Optimizing MD Time Step: A Practical Guide for Accurate Atomic Motion Tracking in Biomedical Research
This article provides a comprehensive guide for researchers and drug development professionals on optimizing the molecular dynamics (MD) time step to balance computational efficiency with physical accuracy.
Optimizing MD Time Step: A Practical Guide for Accurate Atomic Motion Tracking in Biomedical Research
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on optimizing the molecular dynamics (MD) time step to balance computational efficiency with physical accuracy. It covers the foundational principles governing time step selection, explores advanced methodological approaches and their practical applications in fields like drug discovery, outlines troubleshooting and optimization strategies for common pitfalls, and establishes rigorous validation protocols. By synthesizing current research and best practices, this guide aims to empower scientists to generate more reliable simulation data for biomedical applications, from predicting ligand-receptor interactions to designing novel materials.
The Physics Behind the Time Step: Core Principles for Accurate Atomic Motion
Frequently Asked Questions (FAQs)
FAQ 1: What is the Nyquist-Shannon sampling theorem and why is it critical for my MD simulations?
The Nyquist-Shannon sampling theorem is a fundamental principle of signal processing which states that to accurately capture a continuous signal without distortion (aliasing), the sampling frequency must be at least twice the highest frequency present in the signal [1]. In molecular dynamics, this theorem translates to a requirement that your integration time step must be small enough to capture the fastest atomic vibrations in your system. If the time step is too large, you will violate this theorem, leading to simulation instability, aliasing of high-frequency motions, and inaccurate dynamics [2].
FAQ 2: My simulation exploded after running fine for hundreds of picoseconds. Could this be related to the time step choice?
Yes, this is a classic symptom of a time step that is too large. Simulations may appear stable initially but suddenly "blow up" when atoms vibrating at high frequencies accidentally experience a larger-than-usual force, accelerating them to the point where reliable integration is no longer possible [3]. This is particularly problematic for hydrogen atoms due to their low mass, which creates the fastest vibrations in biomolecular systems. The resulting instability can infect neighboring atoms and cause complete simulation failure [3].
FAQ 3: I'm using hydrogen mass repartitioning (HMR) to enable a larger time step. What are the potential drawbacks?
While HMR and other mass-scaling techniques can allow for longer time steps by slowing down the fastest vibrations, they can introduce physical inaccuracies for certain types of simulations. Recent research indicates that HMR can alter ligand recognition kinetics in protein-ligand systems, potentially slowing down the recognition process despite the apparent performance gain [4]. This occurs because the modified masses affect particle diffusion rates, which can reduce the survival probability of metastable intermediates crucial for binding pathways [4].
FAQ 4: How can I quantitatively check if my chosen time step is appropriate?
The most reliable method is to run a constant energy (NVE) simulation and monitor the total energy conservation. A well-chosen time step with a proper symplectic integrator should show minimal energy drift [2]. Quantitative guidelines suggest that for publishable results, the long-term drift in the conserved quantity should be less than 1 meV/atom/ps, while for qualitative results, less than 10 meV/atom/ps may be acceptable [2]. Significant drift indicates your time step is too large or your integration method is inappropriate.
Troubleshooting Guides
Problem: Simulation Instability and "Blow-ups"
Symptoms
- Simulation terminates unexpectedly with error messages about coordinate overflow
- Rapid energy increase leading to unrealistic atomic displacements
- "Flying" atoms or molecules leaving the simulation box
Diagnosis and Solutions
Identify the fastest vibrations in your system
Implement constraint algorithms
- Use LINCS or SHAKE to constrain bond lengths involving hydrogen atoms
- This effectively removes the highest frequency vibrations from numerical integration
- Allows increasing time step from 1 fs to 2 fs while maintaining stability [3]
Consider multiple time-step algorithms
- Use shorter time steps for fast bonded interactions (e.g., 2.5 fs)
- Use longer time steps for slow non-bonded interactions (e.g., 5 fs) [3]
- Implement with care to maintain energy conservation
Problem: Poor Energy Conservation
Symptoms
- Steady drift in total energy in NVE simulations
- Temperature drift in NVT simulations despite proper thermostatting
- Failure to maintain equipartition between different degrees of freedom
Diagnosis and Solutions
Verify time-reversibility
- Run simulation forward, then reverse directions
- A proper symplectic integrator should retrace its path
- Significant deviation indicates time step is too large [2]
Check for resonance effects
- Ensure time step is not commensurate with natural periods of important vibrations
- Avoid time steps that are simple fractions (¼, ⅓, ½) of vibrational periods
- Test multiple time step values in the 0.5-2 fs range
Monitor specific energy components
- Track potential vs. kinetic energy separation
- Look for abnormal energy transfer between degrees of freedom
- Use this information to identify which interactions need more frequent evaluation
Quantitative Data for Time Step Selection
Table 1: Maximum Stable Time Steps for Different Simulation Protocols
| Simulation Method | Maximum Time Step | Key Considerations | Primary Limitations |
|---|---|---|---|
| Unconstrained all-atom | 1 fs | Limited by C-H/O-H bond vibrations (~10 fs period) | Hydrogen vibrations become unstable [3] |
| Bond constraints (SHAKE/LINCS) | 2 fs | Standard for biomolecular simulations; constrains bond lengths | Hydrogen angle vibrations become critical at ~2.5 fs [3] |
| Multiple time-stepping | 2.5 fs (bonds)5 fs (non-bonded) | Different time steps for different interaction types | Requires careful implementation to conserve energy [3] |
| Hydrogen Mass Repartitioning (HMR) | 3-4 fs | Increases hydrogen masses 3-4x; slows fastest vibrations | Alters dynamics; may affect kinetic properties [4] |
Table 2: Characteristic Vibration Periods and Nyquist Time Steps
| Vibration Type | Frequency (cm⁻¹) | Period (fs) | Nyquist Time Step (fs) | Affected Atoms |
|---|---|---|---|---|
| O-H stretch | 3400-3600 | ~9-10 | 4.5-5.0 | Water, hydroxyl groups |
| C-H stretch | 2900-3100 | ~11-12 | 5.5-6.0 | Methyl, methylene groups |
| N-H stretch | 3300-3500 | ~10-11 | 5.0-5.5 | Amine groups, amide backbone |
| C=O stretch | 1600-1800 | ~19-21 | 9.5-10.5 | Peptide backbone, carbonyls |
Experimental Validation Protocols
Protocol 1: Time Step Optimization Procedure
Step-by-Step Methodology
Initial conservative testing
- Begin with a 1 fs time step for all-atom simulations without constraints
- Run a 20 ps NVE simulation monitoring total energy conservation
- Calculate energy drift as meV/atom/ps [2]
Systematic time step increase
- Increase time step by 0.1-0.2 fs increments
- At each step, run a 20-50 ps NVE simulation
- Record the RMS deviation of total energy from mean
- Stop when energy drift exceeds 1 meV/atom/ps for more than 5 consecutive steps
Implementation of constraints
- Once the unconstrained limit is found (typically 1-1.5 fs), implement bond constraints
- Use LINCS or SHAKE to constrain all bonds involving hydrogen atoms
- Retest stability starting at 1.5 fs, increasing to 2 fs
Advanced optimization
- For further time step increases, consider hydrogen mass repartitioning (3-4× mass increase)
- Test kinetic properties to ensure biological relevance is maintained
- Validate against known experimental data or high-quality reference simulations
Protocol 2: Energy Conservation Validation
Procedure
- Run a 100 ps - 1 ns NVE simulation with your production time step
- Calculate the conserved quantity (total energy for NVE)
- Compute the drift rate using linear regression of energy versus time
- Normalize by number of atoms: drift (meV/atom/ps) = slope / (N_atoms × simulation time)
Acceptance Criteria
- Publishable quality: < 1 meV/atom/ps drift [2]
- Qualitative studies: < 10 meV/atom/ps drift [2]
- Unacceptable: > 10 meV/atom/ps or catastrophic energy increase
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool/Solution | Function | Implementation Considerations |
|---|---|---|
| SHAKE Algorithm | Constrains bond lengths to equilibrium values; removes fastest vibrations | Iterative method; can be slow for large systems; requires convergence check [3] |
| LINCS Algorithm | Constrains bond lengths using matrix inversion; faster than SHAKE | More efficient for parallel computation; handles constraints in rings well [3] |
| Velocity Verlet Integrator | Symplectic, time-reversible integration algorithm | Preserves geometric structure of Hamiltonian flow; excellent long-term stability [2] |
| Hydrogen Mass Repartitioning (HMR) | Increases hydrogen masses to slow high-frequency vibrations | Enables 3-4 fs time steps; may alter ligand binding kinetics [4] |
| Multiple Time Stepping | Different time steps for different interaction classes | Can improve efficiency 2-3×; requires careful tuning to avoid resonance [3] |
| NVE Validation Simulation | Gold standard for testing time step stability | Must show minimal energy drift for rigorous results [2] |
Advanced Integration Strategies
Strategy 1: Mixed Multiple Time-Step Algorithm
Advanced implementations can combine different integration strategies for various molecular components. For example, small molecules whose internal degrees of freedom can be completely constrained may be propagated with a large single time step (up to 5 fs), while flexible molecules require multiple time-stepping with shorter intervals for bonded interactions [3]. This hybrid approach maximizes efficiency while maintaining stability.
Strategy 2: Structure-Preserving Machine Learning
Emerging approaches use machine learning to learn structure-preserving (symplectic and time-reversible) maps that can generate long-time-step classical dynamics, effectively learning the mechanical action of the system [5]. These methods show promise for significantly extending integration time steps while preserving the geometric structure of the underlying Hamiltonian flow, though they remain primarily in the research domain.
Critical Limitations and Caveats
When applying the Nyquist-Shannon theorem to molecular dynamics, several important caveats must be considered:
Theoretical vs. practical limits: While Nyquist provides an absolute upper bound (time step ≤ ½ vibration period), practical implementations typically require time steps 5-10× smaller than this limit for accurate integration [2].
Physical accuracy vs. numerical stability: Techniques like hydrogen mass repartitioning may improve numerical stability but can alter physically important dynamics, particularly for processes like ligand binding where diffusion rates are critical [4].
System-dependent optimization: Optimal time steps vary significantly between systems—aqueous solutions with light water molecules require shorter time steps than membrane systems with heavier lipid components.
The Nyquist-Shannon theorem remains the fundamental principle governing time step selection in molecular dynamics, but successful application requires careful validation specific to each biological system and scientific question.
Frequently Asked Questions (FAQs)
Q1: What are the primary factors that limit the time step in my Molecular Dynamics (MD) simulation? The maximum time step is primarily limited by the fastest vibrational frequencies present in your system. This is typically the stretching of bonds involving hydrogen atoms, which oscillate on the order of 10 femtoseconds (fs). To accurately integrate these motions, a time step smaller than the oscillation period is required, often around 0.5-1.0 fs. Using a larger step can lead to numerical instability, causing the simulation to "blow up" as energy incorrectly flows into these fast modes [6].
Q2: How can I increase my time step without causing a simulation to become unstable? There are two main strategies:
- Constraint Algorithms: Methods like SHAKE and LINCS fix the lengths of the fastest bonds (e.g., bonds to hydrogen), effectively removing their high-frequency motion. This allows time steps to be increased to about 2 fs [7] [8].
- Machine-Learned Force Fields (MLFFs): Advanced machine learning models can be trained to predict long-time-step dynamics. Crucially, models that preserve the geometric structure (symplecticity) of Hamiltonian mechanics can use larger steps while conserving energy and maintaining physical fidelity, preventing artifacts like lack of equipartition [5].
Q3: My simulation has a low force error, but the trajectory still becomes unphysical. Why? A low force Mean Absolute Error (MAE) does not guarantee simulation stability. Instability often arises from extrapolation errors, where the model encounters atomic configurations not represented in its training data. This can lead to predictions of extreme, non-physical forces, causing catastrophic trajectory divergence and bond breakage. Stability is better assessed through metrics like the Radial Distribution Function (RDF) or the longevity of the simulation itself [6] [9].
Q4: What is the role of the "physical model" or "force field" in determining the time step? The force field defines the Potential Energy Surface (PES) and the forces acting on atoms. A model with very "stiff" bonds (a steep potential well) creates high-frequency motions that demand a small time step. Conversely, a coarse-grained model, which smoothens the PES by eliminating atomistic detail, removes these fast degrees of freedom, permitting much larger time steps. Therefore, the choice of model directly dictates the system's highest frequency and thus the maximum stable time step.
Q5: How does the numerical integration algorithm influence the time step? The integrator is responsible for propagating the equations of motion. Simple algorithms like the velocity Verlet method require time steps small enough to sample the fastest motion. More sophisticated, symplectic integrators preserve the geometric structure of the Hamiltonian flow, ensuring better long-term energy conservation. This allows for more reliable behavior at the upper limit of stability for a given time step [5] [6].
Troubleshooting Guides
Instability and Energy Drift at Current Time Step
Problem: The total energy of the system drifts significantly over time, or the simulation crashes with atoms flying apart.
| Possible Cause | Diagnostic Steps | Solution |
|---|---|---|
| Time step is too large. | Check the log file for warnings. Monitor the total energy and temperature for a sharp, continuous increase. | Reduce the time step by 50% (e.g., from 2 fs to 1 fs) and test again. |
| System contains stiff bonds (e.g., X-H). | Identify the smallest bond length and its associated force constant in the force field. | Apply a constraint algorithm (e.g., SHAKE, LINCS, or ILVES-PC) to freeze the fastest bond vibrations [7] [8]. |
| Inaccurate force calculations. | For MLFFs, check force MAE on a validation set. Look for configurations where forces are outliers. | Retrain the MLFF with a more diverse dataset or use a pre-trained model to improve generalizability [6] [9]. |
Achieving a Larger Time Step for Improved Efficiency
Goal: Safely increase the time step to sample more configuration space with the same computational resources.
| Strategy | Protocol | Considerations |
|---|---|---|
| Using Constraint Algorithms | 1. In your MD engine (e.g., GROMACS, LAMMPS), enable a constraint algorithm like SHAKE or LINCS.2. Specify which bonds to constrain (typically all bonds to hydrogen).3. Set the time step to 2.0 fs. | Constraint algorithms solve a system of nonlinear equations each step. ILVES-PC, a Newton-method-based algorithm, can be faster and more accurate than SHAKE for proteins [7]. |
| Adopting Machine-Learned Force Fields | 1. Pre-train a Graph Neural Network (e.g., GemNet-T) on a large, diverse dataset (e.g., OC20) [6].2. Fine-tune on a smaller, system-specific dataset.3. Use a symplectic, time-reversible integrator derived from a learned generating function to preserve physics with large steps [5]. | Pre-training is crucial for stability, leading to trajectories three times longer than models trained from scratch, even with similar force MAEs [6]. |
| Validating Stability | 1. Run a benchmark simulation with the new, larger time step.2. Calculate the Radial Distribution Function (RDF) and compare it to a reference (AIMD or experimental data).3. A stable RDF with low MAE (<0.02) confirms structural integrity [9]. | Do not rely solely on force MAE. Always run a short MD simulation and check structural and thermodynamic properties for realism. |
Table 1: Comparison of Constraint Algorithm Performance in MD Simulations. This table summarizes key findings from benchmarking studies, highlighting the trade-offs between speed, accuracy, and applicability of different algorithms.
| Algorithm | Core Methodology | Key Advantage | Reported Speedup | System Tested |
|---|---|---|---|---|
| ILVES-PC | Newton's method with a specialized linear solver | High accuracy and efficiency for polymers | Up to 4.9× (single-thread) and 76× (multi-thread) vs. SHAKE [7] | Proteins of varying sizes |
| SHAKE | Nonlinear Gauss-Seidel method | Robustness; widespread adoption | Baseline | General molecules |
| P-LINCS | Parallelized linear constraint solver | Parallel scalability | Not directly benchmarked | General molecules |
Table 2: Stability and Performance of Selected Graph Neural Network Force Fields. Data adapted from a benchmark study training various GNNs on the Li₁₀GeP₂S₁₂ system for 10 hours, then testing transferability to other materials [9].
| GNN Model | Equivariance | Parameters | MD Performance (ns/day) | Transferability (Forces R² on Li₃PS₄) | Stable RDF? |
|---|---|---|---|---|---|
| CGCNN | E(3)-invariant | 0.25 M | 45.0 | Low | Unstable |
| SchNet | E(3)-invariant | 0.49 M | 22.5 | Low | Unstable |
| DimeNet++ | E(3)-invariant | 1.49 M | 6.0 | >0.96 | Mixed |
| Equiformer | SE(3)/E(3)-equivariant | 7.84 M | 3.0 | >0.96 | Stable |
| AIMD (Reference) | N/A | N/A | 0.034 | N/A | Stable |
Experimental Protocols
Protocol: Implementing a Constraint Algorithm for a Larger Time Step
Objective: Modify an existing MD protocol for a small protein in water to use a 2 fs time step by constraining bond lengths.
Materials:
- Software: GROMACS (or similar MD package).
- Initial Structure: PDB file of the protein solvated in a water box.
- Force Field: CHARMM36 or AMBER.
Methodology:
- Parameter File Modification:
- Edit your MD parameter file (e.g.,
.mdpfile in GROMACS). - Locate the
constraintskeyword and set it toconstraints = h-bondsto constrain all bonds involving hydrogen. - Set the
constraint-algorithmtoLINCS(typically recommended for parallel performance) orSHAKE. - Increase the
dt(time step) parameter from 1.0 to 2.0.
- Edit your MD parameter file (e.g.,
Simulation Execution:
- Process the topology and coordinates using the modified parameter file.
- Run a short energy minimization.
- Initiate the production MD run.
Validation:
Protocol: Assessing MLFF Stability via Radial Distribution Function
Objective: Determine if a trained Machine Learning Force Field produces physically realistic dynamics for a material system.
Materials:
- Software: LAMMPS (with MLFF plugin) or a dedicated MLFF package like GraNNField [9].
- Model: A pre-trained and fine-tuned MLFF (e.g., GemNet-T, Equiformer).
- Reference Data: Ab initio MD trajectory or experimental RDF.
Methodology:
- Run MLFF-MD:
Compute the Radial Distribution Function (RDF):
- For a chosen atom pair (e.g., Li-Li in an ionic conductor), compute the RDF, g(r), from the MLFF-MD trajectory.
- The RDF is calculated as: ( g(r) = \frac{V}{N^2} \left\langle \sumi \sum{j \neq i} \delta(\mathbf{r} - \mathbf{r}{ij}) \right\rangle ), where V is the volume, N is the number of atoms, and rij is the distance between atoms i and j [9].
Comparative Analysis:
- Compute the RDF from the reference AIMD trajectory under identical conditions.
- Visually compare the two RDFs. A stable model will show good agreement in peak positions, heights, and widths.
- Calculate the Mean Absolute Error (MAE) between the two RDFs. An MAE below 0.02 is a strong indicator of stability and physical correctness [9].
System Stiffness and Time Step Logic
Research Reagent Solutions
Table 3: Essential Software and Algorithmic "Reagents" for MD Time Step Optimization.
| Item Name | Type | Function/Benefit |
|---|---|---|
| SHAKE | Constraint Algorithm | The classic algorithm for holonomic constraints. It removes the fastest bond vibrations, allowing a time step of ~2 fs [8]. |
| LINCS / P-LINCS | Constraint Algorithm | A faster, more robust alternative to SHAKE, especially suited for parallel computations and systems with coupled constraints [7]. |
| ILVES-PC | Specialized Constraint Algorithm | Uses Newton's method for higher accuracy and speed, particularly beneficial for polymer and protein simulations [7]. |
| Velocity Verlet | Integrator | A standard, symplectic integrator used with constraint algorithms for stable dynamics [6]. |
| Symplectic ML Integrator | Machine Learning Integrator | A data-driven integrator derived from a learned generating function (S³). It preserves geometric structure, enabling large time steps without energy drift [5]. |
| GemNet-T / Equiformer | Graph Neural Network (GNN) | Advanced GNN architectures for building MLFFs. Pre-training them on diverse datasets (e.g., OC20) is critical for simulation stability [6] [9]. |
| Radial Distribution Function (RDF) | Validation Metric | A critical metric to validate the structural integrity of an MD simulation, especially when using MLFFs or large time steps [9]. |
Frequently Asked Questions
What is the most common time step for all-atom molecular dynamics (AAMD) simulations? In standard AAMD simulations, the time step is typically 1–2 fs [10]. This range is chosen to ensure numerical stability, as it is significantly shorter than the period of the fastest atomic vibrations in the system (e.g., bonds involving hydrogen atoms).
My simulation energy is drifting. Could my time step be too large? Yes, energy drift is a key indicator of an excessively large time step. For publishable results, the long-term drift in the conserved quantity (e.g., total energy in NVE simulations) should be less than 1 meV/atom/ps. For qualitative results, a drift of less than 10 meV/atom/ps may be acceptable [2]. You can test for this by running a constant energy (NVE) simulation and monitoring the total energy over time.
Are there ways to safely use a larger time step? Yes, several methods allow for larger time steps:
- Constraint Algorithms: Using algorithms like SHAKE or LINCS to freeze the fastest bond vibrations (e.g., bonds to hydrogen atoms) is the most common method. This often allows time steps of 2 fs, or even 4 fs in some cases [2].
- Mass Repartitioning: This technique increases the mass of light atoms (like hydrogen) and decreases the mass of bonded heavy atoms, slowing down the highest frequency motions and permitting larger time steps [2].
- Specialized Integrators: Advanced integrators like the geodesic BAOAB (g-BAOAB) are designed for use with constraints and can enable time steps as high as 8 fs for biomolecular systems [2].
How does the presence of light atoms like hydrogen affect time step choice? Light atoms have high-frequency vibrations, which dictate the minimum time step. A C-H bond vibration has a period of about 11 fs. According to the Nyquist theorem, your time step must be at least half of this period to capture the motion. Therefore, for systems with explicit hydrogen dynamics, time steps as low as 0.25 fs may be necessary for accurate sampling [2].
Troubleshooting Guides
Problem: Simulation Energy Blows Up or Crashes
Possible Cause: The time step is too large for the chosen model and integration method.
Solution Steps:
- Immediate Action: Significantly reduce your time step. Try decreasing it by a factor of 2 (e.g., from 2 fs to 1 fs) and restart the simulation.
- Re-evaluate System: Identify the fastest motions in your system. If you have light atoms or fast bonds, consider applying constraint algorithms to those bonds.
- Validate: After implementing constraints, run a short NVE simulation and check for energy conservation. Gradually increase the time step while monitoring stability.
Problem: Energy Drift in NVE Ensemble
Possible Cause: The time step is at the upper limit of stability, or the pair list (neighbor list) is updated too infrequently.
Solution Steps:
- Quantify the Drift: Measure the energy drift in units of meV/atom/ps. Use the thresholds provided in the FAQs to determine the severity [2].
- Adjust Time Step: Slightly reduce the time step and observe if the drift improves.
- Check Neighbor Search: In your MD engine (e.g., GROMACS), you can reduce the
nstlistparameter to update the pair list of interacting atoms more frequently. Modern software can often automatically determine an optimal update frequency to control energy drift [11].
Problem: Inaccurate Physical Sampling (e.g., loss of equipartition)
Possible Cause: Using a non-structure-preserving, machine-learning-based integrator with a large time step.
Solution Steps:
- Diagnose: This is a known artifact when using non-symplectic predictors. Check if your simulation shows a violation of energy equipartition between different degrees of freedom [5].
- Switch Integrators: Consider using structure-preserving (symplectic and time-reversible) integrators, such as those based on learned mechanical action, which are designed to eliminate these pathologies even with extended time steps [5].
Table 1: Common Time Step Ranges and Their Applications
| Time Step | Typical Use Case | Key Considerations |
|---|---|---|
| 0.25 - 0.5 fs | Systems with light nuclei (e.g., quantum dynamics, accurate H dynamics) [2] | Necessary to capture the very fast vibrations of light atoms. Computationally expensive. |
| 1 - 2 fs | Standard for all-atom MD with flexible bonds [10] | A safe default for most biomolecular simulations in explicit solvent. |
| 2 - 4 fs | All-atom MD with constrained bonds to hydrogen (e.g., using SHAKE/LINCS) [2] | Most common for production runs. Offers a good balance of stability and efficiency. |
| 4+ fs | Specialized methods using constraints + mass repartitioning or advanced integrators (e.g., g-BAOAB) [2] | Requires careful validation. Not yet a standard practice. |
Table 2: Experimental Validation Protocols for Time Step Selection
| Validation Method | Protocol Description | Acceptance Criteria |
|---|---|---|
| Energy Drift Test [2] | Run a simulation in the NVE (microcanonical) ensemble and monitor the total energy over time. | Drift < 1 meV/atom/ps for publishable quality; < 10 meV/atom/ps for qualitative work. |
| Time-Reversibility Test [2] | Run a simulation for a number of steps, then reverse the velocities and run for the same number of steps. The system should return to its initial state. | Small deviation from the initial coordinates indicates a good, symplectic integrator and time step. |
| Nyquist Criterion Check [2] | Identify the highest frequency vibration in your system (e.g., a C-H stretch at ~90-100 fs⁻¹). | The time step should be 0.01 to 0.033 of the shortest vibrational period [2]. For an 11 fs period, this means a 0.11-0.36 fs step. Constraints effectively remove these fastest vibrations. |
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for MD Simulations
| Item / "Reagent" | Function in the "Experiment" (Simulation) |
|---|---|
| Force Fields (e.g., AMBER, CHARMM, GROMOS) [10] | Provides the empirical potential energy function that calculates forces between atoms. The foundational model governing interatomic interactions. |
| Constraint Algorithms (SHAKE, LINCS) [2] | "Freeze" the length of specified bonds (usually involving H atoms), allowing for a larger integration time step by removing the highest frequency vibrations. |
| Symplectic Integrators (Velocity Verlet, Leap-frog) [11] [2] | Numerically solve Newton's equations of motion. These algorithms preserve the geometric structure of Hamiltonian flow, ensuring long-term energy stability. |
| Thermostats & Barostats (e.g., Nosé-Hoover, Berendsen) | Maintain constant temperature and pressure during the simulation, mimicking realistic experimental conditions. |
| Neural Network Potentials (NNPs) [5] [12] | Machine-learned models that can predict potential energy and forces, potentially enabling accurate simulations with much larger time steps. |
| Geometry Optimizers (L-BFGS, FIRE, Sella) [13] | Used for energy minimization of initial structures before dynamics, or for optimizing structures using NNPs. |
Experimental Protocol: Validating Time Step via Energy Conservation
The following workflow is a standard method to empirically determine a stable time step for a new system.
Frequently Asked Questions (FAQs)
FAQ 1: What does "symplectic" mean for an integrator, and why is it crucial for MD simulations?
A symplectic integrator is a numerical scheme designed for Hamiltonian systems that preserves the symplectic structure of the phase space. This means the solution exists on a symplectic manifold, which has a direct relationship between position and momentum variables [14] [15].
In practical terms, this property leads to excellent long-term stability and near-conservation of energy, even if the exact energy has small fluctuations. Unlike standard integrators, which may show a quadratic drift in energy over time, symplectic integrators exhibit only a small, bounded error in energy, making them ideal for simulating the long-time evolution of molecular systems [15].
FAQ 2: How does a symplectic integrator differ from a standard integrator like classical Runge-Kutta?
The key difference lies in the long-term behavior rather than single-step accuracy. While both types of integrators can be of high order, a symplectic integrator's solution resides on a symplectic manifold, leading to better preservation of geometric properties and periodicity of the system [15]. Most common methods, like the primitive Euler scheme and the classical Runge-Kutta scheme, are not symplectic integrators [14]. Standard, non-symplectic integrators tend to exhibit significant energy drift over long simulations, meaning the total energy of the system is not conserved, which is undesirable for reliable MD results [15].
FAQ 3: My simulation shows a steady drift in total energy. Is this related to my integrator?
Yes, a steady energy drift is a classic symptom of an issue with the integration algorithm or time step. While a symplectic integrator will show small, bounded fluctuations in energy, a continuous drift often indicates that the integrator is not behaving time-reversibly or that the time step is too large [2]. For non-symplectic integrators, this drift can be quadratic and lead to unrealistic simulation results [15]. To troubleshoot, first verify that you are using a symplectic integrator like Velocity Verlet. Then, check that your time step is appropriate for the fastest vibrations in your system [2].
FAQ 4: Can I use a symplectic integrator with any force field?
Yes, symplectic integrators are a general numerical method for Hamiltonian systems and are compatible with classical molecular mechanics force fields. The core requirement for the standard splitting method is that the Hamiltonian is separable, meaning it can be written as the sum of kinetic energy (a function of momentum) and potential energy (a function of position). This condition is fulfilled by typical force fields used in MD [14].
Troubleshooting Guides
Problem: Energy Drift in NVE Simulation
Symptoms: The total energy of the system in a constant energy (NVE) simulation is not stable but shows a consistent upward or downward trend over time.
Possible Causes and Solutions:
| Cause | Diagnostic Steps | Solution |
|---|---|---|
| Non-symplectic Integrator | Check the documentation of your MD software to confirm which integration algorithm is in use. | Switch to a known symplectic integrator, such as Velocity Verlet or Leapfrog. [15] |
| Time Step Too Large | Check the period of the fastest vibration in your system (e.g., C-H bond stretch). Reduce the time step and re-run the simulation to see if the drift improves. | Reduce the time step so it is at least a factor of 2 smaller than the period of the fastest vibration. A common starting point is 1-2 fs for all-atom systems with flexible bonds. [2] |
| Incorrect Use of Constraints | If constraints (like SHAKE or LINCS) are used to freeze bond vibrations, ensure they are correctly implemented and that the algorithm converges. | Check for SHAKE failures. Ensure the constraint algorithm is compatible with your integrator. [2] [16] |
Problem: Simulation Becomes Unstable (Crashes)
Symptoms: The simulation terminates unexpectedly with errors such as "particle position NaN" or excessively high energy.
Possible Causes and Solutions:
| Cause | Diagnostic Steps | Solution |
|---|---|---|
| Extremely Large Time Step | The time step is too large to accurately capture atomic motions, leading to unphysical forces and instabilities. | Drastically reduce the time step (e.g., to 0.5 fs) and gradually increase it while monitoring stability. [2] |
| Overlapping Atoms in Initial Configuration | Check if the initial structure has atoms placed too close together, resulting in extremely high potential energy. | Use energy minimization before starting the dynamics to relax the initial structure and remove bad contacts. [17] |
Quantitative Data for Time Step Selection
The choice of time step is a critical balance between computational efficiency and simulation accuracy. The following table summarizes key considerations and rules of thumb.
Table 1: Guidelines for MD Time Step Selection
| Factor | Rule of Thumb / Value | Rationale & References |
|---|---|---|
| Nyquist Theorem | Time step ≤ ½ × Period of fastest vibration | To accurately sample a oscillatory motion, you must have at least two points per period. [2] |
| C-H Stretch Vibration | Period ≈ 11 fs | This corresponds to a frequency of ~3000 cm⁻¹. The maximum time step from Nyquist is ~5.5 fs, but in practice, a smaller step is used. [2] |
| Practical Time Step (all-atom, flexible) | 0.5 - 1.0 fs | Required to capture fast hydrogen vibrations. [17] |
| Practical Time Step (all-atom, constrained) | 2 fs | With constraints on bonds involving hydrogen (using SHAKE/LINCS), the fastest motions are removed, allowing a larger step. [2] [16] |
| Energy Drift Tolerance | < 1 meV/atom/ps (for publishable results) | A measure of the conserved quantity's stability to assess integration quality. [2] |
Experimental Protocols
Protocol: Validating Time Step and Integrator Stability
Purpose: To determine the optimal time step and verify the stability of the integration algorithm for a given molecular system.
Methodology:
- System Preparation: Build and equilibrate a small, representative system (e.g., a solvated protein or a sample of the bulk material) at the desired temperature and density.
- NVE Simulation: Run a series of short (10-100 ps) simulations in the NVE (microcanonical) ensemble.
- Independent Variable: Use different time steps (e.g., 0.5, 1.0, 2.0, 4.0 fs).
- Control: Use the same symplectic integrator (e.g., Velocity Verlet) for all runs.
- Data Collection: Monitor the total energy (or the relevant conserved quantity for the ensemble) throughout the simulation.
- Analysis:
- Calculate Drift: Compute the linear drift of the total energy over time, normalized per atom (e.g., in meV/atom/ps).
- Check Periodicity: For closed systems like orbits or periodic vibrations, visually inspect the phase space plot (e.g., position vs. momentum) for long-term stability and periodicity. A symplectic integrator will show a stable, near-periodic trajectory, while a non-symplectic one may spiral in or out. [15]
Research Reagent Solutions
Table 2: Essential Computational "Reagents" for Stable MD Simulations
| Item | Function in Simulation |
|---|---|
| Symplectic Integrator (e.g., Velocity Verlet) | The core algorithm that numerically solves Newton's equations of motion while preserving the geometric structure of the phase space, ensuring long-term stability. [14] [17] |
| Constraint Algorithm (e.g., SHAKE, LINCS) | "Freezes" the fastest vibrational degrees of freedom (e.g., bonds involving hydrogen) by applying holonomic constraints, allowing for a larger integration time step. [16] |
| Thermostat (e.g., Nosé-Hoover, Langevin) | Maintains the simulation at a desired temperature, mimicking a heat bath. Critical for NVT (canonical) simulations. |
| Force Field | Provides the mathematical model and parameters for calculating the potential energy and forces between atoms (e.g., bonded terms, van der Waals, electrostatics). [16] |
Workflow Visualization
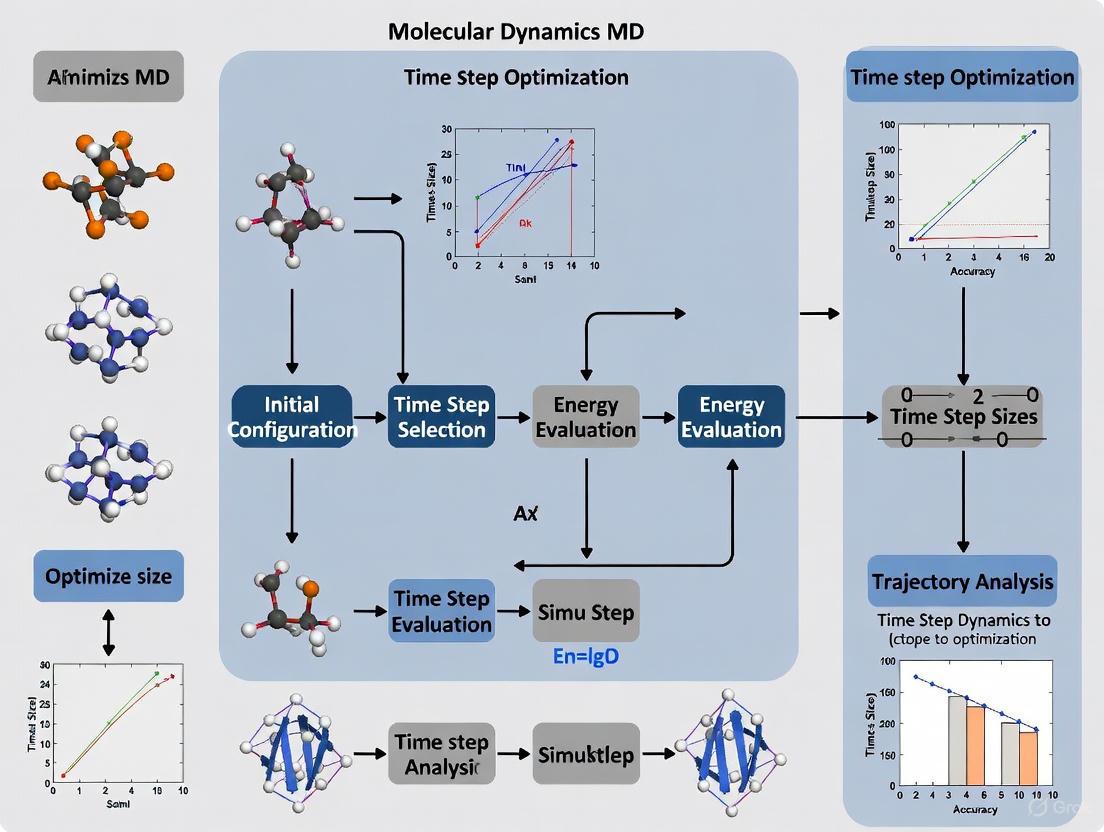
Integrator and Time Step Selection Workflow
Advanced Techniques and Practical Implementations Across Research Fields
FAQs: Understanding and Applying Constraint Algorithms
Q1: Why should I use constraint algorithms in my Molecular Dynamics (MD) simulation?
Constraint algorithms are used to maintain the exact length of specific chemical bonds, particularly those involving hydrogen atoms, which have the highest vibrational frequencies. By "freezing" these fastest motions, the algorithm allows you to use a larger integration time step without the simulation becoming unstable. This is because the time step is limited by the need to accurately sample the fastest motion in the system. Without constraints, the time step would be impractically small (around 0.5 fs). By constraining these bonds, you can often increase the time step to 2 fs or more, making the simulation computationally more efficient [18] [2].
Q2: What is the fundamental difference between SHAKE and LINCS?
SHAKE and LINCS both satisfy distance constraints but use different numerical approaches.
- SHAKE is an iterative algorithm. It works by solving for the Lagrange multipliers that enforce the constraints and continues iterating until all constraints are satisfied within a user-defined relative tolerance [19] [20].
- LINCS is a non-iterative method that typically uses two steps. It first sets the projection of new bonds onto the old bond directions to zero. Then, it applies a correction for the lengthening of bonds due to rotation. LINCS is generally faster and more stable than SHAKE, making it particularly useful for Brownian dynamics. However, it is primarily designed for bond constraints and should not be used with coupled angle constraints [19] [20].
Q3: My simulation failed with a "SHAKE failure" or "LINCS warning". What are the most common causes?
- SHAKE Failure: This usually means the algorithm could not satisfy all constraints within the allowed number of iterations. Common causes are:
- LINCS Warning: These often relate to the appearance of large constraint forces, which can happen if:
Q4: How do I choose the correct time step when using constraints?
The choice is based on the Nyquist sampling theorem. The time step must be smaller than half the period of the fastest remaining vibration in your system.
- A typical rule of thumb is that the time step should be at least 2 times smaller than the period of the fastest vibration [2].
- For a C-H bond stretch (frequency ~3000 cm⁻¹, period ~11 fs), this theoretically allows a time step of about 5 fs. However, in practice, integrator error leads to a commonly used time step of 1-2 fs for all-atom simulations with bond constraints [2].
- You should always validate your choice by running a short simulation in the NVE (constant energy) ensemble and checking for a significant energy drift, which indicates an unstable time step [2].
Q5: How can I validate that my constraints are working properly?
The best practice is to monitor two key aspects:
- Energy Conservation: In an NVE simulation, the total energy should remain constant. A significant drift (e.g., more than 1 meV/atom/ps for publishable results) indicates that your time step is too large or the constraint algorithm is not performing correctly [2].
- Constraint Deviation: Most MD software packages will report the root-mean-square (RMS) deviation of the constrained bonds. This value should be very close to zero (e.g., less than 0.0001 for LINCS) [19] [20].
Troubleshooting Guides
Problem: Large Energy Drift in NVE Simulation
- Symptoms: The total energy of the system consistently increases or decreases over time in an NVE simulation.
- Possible Causes and Solutions:
- Time Step is Too Large:
- Solution: Reduce the time step by 0.5 fs and re-run the test. A good target for energy drift is less than 1 meV/atom/ps [2].
- Constraint Tolerance is Too Lax:
- Incorrect Treatment of Fast Degrees of Freedom:
- Solution: Ensure that all bonds with high vibrational frequencies (especially X-H bonds) are being constrained. Check your molecular topology.
- Time Step is Too Large:
Problem: Simulation Crashes with Constraint Failure
- Symptoms: The simulation terminates prematurely with an error message about SHAKE or LINCS.
- Possible Causes and Solutions:
- Bad Initial Structure:
- Solution: Perform a thorough energy minimization before starting the dynamics. This ensures that the initial bond lengths are close to their target values and the system is not in a highly strained state [17].
- Extremely High Temperature or Pressure:
- Solution: Check the temperature and pressure coupling. The system may be too hot, causing atomic displacements that are too large for the constraint algorithm to handle in one time step. Re-equilibrate at a lower temperature.
- Topology Errors:
- Solution: Verify that all constrained bonds are correctly defined in the molecular topology file and that the reference bond lengths are physically sensible.
- Bad Initial Structure:
Research Reagent Solutions: Essential Components for Constrained MD
The table below lists key "reagents" or parameters essential for setting up and running stable MD simulations with constraints.
| Item/Reagent | Function in the Simulation | Key Considerations |
|---|---|---|
| Constraint Algorithm (SHAKE/LINCS) | Enforces rigid bonds, allowing for a larger MD time step by removing the fastest vibrational frequencies [18]. | SHAKE is iterative; LINCS is faster and non-iterative but not for coupled angles [19] [20]. |
| Integration Time Step (Δt) | The interval at which forces are calculated and positions are updated. Dictates simulation stability and efficiency [17]. | Typically 1-2 fs with constraints. Must be < half the period of the fastest unconstrained vibration [2]. |
| Integration Algorithm (Verlet/Leap-frog) | Numerically solves Newton's equations of motion to update atomic positions and velocities [17]. | Use a symplectic integrator (e.g., Velocity Verlet) for better long-term energy conservation [2]. |
| Constraint Tolerance | The accuracy to which bond lengths are maintained during the simulation [19]. | Tighter tolerance (e.g., 1.0e-6) improves accuracy but may require more iterations (SHAKE) [19]. |
| LINCS Order | The number of terms used in the matrix expansion for solving constraints. Higher order increases accuracy [19] [20]. | Use order 4 for MD; order 6 or 8 may be needed for Brownian dynamics with large time steps [20]. |
Experimental Protocol: Validating Time Step and Constraint Stability
This protocol provides a step-by-step methodology to empirically determine the optimal time step for a new system, as cited in community best practices [2].
- System Preparation:
- Build your system with the desired solvent and ions.
- Generate a topology that includes constraints for all bonds involving hydrogen atoms.
- Initial Minimization and Equilibration:
- Perform steepest descent energy minimization until the maximum force is below a reasonable threshold (e.g., 1000 kJ/mol/nm).
- Equilibrate the system in the NVT and NPT ensembles at the target temperature and pressure for a short time (e.g., 100 ps) using a conservative 1 fs time step.
- Time Step Testing (NVE Production):
- Start from the equilibrated structure and velocities.
- Run a short (e.g., 20-50 ps) simulation in the NVE (microcanonical) ensemble without any thermostat or barostat.
- Repeat this NVE simulation for different time steps (e.g., 1.0, 1.5, 2.0, 2.5 fs).
- Data Analysis and Validation:
- Primary Metric: Energy Drift. For each test, calculate the linear drift in the total energy over the course of the NVE run. A drift of less than 1 meV/atom/ps is a good target for production simulations [2].
- Secondary Metric: Constraint Deviation. Monitor the RMS deviation of constrained bonds reported by your MD engine (e.g., GROMACS). It should be negligible.
- The largest time step that meets the energy drift and constraint stability criteria should be selected for your production runs.
Workflow Diagram: Applying Constraints in an MD Integration Cycle
The following diagram illustrates the logical sequence of how constraint algorithms like SHAKE and LINCS are integrated into a single time step of a Velocity Verlet MD integrator.
Troubleshooting Common HMR Implementation Issues
FAQ: I receive a "LINCS WARNING" and my simulation crashes when using a 4 fs time step with HMR. What should I do?
This is a common stability issue when the integration time step is too large for the system's fastest remaining vibrations.
- Solution 1: Reduce the time step. Try reducing the time step from 4 fs to 3 fs. Empirical evidence shows that a mass repartitioning factor of 3 often allows for a stable 3 fs time step, even if 4 fs produces warnings [21].
- Solution 2: Adjust the LINCS warning threshold. You can change the
lincs-warnangleparameter to a higher value (e.g., 90 degrees) to prevent the simulation from halting due to these warnings. However, this does not address the underlying cause and should be used with caution [21]. - Solution 3: Review your mass repartitioning factor. Ensure the mass repartitioning factor is appropriate for your force field. A factor of 3 is common, but sometimes a factor of 4 is used. Note that setting the factor too high can result in an error if light atoms are bound to atoms with excessively low masses after repartitioning [21].
FAQ: gmx grompp gives me a note about a bond having an "estimated oscillational period" less than 10 times the time step. Is this a problem?
This note is a warning that the chosen time step might be too large for the highest-frequency bond vibration in your system, which could lead to instability.
- Solution: This note can often be ignored if the simulation runs stably. However, if it is accompanied by LINCS warnings or crashes, you should follow the troubleshooting steps above, primarily reducing the time step to 3 fs. The presence of a note (as opposed to an error) typically allows the simulation to proceed, but it indicates a potential risk [21].
FAQ: Do I need to adjust other simulation parameters, like nstlist or tau-t, when switching to a 4 fs time step with HMR?
For most parameters, the defaults or your standard 2 fs values are sufficient.
- Solution: Key insights from developer discussions indicate [21]:
nstlistis optimized automatically bymdrunand does not require manual adjustment.nstcomm(center-of-mass motion removal frequency) can typically be left at its default value (e.g., 100) to avoid performance overhead.nstcalcenergy(energy calculation frequency) only affects the output statistics and has a minor impact on performance; its value can be left unchanged.- Coupling constants like
tau-t(temperature coupling) andtau-p(pressure coupling) are defined in physical units (ps) and are independent of the time step. They do not need to be changed when the time step increases.
FAQ: Should HMR be applied to water molecules in my system?
No, it is generally not recommended.
- Solution: Apply HMR only to the solute (protein, membrane, DNA, etc.). Applying HMR to water molecules has been shown to increase water viscosity, which can artificially slow down protein conformational changes and other dynamic processes [22] [23].
Quantitative Data and Experimental Protocols
HMR Mass Change Examples
The table below illustrates the typical mass changes for atoms in an adenine nucleobase when applying HMR with a common scheme, increasing hydrogen masses to ~3 amu [24].
Table 1: Example Hydrogen Mass Repartitioning for an Adenine Nucleobase
| Atom Name | Atom Type | Mass Before Repartition (amu) | Mass After Repartition (amu) |
|---|---|---|---|
| C5' | Heavy (Carbon) | 12.01 | 7.978 |
| H5' | Hydrogen | 1.008 | 3.024 |
| H5'' | Hydrogen | 1.008 | 3.024 |
| C4' | Heavy (Carbon) | 12.01 | 9.994 |
| H4' | Hydrogen | 1.008 | 3.024 |
| N6 | Heavy (Nitrogen) | 14.01 | 9.978 |
| H61 | Hydrogen | 1.008 | 3.024 |
| H62 | Hydrogen | 1.008 | 3.024 |
Protocol: Creating an HMR Topology for AMBER
This protocol uses CPPTRAJ to create a topology with repartitioned masses [24].
- Load Topology: Start CPPTRAJ and load your original topology file (e.g.,
dna.prmtop). - Apply Mass Repartitioning: Use the
hmassrepartitioncommand to modify masses. This command increases the mass of hydrogen atoms and decreases the mass of the heavy atoms to which they are bonded. - Create New Topology: Save the new, repartitioned topology with a new filename.
- Verify Changes: Use the
atominfocommand on a specific residue to confirm the mass changes have been applied correctly.
Protocol: Running HMR in GROMACS
In GROMACS, HMR can be enabled directly in the molecular dynamics parameter (mdp) file, simplifying the process [21].
- Edit MDP File: Add the following line to your
mdpfile: - Increase Time Step: Change the time step (
dt) to 0.004 (4 fs). - Maintain Constraints: Keep constraints on all bonds involving hydrogen atoms.
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Resources for HMR Molecular Dynamics Simulations
| Item | Function in HMR Research | Example Sources / Force Fields |
|---|---|---|
| Simulation Software | Executes the MD simulations with long time steps. Must support constraints and mass modification. | GROMACS [21], NAMD [22], AMBER [23] [25], OpenMM [25] |
| Force Field Parameters | Defines the starting atomic masses and equilibrium bond lengths/angles. HMR is applied on top of these. | CHARMM36 [22] [25], AMBER force fields (ff14SB, GAFF) [23] [26] |
| System Building Tools | Prepares initial simulation systems, including solvation and ionization. Some now support HMR setup. | CHARMM-GUI [25] |
| Topology Editing Tools | Modifies atomic masses in the topology file to implement HMR. | CPPTRAJ (for AMBER) [24], gmx grompp (for GROMACS) [21] |
Workflow for Implementing and Validating HMR
The following diagram outlines the critical steps and decision points for successfully implementing HMR in a research project.
HMR Implementation and Validation Workflow
Validation: Ensuring HMR Does Not Compromise Scientific Results
A critical final step is to validate that HMR simulations reproduce the key physicochemical properties of interest from standard 2 fs simulations. Research has shown that for many systems, HMR with a 4 fs time step yields nearly identical structural properties but can alter kinetic properties [22].
- Structural Properties to Validate:
- Thermodynamic and Kinetic Properties to Check:
Successful application of HMR across diverse systems, including pure membranes, membrane proteins, and complex lipopolysaccharides, confirms it as a robust method for accelerating sampling when properly validated [22] [25].
Technical Support Center
Frequently Asked Questions (FAQs)
Q1: My machine learning (ML) integrator produces trajectories that do not conserve energy. What could be the cause?
A: This is a common issue when using non-structure-preserving ML predictors. The solution is to use a symplectic and time-reversible map derived from a learned generating function. Unlike direct predictors that learn the trajectory (p, q) -> (p', q'), you should learn a scalar generating function, S3(p̄, q̄), where p̄ and q̄ are the mid-point momenta and coordinates. The symplectic update is then defined by Δp = -∂S3/∂q̄ and Δq = ∂S3/∂p̄ [5]. This ensures the integration is Hamiltonian by construction, eliminating pathological energy drift.
Q2: How can I check if my chosen long time step is acceptable for production simulations?
A: You can perform a two-step validation [2]:
- NVE Test: Run a simulation in the microcanonical (NVE) ensemble and monitor the total energy. A noticeable drift indicates the time step is too large or the integrator is not time-reversible.
- Nyquist Criterion: Ensure your time step is at least 2 times smaller than the period of the fastest vibration in your system. A good rule of thumb is to keep it between 0.0333 to 0.01 of the smallest vibrational period [2]. For systems with C-H bonds (~11 fs period), this typically requires a time step of 1-2 fs with traditional integrators, but ML-enhanced methods can extend this significantly.
Q3: What is a quantitative metric for energy conservation in an NVE simulation?
A: A reasonable rule of thumb is that the long-term drift in the conserved quantity should be:
- Less than 10 meV/atom/ps for qualitative results.
- Less than 1 meV/atom/ps for publishable results [2].
Q4: My ML model for molecular dynamics (MD) requires a massive training set, which is computationally expensive to generate. How can I improve data efficiency?
A: Implement an active learning framework [27] [28]:
- Initialization: Start by training an initial ML interatomic potential (MLIP) on a small dataset from normal mode sampling or short MD trajectories.
- Iterative Refinement: Use the current MLIP to run molecular dynamics (MLMD) simulations. The key is to only select molecular configurations where the model's predicted uncertainty (e.g., high variance in an ensemble model) is highest.
- Acquisition: Add these uncertain configurations to your training set and retrain the MLIP. This strategy enriches the dataset with the most informative structures, allowing you to achieve high accuracy with far fewer data points—in some cases, as few as ~200 structures for molecular crystals [28].
Q5: How do I ensure my ML-enhanced simulation correctly reproduces infrared (IR) spectra?
A: Accurate IR spectra require two ML models working in tandem [27]:
- An ML Interatomic Potential (MLIP): Trained on energies and forces to generate accurate MD trajectories. This model should be refined via active learning.
- A Dipole Moment Model: A separate ML model, trained to predict the dipole moment vector for any atomic configuration along the trajectory. The IR spectrum is computed from the autocorrelation function of these dipole moments, and an accurate dipole model is crucial for correct peak positions and amplitudes [27].
Troubleshooting Guides
Problem: Simulation becomes unstable and energies diverge with a large time step.
- Possible Cause 1: The ML model is predicting non-Hamiltonian dynamics.
- Solution: Switch to a structure-preserving integrator that learns the mechanical action (generating function) instead of directly predicting new coordinates [5].
- Possible Cause 2: The time step violates the Nyquist condition for the underlying physical system.
- Solution: Reduce the time step until it is a fraction (e.g., 1/20th to 1/30th) of the fastest vibrational period and verify energy conservation in an NVE test [2].
Problem: Poor generalization of the ML model to unseen configurations during long-time-step simulation.
- Possible Cause: The training data does not adequately cover the phase space explored with the larger time step.
- Solution: Employ active learning at the target time step. Run simulations, collect configurations where the model is uncertain, compute ab initio energies and forces for these, and add them to the training set. This ensures the model learns the relevant dynamics for large steps [27].
Problem: Loss of equipartition between different degrees of freedom.
- Possible Cause: Using a non-symplectic ML predictor that does not preserve the geometric structure of Hamiltonian flow.
- Solution: This is a key artifact that is eliminated by using a symplectic map derived from a learned generating function. This method ensures the numerically propagated system remains Hamiltonian, preserving equipartition [5].
Data Presentation
Table 1: Comparison of Traditional and ML-Enhanced Integrators
| Feature | Traditional Symplectic Integrator (e.g., Velocity Verlet) | Direct ML Predictor | ML Integrator (Learning the Action) |
|---|---|---|---|
| Time Step | Small (e.g., 0.5-2 fs) due to fast vibrations [2] | Can be large (e.g., 10-100 fs) | Can be large (e.g., 10-100 fs) |
| Energy Conservation | Good for small time steps; has a conserved shadow Hamiltonian [2] | Poor; significant drift and artifacts [5] | Excellent; symplecticity ensures long-time near-conservation [5] |
| Physical Structure | Preserves symplectic structure and time-reversibility [2] | Non-Hamiltonian; lacks underlying geometric structure [5] | Preserves symplectic structure and time-reversibility [5] |
| Key Artifacts | Limited by fastest motion [5] | Energy drift, loss of equipartition [5] | Minimized by design [5] |
| Implementation Complexity | Low | Medium | High (requires learning a generating function and implicit solution) [5] |
Table 2: Key Metrics for Validating Time Step and Model Performance
| Metric | Target Value | How to Measure |
|---|---|---|
| Energy Drift (NVE) | < 1 meV/atom/ps for publication [2] | Linear fit of total energy over time in an NVE simulation. |
| Time Step vs. Period | 0.01 - 0.033 of shortest period [2] | Calculate vibrational frequencies (e.g., from Hessian) to find the shortest period. |
| Harmonic Frequency MAE | As low as possible; benchmark against DFT [27] | Compute harmonic frequencies on a test set using the MLIP and compare to ab initio reference. |
| Force Prediction Uncertainty | Used for data acquisition [27] | Standard deviation of force predictions from an ensemble of ML models. |
Experimental Protocols
Protocol 1: Developing a Structure-Preserving ML Integrator
This protocol outlines the methodology for learning a symplectic map for long-time-step simulations [5].
- Data Generation: Run short, high-fidelity (small time step) ab initio molecular dynamics (AIMD) or MLMD trajectories to collect a dataset of state transitions:
(p(t), q(t)) -> (p(t+h), q(t+h))for the desired large time steph. - Network Architecture: Design a neural network to represent the generating function
S3(p̄, q̄), wherep̄ = (p + p')/2andq̄ = (q + q')/2. - Training: Train the network by minimizing the loss between the predicted
(p', q')(obtained by implicitly solvingΔp = -∂S3/∂q̄andΔq = ∂S3/∂p̄) and the true(p', q')from the reference data. - Validation: Perform NVE simulations with the trained model and check for energy conservation and the absence of equipartition issues over long trajectories.
Protocol 2: Active Learning for Robust ML Interatomic Potentials
This protocol details the iterative refinement of an MLIP for accurate MD simulations [27].
- Initial Dataset Construction: Generate an initial training set by sampling molecular geometries along normal vibrational modes or from short, low-temperature MD runs.
- Model Training: Train an initial MLIP (e.g., an ensemble of MACE models) on energies and forces.
- Active Learning Loop: a. Exploration: Run MLMD simulations at different temperatures (e.g., 300 K, 500 K, 700 K) to explore configurational space. b. Acquisition: From these trajectories, select structures where the model's uncertainty is highest (e.g., highest variance in force predictions among the ensemble). c. Labeling: Compute accurate ab initio energies and forces for the acquired structures. d. Expansion: Add the new structures and their labels to the training dataset. e. Retraining: Retrain the MLIP on the expanded dataset.
- Convergence Check: Repeat the loop until performance on a validation set (e.g., harmonic frequencies) converges.
Workflow and Relationship Diagrams
Active Learning for MLIPs
Structure-Preserving ML Integrator
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software and Models for ML-Enhanced MD
| Item | Function | Reference |
|---|---|---|
| MACE (MP-0b3) | A foundational machine learning interatomic potential architecture that can be fine-tuned for high accuracy on specific systems with minimal data. | [28] |
| PALIRS | An open-source, active learning-based framework designed for efficiently predicting IR spectra using MLIPs and dipole moment models. | [27] |
| DeePMD-kit | A popular open-source package for building deep potential (DeePMD) models, enabling large-scale MD simulations with near-ab initio accuracy. | [29] |
| Symplectic Integrator Code | Custom code to implement the generating function (S3) approach, which is essential for creating structure-preserving, long-time-step ML integrators. | [5] |
| LAMMPS | A widely-used, classical MD simulator that can be integrated with many MLIPs (like MACE and DeePMD) to perform the production MD simulations. | [30] [31] |
Frequently Asked Questions (FAQs)
FAQ 1: Why is predicting binding kinetics (kon and koff) important, beyond just knowing binding affinity (K_d)?
In modern drug discovery, the efficacy of a drug is often better correlated with its binding kinetics, particularly the dissociation rate constant (koff), than with binding affinity alone [32] [33]. The duration for which a drug remains bound to its target (known as residence time, RT = 1/koff) directly influences its pharmacodynamic effects in vivo. A longer residence time can mean a sustained therapeutic effect, even after systemic drug concentrations have declined [34] [33]. While affinity is a thermodynamic property, kinetics provide a dynamic understanding of the drug-target interaction, which is crucial for optimizing lead compounds [32].
FAQ 2: My molecular dynamics (MD) simulations of ligand dissociation are too slow. What enhanced sampling methods can I use?
Standard "brute force" MD simulations are often impractical for directly observing dissociation events, which can occur on timescales from milliseconds to hours [34]. Several enhanced sampling methods have been developed to address this:
- High-Temperature MD: Methods like ModBind run simulations at elevated temperatures (e.g., 600-1000 K) to accelerate unbinding events that would otherwise be rare. The results are then reweighted to recover kinetics at physiological temperatures [34].
- Metadynamics: This method applies a bias potential along predefined collective variables (CVs)—such as the distance between the ligand and the binding pocket—to encourage the system to escape energy wells and explore unbinding pathways [32].
- Path-Sampling and Weighted Ensemble (WE) Methods: These techniques focus computational resources on the low-probability transition regions between bound and unbound states, enabling the calculation of association and dissociation rates [32].
FAQ 3: How do I choose an appropriate time step for my MD simulations of ligand-receptor systems?
The choice of time step is a critical balance between computational efficiency and numerical stability. While conventional MD simulations often use time steps of 1-2 femtoseconds (fs) to maintain energy conservation [35], research has shown that larger time steps can sometimes be used without producing significant errors in observed properties [36]. A good rule-of-thumb for constant-energy simulations is that the time step can be increased as long as there is no appreciable temperature drift [36]. For simulations involving large conformational changes or flexible ligands, a smaller time step (1-2 fs) is generally recommended to ensure accuracy.
FAQ 4: How can I handle large, flexible ligands or major protein conformational changes during binding?
Traditional docking methods often fail with high binding site flexibility [37] [32]. Newer deep learning approaches, such as DynamicBind, use equivariant geometric diffusion networks to construct a smooth energy landscape. This allows for efficient sampling of large conformational changes—like DFG-in/out transitions in kinases or the opening of cryptic pockets—directly from apo (unbound) protein structures, without being computationally prohibitive [37]. For large, flexible ligands like lipo-chitooligosaccharides, hybrid MD/Machine Learning (MD/ML) approaches can predict binding affinity rankings with high accuracy, even from coarse initial structural models [38].
FAQ 5: What are the key differences between the induced-fit and conformational selection binding models?
These are two primary models describing how ligands and receptors interact:
- Induced-Fit: The ligand first binds to the receptor, inducing a conformational change in the receptor to form a stable, active complex (LR*) [33].
- Conformational Selection: The receptor exists in an equilibrium of multiple conformations (e.g., active R* and inactive R) even before the ligand binds. The ligand then selectively binds to and stabilizes a pre-existing compatible conformation, shifting the equilibrium [33]. In practice, these models are now viewed as interconnected, and a ligand's binding mechanism may involve elements of both. This mechanistic interplay is central to phenomena like biased agonism [33].
Troubleshooting Guides
Issue 1: Low Success Rate in Pose Prediction for Flexible Targets
Problem: Predicted ligand binding poses show high root-mean-square deviation (RMSD) from experimentally determined structures, especially when the protein undergoes conformational changes upon binding.
Solutions:
- Incorporate Protein Flexibility: Move beyond rigid docking. Use "dynamic docking" methods like DynamicBind, which can adjust the protein's side-chain and backbone atoms during the pose prediction process to recover holo-like structures from apo inputs [37].
- Validate with Clash Score: Do not rely on ligand RMSD alone. Evaluate success using a combination of ligand RMSD and clash score (e.g., RMSD < 2 Å and clash score < 0.35) to ensure predicted poses are both accurate and physically plausible [37].
- Utilize Specialized Scoring: For methods that generate multiple conformations, employ a reliable scoring function like contact-LDDT (cLDDT) to select the best complex structure from the ensemble of predictions [37].
Recommended Experimental Protocol: Dynamic Docking with DynamicBind
- Input: Provide the protein structure in PDB format (AlphaFold-predicted apo structures are acceptable) and the ligand in SMILES or SDF format [37].
- Process: The model initially randomizes the ligand placement around the protein. Over 20 iterations, it progressively refines the ligand's position, orientation, and torsional angles. After the first few steps, it begins to simultaneously adjust the protein residue positions and side-chain angles [37].
- Output: The output is a predicted protein-ligand complex structure that accommodates substantial protein flexibility. Use the built-in cLDDT score to rank the outputs [37].
Issue 2: Inaccurate Prediction of Ligand Dissociation Rates (k_off)
Problem: Computed k_off values do not correlate with experimental data, or the simulations fail to observe a dissociation event within a feasible simulation time.
Solutions:
- Apply Enhanced Sampling: Use accelerated methods like ModBind or Metadynamics. ModBind, for instance, uses high-temperature MD and a population-based reweighting scheme to predict k_off, achieving speeds ~100 times faster than some conventional MD-based methods [34].
- Use Restraints Judiciously: To prevent non-productive protein unfolding during high-temperature unbinding simulations, apply restraints to the protein backbone, except for residues near the binding site. This focuses the sampling on the relevant unbinding pathway [34].
- Run Multiple Replicates: Conduct multiple independent simulation trajectories (e.g., up to 32 replicates) to obtain sufficient statistics for a reliable calculation of the median unbinding time [34].
Recommended Experimental Protocol: k_off Prediction with ModBind
- System Setup: Prepare the protein-ligand complex structure, ensuring a reasonable binding pose from docking or experimental data. Use tools like OpenMM to generate ligand force-field parameters [34].
- Simulation Parameters:
- Run simulations at high temperatures (600-1000 K) for short durations (1-5 ns).
- Restrain the protein backbone (σ = 3.0 Å) but not residues within 6 Å of the ligand.
- Use a Langevin thermostat and run in the NVT ensemble with a 2 fs time step.
- Execute numerous replicate trajectories [34].
- Analysis: Align trajectory frames and calculate the ligand RMSD over time. Determine the median simulation time for unbinding (using a cutoff, e.g., 5.0 Å, to define the unbound state). Apply the reweighting formula (Eq. 4 in the ModBind theory) to compute k_off at room temperature [34].
Issue 3: Poor Correlation with Experimental Binding Affinity Data
Problem: Calculated binding free energies do not match experimental trends, often due to insufficient sampling of the protein-ligand conformational space.
Solutions:
- Implement Efficient Alchemical Methods: For binding free energy (ΔG) calculations, use methods like the Bennett Acceptance Ratio (BAR) with explicit solvent/membrane models. A re-engineered BAR protocol has shown good correlation with experimental data for challenging targets like GPCRs [39].
- Ensure Extensive Sampling: When using endpoint methods like MM-PBSA/GBSA, run long enough MD simulations of the bound complex to ensure the conformational landscape is adequately sampled. Insufficient sampling is a major source of error [39].
- Leverage MD/ML Hybrids: For large, flexible ligands, combine MD simulations with machine learning. Use MD to generate an ensemble of binding conformations, then train an ML model on structural and interaction features to predict affinity rankings [38].
Data Presentation
Table 1: Comparison of Computational Methods for Studying Binding Kinetics
| Method | Key Principle | Typical Time Scale | Applicability | Key Outputs |
|---|---|---|---|---|
| Conventional MD [35] [32] | Newton's laws of motion on a physical force field. | Nanoseconds to microseconds. | Limited for direct k_off measurement of slow-dissociating ligands. | Atomistic trajectory, mechanistic insights. |
| ModBind [34] | High-temperature MD with population-based reweighting. | 1-5 ns per replica (accelerated). | Absolute k_off predictor for diverse ligands; ~100x faster than some methods. | Dissociation rate constant (k_off). |
| Metadynamics [32] | Bias potential added along Collective Variables to overcome energy barriers. | Accelerated sampling of rare events. | Relative k_off rates; requires prior knowledge of reaction coordinate. | Unbinding pathways, kinetic rates. |
| DynamicBind [37] | Deep equivariant generative model; geometric diffusion. | Efficient state transitions during docking. | Recovering ligand-specific conformations from apo structures; cryptic pocket identification. | Holo-like complex structure, ligand pose. |
| Weighted Ensemble (WE) [32] | Focuses sampling on low-probability transition paths. | Can reach long timescales. | Calculating both kon and koff; pathway analysis. | Association/dissociation rates, binding pathways. |
Table 2: Performance Benchmarks of Recent Methods
| Method | Test System / Benchmark | Reported Performance | Key Metric |
|---|---|---|---|
| DynamicBind [37] | PDBbind test set & Major Drug Target (MDT) set. | 33-39% success rate (ligand RMSD < 2Å, clash score < 0.35). 1.7x higher than best baseline. | Pose Prediction Accuracy |
| ModBind [34] | p38 MAP kinase inhibitors (k_off range: 10⁻¹ to 10⁻⁶ s⁻¹). | Accurate prediction across 5 orders of magnitude in k_off. | k_off Prediction Accuracy |
| BAR (Re-engineered) [39] | β1AR agonists (inactive vs. active states). | R² = 0.7893 correlation with experimental pK_D. | Binding Affinity (ΔG) Correlation |
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Research Reagent Solutions
| Item | Function in Binding Kinetics Studies | Example / Note |
|---|---|---|
| GPCR Structures | The primary target model for many drug discovery projects. Membrane protein preparation is critical [39]. | β1 adrenergic receptor (β1AR), used in BAR method validation [39]. |
| Kinase Inhibitors | Well-characterized ligands for validating methods, often exhibiting complex conformational changes (e.g., DFG-flip) [37] [34]. | p38 MAP kinase inhibitors, used for validating ModBind's k_off predictions [34]. |
| Specialized MD Software | Software engines to run simulations and analyze trajectories [34] [39]. | GROMACS, CHARMM, AMBER, OpenMM. |
| Enhanced Sampling Plugins/Code | Implements advanced algorithms for calculating kinetics and free energies. | Custom code for ModBind [34], Re-engineered BAR method [39]. |
| Ligand Parameterization Tools | Generates consistent force field parameters for small molecules for use in MD simulations [34]. | openmmforcefields package for OpenMM [34]. |
| Trajectory Analysis Tools | Processes and analyzes the large volumes of data produced by MD simulations [34]. | Pytraj (binds to cpptraj) [34]. |
Experimental Protocols & Visualization
Workflow 1: High-Temperature k_off Prediction
This diagram illustrates the ModBind protocol for predicting ligand dissociation rates.
Diagram 1: Workflow for predicting ligand dissociation rates (k_off) using high-temperature molecular dynamics.
Workflow 2: Dynamic Docking for Flexible Receptors
This diagram outlines the DynamicBind process for predicting complex structures with large conformational changes.
Diagram 2: Dynamic docking workflow for predicting ligand-bound structures with protein flexibility.
Technical Support Center
Frequently Asked Questions (FAQs)
Q1: What is the primary challenge with time step selection in MD simulations of polymer-oil systems, and how can I address it? The core challenge is the time scale disparity between fast atomic vibrations (femtoseconds) and slow polymer conformational changes (microseconds or longer). An excessively large time step causes instability or simulation failure, while a too-small step makes capturing slow processes computationally prohibitive [40] [41]. To address this:
- Standard Range: Start with a time step of 1 to 2 femtoseconds (fs) for all-atom simulations using the velocity Verlet algorithm [41].
- Machine Learning Enhancement: For long-time-scale simulations, consider using machine-learning integrators trained to predict long-time-step evolution, which can increase the usable time step by orders of magnitude while aiming to conserve energy [5].
Q2: My simulation shows a persistent energy drift. What could be the cause? A steady energy drift typically indicates that your simulation does not conserve energy, which is a sign of a non-Hamiltonian system. This is a common artifact when using non-structure-preserving, machine-learning-based trajectory predictors with large time steps [5].
- Solution: Implement structure-preserving (symplectic) integrators. These algorithms, often based on generating functions, preserve the geometric properties of the Hamiltonian flow, ensuring long-term stability and near-conservation of energy [5].
Q3: Why does my polymer fail to maintain its viscosity under high-salinity conditions in the simulation? This is a common issue when the force field parameters do not accurately capture polymer behavior in extreme environments.
- Solution: Ensure you are using a potential function that correctly describes the specific interactions in your system.
- For synthetic polymers like HPAM, this may involve accurately modeling carboxylate and amide groups and their interactions with ions [41] [42].
- Dynamic Training: For machine learning potentials, employ a dynamic training (DT) method. This approach integrates the equations of motion into the training process, penalizing the model for errors that accumulate during simulation, thereby enhancing its accuracy and stability under varying conditions like high salinity [43].
Q4: How can I validate that my MD simulation accurately captures atomic motion for polymer-oil interactions? Direct experimental validation at the atomic scale is challenging but emerging.
- Comparison with Advanced Microscopy: While not for polymers in oil, techniques like electron ptychography can now directly image atomic thermal motions in materials. Your simulation predictions of atomic vibrational patterns or diffusion can be qualitatively compared against such experimental data where available [44].
- Benchmarking with Ab Initio Data: A standard practice is to run short, targeted MD simulations and compare the results (e.g., radial distribution functions, diffusion coefficients) against more accurate but computationally expensive ab initio molecular dynamics (AIMD) calculations [43].
Troubleshooting Guides
Issue: Unstable Simulation (Bonds Breaking, System Explodes)
This is often caused by an overly large time step or inaccurate forces.
- Step 1: Verify Time Step Size
- Action: Reduce your time step to 1 fs and test again.
- Principle: The time step must be small enough to resolve the highest frequency vibrations in the system (e.g., C-H bond stretches) [41].
- Step 2: Check Force Field Compatibility
- Action: Ensure the potential functions (force fields) are appropriate for all components in your system (polymer, oil, water, ions). Using a potential designed for metals on a covalent polymer system will yield incorrect forces and instability [40].
- Step 3: Inspect Initial Configuration
- Action: Check for unrealistic atomic overlaps (e.g., atoms placed too close together) in your initial structure, which generate massive repulsive forces. Perform a local energy minimization before starting the dynamics [45].
Issue: Poor Reproduction of Experimental Polymer Rheology
The simulation fails to match lab-measured properties like viscosity or elasticity.
- Step 1: Calibrate with Known Data
- Action: Simulate a simple, well-characterized system (e.g., pure water or a known solvent) to validate that your force field reproduces basic thermodynamic and transport properties [40].
- Step 2: Extend Sampling Time
- Action: Rheological properties are statistical and require long simulation times to converge. Run longer simulations or use enhanced sampling techniques to improve statistics [41].
- Step 3: Re-evaluate Force Field
Experimental Protocols & Data
Detailed Methodology: Simulating Polymer-Oil Interaction via MD
This protocol outlines the key steps for setting up and running an MD simulation to study a polymer (e.g., HPAM) interacting with oil (e.g., decane) in an aqueous environment.
1. System Setup
- Construction: Build an initial simulation box containing:
- One polymer chain (e.g., 30-100 monomers of HPAM).
- Multiple molecules of oil (e.g., decane).
- Water molecules (e.g., SPC or TIP3P model).
- Counter-ions (e.g., Na⁺) to neutralize the system and salt ions (e.g., Na⁺, Cl⁻) to achieve target salinity [41].
- Energy Minimization:
- Purpose: Remove any bad contacts and relieve internal stresses from the initial configuration.
- Method: Use a steepest descent or conjugate gradient algorithm until the maximum force is below a chosen threshold (e.g., 1000 kJ/mol/nm) [45].
2. Equilibrium
- NVT Ensemble (Thermalization):
- Duration: 100-500 ps.
- Goal: Bring the system to the target temperature (e.g., 300-400 K for EOR conditions).
- Thermostat: Use a thermostat like Nosé-Hoover or Berendsen.
- NPT Ensemble (Densification):
- Duration: 1-5 ns.
- Goal: Adjust the system density to the target pressure (e.g., 1 bar).
- Barostat: Use a barostat like Parrinello-Rahman or Berendsen.
3. Production Run
- Ensemble: Typically NVT or NPT.
- Duration: As long as computationally feasible (10s to 100s of ns) to gather sufficient statistics for properties like diffusion and viscosity.
- Integration: Use the velocity Verlet algorithm with a 1-2 fs time step [41].
- Data Saving: Save atomic positions and velocities every 500-1000 steps for analysis.
4. Analysis
- Mean Squared Displacement (MSD): Calculate for polymer segments and oil molecules to determine diffusion coefficients.
- Radial Distribution Function (RDF): Analyze between polymer functional groups and oil molecules to understand interaction sites.
- Viscosity: Compute using Green-Kubo relations or non-equilibrium MD methods.
Quantitative Data for MD Simulations
Table 1: Standard MD Time Step Guidelines for Polymer Systems [40] [41]
| System Type | Recommended Time Step (fs) | Integration Algorithm | Key Considerations |
|---|---|---|---|
| All-Atom, Explicit Solvent | 1 - 2 | Velocity Verlet | Limited by fastest H-atom vibrations. |
| Coarse-Grained | 10 - 40 | Velocity Verlet / Leap-frog | Softer potentials allow larger steps. |
| ML-Structure-Preserving | 50 - 500 (context-dependent) | Symplectic ML Integrator | Requires training; aims to conserve energy [5]. |
Table 2: Key Research Reagent Solutions for Polymer-Oil MD Simulations [40] [41] [42]
| Item (Simulation Component) | Function / Role in the Simulation | Common Examples |
|---|---|---|
| Force Field / Potential | Defines the potential energy function and interatomic forces; critical for accuracy. | EAM (metals), Tersoff (covalent), CHARMM, OPLS-AA (polymers, organics) [40]. |
| Polymer Model | The molecular structure whose interactions and dynamics are the primary study focus. | HPAM, Xanthan Gum, Polyacrylamide [41] [42]. |
| Solvent Model | Represents the bulk fluid environment (typically water) in EOR. | SPC, SPC/E, TIP3P, TIP4P (water models). |
| Oil Model | Represents the hydrocarbon phase being displaced. | n-Alkanes (e.g., decane, hexadecane), Coarse-grained oil models. |
| Software | Performs the numerical integration of equations of motion and analysis. | LAMMPS, GROMACS, AMS [40] [45]. |
Workflow Visualization
Identifying and Solving Common Time Step Problems for Robust Simulations
Frequently Asked Questions
What are the most common symptoms of an excessively large MD time step? The primary symptoms are a significant drift in the conserved quantity (e.g., total energy in NVE simulations) and failures of constraint algorithms like SHAKE. In severe cases, the simulation will become unstable and "blow up," with energies diverging to infinity [2] [47].
How can I quantitatively check for energy drift in my simulation? Run a constant energy (NVE) simulation and monitor the total energy over time. A reasonable rule of thumb is that the long-term drift should be less than 10 meV/atom/ps for qualitative results, and 1 meV/atom/ps for publishable results [2]. Significant, non-random drift indicates an overly large time step.
Why does a larger time step cause SHAKE to fail? The SHAKE algorithm iteratively solves constraint equations to satisfy bond-length conditions. With a larger time step, the initial atomic displacement per step is greater, making it more difficult for the iterative solver to converge on a solution that satisfies all constraints before the preset iteration limit is reached, resulting in failure [48].
Is a 4 fs time step with hydrogen mass repartitioning (HMR) always safe? Not necessarily. Recent research on alchemical free energy calculations suggests that while a 4 fs timestep with HMR is common, it can lead to significant deviations in results compared to shorter timesteps. It is recommended to limit the timestep to a maximum of 2 fs for accurate and reliable free energy calculations, even when using HMR and SHAKE [47].
Troubleshooting Guide: Diagnosing Time Step Issues
Symptom 1: Energy Drift
A clear sign of an inaccurate integration is a steady, non-random drift in the conserved quantity (e.g., total energy for NVE ensembles) over time [2].
- Diagnostic Protocol:
- Configure a short simulation in the NVE (microcanonical) ensemble.
- Plot the total energy of the system as a function of simulation time.
- Observe the long-term trend. A well-conserved energy will fluctuate randomly around a stable mean value. A consistent upward or downward trend signifies energy drift [2].
- Recommended Action: Reduce the time step in increments (e.g., 0.5 fs) and re-run the NVE test until the drift falls within acceptable limits (see FAQ above).
Symptom 2: SHAKE Algorithm Failure
The simulation terminates with an error message indicating that the SHAKE algorithm failed to converge.
- Diagnostic Protocol:
- Check the simulation output log for explicit error messages related to "SHAKE failure," "constraint failure," or the inability to converge constraints.
- Note that without SHAKE, simulations with explicit bonds become unstable at larger timesteps [47].
- Recommended Action:
- The immediate solution is to reduce the integration time step.
- For the long term, ensure that all bonds involving hydrogen atoms are correctly specified in the constraint parameters.
Symptom 3: Catastrophic Instability
The simulation crashes catastrophically, often with atoms acquiring impossibly high velocities and energies, making the trajectory unusable [2].
- Diagnostic Protocol: This is often visually obvious from the trajectory and energy plots. The total energy will diverge to infinity instead of being conserved.
- Recommended Action: This is a severe form of instability. Drastically reduce the time step (e.g., by a factor of 2 or more) and ensure a symplectic (time-reversible) integration algorithm like Velocity Verlet is used [2].
Experimental Data and Protocols
Quantitative Impact of Time Step on Energy Drift
The following table summarizes findings from a study investigating time step impact on energy drift and alchemical free energy calculations, using hydrogen mass repartitioning (HMR) and SHAKE [47].
- Experimental System: The study used cAMP, ethane, and a protein-ligand system (Tyk2).
- Methodology: Energy drift was measured in NVE simulations. Thermodynamic integration (TI) in the NPT ensemble was used to assess accuracy in free energy calculations [47].
| Time Step (fs) | Energy Drift | AFE Calculation Accuracy (TI) |
|---|---|---|
| 0.5 - 2.0 | Low/Acceptable | Consistent, reliable dU/dλ values |
| 4.0 | Significant increase observed | Deviations up to 3 kcal/mol in some cases |
Protocol: Validating Time Step via NVE Energy Conservation
This protocol provides a standardized method to test if your time step is sufficiently small.
- System Preparation: Start with a fully equilibrated system (after NVT and NPT equilibration).
- Simulation Configuration:
- Ensemble: NVE (microcanonical)
- Integrator: A symplectic integrator (e.g., Velocity Verlet)
- Duration: A short simulation (e.g., 10-100 ps) is often sufficient to identify drift.
- Data Analysis:
- Extract the total energy time series from the simulation log.
- Plot the total energy versus time.
- Perform a linear regression on the data. The slope of the fit represents the energy drift rate. Compare this rate to the recommended threshold of 1 meV/atom/ps for high-quality simulations [2].
The Scientist's Toolkit
| Research Reagent / Tool | Function in MD Simulation |
|---|---|
| SHAKE / LINCS | Algorithms that impose holonomic constraints on bond lengths (and optionally angles), allowing for larger time steps by removing the fastest vibrational frequencies [48]. |
| Hydrogen Mass Repartitioning (HMR) | A technique that increases the mass of hydrogen atoms and decreases the mass of the bonded heavy atom, slowing the highest frequency vibrations and permitting larger time steps [2]. |
| Velocity Verlet Integrator | A symplectic (time-reversible) integration algorithm that provides better long-term energy conservation compared to non-symplectic alternatives, allowing for more stable simulations at a given time step [2]. |
| NVE Ensemble | The constant energy ensemble used as a diagnostic tool to measure energy drift and validate the stability and accuracy of the chosen time step and integrator [2]. |
Visualizing the Diagnostic Workflow and Physical Principles
The following diagram illustrates the logical process for diagnosing and resolving time step-related issues in molecular dynamics simulations, integrating the symptoms and actions from this guide.
Diagnosing and Fixing MD Time Step Issues
The physical relationship between time step, atomic vibrations, and numerical stability is foundational. The diagram below illustrates why high-frequency vibrations limit the time step and how techniques like constraints help.
Why High-Frequency Vibrations Limit MD Time Steps
Technical Support Center
Troubleshooting Guides
Guide 1: Slower-than-Expected Biomolecular Recognition
Reported Symptom: "My enhanced sampling simulation using a long time step finds the native binding pocket much slower than a standard simulation, despite the performance boost."
Underlying Cause: The problem is likely rooted in altered ligand diffusion dynamics. Hydrogen Mass Repartitioning (HMR), a common long-time-step protocol, increases ligand diffusion rates. This faster motion reduces the survival probability of crucial on-pathway metastable intermediates—transient, short-lived states that are essential for guiding the ligand to its final binding site. Without sufficient time in these intermediate states, the ligand explores more non-productive pathways, ultimately delaying the final recognition event at the native protein cavity [4] [49].
Diagnosis and Verification:
- Calculate the ligand's Mean Square Displacement (MSD) in both standard and HMR simulations.
- Confirm a higher diffusion coefficient in the HMR simulation.
- Perform a metastable state analysis (e.g., using clustering) on the standard simulation trajectory to identify key intermediate poses.
- Compare the occupancy and lifetime of these intermediates in the HMR simulation. You will likely find significantly reduced occupancy [4].
Solution:
- Primary Fix: For direct simulation of recognition kinetics, revert to a standard time step (1-2 fs). The perceived performance gain of HMR is nullified by its kinetic distortion in this specific context [4].
- Alternative Approach: If a long time step is necessary, carefully validate your results by comparing the kinetics and key intermediates identified against a shorter, standard time-step simulation for the same system [4] [49].
Guide 2: Energy Drift and Instability
Reported Symptom: "My simulation 'blows up' or shows a continuous, unphysical drift in total energy during a constant-energy (NVE) simulation."
Underlying Cause: The time step is too long to accurately integrate the fastest vibrations in the system, violating the Nyquist sampling theorem. This theorem states that the sampling frequency (1/time step) must be at least twice the frequency of the fastest motion. A C-H bond vibration (~3000 cm⁻¹) has a period of about 11 fs, requiring a time step of 5 fs or less just to meet the Nyquist criterion. Real-world integrators introduce error, so the practical maximum is even smaller [2].
Diagnosis and Verification:
- Monitor the "conserved quantity" (e.g., total energy for NVE, or the appropriate Hamiltonian for other ensembles) over the course of the simulation.
- A reasonable rule of thumb is that the long-term drift should be less than 10 meV/atom/ps for qualitative results, and 1 meV/atom/ps for publishable results [2].
Solution:
- Reduce the time step until the energy drift falls within an acceptable range.
- For systems with light atoms (like hydrogen), consider constraining bond vibrations involving hydrogen using algorithms like SHAKE or LINCS, which allows a time step of up to 2 fs [2] [50].
- As a diagnostic step, start with a very short time step (e.g., 0.5 fs) and gradually increase it while monitoring energy conservation [2].
Frequently Asked Questions (FAQs)
FAQ 1: What is the fundamental rule of thumb for choosing an MD time step?
The time step must be shorter than the period of the fastest vibration in your system. A common guideline is that the time step should be about 0.01 to 0.033 times the smallest vibrational period [2]. For typical atomistic systems with explicit hydrogen atoms, this translates to a standard time step of 1-2 fs. This is often enforced by the high frequency of C-H bond stretching vibrations [2].
FAQ 2: What are the primary methods for enabling longer time steps, and what are their trade-offs?
The table below summarizes common protocols, their mechanisms, and associated risks.
Table 1: Comparison of Long-Time-Step Protocols
| Protocol | Mechanism | Typical Time Step | Key Pitfalls and Considerations |
|---|---|---|---|
| Constraint Methods (e.g., SHAKE) [2] [50] | Freezes the fastest bond vibrations (e.g., C-H, O-H) by applying holonomic constraints. | 2 fs | Cannot be used if the constrained degrees of freedom are relevant to the process being studied. |
| Hydrogen Mass Repartitioning (HMR) [4] [49] | Redistributes atomic mass from heavy atoms to bonded hydrogens, slowing the highest frequency motions. | 4 fs | Can artificially accelerate ligand diffusion, retarding biomolecular recognition kinetics and misleading binding pathway analysis. |
| Machine Learning Integrators [5] | Uses a trained model to predict long-time-step trajectories, learning a structure-preserving map. | >10 fs (potentially) | Non-structure-preserving models can violate energy conservation and cause loss of equipartition. |
FAQ 3: My simulation ran without crashing. How can I be sure my time step was acceptable?
A successful run without termination is a low bar. You must validate the physical correctness of your trajectory.
- For NVE ensembles: Check for drift in the total energy [2].
- For all simulations: Monitor system temperature, pressure (if applicable), and structural stability (e.g., root-mean-square deviation of protein core residues).
- For specific processes: If you are studying kinetics (like binding), compare key rates or pathways against known experimental data or a validated, short time-step simulation [4].
FAQ 4: Are there any emerging solutions that avoid these pitfalls?
Yes, research into machine-learning-based, structure-preserving integrators is a promising frontier. These methods aim to learn a symplectic map (a transformation that preserves the geometric structure of Hamiltonian mechanics) equivalent to learning the mechanical action of the system. This approach can, in theory, allow for large time steps while maintaining energy conservation and equipartition, avoiding the physical artifacts of methods like HMR [5].
Experimental Protocols & Data
The following table summarizes key quantitative findings from a landmark study investigating HMR in protein-ligand recognition [4] [49].
Table 2: Key Experimental Findings on HMR Pitfalls
| Metric | Standard MD (2 fs) | HMR MD (4 fs) | Interpretation |
|---|---|---|---|
| Ligand Recognition Time | Baseline reference time | Significantly longer | The purpose of a performance upgrade is defeated, as the physical process is slowed. |
| Ligand Diffusion Coefficient | Baseline diffusion rate | Higher | Faster, less correlated motion reduces productive encounters with the binding site. |
| Survival Probability of Metastable Intermediates | Baseline probability | Reduced | The system has less time to stabilize in crucial on-pathway states, hindering the final step. |
| Total Simulation Time (across 3 protein systems) | 176 µs (cumulative) | The findings are based on extensive sampling, making the conclusions robust. |
Detailed Methodology: Probing HMR Effects on Recognition
This protocol is based on the experiments that uncovered the retarding effect of long-time-step protocols [4] [49].
1. System Selection and Preparation:
- Proteins: Select independent protein systems with buried binding cavities (e.g., T4 lysozyme, sensor domain of MopR, and galectin-3 as used in the study).
- Initial Structure: Obtain protein structures from the Protein Data Bank (PDB). Prepare the protein by adding missing atoms and assigning protonation states.
- System Setup: Solvate the protein in a suitable water box (e.g., TIP3P). Add ions to neutralize the system and achieve a physiologically relevant ionic concentration.
2. Simulation Parameters:
- Control Simulation: Use a standard time step of 1-2 fs.
- Test Simulation: Use the HMR method to repartition masses, enabling a 4 fs time step.
- Common Parameters:
- Integrator: Use a symplectic integrator like Velocity Verlet or leap-frog.
- Constraints: In the control simulation, apply constraints to bonds involving hydrogen.
- Force Field: Choose a modern, appropriate biomolecular force field (e.g., AMBER, CHARMM).
- Ensemble: Use an isothermal-isobaric (NPT) ensemble for equilibration and production.
3. Production Run and Analysis:
- Run multiple, independent long-timescale simulations for both setups to ensure statistical significance.
- Trajectory Analysis:
- Ligand Localization: Monitor the time taken for the ligand to find and bind the native protein cavity in both simulation sets.
- Diffusion Coefficient: Calculate the diffusion coefficient (D) from the slope of the Mean Square Displacement (MSD) vs. time plot using the Einstein relation: ( D = \frac{1}{6} \frac{d(MSD)}{dt} ) [17].
- Metastable Intermediates: Use clustering algorithms (e.g., Principal Component Analysis combined with k-means or density-based clustering) to identify and characterize metastable states. Compare their populations and lifetimes between the two simulation protocols [17].
Workflow Visualization
The diagram below illustrates the logical workflow for diagnosing and troubleshooting long-time-step simulation issues, based on the information in this guide.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Item / Resource | Function / Purpose |
|---|---|
| Protein Data Bank (PDB) | Primary repository for obtaining initial 3D atomic-level protein structures for simulation setup [17]. |
| Molecular Mechanics Force Field | Empirical potential energy function (e.g., AMBER, CHARMM) used to calculate interatomic forces during the simulation [35]. |
| SHAKE / LINCS Algorithm | Numerical algorithm used to apply holonomic constraints to bond lengths, allowing for a larger integration time step [50]. |
| Hydrogen Mass Repartitioning (HMR) | A protocol that increases the mass of hydrogen atoms to slow high-frequency vibrations, enabling a ~2x longer time step (with noted pitfalls) [4] [49]. |
| Velocity Verlet Integrator | A widely used symplectic and time-reversible algorithm for numerically integrating Newton's equations of motion [51]. |
| Principal Component Analysis | Dimensionality reduction technique used to identify the essential large-amplitude motions from a complex MD trajectory [17]. |
A guide to overcoming the trade-off between computational efficiency and physical accuracy in molecular dynamics simulations.
Molecular dynamics (MD) simulations predict how every atom in a protein or other molecular system will move over time, based on a general model of the physics governing interatomic interactions [35]. The choice of time step—the interval at which positions and velocities are updated—is a critical parameter that balances simulation stability, physical accuracy, and computational cost [2] [52]. This guide provides a systematic approach to selecting and validating the optimal time step for your molecular system.
Fundamental principles of time step selection
The time step in MD simulations must be short enough to accurately capture the fastest atomic motions yet long enough to make the simulation computationally feasible [35]. Two key theoretical concepts govern this choice:
Nyquist-Shannon Sampling Theorem: For accurate numerical integration, the time step should be less than half the period of the fastest vibration in your system [2]. In practice, a more conservative ratio of 0.01 to 0.033 of the shortest vibrational period is often recommended [2].
Highest Frequency Vibrations: The fastest motions typically involve bonds with hydrogen atoms. For instance, a C-H bond vibration at approximately 3000 cm⁻¹ corresponds to a period of about 11 femtoseconds, suggesting a maximum time step of roughly 5.5 femtoseconds by the Nyquist criterion [2]. In practice, with proper integration algorithms, time steps of 1-2 femtoseconds are commonly used [2] [35].
Table: Typical Time Step Guidelines for Different Systems
| System Type | Recommended Time Step (fs) | Key Considerations |
|---|---|---|
| All-atom with explicit solvent | 1-2 | Limited by hydrogen-containing bond vibrations [2] |
| All-atom with constraints (SHAKE) | 2 | Constraints allow longer time steps by removing fastest vibrations [2] |
| Coarse-grained models | >2 | Reduced degrees of freedom permit longer time steps |
| Systems with light atoms (H) | 0.25-1 | Require shorter time steps for accurate dynamics [2] |
Practical protocol: Energy conservation test
The most reliable method to validate your time step choice is testing energy conservation in the NVE (microcanonical) ensemble, where total energy should remain constant [2] [53].
Materials and software requirements
Table: Essential Research Reagents and Computational Tools
| Item | Function/Purpose |
|---|---|
| MD Software (GROMACS, LAMMPS, AMBER, etc.) | Performs numerical integration of equations of motion [40] |
| Molecular Viewer (VMD, PyMol) | Visualizes trajectories and identifies structural issues |
| NVE Ensemble | Isolated system with constant Number of particles, Volume, and Energy [40] [53] |
| Thermostat (for equilibration) | Brings system to target temperature before NVE production run |
| Analysis Scripts | Calculate energy drift and other thermodynamic properties |
Step-by-step workflow
System Preparation: Construct your molecular system with appropriate solvation and ionization.
Energy Minimization: Remove steric clashes and bad contacts using steepest descent or conjugate gradient methods.
System Equilibration:
- Run initial equilibration in NVT ensemble for 50-100 ps to stabilize temperature
- Continue equilibration in NPT ensemble for 50-100 ps to stabilize pressure and density
NVE Production Run: Switch to the NVE ensemble and run for a meaningful duration (typically 20-100 ps depending on system size and properties of interest) [2] [53].
Energy Drift Analysis: Calculate the drift in total energy per atom per picosecond using linear regression. A reasonable rule of thumb is that the drift should be less than 10 meV/atom/ps for qualitative results, and 1 meV/atom/ps for publishable results [2].
Advanced optimization strategies
Several advanced techniques can help increase the usable time step while maintaining accuracy:
Constraint Algorithms: Algorithms like SHAKE or LINCS freeze the fastest vibrations (typically bonds involving hydrogen atoms), allowing time steps of 2 femtoseconds or more [2].
Hydrogen Mass Repartitioning (HMR): This technique redistributes mass from heavy atoms to bonded hydrogens, slowing the highest frequency vibrations and enabling time steps up to 4 femtoseconds.
Machine Learning Approaches: Emerging methods use machine learning to learn structure-preserving (symplectic and time-reversible) maps, potentially enabling time steps orders of magnitude longer than conventional methods while maintaining physical fidelity [5].
Frequently asked questions
What are the symptoms of a time step that is too large?
- SHAKE/RATTLE failures: Constraint algorithms fail to converge
- Energy explosion: Total energy increases dramatically, causing simulation crash
- Non-physical artifacts: Atoms passing through each other, abnormal diffusion
- Poor energy conservation: Significant drift in NVE ensemble [2]
How do different integration algorithms affect time step selection?
Symplectic integrators like Velocity Verlet preserve the geometric structure of Hamiltonian dynamics, ensuring good long-term energy conservation even with relatively large time steps [2]. Non-symplectic algorithms may require smaller time steps for equivalent accuracy.
Can I use different time steps for different parts of my system?
While possible in some advanced simulation schemes through multiple time stepping (MST), this requires careful implementation to maintain energy conservation and physical accuracy.
How does system temperature affect time step choice?
Higher temperatures increase atomic velocities and vibrational frequencies, potentially requiring smaller time steps for accurate sampling of the dynamics.
Key takeaways
Selecting the optimal time step requires balancing numerical stability, physical accuracy, and computational efficiency. The energy conservation test in the NVE ensemble provides the most reliable validation of your chosen time step. When in doubt, start with a conservative time step (1-2 fs for all-atom systems) and systematically test based on the protocols outlined above.
Configuring Production MD Parameters in Common Software (GROMACS, LAMMPS)
Frequently Asked Questions (FAQs)
Q1: My energy minimization in GROMACS fails to converge, with maximum forces far above my set emtol. What should I do?
This is a common issue, often stemming from problems in the initial structure. The error message typically states that minimization stopped because the step size became too small, even though forces remain high [54]. Before proceeding, you must address these high forces. Check for structural problems like overlapping atoms, missing atoms, or incorrect residue naming in your initial coordinate file [55]. A system with extremely high forces may require a very conservative approach first: use the steepest descent (integrator = steep) algorithm with a very small emstep (e.g., 0.001) for the first few hundred steps before switching to the more efficient conjugate gradient method [50] [54]. While a negative potential energy is a good sign, a production run should not be started with extremely high, unconverged forces as it will likely crash.
Q2: Why does my LAMMPS simulation crash with "NaN" values for energy or pressure?
The appearance of "NaN" (Not a Number) or "Inf" values indicates a catastrophic numerical failure in your simulation [56] [57]. This is frequently caused by:
- Excessively large time steps: The timestep may be too large to accurately integrate the fastest motions (e.g., bond vibrations involving hydrogen atoms), leading to instability [56] [17].
- Overlapping atoms or bad contacts: If atoms are placed too close together at the start, the calculated forces from the potential can become infinite [56] [57].
- Incorrect force field parameters: Using parameters with the wrong units or that are inconsistent with your
unitsstyle setting can lead to nonsensical forces [56].
Solution: Start by visualizing your system at the point of failure to check for "fast-moving atoms" or obvious explosions [56]. Then, ensure your initial structure is sound by running a thorough energy minimization. For the initial phase of your production run, consider using a softer potential or a smaller timestep to gently relax the system before switching to your final, more efficient parameters [56].
Q3: Why do I get different trajectories when I run the same simulation on a different number of processors?
This is generally not a bug but expected behavior due to numerical round-off [57] [58]. The non-associative nature of floating-point arithmetic means that the order in which forces are summed across processors can cause minute differences. These tiny discrepancies grow exponentially in chaotic Hamiltonian systems, leading to diverged phase space trajectories. Reassuringly, the statistical properties (e.g., average energy, temperature, diffusion coefficients) of the simulations should remain consistent across different runs [57]. To ensure perfectly reproducible initial velocities across processor counts in LAMMPS, use the loop option with the velocity command [57].
Troubleshooting Guides
GROMACS:gromppErrors and Solutions
Many errors occur during the pre-processing stage when grompp assembles the simulation binary (.tpr file). The table below summarizes common errors and their fixes.
| Error Message | Cause | Solution |
|---|---|---|
| "Residue not found in residue topology database" [55] | The force field does not contain parameters for a molecule/residue in your structure. | 1. Check residue names for typos. 2. Rename to match the force field's database. 3. Manually provide a topology for the missing residue [55]. |
| "Found a second defaults directive" [55] | The [defaults] section appears more than once in your topology. |
The [defaults] directive must appear only once, typically in the main force field file. Comment out or remove duplicate entries from other included topology files [55]. |
| "Invalid order for directive..." [55] | The sections in your .top or .itp files are in the wrong order. |
Ensure directives follow the required sequence (e.g., [defaults] first, then [atomtypes], then [moleculetype]). Review the GROMACS reference manual for the correct structure [55]. |
| "Atom index in position_restraints out of bounds" [55] | Position restraint files are included in the wrong order relative to their corresponding molecule definitions. | Place the #include statement for each molecule's position restraint file (posre.itp) immediately after the #include for that molecule's topology [55]. |
LAMMPS: Common Run-Time Errors
These errors often occur during the run or minimize stage of a LAMMPS simulation.
| Error Symptom | Cause | Solution |
|---|---|---|
| "Lost atoms" or "Missing atoms" [56] | Atoms move too far in a single timestep, escaping the processor's sub-domain. | 1. Check for overlapping atoms using delete_atoms overlap. 2. Reduce the timestep. 3. Use fix nve/limit to limit maximum displacement. 4. Avoid using thermo_modify lost ignore as a solution [56]. |
| Segmentation Fault [56] | The process tried to access invalid memory. This can be a bug or due to overly aggressive simulation settings. | 1. First, run a smaller test system to reproduce the issue. 2. Identify the exact code location using a debugger. 3. Check for settings that might cause numerical overflow, like atoms being too close [56]. |
| "Unknown identifier in data file" [56] | A keyword in your data file is mistyped or inconsistent with the atom_style you are using. |
Carefully check the header and section keywords of your data file for typos. Ensure the data file format matches what your chosen atom_style expects [56]. |
| Simulation hangs without error [56] [57] | Often an issue with MPI message buffering in parallel, or an error occurred on only a subset of processors. | 1. Run LAMMPS with the -nonblock or -nb command-line flag to disable I/O buffering. 2. This can reveal error messages that were previously trapped in buffers [56] [57]. |
Production MD Parameter Optimization
Selecting appropriate parameters is crucial for achieving accurate and efficient production simulations. The following tables provide a guideline for key parameters in GROMACS and LAMMPS, framed within the context of time-step optimization research.
GROMACS: Key .mdp Parameters for Production MD
| Parameter | Common Value / Option | Thesis Context & Rationale |
|---|---|---|
integrator |
md (leap-frog), md-vv (velocity Verlet) [50] |
md-vv offers higher accuracy and better energy conservation for NVE ensembles, crucial for studying Hamiltonian dynamics and time-reversibility in time-step studies [5] [50]. |
dt |
1-2 fs (standard), up to 4 fs (with constraints/hydrogen mass repartitioning) [50] | The core trade-off: larger dt improves efficiency but risks instability. The upper limit is dictated by the fastest vibrational mode (e.g., bonds to H). Research focuses on pushing this limit while preserving accuracy [5] [17]. |
nsteps |
e.g., 50,000,000 for 100 ns with dt=2 fs |
Set to achieve the desired total simulation time (nsteps * dt). Long trajectories are needed to capture rare events and ensure statistical significance in properties like diffusion [59] [17]. |
constraints |
h-bonds (all bonds to H), all-bonds |
Constraining the fastest bond vibrations allows for a larger dt. h-bonds is standard; all-bonds is used with constraint-algorithm=lincs [50]. |
mass-repartition-factor |
1 (off), 3 or 4 (on) [50] | Artificially increases hydrogen masses to permit a 4 fs timestep. A key technique for accelerating sampling, but its effect on dynamics must be validated for your system [50]. |
tcoupl |
v-rescale (velocity rescale), nose-hoover |
A thermostat is essential for NVT/NPT ensembles. v-rescale provides a robust and efficient canonical distribution [50]. |
pcoupl |
Parrinello-Rahman, Berendsen |
Berendsen is good for equilibration (weak coupling). Parrinello-Rahman is recommended for production (more accurate pressure control) [50]. |
LAMMPS: Key Commands for Production MD
| Command / Setting | Common Value / Option | Thesis Context & Rationale |
|---|---|---|
timestep |
0.5-1.0 fs (standard), up to 2-4 fs (with fix shake or fix rattle) [17] |
Same principle as in GROMACS. The stability limit is system-dependent. Machine learning integrators are an emerging research area that may allow for significantly longer time steps by learning a structure-preserving map of the dynamics [5]. |
run |
e.g., run 1000000 |
Defines the number of timesteps for the simulation. |
fix nvt / fix npt |
fix 1 all nvt temp 300 300 100.0 |
Applies a thermostat/barostat. The final number (100.0) is the damping parameter tau_t (in time units). A larger value results in weaker coupling to the bath [51]. |
fix shake / fix rattle |
fix 1 all shake 0.0001 20 0 b a a |
Constrains bonds (and angles) involving hydrogen, enabling a larger timestep. rattle is the velocity Verlet equivalent [56]. |
pair_style |
lj/cut/coul/long, eam, etc. |
Defines the non-bonded interaction model. The choice is system-specific (e.g., eam for metals). Cutoffs must be set appropriately to match the force field [56]. |
thermo_modify |
thermo_modify lost ignore NOT RECOMMENDED [56] |
Ignoring lost atoms does not solve the underlying problem, which is typically bad physics (overlapping atoms, timestep too large). This should be avoided in production runs [56]. |
Experimental Protocol: A Workflow for Parameter Optimization
Adopting a systematic workflow is essential for configuring stable and accurate production MD simulations. The following diagram outlines a robust protocol for parameter testing and optimization, directly supporting thesis research on time-step integrity.
Protocol Details:
- Energy Minimization: Always start with extensive energy minimization (e.g., using steepest descent) to remove bad contacts and high forces from the initial structure. This is non-negotiable for a stable simulation [55] [54].
- Equilibration Phases:
- NVT Ensemble: Heat the system to the target temperature using a thermostat (e.g.,
v-rescalein GROMACS,fix nvtin LAMMPS) with a conservative time step. This phase allows the particle velocities to equilibrate correctly [59]. - NPT Ensemble: Apply a barostat (e.g.,
Parrinello-Rahmanin GROMACS,fix nptin LAMMPS) to adjust the system density to the target pressure. Ensure the density has stabilized before proceeding [59].
- NVT Ensemble: Heat the system to the target temperature using a thermostat (e.g.,
- Production Parameter Validation: Before launching a long production run, conduct a short test in the NVE ensemble (microcanonical, no thermostat/barostat). Monitor the total energy conservation. A well-behaved, stable integrator with a correctly chosen time step will show only small fluctuations in total energy. Significant energy drift indicates the time step is too large or the system is not properly equilibrated [5]. This is a critical test for the validity of the chosen parameters in fundamental research.
The Scientist's Toolkit: Essential Research Reagents
This table details key "computational reagents" – the software components and parameters essential for configuring a production MD simulation.
| Item | Function in Production MD |
|---|---|
Integrator (e.g., md, md-vv, verlet) |
The core algorithm that numerically solves Newton's equations of motion to update atomic positions and velocities over time [50] [17]. |
| Force Field (e.g., OPLS-AA, AMBER, CHARMM) | The set of mathematical functions and parameters that define the potential energy of the system, governing all interatomic interactions [55] [17]. |
Thermostat (e.g., v-rescale, nose-hoover, fix nvt) |
A computational algorithm that regulates the system's temperature, mimicking energy exchange with an external heat bath [50] [51]. |
Barostat (e.g., Parrinello-Rahman, Berendsen, fix npt) |
A computational algorithm that regulates the system's pressure by adjusting the simulation box size [50] [51]. |
Constraint Algorithm (e.g., LINCS, SHAKE, RATTLE) |
Allows for bonds involving hydrogen to be constrained to their equilibrium length, which permits the use of a larger integration time step by eliminating the fastest vibrational frequencies [50]. |
Long-Range Electrostatics (e.g., PME, PPPM) |
Method for accurately calculating electrostatic interactions beyond the short-range cutoff, essential for simulating charged systems and polar solvents [50]. |
Troubleshooting Guide: Core Challenges in MD Time-Step Optimization
This guide addresses common issues researchers face when optimizing the molecular dynamics (MD) time step for accurate atomic motion tracking.
Problem: Energy Drift and Non-Conservation
- Symptoms: Total energy of the system is not conserved over long simulations; temperature drifts uncontrollably.
- Underlying Cause: The use of non-structure-preserving integrators, especially with large time steps, can violate the geometric properties of the underlying Hamiltonian flow [5]. This breaks the symplectic structure of the equations of motion.
- Solutions:
- Switch to a Symplectic Integrator: Use algorithms like Velocity Verlet, which preserve the symplectic two-form, ensuring long-term stability and near-conservation of energy [5].
- Adopt a Structure-Preserving ML Integrator: For large time steps, implement machine-learning models that learn a symplectic map via a generating function (e.g., the
S3parametrization), which is equivalent to learning the mechanical action of the system [5]. - Validate with a Shadow Hamiltonian: Monitor the shadow Hamiltonian conserved by your symplectic integrator to verify stability.
Problem: Poor Generalization to Out-of-Distribution Configurations
- Symptoms: The model, trained on pristine structures, performs poorly on configurations with defects, impurities, or under non-equilibrium conditions [60].
- Underlying Cause: Standard machine-learned force fields (MLFFs) are often optimized for performance on in-distribution data and may not generalize well to atomic environments not seen during training.
- Solutions:
- Implement Sharpness-Aware Minimization (SAM): Utilize optimizers like Physics-Informed SAM (PI-SAM) that seek flat minima in the loss landscape. This reduces the model's sensitivity to small parameter perturbations and improves robustness to OOD data [60].
- Incorporate Physics-Informed Regularizations: Enforce physical laws directly into the loss function. Key regularizations include:
Problem: Inaccurate Force Predictions at Large Time Steps
- Symptoms: Forces become noisy or inaccurate when the time step is increased, leading to unphysical atomic trajectories.
- Underlying Cause: Conventional ML models that directly predict
(p', q')from(p, q)for a large time stephdo not inherently respect the Hamiltonian structure and can lead to artifacts like loss of equipartition [5]. - Solutions:
- Learn a Generative Model: Instead of direct prediction, learn a structure-preserving map derived from a generating function. This ensures the predicted
(p', q')is the exact evolution of a Hamiltonian system, preserving properties like equipartition [5]. - Use the Model as a Corrector: The action-derived ML integrator can be applied iteratively as a correction to cheaper, non-structure-preserving predictors, improving accuracy without a full computational overhead [5].
- Learn a Generative Model: Instead of direct prediction, learn a structure-preserving map derived from a generating function. This ensures the predicted
Problem: System Instability and Crash
- Symptoms: Simulation crashes shortly after a time-step increase, often due to atoms coming too close and experiencing excessively high forces.
- Underlying Cause: The time step is too large to accurately capture the fastest motions in the system (e.g., bond vibrations), leading to numerical instability.
- Solutions:
- Apply Constraint Algorithms: Use algorithms like SHAKE or LINCS to constrain the fastest degrees of freedom (e.g., bonds involving hydrogen atoms), allowing for a larger time step for the remaining motions.
- Conduct a Stability Analysis: Systematically increase the time step for your specific system in a controlled environment (NVE ensemble) and monitor the total energy for drift before proceeding to production runs.
Frequently Asked Questions (FAQs)
FAQ 1: What is the fundamental trade-off when selecting a time step in MD simulations? The core trade-off is between computational speed and numerical accuracy/stability. A smaller time step (e.g., 0.5 fs) more accurately integrates the fastest atomic vibrations (e.g., C-H bonds) but requires more steps to simulate the same physical time, increasing computational cost. A larger time step (e.g., 2-5 fs) is computationally faster but risks numerical instability, energy drift, and inaccurate dynamics if it exceeds the stability limit of the integrator for the system's fastest motions [5].
FAQ 2: How can Machine Learning (ML) help overcome the time-step barrier without sacrificing physical fidelity? ML can bypass the traditional time-step barrier by learning to predict the system's state after a long time step. The key is to use structure-preserving ML models. Instead of just learning the trajectory, these models learn the system's mechanical action or a symplectic map, ensuring the predicted evolution conserves energy and respects other physical laws like equipartition, even with time steps orders of magnitude larger than conventional limits [5].
FAQ 3: What are "flat minima" and how do they improve generalization for ML force fields? A "flat minimum" is a region in the model's parameter (weight) space where the loss value remains low even when the parameters are perturbed. In contrast, a "sharp minimum" is a precise point where small perturbations cause the loss to increase significantly. Models converging to flat minima are less sensitive to small changes in input data. This is crucial for MD, as it makes the model more robust to out-of-distribution atomic configurations, such as those with defects or impurities, which are common in real-world semiconductor or drug-target simulations [60].
FAQ 4: What specific physical laws should be embedded into a Machine Learning Force Field (MLFF)? Two critical physical laws to enforce are:
- Energy-Force Consistency: The interatomic forces must be the negative gradient of the total potential energy (
F = -∇E). This is a fundamental relationship that must be hardwired into the model training, often through a joint loss function that penalizes discrepancies in both energy and force predictions [60]. - Potential Energy Surface (PES) Curvature: The model should correctly represent the curvature of the PES, which is related to the vibrational frequencies and stability of atomic configurations. Regularizing for correct curvature ensures the dynamics are physically realistic [60].
Table 1: Trade-offs in Time Step Selection for Classical MD Integrators
| Time Step (fs) | Computational Cost (Relative) | Typical Stability Limit | Key Risks & Considerations |
|---|---|---|---|
| 0.5 - 1.0 | High (Baseline) | Stable for all bonds | Gold standard for accuracy; required for explicit bond vibrations. Computationally expensive for long timescales. |
| 1.0 - 2.0 | Medium | With H-bond constraints | Good balance for all-atom simulations when fast bonds (X-H) are constrained. Most common for production runs. |
| 2.0 - 4.0 | Lower | With multiple constraints | Risk of instability and energy drift. Requires careful testing. May be used for coarse-grained models. |
| > 4.0 | Very Low | Unstable for classical integrators | Leads to catastrophic failure (crash) in classical MD. Only feasible with advanced ML integrators that preserve geometric structure [5]. |
Table 2: Comparison of Traditional and ML-Enhanced Integration Approaches
| Feature | Traditional Symplectic Integrator (e.g., Velocity Verlet) | Direct ML Predictor | Structure-Preserving ML Integrator (Action-Derived) |
|---|---|---|---|
| Max Stable Time Step | ~1-2 fs (with constraints) | 100-200 fs (2 orders of magnitude larger) [5] | 100-200 fs (2 orders of magnitude larger) [5] |
| Energy Conservation | Near-conservation (excellent) | Poor, requires thermostating [5] | Excellent (inherently symplectic) [5] |
| Equipartition | Preserved | Often violated (loss of equipartition) [5] | Preserved [5] |
| Generalization to OOD | N/A (Not an ML model) | Poor | Improved, especially when combined with flat minima search (PI-SAM) [60] |
| Implementation Complexity | Low | Medium | High (requires learning a generating function) |
Experimental Protocol: Implementing a Physics-Informed SAM (PI-SAM) Workflow
This protocol outlines the methodology for training a robust ML force field using the PI-SAM framework to enhance generalization for accurate long-time-step dynamics [60].
Objective: To train a Graph Neural Network (GNN)-based MLFF that maintains high accuracy across diverse and out-of-distribution atomic configurations.
Materials:
- Software: A deep learning framework (PyTorch/TensorFlow) with MD simulation software (e.g., LAMMPS, GROMACS) interface.
- Data: A dataset of atomic configurations with corresponding energies and forces, preferably derived from ab initio calculations (e.g., DFT). The dataset should ideally include a variety of configurations, including some with defects or non-equilibrium structures.
Procedure:
- Data Preparation and Graph Representation:
- Convert your dataset of atomic configurations into graph representations, where nodes represent atoms and edges represent interatomic interactions or bonds [60].
- Split the data into training, validation, and test sets. Ensure the test set contains "out-of-distribution" configurations (e.g., different defect types, surfaces, or impurities) not present in the training data [60].
Model Selection and Initialization:
- Choose an appropriate GNN architecture. Both invariant GNNs (which use scalar features like distances) and equivariant GNNs (which use vector/tensor features for better directional awareness) are suitable choices [60].
- Initialize the model weights.
Define the Composite Loss Function: The loss function
L_totalis the core of PI-SAM and consists of multiple terms:L_energy: Mean Squared Error (MSE) between predicted and true potential energy.L_force: MSE between predicted and true atomic forces.L_physics: Physics-informed regularization terms. This includes:- Energy-Force Consistency Loss: Penalizes the difference between the predicted forces and the negative gradient of the predicted energy with respect to atom positions [60].
- PES Curvature Loss: A regularization term that aligns the model's predicted curvature of the potential energy surface with known physical behavior or ab initio data [60].
L_total = α * L_energy + β * L_force + γ * L_physics(where α, β, γ are weighting hyperparameters).
Sharpness-Aware Minimization (SAM) Optimization: Instead of simply minimizing
L_total(θ)for parametersθ, SAM seeks parameters that lie in a neighborhood with uniformly low loss. The update involves two steps:- 1. Perturbation Step: Calculate the gradient of the loss, then define a perturbation in the direction that maximizes the loss increase within a small radius
ρ:ε̂ = ρ * ∇L(θ) / ||∇L(θ)||. - 2. Update Step: Compute the gradient at the perturbed parameters
θ + ε̂, and use this gradient to update the original parametersθ.
- 1. Perturbation Step: Calculate the gradient of the loss, then define a perturbation in the direction that maximizes the loss increase within a small radius
Validation and Testing:
- Monitor the model's performance on the validation set, paying close attention to both in-distribution and OOD configurations.
- The final model evaluation should be conducted on the held-out test set, specifically analyzing its performance on the OOD atomic configurations to confirm improved generalization [60].
The following workflow diagram illustrates the PI-SAM training procedure:
Research Reagent Solutions
Table 3: Essential Computational Tools for MD Time-Step Optimization
| Item / Software | Category | Primary Function in Research |
|---|---|---|
| Graph Neural Network (GNN) | ML Model Architecture | Learns the relationship between atomic structure and potential energy/forces; the core of modern MLFFs [60]. |
| Symplectic Integrator (e.g., Velocity Verlet) | Simulation Algorithm | Provides numerically stable, long-time energy conservation for traditional MD with small time steps [5]. |
| Physics-Informed SAM (PI-SAM) | Optimization Framework | An optimizer that finds flat minima in the loss landscape and incorporates physical laws as regularization, drastically improving model generalization [60]. |
| Structure-Preserving ML Map | ML-Enhanced Algorithm | A machine-learning model that learns a symplectic and/or time-reversible map, enabling physically-correct dynamics with very large time steps [5]. |
| Electronic Structure Code (e.g., for DFT) | Data Generation Tool | Generates high-quality training data (energies and forces) for the MLFF from first-principles calculations [60]. |
Benchmarking and Validating Your Simulation for Scientific Rigor
FAQ: Understanding Energy Drift in NVE Simulations
Q1: What is energy drift in the context of NVE molecular dynamics simulations?
Energy drift is the gradual change in the total energy of a closed system over the course of a molecular dynamics simulation [61]. According to the laws of mechanics, the total energy in a closed system should be a constant of motion [61]. In practice, numerical integration artifacts arising from the use of a finite time step (Δt) cause the energy to fluctuate over short time scales and increase or decrease over very long time scales [61]. This phenomenon is often used as a key quality metric for assessing simulation reliability [61].
Q2: Why is monitoring energy drift particularly important for NVE ensembles?
In the microcanonical (NVE) ensemble, the number of particles (N), volume (V), and energy (E) are all conserved quantities [61]. Energy drift has much more severe consequences for NVE simulations compared to canonical (NVT) ensembles where temperature is held constant by a thermostat [61]. Significant energy drift indicates that the simulation is no longer accurately representing the intended thermodynamic ensemble, potentially compromising the physical validity of the results [61].
Q3: What are the primary numerical factors that cause energy drift?
The main numerical factors contributing to energy drift include:
- Finite time step (Δt) effects: The use of a discrete time step introduces perturbations to the true velocity [61].
- Non-symplectic integrators: Methods like Runge-Kutta exhibit substantial energy damping, while symplectic integrators (e.g., Verlet) better conserve a "shadow" Hamiltonian [61].
- Force calculation approximations: Cutoff schemes for long-range forces and insufficient smoothing introduce systematic errors with each time step [61].
- Constraint violations: Inaccurate bond constraint algorithms can introduce energy errors [62].
Q4: What level of energy drift is considered acceptable in production simulations?
While specific thresholds may vary by system, energy drift is often measured as a percent increase over time or as time needed to add a given amount of energy to the system [61]. Research has shown that long microcanonical simulations can be performed with "insignificant" energy drift, even for flexible molecules with constraints [61]. As a reference point, one user reported a drift of 4×10⁴ kBT/ns per atom at 400K, which they noted was at least four orders of magnitude higher than values reported in established literature [62].
Troubleshooting Guide: Resolving Excessive Energy Drift
Systematic Diagnosis Procedure
Specific Remediation Protocols
Issue: Time Step Too Large
- Diagnosis: Check for resonance artifacts when the velocity update frequency (2π/Δt) matches natural frequencies of the system (n/m)ω = 2π/Δt [61].
- Solution: Reduce Δt to below 0.225p for motions with natural frequency ω [61]. For biomolecular systems, ensure Δt is appropriate for the fastest vibrational modes.
Issue: Incorrect Neighbor List Parameters
- Diagnosis: Energy drift that improves slightly when increasing Verlet buffer size [62].
- Solution: Adjust
verlet-buffer-toleranceandnstlistparameters. One user achieved improvement by settingverlet-buffer-tolerance = 0.0005withnstlist = 40[62].
Issue: Force Calculation Inaccuracies
- Diagnosis: Systematic errors from electrostatic cutoffs without proper smoothing [61].
- Solution: Implement Particle Mesh Ewald (PME) summation for long-range electrostatics. Ensure system neutrality to avoid PME warnings about non-integer total charge [62].
Issue: Constraint Algorithm Failures
- Diagnosis: LINCS convergence failures or excessive bond length deviations.
- Solution: Adjust constraint tolerances, ensure sufficient equilibration before production runs, and verify initial structures don't contain atomic clashes [63].
Issue: Insufficient Numerical Precision
- Diagnosis: Energy drift persists despite optimized parameters.
- Solution: Compile and use double-precision version of MD software when possible [62].
Experimental Protocols: Measuring and Reporting Energy Drift
Standardized Measurement Methodology
Protocol 1: Baseline Energy Drift Quantification
- System Preparation: Ensure complete energy minimization and thorough equilibration in the desired ensemble.
- Production Simulation: Run extended NVE simulation (typically 100-1000ps depending on system size) [62].
- Data Collection: Record total energy at regular intervals throughout the trajectory.
- Analysis: Calculate the slope of total energy versus time using linear regression. Normalize by number of atoms and simulation time [62].
- Reporting: Express as kJ/mol/ns/atom or kBT/ns/atom at the simulation temperature [62].
Protocol 2: Time Step Optimization Procedure
- Parameter Scanning: Run identical NVE simulations with varying Δt values (e.g., 0.5fs, 1.0fs, 2.0fs).
- Drift Measurement: Quantify energy drift for each Δt using Protocol 1.
- Stability Assessment: Identify the maximum Δt that maintains acceptable energy drift while considering computational efficiency.
- Validation: Verify physical properties (temperature, pressure) remain stable with chosen Δt.
Quantitative Reference Data
Table 1: Typical Energy Drift Values Under Different Simulation Conditions
| System Type | Time Step (fs) | Integration Method | Typical Energy Drift | Reference |
|---|---|---|---|---|
| Biomolecules (59,880 atoms) | Not specified | Verlet | 4×10⁴ kBT/ns/atom @400K | [62] |
| Flexible molecules | Optimized | Symplectic | Insignificant over long timescales | [61] |
| Point-charge models | Standard | Polarizable | Comparable within limited iterations | [64] |
Table 2: Effect of Numerical Parameters on Energy Conservation
| Parameter | Inappropriate Setting | Optimized Setting | Impact on Energy Drift |
|---|---|---|---|
| Verlet buffer tolerance | Too large | 0.0005 | Moderate improvement [62] |
| Neighbor list update | Infrequent | nstlist=40 | Reduces drift accumulation [62] |
| Constraint algorithm | None | LINCS with proper tolerances | Prevents bond energy leakage [63] |
| Numerical precision | Single | Double | Significant improvement [62] |
| Electrostatics | Cutoff | PME | Eliminates systematic errors [61] |
Advanced Research Reagents and Computational Tools
Table 3: Essential Software Tools for Energy Drift Analysis
| Tool Name | Primary Function | Application in Energy Monitoring |
|---|---|---|
| GROMACS | Molecular dynamics simulator | Provides built-in energy drift analysis in NVE ensembles [62] |
| GENESIS | Biomolecular MD simulator | Supports various integrators and constraint methods for energy conservation [63] |
| PyRESP | Parameterization for polarizable models | Optimizes parameters for polarizable force fields to improve energy conservation [64] |
| LIPCG | Preconditioned conjugate gradient | Removes error outliers in induced dipole calculations to maintain energy conservation [64] |
| MOE | Multi-Order Extrapolation | Quickens induction iteration using historical data while preserving energy conservation [64] |
Advanced Topics: Energy Conservation in Polarizable Force Fields
Challenge: Induced dipole models present unique energy conservation challenges, particularly when using historical data for dipole prediction [64].
Solution Implementation:
- Multi-Order Extrapolation (MOE): Accelerates induction iteration while optimizing historical data usage [64].
- Preconditioned Conjugate Gradient with Local Iterations (LIPCG): Refines iteration process and effectively removes error outliers [64].
- Jacobi Under-Relaxation (JUR) "Peek" Step: Provides an additional convergence check for optimal performance [64].
Validation Protocol: For polarizable force fields, ensure energy convergence comparable to point-charge models within a limited number of iterations [64]. Monitor the maximum relative errors as a convergence metric rather than simply relying on historical dipole information [64].
Frequently Asked Questions
Q1: What is the core problem with using large time steps in alchemical free energy calculations? Using excessively large time steps can introduce significant errors, including energy drift and inaccurate free energy estimates. This occurs because large steps can impair the integration of the equations of motion, leading to poor sampling of phase space and ultimately, unreliable results [47] [5].
Q2: I need both accuracy and efficiency. What is the recommended maximum time step? Based on current research, for accurate and reliable alchemical free energy calculations using hydrogen mass repartitioning (HMR) and the SHAKE constraint algorithm, you should limit your time step to a maximum of 2 fs [47]. While 4 fs is sometimes used, it has been shown to cause significant deviations.
Q3: How does increasing the time step quantitatively impact the results? Increasing the time step leads to a measurable increase in energy drift in NVE (constant particle number, volume, and energy) simulations. Furthermore, in the NPT (constant particle number, pressure, and temperature) ensemble, thermodynamic integration (TI) simulations show that a 4 fs time step can cause deviations of up to 3 kcal/mol in some systems compared to a 0.5 fs reference [47].
Q4: Are there advanced methods to overcome time step limitations? Yes, machine learning (ML) methods are being explored to create "learned integrators" that can potentially use much longer time steps. However, early approaches that do not preserve the geometric structure of Hamiltonian mechanics can lead to artifacts like energy non-conservation. Newer, structure-preserving ML integrators are being developed to address these issues [5].
The table below summarizes key findings on how time step selection affects energy conservation and free energy accuracy.
| Time Step (fs) | Energy Drift (NVE) | Deviation in dU/dλ (NPT) | Statistical Significance (p < 0.05) |
|---|---|---|---|
| 0.5 | Low (Reference) | Consistent with reference | Not applicable (Reference) |
| 1 | Increasing | Consistent with reference | No significant difference |
| 2 | Increasing | Consistent with reference | No significant difference |
| 4 | High | Deviations up to 3 kcal/mol | Yes, especially in aqueous solution |
Table 1: Impact of time step on energy drift and alchemical free energy accuracy. Data adapted from [47].
Experimental Protocols for Time Step Validation
This section provides a methodology for validating your chosen time step, ensuring the robustness of your alchemical free energy calculations.
Protocol 1: Assessing Energy Drift in NVE Simulations
- System Preparation: Build your molecular system (e.g., a protein-ligand complex in solvent) as you would for your production run.
- Equilibration: First, equilibrate the system in the NPT ensemble to stabilize the density and temperature.
- NVE Simulation: Switch to the NVE (microcanonical) ensemble. This ensemble is used because, in the absence of external perturbations, the total energy should be conserved. Any significant drift indicates numerical inaccuracies from the time step.
- Multiple Runs: Run a series of short NVE simulations (e.g., 100 ps each) using different time steps (e.g., 0.5, 1, 2, and 4 fs). It is critical to use the same initial coordinates and velocities for all runs.
- Analysis: Plot the total energy (potential + kinetic) as a function of time for each simulation. Calculate the energy drift (the slope of a linear fit to the total energy over time). A strong correlation between increasing time step and energy drift is a clear warning sign [47].
Protocol 2: Validating Free Energy Estimates Across Time Steps
- Thermodynamic Integration (TI) Setup: Set up a standard alchemical free energy calculation, for example, for a relative ligand binding transformation, using a well-tested protocol [65].
- Multi-Timestep TI Simulations: Perform a set of identical TI simulations in the NPT ensemble, where the only variable is the integration time step (e.g., 0.5, 1, 2, and 4 fs).
- Compare dU/dλ: For each λ state, compare the average value of
dU/dλ(the derivative of the Hamiltonian with respect to the alchemical coupling parameter) across the different time steps. - Statistical Testing: Use statistical tests (e.g., a t-test) to determine if the
dU/dλvalues or the final computed free energy differences from the 4 fs simulations are significantly different from those obtained with the 0.5 fs reference. A p-value of less than 0.05 indicates a significant difference [47].
Workflow for Time Step Troubleshooting
The following diagram outlines a logical workflow for diagnosing and resolving time step-related issues in alchemical free energy calculations.
Troubleshooting Workflow for Time Step Validation
The Scientist's Toolkit: Essential Research Reagents
This table details key components and their functions in a typical alchemical free energy simulation setup.
| Item / Reagent | Function / Role in the Simulation |
|---|---|
| Molecular Dynamics Engine | Software (e.g., OpenMM, GROMACS, AMBER) that performs the numerical integration of the equations of motion and calculates forces and energies. |
| Force Field | A set of empirical functions and parameters that define the potential energy surface of the system, governing bonded and non-bonded interactions [65]. |
| Alchemical Intermediate States | Non-physical "bridging" states defined by a coupling parameter (λ) that gradually transform one chemical species into another, enabling efficient free energy calculation [65]. |
| Hydrogen Mass Repartitioning (HMR) | A technique that allows for larger time steps by increasing the mass of hydrogen atoms and decreasing the mass of heavy atoms they are bonded to, keeping the total mass approximately constant [47]. |
| SHAKE / LINCS Algorithm | Constraint algorithms that fix the lengths of bonds involving hydrogen atoms. This is crucial for stability when using time steps larger than 0.5 fs [47]. |
| Thermostat and Barostat | Algorithms (e.g., Langevin, Nosé-Hoover, Berendsen) that maintain constant temperature (NVT/NPT) and pressure (NPT) during simulation, mimicking experimental conditions [65]. |
| Free Energy Estimator | The statistical mechanical method (e.g., MBAR, BAR, TI) used to compute the free energy difference from the ensemble of configurations generated at the different alchemical states [65]. |
Frequently Asked Questions
Q1: My simulation becomes unstable and "explodes" when I increase the time step. Which integrator should I use for larger time steps? Stochastic Dynamics (SD) integrators are often more tolerant of larger time steps for systems coupled to a heat bath. For conservative (NVE) systems, the Velocity Verlet and Leapfrog algorithms are symplectic, meaning they conserve energy well over long simulations, which enhances stability [66]. If you are using constraints (e.g., for bonds with hydrogen atoms), you can also use mass repartitioning to scale the masses of the lightest atoms, which allows for a larger time step [67].
Q2: I need synchronous positions and velocities for analysis. Which integrator provides this? The Velocity Verlet algorithm is the best choice. It calculates positions and velocities at the same time points [66] [68]. In contrast, the basic Leapfrog method calculates positions at integer time steps and velocities at half-integer steps, requiring interpolation to synchronize them [68].
Q3: For accurate sampling in a constant-temperature (NVT) ensemble, should I use a stochastic or deterministic thermostat?
Stochastic integrators like Stochastic Dynamics (SD) or Langevin Dynamics are very effective for canonical (NVT) sampling as they mimic physical collisions with solvent molecules [67] [69]. The friction coefficient (bd-fric or tau-t) controls the strength of coupling to the heat bath [67]. Recent research highlights the "Langevin Impulse" integrator as particularly effective for its stability and accuracy in reproducing statistical averages [69].
Q4: What is the key practical difference between the Velocity Verlet and Leapfrog algorithms? While they are mathematically similar and generate identical trajectory paths [66], they differ in how they handle variables. Velocity Verlet stores and updates positions and velocities synchronously. Leapfrog updates velocities and positions in a staggered fashion. For most practical purposes, especially when requiring direct velocity access, Velocity Verlet is often preferred [68].
Q5: How do I correct for the lack of explicit velocity in the basic Position Verlet method?
In the basic Position Verlet method, velocity is not explicitly stored. You can approximate it at time t using the formula: v(t) ≈ (x(t+Δt) - x(t-Δt)) / (2Δt) [70] [68]. If your simulation requires direct and accurate velocity values (e.g., for calculating kinetic energy or velocity-dependent forces), it is better to use the Velocity Verlet variant [68].
Troubleshooting Guides
Problem: Poor Energy Conservation in NVE Simulation
Description: The total energy of a closed system drifts significantly over time.
Diagnosis and Solution:
- Check the Integrator: Ensure you are using a symplectic integrator like Velocity Verlet (
integrator=md-vvin GROMACS) or Leapfrog (integrator=md) [67] [66]. These are designed for long-term energy conservation in conservative systems. - Reduce the Time Step: A time step that is too large is a common cause of energy drift. A good rule of thumb is that the time step should be about an order of magnitude smaller than the fastest vibration period in your system. For atomistic systems with explicit bonds to hydrogen, a 2 fs timestep is common.
- Consider Mass Repartitioning: To enable a larger, more efficient time step without sacrificing stability, you can use the
mass-repartition-factoroption. This scales the masses of light atoms (like hydrogen), which stiffens the fastest bonds and allows for a timestep of up to 4 fs [67].
Problem: Incorrect Temperature (Kinetic Energy) in NVT Simulation
Description: The simulated system's temperature does not match the target temperature (ref-t).
Diagnosis and Solution:
- Verify Thermostat Parameters: For a Stochastic Dynamics (SD) integrator, the
tau-tparameter is critical. It defines the relaxation time of the thermostat; a value that is too small can cause overly aggressive temperature coupling, while a value that is too large may not maintain temperature effectively. A typical value is 2 ps [67]. - Check your Friction Coefficient: In Langevin/Brownian dynamics, the friction coefficient (
bd-fric) directly influences the kinetic energy distribution. An incorrect value will lead to a deviation from the target temperature [67]. - Validate Integrator Accuracy: Some older or less accurate stochastic integrators may not correctly reproduce the Boltzmann distribution. Consult recent literature or software documentation for recommended integrators, such as the GJF or Langevin Impulse integrators, which are known for their accuracy [69].
Problem: Simulation Instability with Small Time Steps
Description: The simulation crashes even with very small time steps.
Diagnosis and Solution:
- Check Force Calculation: Instability at small time steps is often a sign of a problem in the force calculation, not the integrator itself. Look for:
- Overlapping atoms in the initial structure.
- Incorrectly defined potential functions or parameters in your force field.
- Missing parameters for specific interactions.
- Review System Setup: Ensure your system is properly solvated and neutralized, as large electrostatic forces from unscreened charges can cause instability.
Integrator Comparison and Selection Table
The table below summarizes the core characteristics of the main integrator families to guide your selection.
| Feature | Verlet Family (Position / Velocity) | Leapfrog | Stochastic Dynamics (SD) / Langevin |
|---|---|---|---|
| Mathematical Formulation | Position Verlet: x(t+Δt) = 2x(t) - x(t-Δt) + a(t)Δt² [70]Velocity Verlet: Uses x(t+Δt) = x(t) + v(t)Δt + 0.5a(t)Δt² and v(t+Δt) = v(t) + 0.5(a(t) + a(t+Δt))Δt [68] |
v(t+Δt/2) = v(t-Δt/2) + a(t)Δtx(t+Δt) = x(t) + v(t+Δt/2)Δt [68] |
dp(t) = -∇U(q(t))dt - γp(t)dt + √(2kTγm) dW (Langevin equation) [71] |
| Order of Accuracy | Second-order (global error) [70] | Second-order | Varies by specific algorithm; second-order methods available [69] [71] |
| Numerical Stability | High (Symplectic and time-reversible) [66] | Very High (Symplectic and time-reversible) [66] [68] | Good; often more stable than Newtonian integrators for a given timestep when coupled to a bath [67] [69] |
| Velocity Handling | Position Verlet: Implicit, must be calculated [68]Velocity Verlet: Explicit and synchronous [67] | Explicit, but staggered (half-step offset) [68] | Explicit |
| Primary Use Case | NVE microcanonical ensemble; canonical sampling with a separate thermostat [67] | NVE microcanonical ensemble; efficient large-scale MD [67] | NVT canonical ensemble; implicit solvent simulations; systems coupled to a heat bath [67] [69] |
Key GROMACS integrator option |
md-vv (Velocity Verlet), md (Leapfrog) [67] |
md [67] |
sd (Stochastic Dynamics) [67] |
Experimental Protocol: Benchmarking Integrator Performance
This protocol provides a methodology to compare the accuracy and efficiency of different integrators within the GROMACS package, fitting the context of thesis research on time step optimization.
1. System Preparation
- Software: GROMACS [67] [40]
- Model System: Begin with a well-defined system like a box of water (SPC/E model) or a small, folded protein (e.g., BPTI) in explicit solvent [40].
- Minimization and Equilibration: Use the
steep(steepest descent) integrator to minimize the energy. Subsequently, equilibrate the system in the NVT and NPT ensembles using a standard integrator likemd(Leapfrog) with a Berendsen or velocity-rescale thermostat.
2. Production Runs for Comparison
- Parameter Variation: Run a series of production simulations from the same equilibrated starting structure.
- Integrators: Test
md(Leapfrog),md-vv(Velocity Verlet), andsd(Stochastic Dynamics). - Time Steps: For each integrator, test a range of time steps (e.g., 1 fs, 2 fs, 4 fs). If using 4 fs, ensure the
mass-repartition-factoris set to 3 formdandmd-vv[67]. - Constants: Keep all other parameters (e.g.,
ref-t,tau-tfor SD,nsteps, cut-off schemes, pressure coupling) identical across runs.
- Integrators: Test
3. Data Analysis and Comparison Analyze the resulting trajectories using GROMACS tools and custom scripts to evaluate:
- Energy Conservation: For NVE simulations, calculate the drift in total energy (
energy drift = (E_final - E_initial) / simulation_time). A superior symplectic integrator will show minimal drift [66]. - Temperature Control: For NVT simulations, compute the average and standard deviation of the temperature. Compare it to the target (
ref-t). - Structural Stability: Calculate the root-mean-square deviation (RMSD) of protein backbone atoms relative to the starting structure to assess if the dynamics preserves the native fold.
- Sampling Efficiency: Monitor the evolution of a key observable, such as the radius of gyration or the root-mean-square fluctuation (RMSF) of residues, to see how quickly it converges.
- Computational Performance: Record the simulation time per nanosecond (ns/day) for each run to compare raw efficiency.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below lists key computational "reagents" and parameters essential for running and optimizing molecular dynamics simulations with different integrators.
| Item | Function / Description | Example / Note |
|---|---|---|
GROMACS mdp File |
The parameter file that defines all simulation settings, including the choice of integrator, time step, and force field parameters [67]. | A sample mdp file is provided in the GROMACS documentation [67]. |
Integrator (integrator) |
The core algorithm that solves Newton's equations of motion. The choice dictates the ensemble and stability of the simulation [67]. | md (Leapfrog), md-vv (Velocity Verlet), sd (Stochastic Dynamics) [67]. |
Time Step (dt) |
The discrete interval at which forces are calculated and positions are updated. It is a primary determinant of stability and performance [67]. | Typically 1-2 fs for atomistic systems without mass repartitioning; can be 4 fs with mass repartitioning [67]. |
Friction Coefficient (tau-t, bd-fric) |
In stochastic integrators, this parameter determines the strength of the coupling to the heat bath, influencing how quickly the temperature relaxes to the target value [67]. | For integrator=sd, a tau-t of 2 ps is often appropriate [67]. |
Mass Repartitioning (mass-repartition-factor) |
A technique that scales the masses of light atoms (e.g., hydrogen) to allow for a larger integration time step by reducing the frequency of the fastest bond vibrations [67]. | A factor of 3 is commonly used with constraints=h-bonds to enable a 4 fs timestep [67]. |
| Force Field | A set of empirical potential functions and parameters that describe the potential energy of the system as a function of nuclear coordinates (e.g., AMBER, CHARMM, OPLS) [72]. | The choice of force field (Class I, II, or III) determines the mathematical form of the energy terms and the included physical effects [72]. |
Integrator Selection Workflow
The following diagram illustrates a logical decision pathway for selecting the most appropriate integrator based on your research goals and system properties.
Performance Benchmarking Workflow
This diagram outlines the core steps for designing and executing a comparative benchmark of different integrators, as detailed in the experimental protocol.
Troubleshooting Guides
1. Issue: Non-Physical Energy Drift in Long Simulations
- Problem: The total energy of the system is not conserved and drifts significantly over time, making the simulation physically unrealistic.
- Diagnosis: This is a common artifact of using non-structure-preserving (non-symplectic) machine-learning predictors for integrating equations of motion with large time steps. These methods do not conserve the geometric properties of the underlying Hamiltonian flow [5].
- Solution:
- Implement a structure-preserving integrator. Use a symplectic and time-reversible map derived from a generating function, which is mathematically equivalent to learning the system's mechanical action. This ensures long-time near-conservation of energy [5].
- For conventional integrators (like Velocity Verlet), reduce the time step until energy conservation is acceptable.
- If using a machine-learning model, ensure its architecture is designed to be symplectic.
2. Issue: Incorrect Diffusion Rates from Mean Squared Displacement (MSD) Analysis
- Problem: The calculated diffusion coefficient (D) does not match expected or experimental values.
- Diagnosis: The anomaly could stem from an inaccurate potential function, poor equilibration, or finite-size effects [40] [72].
- Solution:
- Validate the Potential Function: Ensure the chosen force field (e.g., EAM for metals, Tersoff for covalent materials) is appropriate for your system. An inaccurate potential function is a primary source of error [40].
- Extend Equilibration: Confirm that the system has fully equilibrated by monitoring potential energy and temperature until they stabilize before starting production runs.
- Check System Size: If possible, test for finite-size effects by running the simulation in a larger box with periodic boundary conditions [73].
3. Issue: Loss of Equipartition Between Degrees of Freedom
- Problem: Different types of atoms or molecules in the system have different average kinetic energies, violating a key principle of statistical mechanics.
- Diagnosis: This pathological behavior is a direct result of using a non-structure-preserving integrator, which disrupts the underlying Hamiltonian structure [5].
- Solution: Adopt a symplectic ML integrator. This method eliminates the lack of equipartition and other artifacts seen in direct predictors [5].
4. Issue: Kinetic Properties Do Not Reproduce Experimental Trends
- Problem: Simulated kinetic properties, such as ion channel inactivation rates, do not match experimental observations.
- Diagnosis: The Markov model used to represent state transitions (e.g., in a cardiac L-type Ca²⁺ channel) may be inadequately parameterized or may fail to capture coupled gating transitions [74].
- Solution:
- Refine the Markov Model: Develop a more detailed kinetic model that represents discrete ion-channel states and their interactions, moving beyond the independent gating variables of the Hodgkin-Huxley paradigm [74].
- Incorporate Key Interactions: Ensure the model correctly represents critical interactions, such as the calcium-dependent inactivation of CaV1.2 channels, which is sensitive to calcium entering through the channel and from internal stores [74].
Frequently Asked Questions (FAQs)
Q1: What is the maximum time step I can use in my MD simulation without losing accuracy? The maximum stable time step is determined by the highest frequency vibration in your system (e.g., bonds with hydrogen atoms). A common rule of thumb is to use a time step of 1-2 femtoseconds. Machine-learning approaches can potentially extend this to time steps two orders of magnitude longer, but one must use structure-preserving maps to avoid energy drift and other artifacts [5] [73].
Q2: How can I characterize a diffusion process with time-dependent properties? A machine-learning method can be applied at the single-trajectory level to predict properties like the diffusion coefficient or anomalous exponent at every time step. This allows both abrupt and continuous changes in diffusive properties to emerge without prior assumptions about the system [75].
Q3: My simulation lacks a crucial physical property. Where should I look first? The first place to check is the potential energy function (force field). The accuracy of the potential function fundamentally determines the reliability of all simulation results. Using a potential fitted only to solid-state properties, for example, will yield incorrect predictions for phenomena involving solid-liquid interfaces [40].
Q4: What is the difference between NVE, NVT, and NPT ensembles?
- NVE: Constant Number of particles, Volume, and Energy. The system is isolated.
- NVT: Constant Number of particles, Volume, and Temperature. The system is in contact with a thermostat.
- NPT: Constant Number of particles, Pressure, and Temperature. The system is in contact with both a thermostat and a barostat. Most real-world systems are best simulated in the NPT or NVT ensembles [40].
Quantitative Data for MD Validation
Table 1: Common Numerical Integrators for Molecular Dynamics
| Integrator Name | Symplectic? | Time-Reversible? | Key Features / Best Use Cases |
|---|---|---|---|
| Verlet Algorithm [40] | Yes | Yes | Good energy conservation, basic NVE simulations. |
| Velocity Verlet [40] | Yes | Yes | Computes velocities explicitly, widely used. |
| Symplectic ML Map [5] | Yes | Yes | Enables very large time steps; eliminates energy drift and loss of equipartition. |
| Markov Model (for kinetics) [74] | N/A | N/A | Represents discrete channel states; essential for simulating state-specific drug binding or mutations. |
Table 2: Key Parameters for Validating Dynamic Properties
| Property | How to Calculate | What to Look For | Common Force Field Dependencies |
|---|---|---|---|
| Diffusion Coefficient (D) | Slope of the Mean Squared Displacement (MSD) vs. time: ( D = \frac{1}{2dN}\lim_{t \to \infty}\frac{d}{dt}\langle |r(t)-r(0)|^2 \rangle ) (d=dimensions) | Agreement with experimental values; linear MSD plot for normal diffusion. | Strongly dependent on non-bonded interaction parameters (LJ, Coulomb) [72]. |
| Kinetic Rate Constants | From a Markov model by analyzing state transition frequencies [74]. | Reproduction of experimental macroscopic currents and inactivation time courses. | Model parameters are fitted to experimental single-channel and macroscopic-current data [74]. |
| Energy Conservation | Monitor the total energy (potential + kinetic) over time in an NVE simulation. | Small fluctuations around a stable average, with no significant drift. | Integrity of the entire force field; validated by using a symplectic integrator [5] [73]. |
Experimental Protocols
Protocol 1: Setting Up a Standard MD Simulation for Validation [72] [73]
- System Preparation:
- Obtain initial atomic coordinates (e.g., from a PDB file for a protein).
- Define the topology, specifying atom types, bonds, angles, and dihedrals.
- Select an appropriate force field (e.g., AMBER, CHARMM, OPLS).
- Solvation and Ionization:
- Place the molecule in a box of explicit solvent molecules (e.g., TIP3P water model).
- Add ions to neutralize the system's charge and achieve a physiological concentration.
- Energy Minimization:
- Run a steepest descent or conjugate gradient algorithm to remove bad contacts and relieve steric clashes.
- Equilibration:
- NVT Ensemble: Heat the system to the target temperature (e.g., 300 K) using a thermostat (e.g., Nosé-Hoover) for 50-100 ps.
- NPT Ensemble: Adjust the system density to the target pressure (e.g., 1 bar) using a barostat (e.g., Parrinello-Rahman) for 100-200 ps.
- Production Run:
- Run the simulation in the desired ensemble (typically NPT) for the required duration to sample the phenomenon of interest. Use a symplectic integrator like Velocity Verlet with a 1-2 fs time step.
Protocol 2: Benchmarking a Machine-Learning Integrator [5]
- Data Generation: Run a conventional, high-accuracy (small time step) MD simulation to generate a training set of short trajectory snippets
(p(t), q(t)) -> (p(t+h), q(t+h)). - Model Training: Train a machine-learning model to learn a generating function
S³(p̄, q̄)that defines a symplectic and time-reversible map. - Validation:
- Energy Conservation: Run a long simulation with the ML integrator and a large time step. Plot the total energy over time and compare it to the baseline simulation. There should be no pathological drift.
- Equipartition: Check that the average kinetic energy is equal across all degrees of freedom in the system.
- Dynamic Properties: Calculate the diffusion coefficient from the ML-integrated trajectory and ensure it matches the value from the conventional simulation.
Workflow Visualization
Workflow for Validating Dynamic Properties in MD Simulations
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Software and Force Fields for MD Simulations
| Item | Function / Purpose | Reference / Source |
|---|---|---|
| LAMMPS | A highly flexible MD simulator with robust parallel computing capabilities, ideal for large-scale systems like metals and alloys [40]. | https://www.lammps.org/ |
| GROMACS | A high-performance MD package optimized for biomolecular systems like proteins, lipids, and nucleic acids [40]. | https://www.gromacs.org/ |
| EAM Potential | An embedded atom method potential for metals and alloys; accounts for multi-body interactions via electron density [40]. | Included in major MD software like LAMMPS. |
| Tersoff Potential | A bond-order potential for covalent materials (e.g., Si, C); describes bond formation/breaking and bond directionality [40]. | Included in major MD software like LAMMPS. |
| AMBER Force Field | A Class I force field widely used for simulations of proteins, DNA, and RNA [72] [73]. | https://ambermd.org/ |
| CHARMM Force Field | A Class I force field extensively used for biomolecules, with a polarizable variant (CHARMM-Drude) available [72]. | https://www.charmm.org/ |
| Markov Model | A computational model representing discrete states and transitions of a system, crucial for simulating complex kinetics like ion channel gating [74]. | Custom implementation based on experimental data. |
Troubleshooting Guide: Common MD Simulation Issues
SHAKE Algorithm Failures
Problem: The SHAKE algorithm, used to constrain bond lengths, fails during a simulation, causing the simulation to terminate.
- Potential Cause 1: Timestep is too large. A large timestep can prevent the constraint algorithm from converging to a solution.
- Solution: Reduce the timestep. The timestep should be at least a factor of 2 smaller than the period of the fastest vibration in the system [2]. For systems with C-H bonds (~11 fs period), a timestep of 1-2 fs is common.
- Potential Cause 2: Incorrect system topology. Missing bond parameters or incorrect constraints in the molecular topology file can cause failures.
- Solution: Carefully check the system topology to ensure all bonds, angles, and dihedrals are correctly defined and match the chosen force field.
Energy Drift in NVE Simulations
Problem: In a constant energy (NVE) simulation, the total energy is not conserved but shows a significant upward or downward drift over time.
- Potential Cause 1: Timestep is too large. A large timestep can cause numerical instability and break time-reversibility, leading to energy drift [2].
- Solution: A reasonable rule of thumb is that the long-term drift in the conserved quantity should be less than 10 meV/atom/ps for qualitative results, and 1 meV/atom/ps for publishable results [2]. Reduce the timestep until this criterion is met.
- Potential Cause 2: Inaccurate force calculations. This could be due to a too-short nonbonded cutoff or problems with the treatment of long-range electrostatics.
- Solution: Increase the nonbonded cutoff (e.g., to 1.2 nm) and ensure a robust method for long-range electrostatics (e.g., Particle Mesh Ewald) is used.
Poor Convergence of Physical Properties
Problem: Calculated properties (e.g., density, radius of gyration, diffusion coefficient) do not converge even after long simulation times.
- Potential Cause 1: Insufficient sampling. The simulation may not have reached equilibrium, or the production run may be too short to sample the relevant configurational space.
- Solution: Perform at least three independent simulations starting from different initial configurations to demonstrate convergence [76]. Visually inspect the time-course of the property to ensure it has equilibrated before starting production analysis.
- Potential Cause 2: Trapping in a metastable state. The system may be kinetically trapped in a local energy minimum, especially in complex biomolecular systems.
- Solution: If the event of interest is beyond the timescale of unbiased sampling, enhanced sampling methods may be required. Justify the choice of enhanced sampling method and provide its convergence criteria [76].
Frequently Asked Questions (FAQs)
What is the scientific basis for choosing a timestep?
The timestep is primarily governed by the Nyquist-Shannon sampling theorem, which states that to accurately capture a vibrational motion, you must sample it at least twice per period [2]. The timestep must be less than half the period of the fastest vibration in your system. In practice, a more conservative factor of 5-10 is often used to ensure numerical stability and good energy conservation.
How can I validate that my chosen timestep is appropriate?
Run a short simulation in the NVE (microcanonical) ensemble and monitor the total energy. A well-chosen timestep and a symplectic integrator (like Velocity Verlet) will result in minimal long-term energy drift [2]. The drift should ideally be less than 1 meV/atom/ps for publishable results [2].
What are the minimum requirements for publishing a simulation study?
A reliability and reproducibility checklist for MD simulations recommends [76]:
- Convergence Analysis: Show the property of interest has equilibrated; use at least 3 independent replicates per condition.
- Method Justification: Justify the choice of force field, water model, and enhanced sampling methods (if used).
- Connection to Experiment: Where possible, connect simulation results to experimental data (e.g., NMR, SAXS, binding assays).
- Data & Code Availability: Provide initial/final coordinates, input files, and custom code in a public repository.
Can I use a larger timestep than 2 fs?
Yes, but strategies are required to remove the fastest vibrations. Constraint algorithms (like SHAKE or LINCS) that freeze bond vibrations involving hydrogen allow timesteps of 2 fs, sometimes 2.5-4 fs for specific solvents [2]. More advanced methods like hydrogen mass repartitioning (HMR) or treating molecules as rigid bodies can enable even larger timesteps (4-5 fs) by lowering the frequency of the fastest motions [16].
Quantitative Data for Timestep Selection
Table 1: Maximum Timestep Guidelines for Different Simulation Types
| Simulation Type | Recommended Timestep (fs) | Key Considerations |
|---|---|---|
| Unconstrained, all-atom | 0.5 - 1.0 | Must capture C-H/O-H stretch (~10 fs period) [17]. |
| Constrained (SHAKE/LINCS) | 1.5 - 2.0 | Standard for most biomolecular simulations in explicit solvent [2]. |
| Rigid Water Models | 2.0 - 4.0 | Allows a larger timestep by removing internal vibrations of water [16]. |
| Hydrogen Mass Repartitioning | 3.0 - 4.0 | Increases the mass of H atoms, slowing the fastest vibrations [2]. |
Table 2: Validation Criteria for a Production-Ready Simulation
| Validation Metric | Target Value | How to Measure |
|---|---|---|
| NVE Energy Drift | < 1 meV/atom/ps | Linear fit of total energy vs. time over a production simulation [2]. |
| Number of Replicates | ≥ 3 | Independent simulations from different initial conditions [76]. |
| Property Convergence | Stable mean & variance | Time-course analysis (block averaging) of the key output property. |
| Bond Length Drift (SHAKE) | < 0.001 nm | RMSD of constrained bonds from their ideal length over the trajectory. |
Experimental Protocols for Validation
Protocol 1: Timestep Validation via Energy Conservation
Purpose: To determine the optimal timestep by evaluating the stability of total energy in an NVE simulation.
- System Setup: Prepare a small, representative system (e.g., a protein in a water box).
- Equilibration: Equilibrate the system thoroughly in the NPT ensemble at the target temperature and pressure.
- NVE Production Runs: Switch to the NVE ensemble and run multiple short simulations (e.g., 50-100 ps each) using different timesteps (e.g., 0.5, 1.0, 1.5, 2.0, 2.5 fs).
- Data Analysis: For each run, calculate the linear drift of the total energy. The largest timestep that maintains a drift below 1 meV/atom/ps is a suitable candidate for production [2].
Protocol 2: Convergence Assessment via Multiple Replicates
Purpose: To provide evidence that simulation results are reproducible and not dependent on initial conditions [76].
- Generate Replicates: Create at least 3 independent starting structures for the same system. This can be done by:
- Using different random seeds for initial velocities.
- Starting from different conformations (e.g., from different NMR models).
- Sampling initial structures from a long pre-equilibration.
- Run Production Simulations: Simulate each replicate for the same duration using identical parameters.
- Statistical Analysis: Calculate the key properties of interest from each trajectory. Report the mean and standard deviation across replicates. Convergence is supported if the means are similar and the standard deviation is small relative to the mean.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Reproducible MD Simulations
| Resource Category | Specific Examples | Function & Importance |
|---|---|---|
| Force Fields | CHARMM, AMBER, OPLS, GROMOS | Provide the set of parameters (masses, charges, bond constants, etc.) that define the potential energy surface and are critical for model accuracy [16]. |
| Simulation Software | GROMACS, NAMD, AMBER, OpenMM | The core engines that perform the numerical integration of the equations of motion and force calculations [76]. |
| Structure Databases | Protein Data Bank (PDB), Materials Project, PubChem | Repositories for obtaining initial atomic coordinates for proteins, materials, and small molecules [17]. |
| Analysis Tools | MDAnalysis, MDTraj, VMD, CPPTRAJ | Software packages used to process simulation trajectories and compute physical properties (RDF, MSD, etc.) [17]. |
| Validation Checklists | Reliability and Reproducibility Checklist [76] | A structured list of criteria to ensure simulations meet the standards required for publication. |
Workflow Diagram for Timestep Optimization
Timestep Optimization and Validation Workflow
Conclusion
Optimizing the MD time step is not a one-size-fits-all setting but a critical compromise between computational speed and physical fidelity. The key takeaway is that while techniques like hydrogen mass repartitioning and machine learning integrators offer exciting pathways to accelerate simulations, they must be applied and validated with caution to avoid introducing artifacts that distort the system's kinetics and thermodynamics. Future directions will likely involve the wider adoption of structure-preserving ML integrators and multi-scale methods, further closing the gap between simulation efficiency and biological realism. For biomedical research, particularly in drug discovery where predicting accurate binding residence times is crucial, rigorous time step optimization and validation are indispensable for generating reliable, hyper-predictive models that can truly impact clinical outcomes.
References
- 1. Nyquist–Shannon sampling theorem [https://en.wikipedia.org/wiki/Nyquist%E2%80%93Shannon_sampling_theorem]
- 2. How should one choose the time step in a molecular ... [https://mattermodeling.stackexchange.com/questions/498/how-should-one-choose-the-time-step-in-a-molecular-dynamics-integration]
- 3. New ways to boost molecular dynamics simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6680170/]
- 4. Long-time-step molecular dynamics can retard simulation ... [https://www.sciencedirect.com/science/article/pii/S0006349523000681]
- 5. Learning the action for long-time-step simulations of ... [https://arxiv.org/html/2508.01068v1]
- 6. Beyond Force Metrics: Pre-Training MLFFs for Stable MD ... [https://arxiv.org/html/2506.14850v1]
- 7. Accurate and efficient constrained molecular dynamics of ... [https://www.sciencedirect.com/science/article/pii/S0010465523000875]
- 8. Potentials of Mean Force for the Interaction of Blocked Alanine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1366650/]
- 9. Stability and transferability of machine learning force fields for ... [https://pubs.rsc.org/en/content/articlehtml/2024/dd/d4dd00140k]
- 10. Molecular Dynamics Simulations in Drug Discovery and ... [https://www.mdpi.com/2227-9717/9/1/71]
- 11. Molecular Dynamics - GROMACS 2025.1 documentation [https://manual.gromacs.org/documentation/2025.1/reference-manual/algorithms/molecular-dynamics.html]
- 12. Force-free molecular dynamics for fast and accurate long ... [https://research.ibm.com/publications/force-free-molecular-dynamics-for-fast-and-accurate-long-timescale-simulations]
- 13. Which Optimizer Should You Use With NNPs? | Rowan [https://www.rowansci.com/blog/which-optimizer-should-you-use-with-nnps]
- 14. Symplectic integrator [https://en.wikipedia.org/wiki/Symplectic_integrator]
- 15. What does "symplectic" mean in reference to numerical ... [https://scicomp.stackexchange.com/questions/29149/what-does-symplectic-mean-in-reference-to-numerical-integrators-and-does-scip]
- 16. Best Practices for Foundations in Molecular Simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6884151/]
- 17. Molecular Dynamics Simulations for Materials and ... [https://matlantis.com/en/resources/blog/md-intro/]
- 18. Constraint (computational chemistry) - Wikipedia [https://en.wikipedia.org/wiki/Constraint_(computational_chemistry)]
- 19. Constraint algorithms - GROMACS 2025.4 documentation [https://manual.gromacs.org/documentation/current/reference-manual/algorithms/constraint-algorithms.html]
- 20. Constraint algorithms — GROMACS 2019-rc1 documentation [https://manual.gromacs.org/documentation/2019-rc1/reference-manual/algorithms/constraint-algorithms.html]
- 21. How does hydrogen mass repartitioning with an increased ... [https://gromacs.bioexcel.eu/t/how-does-hydrogen-mass-repartitioning-with-an-increased-time-step-of-4-fs-affect-other-mdp-options/9173]
- 22. Accelerating membrane simulations with Hydrogen Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7271963/]
- 23. Long-Time-Step Molecular Dynamics through Hydrogen ... [https://pubmed.ncbi.nlm.nih.gov/26574392/]
- 24. Creating a hydrogen mass repartitioned topology [https://amberhub.chpc.utah.edu/creating-a-repartitioned-topology/]
- 25. CHARMM-GUI Supports Hydrogen Mass Repartitioning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7902386/]
- 26. MOLECULE: Molecular-dynamics and Optimized deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12461829/]
- 27. Leveraging active learning-enhanced machine ... [https://www.nature.com/articles/s41524-025-01827-8]
- 28. Accurate and efficient machine learning interatomic potentials ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc01325a]
- 29. A critical review of machine learning interatomic potentials ... [https://www.oaepublish.com/articles/jmi.2025.17]
- 30. A fine-tuned large language model based molecular ... [https://www.nature.com/articles/s41598-025-92337-6]
- 31. Molecular Dynamics - QuantumATK [https://docs.quantumatk.com/manual/technicalnotes/molecular_dynamics/molecular_dynamics.html]
- 32. New approaches for computing ligand–receptor binding ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X17301094]
- 33. Residence time in drug discovery: current insights and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12241192/]
- 34. ModBind, a Rapid Simulation-Based Predictor of Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11733936/]
- 35. Molecular dynamics simulation for all - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6209097/]
- 36. Choice of time step in Molecular Dynamics simulations [https://lammpstube.com/2021/12/12/choice-of-time-step-in-md-method/]
- 37. DynamicBind: predicting ligand-specific protein ... [https://www.nature.com/articles/s41467-024-45461-2]
- 38. Predicting receptor-ligand pairing preferences in plant ... [https://www.sciencedirect.com/science/article/pii/S2001037025002454]
- 39. Enhancing binding affinity predictions through efficient ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc01030f]
- 40. Research Status of Molecular Dynamics Simulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12565893/]
- 41. Frontier advances in molecular dynamics simulations for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12138808/]
- 42. A comprehensive review of polymers and their derivatives ... [https://www.sciencedirect.com/science/article/abs/pii/S2213343725022973]
- 43. Dynamic Training Enhances Machine Learning Potentials ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12344694/]
- 44. Scientists capture atomic motion on camera for the first time [https://interestingengineering.com/science/scientists-capture-atomic-vibrations-first-time]
- 45. Geometry optimization — AMS 2025.1 documentation - SCM [https://www.scm.com/doc/AMS/Tasks/Geometry_Optimization.html]
- 46. Molecular dynamics enabled data-driven modeling of ... [https://www.sciencedirect.com/science/article/pii/S004578252500502X]
- 47. Impact of Timestep on Energy Drift and Accuracy ... [https://chemrxiv.org/engage/chemrxiv/article-details/67a674aafa469535b9727e10]
- 48. SHAKE parallelization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3285512/]
- 49. Long-time-step molecular dynamics can retard simulation ... [https://pubmed.ncbi.nlm.nih.gov/36726313/]
- 50. Molecular dynamics parameters (.mdp options) [https://manual.gromacs.org/current/user-guide/mdp-options.html]
- 51. Molecular dynamics — AMS 2025.1 documentation - SCM [https://www.scm.com/doc/AMS/Tasks/Molecular_Dynamics.html]
- 52. Issues on the Choice of a Proper Time Step in Molecular ... [https://www.sciencedirect.com/science/article/pii/S1875389214000388]
- 53. Molecular Dynamics simulation [https://docs.matlantis.com/atomistic-simulation-tutorial/en/6_1_md-nve.html]
- 54. MD minimization - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/md-minimization/571]
- 55. Common errors when using GROMACS [https://manual.gromacs.org/current/user-guide/run-time-errors.html]
- 56. 7.2. Errors and warnings details [https://docs.lammps.org/Errors_details.html]
- 57. 7.1. Common issues that are often regarded as bugs [https://docs.lammps.org/Errors_common.html]
- 58. 13.1. Common problems — LAMMPS documentation [https://gensoft.pasteur.fr/docs/lammps/2020.03.03/Errors_common.html]
- 59. Avoiding Common Mistakes When Choosing MD Parameters ... [https://blog.samson-connect.net/avoiding-common-mistakes-when-choosing-md-parameters-in-gromacs-wizard]
- 60. Sharpness-aware minimization with physics-informed ... [https://www.sciencedirect.com/science/article/abs/pii/S0169743925001960]
- 61. Energy drift [https://en.wikipedia.org/wiki/Energy_drift]
- 62. Energy drift in NVE - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/energy-drift-in-nve/4423]
- 63. Frequently Asked Questions - GENESIS [http://mdgenesis.org/docs/FAQ/]
- 64. An Optimal Scheme to Achieve Energy Conservation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10434752/]
- 65. Best Practices for Alchemical Free Energy Calculations [Article ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388617/]
- 66. Comparison of velocity Verlet and leapfrog algorithms [https://scicomp.stackexchange.com/questions/24232/comparison-of-velocity-verlet-and-leapfrog-algorithms]
- 67. Molecular dynamics parameters (.mdp options) [https://manual.gromacs.org/2025.0-beta/user-guide/mdp-options.html]
- 68. Verlet Integration | A Stable and Natural Numerical Method ... [https://barbegenerativediary.com/en/tutorials/verlet-integration/]
- 69. Comparison of effective and stable Langevin dynamics ... [https://www.sciencedirect.com/science/article/abs/pii/S0010465524000754]
- 70. Verlet integration [https://en.wikipedia.org/wiki/Verlet_integration]
- 71. Time Integrators for Molecular Dynamics [https://www.mdpi.com/1099-4300/16/1/138]
- 72. Practical considerations for Molecular Dynamics - GitHub Pages [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/aio/index.html]
- 73. An Introduction to Molecular Dynamics Simulations [https://portal.valencelabs.com/blogs/post/an-introduction-to-molecular-dynamics-simulations-C9nXeGJ8hbNYghL]
- 74. Kinetic Properties of the Cardiac L-Type Ca2+ Channel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1796810/]
- 75. Inferring pointwise diffusion properties of single trajectories ... [https://www.sciencedirect.com/science/article/pii/S0006349523006513]
- 76. Reliability and reproducibility checklist for molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10014944/]