science Research (BMSR) Team - ACG 150 [https://acg150.acg.edu/initiatives/institute-for-hellenic-growth-and-prosperity/research-technology-innovation-network/biomedical-science-research-team/]
- 53. Mathematical optimization: finding minima of functions [https://lectures.scientific-python.org/advanced/mathematical_optimization/index.html]
- 54. The Concept of Conjugate Gradient Descent in Python [https://ilyakuzovkin.com/ml-ai-rl-cs/the-concept-of-conjugate-gradient-descent-in-python/]
- 55. Development of a Robust Design Optimization Algorithm ... [https://www.sciencedirect.com/science/article/pii/S2214716025000314]
- 56. Automated drug design for druggable target identification ... [https://www.nature.com/articles/s41598-025-18091-x]
- 57. Implementing the Steepest Descent Algorithm in Python ... [https://towardsdatascience.com/implementing-the-steepest-descent-algorithm-in-python-from-scratch-d32da2906fe2/]
- 58. Implementing gradient descent in Python to find a local ... [https://www.geeksforgeeks.org/machine-learning/how-to-implement-a-gradient-descent-in-python-to-find-a-local-minimum/]
- 59. Implicit Bias of Steepest Descent in Homogeneous Neural ... [https://arxiv.org/html/2410.22069v4]
- 60. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 61. How AI will reshape pharma in 2025 [https://www.drugtargetreview.com/article/154981/how-ai-will-reshape-pharma-by-2025/]
- 62. Implicit Bias of Steepest Descent in Homogeneous Neural ... [https://proceedings.iclr.cc/paper_files/paper/2025/hash/1c9ee8c9e1a0c7de49e15720c4ce6f78-Abstract-Conference.html]
- 63. Steepest descent method using novel adaptive stepsizes ... [https://optimization-online.org/2025/05/steepest-descent-method-using-novel-adaptive-stepsizes-for-unconstrained-nonlinear-multiobjective-programming/]
- 64. Learning Complexity of Gradient Descent and Conjugate ... [https://ojs.aaai.org/index.php/AAAI/article/view/33943]
- 65. Line search [https://en.wikipedia.org/wiki/Line_search]
- 66. Line search methods [https://optimization.cbe.cornell.edu/index.php?title=Line_search_methods]
- 67. Line-Search methods for Optimization | by Abhijeet Nayak [https://medium.com/@abhijeetknayak/line-search-methods-for-optimization-28eacddd95ec]
- 68. 3.4 Conjugate Gradient | Advanced Statistical Computing [https://bookdown.org/rdpeng/advstatcomp/conjugate-gradient.html]
- 69. AdaLRS: Loss-Guided Adaptive Learning Rate Search for ... [https://www.arxiv.org/abs/2506.13274]
- 70. AMC: Adaptive Learning Rate Adjustment Based on Model ... [https://www.mdpi.com/2227-7390/13/4/650]
- 71. Improving neural network training using dynamic learning ... [https://www.sciencedirect.com/science/article/pii/S2666827025000805]
- 72. learning preconditioners for the conjugate gradient method [https://arxiv.org/html/2305.16368v3]
- 73. Efficient preconditioning strategies for accelerating GMRES in ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0322146]
- 74. Steepest Descent Preconditioning for Nonlinear GMRES ... [https://arxiv.org/abs/1106.4426]
- 75. Prior-Preconditioned Conjugate Gradient Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10977663/]
- 76. Graph Neural Preconditioners for Iterative Solutions of ... [https://research.ibm.com/publications/graph-neural-preconditioners-for-iterative-solutions-of-sparse-linear-systems]
- 77. Comparison of iteration methods: number of iterations vs. ... [https://scicomp.stackexchange.com/questions/7638/comparison-of-iteration-methods-number-of-iterations-vs-cpu-time]
- 78. Big O, Big Efficiency : Recursion vs. Iteration Unveiled [https://medium.com/@ireneselenam/big-o-big-efficiency-recursion-vs-iteration-unveiled-f0707501be5b]
- 79. Big O Time Complexity for a for loop when the number of ... [https://stackoverflow.com/questions/76029142/big-o-time-complexity-for-a-for-loop-when-the-number-of-iterations-isnt-constan]
- 80. Descent method — Steepest descent and conjugate gradient [https://medium.com/data-science/descent-method-steepest-descent-and-conjugate-gradient-math-explained-78601d8df3ce]
- 81. Learning complexity of gradient descent and conjugate ... gradient [https://arxiv.org/html/2412.13473v1]
- 82. The exact worst-case convergence rate of the gradient ... [https://www.semanticscholar.org/paper/The-exact-worst-case-convergence-rate-of-the-method-Abbaszadehpeivasti-Klerk/932d71a78df3d0d398678a2a2a2c92e80c6a6886]
- 83. Global Convergence and Rate Analysis of the Steepest ... [https://arxiv.org/html/2503.06476v1]
- 84. An adaptive restarting RMIL conjugate method and its hybrid... gradient [https://link.springer.com/article/10.1007/s12190-025-02466-4]
- 85. Convergence and complexity guarantees for a wide class ... [https://www.sciencedirect.com/science/article/abs/pii/S0167637724000518]
- 86. A revised MRMIL Riemannian conjugate gradient method ... [https://www.sciencedirect.com/science/article/abs/pii/S0168927425000698]
- 87. Two improved nonlinear conjugate gradient methods with ... [https://www.aimsciences.org/article/doi/10.3934/jimo.2024098]
- 88. A modified three-term of Dai–Yuan type derivative-free ... [https://www.sciencedirect.com/science/article/pii/S2773186325000453]
- 89. Some descent Dai-Liao-type conjugate gradient methods ... [https://www.sciencedirect.com/science/article/pii/S2773186325000210]
- 90. An efficient gradient-based algorithm with descent direction ... [https://peerj.com/articles/cs-2783/]
- 91. (PDF) Comparison Between Steepest Descent Method and ... [https://www.academia.edu/108066513/Comparison_Between_Steepest_Descent_Method_and_Conjugate_Gradient_Method_by_Using_Matlab]
- 92. facebookresearch/SteepGS: Code for CVPR 2025 paper " ... [https://github.com/facebookresearch/SteepGS]
- 93. Color Contrast Accessibility: Complete WCAG 2025 Guide ... [https://www.allaccessible.org/blog/color-contrast-accessibility-wcag-guide-2025]
- 94. Best Practices for Accessible Color Contrast in UX [https://developerux.com/2025/07/28/best-practices-for-accessible-color-contrast-in-ux/]
- 95. Which Optimizer Should You Use With NNPs? | Rowan [https://www.rowansci.com/blog/which-optimizer-should-you-use-with-nnps]
- 96. IOHanalyzer: Detailed Performance Analyses for Iterative ... [https://arxiv.org/abs/2007.03953]
- 97. An efficient preconditioned conjugate-gradient solver for a ... [https://arxiv.org/html/2510.24543v1]
- 98. Performance metric based stopping criteria for iterative ... [https://patents.google.com/patent/US9866954B2/en]
- 99. Energy Minimization - GROMACS .3 documentation [https://manual.gromacs.org/current/reference-manual/algorithms/energy-minimization.html]
- 100. Green adeptness in synthesis of non-toxic copper and cobalt ... [https://cancer-nano.biomedcentral.com/articles/10.1186/s12645-023-00226-2]
- 101. Design of Cobalt Nanoparticles with Tailored Structural ... [https://www.mdpi.com/2073-4344/5/1/442]
- 102. Rich Landscape of Colloidal Semiconductor–Metal Hybrid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10103135/]
- 103. Artificial intelligence-assisted modelling of heavy metal ... [https://www.sciencedirect.com/science/article/pii/S2590123025032025]
- 104. Research progress on the preparation of cobalt-based oxide ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta06101f]
- 105. Cobalt-Based Electrocatalysts for Sustainable Nitrate ... [https://link.springer.com/article/10.1007/s40820-025-01877-z]
- 106. Enhancing Water Splitting Performance via NiFeP-CoP on ... [https://www.mdpi.com/2079-4991/15/12/883]
- 107. Revolutionizing diabetes treatment: computational insights ... [https://www.explorationpub.com/Journals/eds/Article/1008104]
- 108. Investigating the molecular mechanisms of the “Astragalus ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1618575/full]
- 109. A review on computational tools for antidiabetic herbs ... [https://link.springer.com/article/10.1007/s44371-025-00135-w]
- 110. Structure based drug design and machine learning ... [https://www.nature.com/articles/s41598-025-17708-5]
- 111. What is the difference between Gradient Descent and ... [https://stackoverflow.com/questions/12066761/what-is-the-difference-between-gradient-descent-and-newtons-gradient-descent]
- 112. The essential best and average rate of convergence ... [https://link.springer.com/article/10.1007/s11075-025-02244-0]
- 113. A Fundamental Convergence Rate Bound for Gradient ... [https://arxiv.org/html/2508.21335v1]
- 114. Convergence of a steepest descent algorithm in shape ... [https://sussex.figshare.com/articles/journal_contribution/Convergence_of_a_steepest_descent_algorithm_in_shape_optimisation_using_i_W_i_sup_1_sup_functions/28788173]
- 115. Gradient Genetic Algorithm for Drug Molecular Design [https://arxiv.org/html/2502.09860v1]