Predicting Molecular Mechanics Parameters with Graph Neural Networks: A New Paradigm for Force Field Development
This article explores the transformative role of Graph Neural Networks (GNNs) in predicting Molecular Mechanics (MM) force field parameters, a critical task for accurate and efficient molecular dynamics simulations in...
Predicting Molecular Mechanics Parameters with Graph Neural Networks: A New Paradigm for Force Field Development
Abstract
This article explores the transformative role of Graph Neural Networks (GNNs) in predicting Molecular Mechanics (MM) force field parameters, a critical task for accurate and efficient molecular dynamics simulations in drug discovery and materials science. We cover the foundational principles of GNNs and MM, detail cutting-edge methodological architectures and their specific applications, address key challenges and optimization strategies, and provide a comparative analysis of model performance and validation. Aimed at researchers and development professionals, this review synthesizes recent advances to guide the selection, implementation, and future development of GNN-driven force fields, highlighting their potential to achieve near-chemical accuracy at traditional MM computational cost.
From Atoms to Graphs: Foundational Principles of GNNs and Molecular Mechanics
Molecular mechanics (MM) force fields are foundational computational tools that use classical physics to model the potential energy surface of molecular systems. Their application is crucial for molecular dynamics simulations, which are extensively used in drug discovery to study protein-ligand interactions, conformational dynamics, and other phenomena relevant to pharmaceutical development. The accuracy of these simulations is intrinsically tied to the quality of the force field parameters. The core challenge, known as the parameterization problem, involves determining the optimal numerical values for these parameters—which govern bonded interactions (bonds, angles, dihedrals) and non-bonded interactions (van der Waals, electrostatic)—so that the force field reliably reproduces reference data, typically from quantum mechanical (QM) calculations or experimental measurements [1] [2].
This challenge is multifaceted. First, the parameter space is high-dimensional and interdependent, where adjusting one parameter can necessitate recalibration of others. Second, traditional methods often rely on "look-up tables" of parameters based on discrete atom types, which struggle to cover the vastness of synthetically accessible, drug-like chemical space [1]. Furthermore, conventional parameter fitting can become suboptimal when inconsistencies exist between the equilibrium geometries of QM and MM models, a common issue when parameters are transferred from one molecule to another [3]. Finally, the process of refining parameters against experimental data must robustly handle the sparse, noisy, and ensemble-averaged nature of that data [4].
Modern Data-Driven and Machine Learning Approaches
To overcome these limitations, the field is rapidly shifting toward data-driven and machine learning (ML) approaches. These methods leverage large, diverse datasets and advanced algorithms to create more accurate, transferable, and automatable parameterization schemes.
Graph Neural Networks for Parameter Prediction
Graph Neural Networks (GNNs) are particularly well-suited for molecular modeling as they natively operate on graph representations of molecules, where atoms are nodes and bonds are edges. A leading approach involves training GNNs on expansive QM datasets to predict MM parameters directly from molecular structure.
The ByteFF force field exemplifies this paradigm. Its development involved generating a massive dataset containing 2.4 million optimized molecular fragment geometries and 3.2 million torsion profiles at the B3LYP-D3(BJ)/DZVP level of theory [1]. An edge-augmented, symmetry-preserving GNN was then trained on this dataset to simultaneously predict all bonded and non-bonded parameters for drug-like molecules. This end-to-end, data-driven approach allows ByteFF to achieve state-of-the-art accuracy across a broad chemical space, covering geometries, torsional profiles, and conformational energies [1].
Innovations in GNN architecture are further pushing the boundaries of performance and interpretability. Kolmogorov-Arnold GNNs (KA-GNNs) integrate Kolmogorov-Arnold networks (KANs) into the core components of GNNs: node embedding, message passing, and readout [5]. By using learnable, Fourier-series-based univariate functions instead of fixed activation functions, KA-GNNs demonstrate superior expressivity and parameter efficiency. This translates to higher accuracy in molecular property prediction and offers improved interpretability by highlighting chemically meaningful substructures [5].
Automated and Multi-Objective Optimization Algorithms
For systems where high-fidelity parameterization is required, automated iterative optimization against QM data or experimental observables is key. Modern algorithms make this process less tedious and more robust.
One advanced framework combines the Simulated Annealing (SA) and Particle Swarm Optimization (PSO) algorithms, augmented with a Concentrated Attention Mechanism (CAM). This hybrid method performs a multi-objective search of the parameter space, with the CAM strategically weighting key data points (like optimal structures) to enhance accuracy. This approach has been successfully applied to optimize parameters for reactive force fields (ReaxFF), demonstrating higher efficiency and accuracy compared to using either SA or PSO alone [6].
When the goal is to match ensemble-averaged experimental data (e.g., from NMR or free energy measurements), Bayesian methods are highly effective. The Bayesian Inference of Conformational Populations (BICePs) algorithm treats experimental uncertainty as a nuisance parameter sampled alongside conformational populations [4]. It uses a replica-averaged forward model to predict observables and can employ specialized likelihood functions to automatically detect and down-weight outliers or systematic errors. The BICePs score serves as a differentiable objective function, enabling robust variational optimization of force field parameters against complex experimental data [4].
Quantitative Comparisons of Parameterization Methods
The table below summarizes the key performance characteristics of contemporary force field parameterization methods discussed in this note.
Table 1: Comparison of Modern Force Field Parameterization Approaches
| Method / Framework | Core Approach | Key Advantages | Demonstrated Application |
|---|---|---|---|
| ByteFF GNN [1] | Graph Neural Network trained on QM data | Simultaneous parameter prediction; broad coverage of drug-like chemical space; state-of-the-art accuracy on multiple benchmarks. | General organic molecules / drug-like compounds |
| KA-GNNs [5] | GNN with Kolmogorov-Arnold network modules | Enhanced prediction accuracy & parameter efficiency; improved interpretability of learned chemical patterns. | Molecular property prediction |
| SA+PSO+CAM [6] | Hybrid metaheuristic optimization | High accuracy; avoids local minima; more efficient than sequential or single-algorithm methods. | Reactive force fields (ReaxFF) for specific systems (e.g., H/S) |
| BICePs Optimization [4] | Bayesian inference with replica averaging | Robust handling of noisy/sparse experimental data; automatic treatment of uncertainty and outliers. | Lattice models, polymers, and neural network potentials |
| ML Surrogate Models [7] | Replacing MD simulations with ML | Speeds up parameter optimization by a factor of ~20 while retaining force field quality. | Multi-scale parameter optimization (e.g., bulk density) |
Detailed Experimental Protocols
Protocol: GNN-Based Force Field Parameterization (e.g., ByteFF)
This protocol outlines the key steps for developing a GNN-based force field like ByteFF [1].
Dataset Curation:
- Objective: Assemble a expansive and chemically diverse dataset of small molecules and molecular fragments.
- Procedure: a. Select molecules representative of the target chemical space (e.g., drug-like compounds). b. Perform quantum mechanical calculations for all selected molecules. For ByteFF, this involved geometry optimization and frequency calculation at the B3LYP-D3(BJ)/DZVP level of theory to obtain energies, gradients, and Hessian matrices. c. Perform torsional scans for relevant dihedral angles to generate conformational energy profiles.
- Output: A QM dataset containing millions of data points (e.g., optimized geometries with energies/Hessians, torsion profiles).
Model Architecture and Training:
- Objective: Train a GNN to map molecular structures to MM parameters.
- Procedure: a. Representation: Represent each molecule as a graph with atoms as nodes and bonds as edges. Node features can include atom type, hybridization, formal charge, etc. b. Architecture: Implement a symmetry-preserving GNN. The ByteFF approach used an edge-augmented GNN to process structural information. c. Output Layer: Design the final network layer to predict all necessary MM parameters (equilibrium bond lengths, angle values, force constants, partial charges, etc.) simultaneously. d. Training: Train the model by minimizing a loss function that quantifies the difference between MM-calculated energies/properties (using the predicted parameters) and the reference QM data. This requires a differentiable MM engine within the training loop.
Validation and Benchmarking:
- Objective: Assess the performance and transferability of the resulting force field.
- Procedure: a. Evaluate the force field on held-out benchmark molecules. b. Key metrics include accuracy in predicting relaxed molecular geometries, torsional energy profiles, conformational energies, and atomic forces [1]. c. Compare performance against existing traditional and machine-learning force fields.
Protocol: Fine-Tuning a GNN Force Field to Experimental Data
This protocol describes how to refine a pre-trained GNN force field using experimental free energy data, as demonstrated with espaloma [8].
Foundation Model and Data Preparation:
- Objective: Establish a starting model and collect experimental data for fine-tuning.
- Procedure: a. Start with a pre-trained GNN force field (e.g., espaloma-0.3.2) as the foundation model. b. Gather a limited set of experimental free energy measurements, such as hydration free energies from the FreeSolv database. c. Prepare the molecular structures for the compounds in the dataset.
Efficient One-Shot Fine-Tuning:
- Objective: Adjust the model's parameters to better match experiment without costly re-simulation.
- Procedure: a. Low-Rank Projection: To improve data efficiency, project the GNN's high-dimensional atom embedding vectors to a lower-dimensional space for fine-tuning. b. Reweighting: Use an exponential (Zwanzig) reweighting estimator to compute the effect of changing force field parameters on the simulated free energies. This allows for parameter optimization using existing simulation data from the initial model. c. Effective Sample Size (ESS) Regularization: Apply regularization during optimization to ensure the fine-tuned force field retains sufficient overlap with the initial model, maintaining statistical reliability.
Validation:
- Objective: Confirm that fine-tuning improves prediction without degrading other properties.
- Procedure: Test the fine-tuned model on the target experimental property (e.g., hydration free energy) and related free energy predictions to assess transferability and overall improvement [8].
Table 2: Key Computational Tools and Resources for Force Field Parameterization
| Item / Resource | Function / Description | Relevance in Parameterization |
|---|---|---|
| Quantum Chemical Software (e.g., Gaussian, ORCA, PSI4) | Performs electronic structure calculations to generate high-quality reference data. | Provides target energies, forces, and Hessians for fitting bonded parameters and torsional profiles [1] [3]. |
| Differentiable MM Engine | A molecular mechanics engine that allows gradients to be backpropagated from the energy to the parameters. | Essential for the end-to-end training of GNN-based force field parameter predictors [1] [8]. |
| Automation & Optimization Libraries (e.g., for SA, PSO, GA) | Provides algorithms for automated parameter search and multi-objective optimization. | Enables efficient and robust parameter fitting against complex QM or experimental targets [2] [6]. |
| Bayesian Inference Software (e.g., BICePs) | Implements statistical reweighting and uncertainty quantification for ensemble-averaged data. | Used to refine parameters and conformational ensembles against noisy experimental data with unknown error [4]. |
| ML Surrogate Models | A machine learning model trained to predict simulation outcomes. | Dramatically speeds up parameter optimization loops by replacing expensive MD simulations [7]. |
| Amber/CHARMM Parameter Files | Standardized file formats for storing force field parameters. | The output target for new parameterization methods; ensures compatibility with major simulation packages [1] [3]. |
Workflow and Signaling Pathway Diagrams
GNN Parameterization Workflow
Iterative Parameter Refinement
Why Graphs? Representing Molecules as Nodes and Edges
In computational chemistry and drug discovery, the translation of molecular structures into a machine-readable format is a foundational step. The graph-based representation, which models atoms as nodes and bonds as edges, has emerged as a particularly powerful and intuitive method [9]. This approach directly mirrors the fundamental structure of molecules, providing a natural bridge between chemical reality and computational analysis. For researchers focused on predicting molecular mechanics parameters using Graph Neural Networks (GNNs), this representation is indispensable because it preserves the topological and relational information that governs molecular properties and interactions [5] [10]. Unlike simpler string-based representations like SMILES, graph structures natively encode the connectivity and local environments of atoms, which are critical for accurate physical property prediction [11] [9].
Theoretical Foundation: The Molecular Graph
Mathematical Formalism
A molecular graph is formally defined as a tuple ( G = (V, E) ), where:
- ( V ) is the set of nodes (atoms)
- ( E ) is the set of edges (bonds) connecting pairs of nodes [9].
This abstract mathematical structure is implemented computationally using matrices. The adjacency matrix (A) encodes connectivity, where element ( a_{ij} = 1 ) indicates a bond between atoms ( i ) and ( j ). The node feature matrix (X) contains atom-level attributes (e.g., atom type, formal charge), and the edge feature matrix (E) describes bond characteristics (e.g., bond type, bond length) [9]. This structured data format is ideally suited for processing by graph neural networks.
Advantages for Molecular Mechanics
Graph representations offer several distinct advantages for predicting molecular mechanics parameters:
- Structural Fidelity: They explicitly represent the connectivity and topology of a molecule, which directly determines its spatial conformation, vibrational modes, and steric interactions [9].
- Feature Integration: Both atomic properties (e.g., mass, hybridization) and bond properties (e.g., order, conjugation) can be incorporated as features, providing a comprehensive description of the molecular system [5] [10].
- Scale-Invariance: The representation is inherently independent of molecular size and complexity, making it suitable for modeling diverse chemical spaces [9].
Experimental Protocols: From Molecules to Predictions
Protocol 1: Constructing a Molecular Graph from a SMILES String
Objective: Convert a Simplified Molecular-Input Line-Entry System (SMILES) string into a structured molecular graph for GNN-based property prediction [10].
Materials:
- SMILES string of the target molecule.
- Cheminformatics library (e.g., RDKit).
Methodology:
- SMILES Parsing: Input the SMILES string into a parser to generate a 2D molecular structure.
- Node Identification: Identify each atom in the structure. Populate the node feature matrix ( X ) with atomic features (e.g., atom type, degree, formal charge, hybridization state).
- Edge Identification: Identify all covalent bonds between atoms. Construct the adjacency matrix ( A ), and populate the edge feature matrix ( E ) with bond features (e.g., bond type, conjugation).
- Graph Validation: Visually inspect the resulting graph against the original 2D structure to ensure fidelity.
Table 1: Default Node and Edge Features for Molecular Mechanics Studies
| Feature Type | Feature Description | Data Format | Role in Molecular Mechanics |
|---|---|---|---|
| Node Features | Atom type (e.g., C, N, O) | One-hot encoding | Defines element-specific properties |
| Atomic number | Integer | Correlates with van der Waals radius | |
| Hybridization (sp, sp², sp³) | One-hot encoding | Determines bond angles and geometry | |
| Partial charge | Continuous float | Influences electrostatic interactions | |
| Number of bonded hydrogens | Integer | Affects local steric environment | |
| Edge Features | Bond type (single, double, triple, aromatic) | One-hot encoding | Determines bond length and strength |
| Bond length (if 3D data available) | Continuous float (Å) | Critical for strain energy and conformation | |
| Bond stereochemistry | One-hot encoding | Affects chiral centers and isomerism | |
| Graph distance between nodes | Integer | Captures long-range intramolecular interactions |
Protocol 2: Implementing a GNN for Property Prediction
Objective: Train a Graph Neural Network to predict a target molecular mechanics parameter (e.g., dipole moment, HOMO-LUMO gap) [12] [10].
Materials:
- Dataset of molecular graphs (e.g., QM9 [12] [10]).
- Deep learning framework with GNN support (e.g., PyTorch Geometric).
Methodology:
- Data Preparation: Split the dataset into training, validation, and test sets (common ratio: 80/10/10).
- Model Selection: Choose a GNN architecture suitable for the task. For example:
- Message Passing: In each layer, nodes aggregate feature information from their neighbors. For a GCN, the update for node ( i ) is: [ hi^{(l+1)} = \sigma \left( \sum{j \in \mathcal{N}(i) \cup {i}} \frac{1}{\sqrt{\hat{d}i \hat{d}j}} hj^{(l)} W^{(l)} \right) ] where ( \mathcal{N}(i) ) are the neighbors of ( i ), ( \hat{d}i ) is the node degree, ( h_j^{(l)} ) are features, ( W^{(l)} ) is a learnable weight matrix, and ( \sigma ) is a non-linear activation [10].
- Readout / Pooling: After ( L ) message-passing layers, generate a single graph-level embedding ( hG ) from the set of node embeddings ( {h1^{(L)}, ..., h_n^{(L)}} ) for molecular property prediction.
- Training and Evaluation: Train the model using a regression loss (e.g., Mean Squared Error) and evaluate performance on the test set using metrics like Root Mean Square Error (RMSE) and Mean Absolute Error (MAE) [10].
Diagram 1: GNN Workflow for Molecular Property Prediction.
Advanced Architectures and Applications
Enhanced GNN Frameworks
Recent research has developed advanced GNN frameworks that integrate novel components to boost performance in molecular tasks:
Kolmogorov–Arnold GNNs (KA-GNNs): These models integrate Kolmogorov–Arnold networks (KANs) into the core components of GNNs—node embedding, message passing, and readout [5]. KA-GNNs use learnable univariate functions (e.g., based on Fourier series) on edges instead of fixed activation functions on nodes. This leads to improved expressivity, parameter efficiency, and interpretability compared to standard GNNs based on multi-layer perceptrons (MLPs) [5]. Experimental results show that KA-GNNs consistently outperform conventional GNNs in prediction accuracy and computational efficiency across multiple molecular benchmarks [5].
Quantized GNNs: To address the high computational and memory demands of GNNs, quantization techniques represent model parameters (weights, activations) using fewer bits [10]. Applying the DoReFa-Net quantization algorithm to GNN models can significantly reduce memory footprint and inference latency while maintaining predictive performance, making deployment on resource-constrained devices feasible [10]. Performance is maintained well at 8-bit precision for tasks like predicting quantum mechanical properties, though aggressive 2-bit quantization can lead to significant degradation [10].
Table 2: Performance Comparison of GNN Architectures on Molecular Tasks
| Model Architecture | Dataset | Target Property | Key Metric | Reported Performance | Key Advantage |
|---|---|---|---|---|---|
| KA-GNN [5] | Multiple Molecular Benchmarks | Various Properties | Accuracy | Consistent Improvement over baselines | High accuracy & interpretability |
| GNN + Quantization (8-bit) [10] | QM9 | Dipole Moment (μ) | RMSE | Comparable to Full-Precision | High computational efficiency |
| GNN + Quantization (2-bit) [10] | QM9 | Dipole Moment (μ) | RMSE | Significant performance degradation | (High compression) |
| DIDgen (Inverse Design) [12] | QM9 | HOMO-LUMO Gap | Success Rate (within 0.5 eV of target) | Comparable or better than genetic algorithms | Direct molecular generation |
Application: Inverse Molecular Design
A powerful application of GNNs is inverse molecular design, where the goal is to generate novel molecular structures with desired target properties. The DIDgen (Direct Inverse Design Generator) method leverages the differentiability of a pre-trained GNN property predictor [12]. It starts from a random graph or existing molecule and performs gradient ascent on the input graph (adjusting the adjacency and feature matrices) to optimize the target property, while enforcing chemical validity constraints [12]. This approach can generate diverse molecules with specific electronic properties, such as HOMO-LUMO gaps, verified by density functional theory (DFT) calculations [12].
Diagram 2: Inverse Design via GNN Gradient Ascent.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Computational Reagents for Molecular Graph Research
| Item / Resource | Category | Function / Application | Example / Note |
|---|---|---|---|
| RDKit | Cheminformatics Library | Construction, manipulation, and featurization of molecular graphs from SMILES or other formats. | Open-source; provides features for nodes (atoms) and edges (bonds). |
| PyTorch Geometric (PyG) | Deep Learning Library | Implements popular GNN architectures (GCN, GIN, GAT) and provides molecular graph datasets. | Standard library for building and training GNN models. |
| QM9 Dataset | Benchmark Dataset | Contains 130k+ small organic molecules with quantum mechanical properties (e.g., dipole moment, HOMO-LUMO gap). | Used for training and benchmarking models for molecular mechanics prediction [12] [10]. |
| MoleculeNet | Benchmark Suite | A collection of diverse molecular datasets for property prediction tasks. | Includes ESOL (solubility), FreeSolv (hydration energy), Lipophilicity, etc. [10]. |
| DIDgen Framework | Generative Model | Enables inverse molecular design by optimizing graph inputs via gradient ascent on a pre-trained GNN. | Generates molecules with targeted electronic properties [12]. |
| DoReFa-Net Algorithm | Quantization Tool | Reduces the memory footprint and computational cost of GNNs by quantizing weights and activations to low-bit precision. | Enables deployment on resource-constrained hardware [10]. |
Graph Neural Networks (GNNs) have become indispensable tools in computational chemistry and drug discovery, providing a powerful framework for learning from molecular graph structures where atoms represent nodes and chemical bonds represent edges [13]. Among the various GNN architectures, Message Passing Neural Networks (MPNNs), Graph Attention Networks (GATs), and Graph Convolutional Networks (GCNs) constitute the foundational pillars for molecular property prediction [14] [15]. These architectures naturally operate on the graph-structured representation of molecules, enabling end-to-end learning of task-specific molecular representations that capture intricate atomic interactions and topological patterns [13] [14]. The significance of these core architectures lies in their ability to overcome limitations of traditional hand-crafted molecular descriptors by directly learning from molecular topology and features [14] [10].
The MPNN framework provides a universal formulation that generalizes many GNN variants, operating through iterative message exchange between connected nodes [14]. GCNs implement spectral graph convolutions with normalized aggregation of neighbor information [15], while GATs incorporate attention mechanisms to weigh the importance of neighboring nodes dynamically [5] [15]. Recent advances have further enhanced these architectures through Kolmogorov-Arnold network (KAN) integrations, bidirectional message passing, and spatial descriptor incorporations, pushing the boundaries of predictive performance for molecular mechanics parameters [5] [15].
Theoretical Foundations of Core GNN Architectures
Mathematical Formulations
The Message Passing Neural Network (MPNN) framework provides a generalized mathematical structure that encompasses many GNN variants [14]. The core MPNN operates through three fundamental phases: message passing, feature update, and readout. For a molecular graph G with node features (hv) and edge features (e{vw}), the message passing at step (t+1) is defined as:
[mv^{t+1} = \sum{w \in N(v)} Mt(hv^t, hw^t, e{vw})]
where (M_t) is the message function and (N(v)) denotes the neighbors of node (v) [14]. The node update function then computes:
[hv^{t+1} = Ut(hv^t, mv^{t+1})]
where (U_t) is the update function [14]. After T message passing steps, a readout function generates the graph-level representation:
[\hat{y} = R({h_v^T | v \in G})]
where R must be permutation invariant to handle variable node orderings [13] [14].
Graph Convolutional Networks (GCNs) implement a specific instantiation of this framework using a convolution-like aggregation function with normalized feature propagation [15]. The layer-wise propagation rule in GCNs is:
[hv^{t+1} = \sigma \left( \sum{w \in N(v) \cup {v}} \frac{1}{\sqrt{dv dw}} h_w^t W^t \right)]
where (dv) and (dw) are node degrees, and (W^t) is a learnable weight matrix [15]. This normalization balances the contribution of highly connected nodes, preventing over-smoothing while enabling feature propagation across the graph.
Graph Attention Networks (GATs) enhance the basic MPNN framework by incorporating attention mechanisms that assign learnable importance weights to neighbors [15]. The attention mechanism in GATs computes:
[\alpha{vw} = \frac{\exp(\text{LeakyReLU}(a^T[Whv || Whw]))}{\sum{k \in N(v)} \exp(\text{LeakyReLU}(a^T[Whv || Whk]))}]
where (a) is a learnable attention vector, (W) is a weight matrix, and (||) denotes concatenation [15]. The node update then becomes:
[hv^{t+1} = \sigma \left( \sum{w \in N(v)} \alpha{vw} Whw^t \right)]
This attention mechanism enables dynamic, task-specific weighting of neighbor influences, improving model expressivity and interpretability [15].
Architectural Properties and Molecular Applications
These core GNN architectures maintain critical properties essential for molecular modeling. Permutation invariance ensures predictions are unchanged by node reordering, while permutation equivariance guarantees consistent feature transformations across the graph [15]. The relational inductive bias inherent in MPNNs preferentially learns from connected nodes, making them particularly suitable for detecting functional groups associated with chemical properties [15].
For molecular applications, these architectures effectively capture both local connectivity and global structural information. Graph Isomorphism Networks (GIN), a variant of MPNNs, have demonstrated exceptional capability in capturing molecular topology, achieving up to 92.7% accuracy in molecular point group prediction tasks [16]. The bidirectional message passing schemes in modern MPNNs better reflect the symmetric nature of covalent bonds, enhancing molecular representation fidelity [15].
Table 1: Core GNN Architectural Properties for Molecular Applications
| Architecture | Key Mechanism | Molecular Advantage | Computational Consideration |
|---|---|---|---|
| MPNN | Message functions + node updates | Flexible framework for complex atomic interactions | High parameter count, moderate complexity |
| GCN | Normalized neighborhood aggregation | Efficient feature propagation | Fastest among GNNs, potential over-smoothing |
| GAT | Attention-weighted aggregation | Dynamic neighbor importance weighting | Doubled parameters vs GCN, enhanced interpretability |
Advanced Architectural Variants and Performance
Integrated and Enhanced Architectures
Recent research has developed sophisticated integrations that combine strengths across architectural families. The Kolmogorov-Arnold GNN (KA-GNN) framework integrates KAN modules into all three core GNN components: node embedding, message passing, and readout [5]. KA-GNN replaces standard multilayer perceptrons (MLPs) with Fourier-series-based univariate functions, enhancing function approximation capabilities and theoretical expressiveness [5]. This integration has spawned variants like KA-GCN and KA-GAT, which consistently outperform conventional GNNs in prediction accuracy and computational efficiency across multiple molecular benchmarks [5].
The Edge-Set Attention (ESA) architecture represents another significant advancement, treating graphs as sets of edges and employing purely attention-based learning [17]. ESA vertically interleaves masked and vanilla self-attention modules to learn effective edge representations while addressing potential graph misspecifications [17]. Despite its simplicity, ESA outperforms tuned message passing baselines and complex transformer models across more than 70 node and graph-level tasks, demonstrating exceptional scalability and transfer learning capability [17].
Bidirectional message passing with attention mechanisms has emerged as a particularly effective strategy, surpassing more complex models pre-trained on external databases [15]. This approach eliminates artificial directionality in molecular graphs, better reflecting the symmetric nature of covalent bonds. Simpler architectures that exclude redundant self-perception and employ minimalist message formulations have shown higher class separability, challenging the assumption that increased complexity always improves performance [15].
Quantitative Performance Comparison
Table 2: Performance Comparison of GNN Architectures on Molecular Benchmarks
| Architecture | QM9 Accuracy (MAE) | Point Group Prediction | Computational Efficiency | Key Advantage |
|---|---|---|---|---|
| Standard GCN | Baseline | N/A | Fastest | Simplicity, speed |
| Standard GAT | +5-10% vs GCN | N/A | Moderate | Dynamic weighting |
| KA-GNN | +15-20% vs GCN [5] | N/A | High | Expressivity, parameter efficiency |
| GIN | N/A | 92.7% accuracy [16] | Moderate | Captures graph isomorphisms |
| Bidirectional MPNN | Superior to classical MPNN [15] | N/A | Moderate | Molecular symmetry preservation |
| ESA | Outperforms tuned baselines [17] | N/A | High scalability | Transfer learning capability |
Experimental Protocols for Molecular Property Prediction
Protocol 1: KA-GNN Implementation for Molecular Property Prediction
Purpose: To implement a Kolmogorov-Arnold Graph Neural Network for enhanced molecular property prediction accuracy and interpretability [5].
Materials and Reagents:
- Dataset: QM9 dataset (130,831 molecules with 19 quantum mechanical properties) [10]
- Software: PyTorch Geometric, RDKit for molecular featurization
- Hardware: GPU with ≥8GB memory (NVIDIA RTX 2080+ recommended)
- KA-GNN Implementation: Fourier-based KAN layers for node embedding, message passing, and readout
Procedure:
- Data Preprocessing:
- Convert SMILES representations to molecular graphs with node and edge features
- Initialize atom features (atomic number, radius, hybridization) and bond features (bond type, conjugation)
- Split dataset into training (80%), validation (10%), and test (10%) sets
Model Architecture Configuration:
- Implement Fourier-KAN layers with basis functions: (\phi(x) = \sum{k=1}^K (ak \cos(kx) + b_k \sin(kx)))
- Construct KA-GCN variant with KAN-based node embedding initialization
- Configure message passing with 4-6 layers of residual KAN modules
- Implement graph-level readout using attention-based KAN pooling
Training Protocol:
- Initialize parameters using Xavier uniform distribution
- Use Adam optimizer with learning rate 0.001 (exponentially decayed by 0.95 every 50 epochs)
- Train with mean absolute error (MAE) loss function for 500 epochs
- Apply early stopping with patience of 30 epochs based on validation loss
Interpretation and Analysis:
- Extract attention weights from KA-GAT layers to identify chemically significant substructures
- Visualize Fourier basis functions to understand frequency patterns captured
- Compare predictive performance against baseline GCN and GAT models
Troubleshooting: If training instability occurs, reduce learning rate to 0.0005 or decrease KAN network width. For overfitting, apply edge dropout (0.1 rate) during message passing.
Protocol 2: Bidirectional MPNN with Spatial Descriptors
Purpose: To implement a bidirectional message passing network with 3D molecular descriptors for improved molecular mechanics parameter prediction [15].
Materials and Reagents:
- Dataset: Target-specific molecular dataset (e.g., MD17 for molecular dynamics)
- Spatial Descriptors: van der Waals radius, electronegativity, dipole polarizability
- Software: RDKit for 3D conformation generation and descriptor calculation
Procedure:
- Molecular Graph Construction:
- Generate 3D conformations for all molecules using RDKit MMFF94 force field optimization
- Extract element-like 2D features (atomic number, hybridization, bond types)
- Calculate spatial descriptors: van der Waals interactions, electronegativity differences
- Construct bidirectional edges for all covalent bonds
Model Architecture:
- Implement bidirectional message passing without self-perception connections
- Integrate attention mechanism with 4 attention heads
- Exclude convolution normalization factors based on target dataset characteristics
- Add residual connections between MPNN layers to prevent over-smoothing
Training Configuration:
- Use combined loss function: MAE for properties + auxiliary node classification
- Apply gradient clipping with maximum norm of 1.0
- Implement learning rate warmup for first 10 epochs
- Use batch size of 32 with graph-based batching
Validation and Interpretation:
- Visualize node-level predictions using colormaps on molecular structures
- Analyze attention weights to identify key molecular substructures
- Compare performance with and without spatial descriptors
Troubleshooting: If performance plateaus, increase spatial descriptor dimensionality or add multi-hop edges (2-hop, 3-hop) to capture angular information.
Research Reagent Solutions
Table 3: Essential Research Reagents for GNN Molecular Applications
| Reagent / Resource | Specifications | Application Context | Access Source |
|---|---|---|---|
| QM9 Dataset | 130,831 molecules, 19 quantum mechanical properties [10] | Benchmarking quantum property prediction | PyTorch Geometric MoleculeNet |
| ESOL Dataset | 1,128 molecules with water solubility data [10] | Solubility and pharmacokinetic prediction | PyTorch Geometric MoleculeNet |
| FreeSolv | 642 molecules with hydration free energy [10] | Solvation property prediction | PyTorch Geometric MoleculeNet |
| Lipophilicity Dataset | 4,200 molecules with octanol/water distribution coefficient [10] | Drug permeability and ADMET prediction | PyTorch Geometric MoleculeNet |
| RDKit | Cheminformatics library with 3D conformation generation | Molecular featurization and graph construction | Open-source Python package |
| PyTorch Geometric | GNN library with pre-built molecular graph layers | Model implementation and training | Open-source Python library |
| DoReFa-Net Quantization | Algorithm for model compression with flexible bit-widths [10] | Deployment on resource-constrained devices | Research implementation |
Architectural Visualizations
Message Passing Neural Network Framework
GNN Architecture Comparison
The core GNN architectures—Message Passing Neural Networks, Graph Attention Networks, and Graph Convolutional Networks—provide the fundamental building blocks for molecular property prediction in drug discovery and materials science. While each architecture offers distinct advantages, the emerging trend integrates their strengths into hybrid models like KA-GNNs and bidirectional MPNNs with attention mechanisms [5] [15]. These advanced architectures demonstrate superior performance by combining expressive power with computational efficiency while maintaining chemical interpretability.
Future directions point toward simplified yet powerful architectures that eliminate redundant components while incorporating essential molecular descriptors. The integration of 3D spatial information with 2D topological graphs provides a balanced approach that preserves predictive performance while reducing computational costs by over 50% [15], making these architectures particularly advantageous for high-throughput virtual screening campaigns in drug discovery pipelines.
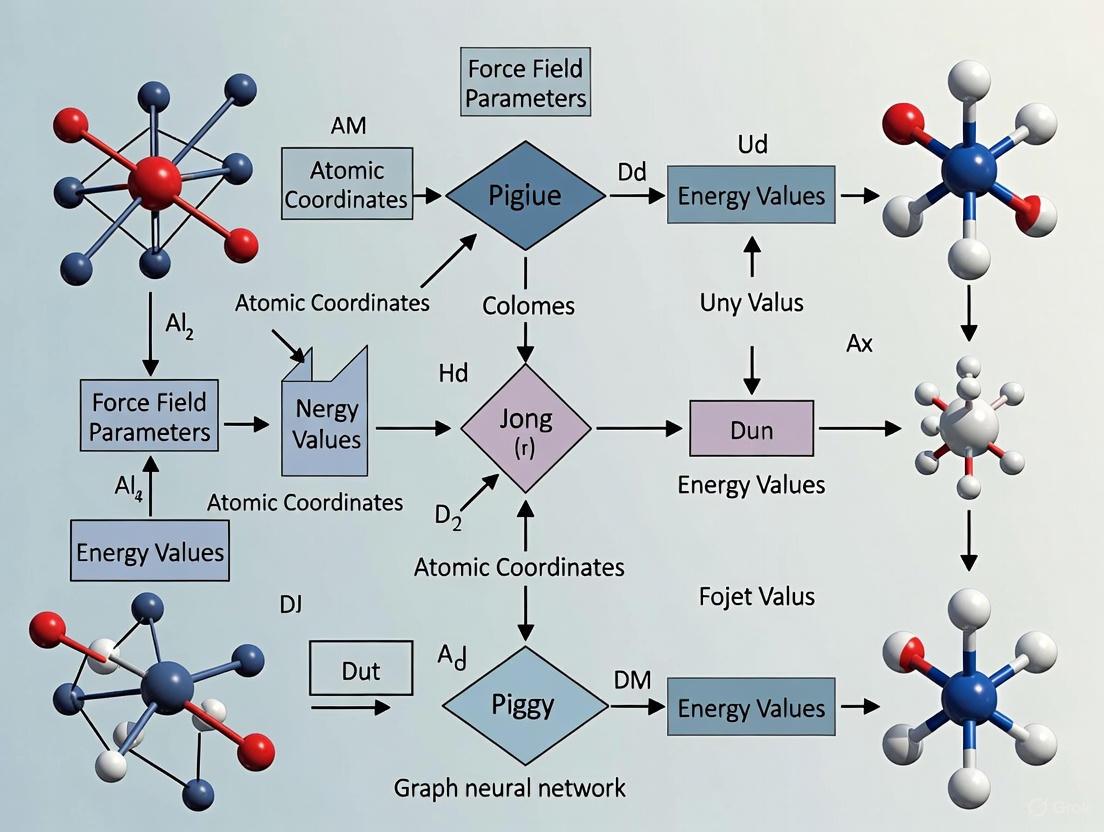
Molecular Mechanics (MM) force fields are the computational cornerstone for simulating large biological systems, such as proteins and nucleic acids, over physiologically relevant timescales. Traditional MM force fields operate by using a pre-defined set of atom types to assign parameters for the energy function via lookup tables. This approach, while computationally efficient, often lacks the accuracy and transferability required for exploring diverse regions of chemical space. The molecular graph—a representation where atoms are nodes and bonds are edges—naturally encodes the topology and connectivity of a molecule. This makes it an ideal foundation for rethinking parameter assignment. Framing MM parameter prediction as a graph learning task harnesses the power of Graph Neural Networks (GNNs) to learn complex, non-linear relationships between a molecule's structure and its optimal force field parameters, creating a synergistic partnership between physical modeling and data-driven learning.
This paradigm shift, exemplified by next-generation force fields like Grappa, moves away from hand-crafted rules and instead uses GNNs to predict MM parameters directly from the molecular graph [18] [19]. The synergy lies in the marriage of MM's computationally efficient energy functional form with the expressive power and accuracy of modern graph learning, enabling simulations that are both fast and highly accurate.
Graph Learning Architectures for Molecular Mechanics
The application of GNNs to molecular graphs relies on a message-passing framework, where nodes iteratively aggregate information from their neighbors to build rich representations of their local chemical environment [20]. Two advanced architectures demonstrate the cutting edge of this approach.
Grappa: A Graph Attention and Transformer-Based Approach
Grappa is a machine-learned molecular mechanics force field that employs a graph attentional neural network to generate atom embeddings from the molecular graph, capturing the local chemical environment without hand-crafted features [18] [19]. A key innovation is its use of a transformer with symmetry-preserving positional encoding to predict the MM parameters (bond force constants k_ij, equilibrium bond lengths r^(0)_ij, etc.) from these embeddings [18].
Critically, the Grappa architecture is designed to respect the inherent permutation symmetries of the MM energy function. For instance, the energy contribution from a bond between atoms i and j must be invariant to the order of the atoms (ξ(bond)_ij = ξ(bond)_ji). Grappa builds these physical constraints directly into the model, ensuring that predicted parameters are physically meaningful [18].
Kolmogorov-Arnold Graph Neural Networks (KA-GNNs)
An emerging alternative enhances GNNs using the Kolmogorov-Arnold representation theorem. KA-GNNs replace standard linear transformations and activation functions in GNNs with learnable univariate functions, often based on Fourier series or B-splines, leading to improved expressivity and parameter efficiency [5].
KA-GNNs can be integrated into all core components of a GNN:
- Node Embedding: Initial atom features are transformed using KAN layers.
- Message Passing: Message aggregation and updating functions are handled by KAN modules.
- Readout: Graph-level representations are constructed using KANs [5].
This architecture has shown superior performance in molecular property prediction tasks, suggesting its potential for capturing the complex functional relationships required for accurate MM parameter assignment [5].
Application Notes: Protocols and Performance
Experimental Workflow for a Grappa-Based Force Field
The following diagram outlines the end-to-end workflow for developing and deploying a machine-learned MM force field like Grappa.
Quantitative Performance Comparison
The table below summarizes the performance of Grappa against traditional and other machine-learned force fields on benchmark tasks. The data demonstrates that Grappa achieves state-of-the-art accuracy while maintaining the computational cost of traditional MM force fields [18] [19].
Table 1: Performance Comparison of Molecular Mechanics Force Fields
| Force Field | Type | Small Molecule Energy Accuracy (RMSE) | Peptide/RNA Performance | Computational Cost | Transferability to Macromolecules |
|---|---|---|---|---|---|
| Grappa | Machine-Learned (GNN) | Outperforms tabulated & ML MM FFs [19] | State-of-the-art MM accuracy; agrees with expt. J-couplings [18] | Same as traditional MM FFs [18] | Demonstrated (proteins, virus particle) [18] |
| Traditional MM (e.g., AMBER) | Tabulated (Atom Types) | Baseline | Good, may require corrections like CMAP [18] | Baseline (Highly efficient) | Excellent |
| Espaloma | Machine-Learned (GNN) | Lower accuracy than Grappa on benchmark [18] | Good | Same as traditional MM FFs | - |
| E(3)-Equivariant NN | Machine-Learned (Geometric) | Very High | N/A | Several orders of magnitude higher than MM [18] | Often limited by cost |
Detailed Experimental Protocol
Protocol 1: Training a Grappa Model for Protein Simulation
This protocol details the steps for training a Grappa force field model to predict energies and forces for peptides and proteins.
Objective: To train a GNN model that predicts MM parameters for a given molecular graph, minimizing the discrepancy between MM-calculated and reference quantum mechanical (QM) energies and forces.
Materials and Input Data:
- Training Dataset: A curated set of molecular graphs and their corresponding conformations with reference QM energies and forces. Example: The Espaloma dataset, containing over 14,000 molecules and more than one million conformations for small molecules, peptides, and RNA [18].
- Model Architecture: Grappa's graph attentional network and symmetric transformer architecture [18].
- Software Framework: A deep learning framework like PyTorch or TensorFlow, and a molecular dynamics engine (GROMACS, OpenMM) for energy evaluation [18].
- Hardware: Modern GPUs for efficient model training.
Procedure:
- Data Preparation:
- Assemble a dataset of molecular graphs and their diverse conformations.
- Generate reference QM energies and atomic forces for each conformation using a QM software package (e.g., Gaussian, ORCA).
Model Configuration:
- Initialize the Grappa model, which consists of a graph attentional network for generating atom embeddings and a symmetric transformer for predicting MM parameters (
ξ) for bonds, angles, and dihedrals [18]. - The model output is a complete set of MM parameters for the input molecular graph.
- Initialize the Grappa model, which consists of a graph attentional network for generating atom embeddings and a symmetric transformer for predicting MM parameters (
End-to-End Training:
- The training loop is as follows:
a. For a given molecular graph and conformation, the model predicts MM parameters
ξ. b. The MM energy functionE_MM(x, ξ)is evaluated using the predicted parameters and the molecular conformationx[18]. c. The loss function is computed by comparing the predicted MM energies and forces to the reference QM energies and forces. d. Model parameters are updated via backpropagation through the entire computational graph, including the differentiable MM energy function.
- The training loop is as follows:
a. For a given molecular graph and conformation, the model predicts MM parameters
Validation and Testing:
- Validate the model on a held-out set of molecules from the training dataset.
- Test the model's transferability on unseen molecular systems, such as peptide radicals or small proteins like chignolin, by assessing its ability to reproduce experimental observables like J-couplings or folding free energies [18].
Deployment in MD Simulations:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Resources for Developing Machine-Learned Force Fields
| Tool / Resource | Function | Application in MM Parameter Prediction |
|---|---|---|
| Graph Neural Network Libraries (PyTorch Geometric, DGL) | Provides pre-built modules for implementing GNN architectures. | Used to construct the core graph learning model (e.g., Grappa's graph attention layers) [18]. |
| Quantum Chemistry Software (Gaussian, ORCA) | Generates high-quality reference data. | Computes QM energies and forces for molecular conformations in the training dataset [18]. |
| Molecular Dynamics Engines (GROMACS, OpenMM) | Performs highly optimized energy and force calculations and MD simulations. | Serves as the backend to evaluate the MM energy function E_MM(x, ξ) with predicted parameters [18] [19]. |
| Benchmark Datasets (e.g., Espaloma Dataset) | Standardized datasets for training and evaluation. | Provides a diverse set of molecules (small molecules, peptides, RNA) for model development and comparison [18]. |
| Kolmogorov-Arnold Network (KAN) Modules | A promising alternative to MLPs for greater expressivity. | Can be integrated into GNNs (KA-GNNs) for node embedding, message passing, and readout, potentially improving accuracy [5]. |
Logical Framework of the Grappa Architecture
The following diagram details the core architecture of Grappa, illustrating how permutation symmetries are maintained during the prediction of different MM parameter types.
Framing MM parameter prediction as a graph learning task represents a paradigm shift with immediate and significant benefits. The synergy between the physical interpretability and efficiency of Molecular Mechanics and the accuracy and adaptability of Graph Neural Networks has been proven technically feasible and scientifically valuable by force fields like Grappa. This approach delivers state-of-the-art accuracy across small molecules, peptides, and RNA while retaining the computational efficiency necessary to simulate massive systems like an entire virus particle [18].
Future research will likely focus on several key areas: extending the graph representation to include non-covalent interactions explicitly, which has been shown to boost performance in other molecular property prediction tasks [5]; integrating geometric information (3D coordinates) alongside the topological graph to better capture steric and electrostatic effects; and the continued development of more expressive and efficient GNN architectures, such as KA-GNNs, to push further toward chemical accuracy. This synergistic framework is poised to remain a driving force in the next generation of biomolecular simulation.
Architectures in Action: Methodological Advances and Real-World Applications
Molecular Mechanics (MM) force fields are the empirical backbone of molecular dynamics (MD) simulations, enabling the study of biomolecular structure, dynamics, and interactions at scales inaccessible to quantum mechanical methods. Traditional MM force fields rely on discrete atom-typing rules and lookup tables for parameter assignment, a process that is both labor-intensive and limited in its ability to cover expansive chemical space [18] [21]. The emergence of graph neural networks (GNNs) has catalyzed a paradigm shift, allowing for the development of end-to-end machine learning frameworks that directly predict MM parameters from molecular graphs.
This Application Note details two pioneering GNN-driven frameworks: Grappa (Graph Attentional Protein Parametrization) and Espaloma (Extensible Surrogate Potential Optimized by Message Passing). These frameworks replace traditional atom-typing schemes with continuous learned representations of chemical environments, enabling accurate, transferable, and automated parameter prediction for diverse molecular classes [18] [21]. We provide a comprehensive technical comparison, detailed protocols for implementation, and resource guidance for researchers seeking to integrate these tools into their computational workflows.
Framework Architectures and Core Methodologies
The Grappa Architecture
Grappa employs a sophisticated two-stage architecture to predict molecular mechanics parameters, leveraging a graph neural network followed by a transformer model with specialized symmetry preservation.
- Stage 1: Atom Embedding Generation. A graph attentional neural network processes the 2D molecular graph to generate d-dimensional atom embeddings (( \nu_i \in \mathbb{R}^d )). These embeddings are designed to represent the local chemical environment of each atom, forming a continuous analogue to discrete atom types used in traditional force fields [18] [22] [23].
- Stage 2: Symmetry-Preserving Parameter Prediction. The atom embeddings are passed to a transformer model with symmetry-preserving positional encoding. This stage uses permutation-equivariant layers followed by symmetric pooling to predict the final MM parameters (( \xi^{(l)}{ij...} )) for each interaction type
l(bonds, angles, torsions, impropers). The architecture is explicitly constrained to respect the required permutation symmetries of the MM energy function, ensuring physical consistency [18] [24]. For instance, bond parameters are symmetric (( \xi^{(bond)}{ij} = \xi^{(bond)}{ji} )), and torsion parameters are symmetric with respect to reversal (( \xi^{(torsion)}{ijkl} = \xi^{(torsion)}_{lkji} )) [18].
A key feature of Grappa is its separation of bonded and non-bonded parameter prediction. The current version of Grappa predicts only bonded parameters (force constants and equilibrium values for bonds, angles, and dihedrals), while non-bonded parameters (partial charges, Lennard-Jones) are sourced from a traditional force field. This hybrid approach combines the accuracy of machine-learned bonded terms with the proven stability of established non-bonded models [24].
The Espaloma Architecture
Espaloma also utilizes graph neural networks to create continuous atomic representations and predict MM parameters in an end-to-end differentiable manner.
- Chemical Perception via GNNs: Espaloma replaces rule-based atom-typing with a GNN that operates on the molecular graph. This network generates atomic representations that capture the chemical environment of each atom [21].
- Differentiable Parameter Assignment: These representations are fed into symmetry-preserving pooling layers and feed-forward neural networks to predict the full set of Class I MM force field parameters. The entire process—from graph to parameters—is differentiable, allowing the model to be trained end-to-end by minimizing the difference between MM and quantum chemical (QM) energies and forces [21].
- Self-Consistent Parametrization: A significant advantage of Espaloma is its ability to self-consistently parametrize heterogeneous systems, such as protein-ligand complexes, using a single unified model. This avoids the potential incompatibilities that can arise from combining separate force fields for different molecule classes [21].
Table 1: Comparative Overview of Grappa and Espaloma Frameworks
| Feature | Grappa | Espaloma |
|---|---|---|
| Core Architecture | Graph Attentional Network + Symmetric Transformer [18] | Graph Neural Networks + Symmetry-Preserving Pooling [21] |
| Parameter Scope | Bonds, Angles, Proper/Improper Dihedrals (Bonded only) [24] | Bonds, Angles, Dihedrals, Non-bonded (Charge, vdW) [21] |
| Symmetry Handling | Explicit permutation constraints via model architecture [18] | Symmetry-preserving pooling layers [21] |
| Key Innovation | High accuracy for peptides/proteins; no hand-crafted input features [18] | Self-consistent parametrization across diverse molecular classes [21] |
| MD Engine Integration | GROMACS, OpenMM [24] [25] | OpenMM, Interoperable via Amber/OpenFF formats [21] |
Diagram 1: Grappa's two-stage prediction architecture.
Performance and Benchmarking
Both Grappa and Espaloma have been rigorously benchmarked against traditional and machine-learned force fields, demonstrating significant advancements in accuracy.
Grappa's Performance: Grappa was evaluated on the Espaloma benchmark dataset, which contains over 14,000 molecules and more than one million conformations. It was shown to outperform traditional MM force fields and the machine-learned Espaloma force field in terms of accuracy [18] [22]. Notably, Grappa closely reproduces quantum mechanical (QM) potential energy landscapes and experimentally measured J-couplings for peptides. It has also demonstrated success in protein folding simulations, with MD simulations recovering the experimentally determined native structure of small proteins like chignolin from an unfolded state [18]. Grappa showcases its extensibility by accurately parametrizing peptide radicals, an area typically outside the scope of traditional force fields [18].
Espaloma's Performance: The espaloma-0.3 model was trained on a massive dataset of over 1.1 million QM energy and force calculations. It accurately reproduces QM energetic properties for small molecules, peptides, and nucleic acids [21]. The force field maintains QM energy-minimized geometries of small molecules and preserves the condensed-phase properties of peptides and folded proteins. Crucially, espaloma-0.3 can self-consistently parametrize proteins and ligands, leading to stable simulations and highly accurate predictions of protein-ligand binding free energies, a critical task in drug discovery [21].
Table 2: Key Quantitative Benchmark Results
| Framework | Training Data | Key Benchmark Result | Reported System Performance |
|---|---|---|---|
| Grappa | >1M conformations (Espaloma dataset) [18] | Outperforms traditional MM & Espaloma on Espaloma benchmark [18] | Folds small protein chignolin; stable MD of a virus particle [18] |
| Espaloma-0.3 | >1.1M QM energy/force calculations [21] | Reproduces QM energetics for small molecules, peptides, nucleic acids [21] | Stable simulations; accurate protein-ligand binding free energies [21] |
| ByteFF | 2.4M optimized fragments, 3.2M torsion profiles [26] | State-of-the-art on relaxed geometries, torsional profiles, conformational energies/forces [26] | Exceptional accuracy for intramolecular conformational PES [26] |
Diagram 2: Espaloma's end-to-end differentiable training process.
Application Protocols
Protocol: Using Grappa in GROMACS and OpenMM
This protocol outlines the steps to parametrize a molecular system with Grappa for subsequent MD simulation.
A. Prerequisites
- A molecular structure file (e.g., PDB).
- A working installation of GROMACS or OpenMM.
- Grappa installed in a CPU-mode environment (GPU not required for inference) [24].
B. GROMACS Workflow
- Initial Parametrization: Use
gmx pdb2gmxwith a traditional force field to generate initial topology and structure files. This step establishes the molecular graph and provides the non-bonded parameters. - Grappa Parameter Prediction: Run the
grappa_gmxcommand-line application to create a new topology file with Grappa-predicted bonded parameters. (The-t grappa-1.4flag specifies the pretrained model version, and-pgenerates a plot of parameters for inspection.) [24] - Proceed with Simulation: Continue the standard GROMACS simulation workflow (solvation, energy minimization, equilibration, production) using the new topology file (
topology_grappa.top).
C. OpenMM Workflow
- Create a Classical System: Parametrize your system using OpenMM's
ForceFieldwith a traditional forcefield to obtain non-bonded parameters. - Apply Grappa: Use the
OpenmmGrappawrapper class to replace the bonded parameters in the system with those predicted by Grappa. Alternatively, use theas_openmmfunction to get aForceFieldobject that calls Grappa automatically. [24]
Protocol: Using Espaloma for Parametrization
While specific command-line protocols for the latest espaloma-0.3 are less detailed in the provided results, the general workflow based on its design is as follows:
- System Preparation: Prepare the molecular system of interest as a chemical graph. Espaloma is designed to handle small molecules, peptides, nucleotides, and their complexes.
- Parameter Prediction: Run the Espaloma model on the molecular graph. The model outputs a complete set of Class I MM parameters, including valence and non-bonded terms, in a format compatible with OpenMM or other engines that support Amber/OpenFF parameters.
- Simulation Execution: Load the Espaloma-generated parameters into your MD engine of choice and run the simulation. The self-consistent nature of the parameters ensures stability across heterogeneous molecular systems [21].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Software Solutions
| Item / Resource | Function / Description | Availability |
|---|---|---|
| Grappa (GitHub) | Primary library for training and applying Grappa models; includes integration code for GROMACS/OpenMM. | github.com/graeter-group/grappa [24] |
| Grappa Pretrained Models (e.g., grappa-1.4) | Off-the-shelf models for parameter prediction, covering peptides, small molecules, RNA, and radicals. | Downloaded automatically via the Grappa API [24] |
| Espaloma Package | Software implementation for training and deploying Espaloma force fields. | Open-source (repository linked in publication) [21] |
| OpenMM | A versatile, high-performance MD simulation toolkit with extensive scripting capabilities and GPU support. | openmm.org [24] |
| GROMACS | A widely used, high-performance MD simulation package. | gromacs.org [18] |
| Espaloma Benchmark Dataset | A public dataset of >14k molecules and >1M conformations for training and benchmarking ML force fields. | Referenced in original Espaloma publication [18] [21] |
| ByteFF Training Dataset | A large-scale, diverse QM dataset of 2.4M optimized molecular fragments and 3.2M torsion profiles. | Described in PMC article; availability likely subject to authors' terms [26] |
Grappa and Espaloma represent a transformative advance in molecular mechanics, moving the field from hand-crafted, discrete rule-based parametrization to automated, continuous, and data-driven frameworks. Grappa excels in its high accuracy for biomolecular systems like proteins and peptides and its seamless integration into established simulation workflows. Espaloma stands out for its comprehensive self-consistent parametrization across diverse chemical domains and its end-to-end differentiability. The choice between them depends on the researcher's specific needs: Grappa for robust, high-accuracy biomolecular simulation with minimal computational overhead, and Espaloma for maximum consistency and coverage in heterogeneous chemical systems. Both frameworks significantly lower the barrier to obtaining accurate force field parameters, promising to accelerate research in drug discovery, materials science, and structural biology.
The accurate prediction of molecular mechanics parameters is a cornerstone of computational drug discovery, directly impacting the reliability of molecular dynamics simulations and virtual screening. Traditional methods often struggle with the expansive coverage of chemical space. The integration of two innovative architectures—Kolmogorov–Arnold Networks (KANs) with Graph Neural Networks (GNNs) and Graph Transformers (GTs)—offers a transformative framework for molecular property prediction. KA-GNNs enhance GNNs by replacing standard linear transformations with learnable, univariate functions, leading to superior parameter efficiency and interpretability [5] [27]. Concurrently, Graph Transformers, with their global self-attention mechanisms, provide a flexible and powerful alternative for modeling molecular structures [28]. This Application Note details the protocols for leveraging these architectures to advance the prediction of molecular mechanics force fields and related properties, providing a practical guide for researchers and scientists in the field.
Theoretical Foundation & Key Components
Kolmogorov-Arnold Networks (KANs) in a Nutshell
Inspired by the Kolmogorov-Arnold representation theorem, KANs present a radical departure from traditional Multi-Layer Perceptrons (MLPs). While MLPs apply fixed, non-linear activation functions on nodes, KANs place learnable univariate functions on edges [5] [27]. A multivariate continuous function can be represented as a composition of these simpler univariate functions and additions:
f(𝐱) = ∑Φq ( ∑ϕq,p (xp) )
The functions (ϕ, Φ) are parameterized using basis functions. Initial implementations used B-splines [29], but recent advances propose Fourier-series-based formulations (equation 4, 5 in [5]) which are particularly effective at capturing both low-frequency and high-frequency patterns in molecular graphs, providing strong theoretical approximation guarantees backed by Carleson’s theorem and Fefferman’s multivariate extension [5].
Graph Neural Networks (GNNs) and Graph Transformers (GTs)
GNNs operate on graph-structured data through a message-passing paradigm, where nodes aggregate information from their neighbors to build meaningful representations [30]. Standard GNNs use MLPs for updating node features during this process.
Graph Transformers adapt the self-attention mechanism to graphs, allowing each node to attend to all other nodes, thereby capturing long-range dependencies that local message-passing might miss [28]. They often incorporate structural and spatial information, such as topological distances or 3D geometries, through bias terms or positional encodings.
Protocol: Implementing KA-GNNs for Molecular Property Prediction
This protocol outlines the steps for constructing and training a KA-GNN model, specifically the KA-GCN variant, for predicting molecular mechanics parameters.
Model Architecture: KA-GCN
The following diagram illustrates the flow of information through the KA-GCN architecture, highlighting the integration of KAN layers into the core components of a Graph Convolutional Network.
Step-by-Step Experimental Procedure
Step 1: Data Preparation and Molecular Graph Construction
- Input Data: Utilize a large-scale, high-quality dataset of molecular structures with associated quantum mechanical properties. The OMol25 dataset is a prime example, containing over 100 million calculations at the ωB97M-V/def2-TZVPD level of theory, covering biomolecules, electrolytes, and metal complexes [31].
- Graph Representation: Represent each molecule as a graph ( G=(V, E) ), where:
- Nodes ((vi \in V)): Represent atoms. Initialize node features ((hi^0)) using atomic properties (e.g., atomic number, charge, number of hydrogens). The protocol from [32] suggests enriching these features using a circular algorithm inspired by Extended-Connectivity Fingerprints (ECFPs) that incorporates information from an atom's r-hop neighborhood.
- Edges ((e_{ij} \in E)): Represent chemical bonds (covalent) or non-covalent interactions (within a cut-off distance, e.g., 5 Å). Initialize edge features with bond type, bond length, or other interatomic spatial information [5] [27].
Step 2: Node and Edge Embedding with KANs
- Pass the initial atom features (e.g., atomic number, radius) through a Fourier-KAN layer to generate the initial node embeddings.
- For models incorporating explicit edge embeddings, pass bond features (e.g., bond type, length) through a separate Fourier-KAN layer [5].
Step 3: KAN-Augmented Message Passing
- For each node, aggregate messages from its neighboring nodes. In KA-GCN, this follows a standard GCN aggregation scheme.
- Update the node features using a residual Fourier-KAN layer instead of an MLP: ( hi^{(l+1)} = hi^{(l)} + \text{KAN}(hi^{(l)} + \sum{j \in N(i)} h_j^{(l)}) ) This formulation helps maintain network expressivity and mitigates oversmoothing in deeper architectures [5] [29].
Step 4: KAN-Based Graph Readout
- After (L) layers of message passing, generate a graph-level representation by pooling all node embeddings (e.g., using a mean or sum operation).
- Pass this pooled representation through a final Fourier-KAN module to produce the prediction for the target molecular property (e.g., energy, force field parameter) [5].
Step 5: Model Training and Evaluation
- Loss Function: Use a mean-squared error (MSE) loss for regression tasks like force field parametrization. For conservative force fields, ensure the loss penalizes deviations in predicted forces and energies [31]. ( \mathcal{L} = \frac{1}{N} \sum{i=1}^N (yi - \hat{y}_i)^2 )
- Training Strategy: Employ a two-phase training strategy if predicting conservative forces: first, train a direct-force model, then use it to initialize a conservative-force model for fine-tuning, which significantly reduces wallclock time [31].
- Benchmarking: Evaluate model performance on held-out test sets and standard benchmarks. Compare against state-of-the-art GNNs and GTs to quantify performance gains.
Protocol: Implementing Graph Transformers for Molecular Representation
This protocol describes the use of Graph Transformer models, which serve as a powerful alternative or complementary approach to KA-GNNs.
Model Architecture: 3D Graph Transformer
The diagram below outlines the architecture of a 3D Graph Transformer, which uses spatial distances to bias the self-attention mechanism.
Step-by-Step Experimental Procedure
Step 1: Data Preparation and Feature Engineering
- Use the same molecular graph construction as in Section 3.2, Step 1, but ensure access to 3D molecular conformers [28].
- For each atom, compute its 3D spatial coordinates.
Step 2: Node Feature Initialization and Structural Encoding
- Project initial atom features into a dense embedding vector.
- Compute the shortest path distance (for 2D-GT) or the real-space Euclidean distance (for 3D-GT) between all atom pairs.
- For 3D-GT, bin these distances (e.g., bin width of 0.5 Å, with a spherical cutoff of 5 Å) to create a discrete spatial bias matrix [28].
Step 3: Graph Transformer Layer with Structural Bias
- The self-attention score between node (i) and (j) is computed as: ( \alpha{ij} = \frac{ \text{Exp}(Qi Kj^T + B{ij}) }{ \sum{k} \text{Exp}(Qi Kk^T + B{ik}) } ) where (B_{ij}) is the bias term derived from the binned spatial distance between atoms (i) and (j). This integrates crucial 3D geometric information directly into the attention mechanism [28].
Step 4: Context-Enriched Training (Optional)
- To boost performance, especially in low-data regimes, employ context-enriched training [28]. This can involve:
- Pre-training the model on auxiliary, quantum-mechanically derived atomic-level properties.
- Multi-task learning with auxiliary losses on related properties during the main task training.
Step 5: Property Prediction and Model Evaluation
- Use a dedicated node or a pooling operation on the final node embeddings to get a graph-level representation.
- Pass this representation through a regression head to predict the target molecular mechanics parameter.
- Benchmark the model's performance against GNN baselines and other state-of-the-art methods.
Performance Benchmarking
The following tables summarize the performance of KA-GNNs and Graph Transformers as reported in recent literature.
Table 1: Performance of KA-GNN variants on molecular property benchmarks.
| Model Variant | Architecture Base | Key Innovation | Reported Performance | Citation |
|---|---|---|---|---|
| KA-GCN | Graph Convolutional Network | Fourier-KAN in embedding, message passing, and readout | Consistently outperforms conventional GNNs in accuracy and efficiency on seven molecular benchmarks | [5] |
| KANG | Message-Passing GNN | Spline-based KAN with data-aligned initialization | Outperforms GCN, GAT, and GIN on node (Cora, PubMed) and graph classification (MUTAG, PROTEINS) tasks | [29] |
| KA-GNN (Fourier) | General GNN | Fourier-series basis functions, non-covalent interactions | Surpasses existing state-of-the-art pre-trained models on several benchmark tests | [27] |
Table 2: Comparative performance of Graph Transformer (GT) and GNN models on specific molecular tasks.
| Dataset | Task | 3D-GT Performance (MAE) | GNN Performance (MAE) | Notes | Citation |
|---|---|---|---|---|---|
| BDE | Binding Energy Estimation | On par with best GNNs | Varies by model (e.g., PaiNN, SchNet) | GT models offer advantages in speed and flexibility | [28] |
| Kraken | Sterimol Parameters | On par with best GNNs | Varies by model (e.g., ChIRo) | Context-enriched training (pretraining) boosts GT performance | [28] |
| tmQMg | Property Prediction for TMCs | Competitive performance | Baseline set by GIN-VN, ChemProp | GT demonstrates strong generalization for transition metal complexes | [28] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key computational tools and datasets for implementing KA-GNNs and Graph Transformers.
| Tool/Resource | Type | Function in Research | Relevance |
|---|---|---|---|
| OMol25 Dataset | Dataset | Provides high-quality, massive-scale QM data for training and benchmarking. | Essential for training robust models on expansive chemical space [31]. |
| ByteFF | Benchmark/Model | An Amber-compatible force field predicted by a GNN; a benchmark for force field accuracy. | Target for property prediction; benchmark for model performance [1]. |
| Fourier-KAN Layer | Software Layer | Learnable activation function using Fourier series to capture complex molecular patterns. | Core component of KA-GNNs for enhanced expressivity [5] [27]. |
| Graphormer | Model Architecture | A leading Graph Transformer implementation that uses spatial biases. | Base architecture for GT protocols; highly flexible [28]. |
| RDKit | Cheminformatics Library | Handles molecule I/O, graph construction, and fingerprint generation. | Foundational for data preprocessing and feature extraction [32]. |
| eSEN / UMA | Model Architecture | State-of-the-art equivariant NNPs; benchmarks for energy and force prediction. | Represents the current state-of-the-art for end-to-end NNP performance [31]. |
The accurate prediction of molecular mechanics parameters is a cornerstone of modern computational chemistry and drug discovery. In this domain, Graph Neural Networks (GNNs) have emerged as transformative tools by natively processing molecular structures represented as graphs, where atoms correspond to nodes and chemical bonds to edges. However, a critical challenge persists: ensuring that these models produce predictions that are consistent with the fundamental laws of physics. This requires the deliberate incorporation of physical symmetries—specifically, E(3)-equivariance for Euclidean transformations (rotations, translations, and reflections) and permutation invariance for the interchange of identical particles.
E(3)-equivariance ensures that a model's predictions for a molecule's energy, forces, or Hamiltonian transform appropriately when the molecule itself is rotated or translated in space. For instance, the forces on atoms, which are vector quantities, should rotate in tandem with the molecular system. Permutation invariance guarantees that the model produces identical outputs for identical molecular structures, regardless of the arbitrary ordering of atoms in the input representation. The integration of these principles is not merely a theoretical exercise; it is essential for developing physically realistic, data-efficient, and generalizable models for molecular property prediction.
Theoretical Foundations and Key Concepts
E(3)-Equivariance in Molecular Systems
In molecular systems, several fundamental symmetries must be respected. A model's architecture must be:
- Translationally invariant: Predictions should not change if every atom in the system is moved by the same displacement vector.
- Rotationally equivariant: The model's vector-valued outputs (e.g., forces or dipole moments) should rotate correspondingly when the input molecule is rotated.
- Permutation invariant: Predictions must be unchanged when identical atoms (e.g., two hydrogen atoms in a methane molecule) are swapped in the input representation.
Formally, a function ( f ) that processes an atomic system is E(3)-equivariant if for any translation ( t ), rotation ( R ), and reflection that are elements of the E(3) group, the following holds: ( f(T{g}(x)) = T'{g}(f(x)) ), where ( Tg ) and ( T'g ) are transformations associated with the group element ( g ) acting on the input and output, respectively. For graph-level predictions such as energy, the requirement is often invariance, a special case of equivariance where ( T'g ) is the identity transformation, meaning ( f(T{g}(x)) = f(x) ) [33].
Permutation Invariance and Equivariance
For graph data, a function ( f ) acting on an adjacency matrix ( A ) satisfies:
- Permutation invariance if ( f(PAP^\top) = f(A) ) for any permutation matrix ( P ).
- Permutation equivariance if ( f(PAP^\top) = Pf(A)P^\top ).
Invariance is typically required for graph-level properties (e.g., total energy), while equivariance is necessary for node-level (e.g., atomic forces) or edge-level predictions. Designing models that inherently possess these properties eliminates the need for data augmentation over all possible permutations and rotations, significantly improving sample efficiency and generalization [34].
Quantitative Performance of Equivariant and Invariant Models
Performance Benchmarks for Molecular Property Prediction
Table 1: Performance comparison of E(3)-equivariant models on molecular property prediction tasks (MAE).
| Model | Dipole Moment | Polarizability | Hessian Matrix | Hyperpolarizability | Reference |
|---|---|---|---|---|---|
| EnviroDetaNet | Lowest MAE | Lowest MAE | 0.016 (MAE) | Lowest MAE | [35] |
| EnviroDetaNet (50% Data) | Slight increase | ~10% error increase | 0.032 (MAE) | Slight increase | [35] |
| DetaNet (Baseline) | Higher MAE | Higher MAE | Baseline MAE | Higher MAE | [35] |
| KA-GNN | Outperforms GCN/GAT | Outperforms GCN/GAT | - | - | [5] |
| NextHAM | - | - | - | - | [36] |
| E2GNN | Outperforms SchNet, MEGNet | Outperforms SchNet, MEGNet | - | - | [37] |
EnviroDetaNet, an E(3)-equivariant message-passing neural network, demonstrates state-of-the-art performance, achieving the lowest Mean Absolute Error (MAE) on properties like dipole moment and polarizability compared to other models like DetaNet. Remarkably, it maintains high accuracy even when trained with only 50% of the original data, showcasing its superior data efficiency and robust generalization capabilities. For instance, on the Hessian matrix prediction task, EnviroDetaNet's error only increased by 0.016 MAE with the halved dataset, remaining 39.64% lower than the original DetaNet model [35].
Table 2: Application-specific benchmarks for Hamiltonian and potential prediction.
| Model | Application | Key Metric | Performance | Reference |
|---|---|---|---|---|
| NextHAM | Materials Hamiltonian | R-space Hamiltonian Error | 1.417 meV | [36] |
| NextHAM | Materials Hamiltonian | SOC Block Error | sub-μeV scale | [36] |
| E2GNN | Interatomic Potentials | MD Simulation Accuracy | Achieves ab initio MD accuracy | [37] |
| Facet | Interatomic Potentials | Training Efficiency | >10x acceleration vs. SOTA | [38] |
| KA-GNN | Molecular Property Prediction | Accuracy & Efficiency | Superior to GCN/GAT baselines | [5] |
For electronic-structure Hamiltonian prediction in materials, NextHAM achieves an error of 1.417 meV in real space (R-space), with spin-orbit coupling (SOC) blocks suppressed to the sub-μeV scale, establishing it as a universal and highly accurate deep learning model [36]. In the realm of interatomic potentials, E2GNN consistently outperforms invariant baselines like SchNet and MEGNet and can achieve the accuracy of ab initio molecular dynamics (MD) across solid, liquid, and gas systems [37].
Efficiency and Data Requirements
A significant advantage of incorporating physical symmetries is the improvement in computational and data efficiency.
- EnviroDetaNet: Shows faster convergence and higher accuracy in the early stages of training compared to non-equivariant baselines, indicating better learning dynamics [35].
- Facet: Incorporates key innovations, such as replacing compute-intensive multi-layer perceptrons (MLPs) with efficient splines for processing interatomic distances. This enables comparable performance to leading approaches with significantly fewer parameters and less than 10% of the training computation, achieving a 2x speedup on crystal relaxation tasks [38].
Experimental Protocols and Application Notes
Protocol 1: Implementing an E(3)-Equivariant Model for Spectral Prediction
This protocol outlines the procedure for training and evaluating the EnviroDetaNet model for molecular spectra prediction, based on the methodology described in its source paper [35].
1. Problem Formulation and Data Preparation
- Objective: Predict molecular spectra (e.g., IR, Raman, UV, NMR) or quantum chemical properties (e.g., dipole moment, polarizability) from 3D molecular structures.
- Input Data: 3D atomic coordinates and atomic numbers for each molecule in the dataset.
- Target Data: Property labels calculated from quantum chemistry methods (e.g., Density Functional Theory).
- Recommended Dataset: QM9S, a standard benchmark for molecular property prediction.
2. Model Architecture: EnviroDetaNet
- Core Component: An E(3)-equivariant message-passing neural network.
- Key Innovation: Integration of intrinsic atomic properties, spatial features, and atomic environment information into a unified atom representation. This is crucial for capturing both local and global molecular features and mitigating issues like message over-smoothing.
- Implementation Details:
- Utilize an equivariant message-passing framework where node (atom) and edge (bond) features are updated in layers.
- Ensure all operations (e.g., convolutions, activations) are designed to be E(3)-equivariant. This often involves representing features as spherical tensors and using Clebsch-Gordan tensor products for their combination [33].
- Incorporate pre-trained atomic environment vectors (e.g., from Uni-Mol) as part of the node feature initialization.
3. Training Procedure
- Loss Function: Mean Absolute Error (MAE) or Mean Squared Error (MSE) between predicted and target properties.
- Training Regime:
- Use a maximum of 200 epochs.
- Employ a reduced dataset (e.g., 50%) to test the model's robustness and data efficiency, a scenario where EnviroDetaNet excels.
- Validation: Monitor validation MAE and R-squared (R²) to assess convergence and fitting performance.
4. Evaluation and Ablation
- Performance Metrics: Report MAE and R² on a held-out test set for all target properties.
- Ablation Study: Crucially, compare the full EnviroDetaNet model against a variant that removes molecular environment information (e.g., DetaNet-Atom). This study confirmed that environmental information is vital for model stability and accuracy, as its removal led to significant performance degradation and increased training loss fluctuation [35].
Protocol 2: Hamiltonian Prediction for Materials with Strict E(3)-Symmetry
This protocol is based on the NextHAM model, designed for predicting the electronic-structure Hamiltonian of complex material systems with high generalization capability [36].
1. Problem Formulation and Data Curation
- Objective: Predict the DFT-level Hamiltonian matrix ( \mathbf{H} ) of a material directly from its atomic configuration, bypassing the expensive self-consistent field loop.
- Dataset Curation (Materials-HAM-SOC):
- Collect a diverse set of material structures (e.g., 17,000 structures).
- Ensure broad coverage of chemical elements (e.g., 68 elements from the first six rows of the periodic table).
- Include high-quality physical effects, such as spin-orbit coupling (SOC), and use extensive orbital basis sets (e.g., up to 4s2p2d1f orbitals).
2. The NextHAM Framework: A Correction Approach
- Zeroth-Step Hamiltonian (( \mathbf{H}^{(0)} )):
- Compute this initial Hamiltonian from the initial electron density of DFT (sum of isolated atomic charge densities). This requires no matrix diagonalization and is computationally efficient.
- Use ( \mathbf{H}^{(0)} ) as a rich physical prior in the input features.
- Regression Target:
- Instead of learning the target Hamiltonian ( \mathbf{H}^{(T)} ) directly, the model learns the correction term ( \Delta\mathbf{H} = \mathbf{H}^{(T)} - \mathbf{H}^{(0)} ). This simplifies the learning task, compresses the output space, and facilitates fine-grained predictions.
3. Network Architecture
- Backbone: A neural Transformer architecture with strict E(3)-symmetry.
- Expressiveness: Extend methods like TraceGrad to the Transformer framework to combine strict symmetry with high non-linear expressiveness. TraceGrad uses an SO(3)-invariant supervision signal (the trace of the Hamiltonian) to guide the learning of non-linear, equivariant features [36].
4. Training Objective: A Joint Optimization
- Multi-Space Loss: To prevent errors in predicted eigenvalues and the appearance of "ghost states," optimize a joint loss function that considers both:
- The real-space (R-space) Hamiltonian.
- The reciprocal-space (k-space) Hamiltonian, from which band structures are derived.
- This ensures accuracy in both the direct Hamiltonian matrix and the resulting electronic properties.
5. Validation
- Primary Metrics: Error in the predicted R-space Hamiltonian (e.g., achieving ~1.4 meV) and k-space band structures.
- Generalization Test: Evaluate on materials containing elements not seen during training to assess the model's universality.
Figure 1: NextHAM Hamiltonian Prediction Workflow.
Protocol 3: Achieving Permutation Invariance in Graph Generative Models
This protocol addresses the specific challenges of building generative models for molecules that are invariant to node ordering [34].
1. Understanding the Permutation Invariance Requirement
- Goal: Ensure the learned probability distribution over graphs satisfies ( p\theta(\hat{A}) = p\theta(P \hat{A} P^\top) ) for any permutation matrix ( P ).
- Challenge: The empirical training data typically contains only one permutation (adjacency matrix) per distinct graph. A theoretically perfect permutation-invariant model would need to learn a distribution with ( O(n!m) ) modes (for n nodes and m training graphs), which is highly complex.
2. Model Design Strategies
- Equivariant Score Networks: For diffusion-based generative models, design the score network ( \mathbf{s}\theta(A) = \nabla{A} \log p\theta(A) ) to be permutation equivariant. Theorem 1 in [34] states that if the score is equivariant, then the implicitly defined log-likelihood ( \log p\theta(A) ) is permutation invariant.
- Alternative Practical Strategy: Empirical evidence suggests that non-invariant models can sometimes achieve better performance, especially when the training data lacks extensive permutation augmentation [34].
3. Post-Processing for Invariant Sampling
- Simple Trick: If a non-invariant model is used for its empirical performance, permutation-invariant sampling can be reclaimed via a simple post-processing step.
- Procedure: For each generated sample (adjacency matrix ( A )), apply a random permutation matrix ( Pr ), drawn uniformly from the set of all permutation matrices. The induced distribution of ( Ar = Pr A Pr^{\top} ) is provably permutation invariant (Lemma 3 in [34]). This ensures physically meaningful generation without compromising sample quality.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key computational tools, datasets, and model components for symmetry-aware GNN research.
| Tool/Resource | Type | Function in Research | Example/Note |
|---|---|---|---|
| QM9/QM9S Dataset | Dataset | Benchmark for molecular property prediction. | Used for training/evaluating models like EnviroDetaNet [35]. |
| Materials-HAM-SOC | Dataset | Benchmark for materials Hamiltonian prediction. | Contains 17k structures, 68 elements, SOC effects [36]. |
| MPTrj Dataset | Dataset | Training machine learning interatomic potentials. | Used for training models like Facet and E2GNN [37] [38]. |
| Zeroth-Step Hamiltonian | Physical Descriptor | Input feature and output correction target. | Provides rich physical prior, simplifies learning [36]. |
| Clebsch-Gordan Tensor Product | Mathematical Operation | Combines spherical tensor features equivariantly. | Core operation in many steerable equivariant networks [33]. |
| Fourier-Series KAN | Network Component | Learnable univariate function for enhanced approximation. | Used in KA-GNNs to capture structural patterns [5]. |
| Radial Basis Functions | Network Component | Encodes interatomic distances in message passing. | Used in models like SchNet and E2GNN [37]. |
| Random Permutation Layer | Post-Processing | Enforces permutation invariance for generative models. | Applied to outputs of non-invariant generative models [34]. |
Figure 2: Post-Processing for Permutation-Invariant Generation.
The deliberate incorporation of E(3)-equivariance and permutation invariance is a paradigm shift that moves molecular GNNs from being mere pattern recognizers to becoming engines of physically grounded prediction. As demonstrated by models like EnviroDetaNet, NextHAM, and KA-GNNs, this approach yields tangible benefits: state-of-the-art accuracy, remarkable data efficiency, and robust generalization to unseen molecular and material systems. The experimental protocols provided offer a practical roadmap for implementing these principles, whether the task is predicting molecular spectra, material Hamiltonians, or generating novel molecular structures. Future work will continue to bridge the gap between theoretical symmetry guarantees and empirical performance, further solidifying the role of symmetry-aware models as indispensable tools in computational chemistry and drug discovery.
Small Molecules: Predicting Quantum Mechanical and Physicochemical Properties
The application of Graph Neural Networks (GNNs) to small molecules represents one of the most mature and successful showcases in computational chemistry. GNNs natively model molecules as graphs, where atoms are nodes and bonds are edges, enabling highly accurate property predictions that accelerate drug discovery [39] [40].
Key Experimental Protocols and Performance
Extensive benchmarking on public datasets demonstrates the capability of GNNs to predict a wide array of molecular properties with high accuracy, often surpassing traditional descriptor-based machine learning methods. The following table summarizes quantitative performance data for various GNN models across standard datasets.
Table 1: GNN Performance on Small Molecule Property Prediction Benchmarks
| Dataset | Property Predicted | Model | Key Metric | Performance | Number of Molecules |
|---|---|---|---|---|---|
| QM9 [41] [10] | Atomization Energy | DTNN_7ib | MAE | 0.34 kcal/mol | ~134,000 |
| QM9 [5] | Various Quantum Properties | KA-GNN (Fourier-based) | Accuracy | Superior to conventional GNNs | 7 Benchmarks |
| ESOL [10] | Water Solubility | Quantized GCN/GIN | RMSE | Varies with bit-width (e.g., INT8: ~0.6 log mol/L) | 1,128 |
| FreeSolv [10] | Hydration Free Energy | Quantized GCN/GIN | RMSE | Varies with bit-width | 642 |
| Lipophilicity [10] | Octanol/Water Partition Coefficient (LogP) | Quantized GCN/GIN | RMSE | Varies with bit-width | 4,200 |
Detailed Methodology: KA-GNN for Molecular Property Prediction
The Kolmogorov-Arnold Graph Neural Network (KA-GNN) exemplifies a recent architectural advancement [5].
- Graph Representation: A molecule is represented as a graph ( G = (V, E) ), where ( V ) is the set of atoms (nodes) and ( E ) is the set of bonds (edges).
- Node and Edge Feature Initialization:
- Node features: Atomic number, radius, hybridization state, etc.
- Edge features: Bond type, bond length, conjugation, etc.
- In KA-GNN, these initial features are processed by a Fourier-based KAN layer instead of a standard MLP for embedding.
- Message Passing with KANs: The core of the GNN involves multiple message-passing layers. In a KA-GNN variant (e.g., KA-GCN), each layer updates node representations by aggregating messages from neighbors. The transformation functions within the message-passing scheme are implemented using Fourier-based KAN layers, which employ learnable activation functions based on Fourier series to capture complex patterns more effectively [5].
- Readout/Global Pooling: After ( L ) message-passing layers, a readout function generates a single graph-level representation vector for the entire molecule. KA-GNN uses a KAN-based readout to replace standard pooling-plus-MLP operations.
- Property Prediction: The graph-level representation is passed through a final output layer (e.g., a linear layer for regression) to predict the target molecular property, such as energy or solubility.
The Scientist's Toolkit: Research Reagents for Small Molecule GNNs
Table 2: Essential Resources for GNN-based Small Molecule Modeling
| Resource Name | Type | Function/Benefit | Reference/Access |
|---|---|---|---|
| QM9 Dataset | Dataset | Benchmark for quantum mechanical property prediction; contains ~134k small molecules with DFT-calculated properties. | MoleculeNet [41] [10] |
| KA-GNN Framework | Model Architecture | Integrates Kolmogorov-Arnold Networks (KANs) into GNNs for enhanced accuracy and interpretability. | [5] |
| ByteFF | Force Field / Dataset | A data-driven force field parameterized using a GNN on a large QM dataset of molecular fragments and torsions. | [42] |
| DoReFa-Net | Algorithm | Enables quantization of GNN models (e.g., INT8) to reduce computational footprint while maintaining performance. | [10] |
Peptides and Proteins: From Force Fields to Interaction Networks
GNN applications for peptides and proteins span from atomic-level force field parameterization to residue-level interaction prediction, addressing critical tasks in structural biology and drug discovery.
Key Experimental Protocols and Performance
2.1.1 Force Field Parametrization with ByteFF A groundbreaking application of GNNs is the development of molecular mechanics force fields, such as ByteFF [42].
- Objective: To predict all bonded (bonds, angles, torsions) and non-bonded (van der Waals, partial charges) parameters for drug-like molecules across an expansive chemical space.
- Dataset: Trained on a massive, diverse QM dataset containing 2.4 million optimized molecular fragment geometries and 3.2 million torsion profiles calculated at the B3LYP-D3(BJ)/DZVP level of theory.
- Model: An edge-augmented, symmetry-preserving molecular GNN.
- Training Strategy: The model was trained using a differentiable partial Hessian loss and an iterative optimization-and-training procedure to ensure high accuracy for conformational energies and forces.
- Performance: ByteFF demonstrates state-of-the-art accuracy in predicting relaxed geometries, torsional energy profiles, and conformational energies, providing expansive coverage of drug-like chemical space [42].
2.1.2 Protein-Protein Interaction (PPI) Prediction GNNs are extensively used to predict whether two proteins will interact, a fundamental question in systems biology [43].
- Data Representation: Proteins are represented as graphs, where nodes are amino acid residues and edges connect residues that are spatially close or sequentially adjacent.
- Node Features: Include sequence embeddings (from LLMs like ProtTrans), evolutionary information, and physicochemical properties.
- Core Architectures:
- Graph Convolutional Networks (GCN): Aggregate neighbor information uniformly.
- Graph Attention Networks (GAT): Use attention mechanisms to weight the importance of different neighboring residues.
- Graph Autoencoders (GAE): Learn compressed representations of protein graphs for interaction inference.
- Performance: Advanced frameworks like AG-GATCN (integrating GAT and Temporal CNNs) and RGCNPPIS (integrating GCN and GraphSAGE) have shown robust performance against noise and enable the extraction of both macro-scale topological patterns and micro-scale structural motifs [43].
Detailed Methodology: GNN for Data-Driven Force Field Development
The protocol for developing a GNN-driven force field like ByteFF involves a sophisticated, end-to-end pipeline [42].
- Dataset Curation: A vast set of drug-like molecules is collected from sources like ChEMBL and ZINC. These molecules are cleaved into smaller fragments to ensure comprehensive coverage of local chemical environments.
- Quantum Mechanical Calculations: Each molecular fragment undergoes geometry optimization and Hessian matrix calculation at a high-level DFT method (e.g., B3LYP-D3(BJ)/DZVP). Torsion scans are performed to capture rotational energy profiles.
- GNN Parameter Prediction:
- The molecule is fed into a symmetry-preserving GNN.
- The GNN processes the molecular graph and, in a single forward pass, predicts all necessary MM parameters: equilibrium bond lengths ((r0)) and force constants ((kr)), angle parameters ((\theta0), (k\theta)), torsion amplitudes ((K_{\phi,n})), and non-bonded parameters ((\sigma), (\varepsilon), (q)).
- Loss Calculation and Training: The loss function compares the molecular mechanics energy (and its derivatives) computed with the predicted parameters against the reference QM data. This includes energy, force, and Hessian-based losses to ensure high fidelity of the resulting force field.
RNA and RNA-Protein Complexes: Predicting Interactions
Predicting RNA-protein interactions (RPI) is a complex challenge due to the limited availability of structural data and the high flexibility of RNA. GNNs, particularly when combined with modern language models, have shown remarkable success in this domain [44].
Key Experimental Protocols and Performance
- Objective: Accurately predict binding between RNA and protein pairs, especially for "orphan" molecules with few or no known interactions.
- Challenge: Classical methods struggle with generalization and are hampered by annotation imbalances in RPI networks.
- Solution - ZHMolGraph: This framework integrates GNNs with unsupervised large language models (LLMs) to address these limitations [44].
- Node Feature Generation: RNA and protein sequences are encoded into rich, contextualized embeddings using RNA-FM (for RNA) and ProtTrans (for protein) LLMs.
- Network Integration: These embeddings are used as node features within an RPI network. A GNN then aggregates and refines these features by propagating information across known interactions within the network.
- Performance: ZHMolGraph achieved an AUROC of 79.8% and an AUPRC of 82.0% on a benchmark dataset of entirely unknown RNAs and proteins, representing a substantial improvement of 7.1–28.7% in AUROC over other state-of-the-art methods [44].
Detailed Methodology: ZHMolGraph for RNA-Protein Interaction Prediction
The workflow for ZHMolGraph provides a protocol for robust biomolecular interaction prediction [44].
- RPI Network Construction: Compile a heterogeneous network from multiple sources (structural databases, high-throughput experiments, literature mining). Nodes represent RNAs and proteins; edges represent confirmed interactions.
- Sequence Embedding with LLMs:
- Input raw RNA and protein sequences.
- Process them through pre-trained foundation models (RNA-FM and ProtTrans) to generate initial node feature vectors. This step captures deep semantic information from the sequences themselves.
- Graph Neural Network Processing:
- The RPI network with LLM-derived node features is input into a GNN.
- The GNN performs message passing, allowing information to flow between connected RNA and protein nodes. This step integrates the topological information of the interaction network with the sequence-level information.
- Feature Concatenation and Prediction: For a given RNA-protein pair, the final LLM embeddings and the GNN-processed features are concatenated. This combined feature vector is fed into a final neural network (VecNN) to predict the binding likelihood.
The Scientist's Toolkit: Research Reagents for Biomolecular Interaction Prediction
Table 3: Essential Resources for Protein and RNA Interaction Modeling
| Resource Name | Type | Function/Benefit | Reference/Access |
|---|---|---|---|
| STRING Database | Database | Comprehensive resource of known and predicted protein-protein interactions. | https://string-db.org/ [43] |
| BioGRID | Database | Open-access repository of genetic and protein interaction data from major model organisms. | https://thebiogrid.org/ [43] |
| ZHMolGraph | Model Framework | Integrates GNNs with RNA-FM and ProtTrans LLMs for highly accurate RNA-protein interaction prediction. | [44] |
| ProtTrans | Language Model | Generates context-aware, deep learning-based embeddings for protein sequences. | [44] |
Navigating Challenges: Scalability, Data Efficiency, and Model Optimization
In the field of computational drug discovery, the accurate prediction of molecular mechanics (MM) force field parameters using Graph Neural Networks (GNNs) is crucial for reliable molecular dynamics simulations. However, researchers and scientists often confront a significant obstacle: the data bottleneck. This challenge manifests in two primary forms—small datasets resulting from the high computational cost of quantum mechanics (QM) calculations, and imbalanced datasets where critical molecular motifs or properties are underrepresented. These data limitations can severely compromise model performance, leading to biased predictions, poor generalization, and reduced reliability in downstream applications. This application note details practical, experimentally-validated strategies to overcome these challenges, with a specific focus on GNN-based force field parametrization. By implementing these protocols, research teams can enhance the robustness and predictive power of their models even under significant data constraints.
The Data Challenge in Molecular Property Prediction
The development of accurate machine learning force fields like ByteFF requires extensive, high-quality QM data. Generating such datasets involves computationally expensive methods like density functional theory (DFT), creating a natural limitation on data volume [1] [45]. Furthermore, the chemical space of drug-like molecules exhibits inherent imbalances; certain functional groups, torsion patterns, or element combinations occur more frequently than others, creating a long-tail distribution problem. When trained on such imbalanced data, GNNs develop prediction biases toward majority patterns, potentially overlooking rare but chemically significant motifs.
Traditional accuracy metrics become misleading under these conditions, as models may achieve high scores by simply predicting majority classes while failing to capture critical minority patterns [46] [47]. For molecular mechanics parameter prediction, this bias could manifest as inaccurate energy profiles for uncommon torsion angles or improper geometry optimization for rare element combinations, ultimately compromising the reliability of molecular dynamics simulations in drug discovery pipelines.
Table 1: Evaluation Metrics for Imbalanced Molecular Datasets
| Metric | Formula | Application Context | Advantages |
|---|---|---|---|
| F1-Score | ( F1 = 2 \times \frac{\text{Precision} \times \text{Recall}}{\text{Precision} + \text{Recall}} ) | Balanced measure for specific molecular class prediction | Harmonizes precision and recall, suitable for imbalanced data [47] |
| Precision-Recall AUC | Area under Precision-Recall curve | Focused evaluation of minority class performance (e.g., rare torsional profiles) | More informative than ROC-AUC for imbalanced data [46] |
| Matthews Correlation Coefficient (MCC) | ( \frac{TP \times TN - FP \times FN}{\sqrt{(TP+FP)(TP+FN)(TN+FP)(TN+FN)}} ) | Overall quality of binary classifications in imbalanced scenarios | Balanced measure even with large class imbalance [46] |
| Balanced Accuracy | ( \frac{1}{2} \left( \frac{TP}{TP+FN} + \frac{TN}{TN+FP} \right) ) | Multi-class scenarios (e.g., element type prediction) | Accounts for imbalance by averaging recall per class [46] |
Strategic Framework and Experimental Protocols
Data-Level Strategies: Augmentation and Resampling
Protocol 3.1.1: Direct Inverse Design for Data Augmentation
The Direct Inverse Design generation (DIDgen) approach addresses data scarcity by leveraging the invertible nature of pre-trained GNNs to generate novel molecular structures with desired properties [12].
- Required Reagents/Tools: Pre-trained GNN property predictor, QM9 or similar dataset, DFT validation pipeline
- Procedure:
- Start with a pre-trained GNN model on available molecular data (e.g., QM9 dataset)
- Initialize with random graph or existing molecular structure
- Perform gradient ascent with fixed GNN weights to optimize input graph toward target property
- Enforce valence rules through constrained graph construction:
- Construct adjacency matrix from weight vector with symmetric, zero-trace properties
- Apply sloped rounding function:
[x]sloped = [x] + a(x-[x])to maintain gradients during rounding [12] - Define atomic elements based on valence constraints (sum of bond orders)
- Validate generated molecules using DFT calculations
- Validation: In HOMO-LUMO gap targeting, DIDgen achieved comparable or better performance than state-of-the-art genetic algorithms while generating more diverse molecules [12]
- Performance Metrics: Success rate (molecules within target property range), diversity (Tanimoto distance), DFT validation error
Protocol 3.1.2: SMOTE for Molecular Feature Space
Synthetic Minority Over-sampling Technique (SMOTE) generates synthetic examples for minority classes by interpolating between existing instances in feature space [47].
- Procedure:
- Represent molecular structures using feature vectors (molecular fingerprints or GNN embeddings)
- For each minority class example, identify k-nearest neighbors (typically k=5)
- Generate synthetic examples by random linear interpolation between the example and its neighbors
- For molecular data, use SMOTE-NC variant for categorical features [46]
- Considerations: For severe imbalance with small datasets, SMOTE or ADASYN are recommended; for large datasets with redundant majority class, undersampling may be preferable [46]
Protocol 3.1.3: Strategic Downsampling with Upweighting
This two-step technique separates learning feature representations from class distribution [48].
- Procedure:
- Downsample majority class: Create balanced subsets by randomly removing majority class examples
- Upweight the downsampled class: Apply higher loss weights to majority class examples (e.g., multiply loss by downsampling factor)
- Benefits: Improves model convergence and representation learning while maintaining awareness of true class distribution [48]
Algorithm-Level Strategies: Model Architecture and Loss Functions
Protocol 3.2.1: Kolmogorov-Arnold GNNs for Enhanced Data Efficiency
KA-GNNs integrate Fourier-based Kolmogorov-Arnold networks into GNN components to improve parameter efficiency and expressivity with limited data [5].
- Architecture Implementation:
- Node Embedding: Replace MLP initialization with Fourier-based KAN modules
- Message Passing: Implement KAN-augmented graph convolutional layers (KA-GCN) or graph attention layers (KA-GAT)
- Readout: Use KAN modules for graph-level representation
- Fourier-KAN Formulation:
- Theoretical Foundation: Based on Kolmogorov-Arnold representation theorem and Fourier convergence theorems [5]
- Performance: KA-GNNs consistently outperform conventional GNNs in accuracy and computational efficiency across multiple molecular benchmarks [5]
Protocol 3.2.2: Cost-Sensitive Learning and Class Weighting
Incorporate imbalance adjustment directly into the learning process without modifying dataset composition.
- Procedure:
- Calculate class weights inversely proportional to class frequencies
- Implement weighted loss function: ( \mathcal{L} = \sum{i} w{yi} \cdot \ell(f(xi), y_i) )
- For severe imbalances, use focal loss to downweight easy examples and focus on hard negatives [46]
- Implementation: Supported in most ML libraries (Scikit-learn, PyTorch, XGBoost) via
class_weightparameters
Protocol 3.2.3: Ensemble Methods with Balanced Sampling
- BalancedBaggingClassifier: Combines bagging with undersampling, creating balanced subsets for training multiple models [47]
- EasyEnsemble: Trains multiple classifiers on different balanced subsets and aggregates predictions [46]
- Balanced Random Forests: Uses class-balanced bootstraps to maintain diversity while addressing imbalance [46]
Validation Framework for Imbalanced Molecular Data
Protocol 3.3.1: Comprehensive Model Validation
- Data Splitting: Use stratified sampling to maintain class distribution in training/validation/test sets
- Validation Metrics: Employ multi-metric evaluation focusing on minority class performance (Table 1)
- Threshold Adjustment: Optimize prediction thresholds based on precision-recall curves rather than default 0.5 [46]
Table 2: Strategic Selection Guide for Imbalanced Molecular Data
| Scenario | Recommended Strategy | Expected Outcome | Validation Focus |
|---|---|---|---|
| Severe imbalance with small dataset | SMOTE/ADASYN + KA-GNN | Improved minority class recall | Precision-Recall AUC, F1-Score |
| Large dataset with redundant majority | Undersampling + BalancedBagging | Reduced bias, maintained accuracy | Balanced Accuracy, MCC |
| High cost of false negatives | Cost-sensitive learning + Focal Loss | Improved minority class detection | Recall, F1-Score |
| Need for model interpretability | Class weighting + threshold adjustment | Transparent trade-offs | Precision-Recall curves |
| Complex molecular patterns | Ensemble methods (EasyEnsemble) | Robust multi-pattern capture | MCC, Balanced Accuracy |
Integrated Workflow and Implementation
The following workflow diagram illustrates the strategic integration of multiple approaches to address data limitations in molecular property prediction:
Workflow Implementation Protocol:
- Initial Assessment: Quantify dataset imbalance and identify critical minority classes
- Strategy Selection: Choose appropriate combination based on scenario analysis (Table 2)
- Iterative Refinement: Implement chosen strategies with cross-validation
- Comprehensive Validation: Apply multiple evaluation metrics and external validation (e.g., DFT calculations)
Research Reagent Solutions
Table 3: Essential Research Tools for GNN Force Field Development
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| ByteFF Dataset | Training data for force field parametrization | Contains 2.4M optimized molecular fragments + 3.2M torsion profiles at B3LYP-D3(BJ)/DZVP level [1] [45] |
| DIDgen Framework | Molecular generation via gradient ascent | Requires pre-trained GNN; enforces valence constraints through specialized graph construction [12] |
| KA-GNN Architecture | Data-efficient graph neural network | Integrates Fourier-KAN modules; improves parameter efficiency and interpretability [5] |
| SMOTE/ADASYN | Synthetic data generation for minority classes | Use SMOTE-NC for mixed categorical-continuous molecular features [46] [47] |
| BalancedBaggingClassifier | Ensemble learning for imbalanced data | Compatible with various base estimators; creates balanced subsets via undersampling [47] |
| Focal Loss | Loss function for class imbalance | Downweights easy examples; focuses training on hard negatives [46] |
The data bottleneck in molecular mechanics force field development presents significant but surmountable challenges. By implementing the integrated strategies outlined in this application note—combining data augmentation techniques like DIDgen, algorithmic improvements through KA-GNNs, and appropriate handling of class imbalance—researchers can develop more accurate and robust GNN models even with limited or skewed data. The provided protocols offer practical, experimentally-validated approaches that maintain scientific rigor while addressing real-world constraints in computational drug discovery. As the field advances, these methodologies will enable more efficient exploration of chemical space and accelerate the development of reliable force fields for drug discovery applications.
The exploration of scaling laws has been a cornerstone of progress in deep learning, driving breakthroughs in domains like natural language processing and computer vision. Until recently, the scalability of Graph Neural Networks (GNNs) for molecular data remained less explored due to challenges including the lower efficiency of sparse operations and substantial data requirements [49] [50]. However, a paradigm shift is now underway. Emerging research demonstrates that GNNs exhibit predictable improvements—characterized by power-law relationships—when scaled across model size, depth, and dataset size and diversity [51] [49] [52]. Understanding these scaling laws is critical for developing foundational models in molecular science that can accurately predict properties, design novel materials, and accelerate drug discovery. This document synthesizes the latest empirical findings and provides detailed protocols for studying scaling behavior in molecular GNNs, framed within a broader research context focused on predicting molecular mechanics parameters.
Quantitative Analysis of Scaling Laws
The performance of molecular GNNs, typically measured by validation loss on key prediction tasks, follows a power-law relationship with respect to model size, dataset size, and compute. This relationship is generally expressed as ( L = \alpha \cdot N^{-\beta} ), where ( L ) is the loss, ( N ) is the relevant scaling variable (e.g., number of model parameters or training data points), and ( \alpha ), ( \beta ) are constants [51]. The tables below consolidate quantitative scaling observations from recent foundational studies.
Table 1: Scaling Laws with Respect to Model and Dataset Size
| Scaling Dimension | Architectures Studied | Observed Impact on Performance | Key Study Findings |
|---|---|---|---|
| Model Size (Parameters) | Message-Passing GNNs, Graph Transformers, Hybrids [49] | Power-law reduction in validation loss [51] [52] | GNNs benefit tremendously from increasing scale of depth and width [49] [50]. |
| Dataset Size | Transformer, EquiformerV2 [51] | Power-law reduction in loss; diminishing returns at large scales [51] | Scaling from millions to billions of data points is crucial for foundational models [52]. |
| Dataset Diversity & Multi-task Labels | MolGPS [49] [50] | Significant performance gains on downstream tasks [49] | Pretraining data with thousands of labels (bio-assays, quantum simulations, imaging) is a major factor [50]. |
Table 2: Scaling Laws for Downstream Task Performance
| Scaling Factor | Impact on Finetuning | Evidence |
|---|---|---|
| Model Scale (Pretrained) | Improved accuracy on diverse downstream tasks [49] [52] | MolGPS, a large-scale graph foundation model, outperformed previous state-of-the-art on 26 out of 38 downstream tasks [49] [50]. |
| Dataset Diversity (Pretraining) | Enhanced generalization and transferability [49] [53] | Multimodal datasets incorporating textual descriptors (IUPAC, properties) alongside graphs improve performance on certain electronic properties [53]. |
Experimental Protocols for Investigating Scaling Laws
Protocol 1: Establishing Baseline Scaling Laws for a Target Task
This protocol outlines the core procedure for empirically determining the power-law relationship between model size, dataset size, and performance for a specific molecular prediction task.
1. Research Question Formulation: Define the target variable (e.g., energy, forces, stress prediction for materials [51] or a specific molecular property for drug discovery [54]).
2. Experimental Setup:
- Dataset: Select a large-scale dataset (e.g., OMat24 for materials [51] or a collection of 2D molecular graphs [49]).
- Model Architectures: Choose a model family (e.g., EquiformerV2, graph Transformer, message-passing GNN).
- Compute: Secure access to high-performance computing clusters with GPU acceleration [51].
3. Controlled Scaling Experiments:
- Vary Model Size: Keep the training dataset constant. Train models with progressively increasing numbers of parameters (e.g., from (10^2) to nearly (10^9) [51]). For GNNs, this involves scaling the depth (number of layers) and/or width (hidden dimensions) [49] [52].
- Vary Dataset Size: Keep the model architecture constant. Train the model on progressively larger random subsets of the full training dataset [51].
4. Data Analysis and Power-Law Fitting:
- For each experiment, record the final validation loss.
- Plot the validation loss against the model size and against the dataset size on a log-log scale.
- Fit a power-law function ( L = \alpha \cdot N^{-\beta} ) to the data points to derive the scaling coefficients ( \alpha ) and ( \beta ) [51].
Protocol 2: Evaluating the Impact of Dataset Diversity
This protocol investigates how the diversity and label richness of pretraining data affect downstream performance, a key factor for foundational models.
1. Dataset Curation:
- Assemble a multimodal dataset. Beyond geometric structures (XYZ coordinates), incorporate diverse labels and textual descriptors such as IUPAC names, molecular formulas, and physicochemical properties from public databases like PubChem [53].
2. Model Training and Fusion:
- Baseline Model: Train a GNN using only geometric graph data.
- Multimodal Model: Implement a model that can process both geometric and textual data. This often involves:
- Feature Extraction: Using separate encoders for graph data and textual data.
- Fusion Mechanism: Employing a gated fusion mechanism [53] or similar technique to adaptively combine the geometric and textual feature vectors.
3. Evaluation:
- Finetune both the baseline and multimodal models on a suite of downstream tasks (e.g., toxicity, solubility, binding affinity prediction [54]).
- Compare the performance gains across tasks to identify where multimodal, diverse data provides the largest benefit [53].
The following workflow diagram illustrates the key steps in these scaling experiments.
Architectural Scaling and Novel Formulations
Scaling GNNs is not solely about increasing parameter counts. Architectural innovations are crucial for enhancing expressivity, parameter efficiency, and ultimately, scaling behavior. The diagram below illustrates the architecture of KA-GNNs, which integrate Kolmogorov-Arnold Networks (KANs) into GNN components to improve performance and efficiency [5].
Key Insight: Replacing standard Multi-Layer Perceptrons (MLPs) in GNNs with KAN modules in the node embedding, message passing, and readout phases can lead to superior prediction accuracy and computational efficiency [5]. The Fourier-series-based functions in KANs enhance the model's ability to capture complex, non-linear patterns in molecular data, contributing to more favorable scaling.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Scaling Molecular GNN Experiments
| Resource Category | Specific Examples | Function & Application |
|---|---|---|
| Large-Scale Datasets | Open Materials 2024 (OMat24) [51]; Largest public collection of 2D molecular graphs [49] [50]; PubChem (for textual descriptors) [53] | Provides millions of data points for pretraining and establishing scaling laws. Multimodal data enhances diversity. |
| Model Architectures | EquiformerV2 (E(3)-equivariant) [51]; Message-Passing Networks; Graph Transformers [49]; KA-GNNs (Kolmogorov-Arnold GNNs) [5] | Serves as the scalable model backbone. Different architectures (constrained vs. unconstrained) are tested for scaling efficacy. |
| Software & Libraries | GPU-Accelerated Deep Learning Frameworks (PyTorch, JAX); LLM-inspired Training Libraries [52]; MD Engines (GROMACS, OpenMM for force fields) [55] | Manages large-scale data and models efficiently. Enables transfer of techniques (e.g., optimized attention) from NLP to GNNs. |
| Compute Infrastructure | High-Performance Computing Clusters (e.g., Savio Cluster [51]); Massive GPU Arrays (1000s of GPUs) [52] | Provides the floating-point operations (FLOPs) required for training models with billions of parameters on terabyte-scale datasets. |
| Force Field Applications | Grappa [55]; Espaloma [55] | Provides a direct application context (molecular mechanics force field prediction) for evaluating the real-world impact of scaled GNNs. |
Enhancing Generalization and Transferability to Uncharted Chemical Space
The application of Graph Neural Networks (GNNs) in molecular sciences has revolutionized property prediction and materials design. However, a significant challenge persists: models often fail to generalize reliably to regions of chemical space not represented in their training data. This limitation severely impacts real-world applications in drug discovery, where models must make accurate predictions for novel molecular scaffolds. The ability to build predictive models that maintain accuracy on out-of-distribution compounds represents the next frontier in computational molecular science.
This application note synthesizes recent methodological advances that enhance the generalization and transferability of GNNs for molecular property prediction, with particular focus on applications in molecular mechanics parameterization. We provide a structured overview of techniques, quantitative performance comparisons, and detailed experimental protocols to guide researchers in implementing these approaches.
Key Challenges in Molecular Generalization
Molecular property prediction models face several interconnected challenges when deployed to uncharted chemical space. The primary issue is the fundamental distribution shift between training data and real-world application scenarios. For example, a model trained on the QM9 dataset (comprising small organic molecules) may perform poorly on complex drug-like molecules or materials with extended conformational landscapes [12] [13]. This problem is compounded by the sparse and noisy nature of experimental chemical data, where complete property information is often unavailable across diverse chemical classes [56].
Additionally, traditional GNN architectures suffer from expressivity limitations in capturing complex physical interactions and long-range dependencies in molecular systems. Recent theoretical work has shown that standard message-passing GNNs struggle with over-smoothing and over-squashing, which limits their ability to propagate information across large molecular graphs [13]. These architectural constraints directly impact predictive performance on complex molecular systems not seen during training.
Methodological Approaches for Enhanced Generalization
Advanced Architecture Designs
Kolmogorov-Arnold GNNs (KA-GNNs) represent a significant architectural innovation that replaces traditional multilayer perceptrons (MLPs) in GNNs with learnable univariate functions based on the Kolmogorov-Arnold representation theorem. By integrating Fourier-series-based univariate functions, KA-GNNs enhance function approximation capabilities and theoretical expressiveness. These networks can be incorporated across all fundamental GNN components: node embedding, message passing, and readout phases. Experimental results across seven molecular benchmarks demonstrate that KA-GNN variants (KA-GCN and KA-GAT) consistently outperform conventional GNNs in both prediction accuracy and computational efficiency while offering improved interpretability through highlighting of chemically meaningful substructures [5].
Transferable Coarse-Grained Models address generalization through a multi-scale approach. As demonstrated in protein modeling, machine-learned coarse-grained force fields can be trained on diverse all-atom simulation data then transferred to novel sequences with low (16-40%) similarity to training examples. These models successfully predict metastable states of folded, unfolded, and intermediate structures while being several orders of magnitude faster than all-atom models, enabling exploration of previously inaccessible chemical spaces [57].
Data-Centric Strategies
Multi-Task Learning provides an effective framework for leveraging additional molecular data – even when sparse or weakly related – to enhance prediction quality in data-limited regimes. Controlled experiments demonstrate that multi-task GNNs systematically outperform single-task models, particularly when auxiliary tasks are chemically relevant to the primary prediction target. This approach enables knowledge transfer across property domains, effectively expanding the model's understanding of structure-property relationships [56].
Systematic Data Augmentation techniques, particularly SMILES enumeration, significantly improve model robustness. Studies using molecular transformer models show that 40-level data augmentation (where each compound is represented by 40 different SMILES strings) combined with normalization preprocessing increases top-1 accuracy in forward reaction prediction from 71.6% to 84.2%. This approach forces models to learn invariant representations regardless of molecular representation syntax, enhancing generalization capability [58].
Table 1: Quantitative Performance of Generalization Techniques
| Method | Architecture | Dataset | Performance Gain | Generalization Metric |
|---|---|---|---|---|
| KA-GNN | Fourier-based KAN modules | 7 molecular benchmarks | Consistent outperformance vs. conventional GNNs | Prediction accuracy & computational efficiency |
| Multi-task GNN | Graph Isomorphism Network | QM9 subsets | Outperforms single-task in low-data regimes | Prediction quality with sparse data |
| Data Augmentation | Molecular Transformer | USPTO-50K | Top-1 accuracy: 71.6% → 84.2% | Forward reaction prediction accuracy |
| Transferable CG Model | CGSchNet | Unseen proteins (16-40% similarity) | Predicts metastable states accurately | Transferability to novel sequences |
Efficiency Optimization for Expanded Coverage
Quantization Approaches enable more efficient exploration of chemical space by reducing computational barriers. Research shows that INT8 quantization of GNN models maintains strong performance on quantum mechanical property prediction (e.g., dipole moment) while significantly reducing memory footprint and computational requirements. This allows for broader hyperparameter exploration and model ensemble techniques that improve generalization [10].
Application to Molecular Mechanics Force Fields
The development of transferable molecular mechanics force fields exemplifies the critical importance of generalization in computational chemistry. Traditional look-up table approaches for force field parameterization face significant challenges with the rapid expansion of synthetically accessible chemical space. ByteFF addresses this limitation through a modern data-driven approach using an edge-augmented, symmetry-preserving molecular GNN trained on an expansive dataset of 2.4 million optimized molecular fragment geometries and 3.2 million torsion profiles [1].
This approach demonstrates state-of-the-art performance across various benchmarks, excelling in predicting relaxed geometries, torsional energy profiles, and conformational energies and forces. The GNN learns to predict all bonded and non-bonded molecular mechanics parameters simultaneously across broad chemical space, enabling accurate parameterization for drug-like molecules not present in the training data. The exceptional accuracy and expansive chemical space coverage make such data-driven force fields valuable tools for multiple stages of computational drug discovery [1].
Table 2: Molecular Mechanics Force Field Generalization Performance
| Model | Training Data | Chemical Coverage | Application | Performance |
|---|---|---|---|---|
| ByteFF | 2.4M molecular fragments, 3.2M torsion profiles | Drug-like molecules | Molecular dynamics | State-of-the-art on geometry, torsion, and energy prediction |
| CGSchNet | All-atom protein simulations | Proteins with <40% sequence similarity | Protein folding dynamics | Predicts metastable states, relative folding free energies |
Experimental Protocols
Protocol: Implementing KA-GNNs for Molecular Property Prediction
Objective: Enhance GNN generalization using Kolmogorov-Arnold network modules integrated into graph neural networks.
Materials:
- Molecular datasets (QM9, ESOL, FreeSolv, Lipophilicity)
- Deep learning framework (PyTorch or TensorFlow)
- KA-GNN implementation (Fourier-based KAN layers)
Procedure:
- Data Preparation:
- Convert molecular structures to graph representations with node features (atom type, hybridization, valence) and edge features (bond type, distance).
- Split data into training/validation/test sets ensuring no structural overlap between sets.
Model Architecture:
- Implement Fourier-based KAN layers using the formulation:
- Replace standard MLP transformations in node embedding, message passing, and readout components with Fourier-KAN layers.
Training Configuration:
- Use Adam optimizer with learning rate 0.001
- Apply multi-task loss function when auxiliary properties are available
- Regularize Fourier coefficients to prevent overfitting
Interpretation Analysis:
- Visualize learned Fourier functions to identify important molecular substructures
- Analyze frequency components to understand captured molecular patterns
Validation: Evaluate on held-out test sets containing structurally novel compounds and measure performance degradation compared to conventional GNNs [5].
Protocol: Direct Inverse Design for Targeted Molecular Generation
Objective: Generate novel molecular structures with specific target properties by inverting pre-trained GNN predictors.
Materials:
- Pre-trained property prediction GNN
- Chemical constraint handler (valence rules, bond constraints)
- Gradient-based optimization framework
Procedure:
- Model Preparation:
- Train or obtain a GNN property predictor on available molecular data (e.g., HOMO-LUMO gap, solubility, binding affinity)
- Verify predictor accuracy on validation benchmarks
Graph Representation Initialization:
- Initialize adjacency matrix from random weights or existing molecular template
- Construct feature matrix using one-hot atom representations
Constrained Optimization:
- Implement gradient ascent on molecular graph with respect to target property
- Apply sloped rounding function for discrete adjacency matrix optimization:
- Enforce valence constraints through penalty terms and gradient masking
- Update both adjacency matrix and feature matrix iteratively
Validity Enforcement:
- Apply chemical validity rules after each optimization step
- Limit maximum atom valence to 4 with gradient blocking
- Ensure molecular symmetry in adjacency matrix
Convergence Checking:
- Stop optimization when target property threshold is reached
- Verify molecular structure validity using RDKit or Open Babel
Validation: Confirm generated molecular properties using independent calculation methods (e.g., DFT verification for energy gaps) [12].
Visualization of Workflows
KA-GNN Molecular Property Prediction Architecture
Direct Inverse Design Optimization Process
Research Reagent Solutions
Table 3: Essential Research Tools for Molecular Generalization Research
| Tool/Resource | Type | Function | Application Example |
|---|---|---|---|
| KA-GNN Framework | Software Library | Implements Kolmogorov-Arnold networks in GNNs | Molecular property prediction with improved generalization [5] |
| ByteFF Training Dataset | Chemical Dataset | 2.4M molecular fragments with geometries and Hessians | Data-driven force field parametrization [1] |
| DoReFa-Net Quantization | Algorithm | Reduces model precision while maintaining performance | Efficient GNN deployment for chemical space exploration [10] |
| SMILES Enumeration | Data Augmentation | Generates multiple representations of molecules | Improved model robustness in molecular transformers [58] |
| CGSchNet Architecture | Model Framework | Transferable coarse-grained molecular dynamics | Protein folding prediction on novel sequences [57] |
| Multi-task GNN Framework | Training Methodology | Joint learning across multiple property domains | Enhanced performance in low-data regimes [56] |
Enhancing the generalization and transferability of graph neural networks to uncharted chemical space requires a multi-faceted approach combining novel architectures, data-centric strategies, and efficient implementation. The methods outlined in this application note – including Kolmogorov-Arnold networks, multi-task learning, data augmentation, and direct inverse design – provide a comprehensive toolkit for researchers addressing this fundamental challenge. As molecular mechanics and drug discovery increasingly rely on computational predictions, these advances in generalization capability will play a crucial role in accelerating the design of novel molecules with tailored properties.
In the field of molecular property prediction using Graph Neural Networks (GNNs), a fundamental challenge is balancing high predictive accuracy with computational efficiency. While advanced GNNs achieve remarkable accuracy, their resource intensity can hinder application in real-world, resource-constrained settings like automated discovery pipelines. This document details protocols and application notes for researchers, framing the trade-offs and solutions within a broader thesis on molecular mechanics research. We explore three cutting-edge strategies: integrating novel network architectures like Kolmogorov-Arnold Networks (KANs), employing inverse design via gradient ascent, and applying model quantization.
Experimental Protocols & Data
The following table summarizes the core quantitative findings from recent studies that directly address the efficiency-accuracy balance.
Table 1: Comparative Performance of Efficiency-Focused GNN Approaches
| Methodology | Model / Dataset | Key Accuracy Metric | Key Efficiency Metric | Key Finding |
|---|---|---|---|---|
| KAN Integration [5] | KA-GNNs across 7 molecular benchmarks | Consistently outperformed conventional GNNs | Improved computational efficiency | Unifies high accuracy with efficiency and improved interpretability. |
| Inverse Design (Gradient Ascent) [12] [59] | DIDgen on QM9 (HOMO-LUMO gap) | Hit target property with comparable/better rate than JANUS | 2.1 - 12.0 seconds per in-target molecule | Achieves targeted generation without additional model training. |
| Model Quantization [60] | GNNs with DoReFa-Net on QM9 (dipole moment) | Maintained strong performance up to 8-bit precision | Reduced memory footprint & computational cost | 8-bit is a "sweet spot"; 2-bit quantization severely degrades performance. |
Protocol 1: Implementing KA-GNNs for Molecular Property Prediction
This protocol outlines the steps for developing a Kolmogorov-Arnold Graph Neural Network (KA-GNN) to enhance both expressivity and parameter efficiency [5].
- 1.1 Model Architecture Selection: Choose a base GNN architecture (e.g., Graph Convolutional Network (GCN) or Graph Attention Network (GAT)) to serve as the backbone for message passing.
- 1.2 KAN Module Integration: Replace the standard Multi-Layer Perceptrons (MLPs) in the base GNN with Fourier-series-based KAN layers in three key components:
- Node Embedding: Initialize node features using a KAN layer that processes atomic features and local bond context.
- Message Passing: Update node representations using residual KAN layers instead of standard activation functions.
- Readout Function: Generate the graph-level representation using a KAN layer for a more expressive aggregation of node embeddings.
- 1.3 Training and Evaluation: Train the model on molecular graph data (e.g., from benchmarks like QM9) using standard loss functions (e.g., Mean Squared Error for regression). Evaluate against conventional GNNs on prediction accuracy and computational cost (training/inference time, parameter count).
Protocol 2: Inverse Molecular Design using a Pre-trained GNN Predictor
This protocol describes a "Direct Inverse Design" (DID) method to generate molecules with desired properties by optimizing the input to a fixed, pre-trained GNN [12] [59].
- 2.1 Pre-trained Predictor: Start with a GNN model that has been pre-trained to accurately predict a target molecular property (e.g., HOMO-LUMO gap).
- 2.2 Input Graph Construction & Constraint Setup:
- Adjacency Matrix: Construct a differentiable adjacency matrix from a weight vector
w_adj. Use a sloped rounding function,[x]_sloped = [x] + a(x-[x]), to enable gradient flow through the rounding operation while enforcing symmetry and zero trace [12] [59]. - Feature Matrix: Define the atom types based on the valence (sum of bond orders) from the adjacency matrix. Use an additional weight matrix
w_feato differentiate between elements with the same valence. - Valence Penalization: Apply a soft penalty in the loss function for atoms exceeding a valence of 4 and block gradient updates that would increase bonds beyond this limit [12] [59].
- Adjacency Matrix: Construct a differentiable adjacency matrix from a weight vector
- 2.3 Gradient Ascent Optimization:
- Initialize a random graph or an existing molecular graph.
- Perform gradient ascent on the graph's
w_adjandw_feawith respect to the target property prediction from the fixed GNN. - Iterate until the predicted property meets the target criterion. The constraints ensure the optimized input remains a valid molecule.
Protocol 3: Quantizing GNNs with DoReFa-Net for Efficient Inference
This protocol applies Post-Training Quantization (PTQ) to reduce the memory and computational demands of a trained GNN model without extensive retraining [60].
- 3.1 Model and Dataset Preparation: Select a pre-trained, full-precision GNN model (e.g., a GCN or GAT) and the corresponding molecular dataset (e.g., QM9, ESOL).
- 3.2 Bit-Width Selection: Choose the level of quantization for weights and activations (e.g., 8-bit, 4-bit). Note that 8-bit is often a safe starting point, while 2-bit may cause significant performance loss [60].
- 3.3 Application of DoReFa-Net Algorithm:
- Quantize the model's weights and activations using the DoReFa-Net algorithm, which maps full-precision values to lower-bit representations in a differentiable manner.
- For a given full-precision value ( r \in [0, 1] ), its k-bit quantized version is: ( r_q = \frac{1}{2^k - 1} \text{round}((2^k - 1)r) ) [60].
- 3.4 Validation: Evaluate the quantized model's predictive performance (e.g., using RMSE, MAE) on a test set and compare it to the full-precision model. Benchmark the reduction in model size and inference latency.
The Scientist's Toolkit
This section lists key computational reagents and their functions for the experiments detailed in the protocols above.
Table 2: Essential Research Reagents and Computational Tools
| Item / Solution | Function / Application | Relevant Protocol |
|---|---|---|
| Fourier-KAN Layer | A learnable activation function using Fourier series to capture complex, oscillatory patterns in data, improving expressivity and interpretability [5]. | Protocol 1 |
| Sloped Rounding Function | A differentiable approximation of the rounding operation, allowing gradients to flow through the discrete structure of a graph during optimization [12] [59]. | Protocol 2 |
| DoReFa-Net Algorithm | A quantization method that converts full-precision (32-bit) model weights and activations into lower bit-widths (e.g., 8-bit), reducing computational load [60]. | Protocol 3 |
| Valence Constraint Module | A set of rules applied during graph optimization that penalizes chemically invalid atom valences, ensuring generated molecules are synthetically plausible [12] [59]. | Protocol 2 |
| Molecular Graph (Adjacency & Feature Matrices) | The fundamental data structure representing a molecule, where atoms are nodes and bonds are edges, serving as the direct input to the GNN [12] [59]. | Protocols 1, 2 |
Workflow Visualization
The following diagram illustrates the logical relationship and workflow between the three core methodologies discussed in this document.
Diagram 1: Three pathways to balance GNN performance, connecting the core challenge to the ultimate objective via distinct methodological approaches.
Benchmarks and Validation: Assessing Performance Against Traditional and QM Methods
The accurate prediction of molecular energies and forces using Graph Neural Networks (GNNs) is revolutionizing computational chemistry and drug discovery. These surrogate models bridge the gap between computationally expensive quantum mechanical methods like Density Functional Theory (DFT) and faster but less accurate classical approaches [61]. However, the reliability of these predictions hinges on robust validation metrics and protocols. This document establishes comprehensive application notes and protocols for validating GNN-based predictions of molecular mechanics parameters, providing researchers with a standardized framework for assessing model performance.
Core Validation Metrics for Energy and Force Predictions
A multi-faceted approach to validation is essential for thoroughly evaluating GNN performance. The metrics below form the foundation of a robust validation protocol, addressing different aspects of predictive accuracy and uncertainty.
Table 1: Core Quantitative Metrics for Energy and Force Predictions
| Metric Category | Specific Metric | Target Property | Interpretation & Ideal Value |
|---|---|---|---|
| Error Metrics | Mean Absolute Error (MAE) | Energy, Forces | Average magnitude of errors. Lower is better (e.g., ~10 meV/atom for energy [61]). |
| Root Mean Square Error (RMSE) | Energy, Forces | Penalizes larger errors more heavily. Lower is better [10]. | |
| Uncertainty Calibration | Error-based Calibration Plot | Energy, Forces | Measures if predicted uncertainties match actual error distributions. A well-calibrated plot should follow the y=x line [62]. |
| Miscalibration Area | Energy, Forces | Quantitative summary of calibration plot; area between the curve and y=x line. Lower is better [62]. | |
| Relaxed Property Validation | DFT-Verified Success Rate | Relaxed Energy | Percentage of generated molecules whose DFT-calculated properties hit the target. Higher is better [12]. |
| Mean Absolute Distance from Target | Relaxed Energy | Average error relative to a specific target property (e.g., HOMO-LUMO gap). Lower is better [12]. |
Beyond these quantitative metrics, the diversity of generated molecular structures is a critical, often overlooked, validation aspect. A successful model should not produce a narrow set of similar molecules but should explore the chemical space effectively [12]. Furthermore, validation must extend beyond the training distribution. Performance should be rigorously tested on out-of-distribution datasets to assess generalizability [12].
Experimental Protocols for Benchmarking
Protocol 1: Validating a Pre-trained GNN Force Field
This protocol outlines the steps to benchmark a pre-trained GNN model on a novel dataset, a common task for researchers adopting existing models.
- Data Preparation & Preprocessing: Compose a test set of molecules relevant to your application. The ChEMBL database is a common source for drug-like molecules [63]. Format the data into a comma-separated value (.csv) file containing at minimum the SMILES string and a unique identifier for each compound [63].
- Model Inference & Prediction: Run the pre-trained model on the prepared test set to obtain predictions for the target properties (e.g., energy, forces).
- Ground Truth Calculation: Compute reference values for the target properties using a high-fidelity method like DFT at an appropriate level of theory (e.g., B3LYP-D3(BJ)/DZVP [42] [45]).
- Metric Calculation & Analysis: Calculate the core metrics from Table 1 (MAE, RMSE) by comparing the model's predictions against the DFT-calculated ground truth.
- Uncertainty Quantification (UQ): Employ a UQ method such as the latent distance technique. Extract a rotationally invariant latent representation for each test molecule and compute its L2-distance to the nearest neighbors in the training set. Use this distance to estimate prediction uncertainty and generate an error-based calibration plot [62].
Protocol 2: Conditional Molecular Generation with Inverse Design
This protocol describes how to validate a GNN used for inverse design—generating molecules with specific target properties.
- Proxy Model Training: Train a GNN property predictor on a relevant dataset (e.g., QM9 for HOMO-LUMO gaps [12]).
- Inverse Design via Gradient Ascent: Starting from a random graph or an existing molecule, perform gradient ascent on the molecular graph. Hold the GNN weights fixed and optimize the input graph towards the target property [12].
- Valence Constraint Enforcement: During optimization, enforce chemical validity by constructing the adjacency matrix from a weight vector that respects symmetry and valence rules. Use a sloped rounding function to maintain non-zero gradients while pushing bond orders to integers [12].
- Proxy Validation & Filtering: Collect generated molecules that meet the target property according to the proxy GNN.
- DFT Verification: Perform DFT calculations on the generated molecules to verify that their true properties (e.g., HOMO-LUMO gap) match the target. This step is critical, as proxy model performance can be significantly worse on generated molecules than on its test set [12].
- Success & Diversity Metrics: Calculate the success rate (e.g., molecules within 0.5 eV of the target) and the diversity (e.g., average Tanimoto distance between molecular fingerprints) of the validated set [12].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Research Reagent Solutions for GNN Validation
| Tool Name | Type | Primary Function in Validation |
|---|---|---|
| Amber/GAFF | Molecular Mechanics Force Field | Provides standard analytical forms and reference parameters for benchmarking bonded and non-bonded interaction predictions [42] [64]. |
| DFT (e.g., B3LYP-D3(BJ)/DZVP) | Quantum Mechanics Method | Serves as the high-fidelity "ground truth" for validating energies, forces, and other quantum properties predicted by GNNs [42] [45]. |
| QM9, Open Catalyst Project | Benchmark Datasets | Standardized datasets for training and benchmarking GNNs on quantum mechanical properties, enabling fair comparison between different models [12] [62]. |
| EdgeSHAPer | GNN Explainability Tool | Explains GNN predictions by approximating Shapley values via Monte Carlo sampling, helping to identify which molecular sub-structures drive a prediction [63]. |
| ByteFF/Espaloma | Data-Driven Force Field | GNN-based force fields that predict parameters for a classical MM functional form; used to validate the parameterization workflow itself [42] [45]. |
| RDKit | Cheminformatics Toolkit | Handles molecular I/O, fingerprint generation (for diversity metrics), and basic cheminformatics operations essential for data preprocessing and analysis [63]. |
| PyTorch Geometric | Deep Learning Library | Provides standardized data loaders for molecular datasets (e.g., ESOL, FreeSolv, QM9) and implementations of common GNN architectures [10]. |
| AGNI | ML Platform | A paradigm for the independent prediction of energy, atomic forces, and stresses using separate ML models, preventing error propagation [61]. |
Molecular dynamics (MD) simulations are indispensable for studying material properties and biomolecular processes at the atomic level. The accuracy of these simulations hinges on the force field—a mathematical model describing the potential energy of a system as a function of its atomic coordinates. For decades, traditional molecular mechanics (MM) force fields have been the workhorse of MD simulations, but recent advances in geometric deep learning have introduced graph neural network (GNN)-based force fields that learn directly from quantum mechanical data. This application note provides a comprehensive technical comparison of these approaches, detailing their respective methodologies, performance benchmarks, and protocols for implementation.
Force Field Classification and Mechanisms
The landscape of force fields can be categorized into three distinct classes based on their functional forms and parameter derivation strategies.
Table 1: Fundamental Characteristics of Force Field Types
| Force Field Type | Parameter Source | Computational Cost | Accuracy | Reactive Capability | Primary Applications |
|---|---|---|---|---|---|
| Traditional MM | Lookup tables & hand-crafted rules [55] [65] | Lowest | Lower for uncharted chemical space | No (fixed bonds) | Biomolecular simulations [55] |
| Machine-Learned MM (e.g., Espaloma) | ML on graph with expert features [55] | Low (equivalent to MM) | Improved over traditional MM | No | Small molecules, peptides, RNA [55] |
| GNN-Based Force Fields (e.g., Grappa) | End-to-end ML from molecular graph [55] | Low (equivalent to MM) | State-of-the-art MM accuracy [55] | No | Broad, including peptide radicals [55] |
| GNN-Based NNPs (e.g., EMFF-2025) | End-to-end ML from QM data [66] | Higher (than MM) | Near-DFT accuracy [66] | Yes | Reactive systems, energetic materials [66] |
Traditional and Machine-Learned Molecular Mechanics Force Fields
Traditional MM force fields employ a physics-inspired functional form that decomposes the total potential energy into bonded terms (bonds, angles, dihedrals) and non-bonded terms (van der Waals, electrostatic) [65]. Parameters for these equations are assigned based on a finite set of atom types defined by expert-crafted rules and stored in lookup tables [55]. This makes them highly computationally efficient but limits their accuracy and transferability to chemical environments not predefined in the atom type list.
Machine-learned MM force fields like Espaloma and Grappa represent an evolution. They retain the computationally efficient functional form of traditional MM but use machine learning to assign parameters directly from the molecular graph. Grappa utilizes a graph attentional neural network and a transformer to predict MM parameters, eliminating the need for hand-crafted chemical features and enabling accurate parametrization for novel chemical species like peptide radicals [55]. A key advantage is that the ML model is invoked only once during parameter assignment; subsequent MD simulations run at the same speed as traditional MM [55].
GNN-Based Neural Network Potentials (NNPs)
In contrast, GNN-based NNPs do not use a pre-defined MM functional form. Instead, they represent the potential energy as a complex function learned entirely by a GNN from quantum mechanical (QM) data. Architectures like KA-GNNs (Kolmogorov-Arnold Graph Neural Networks) integrate novel function approximators into all core GNN components—node embedding, message passing, and readout—to enhance expressivity and interpretability [5]. Models such as EMFF-2025 are trained on QM data and can describe bond formation and breaking, making them reactive force fields suitable for simulating chemical reactions [66]. While more computationally expensive per energy evaluation than MM-based force fields, they offer near-DFT accuracy at a fraction of the cost of full QM calculations.
Quantitative Performance Comparison
Benchmarking across diverse molecular systems reveals the distinct performance profiles of each force field class.
Table 2: Performance Benchmarking Across Molecular Systems
| Force Field / Model | Test System/Metric | Reported Performance | Reference |
|---|---|---|---|
| Grappa (GNN-MM) | Small molecules, peptides, RNA (Energy/Forces) | Outperforms tabulated & machine-learned MM (Espaloma) on benchmark dataset [55] | [55] |
| Grappa (GNN-MM) | Peptide Dihedral Angles | Matches performance of AMBER FF19SB without requiring CMAP corrections [55] | [55] |
| Grappa (GNN-MM) | J-Couplings | Closely reproduces experimentally measured values [55] | [55] |
| EMFF-2025 (GNN-NNP) | 20 CHNO HEMs (Energy/Forces) | MAE for energy within ± 0.1 eV/atom; MAE for force within ± 2 eV/Å [66] | [66] |
| ByteFF-Pol (GNN-NNP) | Organic Liquids (Property Prediction) | Outperforms state-of-the-art classical and ML force fields in predicting thermodynamic/transport properties [67] | [67] |
| Fused Data ML Potential | Titanium (Elastic Constants, Lattice Parameters) | Achieves higher accuracy vs. models trained only on DFT or experiment [68] | [68] |
Experimental and Computational Protocols
Protocol 1: Developing a GNN-MM Force Field (Grappa)
This protocol outlines the procedure for developing a GNN-based molecular mechanics force field as described for Grappa [55].
Workflow Overview
Step-by-Step Procedure
- Input Representation: Represent the molecule as a 2D molecular graph ( G(V, E) ), where nodes ( V ) represent atoms and edges ( E ) represent bonds. No hand-crafted chemical features are required [55].
- Atom Embedding: Process the graph using a graph attentional neural network to generate a d-dimensional embedding vector ( \nu_i ) for each atom ( i ). This embedding captures the atom's chemical environment [55].
- Parameter Prediction: For each molecular mechanics interaction ( l ) (bond, angle, torsion, improper), predict its parameters ( \xi^{(l)}{ij...} ) using a transformer ( \psi^{(l)} ) that acts on the embeddings of the participating atoms: ( \xi^{(l)}{ij...} = \psi^{(l)}(\nui, \nuj, ...) ). The transformer architecture must respect the permutation symmetries inherent to each interaction type (e.g., ( \xi^{\text{(bond)}}{ij} = \xi^{\text{(bond)}}{ji} )) [55].
- Energy Evaluation: The predicted parameter set ( \xi ) defines the potential energy surface for any spatial conformation ( x ) of the molecule using the standard MM energy functional: ( E(x) = E_{\text{MM}}(x, \xi) ) [55].
- Model Training: Train the model end-to-end by minimizing the loss between the forces and energies predicted by the Grappa-defined energy surface and those obtained from reference quantum mechanical (QM) calculations. The model is trained on a diverse dataset of molecular conformations [55].
- MD Simulation Integration: Once trained, use Grappa to predict parameters for a new molecule of interest. The final force field, comprising all bonded parameters, can be used with standard non-bonded parameters in conventional MD engines like GROMACS or OpenMM for highly efficient simulation [55].
Protocol 2: Building a Reactive GNN-NNP with Data Fusion
This protocol describes the creation of a reactive neural network potential using a fusion of simulation and experimental data, a method shown to enhance accuracy [68].
Workflow Overview
Step-by-Step Procedure
- Data Curation:
- DFT Database Generation: Perform ab initio MD and static calculations to create a database of diverse atomic configurations. For each configuration, extract the total energy, atomic forces, and virial stress tensor [68].
- Experimental Database Compilation: Collate experimentally measured properties, such as temperature-dependent elastic constants and lattice parameters. These properties serve as macroscopic constraints [68].
- Model Pre-training: Initialize the GNN potential (e.g., using a architecture like NequIP or a Fourier-based KA-GNN [5] [68]) by training it solely on the DFT database. This provides a physically reasonable starting point for subsequent training [68].
- Fused Training Loop: Employ an iterative, alternating training strategy:
- DFT Trainer Step: For one epoch, update the model parameters ( \theta ) using a standard regression loss to match the model's predictions of energy, forces, and stress with the DFT reference data [68].
- EXP Trainer Step: For one epoch, update the model parameters ( \theta ) to minimize the difference between simulation-predicted and experimentally measured properties. Use the Differentiable Trajectory Reweighting (DiffTRe) method to efficiently compute gradients without backpropagating through the entire MD trajectory [68].
- Validation and Testing: Rigorously test the final model on out-of-target properties not included in the training, such as phonon spectra or properties of different phases, to assess its transferability and generalizability [68].
Table 3: Key Computational Tools for GNN Force Field Development and Application
| Resource Name | Type/Category | Primary Function | Application Context |
|---|---|---|---|
| GROMACS [55] | MD Simulation Software | High-performance molecular dynamics engine. | Running production simulations with MM-compatible force fields like Grappa. |
| OpenMM [55] [69] | MD Simulation Toolkit | A highly flexible toolkit for molecular simulation. | Used as a platform for running simulations with both MM and GNN-based force fields. |
| DP-GEN [66] | Computational Workflow | Deep Potential Generator for automated training data generation and active learning. | Building robust and generalizable Neural Network Potentials (NNPs). |
| DiffTRe [68] | Algorithm/Method | Differentiable Trajectory Reweighting for efficient gradient calculation. | Training ML potentials directly against experimental observables. |
| QM9 Dataset [16] | Benchmark Dataset | A public dataset of quantum mechanical properties for ~134k small organic molecules. | Training and benchmarking models for molecular property prediction. |
The emergence of GNN-based force fields marks a significant evolution in molecular simulation. GNN-MM force fields like Grappa offer a powerful drop-in replacement for traditional MM, providing superior accuracy and transferability without sacrificing the computational efficiency that enables large-scale biomolecular simulations. In contrast, GNN-NNPs provide a fundamentally different approach, learning the potential energy surface directly from QM data to achieve near-DFT accuracy for reactive systems, albeit at a higher computational cost. The choice between these approaches is not one of superiority but of application fit. For large-scale simulations of proteins, nucleic acids, and materials where chemical bonds remain intact, GNN-MM force fields are an excellent choice. For studying chemical reactions, complex catalysis, or systems where electronic effects are critical, GNN-NNPs are indispensable. The emerging paradigm of fused data learning, which integrates both QM and experimental data, promises to further elevate the accuracy and reliability of both classes of GNN force fields, paving the way for more predictive simulations in drug discovery and materials science.
The pursuit of quantum chemical accuracy in molecular modeling is a central goal in computational chemistry and drug discovery. Density functional theory (DFT) has long served as a benchmark for accuracy in predicting molecular properties and reaction mechanisms, yet its computational expense limits application in high-throughput settings. The emergence of graph neural networks (GNNs) offers a promising path to achieving DFT-level accuracy at significantly reduced computational cost. GNNs naturally operate on graph-structured representations of molecules, where atoms correspond to nodes and bonds to edges, enabling end-to-end learning of structure-property relationships without relying on hand-crafted descriptors [13] [70]. This application note details protocols and benchmarks for leveraging GNNs to reach DFT-level accuracy in predicting molecular properties and reaction mechanisms, contextualized within broader research on molecular mechanics parameters.
Quantitative Benchmarking of GNN Performance
Extensive benchmarking across diverse molecular datasets reveals that advanced GNN architectures can match or approach DFT-level accuracy for numerous chemical properties. The following tables summarize key quantitative results from recent studies.
Table 1: Performance of GNNs on Molecular Property Prediction Tasks
| Model Architecture | Dataset | Key Property/Prediction Task | Reported Accuracy/Metric | Reference/Notes |
|---|---|---|---|---|
| KA-GNN (Kolmogorov-Arnold GNN) | Seven molecular benchmarks | General molecular property prediction | Superior prediction accuracy vs. conventional GNNs; Improved computational efficiency | [5] |
| Δ-DFT Framework | Water, Ethanol, Benzene, Resorcinol | Coupled-Cluster (CC) Energy from DFT density | Quantum chemical accuracy (errors < 1 kcal·mol⁻¹) | Corrects DFT failures in strained geometries/conformer changes [71] |
| SEMG-MIGNN (Steric/Electronic Mol. Graph) | Doyle's Pd-catalyzed C–N coupling | Reaction yield prediction | Excellent predictive ability | Benchmarked on high-quality datasets [72] |
| SEMG-MIGNN | Denmark's CPA-catalyzed thiol addition | Enantioselectivity prediction | Excellent predictive ability | Benchmarked on high-quality datasets [72] |
| ReactAIvate | Novel CRM dataset | Elementary reaction step classification | Near-unity accuracy (~100%) | For 7 distinct elementary step classes [73] |
| ReactAIvate | Novel CRM dataset | Reactive atom prediction | 96% accuracy | Identifies atoms involved in reaction step [73] |
Table 2: Key Datasets for Training and Benchmarking GNNs
| Dataset Name | Scale and Content | Key Features/Labels | Utility for DFT-Accuracy GNNs |
|---|---|---|---|
| PubChemQCR [74] | ~3.5M relaxation trajectories, >300M conformations (105M from DFT) | Total energy, atomic forces for intermediate and stable geometries | Training MLIPs for molecular dynamics and geometry optimization |
| QM9 [74] | ~130,000 small molecules | 19 quantum chemical properties per molecule (single conformation) | Benchmarking property prediction models |
| GMTKN55 [75] | 55 subsets for general main-group thermochemistry, kinetics, non-covalent interactions | Comprehensive benchmark for reaction energies, barrier heights | Guiding DFT protocol development and validation |
| CRM Dataset [73] | Elementary steps for transition metal-catalyzed reactions | Reaction class, reactive atom labels, reaction templates | Training and evaluating mechanism prediction models (ReactAIvate) |
Experimental Protocols for Key Applications
Protocol: Predicting Molecular Properties with KA-GNNs
This protocol outlines the procedure for utilizing Kolmogorov-Arnold Graph Neural Networks (KA-GNNs) to predict molecular properties with high accuracy and interpretability [5].
- Molecular Graph Construction: Generate a graph representation from the molecular structure (e.g., from SMILES string), where atoms are nodes and bonds are edges.
- Feature Initialization with KANs:
- Initialize node features by passing atomic features (e.g., atomic number, radius) through a Fourier-based KAN layer.
- Initialize edge features by processing bond features (e.g., bond type, length) through a separate Fourier-based KAN layer.
- Message Passing with KAN Modules: For a predetermined number of steps (K), perform message passing:
- Message Function (Mt): For each node, aggregate messages from its neighboring nodes using a KAN-based function applied to the node and edge features.
- Update Function (Ut): Update each node's embedding using another KAN module that combines its current state and the aggregated message. Residual KAN connections can be used.
- Global Readout: Generate a single graph-level representation from the updated node embeddings using a permutation-invariant readout function (e.g., sum, mean, or attention-based pooling).
- Property Prediction: Pass the graph-level representation through a final KAN layer or a simple linear layer to predict the target molecular property.
- Model Interpretation: Leverage the inherent interpretability of KANs to examine the learned univariate functions and identify chemically meaningful substructures that influence the prediction.
Protocol: Correcting DFT Energies to Coupled-Cluster Accuracy (Δ-DFT)
This protocol describes using machine learning, specifically kernel ridge regression (KRR), to predict high-level coupled-cluster (CC) energies from DFT densities, achieving quantum chemical accuracy [71].
- Training Set Generation:
- Select a diverse set of molecular geometries relevant to the system of interest (e.g., via MD sampling).
- For each geometry, perform: a. A standard DFT calculation (e.g., using the PBE functional) to obtain the self-consistent electron density (nDFT) and DFT energy (EDFT). b. A highly accurate (but expensive) CCSD(T) calculation to obtain the reference energy (ECC).
- Compute the target correction for each molecule: ΔE = ECC - EDFT.
- Descriptor Preparation: Use the DFT electron density (nDFT) as the primary descriptor (input feature) for the ML model. Exploiting molecular point group symmetries can augment the training data.
- Model Training: Train a Kernel Ridge Regression (KRR) model to learn the mapping from the DFT density (nDFT) to the energy correction (ΔE).
- Prediction (Inference):
- For a new molecule, perform only the standard DFT calculation to obtain nDFT.
- Feed nDFT into the trained KRR model to predict ΔE.
- Obtain the predicted CCSD(T)-level energy: E = EDFT + ΔE.
Protocol: Predicting Reaction Mechanisms with ReactAIvate
This protocol employs the ReactAIvate model, an interpretable attention-based GNN, to predict elementary reaction steps and identify reactive atoms in a chemical reaction mechanism (CRM) [73].
- Reaction Representation: Represent the reaction system (reactants, reagents, catalyst) as a set of molecular graphs.
- Graph Processing with Attention: Process the molecular graphs through a graph attention network (GAT) to generate enriched atom-level embeddings.
- Multi-Task Learning for Classification and Interpretation:
- Graph-Level Loss (Classification): Aggregate atom embeddings to a graph-level representation and pass it through a classifier to predict the elementary reaction step (e.g., oxidative addition, reductive elimination).
- Node-Level Loss (Reactive Atom Identification): Simultaneously, use the atom-level embeddings to classify each atom as reactive or non-reactive in the current step.
- Mechanism Assembly: Iteratively apply the trained model to the reactants and subsequent intermediates to predict a sequence of elementary steps, thereby constructing the full CRM.
- Interpretation and Validation: Visualize the model's attention scores to identify reactive centers (reactivity hotspots) in the molecules, providing atomic-level insights and validating the prediction against chemical intuition.
Workflow Visualization
The following diagrams illustrate the logical workflows for the key protocols described above.
Figure 1: KA-GNN Property Prediction Workflow
Figure 2: Δ-DFT Correction Workflow
Figure 3: Reaction Mechanism Prediction Workflow
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Computational Tools and Datasets for GNN Research
| Tool/Resource Name | Type | Primary Function | Relevance to DFT-Accuracy GNNs |
|---|---|---|---|
| Fourier-KAN Layer [5] | Algorithmic Module | Learnable activation function using Fourier series | Captures high/low-frequency patterns in graphs; enhances expressivity in KA-GNNs. |
| Steric & Electronic Embedding (SEMG) [72] | Molecular Representation | Encodes local steric (via SPMS) and electronic (via cube of electron density) environments | Provides rich, quantum-mechanics-informed input features for GNNs. |
| Molecular Interaction Module [72] | GNN Architectural Component | Enables information exchange between graphs of different reaction components | Crucial for modeling synergistic effects in multi-component reaction systems. |
| Message Passing Neural Network (MPNN) [13] | GNN Framework | General blueprint for building GNNs (message, update, readout phases) | Foundational architecture for many molecular GNNs. |
| PubChemQCR Dataset [74] | Benchmark Data | Large-scale DFT relaxation trajectories with energies/forces | Essential for training Machine Learning Interatomic Potentials (MLIPs) to achieve DFT-level dynamics. |
| Kernel Ridge Regression (KRR) [71] | Machine Learning Model | Non-linear regression for learning energy functionals | Core model in the Δ-DFT protocol for correcting DFT to CCSD(T) accuracy. |
The accurate prediction of molecular mechanics parameters is a cornerstone in computational chemistry and drug discovery, enabling researchers to simulate molecular behavior and interactions with high fidelity. Graph Neural Networks (GNNs) have emerged as powerful tools for this task, as they naturally represent molecules as graphs where atoms are nodes and bonds are edges. Among the various GNN architectures, Graph Convolutional Networks (GCNs), Graph Attention Networks (GATs), Message-Passing Neural Networks (MPNNs), and Graph Transformers have each demonstrated unique strengths and limitations. This application note provides a comparative analysis of these architectures within the context of predicting molecular mechanics parameters, offering structured data, detailed experimental protocols, and practical toolkits to guide researchers and development professionals in selecting and implementing the most suitable models for their specific applications.
Key Architectures and Their Mechanisms
- Graph Convolutional Networks (GCNs): Utilize a convolution-like aggregation function across node neighborhoods. Features of connected nodes are normalized using the factor ( \frac{1}{\sqrt{di dj}} ), where ( di ) and ( dj ) denote the degrees of nodes i and j, respectively. This escalation balances node contributions and makes GCNs among the fastest GNN categories [15].
- Graph Attention Networks (GATs): Incorporate self-attention mechanisms to compute attention coefficients between adjacent nodes, allowing the model to assign varying importance to a node's neighbors during feature aggregation. The attention coefficient ( a(xi, xj) ) is typically computed as ( a(xi, xj) = \text{SoftMax}(\text{LeakyReLU}(\mathbf{a}^T [\mathbf{W}xi \| \mathbf{W}xj])) ), where ( \mathbf{a} ) is a learnable attention vector and ( \mathbf{W} ) is a weight matrix [15].
- Message-Passing Neural Networks (MPNNs): Operate through a framework where nodes generate messages based on their own features and those of their neighbors, aggregate incoming messages, and update their representations using learnable functions. A bidirectional message-passing strategy can circumvent the inherent directionality of classical MPNNs, which may be counterintuitive for molecules [15].
- Graph Transformers: Adapt the transformer architecture to graph-structured data, often using self-attention mechanisms that can capture both local and global node relationships more effectively than traditional GNNs. Recent approaches, such as the Edge-Set Attention (ESA) architecture, consider graphs as sets of edges and use an encoder that interleaves masked and self-attention modules to learn effective representations without relying on hand-crafted operators [17].
Quantitative Performance Comparison
Table 1: Comparative performance of GNN architectures across various molecular tasks
| Architecture | Task/Dataset | Performance Metric | Score | Key Advantage |
|---|---|---|---|---|
| MPNN [76] | Cross-coupling reaction yield prediction | R² | 0.75 | Best predictive performance for reaction yields |
| ESA (Transformer) [17] | Multiple node/graph-level tasks (70+ benchmarks) | Performance vs. tuned GNNs | Outperforms baselines | Superior transfer learning, scalability |
| GIN [16] | Molecular point group prediction (QM9) | Accuracy | 92.7% | Effective capture of local/global structures |
| KA-GNN (KAN-augmented) [5] | Molecular property prediction | Accuracy & Efficiency | Consistently outperforms conventional GNNs | Improved interpretability, parameter efficiency |
| GAT [77] | Activity cliff prediction (MoleculeACE) | Sensitivity to local changes | Underperforms ECFPs | Adaptive attention on neighbor nodes |
| Quantized GNN [10] | Dipole moment prediction (QM9) | Performance at 8-bit | Maintains strong performance | Reduced memory & computational cost |
Table 2: Computational efficiency and resource requirements
| Architecture | Scalability | Memory Footprint | Inference Latency | Ideal Use Case |
|---|---|---|---|---|
| GCN [15] | High | Low | Low | Large-scale screening, resource-constrained environments |
| GAT/GATv2 [15] [76] | Medium | Medium | Medium | Tasks requiring differentiation of neighbor importance |
| MPNN [76] | Medium | Medium | Medium | Reaction yield prediction, molecular property prediction |
| Graph Transformer [17] | Medium to High | High | Medium to High | Transfer learning, tasks requiring long-range dependencies |
| ESA Transformer [17] | High | Medium | Medium | Large-scale molecular graphs, various graph-level tasks |
| Quantized GNN [10] | High | Very Low | Low | Edge devices, real-time applications |
Experimental Protocols
Protocol 1: Evaluating GNN Architectures for Molecular Property Prediction
Objective: To systematically assess and compare the performance of GCN, GAT, MPNN, and Graph Transformer architectures for predicting molecular mechanics parameters.
Materials:
- Datasets: QM9 (dipole moment, HOMO-LUMO gap), ESOL (solubility), FreeSolv (hydration free energy) [10]
- Software: PyTorch Geometric, RDKit
- Hardware: GPU-enabled system (recommended)
Procedure:
- Data Preprocessing:
- For each dataset, load molecular structures and convert them to graph representations with nodes (atoms) and edges (bonds).
- Initialize node features using atomic properties (e.g., atomic number, hybridization, number of hydrogens).
- Initialize edge features using bond properties (e.g., bond type, conjugation status) [15].
- Split data into training (80%), validation (10%), and test (10%) sets.
Model Configuration:
- Implement GCN, GAT, MPNN, and Graph Transformer models with comparable parameter counts.
- For GCN: Use 2-3 layers with the convolution normalization factor ( \frac{1}{\sqrt{di dj}} ) [15].
- For GAT: Implement multi-head attention (4-8 heads) with LeakyReLU activation [15].
- For MPNN: Use bidirectional message passing with a gated aggregation function [15].
- For Graph Transformer: Implement edge-set attention (ESA) with masked and vanilla self-attention modules [17].
Training:
- Train each model using the Adam optimizer with a learning rate of 0.001.
- Use Mean Squared Error (MSE) loss for regression tasks or Cross-Entropy loss for classification tasks.
- Implement early stopping with a patience of 50 epochs based on validation loss.
Evaluation:
- Assess model performance on the test set using metrics: RMSE, MAE for regression; Accuracy, F1-score for classification.
- Compare computational efficiency: training time, inference latency, and memory usage.
Protocol 2: Assessing Sensitivity to Activity Cliffs
Objective: To evaluate the capability of GNN architectures to distinguish structurally similar molecules with large potency differences (activity cliffs).
Materials:
- Dataset: MoleculeACE (curated from ChEMBL) [77]
- Software: RDKit for fingerprint generation, PyTorch Geometric
Procedure:
- Data Preparation:
- Identify activity cliff pairs in the dataset defined as structurally similar compounds (>0.7 Tanimoto similarity based on ECFPs) with significant potency difference (>10-fold difference in Ki) [77].
- For each molecule, generate both graph representations and ECFP fingerprints (radius=2, 1024 bits).
Model Training:
- Train GCN, GAT, MPNN, and GraphCliff models on the activity cliff dataset.
- For GraphCliff, implement short- and long-range gating mechanisms to capture both local and global structural information [77].
Analysis:
- Extract molecular embeddings from the trained models for each activity cliff pair.
- Calculate Euclidean distances between embeddings of cliff pairs.
- Compare with ECFP dissimilarities (1 - Tanimoto similarity) to assess sensitivity to local structural changes.
- Evaluate model performance on both cliff and non-cliff compounds using ROC-AUC and Precision-Recall metrics.
Protocol 3: Implementing Quantized GNNs for Efficient Inference
Objective: To reduce memory footprint and computational demands of GNN models through quantization while maintaining predictive performance.
Materials:
- Datasets: QM9 (dipole moment), ESOL, FreeSolv [10]
- Software: PyTorch, DoReFa-Net quantization algorithm [10]
Procedure:
- Model Selection and Training:
- Train GCN and GIN models in full precision (FP32) on the target datasets following standard protocols.
- Save the trained models for quantization.
Quantization:
- Apply the DoReFa-Net algorithm to quantize weights and activations to different bit-widths (INT8, INT4, INT2).
- For INT8 quantization, use the following scheme:
- Quantize weights: ( wq = \text{quantize}(w, k) ) where k=8
- Quantize activations: ( aq = \text{quantize}(a, k) ) where k=8
- Implement custom layers to handle the quantization during forward pass.
Evaluation:
- Compare the performance of quantized models with full-precision models using RMSE and MAE.
- Measure memory footprint reduction and inference speedup across different bit-widths.
- Identify the optimal bit-width that maintains performance while maximizing efficiency.
Visualization of Architectural Workflows
Diagram 1: Comparative workflow for molecular mechanics prediction using different GNN architectures
Diagram 2: Architectural mechanisms of different GNN approaches
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential tools and resources for GNN implementation in molecular mechanics
| Tool/Resource | Function | Application Context |
|---|---|---|
| PyTorch Geometric [10] | Library for graph deep learning | Implements GCN, GAT, GIN, MPNN, and Transformer architectures; standard molecular datasets |
| RDKit [15] | Cheminformatics toolkit | Molecular graph generation; feature extraction (atomic, bond, molecular descriptors) |
| QM9 Dataset [10] [16] | Quantum chemical properties for 130k small organic molecules | Training and benchmarking GNNs for molecular property prediction |
| MoleculeACE Dataset [77] | Curated activity cliff pairs from ChEMBL | Evaluating sensitivity to structurally similar molecules with different potencies |
| DoReFa-Net Algorithm [10] | Quantization method for neural networks | Reducing memory footprint and computational demands of GNN models |
| Grappa Force Field [78] | Machine-learned molecular mechanics force field | Predicting MM parameters from molecular graphs using GNNs |
| GraphCliff Architecture [77] | GNN with short-long range gating | Handling activity cliffs by integrating local and global molecular context |
This comparative analysis demonstrates that each GNN architecture offers distinct advantages for predicting molecular mechanics parameters. GCNs provide computational efficiency for large-scale screening, GATs offer adaptive attention mechanisms for differentiating molecular regions, MPNNs deliver strong performance particularly for reaction yield prediction, and Graph Transformers excel in transfer learning and capturing long-range dependencies. The integration of novel approaches such as Kolmogorov-Arnold networks, edge-set attention, and quantization techniques further enhances the capabilities of GNNs. Researchers should select architectures based on their specific requirements regarding accuracy, interpretability, computational efficiency, and sensitivity to subtle molecular changes. As GNN methodologies continue to evolve, they promise to further bridge the gap between computational predictions and experimental accuracy in molecular mechanics.
Conclusion
The integration of Graph Neural Networks into molecular mechanics parameter prediction marks a significant leap forward, enabling the development of accurate, efficient, and highly transferable force fields. Frameworks like Grappa demonstrate that GNNs can predict parameters directly from molecular graphs, outperforming traditional methods and rivaling the accuracy of quantum mechanics at a fraction of the computational cost. Key to this progress are architectural innovations—from message-passing networks to Graph Transformers and KA-GNNs—coupled with strategies to overcome data limitations and ensure model scalability. Looking ahead, the future of GNN-driven force fields lies in expanding their reach to more complex biomolecular systems, improving their ability to model reactive processes, and fully integrating them into automated, high-throughput discovery pipelines. This progress promises to accelerate drug discovery and materials science by providing a robust computational foundation for reliable in-silico experimentation.
References
- 1. Data-driven parametrization of molecular mechanics force ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc06640e]
- 2. Revisiting iterative molecular mechanics force field ... [https://chemrxiv.org/engage/chemrxiv/article-details/6882c20e23be8e43d6e0bca5]
- 3. Parametrization of Force Field Bonded Terms under ... [https://pubmed.ncbi.nlm.nih.gov/36112364/]
- 4. Automated optimization of force field parameters against ... [https://arxiv.org/html/2402.11169v2]
- 5. Kolmogorov–Arnold graph neural networks for molecular ... [https://www.nature.com/articles/s42256-025-01087-7]
- 6. An improved reactive force field parameter optimization ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025625001193]
- 7. Speed up Multi-Scale Force-Field Parameter Optimization ... [https://pubmed.ncbi.nlm.nih.gov/40911030/]
- 8. Fine-tuning molecular mechanics force fields to experimental ... [https://pubmed.ncbi.nlm.nih.gov/39829785/]
- 9. Molecular representations in AI-driven drug discovery [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-00460-5]
- 10. Enhancing molecular property prediction with quantized GNN ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-00989-3]
- 11. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 12. Using GNN property predictors as molecule generators [https://www.nature.com/articles/s41467-025-59439-1]
- 13. Graph neural networks for materials science and chemistry [https://www.nature.com/articles/s43246-022-00315-6]
- 14. Application of message passing neural networks for ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X23000908]
- 15. Optimal message passing for molecular prediction is simple ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00193e]
- 16. A Method For Predicting Molecular Point Group Based On ... [https://www.sciencedirect.com/science/article/pii/S2949747725000144]
- 17. An end-to-end attention-based approach for learning on ... [https://www.nature.com/articles/s41467-025-60252-z]
- 18. Grappa – a machine learned molecular mechanics force field [https://pmc.ncbi.nlm.nih.gov/articles/PMC11734696/]
- 19. Grappa -- A Machine Learned Molecular Mechanics Force ... [https://arxiv.org/abs/2404.00050]
- 20. [2209.05582] Graph Neural Networks for Molecules [https://arxiv.org/abs/2209.05582]
- 21. Machine-learned molecular mechanics force fields from large ... [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d4sc00690a]
- 22. A Machine Learned Molecular Mechanics Force Field [https://arxiv.org/html/2404.00050v1]
- 23. A Machine Learned Molecular Mechanics Force Field [https://arxiv.org/html/2404.00050v2]
- 24. graeter-group/grappa: A machine learned Molecular ... [https://github.com/graeter-group/grappa]
- 25. GRAPPA - mpip-mainz.mpg.de [https://www.mpip-mainz.mpg.de/en/graeter/software/grappa]
- 26. Data-driven parametrization of molecular mechanics force ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11721737/]
- 27. KA-GNN: Kolmogorov-Arnold Graph Neural Networks for ... [https://arxiv.org/html/2410.11323v1]
- 28. graph-based transformer versus GNN models | Journal of ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01004-5]
- 29. Kolmogorov–Arnold Graph Neural Networks [https://arxiv.org/html/2406.18354v2]
- 30. Utilizing graph machine learning within drug discovery and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8574649/]
- 31. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 32. Drug discovery and mechanism prediction with explainable ... [https://www.nature.com/articles/s41598-024-83090-3]
- 33. The principles behind equivariant neural networks for physics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12541325/]
- 34. On the Permutation Invariance of Graph Generative Models [https://qiyan98.github.io/blog/2025/gen-graph/]
- 35. Pretrained E(3)-equivariant message-passing neural ... [https://www.nature.com/articles/s41524-025-01698-z]
- 36. Advancing Universal Deep Learning for Electronic ... [https://arxiv.org/html/2509.19877v2]
- 37. Efficient equivariant model for machine learning ... [https://www.nature.com/articles/s41524-025-01535-3]
- 38. Facet: highly efficient 𝐸(3)-equivariant networks for ... [https://arxiv.org/html/2509.08418v1]
- 39. A Survey of Graph Neural Networks for Drug Discovery [https://arxiv.org/html/2509.07887v1]
- 40. Graph neural networks driven acceleration in drug discovery [https://www.sciencedirect.com/science/article/pii/S2211383525006884]
- 41. Predicting Molecular Energy using Force-Field Optimized ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6615995/]
- 42. Data-driven parametrization of molecular mechanics force ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06640e]
- 43. Recent advances in deep learning for protein-protein interaction [https://pmc.ncbi.nlm.nih.gov/articles/PMC12168265/]
- 44. RNA-protein interaction prediction using network-guided deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11830795/]
- 45. Data-Driven Parametrization of Molecular Mechanics Force ... [https://arxiv.org/html/2408.12817v1]
- 46. Imbalanced Dataset: Strategies to Fix Skewed Class ... [https://labelyourdata.com/articles/imbalanced-dataset]
- 47. How to Handle Imbalanced Data? [https://www.analyticsvidhya.com/blog/2021/06/5-techniques-to-handle-imbalanced-data-for-a-classification-problem/]
- 48. Class-imbalanced datasets | Machine Learning [https://developers.google.com/machine-learning/crash-course/overfitting/imbalanced-datasets]
- 49. On the Scalability of GNNs for Molecular Graphs [https://arxiv.org/abs/2404.11568]
- 50. On the Scalability of GNNs for Molecular Graphs [https://openreview.net/forum?id=klqhrq7fvB¬eId=4Yy3RGUYaP]
- 51. Scaling Laws for Neural Material Models [https://arxiv.org/html/2509.21811v1]
- 52. Scaling Laws of Graph Neural Networks for Atomistic ... [https://arxiv.org/abs/2504.08112]
- 53. Understanding the Capabilities of Molecular Graph Neural ... [https://arxiv.org/html/2505.12137v1]
- 54. Recent Developments in GNNs for Drug Discovery [https://arxiv.org/html/2506.01302v1]
- 55. Grappa – a machine learned molecular mechanics force field [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc05465b]
- 56. Multi-task methods for molecular property prediction [https://www.sciencedirect.com/science/article/pii/S0098135425002571]
- 57. Navigating protein landscapes with a machine-learned ... [https://www.nature.com/articles/s41557-025-01874-0]
- 58. Harnessing Data Augmentation and Normalization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10180765/]
- 59. Using GNN property predictors as molecule generators [https://pmc.ncbi.nlm.nih.gov/articles/PMC12062463/]
- 60. Enhancing molecular property prediction with quantized GNN ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12108020/]
- 61. Machine learning models for the prediction of energy ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025619307827]
- 62. Rotationally Invariant Latent Distances for Uncertainty ... [https://arxiv.org/html/2407.10844v1]
- 63. Protocol to explain graph neural network predictions using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9700376/]
- 64. Statistical Prediction and Molecular Dynamics Simulation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2576400/]
- 65. Modeling the potential energy surface by force fields for ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc02715b?page=search]
- 66. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 67. Bridging Quantum Mechanics to Organic Liquid Properties ... [https://arxiv.org/abs/2508.08575]
- 68. Accurate machine learning force fields via experimental ... [https://www.nature.com/articles/s41524-024-01251-4]
- 69. Evaluating the transferability of machine-learned force ... [https://www.sciencedirect.com/science/article/abs/pii/S0010465523000681]
- 70. Graph neural networks for the prediction of molecular ... [https://arxiv.org/abs/2208.04852]
- 71. Quantum chemical accuracy from density functional ... [https://www.nature.com/articles/s41467-020-19093-1]
- 72. Reaction performance prediction with an extrapolative and ... [https://www.nature.com/articles/s41467-023-39283-x]
- 73. ReactAIvate: A Deep Learning Approach to Predicting ... [https://arxiv.org/html/2407.10090v1]
- 74. A Benchmark for Quantum Chemistry Relaxations via ... [https://arxiv.org/html/2506.23008v1]
- 75. Best‐Practice DFT Protocols for Basic Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9826355/]
- 76. Performance assessment of various graph neural network ... [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp01072a]
- 77. GraphCliff: Short-Long Range Gating for Subtle Differences ... [https://arxiv.org/html/2511.03170v1]
- 78. Grappa – a machine learned molecular mechanics force field [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc05465b]