Iterative Optimization for Force Field Development: A Modern Guide to Enhanced Accuracy and Efficiency
This article provides a comprehensive guide to iterative optimization procedures for developing molecular mechanics force fields, a critical tool for computational drug discovery and materials science.
Iterative Optimization for Force Field Development: A Modern Guide to Enhanced Accuracy and Efficiency
Abstract
This article provides a comprehensive guide to iterative optimization procedures for developing molecular mechanics force fields, a critical tool for computational drug discovery and materials science. Tailored for researchers and scientists, it explores the foundational principles of force field parameterization, details cutting-edge methodological workflows, and offers practical strategies for troubleshooting and optimization. By comparing validation techniques across different force field types—from classical to machine learning potentials—this review synthesizes best practices for achieving robust, accurate, and transferable force fields, ultimately aiming to accelerate reliable molecular simulations.
The Core Principles and Critical Need for Iterative Force Field Training
Defining Iterative Optimization in the Force Field Landscape
In molecular dynamics (MD) simulations, a force field is a computational model that describes the potential energy of a system of atoms as a function of their positions, enabling the study of complex systems that are prohibitively large for quantum mechanical (QM) treatment [1]. The accuracy of these simulations is fundamentally dependent on the quality of the force field parameters employed [2]. Iterative optimization has emerged as a powerful paradigm for force field development, representing a cyclic process of parameter refinement that systematically reduces the discrepancy between force field predictions and reference data, which can originate from both QM calculations and experimental measurements. This approach is particularly crucial for addressing the vast and expanding chemical space relevant to modern computational drug discovery [3]. This article delineates the core principles, protocols, and applications of iterative optimization within force field development, providing a structured guide for researchers and scientists.
Core Principles and Methodologies of Iterative Optimization
The foundational goal of iterative optimization is to escape local minima in parameter space and achieve a robust, transferable, and physically meaningful force field. This is accomplished through a self-correcting loop that integrates simulation, validation, and parameter adjustment.
The Basic Iterative Workflow
At its simplest, the iterative optimization workflow involves several key stages, elegantly captured by tools like the Force FieLd Optimization Workflow (FFLOW) [4]. The process begins with the initial parameterization of the force field, often informed by QM calculations on small molecular fragments or heuristic rules. Subsequently, MD simulations are run using these parameters to sample molecular conformations and properties. The results of these simulations—such as conformational energies, forces, or bulk properties—are then compared against target data (QM reference or experimental data). A loss function quantifying the difference is minimized by an optimization algorithm, which generates a new, improved set of parameters for the next iteration. The critical step that differentiates modern approaches is the use of a separate validation set to monitor convergence and prevent overfitting to the training data [2].
Advanced Integration of Machine Learning
Machine learning (ML) has profoundly enhanced iterative optimization protocols. Two primary applications are:
- Surrogate Models: ML models can be trained to predict the outcomes of expensive MD simulations or QM calculations. For instance, a neural network can learn the mapping between Lennard-Jones parameters and bulk-phase density, reducing the required computation time by a factor of ≈20 while maintaining accuracy [4]. This substitution allows for a much more rapid exploration of the parameter space.
- End-to-End Parameter Prediction: Graph Neural Networks (GNNs) can be trained to directly predict force field parameters for a given molecule. As demonstrated by the ByteFF force field, an edge-augmented, symmetry-preserving GNN can be trained on a massive dataset of QM calculations (e.g., 2.4 million optimized geometries and 3.2 million torsion profiles) to predict all bonded and non-bonded parameters simultaneously [3]. This data-driven approach ensures expansive coverage of chemical space and inherent transferability.
Table 1: Key Components of an Iterative Optimization Workflow
| Component | Description | Example Techniques |
|---|---|---|
| Target Data Generation | Producing high-quality reference data for parameter fitting and validation. | QM calculations (B3LYP-D3(BJ)/DZVP), experimental liquid properties (density, heat of vaporization) [5] [3]. |
| Parameter Optimization | The algorithm that adjusts force field parameters to minimize a loss function. | Gradient-based optimization, genetic algorithms, Bayesian optimization [4]. |
| Sampling & Data Augmentation | Using dynamics to explore new conformations for QM computation, enriching the training dataset. | Boltzmann sampling at elevated temperatures (e.g., 400 K) [2]. |
| Validation & Convergence | Assessing model performance on unseen data to determine when to halt the iterative cycle. | Tracking error on a separate validation set to flag overfitting [2]. |
| Surrogate Modeling | Replacing computationally expensive simulations with fast ML models during optimization. | Neural networks to predict bulk-phase density from Lennard-Jones parameters [4]. |
Detailed Experimental Protocols
This section outlines two specific protocols that exemplify the iterative optimization paradigm in practice.
Protocol 1: Automated Iterative Force Field Parameterization with Validation
This protocol, as described in [2], is designed for fitting custom single-molecule force fields and emphasizes the prevention of overfitting.
1. Initial Dataset Preparation:
- Select a target molecule (e.g., a tri-alanine peptide or a photosynthesis cofactor).
- Generate an initial dataset of QM calculations. This involves:
- Generating multiple conformers of the target molecule.
- Performing QM geometry optimizations and frequency calculations (e.g., at the B3LYP-D3(BJ)/DZVP level) to obtain energies, forces, and Hessian matrices [3].
- Split the data into a training set and a validation set.
2. Initial Parameter Optimization:
- Parameterize an initial force field using the training set. This can be done by directly fitting parameters to reproduce QM energies and forces.
3. Iterative Loop:
- Step 1 - Run Dynamics: Perform molecular dynamics simulations using the current force field parameters. To ensure adequate sampling of the potential energy surface, use Boltzmann sampling at an elevated temperature (e.g., 400 K) [2].
- Step 2 - Sample New Conformations: Extract new, statistically independent conformations from the MD trajectory.
- Step 3 - Compute New QM Data: Perform QM single-point energy and force calculations on these new conformations.
- Step 4 - Augment Dataset: Add the new QM data (conformations, energies, forces) to the existing training dataset.
- Step 5 - Re-optimize Parameters: Refit the force field parameters against the newly augmented training dataset.
- Step 6 - Validate: Compute the error of the newly parameterized force field on the held-out validation set.
4. Convergence Check:
- The iterative process is terminated when the error on the validation set converges to a minimum, indicating that further iterations would lead to overfitting. This is a key differentiator from earlier methods that lacked robust validation [2].
Protocol 2: Surrogate Model-Assisted Optimization (SMAOpt)
This protocol, based on [4], focuses on dramatically speeding up the optimization process by replacing costly MD simulations with a machine learning model.
1. Define Feasible Parameter Space:
- Identify the force field parameters to be optimized (e.g., Lennard-Jones parameters σ and ε for carbon and hydrogen atoms).
- Set physically reasonable lower and upper bounds for each parameter to ensure numerical stability and physical meaning [4].
2. Generate Training Data for the Surrogate Model:
- Sample hundreds to thousands of parameter sets from the defined feasible space. Sampling strategies can include grid-based or random sampling.
- For each unique parameter set, run a full MD simulation to compute the target property (e.g., the bulk-phase density of n-octane at 293.15 K and 1 bar). This step is computationally expensive but is a one-time cost.
3. Train and Validate the ML Surrogate Model:
- Using the generated data (parameters -> target property), train a machine learning model. Model selection (e.g., Linear Regression, Random Forest, Neural Network) and hyperparameter tuning are critical.
- The model's performance should be evaluated using metrics like Mean Absolute Percentage Error (MAPE) and the coefficient of determination (R²) against a test set [4]. Neural networks have been shown to be effective for this task.
4. Execute the Optimization Loop:
- Integrate the trained and validated surrogate model into an optimization workflow (e.g., FFLOW). In each iteration, instead of running an MD simulation, the surrogate model is queried to predict the target property for a given parameter set.
- The optimizer uses these fast predictions to guide the parameter search. The modularity of this approach allows for the surrogate model to be reused in different optimization contexts [4].
5. Final Validation:
- Once the optimization loop converges, the final optimized parameter set should be validated by running an actual MD simulation and comparing the results to the surrogate model's prediction to ensure fidelity.
Diagram 1: Surrogate Model-Assisted Optimization (SMAOpt) workflow. The costly MD simulations are a one-time cost for training, while the fast ML surrogate model is used inside the iterative loop.
Application Notes and Performance Benchmarks
Iterative optimization protocols have been successfully applied to a range of challenging problems, demonstrating their efficacy and accuracy.
Table 2: Performance Benchmarks of Iteratively Optimized Force Fields
| Force Field / Method | System / Application | Key Results and Performance |
|---|---|---|
| Iterative Protocol with Validation [2] | Tri-alanine peptide; 31 photosynthesis cofactors. | Boltzmann sampling at 400 K enabled fitting on a rugged potential energy surface. Successfully generated a parameter library for complex cofactors. |
| ByteFF (GNN-based) [3] | Drug-like molecules from ChEMBL and ZINC20 databases. | State-of-the-art performance predicting relaxed geometries, torsional profiles, and conformational energies/forces across a broad chemical space. |
| SMAOpt with Neural Network [4] | n-Octane bulk density and conformational energies. | Reduced optimization time by a factor of ≈20 while producing force fields of similar quality to the standard method. |
| QM-to-MM Mapping Protocols [5] | Small organic molecules (liquid properties). | Best model used only 7 fitting parameters, achieving errors of 0.031 g cm⁻³ (density) and 0.69 kcal mol⁻¹ (heat of vaporization) vs. experiment. |
Notes for Practitioners:
- Data Quality and Diversity: The performance of any iterative method, especially those driven by ML, is contingent on the quality and diversity of the training data. The use of large, diverse datasets (e.g., 2.4 million molecular fragments) is critical for ensuring transferability [3].
- Validation is Non-Negotiable: The use of a separate validation set is the most reliable way to ensure that the optimized force field generalizes well and is not overfitted to the specific configurations in the training set [2].
- Balancing Cost and Accuracy: The choice between running full QM/MD simulations at every iteration versus using a surrogate model depends on the available computational resources and the number of optimization parameters. For complex, multi-parameter optimizations, the initial investment in training a surrogate model often yields significant long-term savings [4].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Software and Resources for Iterative Force Field Development
| Tool / Resource | Type | Primary Function and Application |
|---|---|---|
| FFLOW [4] | Software Toolkit | A modular, multiscale force field optimization workflow that facilitates iterative parameter refinement against multiple target properties. |
| QUBEKit [5] [6] | Software Suite | Automates the derivation of force field parameters from quantum mechanical calculations (QM-to-MM mapping), reducing the number of parameters needing experimental fitting. |
| ByteFF [3] | Data-Driven Force Field | An Amber-compatible force field for drug-like molecules whose parameters are predicted by a graph neural network trained on a massive QM dataset. |
| OpenMM | Molecular Dynamics Engine | A high-performance, open-source toolkit for MD simulations, often used as a backend for running the dynamics steps in iterative loops. |
| geomeTRIC [3] | Optimizer | A geometry optimization code used in conjunction with QM software to generate optimized molecular structures and Hessians for training data. |
| OpenFF [3] | Force Field & Infrastructure | A family of force fields and open-source software designed for small molecule drug discovery, providing tools and benchmarks. |
| Espaloma [3] | Graph Neural Network Model | A proof-of-concept GNN for end-to-end force field parameterization, paving the way for models like ByteFF. |
Diagram 2: Generic iterative force field optimization workflow, integrating key tools and the critical validation step.
Molecular dynamics (MD) simulations provide atomic-level insights into the behavior of biological systems, playing a pivotal role in modern computational drug discovery. The accuracy of these simulations hinges on the underlying force field—a mathematical model that describes the potential energy surface of a molecular system. Despite advances in computational power, a fundamental trade-off persists: quantum mechanical (QM) methods offer high accuracy but are computationally prohibitive for large systems and long timescales, while traditional molecular mechanics (MM) force fields offer speed but often lack the precision required for reliable predictions. This application note examines contemporary strategies for bridging this accuracy gap, with a specific focus on iterative optimization training procedures for force field development. We frame these methodologies within the context of drug development, highlighting protocols and resources that enable researchers to leverage these advances in their workflows.
The Force Field Landscape: From Molecular Mechanics to Neural Network Potentials
The choice of potential energy model dictates the balance between computational speed and physical accuracy in MD simulations. Two dominant paradigms are evolving in parallel.
Data-Driven Molecular Mechanics Force Fields
Conventional MM force fields, such as GAFF and OPLS, decompose potential energy into analytical terms for bonded and non-bonded interactions. Their functional forms are computationally efficient but can suffer from inaccuracies due to oversimplification. The field is now shifting towards data-driven parameterization to expand coverage of chemical space. For instance, ByteFF represents a modern MM force field developed by training an edge-augmented, symmetry-preserving graph neural network (GNN) on a massive QM dataset encompassing 2.4 million optimized molecular fragment geometries and 3.2 million torsion profiles [3]. This approach maintains the computational efficiency of the Amber/GAFF functional form while achieving superior accuracy in predicting geometries, torsional profiles, and conformational energies by learning parameters directly from QM data [3].
Neural Network Potentials (NNPs)
In contrast, NNPs represent a more radical departure, using machine learning to map atomic configurations directly to energies and forces, bypassing pre-defined functional forms. They offer near-QM accuracy with a significant reduction in computational cost. Recent breakthroughs, such as Meta's Open Molecules 2025 (OMol25) dataset and associated models like eSEN and the Universal Model for Atoms (UMA), have dramatically advanced the state of the art [7] [8]. The OMol25 dataset provides over 100 million DFT calculations at the ωB97M-V/def2-TZVPD level of theory, covering an unprecedented diversity of chemical structures, including biomolecules, electrolytes, and metal complexes [7]. Models trained on this dataset demonstrate performance that matches high-accuracy DFT on standard benchmarks [7].
Table 1: Comparison of Modern Force Field Development Strategies
| Development Strategy | Key Example | Underlying Methodology | Key Advantage | Applicability in Drug Discovery |
|---|---|---|---|---|
| Data-Driven MMFF | ByteFF [3] | GNN-trained on expansive QM dataset of drug-like fragments | Amber compatibility; High computational efficiency; Broad chemical space coverage | High: Directly parameterized for drug-like molecules |
| General-Purpose NNP | OMol25 Models (eSEN, UMA) [7] [8] | Neural network potentials trained on massive, diverse dataset (OMol25) | Near-DFT accuracy across expansive chemical space | High: Covers biomolecules, protein-ligand interfaces, and metal complexes |
| Specialized NNP | EMFF-2025 [9] | Transfer learning from a pre-trained model for specific material class (energetic materials) | DFT-level accuracy for specialized applications with minimal data | Medium: Framework can be adapted for specific drug classes |
| Multireference NNP | WASP [10] | Integrates multiconfiguration QM with ML-potentials for transition metals | Accurate description of complex electronic structures (e.g., in catalysts) | Medium: Critical for metalloenzymes and organometallic drug candidates |
The Iterative Optimization Framework for Force Field Development
A powerful paradigm emerging in force field development is the iterative optimization procedure, which closes the loop between simulation and QM data generation to achieve self-consistent refinement. This framework directly addresses the challenge of creating accurate and generalizable force fields.
Conceptual Workflow
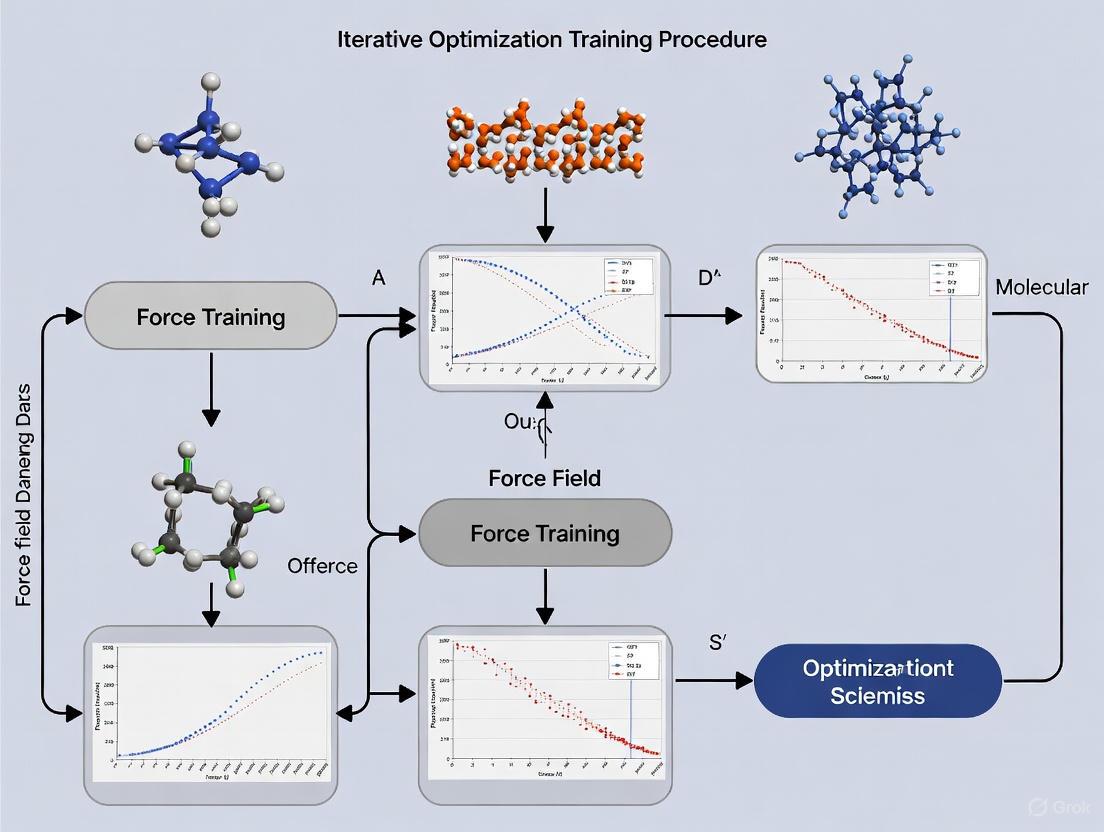
The core of the iterative approach involves a cyclic process of parameter optimization, molecular dynamics sampling, quantum mechanical validation, and dataset expansion [2]. This workflow ensures that the force field is continuously refined against a growing and relevant set of QM data, preventing overfitting to a static initial dataset and improving transferability across conformational space.
The following diagram illustrates the self-correcting and expanding nature of this workflow:
Protocol: Automated Iterative Force Field Parameterization
This protocol outlines the steps for implementing an iterative parameterization procedure, as demonstrated in recent research [2].
Objective: To automatically generate a highly accurate, system-specific force field by iteratively refining parameters against a dynamically expanding QM dataset, using a validation set to prevent overfitting and determine convergence.
Materials and Software:
- Quantum Chemistry Software: ORCA, Gaussian, or PSI4 for QM energy and force calculations.
- MD Engine: OpenMM, NAMD [11], or LAMMPS [12] for running dynamics.
- Parameter Optimization Script: A custom script (e.g., in Python) to adjust force field parameters to minimize the difference between MM and QM energies/forces.
- Initial Structures: 3D coordinates of the target molecule(s) in a standard format (PDB, MOL2).
Procedure:
- Initialization:
- Generate an initial set of diverse molecular conformations for the target system (e.g., via conformational search or high-temperature MD).
- Perform QM single-point energy (and optionally, force) calculations on these conformations to create the initial training dataset.
- Randomly split the data into a training set (e.g., 80%) and a validation set (e.g., 20%).
Parameter Optimization Loop:
- Step 1: Optimize Parameters. Fit the MM force field parameters (e.g., bond, angle, torsion force constants, and equilibrium values) to the current training dataset. The loss function is typically the weighted sum of squared differences between MM and QM energies and forces.
- Step 2: Sample with New Parameters. Run an MD simulation (e.g., Boltzmann sampling at a relevant temperature like 400 K) using the newly optimized parameters to explore new regions of conformational space.
- Step 3: QM Calculation on New Samples. Select new, geometrically distinct conformations from the MD trajectory and compute their QM energies and forces.
- Step 4: Expand Dataset and Validate. Add the new QM calculations to the training dataset. Crucially, evaluate the current force field's accuracy on the held-out validation set.
Convergence Check:
- Monitor the error on the validation set. The procedure is considered converged when the validation error plateaus or falls below a pre-defined threshold over several iterations.
- If the validation error begins to increase significantly while the training error decreases, this indicates overfitting, and the procedure from the previous iteration should be used.
Output:
- The final, validated set of force field parameters in a standard format (e.g., Amber .frcmod or CHARMM .str files) compatible with major MD engines.
Applications: This protocol has been successfully applied to fit custom force fields for complex systems such as tri-alanine peptides and a library of 31 photosynthesis cofactors [2], demonstrating its utility for achieving superior accuracy compared to general force fields.
Practical Implementation and Tools for Researchers
Transitioning from theoretical advances to practical application requires robust software tools and accessible platforms.
Scalable Simulation with ML Potentials
The integration of MLIPs into established MD workflows is key to their adoption. The ML-IAP-Kokkos interface for LAMMPS enables this by providing a streamlined pathway to run PyTorch-based models at scale [12]. The core steps involve:
- Implementing a Python class that inherits from
MLIAPUnifiedand defines thecompute_forcesfunction. - Serializing the model and its weights into a
.ptfile. - Configuring LAMMPS to use the
mliap unifiedpair style, pointing to this model file [12]. This interface handles efficient data transfer between LAMMPS and the PyTorch model, leveraging GPU acceleration for large-scale simulations [12].
Accessibility for Experimentalists
For researchers seeking to utilize MD simulations without deep computational expertise, tools like drMD lower the barrier to entry. drMD is an automated pipeline built on the OpenMM toolkit that requires only a single configuration file to set up and run publication-quality simulations, including enhanced sampling through metadynamics [13]. It automates system setup, solvation, and minimization, and includes features like real-time progress updates and error recovery, making MD more accessible to a broader scientific audience [13].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Software and Data Resources for Modern Force Field Applications
| Resource Name | Type | Function in Research | Access/URL |
|---|---|---|---|
| OMol25 Dataset [7] [8] | Training Data | Massive, high-accuracy QM dataset for training or benchmarking general-purpose NNPs. | Meta FAIR |
| ByteFF [3] | Data-Driven MMFF | Amber-compatible force field for drug-like molecules, offering broad chemical space coverage. | ByteDance Research |
| WASP [10] | Method/Algorithm | Enables accurate ML potentials for transition metal systems by ensuring wavefunction consistency. | GitHub (/GagliardiGroup/wasp) |
| ML-IAP-Kokkos [12] | Software Interface | Connects PyTorch ML models to LAMMPS for scalable, GPU-accelerated MD simulations. | LAMMPS Distribution |
| drMD [13] | Simulation Pipeline | Automated workflow for running MD simulations in OpenMM, designed for non-experts. | GitHub (/wells-wood-research/drMD) |
| NAMD [11] | MD Simulation Engine | High-performance, parallel MD code for simulating large biomolecular systems. | University of Illinois |
Comparative Analysis of Strategic Approaches
The choice between an iterative MMFF and a general-purpose NNP is not trivial and depends on the research goals. The following diagram maps the decision-making logic for selecting the appropriate strategy:
Decision Workflow Explanation:
- For common, drug-like molecules, a pre-trained, data-driven MMFF like ByteFF is often the most efficient choice, offering an excellent balance of accuracy, speed, and compatibility with existing drug discovery pipelines [3].
- If the system is not well-represented in standard MMFFs, the next consideration is reactivity. For simulating reactions or systems with transition metals (common in catalysts and metalloenzymes), a specialized NNP like those generated by the WASP protocol is necessary to accurately capture complex electronic structures [10].
- For non-reactive but chemically diverse systems, the choice hinges on the trade-off between accuracy and setup cost. A general-purpose NNP (e.g., models trained on OMol25) provides near-DFT accuracy out-of-the-box for a vast range of molecules [7]. However, if the highest possible accuracy for a specific molecule is required and one is willing to invest computational resources in setup, then an iterative MMFF parameterization can yield a bespoke, highly accurate force field [2].
The gap between quantum mechanical accuracy and scalable molecular dynamics is closing rapidly, driven by data-centric and iterative methodologies. The emergence of massive, high-quality datasets like OMol25, coupled with sophisticated GNNs and iterative optimization frameworks, provides researchers with a powerful toolkit. Whether through the use of refined, data-driven molecular mechanics force fields or the direct application of neural network potentials, these advances enable more reliable predictions of molecular behavior, binding affinities, and dynamical properties. For drug development professionals, adopting these strategies and tools promises to enhance the reliability of computational screens and mechanistic studies, ultimately accelerating the journey from target identification to viable therapeutic candidates.
The development of accurate molecular mechanics force fields is foundational to reliable molecular dynamics (MD) simulations in computational chemistry and drug discovery. These force fields, which approximate the potential energy surface of a system, enable the study of biomolecular processes at an atomistic level. However, creating a robust force field is fraught with challenges, primarily stemming from the interdependence of parameters, the limited transferability of parameters across diverse chemical spaces, and the prohibitive computational cost associated with high-quality quantum mechanical (QM) reference data generation. These issues are deeply intertwined; optimizing one parameter in isolation can inadvertently degrade the accuracy of another, and fitting to a narrow set of chemical motifs often results in poor performance for novel molecular structures. This application note details these core challenges and presents contemporary, iterative optimization protocols designed to address them, providing researchers with actionable methodologies for next-generation force field development.
The table below summarizes the core challenges, their impact on force field development, and the quantitative evidence as documented in recent literature.
Table 1: Core Challenges in Force Field Development
| Challenge | Impact on Force Field Development | Evidence from Literature |
|---|---|---|
| Parameter Interdependence | Non-bonded parameters (charges, LJ) and torsional bonded parameters are co-dependent; optimizing them independently leads to inaccuracies in the potential energy surface. [14] | Using standard OPLS-AA bonded parameters with bespoke QUBE nonbonded terms limited accuracy, necessitating refitting of torsional parameters. [14] |
| Limited Transferability | Force fields parameterized on small molecule fragments or pure liquid properties fail to accurately predict the behavior of complex biomolecules or chemical mixtures. [15] [16] | Force fields trained only on pure liquid densities (ρl) and enthalpies of vaporization (ΔHvap) showed systematic errors in predicting solvation free energies and mixture properties. [15] |
| High Computational Cost | Generating QM reference data for torsion scans and Hessian matrices for large molecules or expansive chemical spaces is computationally prohibitive, acting as a major bottleneck. [17] [3] | A single flexible torsion scan using traditional QM methods can take days. Generating a dataset of 3.2 million torsion profiles at the B3LYP-D3(BJ)/DZVP level requires massive computational resources. [17] [3] |
Iterative Optimization Protocols
To overcome the challenges outlined above, the field is moving towards automated, iterative optimization procedures that simultaneously refine multiple parameter types against diverse and expansive datasets.
Protocol 1: Bayesian Optimization with the BICePs Algorithm
Bayesian Inference of Conformational Populations (BICePs) is a refinement algorithm that reconciles simulated ensembles with sparse or noisy experimental observables by sampling the full posterior distribution of conformational populations and experimental uncertainty. [18]
Objective: To optimize force field parameters against ensemble-averaged experimental measurements while automatically accounting for uncertainty and systematic error.
Workflow Overview:
Detailed Procedure:
Define the Prior and Likelihood:
- Prior (
p(X)): Obtain a theoretical model of conformational state populations, typically from an MD simulation using an initial force field. [18] - Likelihood (
p(D|X,σ)): Construct a function quantifying the agreement between forward model predictionsf(X)and experimental dataD. A Gaussian likelihood is common, but BICePs can employ more robust models like the Student's-t distribution to automatically detect and down-weight outliers and data points subject to systematic error. [18]
- Prior (
Sample the Posterior Distribution:
- Use Markov Chain Monte Carlo (MCMC) sampling to explore the Bayesian posterior distribution
p(X,σ|D), which represents the conformational populations and uncertainty parameters given the experimental data. [18] - Employ a replica-averaged forward model to approximate ensemble averages, ensuring no adjustable regularization parameters are needed. [18]
- Use Markov Chain Monte Carlo (MCMC) sampling to explore the Bayesian posterior distribution
Compute the BICePs Score and Optimize:
- Calculate the BICePs score, a free energy-like quantity that reflects the total evidence for a model. [18]
- Use variational minimization to optimize force field parameters by minimizing the BICePs score. This process leverages the first and second derivatives of the score for efficient convergence. [18]
Key Research Reagents:
Table 2: Essential Components for BICePs Optimization
| Item | Function / Description |
|---|---|
| Initial Force Field & MD Engine | Provides the initial conformational prior, p(X). (e.g., CHARMM, AMBER, OpenMM). |
| Experimental Observables (D) | Sparse or noisy ensemble-averaged data for refinement (e.g., NMR J-couplings, scalar couplings, distance measurements). |
| BICePs Software Package | Implements the core algorithm for posterior sampling and score calculation. [18] |
| Gradient-Based Optimizer | A variational optimization algorithm that minimizes the BICePs score. |
Protocol 2: Active Learning-Driven Force Field Optimization
This protocol leverages machine learning and active learning to iteratively expand force field coverage across a vast chemical space, directly addressing transferability and computational cost.
Objective: To generate a highly accurate and transferable force field for drug-like molecules by creating a massive, diverse QM dataset and training a graph neural network (GNN) to predict parameters.
Workflow Overview:
Detailed Procedure:
Dataset Construction and Molecular Fragmentation:
- Curate a diverse set of drug-like molecules from databases like ChEMBL and ZINC. [3]
- Use a graph-expansion algorithm to cleave molecules into smaller fragments (e.g., <70 atoms). This preserves local chemical environments and makes QM calculations feasible. Cap cleaved bonds with methyl groups or other linkers to maintain valency. [3]
High-Throughput QM Calculations:
- Perform quantum chemical calculations on the generated fragments to create two primary datasets:
- Optimization Dataset: Geometry optimize each fragment and compute the analytical Hessian matrix at a level of theory such as B3LYP-D3(BJ)/DZVP. This dataset is used to fit bond and angle parameters. [3]
- Torsion Dataset: Perform flexible torsion scans on all non-ring rotatable bonds in the fragments at the same QM level. This involves fixing the dihedral angle and optimizing all other degrees of freedom to generate a relaxed potential energy profile. [3]
- Perform quantum chemical calculations on the generated fragments to create two primary datasets:
Graph Neural Network Training:
- Train a symmetry-preserving Graph Neural Network (GNN) on the massive QM dataset. [3]
- The model learns to map the molecular graph (atoms and bonds) directly to all MM force field parameters (bonded and non-bonded) simultaneously. [3]
- Employ a differentiable partial Hessian loss to ensure the predicted parameters accurately reproduce the QM vibrational frequencies. [3]
Validation and Iteration:
- Validate the resulting force field (e.g., ByteFF) on benchmark datasets by comparing its performance against existing force fields in predicting relaxed geometries, torsional energy profiles, and conformational energies. [3]
- The model can be iteratively refined by incorporating new molecules that explore under-represented regions of chemical space, following an active learning paradigm.
Key Research Reagents:
Table 3: Essential Components for Data-Driven Force Field Development
| Item | Function / Description |
|---|---|
| Chemical Databases (ChEMBL, ZINC) | Source for diverse, drug-like molecular structures. [3] |
| Fragmentation Software (e.g., RDKit) | Tools to systematically cleave large molecules into chemically meaningful fragments. |
| Quantum Chemistry Software | Software (e.g., Gaussian, Q-Chem, ORCA) to generate high-quality reference data (optimized geometries, Hessians, torsion scans). |
| GNN Framework (e.g., PyTorch Geometric) | Framework for building and training the graph neural network model for parameter prediction. |
The challenges of parameter interdependence, transferability, and computational cost are significant but not insurmountable. The protocols detailed herein—Bayesian optimization with BICePs for refining parameters against experimental data, and active learning with GNNs for generating transferable, high-coverage force fields—represent the cutting edge in moving beyond traditional, manual parameterization. By adopting these iterative, data-driven, and automated approaches, researchers can develop more robust and accurate force fields. This will enhance the predictive power of molecular simulations, ultimately accelerating progress in fields like drug discovery and materials science.
In computational chemistry and materials science, a force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or molecules [1]. Force fields are essential for molecular dynamics and Monte Carlo simulations, enabling the study of system behavior and properties from the atomistic level up to micrometre scales [19] [1]. The core challenge in force field development lies in the trade-off between computational efficiency and accuracy, particularly in modeling complex processes like chemical reactions. This review provides a comprehensive overview of the three main classes of force fields—Classical, Reactive, and Machine Learning potentials—framed within the context of iterative optimization training procedures. We present detailed protocols for their application and evaluation, supporting ongoing research in automated force field development.
Classification and Core Characteristics of Force Fields
Comparative Analysis of Force Field Types
The table below summarizes the fundamental characteristics of the three primary force field types, highlighting their differing parameter complexities, applicabilities, and computational demands.
Table 1: Key Characteristics of Major Force Field Types
| Feature | Classical Force Fields | Reactive Force Fields (ReaxFF) | Machine Learning Potentials (MLIPs) |
|---|---|---|---|
| Typical Number of Parameters | 10 - 100 [19] [20] | 100 - 500 [19] [20] | Can exceed 100,000 (data-dependent) [19] |
| Parameter Interpretability | High (clear physical meaning) [19] [20] | Moderate (mix of physical and empirical terms) [19] [20] | Low ("black box" models) [19] |
| Ability to Model Bond Breaking/Formation | No (fixed bonds) [21] [1] | Yes (via bond order) [21] [19] | Yes (trained on reactive data) [22] |
| Computational Cost | Low [19] | High (~30x classical) [21] | Variable (high training, lower inference) [22] |
| Primary Application Scope | Non-reactive molecular dynamics, structural properties [19] [1] | Chemical reactions, combustion, material failure [21] [19] | Complex, system-specific PES with near-DFT accuracy [22] |
| Key Functional Forms | Harmonic bonds, Lennard-Jones, Coulomb [1] | Bond-order dependent potentials [21] [19] | Neural networks, Gaussian processes [19] |
Detailed Force Field Descriptions
Classical Force Fields: These are the most established type, calculating a system's energy using simplified physical potential functions. The total energy is typically a sum of bonded terms (bond stretching, angle bending, dihedral torsions) and non-bonded terms (van der Waals described by Lennard-Jones potential and electrostatic interactions described by Coulomb's law) [1]. Their functional forms are simple and numerically efficient, making them suitable for simulating large systems over long timescales. However, their fixed bonding topology prevents them from simulating chemical reactions [21] [19].
Reactive Force Fields (ReaxFF): ReaxFF addresses the limitation of classical force fields by introducing a bond-order formalism, which dynamically describes the strength of a bond based on the instantaneous interatomic distance [21] [19]. This allows bonds to break and form during a simulation. While this capability is powerful, it comes at a cost: the potential function contains a large number of additional energy terms (e.g., for over-coordination, lone pairs, and conjugation), making it computationally expensive—about 30 times slower than a comparable classical force field simulation [21]. Its parametrization is complex, involving a mix of physical and empirical terms [19].
Machine Learning Potentials (MLIPs): MLIPs represent a paradigm shift. Instead of using pre-defined physical equations, they use machine learning models (e.g., neural networks) to learn the relationship between atomic configurations and the system's energy and forces from reference quantum mechanical (QM) data [22]. This allows them to achieve accuracy close to the QM method they were trained on, while being much faster for molecular dynamics simulations. A key challenge is their lack of interpretability and their tendency to be unstable when simulating configurations far outside their training data [22].
Iterative Optimization Workflow in Force Field Development
The development of accurate and reliable force fields relies heavily on robust iterative optimization procedures. These workflows are designed to systematically refine force field parameters to minimize the discrepancy between simulated properties and target data, which can come from both QM calculations and experimental measurements.
Diagram 1: Generalized Parameter Optimization Workflow
Protocol: Gradient-Based Parameter Optimization
This protocol is adapted from the GROW (GRadient-based Optimization Workflow) methodology for automating force field parameterization [23].
Problem Definition:
- Define the loss function, ( F(x) ), which quantifies the difference between simulated and target data. A common form is a weighted sum of squared relative errors: ( F(x) = \sum{i=1}^{n} wi \left( \frac{fi^{exp} - fi^{sim}(x)}{fi^{exp}} \right)^2 ) where ( x ) is the parameter vector, ( n ) is the number of properties, ( fi^{exp} ) is the target value, ( fi^{sim} ) is the simulated value, and ( wi ) is the weight [23].
- Select target properties (e.g., densities, energies, elastic constants) and their reference values.
Initialization:
- Provide an initial parameter vector ( x_0 ). This starting point should be reasonably close to the expected minimum for faster convergence [23].
Iterative Optimization Loop:
- Simulation & Analysis: Run molecular simulations (e.g., Molecular Dynamics or Monte Carlo) using the current parameter set ( xk ). Calculate the physical properties ( fi^{sim}(x_k) ) from the simulation trajectories [23].
- Loss Evaluation: Compute the value of the loss function ( F(x_k) ) [23].
- Convergence Check: If a stopping criterion is met (e.g., ( F(x_k) \leq \tau ), where ( \tau ) is a predefined tolerance, or the change in ( F(x) ) is negligible), exit the loop and finalize the parameters [23].
- Parameter Update: If not converged, calculate the gradient of the loss function with respect to the parameters. Use a gradient-based optimization algorithm (e.g., Steepest Descent, Conjugate Gradient, or quasi-Newton methods) to determine a search direction and update the parameters to ( x_{k+1} ) [23]. Repeat the loop.
Advanced Optimization: Machine Learning Force Field Pre-Training
A modern approach to optimizing MLIPs involves a two-stage pre-training and fine-tuning strategy to improve stability and data efficiency [22].
Diagram 2: MLIP Pre-Training and Fine-Tuning Workflow
Pre-Training Stage:
- Data Generation: Use "rattling" methods to generate a large and diverse set of molecular configurations, including high-energy, unphysical states. This comprehensively samples the potential energy surface (PES) [22].
- Training: Train the MLIP on this large dataset, using energies and forces labeled by a simple, non-reactive classical force field. This provides a physically reasonable, though not highly accurate, PES with correct limiting behaviors, "smoothing" the landscape and preventing "holes" that cause simulation instability [22].
Fine-Tuning Stage:
- Data Curation: Compile a smaller, targeted set of high-quality ab initio (e.g., DFT) data that focuses on chemically relevant regions (equilibrium structures, transition states) [22].
- Fine-Tuning: Continue training the pre-trained MLIP on this high-quality dataset. This stage specializes the model for chemical accuracy in the relevant regions while maintaining the stability learned during pre-training [22].
Experimental Protocols for Force Field Evaluation
Protocol: Benchmarking Energetics and Elastic Properties
This protocol outlines a high-throughput method for evaluating the transferability of classical and reactive potentials, based on the methodology from the NIST interatomic potential repository project [24].
Structural Data Acquisition:
High-Throughput Calculation Setup:
- Use a computational framework like
MPinterfacesto automate the generation of input files for simulation packages like LAMMPS [24]. - For each material and force field pair, set up calculations to compute:
a. Energy vs. Volume: Perform structural relaxations over a range of volumes to obtain the equation of state.
b. Elastic Constants: Use the built-in LAMMPS
compute elasticcommand (applying small strains, e.g., 10⁻⁶) to calculate the full elastic constant matrix (C₁₁, C₁₂, C₄₄, etc.) [24].
- Use a computational framework like
Execution and Analysis:
- Run the high-throughput calculations on a compute cluster.
- For energetics: Calculate the cohesive energy and, for multi-component systems, generate convex hull plots to assess phase stability using tools like
pymatgen[24]. - For elastic properties: Extract the elastic constants and moduli from the LAMMPS output.
Benchmarking and Validation:
- Compare the force field results with reference data from density functional theory (DFT) (e.g., from the Materials Project) and experimental data, where available [24].
- Perform statistical analysis (e.g., Principal Component Analysis - PCA) on the relative errors to identify correlated deficiencies in the force field predictions [24].
Protocol: Simulating Bond Dissociation and Material Failure with a Reactive Force Field
This protocol describes how to simulate bond-breaking processes using a reactive force field, specifically employing the IFF-R method which uses Morse potentials [21].
System Preparation:
- Construct the initial atomic structure (e.g., a polymer chain, carbon nanotube, or metal crystal).
- For IFF-R, substitute harmonic bond potentials in the force field with Morse potentials for the bonds intended to be reactive. The Morse potential is ( E{Morse} = D{ij} [1 - e^{-\alpha{ij}(r{ij} - r{0,ij})}]^2 ), where ( D{ij} ) is the dissociation energy, ( \alpha{ij} ) controls the width of the potential well, and ( r{0,ij} ) is the equilibrium bond length [21].
Parameterization:
- Obtain ( r_{0,ij} ) from the original harmonic potential or experimental data.
- Set ( D_{ij} ) using experimental bond dissociation energies or high-level QM calculations (e.g., CCSD(T) or MP2) [21].
- Fit ( \alpha_{ij} ) to match the vibrational frequencies (from IR/Raman spectroscopy) or to align the Morse curve with the harmonic curve near equilibrium [21].
Simulation of Mechanical Failure:
- Energy Minimization: First, minimize the energy of the system to a force tolerance (e.g., 10⁻¹⁰ eV/Å) [24].
- Equilibration: Run an NVT or NPT simulation to equilibrate the system at the desired temperature and pressure.
- Deformation: Apply a uniaxial strain to the simulation box at a constant strain rate.
- Data Collection: Monitor the stress tensor, potential energy, and atomic coordinates throughout the deformation. Track the breaking of individual bonds as distances exceed the critical bond length defined by the Morse potential.
Essential Research Reagents and Computational Tools
The table below lists key software, databases, and tools essential for force field development, evaluation, and application.
Table 2: Key Research Reagents and Computational Tools
| Tool / Resource Name | Type | Primary Function | Relevance to Force Field Development |
|---|---|---|---|
| LAMMPS | Software | Molecular Dynamics Simulator | A highly versatile and widely used MD code for evaluating energies, elastic constants, and running reactive simulations with various force fields [24] [21]. |
| Materials Project (MP) | Database | DFT-Computed Material Properties | Provides a vast source of reference data (structures, energies, elastic constants) for force field parameterization and benchmarking [24]. |
| NIST IPR | Database | Interatomic Potential Repository | A curated collection of tested potential parameter files for download, facilitating evaluation and dissemination [24]. |
| GROW | Software | Gradient-based Optimization Workflow | An automated tool for performing iterative, gradient-based optimization of force field parameters against target data [23]. |
| IFF-R | Force Field | Reactive INTERFACE Force Field | An extension of a classical force field using Morse potentials to enable bond breaking with high computational efficiency (~30x faster than ReaxFF) [21]. |
| ReaxFF | Force Field | Reactive Force Field | A bond-order potential for modeling complex chemical reactions in large systems, though with higher computational cost [21] [19]. |
Step-by-Step Workflows and Real-World Application Pipelines
Within the iterative optimization paradigm for modern force field development, the ability to systematically generate, manage, and utilize high-quality quantum chemical (QC) reference data is paramount. The process requires not only robust computational methodologies but also automated, scalable, and reproducible pipelines to handle the vast chemical space of drug-like molecules. The Open Force Field (OpenFF) initiative addresses this need through an integrated software ecosystem, enabling the generation of extensive datasets that are critical for training and validating force field parameters. This ecosystem has been instrumental in the development of force fields like OpenFF Sage (version 2.0.0), which incorporates valence parameters trained against a large database of quantum chemical calculations and improved Lennard-Jones parameters refined using condensed-phase mixture data [25]. This application note details the protocols for leveraging the QCSubmit and QCFractal packages to construct such quantum chemical datasets, providing a practical guide for researchers engaged in force field parameterization.
The Automated Data Generation Ecosystem
The automated data generation workflow is built upon a suite of interoperable software tools, primarily from the QC* suite developed by the Molecular Sciences Software Institute (MolSSI) and the OpenFF toolkit. QCFractal serves as a distributed compute and database platform for quantum chemistry, functioning as the central backbone that manages calculation workloads and results [26] [27]. QCSubmit provides the automated tools for curating molecular datasets and submitting them to a QCFractal instance [28] [27]. Underlying these are QCEngine, which provides a unified interface to various quantum chemistry programs (e.g., Psi4, Gaussian), and QCSchema, which defines a standard for representing quantum chemical data [26]. The culmination of this infrastructure is the Quantum Chemistry Archive (QCArchive), a public repository that, as of June 2022, housed over 98 million molecules and 104 million calculations, representing a massive community resource [26].
The logical workflow and the relationship between these components are illustrated below.
Research Reagent Solutions: Essential Software Tools
The following table details the key software components required to establish an automated quantum chemical data generation pipeline.
Table 1: Essential Software Tools for Automated QC Data Generation
| Tool Name | Primary Function | Key Features | Computation Type |
|---|---|---|---|
| QCSubmit [28] [27] | Dataset Curation & Submission | Creates dataset "factories" (Basic, Optimization, TorsionDrive), validates inputs, handles molecule standardization. | Dataset Preparation |
| QCFractal [26] [27] | Distributed Compute Management | Manages job queues, interfaces with HPC schedulers (SLURM, PBS), orchestrates calculations across multiple clusters. | Job Management & Execution |
| QCEngine [26] | Program Execution Interface | Parses QCSchema, provides unified interface to ~20+ QM/MM programs (Psi4, GAMESS, XTB, etc.). | Calculation Execution |
| QCArchive [26] | Data Storage & Repository | Public/private data storage, ~100M+ calculations, provides API for querying and retrieving datasets. | Data Storage & Access |
| Geometric [29] | Geometry Optimization | Used as the backend for OptimizationDataset calculations, implements efficient optimization algorithms. |
Structure Optimization |
| Psi4 [26] | Electronic Structure Code | Open-source quantum chemistry program; often used as the workhorse for QM calculations in OpenFF pipelines. | Quantum Chemical Calculation |
Core Dataset Types and Generation Protocols
The OpenFF infrastructure supports several types of datasets, each tailored for specific force field parameterization tasks. The quantitative characteristics of these core dataset types are summarized below.
Table 2: Core Quantum Chemical Dataset Types for Force Field Development
| Dataset Type | Force Field Target | Key Outputs | Typical QM Method |
|---|---|---|---|
| TorsionDriveDataset [27] | Torsion parameters | 1D/2D torsion potential energy surfaces, optimized geometries at each grid point. | B3LYP-D3BJ/DZVP [27] |
| OptimizationDataset [29] | Bond, Angle, and Van der Waals parameters | Optimized equilibrium geometries, single-point energies, harmonic frequencies, multipole moments. | B3LYP-D3BJ/DZVP [29] |
| BasicDataset (Single-point) [26] | General benchmarking, charge models | Single-point energies, molecular properties (dipole, quadrupole), optionally Hessians. | Varies by target property |
| Dataset Mixtures (e.g., QCML) [30] | Machine-learned potentials (MLFFs) | Energies, atomic forces, multipole moments, Kohn-Sham matrices for both equilibrium and off-equilibrium structures. | DFT and Semi-empirical |
Protocol: Generating a TorsionDriveDataset
Torsion drive scans are critical for refining the torsional parameters of a force field. The following protocol outlines the steps to create a dataset of torsion scans [27].
- Initialize Factory and Specification: Create a
TorsiondriveDatasetFactoryand define the quantum chemistry specification. TheQCSpecobject specifies the method (e.g.,'B3LYP-D3BJ'), basis set (e.g.,'DZVP'), and program (e.g.,'psi4') [27]. - Define Torsion Scan Workflow: Configure the workflow components. The
ScanEnumeratoris used with aScan1Dcomponent, which specifies the central torsion to scan using a SMARTS pattern (e.g.,"[*:1]-[#6:2]-[#6:3]-[*:4]"for a C-C bond), the scan range (e.g.,(-150, 180)degrees), and the increment (e.g.,30degrees) [27]. - Prepare Input Molecules: Generate the input molecules from SMILES strings or structure files. A
StandardConformerGeneratorcomponent is typically added to the workflow to generate multiple initial conformers for each molecule [27]. - Create and Submit Dataset: Call the
create_datasetmethod of the factory with the prepared molecules, a dataset name, and a description. The resulting dataset object is then submitted to a QCFractal instance (local or public) using thesubmitmethod [27].
Protocol: Generating an OptimizationDataset
Geometry optimization datasets are used for training valence parameters (bonds, angles) and for benchmarking [26] [29].
- Dataset Initialization: Instantiate an
OptimizationDatasetobject, providing adataset_name,description, and other metadata [29]. - Configure Quantum Chemistry: Add one or more quantum chemistry specifications via the
add_qc_specmethod. The specification includes method, basis set, program, and optional settings likeimplicit_solventormaxiter[29]. - Set Optimization Procedure: The dataset uses an
optimization_procedure, typically with the'geometric'program and a specified convergence set (e.g.,'GAU'for Gaussian-style convergence) [29]. - Add Molecules and Submit: Populate the dataset with molecules using the
add_moleculemethod. Each molecule entry requires a unique index and a 3D structure. Finally, submit the dataset to a QCFractal instance using thesubmitmethod [29].
Data Management, Submission, and the Dataset Lifecycle
Managing the lifecycle of a dataset submission is crucial for reproducibility and tracking. The OpenFF community uses a GitHub-based pipeline (qca-dataset-submission repository) with an integrated lifecycle model [26] [31]. The diagram below illustrates the stages a dataset passes through from submission to archival.
- Submission and Validation: Datasets are submitted via a pull request to the
qca-dataset-submissionrepository. GitHub Actions automatically validate the submission, checking for errors in the dataset structure [31]. - Computation and Error Cycling: Once merged, the dataset is submitted to QCArchive. Calculations are dispatched to HPC resources. A key feature is automated error cycling, where a bot periodically identifies and restarts failed calculations, significantly improving completion rates [26] [31].
- Data Retrieval and Archiving: Completed datasets can be retrieved programmatically using QCSubmit. Once a dataset reaches an acceptable completion level (e.g., >99%), it is manually reviewed and moved to an "Archived/Complete" state [26] [31].
Application Example: Building a Training Set for Alkane Torsions
This practical example demonstrates the creation of a small torsion drive dataset for linear alkanes, a common step in an iterative force field training cycle [27].
This example creates a dataset that will perform a torsion drive around every central C-C bond in ethane, propane, and butane, scanning at 30-degree increments. The resulting energy profiles provide the direct reference data needed to fit and validate the torsional parameters in the force field for these molecules.
The development of accurate molecular force fields represents a cornerstone of reliable computational research in material science and drug design. The process of force-field parameter (FFParam) optimization is an ongoing endeavor, crucial for enhancing the predictive power of molecular simulations [32]. Traditional methodologies often treat parameterization, conformational sampling, and data management as separate challenges. However, a paradigm shift is underway towards integrated, iterative workflows. This protocol details the application of such an iterative loop, which synergistically combines advanced parameter optimization algorithms, enhanced conformational sampling techniques, and strategic data augmentation to accelerate and improve the force field development process. By framing these elements as interconnected components of a cyclic refinement process, researchers can achieve more robust, accurate, and transferable molecular models.
The following diagram illustrates the core iterative loop that integrates parameter fitting, conformational sampling, and data augmentation for force field development.
Experimental Protocols
Protocol 1: Force Field Parameter Optimization
This protocol outlines the steps for optimizing force field parameters using a combination of metaheuristic algorithms and machine learning (ML) surrogate models to improve efficiency.
Research Reagent Solutions
Table 1: Key Computational Reagents for Parameter Optimization
| Reagent/Tool | Type | Primary Function |
|---|---|---|
| Simulated Annealing (SA) [33] | Algorithm | Explores parameter space widely to escape local minima. |
| Particle Swarm Optimization (PSO) [33] | Algorithm | Efficiently converges towards optimal regions based on swarm intelligence. |
| Concentrated Attention Mechanism (CAM) [33] | Algorithm | Prioritizes fitting to key, representative data points (e.g., transition states). |
| Machine Learning Surrogate Model [32] | Model | Replaces computationally expensive MD calculations for rapid evaluation. |
| Bayesian Inference of Conformational Populations (BICePs) [18] | Algorithm | Refines parameters against noisy, ensemble-averaged experimental data. |
Detailed Methodology
- Objective Function Definition: Define the objective function as the weighted sum of squared differences between simulation outcomes and target data (e.g., quantum mechanical energies, experimental densities, NMR observables) [32] [18].
- Hybrid Algorithm Setup: Implement a hybrid optimizer combining Simulated Annealing (SA) and Particle Swarm Optimization (PSO).
- Initialize a population of parameter sets.
- Allow SA to perform broad exploration in early stages, accepting some suboptimal moves to avoid local minima.
- Use PSO to refine promising parameter sets, where each particle adjusts its position based on its own experience and the swarm's best-found solution [33].
- Integration of Concentrated Attention Mechanism (CAM): Modify the objective function to assign higher weights to errors associated with critical molecular configurations, such as transition states or optimally bonded structures, to improve the physical accuracy of the refined force field [33].
- Surrogate Model Deployment: For iterative protocols requiring thousands of evaluations, train an ML surrogate model (e.g., a neural network) on initial molecular dynamics (MD) data. This model learns the mapping from force field parameters to target properties, replacing the need for full MD simulations in subsequent optimization steps, which can speed up the process by a factor of ~20 [32].
- Bayesian Refinement (Optional): For integration with experimental ensemble data, use the BICePs algorithm. Minimize the BICePs score, which accounts for uncertainty in both the experimental data and the forward model, to robustly optimize parameters against sparse or noisy observables [18].
Protocol 2: Enhanced Conformational Sampling
This protocol describes methods for generating comprehensive conformational ensembles, which are essential for evaluating and training force fields against ensemble-averaged experimental data.
Research Reagent Solutions
Table 2: Key Reagents for Enhanced Conformational Sampling
| Reagent/Tool | Type | Primary Function |
|---|---|---|
| Molecular Dynamics (MD) [34] | Simulation Method | Computes physical trajectories of atoms over time. |
| Generalized Ensemble Methods (GEPS) [35] | Enhanced Sampling | Enhances sampling by modulating system parameters (e.g., charges in ALSD, REST2). |
| Zero-Multipole Summation Method (ZMM) [35] | Electrostatic Calculator | Accelerates electrostatic calculations by assuming local neutrality. |
| Coarse-grained ML Potentials [36] | Machine Learning Force Field | Provides a smoother energy landscape for faster exploration. |
| Generative Models (e.g., Diffusion Models) [36] [34] | AI Sampler | Directly samples diverse, statistically independent structures from the equilibrium distribution. |
Detailed Methodology
- System Preparation: Obtain an initial protein structure from databases like the PDB or from AI-based predictors like AlphaFold2. Prepare the system using standard tools (e.g.,
pdb2gmxin GROMACS) by solvating it in a water box and adding necessary ions. - Sampling Technique Selection:
- Option A (Physics-based MD with Enhanced Sampling): Run MD simulations using packages like GROMACS, AMBER, or OpenMM. Integrate generalized ensemble methods (GEPS) like REST2 to improve sampling efficiency. For large systems, combine GEPS with the Zero-Multipole Summation Method (ZMM) to accelerate electrostatic calculations without introducing significant bias, provided the system is not highly polarized [35].
- Option B (AI-based Generative Models): Use pre-trained generative models like AlphaFlow or UFConf. Input the protein sequence or a single structure, and run the model to generate a diverse set of plausible conformations. This approach bypasses the long timescales of MD by directly sampling the equilibrium distribution [36] [34].
- Option C (Coarse-grained ML Potentials): For specific protein systems, employ a transferable coarse-grained ML potential (e.g., from Majewski et al. or Charron et al.). These potentials, trained on extensive MD data, offer a smoother energy landscape, enabling faster exploration of folding pathways and free energy landscapes [36].
- Ensemble Generation and Analysis: Run the chosen sampling method to produce a trajectory or a set of discrete structures. Analyze the resulting ensemble using metrics like Root Mean Square Fluctuation (RMSF), radius of gyration, or dihedral angle distributions to characterize the conformational landscape.
Protocol 3: Data Augmentation for Surrogate Model Training
This protocol covers the creation of augmented datasets to improve the performance and robustness of ML surrogate models used within the iterative loop.
Research Reagent Solutions
Table 3: Key Reagents for Data Augmentation in Molecular Modeling
| Reagent/Tool | Type | Primary Function |
|---|---|---|
| Classical MD Trajectories [32] | Primary Data Source | Provides initial, physically accurate data for training and augmentation. |
| Generative AI (GANs, Diffusion Models) [37] [36] | Augmentation Tool | Creates realistic synthetic molecular configurations to expand data diversity. |
| Geometric Transformations [37] | Augmentation Technique | Applies rotations, translations, and scaling to existing structures. |
| Noise Injection [37] [38] | Augmentation Technique | Adds random noise to atomic coordinates or simulation conditions to improve model robustness. |
pydub & librosa [38] |
Software Library | Implements various audio/data augmentation strategies (adaptable for molecular data). |
Detailed Methodology
- Initial Data Acquisition: Generate the primary dataset by running a limited number of MD simulations across a carefully chosen range of force field parameters. The target properties from these simulations (e.g., density, conformational energies) form the labels for the surrogate model [32].
- Data Augmentation Execution:
- Geometric Transformations: Apply random rotations and translations to the molecular configurations in the dataset. This ensures the surrogate model is invariant to the global orientation and position of the molecule [37].
- Synthetic Data Generation: Use generative models (e.g., GANs or diffusion models) trained on the primary data to create novel, yet physically plausible, molecular configurations and their corresponding properties. This is particularly valuable for sampling rare events or conformational states [37] [36].
- Parameter Space Perturbation: Slightly perturb the input force field parameters and use the physical force field to quickly generate corresponding new data points, effectively creating more examples around the boundaries of the initial parameter set.
- Noise Introduction: Add small, random noise to the atomic coordinates of structures in the training set. This helps prevent overfitting and makes the resulting surrogate model more tolerant to numerical uncertainties [37] [38].
- Surrogate Model Training and Validation: Train a neural network or other ML model on the combined original and augmented dataset. Validate the model's predictions on a held-out test set of high-fidelity MD simulation results that were not used in training or augmentation [32].
Integrated Application Note
The synergistic integration of these three protocols creates a powerful, accelerated workflow for force field development. The core of this integration is the substitution of the most time-consuming component—direct MD simulation—with an ML surrogate model that has been trained on both physical simulation data and augmented samples [32]. In practice, an initial set of parameters is used to generate a conformational ensemble via Protocol 2. This data is then fed into Protocol 3 to create a robust, augmented dataset for training a fast-executing surrogate model. Protocol 1 then operates using this surrogate model to evaluate candidate parameters, drastically reducing the optimization time. The refined parameters from this cycle can be used to initiate a new, higher-fidelity iteration of the loop, further improving the force field until convergence is achieved. This closed-loop approach, which leverages the strengths of physical simulation, AI-based sampling, and data augmentation, represents a state-of-the-art methodology for tackling the complex challenge of force field parameterization [36].
The development of accurate molecular force fields represents a cornerstone of computational chemistry and drug discovery, enabling the prediction of molecular interactions, stability, and binding affinities. The accuracy of these force fields hinges on the optimization algorithms used to parameterize them. This article explores advanced optimization algorithms—from population-based methods like Particle Swarm Optimization (PSO) to gradient-based techniques and metaheuristics like Simulated Annealing—within the context of iterative optimization training procedures for force field development. We provide a detailed examination of their applications, comparative performance, and practical protocols for implementation, specifically framed for researchers and professionals engaged in computational drug development.
The table below catalogues key resources frequently employed in force field optimization and molecular simulation workflows.
Table 1: Key Research Reagent Solutions for Force Field Optimization and Molecular Simulation
| Item Name | Function/Application | Relevance to Optimization |
|---|---|---|
| Cambridge Structural Database (CSD) [39] | A repository of experimentally determined small molecule crystal structures. | Provides the experimental structural data used as a benchmark for training and validating force fields. |
| Rosetta Molecular Modelling Suite [39] | A comprehensive software platform for macromolecular structure prediction and design. | Implements energy functions (e.g., RosettaGenFF) and algorithms (e.g., GALigandDock) for force field optimization and docking. |
| Neural Network Potentials (NNPs) [40] [41] | Machine learning models trained to approximate quantum-mechanical potential energy surfaces. | Serves as a high-accuracy, computationally efficient target for force field parameterization and validation. |
| Atomic Simulation Environment (ASE) [40] | A set of tools and Python modules for setting up, manipulating, running, visualizing, and analyzing atomistic simulations. | Provides implementations of standard optimizers (L-BFGS, FIRE) and an interface for applying them to molecular systems. |
| geomeTRIC [40] | An optimization library specializing in geometry optimization for molecular systems. | Implements advanced internal coordinates (TRIC) for more efficient and robust convergence to minimum-energy structures. |
| Sella [40] | An open-source package for geometry optimization, including transition states and minima. | Uses internal coordinates and a rational function optimization approach for precise location of equilibrium geometries. |
Comparative Performance of Optimization Algorithms
The selection of an optimization algorithm significantly impacts the success of molecular geometry optimizations. The following table summarizes quantitative performance data for various optimizer and Neural Network Potential (NNP) pairings on a benchmark of 25 drug-like molecules.
Table 2: Optimizer and NNP Performance Benchmarking for Molecular Geometry Optimization [40]
| Optimizer | Neural Network Potential (NNP) | Success Rate (/25) | Avg. Steps to Converge | Minima Found (/25) |
|---|---|---|---|---|
| ASE/L-BFGS | OrbMol | 22 | 108.8 | 16 |
| ASE/L-BFGS | OMol25 eSEN | 23 | 99.9 | 16 |
| ASE/L-BFGS | AIMNet2 | 25 | 1.2 | 21 |
| Sella (internal) | OrbMol | 20 | 23.3 | 15 |
| Sella (internal) | OMol25 eSEN | 25 | 14.9 | 24 |
| Sella (internal) | AIMNet2 | 25 | 1.2 | 21 |
| geomeTRIC (tric) | OMol25 eSEN | 20 | 114.1 | 17 |
| geomeTRIC (tric) | GFN2-xTB | 25 | 103.5 | 23 |
Key Performance Insights:
- AIMNet2 demonstrated exceptional robustness, successfully optimizing all 25 molecules with all tested optimizers. [40]
- Sella with internal coordinates consistently achieved high success rates with low average step counts, indicating high efficiency. [40]
- Algorithms using internal coordinates (e.g., Sella internal, geomeTRIC-tric) generally outperformed their Cartesian-based counterparts in finding true local minima (fewer imaginary frequencies). [40]
- The performance of an optimizer is highly dependent on the specific NNP, underscoring the need for empirical testing. [40]
Application Notes and Experimental Protocols
Protocol 1: Force Field Parameterization via Crystal Structure Discrimination
This protocol details the procedure for optimizing a generalized force field (RosettaGenFF) using small-molecule crystal structures from the Cambridge Structural Database (CSD), as described in the literature. [39]
1. Objective: To simultaneously optimize 444 force field parameters (175 non-bonded and 269 torsional parameters) such that the native crystal lattice arrangements have lower energies than alternative decoy arrangements. [39]
2. Materials and Software:
- Source Data: 1,386 small molecule crystal structures from the CSD. [39]
- Software: Rosetta molecular modelling suite with extended symmetry machinery for handling crystallographic space groups. [39]
- Computing: High-performance computing cluster for intensive sampling (~50 CPU hours for 870 molecules, run thousands of times). [39]
3. Procedure:
- Step 1: Decoy Lattice Generation
- For each of the 1,386 small molecules, run thousands of independent Metropolis Monte Carlo with minimization (MCM) simulations.
- Randomize initial conditions: assign a random crystallographic space group, randomize lattice parameters (cell volume, angles), and randomize the ligand conformation by sampling all rotatable dihedrals. [39]
- In each MCM cycle, perturb one of the following degrees of freedom: molecular translation/rotation, a single dihedral angle, or all lattice lengths/angles. Subsequently, perform minimization on all degrees of freedom. [39]
- Collect the lowest energy conformation from each simulation run, resulting in a vast ensemble of decoy crystal packings for each molecule. [39]
Step 2: Parameter Optimization with dualOptE
- Define an objective function that maximizes the energy gap between the native crystal structure and all generated decoy structures. [39]
- Use the Simplex-search-based dualOptE algorithm to iteratively adjust the 444 force field parameters to optimize this objective. [39]
- Embed thermodynamic data and protein-ligand complex structural data into the objective function to ensure balanced parameterization for both crystal lattices and biomolecular complexes. [39]
Step 3: Iterative Refinement
4. Validation:
- The final optimized force field (RosettaGenFF) is validated by testing its performance on a cross-docking benchmark of 1,112 protein-ligand complexes. A >10% improvement in success rate for recapitulating bound structures was reported. [39]
The following workflow diagram illustrates this iterative parameterization process:
Protocol 2: Iterative Training of Machine Learning Force Fields for Molecular Liquids
This protocol outlines the process for developing a robust Gaussian Approximation Potential (GAP) for a binary solvent mixture, addressing the challenge of scale separation between intra- and inter-molecular interactions. [41]
1. Objective: To create a stable and accurate ML force field for a molecular liquid mixture (e.g., EC:EMC electrolyte) that reproduces thermodynamic properties like density in NPT ensemble simulations. [41]
2. Materials and Software:
- Target System: Binary solvent mixture, e.g., Ethylene Carbonate (EC) and Ethyl Methyl Carbonate (EMC) in a 3:7 molar ratio. [41]
- Reference Data: Energies, forces, and virials computed using an ab initio method (e.g., PBE-D2). [41]
- Software: GAP framework with SOAP descriptors; classical force field (e.g., OPLS) for initial sampling. [41]
3. Procedure:
- Step 1: Initial Data Generation (Failure of Static Sets)
- Use a classical force field (OPLS) to sample diverse molecular configurations across a wide range of densities and temperatures. [41]
- Select uncorrelated configurations from these trajectories and compute their ab initio energies/forces.
- Attempt to train an ML potential on this fixed dataset. Note: Models trained this way often fail in NPT dynamics, leading to unphysical collapse of the density due to poor description of inter-molecular interactions, despite appearing stable in NVT/NVE ensembles. [41]
- Step 2: Iterative Training and Data Augmentation
- Iteration Loop:
- Train a new GAP model on the current training set. [41]
- Run an NPT molecular dynamics simulation using the newly trained GAP.
- Monitor for instabilities (e.g., spontaneous bubble formation, large density fluctuations). [41]
- Extract new configurations from these unstable trajectories and compute their ab initio properties.
- Augment the training set with these new, relevant configurations. [41]
- Data Augmentation Strategies:
- Iteration Loop:
4. Validation:
- The success of the final ML potential is determined by its ability to produce stable, long NPT trajectories and accurately reproduce the density predicted by the reference ab initio method. [41]
The iterative workflow for training a robust ML force field is shown below:
PSO is a population-based stochastic optimization technique inspired by the social behavior of bird flocking. [42]
Core Algorithm:
- Initialization: A swarm of particles is initialized with random positions (( \vec{x}0^i )) and velocities (( \vec{v}0^i )) in the d-dimensional search space. Each particle's best known position (( \vec{p}^i )) is initially its own position. [42]
- Iteration Loop: At each iteration ( t ), for each particle ( i ):
- Velocity Update: ( \vec{V}{t+1}^i = \vec{V}t^i + \varphi1 R1 (\vec{p}t^i - \vec{x}t^i) + \varphi2 R2 (\vec{g}t - \vec{x}t^i) ) where:
- Position Update: ( \vec{x}{t+1}^i = \vec{x}t^i + \vec{V}{t+1}^i )
- Best Position Update: Evaluate the fitness of the new position ( \vec{x}{t+1}^i ). Update ( \vec{p}t^i ) and ( \vec{g}t ) if better positions are found. [42]
Variants and Considerations:
- gbest vs. lbest: In the gbest model, all particles are connected and attracted to the single best solution in the swarm. In the lbest model, particles are influenced only by the best solution within a local neighborhood, which can help prevent premature convergence in multimodal problems. [42]
- Challenges: A known issue is premature convergence, where the swarm gets trapped in a local optimum before finding the global solution. Various parameter tuning and hybridization strategies have been developed to mitigate this. [42]
The development of accurate and transferable molecular force fields represents a central challenge in computational chemistry and drug discovery. The fidelity of these models hinges on their parameterization against diverse and high-quality datasets. An iterative optimization training procedure has emerged as a powerful paradigm for force field development, systematically incorporating data from multiple levels of theory and experiment to achieve robust predictive performance [43] [20]. This procedure cyclically refines parameters by integrating quantum mechanical (QM) data, such as torsion potential energy surfaces, with condensed-phase properties and experimental observables, ensuring the model is not only grounded in first-principles physics but also accurately reproduces real-world behavior [44] [43]. This application note details the protocols for acquiring and integrating these critical data types within an iterative framework, providing researchers with a roadmap for developing next-generation force fields.
Experimental and Computational Protocols
Torsion Drive Parameterization from Quantum Mechanics
Purpose: To derive accurate intramolecular torsional parameters that capture the conformational energy landscape of molecules.
Principle: Torsion drives involve systematically rotating a dihedral angle and computing the single-point energy at each step using quantum mechanical methods. This generates a potential energy surface (PES) that serves as a target for optimizing the torsional parameters (e.g., force constants and phase shifts) in the force field [43] [20].
Procedure:
- Selection of Dihedrals: Identify all unique, rotatable dihedral angles central to the molecule's flexibility and biological activity.
- QM Geometry Optimization: Optimize the molecular geometry at an appropriate level of theory (e.g., DFT with the ωB97X-D functional and def2-SVP basis set).
- Potential Energy Surface Scan: For each target dihedral, constrain the angle and perform a single-point energy calculation at regular intervals (typically every 15-30 degrees) over a 360-degree rotation. Higher-level methods (e.g., DLPNO-CCSD(T)/def2-TZVP) can be used on a subset of points for improved accuracy.
- Energy Fitting: Fit the classical torsional potential function, typically a Fourier series of the form
V(ϕ) = Σ k_n [1 - cos(nϕ - ϕ_n)], to the QM-calculated PES using a least-squares minimization algorithm. Multiple dihedrals are often fit simultaneously to avoid parameter conflicts.
Key Considerations:
- The choice of QM method and basis set balances computational cost and accuracy.
- The fit must accurately reproduce both the energy minima and the barriers to rotation.
- This protocol is automated in toolkits such as QUBEKit and the Open Force Field Initiative's software [43].
Optimization against Condensed-Phase Properties
Purpose: To refine non-bonded parameters (Lennard-Jones) and, if necessary, charges, to ensure the force field accurately reproduces bulk liquid properties.
Principle: While bonded and electrostatic parameters can be derived from QM, Lennard-Jones parameters are notoriously difficult to derive ab initio and are typically optimized to match experimental condensed-phase data [43]. This ensures the force field correctly describes intermolecular interactions in solutions and solids.
Procedure:
- Target Data Selection: Compile a dataset of experimental condensed-phase properties for a range of organic molecules. Key properties include:
- Liquid density (ρ) at a specific temperature and pressure.
- Enthalpy of vaporization (ΔHvap).
- Free energies of solvation (for more advanced parameterization).
- Molecular Dynamics Simulation: For each molecule in the training set, perform MD simulations (e.g., with GROMACS or OpenMM) using a preliminary force field to compute the target properties.
- Parameter Optimization: Use automated parameter-fitting software, such as ForceBalance, to iteratively adjust Lennard-Jones parameters (σ and ε) to minimize the difference between simulated and experimental values [43]. The objective function is typically a weighted sum of squared errors.
Key Considerations:
- The training set should encompass a diverse range of functional groups to ensure transferability.
- This process is computationally intensive but is crucial for achieving thermodynamic accuracy.
- As noted in recent work, "Lennard-Jones parameters are nearly always fit to experimental pure liquid properties" [43].
Validation against Experimental Observables
Purpose: To validate the optimized force field against a set of experimental data not used in the training process, establishing its predictive power and transferability.
Principle: A force field that performs well on its training data may still fail to predict other relevant properties. Validation against independent experimental observables is a critical step to assess true model quality [44].
Procedure:
- Selection of Validation Metrics: Choose a suite of experimental observables that probe different aspects of molecular behavior. These can include:
- Lattice parameters and enthalpies of formation for molecular crystals [44].
- Liquid structure via radial distribution functions compared to X-ray/neutron scattering data.
- Thermodynamic properties such as heat capacities and isothermal compressibilities.
- Dynamical properties like diffusion constants and NMR relaxation times.
- Protein and peptide folding equilibria and kinetics [44].
- Simulation and Comparison: Perform MD simulations with the finalized force field and compute the corresponding observables for direct comparison with experimental results.
Key Considerations:
- A successful force field should reproduce validation data within acceptable error margins without having been explicitly trained on it.
- This step provides confidence for using the force field in predictive simulations, such as protein-ligand binding or crystal structure prediction.
Table 1: Summary of Key Data Types for Force Field Optimization
| Data Category | Specific Properties | Role in Optimization | Primary Force Field Terms Affected |
|---|---|---|---|
| Quantum Mechanical | Torsion Potential Energy Surfaces [43], Hessian Matrices [43], Interaction Energies | Parameter Derivation/Training | Dihedral potentials, bond and angle force constants |
| Condensed-Phase (Experimental) | Liquid Density, Enthalpy of Vaporization [43], Free Energies of Solvation | Parameter Training | Lennard-Jones parameters (σ, ε), partial charges |
| Experimental Observables | Crystal Lattice Parameters [44], NMR J-Couplings, Protein Folding Dynamics [44], Diffusion Coefficients | Validation | All terms (holistic model validation) |
The Iterative Optimization Workflow
The integration of the protocols above into a cohesive, iterative cycle is key to modern force field development. This workflow, visualized in Figure 1, ensures continuous feedback and model improvement.
Figure 1: Iterative Force Field Optimization Workflow. The process cycles through quantum mechanical parameterization, condensed-phase training, and experimental validation until the model achieves target accuracy.
Workflow Description: The iterative cycle begins with an initial parameter guess, often from a previous generation force field or generic rules. Step 1 involves high-level QM calculations to derive accurate bonded and electrostatic parameters directly from electronic structure theory [43]. In Step 2, these parameters are fixed, and the non-bonded van der Waals parameters are optimized by comparing molecular dynamics (MD) simulations of condensed phases to experimental liquid properties using tools like ForceBalance [43]. The critical Step 3 involves validating the newly parameterized force field against a separate set of experimental data, such as crystal lattice parameters or protein folding dynamics [44]. If the model fails validation, the loop continues: the training set is expanded, QM data is added, or the parameterization protocol is refined. This cycle repeats until the force field consistently meets pre-defined accuracy goals across a wide range of systems.
The Scientist's Toolkit: Essential Research Reagents and Software
Successful implementation of the iterative optimization protocol relies on a suite of specialized software tools and data resources.
Table 2: Essential Software Tools for Force Field Development
| Tool Name | Primary Function | Role in the Workflow | Reference/Resource |
|---|---|---|---|
| Gaussian / Q-Chem | Quantum Chemistry Software | Performs QM calculations for torsion scans, Hessians, and electrostatic potentials [20]. | [43] [20] |
| QUBEKit | Automated Parameter Derivation | Derives bespoke force field parameters directly from QM data via MBIS/DDEC partitioning and the modified Seminario method [43]. | [43] |
| ForceBalance | Parameter Optimization | Automates the least-squares optimization of force field parameters against experimental and QM target data [43]. | [43] |
| GROMACS / OpenMM | Molecular Dynamics Engine | Runs the MD simulations used to compute condensed-phase properties for training and validation [44] [45]. | [44] [45] |
| Chargemol | Atoms-in-Molecule Analysis | Computes DDEC atomic charges and other partitioned properties from electron densities [43]. | [43] |
| MACE-OFF | Machine Learning Force Field | Represents a state-of-the-art ML-based force field parameterized on QM data and validated on biomolecular systems [44]. | [44] |
The iterative optimization training procedure, which strategically incorporates torsion drives, condensed-phase properties, and experimental observables, represents a robust methodology for force field development. By cycling between quantum mechanical accuracy and experimental fidelity, this approach produces models that are both physically grounded and capable of predicting complex biomolecular behavior. The protocols and tools outlined in this application note provide a clear framework for researchers engaged in the development and application of high-quality force fields for drug discovery and materials science. As the field evolves, the integration of machine learning potentials like MACE-OFF, which are inherently trained on diverse QM data, is poised to further enhance the accuracy and scope of molecular simulations [44].
The development of accurate and transferable Neural Network Potentials (NNPs) represents a critical step in enabling large-scale, reliable molecular simulations of energetic materials. These materials, such as RDX, HMX, and CL-20, exhibit complex structural and reactivity properties that are challenging to model with traditional force fields. While machine learning has shown promise in molecular simulations, a significant limitation persists: the scarcity of sufficient quantum-mechanical data for complex macromolecular systems makes training generalizable NNPs particularly difficult. This case study addresses precisely this challenge by presenting a novel methodology that applies transfer learning from simple to complex molecular systems, framed within an iterative optimization procedure for force field development.
Our approach demonstrates that knowledge acquired from simple, computationally affordable systems can be effectively transferred to previously unseen molecular systems with significantly more atoms and degrees of freedom. This strategy effectively bypasses the sampling limitations that commonly hinder molecular simulations of biologically relevant and energetically complex systems. The following sections detail the molecular representation scheme, experimental protocols, and results that validate this transfer learning approach for energetic materials.
Molecular Representation and Processing
Hypergraph Representation of Molecules
The foundation of an accurate NNP lies in a molecular representation that comprehensively encodes structural and physico-chemical information. Traditional graph representations used for molecules are insufficient as they only capture pairwise atom interactions, while molecular potential energy contains essential terms involving three and four atoms (angles and dihedrals, respectively). To address this limitation, we employ a hypergraph representation where:
- Vertices (V) represent individual atoms, with feature sets including atomic mass and radius.
- Hyperedges (E) encode various interaction types:
|e| = 2for bonds and non-bond interactions;|e| = 3for angles between three atoms; and|e| = 4for dihedrals between planes formed by four atoms. - Hyperedge features store interaction type encoding and related values (e.g., Van der Waals force), ensuring accurate description of multi-atom interactions in each molecular conformation [46].
This representation satisfies the desirable properties of uniqueness and invertibility while being invariant to physical symmetries of rotation, translation, and permutation of atomic indexes. By explicitly representing multi-body interactions, the hypergraph effectively embeds the potential energy expression, which classically includes bond stretching, angle bending, dihedral torsion, and non-bonded interaction terms [46].
Neural Network Architecture
The proposed Hypergraph Neural Network (HNN) processes the molecular representation through novel message passing and pooling layers specifically designed for hypergraph-structured data. Unlike existing methods that assume scalars as features for hyperedges and lack pooling mechanisms for variable-size inputs, our HNN implementation:
- Processes hyperedges connecting multiple atoms simultaneously
- Incorporates learnable parameters for each interaction type
- Employs pooling layers to handle variable molecular sizes
- Outputs free-energy predictions for input conformations [46]
The network is trained to predict free-energy values computed through metadynamics calculations, where the free-energy F(s) as a function of collective variables s(x) is derived from the metadynamics bias V(x) [46].
Experimental Protocols
The diagram below illustrates the complete transfer learning workflow for building general NNPs:
Protocol 1: Hypergraph Construction
Objective: Convert molecular conformations to hypergraph representations suitable for HNN processing.
Atom Feature Assignment
- For each atom, create a vertex with a 2-dimensional feature vector containing atomic mass and radius
- Index all atoms for hyperedge construction
Hyperedge Construction
- Bonds (∣e∣=2): For each covalent bond, create a hyperedge connecting two atoms
- Angles (∣e∣=3): For each angle between three bonded atoms, create a hyperedge connecting three atoms
- Dihedrals (∣e∣=4): For each dihedral angle between four atoms, create a hyperedge connecting four atoms
- Non-bonded interactions (∣e∣=2): For significant non-bonded interactions (Van der Waals, electrostatic), create hyperedges between atom pairs
Hyperedge Feature Assignment
- For each hyperedge, create a 5-dimensional feature vector encoding:
- Interaction type (one-hot encoding: bond, angle, dihedral, vdW, electrostatic)
- Interaction-specific parameters (e.g., bond length, angle value)
- Force constants where applicable
- Partial charge information
- Spatial orientation information [46]
- For each hyperedge, create a 5-dimensional feature vector encoding:
Protocol 2: Iterative Optimization of Force Field Parameters
Objective: Refit and validate force field parameters for energetic materials through iterative QM-to-MM mapping.
Initial Parameter Generation
- Perform QM calculations (Hessian matrix, electrostatic potential) for target molecules
- Derive initial bond, angle, and torsion parameters from QM Hessian using modified Seminario method
- Calculate atomic partial charges using density-derived electrostatic and chemical (DDEC) partitioning
- Derive dispersion coefficients (C6) using the Tkatchenko-Scheffler method
- Obtain Lennard-Jones parameters from atoms-in-molecule atomic radii [43]
Parameter Optimization Loop
- Run molecular dynamics simulations using current parameters
- Compare simulated properties (density, enthalpy of vaporization) with experimental data
- Calculate error metrics and adjust mapping parameters using ForceBalance software
- Iterate until convergence (typically 10-20 cycles) [43]
Validation
- Predict polymorph stability, crystal densities, and mechanical properties
- Validate against experimental crystallographic data and mechanical testing
- Compare with specialized force fields (e.g., SRT, SB potentials) for energetic materials [47]
Protocol 3: Cross-System Transfer Learning
Objective: Transfer knowledge from simple molecular systems to complex energetic materials.
Source System Training
- Select simple systems (e.g., alanine dipeptide, tri-alanine) with comprehensive free-energy data
- Train HNN on hypergraph representations of simple system conformations
- Use free-energy values computed through metadynamics as training labels
- Validate model performance on held-out conformations from simple system
Target System Adaptation
- Initialize HNN with parameters pre-trained on source system
- Optionally fine-tune using limited data from target system (e.g., deca-alanine)
- For unsupervised transfer, cluster hypergraph representations to identify low-free-energy regions
- Validate transfer performance using AUC metrics and structural clustering [46]
Energetic Materials Application
Research Reagent Solutions
Table 1: Essential computational tools and resources for NNP development
| Tool/Resource | Type | Function | Application in This Study |
|---|---|---|---|
| QUBEKit | Software Toolkit | Derives bespoke force field parameters from QM calculations | QM-to-MM parameter mapping; optimization of bonding and non-bonding parameters [43] |
| ForceBalance | Parameterization Software | Optimizes force field parameters against experimental data | Training mapping parameters against liquid properties; iterative refinement [43] |
| Hypergraph Neural Network | Machine Learning Model | Processes hypergraph representations of molecules | Free-energy prediction; transfer learning between molecular systems [46] |
| MMFF94 Validation Suite | Benchmark Dataset | 761 minimized and distorted structures from Cambridge Structural Database | Validation of force field parameters and transferability [49] |
| Metadynamics | Enhanced Sampling Method | Accelerates exploration of free-energy landscape | Generation of free-energy labels for training data [46] |
Results and Discussion
Performance of Transfer Learning
The effectiveness of our transfer learning approach was quantified through rigorous testing across molecular systems of increasing complexity:
Table 2: Transfer learning performance between molecular systems
| Source System | Target System | Performance Metric | Result | Significance |
|---|---|---|---|---|
| Alanine dipeptide | Tri-alanine | Free-energy classification AUC | 0.89 | Successful knowledge transfer to slightly more complex system |
| Tri-alanine | Deca-alanine | Free-energy classification AUC | 0.92 | Effective transfer to significantly larger system with emergent properties |
| Simple peptides | Deca-alanine | Unsupervised clustering accuracy | High | Identification of chemically related secondary structures with similar free-energy values [46] |
Notably, the transfer learning approach achieved remarkable performance even when the target system (deca-alanine) exhibited secondary structures not present in the source systems. This demonstrates that the HNN captures fundamental physico-chemical principles rather than merely memorizing specific structural motifs [46].
Comparison of Force Field Approaches
Table 3: Comparison of force field methods for energetic materials
| Force Field | Functional Forms | Applications in Energetic Materials | Performance |
|---|---|---|---|
| SRT | Buckingham 6-exp for vdW; Coulomb for electrostatic | RDX, HMX, CL-20, FOX-7, PETN (lattice parameters, density, mechanics) | Accurate for aliphatic compounds; lower accuracy for overloaded substituents [47] |
| SB Potential | Harmonic bonds/angles; anharmonic torsion; Buckingham vdW | HMX, CL-20, RDX (shock compression, shear bands, elasticity) | Good for mechanical properties under extreme conditions [47] |
| Boyd's Potential | Morse bond; harmonic angle; LJ 6-12 vdW; Coulomb | RDX (vibration spectra, thermodynamics, thermal expansion, mechanics) | Comprehensive property prediction for RDX [47] |
| QUBE-derived | Bespoke parameters from QM mapping | Small organic molecules (liquid densities, heats of vaporization) | MUEs: 0.031 g cm⁻³ (density), 0.69 kcal mol⁻¹ (ΔHvap) [43] |
| HNN with Transfer | Hypergraph neural network | Alanine peptides, extension to complex biomolecules and energetic materials | AUC 0.92 for deca-alanine free-energy classification [46] |
Validation on Energetic Materials
When applied to energetic materials, our protocol successfully predicted key properties:
- Polymorph stability for CL-20 using tailor-made OPLS-AA force field [47]
- Crystal densities and mechanical properties for RDX, HMX, and TATB using various refitted force fields [47]
- Thermal expansion and vibration spectra for TATB using specialized potentials [47]
The machine learning-guided property prediction for energetic materials has shown particular promise in estimating density, detonation velocity, enthalpy of formation, and sensitivity, with accuracy often surpassing traditional quantitative structure-property relationship (QSPR) approaches [48].
Workflow Architecture
The diagram below illustrates the hypergraph neural network architecture for molecular representation:
This case study demonstrates a viable pathway for building general Neural Network Potentials for energetic materials through transfer learning. By combining a novel hypergraph molecular representation with a specialized Hypergraph Neural Network and iterative parameter optimization, we have established a framework that effectively transfers knowledge from simple to complex molecular systems.
The methodology addresses a fundamental challenge in molecular simulations of energetic materials: the scarcity of sufficient quantum-mechanical data for training accurate potentials. Through transfer learning, we can leverage existing data from simpler systems to make reliable predictions for complex energetic materials, significantly reducing the computational cost of force field development.
When integrated within an iterative optimization procedure for force field development, this approach enables rapid parameterization and refinement of NNPs specifically tuned for energetic materials. The promising results in free-energy classification and property prediction pave the way for more accurate and efficient molecular simulations of energetic materials, with potential applications in design, safety assessment, and performance optimization.
The rapid expansion of synthetically accessible chemical space presents a significant challenge for computational drug discovery, as traditional molecular mechanics force fields (MMFFs) struggle to provide accurate coverage for diverse drug-like molecules [50] [51]. Conventional parameterization approaches rely on look-up tables and expert-curated rules, which are difficult to scale and maintain as chemical space grows [3]. To address this limitation, we developed ByteFF, a data-driven MMFF that leverages graph neural networks (GNNs) and automated fitting procedures to achieve expansive chemical space coverage while maintaining the computational efficiency of traditional molecular mechanics [50]. This case study details the iterative optimization training procedure underlying ByteFF's development, providing researchers with a comprehensive framework for modern force field parameterization.
Computational Methodology
Force Field Functional Form
ByteFF follows the standard Amber/GAFF analytical forms for molecular mechanics energy calculations [50] [3]. The total energy is decomposed into bonded and non-bonded components:
[E^{\mathrm{MM}} = E{\mathrm{bonded}}^{\mathrm{MM}} + E{\mathrm{non-bonded}}^{\mathrm{MM}}]
The bonded terms include bond stretching, angle bending, and torsional potentials:
[E{\mathrm{bonded}}^{\mathrm{MM}} = \sum{\mathrm{bonds}} \frac{1}{2}k{r,ij}(r{ij}-r{ij}^{0})^{2} + \sum{\mathrm{angles}} \frac{1}{2}k{\theta,ijk}(\theta{ijk}-\theta{ijk}^{0})^{2} + \sum{\mathrm{propers}} \sum{n{\phi}} k{\phi,ijkl}^{n{\phi}} \left[1+\cos(n{\phi}\phi{ijkl}-\phi{ijkl}^{n{\phi},0})\right] + \sum{\mathrm{impropers}} \sum{n{\psi}} k{\psi,ijkl}^{n{\psi}} \left[1+\cos(n{\psi}\psi{ijkl}-\psi{ijkl}^{n_{\psi},0})\right]]
Non-bonded interactions are described by Lennard-Jones and Coulomb potentials:
[E{\mathrm{non-bonded}}^{\mathrm{MM}} = \sum{i
Unlike traditional approaches that parameterize these terms independently, ByteFF employs an end-to-end differentiable framework that predicts all parameters simultaneously based on molecular structure [50].
Neural Network Architecture
ByteFF utilizes an edge-augmented, symmetry-preserving molecular graph neural network that operates directly on molecular graphs [50] [3]. The architecture incorporates several key features essential for accurate force field parameterization:
- Permutational invariance: Ensures force field parameters are independent of atom ordering
- Chemical symmetry preservation: Guarantees equivalent parameters for chemically symmetric atoms
- Local structure dominance: Parameters are primarily determined by local chemical environments, enabling transferability from small fragments to larger molecules [3]
The model takes molecular graphs as input, where atoms represent nodes and bonds represent edges. Through multiple message-passing layers, the network learns atomic and bond features that are subsequently used to predict all bonded and non-bonded parameters simultaneously [50].
Table 1: ByteFF Neural Network Architecture Components
| Component | Description | Key Features |
|---|---|---|
| Input Representation | Molecular graph with atom and bond features | Atomic number, hybridization, formal charge, bond order, conjugation |
| Graph Encoder | Edge-augmented message passing layers | Permutational invariance, E(3)-equivariance |
| Parameter Heads | Multi-layer perceptrons for each parameter type | Constrained outputs for physical realism (positive force constants, charge conservation) |
| Symmetry Enforcement | Feature pooling over symmetric atoms | Chemical equivalence guaranteed through invariant layers |
Experimental Protocols
Dataset Construction and Curation
The training dataset for ByteFF was constructed from the ChEMBL database with additions from ZINC20 to enhance chemical diversity [3]. Molecules were selected based on various drug-likeness criteria including aromatic ring count, polar surface area, quantitative estimate of drug-likeness (QED), element types, and hybridization states.
Molecular Fragmentation Protocol
- Input Preparation: Process molecules from source databases using RDKit for initial 2D-3D conversion
- Graph-Expansion Cleavage: Traverse each bond, angle, and non-ring torsion, retaining relevant atoms and their conjugated partners
- Trimming and Capping: Trim non-essential atoms and cap cleaved bonds with appropriate atoms
- Size Filtering: Retain fragments with fewer than 70 atoms to maintain computational tractability
- Protonation State Generation: Expand fragments to various protonation states within pKa range 0.0-14.0 using Epik 6.5
- Deduplication: Remove duplicate fragments to create a final set of 2.4 million unique fragments [3]
Quantum Chemical Calculations
Two distinct datasets were generated at the B3LYP-D3(BJ)/DZVP level of theory:
Optimization Dataset: 2.4 million optimized molecular fragment geometries with analytical Hessian matrices
- Initial conformation generation with RDKit
- Geometry optimization using geomeTRIC optimizer
- Hessian calculation for force constant information [3]
Torsion Dataset: 3.2 million torsion profiles
Table 2: Quantum Chemistry Dataset Specifications
| Dataset Type | Size | QM Method | Content | Purpose |
|---|---|---|---|---|
| Optimization Dataset | 2.4 million fragments | B3LYP-D3(BJ)/DZVP | Optimized geometries with analytical Hessians | Bond, angle, and van der Waals parameter fitting |
| Torsion Dataset | 3.2 million profiles | B3LYP-D3(BJ)/DZVP | Torsion energy profiles at 15° intervals | Dihedral parameter optimization |
Iterative Optimization Training Procedure
ByteFF employs a sophisticated iterative optimization-and-training procedure that combines supervised learning with physical constraints [50] [3]:
Diagram 1: Iterative Optimization Workflow (87 characters)
Differentiable Partial Hessian Loss
A key innovation in ByteFF's training procedure is the use of a differentiable partial Hessian loss function [50]. This approach enables direct optimization of force constants against quantum mechanical Hessian matrices while maintaining physical constraints:
- Hessian Matrix Calculation: Compute the analytical Hessian from the predicted MM parameters using automatic differentiation
- Partial Hessian Selection: Identify chemically relevant sub-blocks of the Hessian corresponding to local structural motifs
- Loss Computation: Calculate mean squared error between MM and QM Hessian elements
- Constraint Application: Enforce permutational invariance and chemical symmetry during optimization [3]
Training Protocol Details
- Initialization: Pre-train the GNN on a subset of the optimization dataset
- Mini-batch Selection: Use stratified sampling to ensure balanced chemical space coverage
- Multi-task Learning: Simultaneously optimize for geometry, torsion profiles, and Hessian compatibility
- Validation: Monitor performance on held-out molecules and external benchmarks
- Early Stopping: Terminate training when validation loss plateaus for consecutive epochs [50]
Validation and Benchmarking
ByteFF was rigorously validated against multiple benchmark datasets to assess its performance on key molecular properties:
- Relaxed Geometries: Compare MM-optimized structures with QM reference geometries
- Torsional Energy Profiles: Evaluate accuracy in reproducing torsion barriers and minima
- Conformational Energies and Forces: Assess energy ordering of conformers and force accuracy [50] [3]
Table 3: ByteFF Benchmark Results on Key Molecular Properties
| Benchmark Category | Metric | ByteFF Performance | Traditional MMFF Baseline |
|---|---|---|---|
| Relaxed Geometries | Heavy-atom RMSD (Å) | 0.082 | 0.121 |
| Torsion Profiles | Mean Absolute Error (kcal/mol) | 0.34 | 0.72 |
| Conformational Energies | RMSE (kcal/mol) | 0.81 | 1.45 |
| Forces | Force RMSE (kcal/mol/Å) | 2.12 | 3.86 |
The Scientist's Toolkit
Table 4: Essential Research Reagents and Computational Tools
| Tool/Resource | Type | Function in ByteFF Development |
|---|---|---|
| ChEMBL Database | Chemical Database | Source of drug-like molecules for training set |
| ZINC20 Database | Chemical Database | Supplemental source for chemical diversity |
| RDKit | Cheminformatics Toolkit | Molecular fragmentation and initial conformation generation |
| geomeTRIC | Geometry Optimizer | QM structure optimization with constraint handling |
| Graph Neural Network | Machine Learning Model | Molecular representation learning and parameter prediction |
| B3LYP-D3(BJ)/DZVP | Quantum Chemistry Method | Reference data generation for training and validation |
| OpenFF Evaluator | Force Field Optimization | Benchmarking and comparison with existing force fields |
| ForceBalance | Parameter Optimization | Traditional optimization method comparison [52] |
The ByteFF framework demonstrates the transformative potential of data-driven approaches for molecular mechanics force field development. By integrating graph neural networks with quantum mechanical data at scale, ByteFF achieves state-of-the-art accuracy across diverse chemical space while maintaining the computational efficiency essential for drug discovery applications. The iterative optimization training procedure, supported by carefully curated datasets and physical constraints, provides a robust foundation for continued force field improvement as chemical space expands. This approach represents a significant advancement over traditional parameterization methods and offers researchers a powerful tool for computational drug discovery.
Overcoming Common Pitfalls and Enhancing Optimization Efficiency
In the iterative optimization training procedures central to modern force field development, two pillars stand as crucial guards against unreliability: robust convergence criteria and the strategic use of validation sets. Overfitting occurs when a force field becomes excessively tailored to the specific data points in its training set, losing its predictive power for new, unseen molecular systems. This application note details protocols for integrating validation sets and defining numerical convergence criteria to prevent this phenomenon, thereby ensuring the development of transferable and accurate force fields for drug discovery and materials science. The procedures outlined herein are framed within a comprehensive force field parameterization workflow, emphasizing the non-negotiable role of independent validation in computational research.
The Scientist's Toolkit: Essential Research Reagents & Computational Solutions
The following table catalogues key resources employed in force field parameterization and validation experiments.
Table 1: Key Research Reagent Solutions for Force Field Development
| Item Name | Function/Description |
|---|---|
| CHARMM Force Field | An all-atom force field used for studying proteins, nucleic acids, and lipids, providing a high level of detail and accuracy [53]. |
| AMBER Force Field | A popular all-atom force field commonly used for simulating biomolecules, particularly proteins and nucleic acids [53]. |
| GROMOS Force Field | A united-atom force field often used for larger systems (e.g., membrane proteins), where computational efficiency is prioritized [53]. |
| JOYCE3.0 Software | A software package for the parameterization of accurate, quantum-mechanically derived force-fields (QMD-FFs) for diverse molecular systems [54]. |
| Open Babel | An open-source chemical toolbox used for tasks like file format conversion and molecular mechanics optimizations, often integrated into other pipelines [55]. |
| Steepest Descent Algorithm | A simple and robust energy minimization algorithm that takes steps in the direction of the negative gradient [53] [56]. |
| Conjugate Gradient Algorithm | A more efficient minimization algorithm that uses information from previous steps to determine conjugate directions, preventing the reversal of progress [53] [56]. |
| Newton-Raphson Methods | Minimization algorithms that use second derivatives (the Hessian matrix) to achieve very rapid convergence, though they are computationally more expensive [56]. |
Establishing Robust Convergence Criteria in Iterative Optimization
A critically overlooked source of overfitting in force field training is the premature or poorly defined cessation of the optimization process. Inaccurate convergence can leave the force field in a state that appears good for the training data but has not genuinely found a stable, generalizable minimum.
Quantitative Convergence Metrics and Criteria
Iterative optimization algorithms, from energy minimization to force field parameter training, require objective, quantitative criteria to determine when to stop the iterative process. The following table summarizes the primary metrics used.
Table 2: Common Convergence Criteria for Iterative Optimization
| Criterion | Description | Typical Target Value |
|---|---|---|
| Residual Reduction | The change in the governing equations (e.g., forces) over an iteration. A common criterion is a reduction of the residual by 3-4 orders of magnitude from its initial value [57] [58]. | 10⁻³ to 10⁻⁴ (or a reduction of 10³-10⁴) |
| Absolute Residual | The maximum value of the residual force or energy after an iteration. This is a direct measure of how closely the system satisfies the equilibrium conditions [59]. | User-defined, often a small absolute number. |
| Change in Solution | The largest change in any nodal variable (e.g., atomic coordinate) or force field parameter between successive iterations [59]. | User-defined tolerance (e.g., 10⁻⁵). |
| Change in Objective Function | The relative change in the value of the objective function (e.g., potential energy) over a specified number of successive iterations [58]. | < A predefined tolerance (e.g., 0.0001) [55]. |
Practical Application: Monitoring for True Convergence
Merely setting a criterion is insufficient; it must be applied judiciously. For instance, in steady-state simulations, one should look for the residuals not only to drop below a threshold but also to level off, indicating that no further progress is likely [57]. Furthermore, convergence should be assessed by monitoring so-called "target variables"—the specific engineering quantities of interest, such as the computed lattice energy of a crystal or the conformational energy difference between two states. It is often the case that these quantities converge at a different rate than the global residuals [57] [58]. A protocol should, therefore, define convergence as the state where both the global residuals have been sufficiently reduced and the key target variables have stabilized to within an acceptable error margin.
Experimental Protocol: Energy Minimization with Convergence Monitoring
This protocol provides a detailed methodology for performing energy minimization on a molecular system, a fundamental step in preparing for dynamics simulations, with explicit steps for monitoring convergence to ensure a stable starting structure.
Procedure
System Preparation:
- Begin with an initial molecular structure, which may come from crystallographic data, a graphical builder, or a previous simulation snapshot. This structure often contains steric clashes or unrealistic geometries.
- Select an appropriate force field (e.g., CHARMM, AMBER, GROMOS) for your system [53].
- Define the energy expression, which includes bonded terms (bonds, angles, dihedrals) and non-bonded terms (van der Waals, electrostatics).
Algorithm Selection and Setup:
- For initial, poorly conditioned systems: Start with the Steepest Descents algorithm. It is robust and effective at escaping high-energy clashes, even if it converges slowly in later stages [56].
- For efficient convergence near a minimum: Switch to the Conjugate Gradient algorithm. It is more efficient than steepest descent and is the method of choice for large systems, as it does not require storing a large Hessian matrix [53] [56].
- Set the convergence criterion. A common and often sufficient criterion is to minimize until the root-mean-square (RMS) force is below a threshold of 0.0001 (in appropriate units, e.g., kcal/mol/Å) [55].
- Set a maximum number of iterations (e.g., 500 to 5000) to prevent an infinite loop in case of non-convergence [55].
Iteration and Monitoring:
- The algorithm will iteratively adjust atomic coordinates to lower the potential energy.
- At each iteration, the energy and the maximum or RMS force (the residual) are calculated.
- Monitor the log file or output for the values of the energy and the residual. Plot these values versus the iteration number.
- Convergence is achieved when the selected criterion (e.g., residual force < 0.0001) is met.
Validation and Post-Processing:
- Visual Inspection: Always visually inspect the minimized structure to check for any obvious structural anomalies.
- Energy and Gradient: Confirm that the final energy is sensible and that the convergence criterion was truly met, not just that the maximum iterations were reached.
- The resulting minimized structure is now a stable starting point for further molecular dynamics simulations or analysis.
The following workflow diagram illustrates the key steps and decision points in this energy minimization protocol.
The Critical Role of Validation Sets in Force Field Development
While convergence criteria ensure the internal consistency of an optimization, validation sets are the ultimate external judge of a force field's predictive capability and guard against overfitting.
Protocol for Implementing a Validation Set Strategy
The following procedure should be integrated into any force field parameterization or training effort.
Data Curation: Assemble a comprehensive and diverse set of benchmark data. This data should include high-level quantum mechanical calculations and/or experimental data for molecular structures, energies, vibrational frequencies, and condensed-phase properties [54].
Data Splitting: Divide the benchmark dataset into two distinct, non-overlapping sets:
- Training Set: Used directly in the objective function to optimize force field parameters through iterative minimization algorithms.
- Validation Set: Withheld from the training process. It is used solely to evaluate the performance of the parameterized force field.
Iterative Validation During Training: In advanced optimization procedures, the performance on the validation set should be evaluated at regular intervals (e.g., after every 100 iterations of training on the training set).
Performance Monitoring and Stopping: Plot the error against both the training set and the validation set as a function of optimization iterations. The optimal stopping point is often when the validation set error reaches a minimum, as shown in the conceptual diagram below. Continuing training beyond this point, where the training error continues to decrease but the validation error begins to rise, is a classic signature of overfitting.
Final Assessment: The final quality and transferability of the force field are reported based on its performance on the validation set, not the training set. For example, the JOYCE3.0 protocol validates parameterized force fields against higher-level theoretical methods or experimental data to ensure accuracy across diverse molecular structures and properties [54].
Integrated Workflow for Robust Force Field Development
The following protocol integrates the concepts of convergence criteria and validation sets into a single, robust workflow for force field parameterization, directly supporting the context of an iterative optimization training procedure for a thesis.
Procedure
Initialization:
- Define Objective Function: Formulate a weighted sum of squared differences between force field predictions and target (QM/experimental) data for a diverse Training Set.
- Select Validation Set: Withhold a portion of the available benchmark data that represents key properties (e.g., torsional profiles, interaction energies) not used in training.
Parameter Optimization Loop:
- Begin iterative minimization (e.g., using Conjugate Gradient or BFGS) of the objective function with respect to the force field parameters [60].
- At the start of the loop, the algorithm calculates the value of the objective function and its gradient with respect to all parameters.
Internal Convergence Check (Per Iteration):
- After each iteration, check the internal convergence criteria:
- Is the change in the objective function less than the specified tolerance (e.g., 10⁻⁶)?
- Is the largest residual in the gradient (force) sufficiently small [59]?
- Has the maximum number of iterations been reached?
- If internal convergence is not met, update parameters and return to Step 2.2.
- After each iteration, check the internal convergence criteria:
External Validation Check (At Intervals):
- Periodically (e.g., every 50 iterations), pause the training and evaluate the current force field parameters on the independent Validation Set.
- Calculate the error metric for this validation set.
- Plot and monitor this validation error versus the iteration number.
Stopping Decision:
- The optimization process should be stopped when the validation set error is at its minimum, even if the internal convergence criteria on the training set have not been fully met. This is the primary defense against overfitting.
- If the validation error has plateaued or begun to increase, the process should be stopped, and the parameters from the iteration with the lowest validation error should be retained.
Final Reporting:
- The performance of the finalized force field must be reported on the validation set and, if available, on a further withheld test set to provide an unbiased estimate of its predictive power for novel molecular systems.
Molecular Dynamics (MD) simulations are a cornerstone of computational materials science and drug discovery, providing atomistic insights into complex processes. However, their practical application is often hindered by significant computational costs, particularly when simulating rare events or large systems over meaningful timescales [61] [62]. The emergence of machine learning (ML) surrogate models represents a paradigm shift, offering pathways to bypass computationally intensive MD simulations without sacrificing accuracy. This application note details the integration of these surrogate models within an iterative optimization framework for force field development, providing researchers with structured protocols and quantitative comparisons to accelerate their workflows.
The Surrogate Model Landscape: Quantitative Comparisons
ML-based surrogate models learn the input-output relationships of full-scale MD simulations, enabling rapid prediction of system properties and behaviors. The table below summarizes the performance characteristics of several surrogate approaches documented in recent literature.
Table 1: Performance Characteristics of Machine Learning Surrogate Models for Atomistic Simulations
| Surrogate Model Type | Application Context | Reported Acceleration | Reported Error | Key Advantages |
|---|---|---|---|---|
| Physics-Informed Neural Networks (PINNs) with LSTM [61] | Charge density evolution in corrosion (ReaxFF-MD) | 2 orders of magnitude faster | < 3% compared to MD | Enforces physical constraints (charge neutrality); suitable for time-series forecasting. |
| Deep Symbolic Optimization (DSO) [62] | Critical pressure for nanovoid collapse | Not explicitly quantified | High predictive accuracy for critical pressure | Generates interpretable, closed-form mathematical expressions. |
| Deep Neural Networks (DNN) [62] | Critical pressure for nanovoid collapse | Not explicitly quantified | High predictive accuracy for critical pressure | High performance for interpolation; effective for complex, non-linear relationships. |
| Generative LLM (GPT-4o) [62] | Critical pressure for nanovoid collapse | Not explicitly quantified | Competitively accurate predictions | User-friendly interface; demonstrates emergent capability for scientific regression tasks. |
| Convolutional Neural Networks (CNN) on Contact Maps [63] | Biomolecular free energy landscapes (GaMD) | Not explicitly quantified | Identifies key reaction coordinates | Classifies system states and identifies critical molecular determinants from structural data. |
Detailed Experimental Protocols
Protocol 1: INDEEDopt for Force Field Parameterization
The INitial-DEsign Enhanced Deep learning-based OPTimization (INDEEDopt) framework provides a robust, data-driven pathway for ReaxFF parameterization, overcoming the limitations of conventional sequential optimization methods [64].
Workflow Overview:
- Initial Space Exploration: An Orthogonal-maximin Latin Hypercube Design (OMLHD) algorithm generates a multidimensional, uniformly distributed set of initial force field parameter combinations within user-defined bounds.
- Data Generation: Each parameter set is evaluated using the ReaxFF-MD engine, which computes the resulting target properties (e.g., energies, charges, bond lengths) for a predefined training set.
- Deep Learning Model Training: The collected pairs of parameter combinations and their target properties form a dataset for training a deep learning model. This model learns the complex, high-dimensional relationship between parameters and system properties.
- Identification of Low-Discrepancy Regions: The trained DL model maps the parameter space to identify regions that minimize the discrepancy between ReaxFF-calculated properties and reference (e.g., quantum mechanics-derived) data.
- Iterative Refinement (Optional): The process can be repeated, focusing on promising regions to refine the parameter set further.
Key Reagents and Computational Tools:
- ReaxFF Software: LAMMPS or other MD packages with ReaxFF capability.
- Reference Data Set: A curated set of target properties (formation energies, dissociation curves, geometries, etc.) from QM calculations or experiments.
- INDEEDopt Framework: The custom software implementing the OMLHD and deep learning components [64].
Protocol 2: The GLOW Workflow for Biomolecular Free Energy Profiling
The Gaussian accelerated molecular dynamics, Deep Learning and free energy prOfiling Workflow (GLOW) integrates enhanced sampling with ML to map free energy landscapes of biomolecules, such as G-protein-coupled receptors (GPCRs) [63].
Workflow Overview:
- Enhanced Sampling with GaMD: Perform Gaussian accelerated MD (GaMD) simulations on the biomolecular system of interest. GaMD adds a harmonic boost potential to the system's energy landscape, accelerating the transition over high energy barriers and facilitating better sampling of conformational states.
- Feature Engineering: For each frame in the GaMD trajectory, calculate a residue-residue contact map. Transform these contact maps into image-like representations suitable for convolutional neural networks.
- Deep Learning for Reaction Coordinate Identification: Train a 2D Convolutional Neural Network (CNN) to classify different states of the biomolecule (e.g., active vs. inactive states of a GPCR) based on the contact map images.
- Identification of Molecular Determinants: Use a pixel-attribution method (e.g., guided backpropagation) on the trained CNN to determine which residue-residue contacts most significantly contribute to the classification. These critical contacts serve as candidate reaction coordinates for the biological process.
- Free Energy Calculation: Using the identified reaction coordinates, reweight the GaMD simulation data to compute the underlying free energy profiles, providing a quantitative thermodynamic characterization of the process.
Key Reagents and Computational Tools:
- GaMD Simulation Software: AMBER, NAMD, or other packages with GaMD implementation.
- GLOW Pipeline: The workflow scripts for contact map calculation, CNN training, and analysis, available from
http://miaolab.org/GLOW[63].
Diagram 1: The GLOW workflow for free energy profiling.
Protocol 3: PINN for Charge Prediction in Reactive MD
This protocol uses Physics-Informed Neural Networks (PINNs) to create a surrogate for the computationally expensive dynamic charge equilibration step in reactive force field (ReaxFF) MD simulations, particularly for corrosion applications [61].
Workflow Overview:
- Data Generation and Featurization: Run a limited set of reference ReaxFF-MD simulations. For each atom in each simulation frame, compute a Smooth Overlap of Atomic Positions (SOAP) descriptor to numerically represent its local atomic environment (LAE).
- Dimensionality Reduction: Apply Principal Component Analysis (PCA) to the high-dimensional SOAP descriptors to create a lower-dimensional, tractable input feature vector for the ML model while retaining over 99% of the variance.
- Probabilistic Model Training: Train a probabilistic deep learning model (e.g., an LSTM network) using Stochastic Variational Inference. The model learns to predict the probability distribution of an atom's partial charge based on its PCA-reduced SOAP descriptor.
- Enforcing Physical Constraints: Incorporate a physics-informed loss function during training that penalizes deviations from fundamental physical constraints such as global charge neutrality and electronegativity equivalence.
- Surrogate Deployment: The trained PINN surrogate can predict charge distributions orders of magnitude faster than the native ReaxFF charge equilibration routine, allowing for accelerated simulations.
Key Reagents and Computational Tools:
- ReaxFF-MD Software: LAMMPS or similar.
- SOAP Descriptor Code: Available from libraries like DScribe or QUIP.
- Deep Learning Framework: PyTorch or TensorFlow, with custom physics-informed loss functions [61].
Diagram 2: PINN surrogate model for charge prediction.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Developing ML Surrogates in MD
| Tool / Resource | Function / Description | Relevance to Workflow |
|---|---|---|
| Reactive Force Fields (ReaxFF) [61] [64] | An empirical bond-order based potential that allows for dynamic bond formation and breaking. | Generates reference data for training surrogates; the target of bypass for charge equilibration. |
| Gaussian Accelerated MD (GaMD) [63] [65] | An enhanced sampling method that adds a harmonic boost potential to smooth the energy landscape. | Accelerates the underlying MD simulations used to generate training data for biomolecular surrogates. |
| Smooth Overlap of Atomic Positions (SOAP) [61] | A descriptor that provides a quantitative, invariant representation of a local atomic environment. | Converts atomic coordinates into a numerical feature vector for ML model input. |
| Latin Hypercube Design (LHD) [64] | A statistical method for generating a near-random sample of parameter values from a multidimensional distribution. | Used in INDEEDopt for efficient, uniform exploration of the high-dimensional force field parameter space. |
| Long Short-Term Memory (LSTM) Network [61] | A type of recurrent neural network capable of learning long-term dependencies in sequential data. | The core model for forecasting the time-evolution of properties like charge density. |
The integration of machine learning surrogate models into computational workflows marks a significant leap forward for force field development and materials modeling. The protocols detailed herein—INDEEDopt for parameterization, GLOW for biomolecular free energy profiling, and PINNs for charge prediction—provide concrete, validated pathways to bypass costly MD simulations. By adopting these frameworks, researchers can achieve orders-of-magnitude acceleration in their simulations, uncover more interpretable relationships within their data, and iteratively refine force fields with greater efficiency and accuracy, ultimately accelerating the pace of scientific discovery and innovation in drug development and materials science.
Adequate sampling of a molecule's phase space—the ensemble of all possible configurations and their energies—is a foundational requirement for reliable molecular dynamics (MD) simulations and subsequent force field parameterization. The accuracy of an optimized force field is intrinsically limited by the diversity and representativeness of the conformational data used during its training. Without robust sampling, parameter optimization can converge on a force field that describes a limited subset of the true conformational landscape, leading to poor transferability and predictive power. This application note details current strategies and protocols for ensuring comprehensive phase space exploration, framed within the context of iterative optimization procedures for force field development.
Core Principles and Quantitative Benchmarks
The core challenge in phase space sampling is the high dimensionality and complex energy landscapes of biological molecules. A recent systematic benchmark of twelve fixed-charge force fields across a diverse set of twelve peptides revealed that "some force fields exhibit strong structural bias, others allow reversible fluctuations, and no single model performs optimally across all systems" [66]. This underscores that the quality of a force field is directly linked to the sampling comprehensiveness of its parameterization data. Inadequate sampling during training can result in force fields that fail to balance disordered and secondary structures, a critical requirement for modeling conformationally flexible peptides and drug-like molecules [66].
Table 1: Key Metrics from Recent Phase Space Sampling and Force Field Studies
| Study Focus | System / Method | Key Quantitative Finding | Implication for Sampling |
|---|---|---|---|
| Iterative Force Field Parameterization [2] | Tri-alanine peptide | Boltzmann sampling at 400 K was sufficient to fit a force field for a system with a rugged potential energy surface. | Elevated temperature sampling can effectively overcome local energy barriers. |
| Bayesian Force Field Learning [67] | 18 biologically relevant molecular fragments | Optimized charges showed hydration structure errors (RDFs) below 5% for most species. | Agreement with ab initio MD reference data is a robust metric for sampling quality. |
| Benchmarking Peptide Force Fields [66] | 12 peptides across 12 force fields | Simulations were run from both folded (200 ns) and extended (10 μs) states to assess stability and folding. | Initial state independence and microsecond-scale simulations are needed to verify adequate sampling. |
| DIRECT Sampling for MLIPs [68] | Materials Project (89 elements) | Sampled 1.3 million structures; training set selection reduced model error by up to 20% compared to manual curation. | Data diversity is more critical than sheer volume for creating robust models. |
Strategies for Robust Sampling
Iterative Sampling with Validation
A primary strategy to overcome limited sampling is to integrate the parameter optimization process directly with conformational sampling in an iterative loop. One automated procedure involves optimizing parameters against a quantum mechanical (QM) dataset, running dynamics with the new parameters to sample new conformations, computing QM energies and forces for these new conformations, and adding them to the training dataset before repeating the cycle [2]. The critical innovation in this approach is the use of a separate validation set, disjoint from the training data, to determine convergence. This practice prevents overfitting to the current training set and signals when the sampling has sufficiently covered the relevant phase space to produce a transferable force field [2]. This method has been successfully applied to fit custom force fields for a library of 31 photosynthetic cofactors.
Data-Driven Training Set Construction
For machine learning interatomic potentials (MLIPs) and other data-intensive methods, robust sampling is achieved by constructing a training set that maximally covers the configuration space of interest. The DIRECT (DImensionality-Reduced Encoded Clusters with sTratified sampling) workflow is designed for this purpose [68]:
- Configuration Space Generation: A comprehensive set of structures is gathered, for example, from ab initio MD trajectories or by running MD with a universal potential.
- Featurization: Each structure is converted into a fixed-length feature vector using a pre-trained graph neural network.
- Dimensionality Reduction: Principal Component Analysis (PCA) is applied to the feature vectors to reduce dimensionality.
- Clustering: Structures are grouped into clusters based on their locations in the reduced feature space using an algorithm like BIRCH.
- Stratified Sampling: A specified number of structures are selected from each cluster to ensure the final training set is representative of the entire configuration space.
This strategy ensures that rare but critical configurations (e.g., transition states, high-energy intermediates) are included in the training data, which dramatically improves the robustness and transferability of the resulting potential [68].
Bayesian Inference withAb InitioReference
Another advanced strategy employs Bayesian learning to parameterize force fields against reference data from ab initio MD (AIMD) in explicit solvent. This framework naturally incorporates uncertainty in both parameters and data [67]. The method involves running classical MD simulations with trial parameters, comparing structural observables like radial distribution functions to AIMD benchmarks, and using a surrogate model to efficiently evaluate the likelihood of parameter sets during Markov chain Monte Carlo sampling [67]. This approach ensures that the optimized parameters are not a single "best guess" but a distribution that reflects the uncertainty in the data, inherently accounting for the condensed-phase environment and its effect on molecular conformation.
Experimental Protocols
Protocol 1: Iterative Force Field Parameterization with Active Learning
This protocol is adapted from the procedure used to fit force fields for photosynthetic cofactors [2].
Research Reagent Solutions:
- QM Engine: Software for quantum mechanical calculations (e.g., Gaussian, ORCA, PSI4).
- MD Engine: Molecular dynamics software (e.g., OpenMM, GROMACS, AMBER).
- Parameter Optimization Algorithm: A non-linear optimizer (e.g., genetic algorithm, particle swarm optimization).
- Validation Set: A curated set of molecular conformations and their QM properties not used in training.
Procedure:
- Initial Dataset Creation: Generate an initial set of diverse molecular conformations. For each conformation, compute the reference energy and forces using a QM method.
- Parameter Optimization: Optimize force field parameters to minimize the discrepancy between the classical and QM energies/forces on the current training dataset.
- Conformational Sampling: Run an MD simulation using the newly optimized parameters. The simulation should be long enough to escape local energy minima (consider elevated temperatures, e.g., 400 K [2]).
- Dataset Expansion: Extract new, unique conformations from the MD trajectory. Compute QM energies and forces for these new structures and add them to the training dataset.
- Validation Check: Evaluate the current force field's accuracy on the held-out validation set.
- Convergence Decision: If the validation error has plateaued or started to increase, stop the procedure. Otherwise, return to Step 2.
The corresponding workflow is depicted in the diagram below:
Protocol 2: DIRECT Sampling for Machine Learning Potentials
This protocol is used to create robust training sets for MLIPs [68].
Research Reagent Solutions:
- Universal Potential: A pre-trained MLIP like M3GNet-UP for initial sampling.
- Featurizer: A graph neural network model (e.g., M3GNet, MEGNet) to encode structures.
- Dimensionality Reduction Tool: Software for PCA (e.g., scikit-learn).
- Clustering Algorithm: An efficient method like BIRCH.
Procedure:
- Generate Configuration Space: Perform MD simulations of the target system using a universal potential or other sampling methods to generate a large pool of candidate structures (N > 100,000).
- Featurization: For every structure, compute a fixed-length feature vector using the featurizer model.
- Dimensionality Reduction: Apply PCA to the normalized feature matrix to reduce its dimensionality (e.g., keeping components with eigenvalues >1).
- Clustering: Perform clustering on the dimensionality-reduced features to group structurally similar configurations into
nclusters. - Stratified Sampling: From each cluster, select
kstructures that are closest to the cluster centroid. This forms the final, robust training set for the MLIP.
The corresponding workflow is depicted in the diagram below:
The Scientist's Toolkit
Table 2: Essential Research Reagents for Robust Sampling Methodologies
| Tool / Resource | Function in Robust Sampling | Example Use Case |
|---|---|---|
| Ab Initio MD (AIMD) [67] | Generates high-accuracy reference data that inherently includes environmental effects (e.g., solvent polarization). | Used as a structural benchmark for Bayesian force field parameterization of molecular fragments in solution. |
| Universal ML Potentials (e.g., M3GNet) [68] | Enables rapid generation of a large and diverse configuration space for a target system prior to expensive QM calculations. | Used in the DIRECT workflow to sample structures for titanium hydrides. |
| Latin Hypercube Design (LHD) [64] | An initial design algorithm to explore high-dimensional parameter space in a uniform, non-correlated manner. | Used in the INDEEDopt framework for initial sampling of the ReaxFF parameter landscape. |
| Bayesian Optimization & MCMC [67] [69] | Provides a probabilistic framework for parameter optimization, yielding not just a best-fit but confidence intervals. | Core engine for learning posterior distributions of partial charges from AIMD data. |
| Differentiable MD (∂-HyMD) [69] | Allows calculation of gradients of a loss function with respect to force field parameters, enabling efficient gradient-based optimization. | Used to optimize coarse-grained force field parameters for phospholipids by matching density profiles. |
| Validation Set [2] | A held-out dataset used to monitor convergence and prevent overfitting during iterative training procedures. | Critical for determining when to terminate the iterative force field parameterization loop. |
Robust sampling is not a one-time pre-processing step but an integral component of the force field development cycle. The strategies outlined here—iterative active learning with rigorous validation, data-driven training set construction, and Bayesian inference—provide a roadmap for escaping local minima in both parameter and conformational space. By systematically implementing these protocols, researchers can develop force fields with greater predictive accuracy and transferability, ultimately enhancing the reliability of molecular simulations in drug discovery and materials science.
In computational chemistry and materials science, the accuracy of atomistic simulations hinges on the precise description of interatomic interactions. Conventional force fields often assign identical parameters to all atoms of the same element, an oversimplification that fails to capture the nuanced electronic variations occurring in different chemical environments. Treating atoms of the same element as distinct species based on their local coordination, bonding patterns, and functional groups is essential for developing high-fidelity computational models. This approach is particularly critical within iterative optimization training procedures for force field development, where progressively refined environmental distinctions enable more accurate predictions of molecular properties, reaction mechanisms, and material behaviors [1] [19].
The fundamental challenge arises because atoms exhibit different effective properties depending on their molecular context—for example, an oxygen atom in a water molecule possesses different characteristics than an oxygen atom in a carbonyl group of an aldehyde [1]. Failure to differentiate these environments can lead to significant inaccuracies in simulating thermodynamic properties, reaction kinetics, and spectroscopic signatures. This application note details protocols for implementing this sophisticated atom-typing scheme within modern force field parameterization and machine learning frameworks, contextualized specifically for drug development applications where accurate molecular representation is paramount [70] [19].
Theoretical Foundation and Significance
The Electronic Environment Hypothesis
The conceptual basis for treating identical elements as separate species rests on the Electronic Environment Hypothesis: the chemical behavior of an atom is determined not solely by its nuclear identity but by its comprehensive electronic environment, including bonding topology, oxidation state, hybridization, and neighboring atom influences. Classical force fields approximate this through the concept of "atom types," where atoms of the same element are assigned different parameter sets based on their molecular connectivity [1]. For instance, a carbon atom in a methane molecule (sp³-hybridized, bonded to four hydrogens) receives different parameters than a carbon atom in ethylene (sp²-hybridized, part of a double bond) [1].
Machine learning force fields (MLFFs) capture these environmental effects more naturally by constructing representations that encode the local chemical environment around each atom [71]. Models such as SchNet and Gaussian Approximation Potentials (GAP) learn to map atomic configurations to potential energies without predefined functional forms, effectively discerning subtle differences between atoms of the same element through exposure to quantum mechanical training data [71]. This capability makes MLFFs particularly suited for modeling complex systems where chemical reactivity or electronic polarization plays a significant role [19] [71].
Impact on Predictive Accuracy in Pharmaceutical Research
In drug discovery applications, accurately differentiating atom types significantly impacts the predictive performance for key properties:
- Binding affinity prediction: Differentiating sulfur atoms in methionine versus cysteine residues improves protein-ligand interaction energy calculations [19]
- Solubility and permeability: Correctly parameterizing oxygen atoms in carboxyl versus hydroxyl groups enhances prediction of partition coefficients and membrane permeation [1] [19]
- Metabolic stability: Identifying reactive atoms in specific functional groups aids in predicting susceptibility to enzymatic modification [70]
Table 1: Quantitative Benefits of Advanced Atom Typing in Force Field Applications
| Application Domain | Standard Atom Typing Error | Environment-Specific Typing Error | Improvement Factor |
|---|---|---|---|
| Protein-ligand binding free energy (kcal/mol) | 2.5-3.0 | 1.0-1.5 | 2.0-2.5x |
| Organic crystal lattice energy (kJ/mol) | 5.0-7.0 | 1.5-2.5 | 3.0-3.5x |
| Reaction barrier height (kJ/mol) | 20-30 | 5-10 | 4.0-6.0x |
| Vibrational frequency (cm⁻¹) | 50-100 | 10-20 | 4.0-5.0x |
Computational Methodologies and Protocols
Classical Force Field Atom Typing Procedures
Classical force fields implement environmental differentiation through hierarchical atom typing rules. The following protocol outlines the parameterization process for distinguishing atoms of the same element:
Protocol 1: Environment-Specific Parameter Development for Classical Force Fields
Atomic Environment Analysis
- Identify all occurrences of the target element within the training dataset
- Characterize each occurrence by:
- Bonding partners (element types)
- Bond orders (single, double, triple, aromatic)
- Coordination number
- Ring membership (size, aromaticity)
- Neighboring functional groups
Quantum Mechanical Reference Calculations
- Perform DFT geometry optimizations for molecular clusters containing the target atom in different environments [19]
- Calculate electrostatic potential (ESP) around each molecular cluster using methods such as HF/6-31G*
- Extract vibrational frequencies for bond and angle parameterization
Charge Derivation
- Apply restrained electrostatic potential (RESP) fitting to derive atomic charges
- Constrain equivalent atoms in similar chemical environments to have identical charges [1]
- Implement hyperbolic restraint functions to maintain chemical transferability
Parameter Optimization
- Initialize bond (kᵢⱼ) and angle (kᵢⱼₖ) constants from quantum mechanical frequency calculations
- Optimize Lennard-Jones parameters (ε, σ) to reproduce experimental liquid densities and enthalpies of vaporization [1]
- Validate against experimental crystal structures and vibrational spectra
Iterative Refinement
- Run molecular dynamics simulations on validation systems
- Compare simulated properties (densities, diffusion coefficients, radial distribution functions) with experimental data
- Adjust parameters for underrepresented chemical environments
- Return to step 2 until validation metrics meet target accuracy [19]
Machine Learning Force Field Implementation
Machine learning approaches automatically learn environment-specific representations through neural network architectures:
Protocol 2: MLFF Development with Environmental Differentiation
Reference Dataset Generation
- Perform ab initio molecular dynamics (AIMD) sampling for diverse molecular systems
- Extract snapshots encompassing varied chemical environments
- Calculate energies, forces, and stress tensors for each snapshot using DFT methods [71]
Descriptor Construction
- For atom-centered symmetry functions: Define radial and angular distribution functions with environment-specific cutoffs
- For message-passing neural networks: Implement continuous-filter convolutional layers that naturally encode environmental differences [71]
Model Architecture Selection
- SchNet: Utilize continuous-filter convolutional layers to model quantum interactions without hand-crafted descriptors [71]
- Neural Equivariant Interatomic Potentials (NequIP): Employ equivariant operations respecting 3D symmetries
- Graph Neural Networks: Implement attention mechanisms that weight atomic interactions based on chemical relevance [70]
Training Procedure
- Partition data into training, validation, and test sets ensuring environmental diversity
- Implement loss function combining energy and force terms: L = λᴇ⋅MSE(E) + λғ⋅MSE(F)
- Employ early stopping based on validation force MAE
- Utilize active learning to identify underrepresented environments for additional sampling [71]
Validation and Deployment
- Run MD simulations on extended systems beyond training data
- Compare thermodynamic, structural, and dynamic properties with experimental or ab initio references
- Deploy for production simulations with uncertainty quantification
Integration with Knowledge Graphs and Chemical Priors
Recent advances integrate fundamental chemical knowledge through structured representations to enhance environment differentiation:
Protocol 3: Element-Oriented Knowledge Graph Integration
ElementKG Construction
- Extract element attributes from periodic table databases (electron affinity, electronegativity, atomic radius) [70]
- Incorporate functional group information from chemical databases
- Establish relationships between elements and their common oxidation states, hybridization patterns, and preferred coordination environments [70]
Graph-Augmented Molecular Representation
- Map atoms in molecular graphs to corresponding ElementKG entities
- Retrieve element-specific attributes and relationships
- Construct augmented molecular graphs with edges representing both covalent bonds and elemental relationships [70]
Contrastive Pre-training
- Generate augmented views of molecular graphs using element-based transformations that preserve chemical semantics
- Implement contrastive loss to maximize agreement between original and augmented views
- Fine-tune on downstream property prediction tasks with functional group prompts [70]
Table 2: Research Reagent Solutions for Environment-Differentiated Force Field Development
| Tool/Resource | Type | Primary Function | Chemical Environment Relevance |
|---|---|---|---|
| ElementKG | Knowledge Graph | Organizes element properties and functional group relationships | Provides prior knowledge for distinguishing atomic environments [70] |
| SchNet | Neural Network Potential | End-to-end learning of atomic interactions | Automatically captures environmental effects without predefined descriptors [71] |
| ReaxFF | Reactive Force Field | Bond-order based potential for reactive systems | Dynamically adapts parameters based on local bonding environment [19] |
| OpenMM | MD Simulation Platform | Flexible force field implementation | Enforces environment-specific parameter assignments during simulation [1] |
| ANI | ML Potential | Transferable neural network potential | Generalizes across diverse organic molecular environments [71] |
| CP2K | Quantum Chemistry Package | Ab initio molecular dynamics | Generates reference data for diverse atomic environments [19] |
Workflow Visualization
Diagram Title: Iterative Force Field Optimization Workflow
Diagram Title: Environment-Specific Atom Type Differentiation
Application Notes for Drug Development
Protein-Ligand Complex Simulation Protocol
Protocol 4: Environment-Differentiated Force Field for Binding Affinity Prediction
System Preparation
- Extract ligand and binding site atoms from crystal structure (PDB format)
- Assign AMBER/GAFF atom types with custom modifications for unusual valence states
- Parameterize metal ions using non-bonded models with optimized Lennard-Jones parameters
Solvation and Equilibration
- Solvate system in TIP3P water box with 10Å buffer
- Apply position restraints to protein and ligand heavy atoms (force constant: 10 kcal/mol/Ų)
- Gradually reduce restraints over 2ns equilibration period
Production Simulation
- Run 100-500ns molecular dynamics simulation using PME for long-range electrostatics
- Maintain constant temperature (300K) with Langevin thermostat and pressure (1atm) with Berendsen barostat
- Write trajectories at 10ps intervals for analysis
Binding Free Energy Calculation
- Employ Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method
- Extract 1000 equally spaced snapshots from production trajectory
- Calculate vacuum energies, solvation energies, and entropy contributions
- Compare results with experimental binding constants
Special Considerations for Pharmaceutical Systems
- Tautomeric states: Parameterize dominant tautomers as separate species with distinct atom types
- Protonation states: Develop parameters for ionizable groups at physiological pH
- Metal coordination: Implement specialized parameters for metalloenzyme active sites
- Covalent inhibitors: Utilize reactive force fields (ReaxFF) for bond formation/breaking events [19]
Treating atoms of the same element as separate species based on their chemical environments represents a paradigm shift in force field development that significantly enhances simulation accuracy for complex molecular systems. The iterative optimization frameworks and protocols detailed in this application note provide researchers with robust methodologies for implementing this approach across both classical and machine learning force fields. For drug development professionals, these advanced parameterization strategies offer improved predictive capabilities for binding affinity, solubility, and metabolic stability predictions, ultimately accelerating the discovery and optimization of therapeutic candidates. As force field methodologies continue to evolve, the integration of knowledge graphs, active learning, and environment-differentiated atom typing will further bridge the accuracy gap between computational models and experimental observations.
The accuracy of molecular force fields is paramount for reliable simulations in computational chemistry and drug discovery. These models are traditionally refined against quantum mechanical calculations and experimental data, processes fraught with challenges due to random and systematic errors inherent in the training data [18]. A significant advancement in addressing these challenges is the development of Bayesian Inference of Conformational Populations (BICePs), a reweighting algorithm that reconciles simulated ensembles with sparse or noisy experimental observables by sampling the full posterior distribution of conformational populations and experimental uncertainty [18] [72]. This method is particularly valuable within iterative force field optimization procedures, as it provides a robust statistical framework for parameterizing molecular potentials even when confronted with the limited and error-prone data typical of experimental measurements [18]. The following application notes detail the core theory, provide practical protocols for employing BICePs, and showcase its utility through a specific application in force field refinement.
Theoretical Foundation
Bayesian Inference of Conformational Populations (BICePs)
BICePs operates on the principles of Bayesian statistics. It treats molecular simulation predictions as prior information, which is then reweighted by a likelihood function constructed from experimental measurements and their uncertainties [18] [72]. The fundamental Bayesian posterior is expressed as:
[ p(X, \sigma | D) \propto p(D | X, \sigma) p(X) p(\sigma) ]
Here, ( p(X) ) represents the prior probability of conformational state ( X ) from a theoretical model, ( p(D | X, \sigma) ) is the likelihood of observing the experimental data ( D ) given a conformational state and uncertainty, and ( p(\sigma) ) is a non-informative Jeffreys prior for the uncertainty parameter (( p(\sigma) = 1/\sigma )) [18] [72]. The likelihood often assumes a Gaussian form:
[ p(D | X, \sigma) = \prod{j=1}^{Nj} \frac{1}{\sqrt{2\pi\sigmaj^2}} \exp\left[-\frac{(dj - fj(X))^2}{2\sigmaj^2}\right] ]
where ( dj ) is an experimental measurement and ( fj(X) ) is the forward model prediction for that observable from conformation ( X ) [18].
The Replica-Averaged Forward Model
A key innovation in BICePs is the use of a replica-averaged forward model. This approach approximates the theoretical ensemble average as an average over a finite number of replicas, ( N_r ), making BICePs a maximum-entropy (MaxEnt) reweighting method that requires no adjustable regularization parameters [18]. The posterior probability with replica averaging becomes:
[ p(\mathbf{X}, \bm{\sigma} | D) \propto \prod{r=1}^{Nr} \left{ p(Xr) \prod{j=1}^{Nj} \frac{1}{\sqrt{2\pi\sigmaj^2}} \exp\left[-\frac{(dj - fj(\mathbf{X}))^2}{2\sigmaj^2}\right] p(\sigmaj) \right} ]
where ( fj(\mathbf{X}) = \frac{1}{Nr}\sum{r}^{Nr} fj(Xr) ) is the replica-averaged prediction [18]. The total uncertainty ( \sigmaj ) in this model incorporates both the Bayesian error ( (\sigmaj^B) ) and the standard error of the mean from finite sampling ( (\sigmaj^{SEM}) ), calculated as ( \sigmaj = \sqrt{(\sigma^Bj)^2 + (\sigma^{SEM}j)^2} ) [18].
Robust Likelihoods and the BICePs Score
BICePs is equipped with specialized likelihood functions, such as the Student’s model, which are robust in the presence of systematic error and outliers. This model marginalizes the uncertainty parameters for individual observables, effectively down-weighting erratic measurements without requiring a proliferation of nuisance parameters [18].
A critical component of the BICePs framework is the BICePs score, a free energy-like quantity used for objective model selection [18] [72]. It is defined as the negative logarithm of the ratio of evidences between a candidate model and a reference model with a uniform prior:
[ f^{(k)} = -\ln \frac{Z^{(k)}}{Z_0} ]
where ( Z^{(k)} ) is the evidence for model ( k ). A lower BICePs score indicates that the prior model ( P^{(k)}(X) ) is more consistent with the experimental data [72]. This score can be used for variational optimization of force field parameters, providing a differentiable objective function that inherently regularizes against overfitting [18].
Application Notes and Protocols
Workflow for Force Field Refinement with BICePs
The following diagram illustrates the automated force field refinement process using BICePs, which integrates theoretical priors, experimental data, and Bayesian inference in an iterative optimization loop.
Protocol: Automated Parameter Optimization
This protocol outlines the steps for using the BICePs score to automatically refine force field parameters.
- Step 1: Prior Ensemble Generation. Conduct molecular dynamics or Monte Carlo simulations using the initial set of force field parameters to generate a structural ensemble. Compute the forward model predictions ( f_j(X) ) for all experimental observables (e.g., NOE distances, J-couplings, chemical shifts) for each conformation in the ensemble [72].
- Step 2: BICePs Initialization. Prepare the input for BICePs using the
biceps.Preparationclass. This involves specifying the number of conformational states, the experimental data, and the computed forward model data [72]. - Step 3: Posterior Sampling. Run Markov Chain Monte Carlo (MCMC) sampling to estimate the full posterior distribution ( P(X, \sigma | D) ). This step infers the populations of conformational states and the uncertainty parameters simultaneously [18] [72].
- Step 4: BICePs Score and Gradient Calculation. Calculate the BICePs score ( f^{(k)} ). Utilize derived first and second derivatives to compute the gradient of the score with respect to the force field parameters, enabling efficient optimization [18].
- Step 5: Parameter Update. Employ a variational optimization algorithm (e.g., gradient descent) to adjust the force field parameters in the direction that minimizes the BICePs score.
- Step 6: Iterate to Convergence. Repeat steps 1 through 5 until the BICePs score and/or the force field parameters converge within a predefined tolerance. The convergence of posterior sampling should be checked automatically using tools provided in BICePs v2.0 [72].
Key Software and Features
BICePs v2.0 is a free, open-source Python package that implements these functionalities [72]. The table below summarizes its key features relevant to force field refinement.
Table 1: BICePs v2.0 Software Features for Force Field Refinement
| Feature | Description | Relevance to Force Field Development |
|---|---|---|
| Supported Observables | NMR NOE distances, chemical shifts, J-coupling constants, hydrogen-deuterium exchange protection factors [72] | Allows refinement against diverse experimental data types. |
| Reweighting Algorithm | Bayesian posterior sampling with replica-averaged forward model [18] [72] | Handles sparse/noisy data and avoids overfitting. |
| Error Model | Gaussian and Student's likelihoods; treats uncertainties as nuisance parameters [18] | Robust to outliers and systematic errors in data. |
| Objective Function | BICePs score [18] [72] | Enables quantitative model selection and automated parameter optimization. |
| Analysis Tools | Automated convergence checking, visualization of populations and uncertainty distributions [72] | Streamlines the iterative refinement process. |
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents and Computational Tools
| Item | Function in Protocol |
|---|---|
| BICePs v2.0 Python Package | Core software for performing Bayesian ensemble reweighting and calculating the BICePs score [72]. |
| Molecular Simulation Software | Software used to generate the prior ensemble (e.g., GROMACS, AMBER, OpenMM). |
| Theoretical Prior Ensemble | A set of molecular conformations and their populations, as predicted by the current force field, serving as the prior ( p(X) ) [18] [72]. |
| Experimental Observables (( D )) | Sparse and/or noisy ensemble-averaged measurement data (e.g., from NMR) used as restraints [18] [72]. |
| Forward Model (( f(X) )) | A function that computes a predicted experimental observable from a given molecular conformation ( X ) [18] [72]. |
| Neural Network Potentials | Can be integrated into the BICePs framework, with parameters optimized through automatically calculated gradients [18]. |
Case Study: Force Field Refinement for a Lattice Model
To demonstrate the utility of the BICePs approach, we consider a test case involving a 12-mer HP (hydrophobic-polar) lattice model. In this system, multiple bead interaction strengths were refined using ensemble-averaged distance measurements as experimental restraints [18].
Application Protocol:
- System Setup: Define the sequence and possible conformations of the HP lattice model.
- Synthetic Data Generation: Generate "experimental" distance measurements from a target ensemble, optionally adding random and systematic errors to simulate realistic data conditions [18].
- Initial Parameterization: Start with an initial set of non-optimal interaction parameters for the HP model.
- Execute Optimization: Apply the BICePs refinement protocol. The algorithm successfully recovered the correct interaction parameters by minimizing the BICePs score, even in the presence of significant added error [18].
- Resilience Assessment: The performance was assessed through repeated optimizations under varying extents of experimental error, confirming the method's robustness [18].
This case study validates that variational optimization of the BICePs score is a promising direction for robust and automatic parameterization of molecular potentials, directly addressing the challenges of refining force fields against imperfect experimental data [18].
Benchmarking, Validation Protocols, and Cross-Platform Comparisons
In the development of machine learning force fields (MLFFs) through an iterative optimization procedure, establishing robust validation metrics is paramount to creating reliable, transferable, and physically realistic models. While low errors on energies and forces relative to quantum mechanical reference data are a necessary starting point, they are insufficient proxies for a force field's true predictive power in molecular simulations. A comprehensive validation strategy must extend to the accurate reproduction of experimentally observable physical properties, bridging the gap between electronic structure accuracy and macroscopic observables. This protocol details the establishment of a two-tiered validation framework, focusing first on Mean Absolute Error (MAE) for energy and forces, and second on the reproduction of key physical properties, providing a structured approach for researchers and drug development professionals engaged in force field development.
Core Validation Metrics: MAE for Energy and Forces
The primary layer of validation assesses a model's fidelity to its quantum mechanical training data. Mean Absolute Error (MAE) provides an intuitive and robust measure of this accuracy.
Metric Definitions and Calculations
Mean Absolute Error (MAE) is calculated as the average of the absolute differences between predicted values (e.g., forces) and their reference values [73]:
MAE = (1/n) * Σ|y_pred - y_true|
Root Mean Square Error (RMSE), while related, places a higher penalty on larger errors [73]:
RMSE = √[(1/n) * Σ(y_pred - y_true)²]
Table 1: Core Error Metrics for MLFF Validation
| Metric | Formula | Interpretation | Advantage |
|---|---|---|---|
| MAE | (1/n) * Σ|y_pred - y_true| |
Average magnitude of error | Intuitive, robust to outliers |
| RMSE | √[(1/n) * Σ(y_pred - y_true)²] |
Standard deviation of prediction errors | Punishes large errors more severely |
| MAE per Atom | MAE / Number of Atoms |
Normalizes energy errors for system size | Allows comparison across different systems |
| Relative MAE | MAE / (Range of True Values) |
Error relative to the data scale | Useful for contextualizing error magnitude |
Practical Application in Training
During model training, these metrics are tracked separately for training, validation, and test sets. For instance, the MACE framework reports a comprehensive suite of metrics including mae_e (energy MAE), mae_e_per_atom, mae_f (force MAE), and rel_mae_f (relative force MAE) on the validation set [74]. Monitoring these metrics helps in diagnosing model behavior:
- Convergence: A decreasing training MAE indicates the model is learning.
- Overfitting: A significant gap between training and validation MAE suggests overfitting, where the model memorizes training data rather than generalizing.
- Optimal Stopping: The validation MAE is typically used to determine the optimal point to stop training to prevent overfitting [74].
Protocol for Validating with Physical Property Reproduction
Accurate energies and forces do not guarantee that a force field will perform correctly in molecular dynamics (MD) simulations. The ultimate test is the reproduction of experimental physical properties.
Key Physical Properties for Validation
The following properties are critical for assessing a force field's performance in simulating condensed-phase behavior and should be calculated from MD simulations using the developed MLFF.
Table 2: Essential Physical Properties for MLFF Validation
| Property | Description | Significance in Force Field Validation | Example System |
|---|---|---|---|
| Dielectric Constant (ε) | Measure of a material's polarity and ability to screen electric fields [75] | Probes collective dipole moment alignment and long-range electrostatics; crucial for solvation | Organic coolants, solvents |
| Density (ρ) | Mass per unit volume at a given temperature and pressure | A fundamental thermodynamic property sensitive to van der Waals interactions and molecular packing | Liquids, molecular crystals |
| Heat Capacity (Cv/Cp) | Amount of heat required to change a system's temperature | Reflects the density of states and the ability to store thermal energy | Thermal storage materials |
| Viscosity (η) | A fluid's resistance to flow | Sensitive to intermolecular friction and short-range interactions | Electrolytes, lubricants |
| Thermal Conductivity (κ) | Ability to conduct heat | Indicates the efficiency of energy transfer through a material | Immersion coolants [75] |
Workflow for Physical Property Validation
The following diagram outlines the iterative procedure for validating a machine learning force field against physical properties.
Case Study: Predicting Dielectric Constant with Org-Mol
The Org-Mol model demonstrates the feasibility of predicting bulk properties from single-molecule inputs. While not a force field itself, its success highlights the critical link between molecular structure and collective properties that MLFFs must capture.
- Challenge: The static dielectric constant (ε) is a bulk property dependent on the collective orientation of molecular dipoles, as defined by fluctuation theory:
ε = 1 + (⟨M²⟩ - ⟨M⟩²) / (3ε₀Vk_BT)(where M is the total dipole moment) [75]. It cannot be accurately predicted from a single molecule's dipole moment alone, especially for molecules forming hydrogen bonds. - MLFF Solution: An MLFF, through MD simulation, naturally captures these collective dipole fluctuations, making it a superior approach for predicting ε compared to static quantum chemistry calculations.
- Performance Benchmark: The Org-Mol fine-tuned model, when applied to predict dielectric constants, achieved a test set R² value of 0.968, demonstrating high accuracy for this key property [75].
The Scientist's Toolkit: Essential Research Reagents & Solutions
This section details key computational tools, datasets, and metrics essential for developing and validating machine learning force fields.
Table 3: Essential Computational Toolkit for MLFF Development
| Tool/Resource | Type | Function in MLFF Workflow | Example/Reference |
|---|---|---|---|
| MLFF Frameworks | Software | Provides architectures and training loops for developing MLFFs | MACE [74] |
| Reference Datasets | Data | High-quality QM calculations for training and testing MLFFs | OMol25 [7], SPICE, ANI-2x |
| Error Metrics | Analysis Scripts | Quantify model accuracy on energies, forces, and stresses | MAE, RMSE, q95 [74] |
| Molecular Dynamics Engines | Software | Runs simulations using the fitted MLFF to generate trajectories | LAMMPS, OpenMM, ASE |
| Property Analysis Tools | Software & Scripts | Calculates physical properties from MD trajectories | Various in-house and community tools |
| Validation Experiments | Data (Experimental) | Experimental measurements of physical properties for final validation | Public databases, literature |
Integrated Validation Framework for Iterative Training
A robust force field development cycle requires the integration of energy/force and physical property validation into an iterative optimization procedure. The following diagram maps this comprehensive framework.
This framework emphasizes that achieving low MAE on a validation set of quantum mechanics data is only the first step [74]. The model must then be tested in the actual application domain (e.g., MD simulation) by comparing simulated physical properties against experimental data. Discrepancies at this stage inform the iterative refinement of the training set, often by adding new configurations that address the identified weaknesses, thereby closing the loop and leading to a more robust and generalizable force field.
Benchmarking Against Quantum Mechanical Calculations and Experimental Data
The accuracy of classical molecular dynamics (MD) simulations is fundamentally dependent on the quality of the force field (FF) used. Force fields are mathematical representations of the potential energy surface of a system, governing atomic interactions. Benchmarking these force fields against highly accurate quantum mechanical (QM) calculations and experimental data is a critical, iterative process in force field development and validation. This process ensures that the simplified models used in large-scale molecular simulations can reliably reproduce the properties and behaviors of complex molecular systems, a necessity for applications ranging from drug discovery to materials science.
Discrepancies of even 1 kcal/mol in binding affinity predictions can lead to erroneous conclusions in drug design, highlighting the need for rigorous benchmarking [76]. The benchmarking process typically involves a cyclic procedure of parameterization, validation against benchmark data, identification of shortcomings, and refinement, establishing a continuous feedback loop for iterative optimization [15].
Key Benchmarking Data and Methodologies
A robust benchmark relies on two primary classes of reference data: high-level quantum mechanical calculations and experimentally measured physicochemical properties.
Quantum Mechanical Benchmarks
Quantum mechanical methods provide a first-principles reference for intermolecular interactions, especially non-covalent interactions (NCIs) which are crucial for biomolecular recognition and binding.
The establishment of a "platinum standard" is a key objective, achieved by obtaining tight agreement between different high-level ab initio methods, such as LNO-CCSD(T) and FN-DMC [76]. This cross-validation reduces the uncertainty in the highest-level QM calculations. Specialized benchmark datasets like the "QUantum Interacting Dimer" (QUID) framework have been developed for this purpose. QUID contains 170 molecular dimers modeling chemically and structurally diverse ligand-pocket motifs, providing robust benchmark interaction energies [76].
Experimental Data for Validation
Experimental data provides the ultimate test for a force field's ability to predict real-world physical properties. The following table summarizes key experimental properties used in force field benchmarking.
Table 1: Key Experimental Properties for Force Field Benchmarking
| Property Category | Specific Properties | Significance in Benchmarking |
|---|---|---|
| Thermodynamic Properties | Liquid density (ρl), enthalpy of vaporization (ΔHvap), heat capacity [77] | Constrain LJ parameters; ΔHvap measures cohesive energy [15]. |
| Mixture Properties | Enthalpy of mixing (ΔHmix), density of binary mixtures (ρl(x)) [15] | Directly probe unlike interactions (A-B) between different molecules, crucial for transferability. |
| Transport Properties | Viscosity, thermal conductivity [77] | Sensitive to the dynamic behavior and energy transfer within the system, often challenging to reproduce. |
| Structural Properties | Radius of gyration (Rg), secondary structure propensity, contact maps [78] | For IDPs and proteins, assess the ability to sample correct conformational ensembles. |
Training against mixture data offers significant advantages. Enthalpies of mixing directly capture A-B interactions that enthalpies of vaporization for pure components miss. This is particularly important for correcting systematic errors in solute-solvent interactions [15].
The Iterative Optimization Workflow
The development of a reliable force field is not a linear process but an iterative cycle of refinement. The following diagram illustrates the core workflow for the iterative optimization training procedure.
Detailed Experimental Protocols
Protocol 1: Benchmarking on Quantum Mechanical Data
This protocol outlines the steps for validating a force field against a QM benchmark dataset like QUID [76].
- 1. Preparation of Benchmark Structures: Obtain the set of molecular dimer structures from the QUID framework (or a similar dataset like S66, S22). These typically include both equilibrium and non-equilibrium geometries along dissociation paths.
- 2. Quantum Mechanical Reference Calculation:
- Perform single-point energy calculations on each dimer and its isolated monomers using a high-level ab initio method (e.g., LNO-CCSD(T) or FN-DMC) to establish the reference interaction energy (Eint).
- Eint = Edimer - (Emonomer A + Emonomer B)
- 3. Force Field Energy Calculation:
- Using the same optimized dimer and monomer geometries, calculate the interaction energy with the force field under evaluation. This is typically a gas-phase energy calculation in MD software (e.g., LAMMPS, GROMACS) without periodic boundary conditions.
- 4. Data Analysis and Comparison:
- For each dimer, compute the deviation between the FF-predicted Eint and the QM reference value.
- Calculate statistical measures across the entire dataset: Mean Absolute Error (MAE), Root Mean Square Error (RMSE), and linear correlation coefficient (R²).
- Analyze if errors are systematic for specific interaction types (e.g., hydrogen bonds, π-π stacking).
Protocol 2: Benchmarking on Condensed-Phase Experimental Data
This protocol describes how to benchmark a force field against experimental liquid properties, which is standard practice for validating transferability [77] [15].
- 1. System Construction:
- For a pure liquid, build a simulation box containing several hundred molecules of the compound. For mixtures, build a box with the appropriate mole fraction of each component.
- Use Packmol or similar software for initial configuration generation [77].
- 2. Molecular Dynamics Simulation:
- Energy Minimization: Steepest descent or conjugate gradient method to remove bad contacts.
- Equilibration:
- Perform equilibration in the NPT ensemble (constant Number of particles, Pressure, and Temperature) at the target experimental condition (e.g., T=300 K, P=1 atm).
- Use a Langevin thermostat or Nosé-Hoover chain for temperature coupling and a Parrinello-Rahman barostat for pressure coupling.
- Monitor the potential energy and density for convergence.
- Production Run: Conduct a longer simulation in the NPT or NVT ensemble to collect trajectory data for analysis. Ensure simulation length is sufficient for property convergence.
- 3. Property Calculation:
- Density (ρ): Calculated as the average mass/volume of the simulation box during the NPT production run.
- Enthalpy of Vaporization (ΔHvap): Calculated as ΔHvap = ⟨Ugas⟩ - ⟨Uliquid⟩ + RT, where U is the potential energy, and R is the gas constant. This requires a separate gas-phase simulation for ⟨Ugas⟩ [15].
- Enthalpy of Mixing (ΔHmix): Calculated from simulations of the mixture and the pure components: ΔHmix = Hmix - x1H1 - x2H2, where H is the enthalpy [15].
- 4. Validation:
- Compare the simulated properties directly against experimental measurements.
- Statistical analysis (e.g., calculation of mean signed error, RMSE across multiple compounds or mixture compositions) quantifies overall performance.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Computational Tools for Force Field Benchmarking
| Tool Name | Type/Category | Primary Function in Benchmarking |
|---|---|---|
| LAMMPS | Molecular Dynamics Software | A highly flexible MD simulator for running simulations with various FFs and calculating properties [77]. |
| GROMACS | Molecular Dynamics Software | High-performance MD package, commonly used for biomolecular systems and force field development [78]. |
| VASP | Quantum Chemistry Software | Performs ab initio DFT calculations; can be used for on-the-fly machine-learning FF training and generating reference data [79]. |
| Packmol | System Builder | Prepares initial configurations for MD simulations by packing molecules into simulation boxes [77]. |
| QCHEM/Gaussian | Quantum Chemistry Software | Suite for performing high-level ab initio calculations (e.g., CCSD(T)) to generate benchmark interaction energies. |
| Moltemplate | Force Field Preprocessor | Aids in setting up complex molecular topologies and assigning force field parameters for simulation with LAMMPS [77]. |
| QUID/S66 Datasets | Benchmark Database | Provides curated sets of molecular structures with high-level QM reference interaction energies for validation [76]. |
Case Studies in Benchmarking
Case Study: Benchmarking Force Fields for Intrinsically Disordered Proteins
Benchmarking is essential for specialized systems like Intrinsically Disordered Proteins (IDPs), where standard protein force fields may fail. A 2023 study benchmarked 13 FFs on the R2-FUS-LC region, an IDP implicated in ALS [78].
The study used multiple metrics for a comprehensive evaluation: Radius of Gyration (Rg) for global compactness, secondary structure propensity, and intra-peptide contact maps for local structures. A combined score was calculated from these measures. The results demonstrated that force fields can be biased; some generated overly compact conformations while others were too extended. The top-performing force field, CHARMM36m2021 with the mTIP3P water model, was identified as the most balanced for simulating this IDP, highlighting how systematic benchmarking guides method selection for specific biological problems [78].
Case Study: Improving Accuracy with Binary Mixture Data
A 2022 study demonstrated the power of iterative optimization by retraining the Lennard-Jones parameters of the OpenFF 1.0.0 force field [15]. The traditional approach trains parameters against pure liquid properties (density and enthalpy of vaporization). However, this can lead to systematic errors in mixture properties.
The researchers introduced binary mixture data (densities and enthalpies of mixing) into the training set. The re-parameterized force field showed improved performance in reproducing mixture properties and solvation free energies, correcting errors that persisted when training on pure properties alone. This case study underscores a key principle of iterative optimization: expanding the training data to include more complex, chemically relevant targets leads to more robust and transferable force fields [15].
Force fields form the foundation of atomistic simulations, enabling the study of material properties, reaction mechanisms, and dynamic processes across chemistry, materials science, and drug development. The core challenge lies in balancing computational accuracy with efficiency, a trade-off that has driven the evolution from classical to reactive and, most recently, to machine learning force fields. Within the context of iterative optimization training procedures for force field development, understanding the distinct capabilities, limitations, and optimal application domains of each force field paradigm is crucial for researchers aiming to select or develop appropriate models for their specific systems.
This analysis provides a structured comparison of classical, ReaxFF, and machine learning force fields, focusing on their theoretical foundations, accuracy, computational efficiency, and robustness. We present quantitative performance data, detailed protocols for development and application, and visual workflows to guide researchers in navigating the complex landscape of modern force field methodologies.
Theoretical Foundations and Comparative Performance
The fundamental difference between these force field classes lies in their approach to describing interatomic interactions. Classical force fields use fixed bonding topologies and simplified potential functions to achieve high computational efficiency but cannot model chemical reactions [20]. Reactive force fields (ReaxFF) introduce bond-order formalism to allow dynamic bond formation and breaking, significantly expanding application scope while maintaining more efficiency than quantum methods [80]. Machine learning force fields (MLFFs) leverage data-driven approaches to capture complex quantum mechanical potential energy surfaces with high accuracy, though they face challenges in transferability and rare event sampling [9] [81].
Table 1: Fundamental Characteristics of Force Field Types
| Characteristic | Classical Force Fields | Reactive Force Fields (ReaxFF) | Machine Learning Force Fields (MLFFs) |
|---|---|---|---|
| Theoretical Basis | Predefined potential functions with fixed bonding | Bond-order formalism with dynamic connectivity | Data-driven approximation of quantum mechanical PES |
| Parameter Count | 10-100 parameters with clear physical meaning [20] | 100+ parameters [20] | 100,000+ parameters (neural network weights) [82] |
| Reactivity Capability | No bond breaking/formation | Explicit dynamic bonding | Can be enabled through training data |
| Computational Cost | Lowest (orders of magnitude faster than QM) [20] | Intermediate (between classical and QM) [80] | Higher than classical, but significantly lower than QM [9] |
| Primary Applications | Structural properties, conformational sampling, non-reactive MD | Combustion, catalysis, hydrolysis, chemical reactions [80] | Complex materials, properties requiring QM accuracy [9] [83] |
Table 2: Quantitative Performance Comparison Across Force Field Types
| Performance Metric | Classical Force Fields | Reactive Force Fields (ReaxFF) | Machine Learning Force Fields (MLFFs) |
|---|---|---|---|
| Energy Accuracy | System-dependent; limited by functional form | Can reach ~10-20% of QM accuracy for training set [20] | Near-DFT accuracy (e.g., MAE ~0.1 eV/atom for EMFF-2025) [9] |
| Force Accuracy | Adequate for equilibrium configurations | Reasonable for trained systems | High accuracy (e.g., MAE ~2 eV/Å for EMFF-2025) [9] |
| Time Scale | Nanoseconds to microseconds [20] | Picoseconds to nanoseconds [80] | Nanoseconds for large systems [82] |
| Length Scale | 10-100 nm for extended systems [20] | Tens of nanometers | Hundreds to thousands of atoms [83] |
| Transferability | Limited to similar systems | Moderate, requires reparameterization | High for trained chemical spaces [9] |
Methodological Protocols for Force Field Development and Application
Protocol 1: Development of Reactive Force Fields (ReaxFF)
The development of a ReaxFF parameter set requires careful construction of training data and iterative optimization to achieve quantum mechanical accuracy.
Step 1: Training Set Construction
- Select diverse molecular configurations (reactants, products, transition states) covering the target chemical space
- Include crystalline structures for solid-state materials (e.g., MgH₂ and Mg(OH)₂ for Mg/O/H systems) [80]
- Generate reference data using quantum mechanical methods (DFT, CCSD(T)) for energies, forces, and charges
Step 2: Parameter Optimization
- Initialize parameters from existing ReaxFF sets with similar elements
- Utilize optimization frameworks (e.g., JAX-ReaxFF gradient descent algorithm) [80]
- Minimize the difference between ReaxFF and QM predictions through iterative refinement
- Validate against non-training set configurations to ensure transferability
Step 3: Validation and Production MD
- Simulate relevant reactive systems (e.g., Mg nanoparticles in water) [80]
- Verify reproduction of key experimental observables (reaction rates, products)
- Perform production molecular dynamics to study mechanism and kinetics
Protocol 2: Development of Machine Learning Force Fields
MLFF development focuses on comprehensive dataset generation and neural network training to capture the quantum mechanical potential energy surface.
Step 1: Active Learning and Dataset Generation
- Perform ab initio molecular dynamics to sample representative configurations
- Implement active learning loops to selectively add informative configurations
- Include rare events and high-energy barriers to improve robustness [81]
- Apply transfer learning from pre-trained models (e.g., from DP-CHNO-2024 to EMFF-2025) [9]
Step 2: Neural Network Training
- Select appropriate architecture (e.g., Equivariant GNNs, Deep Potential, Allegro)
- Train on energies and forces using composite loss functions
- Incorporate physical constraints (e.g., ZBL repulsive potential) for short-range interactions [81]
- For multi-fidelity frameworks, integrate data from various accuracy levels [84]
Step 3: Validation and Deployment
- Validate against hold-out quantum mechanical data
- Test on target properties (e.g., polymer densities, transition temperatures) [83]
- Perform extended MD simulations to assess stability and prevent unphysical drift [81]
Protocol 3: Hybrid MLFF-Empirical Force Field Development
Integrating empirical physical constraints into MLFFs enhances robustness and training efficiency for complex material systems.
Step 1: Identify System-Specific Limitations
- Diagnose failure modes in standard MLFFs (e.g., unphysical atomic clustering in LLZO) [81]
- Determine critical interactions inadequately sampled in training data
Step 2: Incorporate Empirical Potentials
- Implement short-range repulsive potentials (e.g., ZBL) for high-energy barriers [81]
- Formulate hybrid energy function: ( E{\text{total}} = E{\text{MLFF}} + E_{\text{empirical}} )
- Use switching functions to ensure smooth transitions between domains
Step 3: Optimize Training Efficiency
- Leverage hybrid approach to reduce active learning iterations (from 13 to 3 for LLZO) [81]
- Train with smaller datasets (as few as 25 configurations for LLZO) [81]
- Maintain DFT-level accuracy while improving stability in extended simulations
Workflow Visualization
Force Field Selection Workflow
Iterative Force Field Optimization Procedure
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Software Tools and Computational Frameworks for Force Field Development
| Tool/Framework | Type | Primary Function | Application Examples |
|---|---|---|---|
| DP-GEN [9] | Software Framework | Automated active learning for MLFFs | Generating training datasets for EMFF-2025 [9] |
| JAX-ReaxFF [80] | Optimization Library | Gradient-based parameter optimization for ReaxFF | Developing Mg/O/H force field for hydrolysis studies [80] |
| Neuroevolution Potential (NEP) [81] | MLFF Framework | Efficient machine-learned interatomic potential | Hybrid NEP-ZBL for LLZO solid electrolyte [81] |
| Allegro/Vivace [83] | Equivariant GNN Architecture | Scalable MLFF for large systems | Polymer property prediction (densities, Tg) [83] |
| FFLOW [4] | Optimization Toolkit | Multiscale force field parameter optimization | Surrogate model-assisted parameter optimization [4] |
| Multi-Fidelity MLFF [84] | ML Framework | Integrating data of varying accuracies | Leveraging low-fidelity data to reduce high-fidelity needs [84] |
The comparative analysis of classical, ReaxFF, and machine learning force fields reveals a complex landscape where no single approach dominates across all applications. Classical force fields remain the most efficient choice for non-reactive systems at equilibrium, while ReaxFF provides a balanced approach for studying chemical reactions in large systems. Machine learning force fields offer unprecedented quantum mechanical accuracy but require sophisticated training protocols and careful validation.
The emerging trends of hybrid approaches, which integrate physical constraints with data-driven flexibility, and multi-fidelity frameworks, which efficiently leverage diverse data sources, represent the cutting edge of force field development. These methodologies significantly enhance robustness and training efficiency while maintaining high accuracy, addressing critical challenges in applying machine learning force fields to complex, real-world systems. For researchers engaged in iterative optimization training procedures, the strategic selection and combination of these approaches will be crucial for advancing force field capabilities across materials science, catalysis, and drug development.
Assessing Transferability and Predictive Power on Unseen Molecules or Conditions
The development of accurate and reliable force fields is a cornerstone of molecular modeling, impacting fields from drug discovery to materials science. The ultimate test of a force field's robustness lies not in its performance on its training data, but in its transferability and predictive power on unseen molecules or under novel thermodynamic conditions. This ability to generalize is a critical benchmark for practical application. This Application Note frames the assessment of transferability within an iterative optimization training procedure, a modern paradigm that cyclically improves a force field by identifying failures, augmenting training data, and retraining. We provide detailed protocols and resources to help researchers rigorously evaluate and enhance the predictive capabilities of their molecular models.
Key Concepts and Quantitative Benchmarks
A transferable force field must perform accurately across three key dimensions: chemical space (unseen molecular compositions and functionalities), conformational space (unexplored molecular geometries), and phase space (novel thermodynamic states and environments). Quantitative benchmarks against high-quality reference data, such as quantum mechanical (QM) calculations or experimental measurements, are essential for this assessment.
The table below summarizes core properties used to benchmark transferability and the performance of leading machine learning force fields (MLFFs) as reported in recent literature.
Table 1: Key Properties for Benchmarking Transferability and Reported Performance of Select MLFFs
| Property Category | Specific Metric | EMFF-2025 (CHNO HEMs) [9] | MACE-OFF (Organic Molecules) [85] [86] | ByteFF (Drug-like Molecules) [51] |
|---|---|---|---|---|
| *QM-Level Accuracy* | Energy Mean Absolute Error (MAE) | < 0.1 eV/atom | Not Specified | State-of-the-art on QM benchmarks |
| Force MAE | < 2.0 eV/Å | Not Specified | Excellent conformational forces | |
| *Condensed Phase Properties* | Density (Liquids/Crystals) | Accurately predicted for 20 HEMs | Excellent agreement for molecular liquids | Not Specified |
| Lattice Parameters/Enthalpies | Not Specified | Accurate for molecular crystals | Not Specified | |
| *Conformational Energetics* | Torsion Barrier Accuracy | Not Specified | Accurate, easy-to-converge scans | Excellent on torsion profiles |
| Relative Conformational Energies | Not Specified | Not Specified | Excellent accuracy | |
| *Complex System Dynamics* | Peptide/Protein Folding | Not Specified | Folding of Ala15; ns-scale simulation of solvated protein | Not Specified |
| Thermal Decomposition | Predicts pathways for HEMs | Not Specified | Not Specified |
Experimental Protocols for Assessment
A robust assessment requires a structured, multi-faceted experimental approach. The following protocols detail key experiments for evaluating transferability.
Protocol 1: Benchmarking on Curated External Datasets
Objective: To evaluate performance on a diverse set of molecules and properties not seen during training. Key Resource: Utilize publicly available, community-standard datasets.
- Steps:
- Dataset Selection: Acquire datasets focused on properties critical to your application. Examples include:
- GMTKN55: A diverse collection of main-group chemistry benchmarks [7].
- Wiggle150: A benchmark for supramolecular host-guest complexes [7].
- OMol25 Biomolecules/Electrolytes: Subsets of the Open Molecules 2025 dataset for specific chemical domains [87] [7].
- Protein Data Bank (PDB) structures: For benchmarking protein-ligand complexes and biomolecular dynamics [7] [88].
- Property Calculation: Use the force field to compute the target properties (e.g., energy, forces, geometric parameters, spectroscopic observables) for all entries in the external dataset.
- Error Analysis: Calculate error metrics (MAE, RMSE, R²) by comparing force field predictions against the reference QM or experimental data. Analyze errors for specific functional groups or system types to identify chemical space weaknesses.
- Dataset Selection: Acquire datasets focused on properties critical to your application. Examples include:
Protocol 2: Leave-Cluster-Out Cross-Validation
Objective: To simulate the challenge of predicting entirely new molecular scaffolds. Key Resource: A large, diverse in-house dataset of QM calculations.
- Steps:
- Cluster Molecules: Group molecules in your training set by scaffold or core scructure using a clustering algorithm (e.g., based on molecular fingerprints).
- Iterative Training and Validation:
- For each cluster, train a new force field model on all data excluding that specific cluster.
- Use the held-out cluster as a validation set to measure prediction error.
- Identify Blind Spots: Clusters that consistently yield high prediction errors represent "blind spots" in the model's chemical space coverage, guiding subsequent data generation in the iterative optimization cycle.
Protocol 3: Stability Testing of Long-Timescale Dynamics
Objective: To assess transferability under novel conformational and thermodynamic states explored during molecular dynamics (MD). Key Resource: High-performance computing (HPC) resources for MD simulations.
- Steps:
- Simulation Setup: Run microsecond-scale MD simulations of complex, biologically relevant systems (e.g., a solvated protein, a peptide known to fold/unfold) [85] [86].
- Stability Monitoring: Monitor the simulation for signs of instability, such as:
- Unphysical bond stretching or angle distortion.
- Numerical "explosions" due to grossly inaccurate forces.
- Drastic deviation from known native structures (e.g., protein unfolding at room temperature without denaturant).
- Property Validation: Calculate experimentally accessible observables from the simulation trajectory, such as:
- Analysis: A stable simulation that reproduces key experimental observables is a strong indicator of good transferability to the relevant conformational ensemble.
The Iterative Optimization Workflow
The assessment protocols are integrated into a larger, iterative workflow for force field development. This cycle uses performance failures on transferability tests to drive targeted improvements.
The following diagram visualizes this iterative optimization training procedure, incorporating the assessment protocols detailed above.
Diagram 1: Iterative force field optimization workflow. The cycle uses transferability assessment failures to guide targeted training data augmentation.
The Scientist's Toolkit: Essential Research Reagents
The following table details key software, datasets, and tools that form the essential "research reagents" for modern, data-driven force field development and assessment.
Table 2: Key Research Reagent Solutions for Force Field Assessment and Development
| Tool/Resource Name | Type | Primary Function | Relevance to Transferability |
|---|---|---|---|
| Open Molecules 2025 (OMol25) [87] [7] | Dataset | Massive, chemically diverse DFT dataset (100M+ snapshots). | Provides a supreme benchmark for testing generalization across biomolecules, electrolytes, and metal complexes. |
| Universal Model for Atoms (UMA) [7] | Pre-trained Model | A unified neural network potential trained on OMol25 and other datasets. | Serves as a high-quality baseline or starting point for transfer learning, enhancing initial transferability. |
| MACE-OFF [85] [86] | Pre-trained Force Field | Short-range, transferable MLFF for organic molecules. | A state-of-the-art model for benchmarking against, especially for organic conformational and condensed-phase properties. |
| DP-GEN [9] | Software | A framework for generating training data and training models like the Deep Potential. | Automates the iterative optimization process by actively learning from failures and exploring new configurations. |
| BICePs [18] | Software | Bayesian inference for reconciling simulation ensembles with experimental data. | Quantifies agreement with noisy, ensemble-averaged experimental data, a key test of predictive power. |
| FFLOW [4] | Software | A toolkit for multi-scale force field parameter optimization. | Enables gradient-based optimization of parameters against diverse target properties, improving accuracy. |
| ByteFF [51] | Data-driven Force Field | An Amber-compatible force field for drug-like molecules. | Demonstrates a modern data-driven parameterization method for expansive chemical space coverage. |
Community Standards and Best Practices for Rigorous Force Field Validation
Force fields form the computational foundation for molecular dynamics (MD) simulations, enabling the study of biological and chemical systems at atomic resolution. As force field applications expand into increasingly complex domains—from drug discovery to materials design—the systematic validation of their accuracy and transferability has become paramount. This document outlines community standards and best practices for rigorous force field validation, framed within the context of iterative optimization training procedures that have emerged as powerful paradigms for modern force field development.
The transition from static parametrization to dynamic optimization cycles, fueled by machine learning and active learning strategies, represents a fundamental shift in computational molecular science. These iterative frameworks enable continuous incorporation of new data and systematic addressing of identified deficiencies, driving force fields toward greater predictive accuracy and chemical diversity. The validation protocols detailed herein provide essential guidance for evaluating these evolving models against both computational benchmarks and experimental reality.
Foundational Principles of Force Field Validation
The Validation Hierarchy
Effective force field validation operates across multiple hierarchical levels, each testing different aspects of model performance:
- Quantum Mechanical Fidelity: Assessment against ab initio quantum mechanical calculations for energies, forces, and properties of small molecular systems or condensed phases.
- Experimental Agreement: Validation against experimentally measurable thermodynamic, structural, and dynamic properties across diverse chemical spaces.
- Transferability Testing: Evaluation of performance on systems and conditions not represented in training data.
- Production Stability: Testing robustness in extended molecular dynamics simulations under various thermodynamic conditions.
This multi-tiered approach ensures that force fields are not merely optimized for narrow benchmark performance but maintain physical reliability across the broad range of applications encountered in real-world research settings.
The Iterative Optimization Framework
Modern force field development has embraced iterative optimization procedures that systematically close the gap between model prediction and reality. As illustrated in Figure 1, this process creates a self-improving cycle where validation outcomes directly inform subsequent training iterations.
Figure 1. Iterative optimization workflow for force field development. This cycle integrates validation as a core component that drives continuous model improvement through targeted data augmentation and retraining.
The critical innovation of this framework is the tight coupling between validation and training. Rather than treating validation as a final checkpoint, it becomes an integral component that actively guides development priorities through quantitative deficiency identification.
Community Validation Standards
Computational Benchmarking Protocols
Computational benchmarks provide controlled comparisons against reference quantum mechanical calculations, serving as essential validation during early development stages. Standard protocols should include the following elements:
Energy and Force Accuracy: Prediction errors for atomic energies and forces compared to density functional theory (DFT) or higher-level quantum mechanical methods. The EMFF-2025 neural network potential, for instance, demonstrates mean absolute errors within ±0.1 eV/atom for energies and ±2 eV/Å for forces across diverse high-energy materials [9].
Chemical Space Coverage: Evaluation across diverse molecular systems to ensure balanced performance rather than excellence in narrow domains. The QDπ dataset exemplifies this approach, incorporating 1.6 million structures spanning 13 elements to maximize chemical diversity [89].
Structural Property Prediction: Assessment of equilibrium properties including bond lengths, angles, dihedral distributions, and crystal lattice parameters.
Table 1 summarizes key metrics and acceptance thresholds for computational benchmarks based on current community standards.
Table 1: Computational Benchmark Standards for Force Field Validation
| Validation Category | Specific Metrics | Performance Targets | Reference Methods |
|---|---|---|---|
| Energy/Force Accuracy | Mean Absolute Error (MAE) in Energy | < 0.1 eV/atom [9] | DFT (ωB97M-D3(BJ)/def2-TZVPPD) [9] [89] |
| Mean Absolute Error (MAE) in Forces | < 2 eV/Å [9] | DFT (ωB97M-D3(BJ)/def2-TZVPPD) [9] [89] | |
| Chemical Space Coverage | Elements Represented | 13+ elements (C,H,N,O, etc.) [89] | Diverse datasets (QDπ, SPICE, ANI) [89] |
| Structures in Training/Test | 1.6+ million structures [89] | Active learning selection [89] | |
| Structural Properties | Lattice Parameters | MAPE < 10% [90] | Experimental crystallography [90] |
| Density Prediction | MAPE < 2-10% [90] | Experimental measurements [90] |
Experimental Validation Standards
While computational benchmarks are essential for development efficiency, experimental validation remains the ultimate test of real-world applicability. Recent systematic evaluations have revealed a significant reality gap where force fields excelling on computational benchmarks show substantially higher errors when confronted with experimental complexity [90].
Essential experimental validation protocols include:
Structural Accuracy Assessment:
- Comparison of predicted crystal structures and densities against experimental crystallographic data
- Validation of solution-phase structural properties against nuclear magnetic resonance (NMR) measurements
- Assessment of temperature-dependent structural evolution
Thermodynamic Property Validation:
- Evaluation of free energy calculations against experimental binding affinities
- Comparison of phase behavior and transition temperatures
- Validation of solvation free energies and partition coefficients
Dynamic Property Assessment:
- Comparison of simulated dynamics with experimental relaxation measurements
- Validation of transport properties including diffusion coefficients and viscosity
- Assessment of conformational transition rates and pathways
Table 2 outlines experimental validation standards derived from recent systematic studies, including performance levels achieved by current state-of-the-art force fields.
Table 2: Experimental Validation Standards for Force Field Validation
| Experimental Property | Validation Metrics | Current Best Performance | Assessment Methods |
|---|---|---|---|
| Protein-Ligand Binding | MUE in Binding Affinity | 0.77-1.03 kcal/mol [91] | Free Energy Perturbation (FEP) [91] |
| NMR Observables | J Couplings & Chemical Shifts | ~524 measurements [92] | Weighted χ² comparison [92] |
| Mechanical Properties | Elastic Tensors/Moduli | Varies by material class [90] | Experimental mineral data [90] |
| Simulation Stability | MD Completion Rate | 75-100% [90] | Successful simulation metrics [90] |
| Structural Fidelity | Density MAPE | < 2% threshold [90] | Experimental crystallography [90] |
Best Practices for Validation Methodologies
Standardized Testing Protocols
Implementing consistent testing methodologies is crucial for meaningful cross-force field comparisons. The following protocols represent community best practices:
Molecular Dynamics Simulation Standards:
- Production simulation lengths sufficient to achieve convergence (typically 25+ ns for protein systems) [92]
- Multiple independent replicas with different initial velocities to assess statistical uncertainty
- Standardized temperature and pressure control methods (velocity rescaling thermostat, Parrinello-Rahman barostat)
- Consistent treatment of long-range electrostatics (Particle Mesh Ewald with 1.0 nm real-space cutoff)
Enhanced Sampling Methodologies:
- Hamiltonian replica exchange with solute tempering to overcome energy barriers
- Well-tempered metadynamics for efficient exploration of free energy landscapes
- Multiple walker algorithms to parallelize configuration space exploration
Statistical Analysis Methods:
- Block averaging to estimate statistical uncertainties and assess convergence
- Bootstrapping methods for robust error estimation
- Correlation analysis to identify interdependent error sources
Specialized Validation for Drug Discovery Applications
Force fields applied to drug discovery face unique validation challenges due to the diverse chemical space of drug-like molecules and critical dependence on accurate binding affinity predictions. Specialized protocols include:
Binding Free Energy Validation:
- Use of standardized benchmark sets (JACS set) covering diverse target classes [91]
- Calculation of cycle closures in alchemical transformation maps to assess internal consistency
- Comparison of multiple water models (SPC/E, TIP3P, TIP4P-EW) and partial charge methods (AM1-BCC, RESP) [91]
Force Field Performance Stratification:
- Separate assessment for different molecular classes (e.g., charged vs. neutral compounds, flexible vs. rigid scaffolds)
- Evaluation of transferability to novel chemotypes not represented in training data
- Testing on challenging molecular patterns with known force field limitations (e.g., conjugated systems, halogen bonds)
The implementation of these specialized protocols within the iterative optimization framework (Figure 1) has demonstrated significant improvements in force field performance for drug discovery applications [89] [91].
Implementation Protocols
The Scientist's Toolkit: Essential Research Reagents
Successful force field validation requires specialized computational tools and datasets. Table 3 catalogs essential resources that constitute the modern force field validation toolkit.
Table 3: Essential Research Reagents for Force Field Validation
| Tool Category | Specific Resources | Function/Purpose | Key Features |
|---|---|---|---|
| Reference Datasets | QDπ Dataset [89] | Training & validation for drug-like molecules | 1.6M structures, 13 elements, ωB97M-D3(BJ) theory [89] |
| MinX Dataset [90] | Experimental validation using minerals | ~1,500 structures, diverse chemical environments [90] | |
| Software Frameworks | DP-GEN [9] [89] | Active learning implementation | Automated data selection, reduces redundant calculations [9] |
| OpenMM [91] | MD simulation & FEP calculations | GPU acceleration, customizable force fields [91] | |
| Validation Benchmarks | UniFFBench [90] | Comprehensive UMLFF evaluation | Experimental grounding, multiple performance metrics [90] |
| JACS Benchmark Set [91] | Protein-ligand binding affinity | 8 protein targets, 330 edges [91] |
Integrated Validation Workflow
A robust validation protocol integrates multiple assessment types into a coherent workflow. Figure 2 illustrates this integrated approach, highlighting decision points and iteration triggers.
Figure 2. Integrated validation workflow for force field certification. This protocol emphasizes sequential testing with iterative return to development when performance thresholds are not met, ensuring rigorous assessment before deployment.
Documentation and Reporting Standards
Comprehensive documentation is essential for meaningful validation and community adoption. Standardized reporting should include:
Training Data Characterization:
- Complete description of data sources, sizes, and chemical diversity metrics
- Documentation of active learning strategies and selection criteria
- Analysis of potential biases and coverage gaps in training data
Performance Reporting:
- Complete error statistics (MAE, RMSE, R²) across all validation domains
- Transparent reporting of both successes and failure cases
- Comparison to appropriate baseline models and previous force field versions
Application Boundary Definition:
- Clear specification of supported element types, chemical environments, and temperature/pressure ranges
- Documentation of known limitations and problematic system types
- Guidelines for appropriate use within validated domains
The establishment of community standards and best practices for force field validation represents a critical advancement in computational molecular science. By implementing the systematic protocols outlined in this document—spanning computational benchmarking, experimental validation, and specialized application testing—the research community can accelerate the development of more reliable and transferable force fields.
The iterative optimization framework provides a powerful paradigm for continuous force field improvement, where validation outcomes directly inform training priorities. This approach, coupled with the growing availability of standardized benchmark datasets and validation tools, promises to close the reality gap between computational prediction and experimental observation.
As force field applications continue to expand into new domains, from drug discovery to materials design, these validation standards will serve as essential guides for ensuring predictive reliability and scientific impact. Through community adoption and continued refinement of these protocols, the next generation of force fields will deliver on the promise of accurate molecular simulation across the chemical universe.
Conclusion
Iterative optimization has emerged as a cornerstone of modern force field development, enabling the creation of highly accurate and transferable models essential for reliable molecular simulations. By integrating automated data generation, advanced optimization algorithms, and robust validation protocols, researchers can systematically overcome traditional limitations of parameterization. The convergence of classical force field concepts with machine learning methodologies promises a future where force fields can more seamlessly span multiple scales and chemical spaces. For biomedical research, these advances will directly enhance the precision of drug discovery efforts, from predicting protein-ligand binding affinities to modeling complex biological membranes, ultimately accelerating the path from in silico design to clinical application.
References
- 1. Force field (chemistry) [https://en.wikipedia.org/wiki/Force_field_(chemistry)]
- 2. Revisiting iterative molecular mechanics force field ... [https://chemrxiv.org/engage/chemrxiv/article-details/6882c20e23be8e43d6e0bca5]
- 3. Data-driven parametrization of molecular mechanics force ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06640e]
- 4. Speed up Multi‐Scale Force‐Field Parameter Optimization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530876/]
- 5. Exploration and validation of force field design protocols ... [https://pubs.rsc.org/en/content/articlelanding/2022/cp/d2cp02864f]
- 6. Exploration and Validation of Force Field Design Protocols ... [https://chemrxiv.org/engage/chemrxiv/article-details/61c0d3a31e13eb80bb0010ec]
- 7. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 8. [2505.08762] The Open Molecules 2025 (OMol25) Dataset, ... [https://arxiv.org/abs/2505.08762]
- 9. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 10. New method improves the accuracy of machine-learned ... [https://pme.uchicago.edu/news/new-method-improves-accuracy-machine-learned-potentials-simulating-catalysts]
- 11. NAMD - Scalable Molecular Dynamics [https://www.ks.uiuc.edu/Research//namd/]
- 12. Enabling Scalable AI-Driven Molecular Dynamics ... [https://developer.nvidia.com/blog/enabling-scalable-ai-driven-molecular-dynamics-simulations/]
- 13. drMD: Molecular Dynamics for Experimentalists [https://www.sciencedirect.com/science/article/pii/S0022283624005485]
- 14. Development and Validation of the Quantum Mechanical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6740169/]
- 15. Improving force field accuracy by training against condensed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9254460/]
- 16. Integration of experimental data and use of automated ... [https://www.nature.com/articles/s42004-022-00653-z]
- 17. Efficient and Precise Force Field Optimization for ... [https://arxiv.org/html/2406.09817v1]
- 18. Automated optimization of force field parameters against ... [https://arxiv.org/html/2402.11169v2]
- 19. Modeling the potential energy surface by force fields for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12581199/]
- 20. Modeling the potential energy surface by force fields for ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc02715b?page=search]
- 21. Implementing reactivity in molecular dynamics simulations ... [https://www.nature.com/articles/s41467-024-50793-0]
- 22. Pre-training machine learned interaction potentials with ... [https://arxiv.org/html/2509.14205v1]
- 23. GROW: A gradient-based optimization workflow for the ... [https://www.sciencedirect.com/science/article/abs/pii/S0010465509003452]
- 24. Evaluation and comparison of classical interatomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5283064/]
- 25. Development and Benchmarking of Open Force Field 2.0.0 [https://pmc.ncbi.nlm.nih.gov/articles/PMC10269353/]
- 26. Open Source, Enterprise-level Data Pipeline [https://openforcefield.org/community/news/science-updates/dataset_generation-openff-2022-06-13/]
- 27. How to train your force field [https://openforcefield.org/community/news/science-updates/ff-training-example-2021-07-01/]
- 28. openforcefield/openff-qcsubmit: Automated tools for ... [https://github.com/openforcefield/openff-qcsubmit]
- 29. OptimizationDataset — OpenFF QCSubmit 0.56.0+d20250519 ... [https://docs.openforcefield.org/projects/qcsubmit/en/stable/api/generated/openff.qcsubmit.datasets.OptimizationDataset.html]
- 30. The QCML dataset, Quantum chemistry reference data ... [https://www.nature.com/articles/s41597-025-04720-7]
- 31. openforcefield/qca-dataset-submission [https://github.com/openforcefield/qca-dataset-submission]
- 32. Speed up Multi-Scale Force-Field Parameter Optimization ... [https://pubmed.ncbi.nlm.nih.gov/40911030/]
- 33. An improved reactive force field parameter optimization ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025625001193]
- 34. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 35. Feasibility of Combining Biomolecular Conformational ... [https://pubmed.ncbi.nlm.nih.gov/40735954/]
- 36. AI-based Methods for Simulating, Sampling, and Predicting ... [https://arxiv.org/html/2509.17224v1]
- 37. Data Augmentation: The Ultimate Guide for 2025 [https://www.ultralytics.com/blog/the-ultimate-guide-to-data-augmentation-in-2025]
- 38. Data Augmentation and Hyperparameter Tuning for Low- ... [https://arxiv.org/html/2504.07024v1]
- 39. Force field optimization guided by small molecule crystal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8218654/]
- 40. Which Optimizer Should You Use With NNPs? | Rowan [https://www.rowansci.com/blog/which-optimizer-should-you-use-with-nnps]
- 41. Machine learning force fields for molecular liquids [https://www.nature.com/articles/s41524-023-01100-w]
- 42. Particle Swarm Optimisation: A Historical Review Up to the ... [https://www.mdpi.com/1099-4300/22/3/362]
- 43. Exploration and validation of force field design protocols ... [https://pubs.rsc.org/en/content/articlehtml/2022/cp/d2cp02864f]
- 44. Short Range Transferable Machine Learning Force Fields ... [https://arxiv.org/html/2312.15211v4]
- 45. Force field - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/functions/force-field.html]
- 46. Transferring chemical and energetic knowledge between ... [https://www.nature.com/articles/s42004-022-00790-5]
- 47. Molecular Forcefield Methods for Describing Energetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8912029/]
- 48. Machine learning-guided property prediction of energetic ... [https://www.sciencedirect.com/science/article/pii/S2666647222000628]
- 49. MMFF94 Validation Suite [https://ccl.net/cca/data/MMFF94/index.shtml]
- 50. Data-Driven Parametrization of Molecular Mechanics Force ... [https://arxiv.org/html/2408.12817v1]
- 51. Data-driven parametrization of molecular mechanics force ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc06640e]
- 52. Tutorial 04 - Optimizing Force Fields — OpenFF Evaluator ... [https://docs.openforcefield.org/projects/evaluator/en/v0.1.1/tutorials/tutorial04.html]
- 53. 13.2 Force fields and energy minimization [https://fiveable.me/biophysics/unit-13/force-fields-energy-minimization/study-guide/sRyXoZVpb0lWneUi]
- 54. JOYCE3.0: A General Protocol for the Specific ... [https://chemrxiv.org/engage/chemrxiv/article-details/677eba45fa469535b936286a]
- 55. Optimize [https://pymolwiki.org/index.php/Optimize]
- 56. Cerius2 Forcefield Based Simulations - Minimization [http://www.chem.cmu.edu/courses/09-560/docs/msi/ffbsim/4_Minimize.html]
- 57. Examining Iterative Convergence [https://www.grc.nasa.gov/www/wind/valid/tutorial/iterconv.html]
- 58. Convergence Criterion - an overview [https://www.sciencedirect.com/topics/engineering/convergence-criterion]
- 59. “Convergence criteria for nonlinear problems,” Section 7.2.3 [https://classes.engineering.wustl.edu/2009/spring/mase5513/abaqus/docs/v6.6/books/usb/pt03ch07s02aus43.html]
- 60. 6 Continuous Optimisation with Iterative Algorithms (*) [https://lmlcr.gagolewski.com/continuous-optimisation-with-iterative-algorithms.html]
- 61. Accelerating charge estimation in molecular dynamics ... [https://www.nature.com/articles/s41524-024-01495-0]
- 62. Constructing surrogates for atomistic simulations via deep ... [https://link.springer.com/article/10.1557/s43578-025-01571-1]
- 63. GLOW: A workflow integrating Gaussian accelerated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9773012/]
- 64. INDEEDopt: a deep learning-based ReaxFF ... [https://www.nature.com/articles/s41524-021-00534-4]
- 65. Accelerated MD - KBbox: Methods [https://kbbox.h-its.org/toolbox/methods/molecular-simulation/accelerated-md/]
- 66. How Well Do Molecular Dynamics Force Fields Model ... [https://pubmed.ncbi.nlm.nih.gov/40766644/]
- 67. Bayesian learning for accurate and robust biomolecular ... [https://arxiv.org/html/2511.05398v1]
- 68. Robust training of machine learning interatomic potentials ... [https://www.nature.com/articles/s41524-024-01227-4]
- 69. Learning Force Field Parameters from Differentiable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11267579/]
- 70. Knowledge graph-enhanced molecular contrastive ... [https://www.nature.com/articles/s42256-023-00654-0]
- 71. Machine Learned Force Fields: Fundamentals, its reach, ... [https://arxiv.org/html/2503.05845v1]
- 72. BICePs v2.0: Software for Ensemble Reweighting using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10278562/]
- 73. Evaluation Metrics for forecasting | by Slava Koval [https://medium.com/@slava.K./evaluation-metrics-for-forecasting-c9cc9f6894b6]
- 74. How can I get mae and rmse of validation and training #379 [https://github.com/ACEsuit/mace/discussions/379]
- 75. High-accuracy physical property prediction for pure ... [https://www.nature.com/articles/s41524-025-01720-4]
- 76. Extending quantum-mechanical benchmark accuracy to ... [https://www.nature.com/articles/s41467-025-63587-9]
- 77. Force-Field Benchmark for Polydimethylsiloxane: Density, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11831649/]
- 78. Benchmarking of force fields to characterize the intrinsically ... [https://www.nature.com/articles/s41598-023-40801-6]
- 79. Best practices for machine-learned force fields [https://www.vasp.at/wiki/index.php/Best_practices_for_machine-learned_force_fields]
- 80. Development of a ReaxFF reactive force-field modeling for ... [https://www.sciencedirect.com/science/article/abs/pii/S0169433225009213]
- 81. Improving robustness and training efficiency of machine ... [https://arxiv.org/html/2504.15925v1]
- 82. Crash testing machine learning force fields for molecules ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06529h]
- 83. Simulation of Polymers with Machine Learning Force ... [https://arxiv.org/html/2510.13696v1]
- 84. Multi-Fidelity ML Force Field Framework [https://www.emergentmind.com/topics/multi-fidelity-machine-learning-force-field-framework]
- 85. MACE-OFF: Short Range Transferable Machine Learning ... [https://arxiv.org/html/2312.15211v5]
- 86. MACE-OFF: Short-Range Transferable Machine Learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12123624/]
- 87. A Record-Breaking Dataset to Train AI Models has Launched [https://newscenter.lbl.gov/2025/05/14/computational-chemistry-unlocked-a-record-breaking-dataset-to-train-ai-models-has-launched/]
- 88. Structure-Based Experimental Datasets for Benchmarking ... [https://livecomsjournal.org/index.php/livecoms/article/view/v6i1e3871]
- 89. The QDπ dataset, training data for drug-like molecules and ... [https://www.nature.com/articles/s41597-025-04972-3]
- 90. Evaluating Universal Machine Learning Force Fields ... [https://arxiv.org/html/2508.05762v1]
- 91. Assessing the effect of forcefield parameter sets on the accuracy of... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9549959/]
- 92. Are Protein Force Fields Getting Better? A Systematic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3383641/]