Implicit vs Explicit Solvent Models for Small Protein Simulations: A Practical Guide for Computational Researchers
This article provides a comprehensive analysis of implicit and explicit solvent models for molecular dynamics simulations of small proteins, targeting researchers and drug development professionals.
Implicit vs Explicit Solvent Models for Small Protein Simulations: A Practical Guide for Computational Researchers
Abstract
This article provides a comprehensive analysis of implicit and explicit solvent models for molecular dynamics simulations of small proteins, targeting researchers and drug development professionals. It covers foundational principles of both approaches, explores methodological implementations and specific applications like protein folding and structure refinement, addresses common troubleshooting and optimization strategies, and validates methods through comparative performance analysis. By synthesizing recent advances and practical considerations, this guide aims to equip scientists with the knowledge to select appropriate solvent models for their specific research objectives in computational biophysics and drug discovery.
Fundamental Principles: Understanding Solvent Representation in Biomolecular Simulations
In molecular dynamics (MD) simulations, the choice of how to represent the solvent environment is pivotal, placing researchers at a crossroads between computational efficiency and physical realism. This document frames this critical decision within the broader thesis of implicit versus explicit solvent models for small protein research. While implicit models approximate the solvent as a continuous dielectric medium, explicit solvent models represent water molecules at an atomic level, offering a more physically realistic, albeit computationally demanding, depiction of the solvated environment [1]. For research and drug development applications where accurate portrayal of specific solute-solvent interactions—such as hydrogen bonding, hydrophobic effects, and solvent structure—is non-negotiable, explicit models provide an indispensable level of detail [2] [3]. This application note details the protocols, resources, and quantitative data necessary for the effective deployment of explicit solvent models in the study of small proteins.
Comparative Analysis: Explicit vs. Implicit Solvent Models
The core distinction between solvent models lies in their representation of solvent molecules. Implicit solvent models, also known as continuum models, replace explicit solvent molecules with a homogeneously polarizable medium characterized primarily by its dielectric constant [4] [1]. In contrast, explicit solvent models incorporate individual, atomistically detailed solvent molecules, allowing for direct and specific interactions with the solute [1].
The table below summarizes the key characteristics and comparative performance of these two approaches.
Table 1: Quantitative Comparison of Explicit and Implicit Solvent Models for Protein Simulations
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Solvent Representation | Individual, atomistic water molecules (e.g., TIP3P, SPC) [1] | Continuum dielectric medium (e.g., PCM, GB, COSMO) [4] [1] |
| Physical Realism | High; captures specific H-bonding, solvation shells, and hydrophobic effect [2] [3] | Lower; misses atomic-level details and solvent structure [2] |
| Computational Cost | High (often >80% of computation spent on solvent) [3] | Low (no solvent degrees of freedom) [3] |
| Sampling Speed | Slower due to solvent viscosity [3] | Faster; conformational search is accelerated [3] |
| Handling of Non-Covalent Interactions | Directly models specific solute-solvent interactions [5] | Approximated; can fail to capture local fluctuations [1] |
| Performance in Aggregation Prediction | 97% success rate in identifying small molecule aggregators [5] | Less successful; Generalized Born model misclassified many compounds [5] |
| Application Example | Predicting aggregation propensity of diverse chemical structures [5] | Calculating solvation energies of small molecules and proteins [4] |
The choice between these models is not merely a trade-off between cost and accuracy but should be guided by the specific scientific question. Explicit solvents are paramount for processes where the atomic details of hydration are critical, while implicit solvents can be sufficient for large-scale conformational sampling where specific solvent effects are secondary.
Application Notes: Key Use Cases for Explicit Solvents
Predicting Small Molecule Aggregation Propensity
Small colloidally aggregating molecules (SCAMs) are a major source of false positives in drug discovery. Molecular dynamics simulations with explicit solvent have proven highly effective in predicting this aggregation propensity. A 2024 study demonstrated that explicit solvent MD simulations achieved a 97% success rate (31/32 molecules correctly classified) in distinguishing aggregators from non-aggregators. This performance surpassed cheminformatics-based filters like Aggregator Advisor (75%) and ChemAGG (72%) [5]. The key advantage of the explicit solvent approach is its fundamental, physics-based route to prediction, which does not rely on fitting to existing aggregation data and can thus be applied to novel chemical spaces. Furthermore, these simulations provide dynamic information on the scale and nature of aggregate formation [5].
Enhancing Realism in Constant-pH Molecular Dynamics
The incorporation of explicit solvent is crucial for advancing constant-pH molecular dynamics techniques, which aim to model the protonation state changes of biomolecules in response to their environment. A landmark study introduced titratable water, a method that directly couples proton titration of the solute to the interconversion between water and hydroxide/hydronium ions [6]. This approach, which requires an explicit solvent representation, offers a more physically realistic model of proton exchange between solute and solvent. The technique demonstrated impressive accuracy, calculating pKa values for three proteins with average absolute errors of just 0.7 pH units [6], opening new avenues for studying biological phenomena involving proton translocation.
Machine Learning Potentials for Chemical Reactions in Solution
Machine learning potentials (MLPs) are emerging as powerful tools for modeling complex chemical processes, and their application in explicit solvent is a frontier of research. A 2024 study presented a general active learning strategy for generating MLPs to model reactions like the Diels-Alder cycloaddition in explicit water and methanol [2]. This approach combines active learning with descriptor-based selectors to efficiently build training sets that capture the relevant chemical and conformational space. The generated MLPs provided reaction rates in agreement with experimental data and allowed for a detailed analysis of solvent effects on the reaction mechanism, showcasing the power of combining explicit solvent representation with advanced ML techniques for studying solution-phase chemistry [2].
Experimental Protocols
Protocol 1: Predicting Small Molecule Aggregation
This protocol outlines the procedure for using explicit solvent MD simulations to assess the aggregation propensity of small molecules, as validated in recent literature [5].
Workflow Overview:
Detailed Methodology:
System Construction
- Force Field Assignment: Assign parameters to the solute compounds using the general AMBER force field (GAFF2) with AM1-BCC partial charges, for example using the
antechamberprogram [5]. - Simulation Box: Place 11-12 solute molecules in an octahedral box with an approximate edge length of 180 Å. This corresponds to millimolar concentrations, ensuring conditions above the critical aggregation concentration [5].
- Solvation and Ions: Solvate the system with explicit TIP3P water molecules and add counterions to neutralize the system's net charge [5].
- Mimicking Experimental Conditions: Add 5% v/v dimethyl sulfoxide (DMSO) and 50 mM sodium chloride to the solvated system to accurately model common experimental assay conditions [5].
- Force Field Assignment: Assign parameters to the solute compounds using the general AMBER force field (GAFF2) with AM1-BCC partial charges, for example using the
MD Simulation Protocol
- Energy Minimization: Perform an initial energy minimization to remove any steric clashes.
- Heating: Heat the system in two phases under NVT conditions using a Langevin thermostat. First, heat from 0 to 500 K over 20 ps to ensure proper solute distribution, then cool from 500 K down to the target temperature of 300 K over another 20 ps [5].
- Equilibration: Equilibrate the system for 2 ns in the NPT ensemble (constant number of particles, pressure, and temperature) at 300 K and 1 atm [5].
- Production Simulation: Run the production simulation for a duration of 100 ns to 1 µs at 300 K, saving trajectory frames every 20 ps for subsequent analysis [5].
Analysis of Aggregation
- Cluster Analysis: Use tools like
cpptrajor custom scripts (e.g., in Python) to perform clustering analysis on the production trajectory. A molecule is assigned to a cluster if any of its heavy or hydrogen atoms are within a 3.0 Å cutoff of an atom from another molecule in the same cluster [5]. - Propensity Determination: Analyze the population distributions as a function of cluster size (Nc) across the trajectory. A molecule is classified as an aggregator if stable, multi-molecule clusters (Nc ≥ 2) form and persist throughout the simulation [5].
- Cluster Analysis: Use tools like
Protocol 2: Modeling Reactions with Machine Learning Potentials
This protocol describes a general active learning (AL) strategy for generating machine learning potentials to model chemical processes in explicit solvents [2].
Workflow Overview:
Detailed Methodology:
Initial Data Set Generation
- Create two initial training sets:
- Gas Phase/Implicit Set: Generate configurations of the reacting substrates by randomly displacing atomic coordinates, starting from relevant states like the transition state (TS) [2].
- Explicit Solvent Set: Generate configurations containing the substrate and explicit solvent molecules. This can be done using cluster models (a handful of solvent molecules placed at relevant positions) or periodic boundary conditions (PBC). Cluster data is often more feasible and offers good transferability to bulk systems [2].
- Create two initial training sets:
Active Learning Loop
- Initial MLP Training: Train the initial machine learning potential (e.g., using Atomic Cluster Expansion - ACE) on the small, initially labeled dataset [2].
- ML-MD Simulation: Use the current MLP to run molecular dynamics simulations, generating new configurations.
- Structure Selection: Employ descriptor-based selectors (e.g., Smooth Overlap of Atomic Positions - SOAP) to identify structures generated by the ML-MD that are underrepresented in the current training set. These selectors evaluate the coverage of the training set in the chemical space of interest [2].
- QM Reference Calculation: Perform accurate quantum mechanical (QM) calculations to obtain reference energies and forces for the newly selected structures.
- Data Set Augmentation and Retraining: Add the new labeled structures to the training set and retrain the MLP. This loop is repeated until the MLP's performance converges and no new, critically under-sampled regions are found [2].
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools for Explicit Solvent Simulations
| Item / Resource | Function / Description | Example Applications |
|---|---|---|
| AMBER Software Suite | A comprehensive package for MD simulations, including tools for system setup (e.g., antechamber), simulation, and analysis [5]. |
Energy minimization, heating, equilibration, and production MD simulations [5]. |
| General AMBER Force Field (GAFF2) | A force field providing parameters for organic molecules, enabling simulation of a wide range of drug-like compounds [5]. | Parametrization of small molecule solutes for aggregation studies [5]. |
| TIP3P Water Model | A widely used 3-site explicit water model offering a good balance of accuracy and computational efficiency [5] [1]. | Solvation of proteins and small molecules in aqueous solution [5]. |
| Active Learning (AL) Workflow | A strategy for efficiently building training sets for Machine Learning Potentials by iteratively identifying and labeling the most informative data points [2]. | Developing accurate MLPs for chemical reactions in explicit solvent [2]. |
| Cluster Analysis Scripts | Tools for analyzing MD trajectories to identify and characterize molecular clusters based on intermolecular distances (e.g., cpptraj, custom Python scripts) [5]. |
Determining the size and population of small molecule aggregates in solution [5]. |
| Machine Learning Potentials (MLPs) | Surrogates for QM methods that offer near-quantum accuracy at a fraction of the computational cost, enabling high-fidelity reactive simulations [2]. | Modeling bond formation/breaking and detailed reaction mechanisms in explicit solvent [2]. |
Explicit solvent models remain the gold standard for molecular dynamics simulations where atomic-level detail of hydration and specific solute-solvent interactions is critical. While computationally intensive, their superior physical realism, as evidenced by high accuracy in predicting phenomena like small molecule aggregation and enabling advanced techniques like constant-pH MD with titratable water, justifies their application in targeted research. The emergence of machine learning potentials, trained through efficient active learning workflows, is poised to further expand the boundaries of what is possible with explicit solvent simulations, making the routine and accurate modeling of complex chemical processes in solution an increasingly attainable goal for researchers and drug developers.
Implicit solvation, often termed continuum solvation, is a computational method that represents the solvent as a continuous medium rather than individual explicit solvent molecules [7]. This approach is fundamentally based on applying the potential of mean force to approximate the averaged behavior of highly dynamic solvent molecules in liquids [7] [8]. For researchers investigating small protein dynamics, these models offer a strategic compromise, replacing the computational burden of simulating thousands of explicit water molecules with a streamlined representation of solvation effects [9] [10].
The core justification for continuum models lies in their treatment of solvent as a dielectric continuum characterized by macroscopic properties such as dielectric constant, enabling efficient estimation of solvation energies [9] [10]. This methodology has become indispensable in molecular dynamics simulations and other molecular mechanics applications, particularly for estimating free energies of solute-solvent interactions in structural and chemical processes including protein folding, conformational transitions, and ligand association [7] [8]. By eliminating the need for explicit solvent degrees of freedom, implicit models significantly accelerate conformational sampling, a critical advantage for studying processes that occur on microsecond to millisecond timescales [11] [12].
Theoretical Foundations of Implicit Solvation
Fundamental Energy Components
The solvation free energy (ΔGsolv) is the central thermodynamic quantity in implicit modeling, representing the free energy change associated with transferring a solute molecule from a vacuum to a solvent [7] [8]. This energy is traditionally partitioned into physically distinct components, though the exact partitioning varies across different theoretical frameworks [9] [10].
Table 1: Components of Solvation Free Energy in Implicit Models
| Component | Symbol | Physical Meaning | Common Calculation Method |
|---|---|---|---|
| Cavitation | ΔGcav | Energy to create cavity in solvent for solute | Solvent-accessible surface area (SASA) |
| van der Waals | ΔGvdW | Dispersion and repulsion interactions between solute and solvent | SASA or volume-based terms |
| Electrostatic | ΔGele | Polar interactions between solute charge distribution and dielectric solvent | Poisson-Boltzmann, Generalized Born |
| Nonpolar (Composite) | ΔGnp | Combined cavitation and van der Waals contributions | SASA with atomic solvation parameters |
The most comprehensive partition separates ΔGsolv into three components [8] [9]: [ \Delta G{solv} = \Delta G{cav} + \Delta G{ele} + \Delta G{vdW} ] where ΔGcav represents the energy cost of creating a cavity in the solvent to accommodate the solute molecule, ΔGele accounts for electrostatic interactions between the solute and solvent, and ΔGvdW describes van der Waals interactions [8].
Alternatively, a simpler two-component framework combines the non-electrostatic terms [9] [10]: [ \Delta G{solv} = \Delta G{np} + \Delta G_{ele} ] where ΔGnp encompasses both cavitation and van der Waals contributions [9].
Key Implicit Solvent Methodologies
Poisson-Boltzmann Equation
The Poisson-Boltzmann (PB) equation provides a rigorous continuum description of electrostatic interactions between solutes and their dielectric environment, incorporating spatial variations in dielectric properties and ionic strength [7] [9]. The general form of the PB equation in cgs units is: [ \vec{\nabla} \cdot \left[\epsilon(\vec{r})\vec{\nabla}\Psi(\vec{r})\right] = -4\pi\rho^{f}(\vec{r}) - 4\pi\sumi ci^\infty zi q \lambda(\vec{r}) e^{\frac{-zi q\Psi(\vec{r})}{kT}} ] where ε((\vec{r})) is the dielectric constant, Ψ((\vec{r})) is the electrostatic potential, ρf((\vec{r})) is the fixed charge density, ci∞ is the bulk concentration of ion i, zi is its valence, and λ((\vec{r})) is a masking function for the position-dependent accessibility of position (\vec{r}) to ions [7].
Although the PB equation has solid theoretical justification, it is computationally expensive to solve without approximations, prompting the development of various numerical solvers [7]. Despite advancements, PB solvers generally do not yet match the computational efficiency of the more approximate Generalized Born method [7].
Generalized Born Model
The Generalized Born (GB) model is an efficient approximation to the linearized Poisson-Boltzmann equation that models the solute as a set of spheres with internal dielectric constants differing from the external solvent [7] [8]. The fundamental functional form of the GB electrostatic solvation energy is: [ Gs = -\frac{1}{8\pi\epsilon0}\left(1-\frac{1}{\epsilon}\right)\sum{i,j}^{N}\frac{qi qj}{f{GB}} ] where [ f{GB} = \sqrt{r{ij}^2 + a{ij}^2 e^{-D}} \quad \text{and} \quad D = \left(\frac{r{ij}}{2a{ij}}\right)^2, \quad a{ij} = \sqrt{ai aj} ] Here, ε0 is the vacuum permittivity, ε is the solvent dielectric constant, qi are atomic charges, rij is the distance between atoms i and j, and ai is the Born radius of atom i [7].
The GB model is particularly attractive for molecular dynamics simulations due to its computational efficiency, often providing substantial speedups in conformational sampling compared to explicit solvent simulations [7] [12]. This efficiency makes GB especially valuable for studying protein folding and large-scale conformational changes [12].
Solvent-Accessible Surface Area Methods
Accessible Surface Area (ASA) methods employ a empirically-derived linear relationship between solvation free energy and the solvent-accessible surface area of solute atoms [7] [8]. The basic formulation is: [ \Delta G{solv} = \sumi \sigmai ASAi ] where ASAi is the solvent-accessible surface area of atom i, and σi is an atom-specific solvation parameter [7]. These parameters were initially derived from water-gas partition data but newer parameters have been developed from octanol-water partition coefficients or other similar data, making them suitable for describing transfer energies between condensed media [7].
SASA methods operate directly with free energy of solvation, unlike molecular mechanics or electrostatic methods that include only the enthalpic component [7]. These models are frequently combined with GB or PB electrostatics in what are termed GB/SASA or PB/SASA approaches [7] [8].
Practical Implementation and Protocols
Experimental Protocol: GBSA for Protein Folding Studies
Objective: Utilize the Generalized Born model augmented with surface area terms (GBSA) to simulate the folding pathway of a small protein or miniprotein.
Materials and Computational Setup:
- Molecular System: Protein structure in PDB format (experimental or predicted)
- Software Requirements: MD package with implicit solvent capability (e.g., AMBER, CHARMM, GROMACS)
- Force Field: Compatible protein force field (e.g., AMBER ff19SB, CHARMM36m)
- Implicit Solvent Model: Generalized Born with appropriate parameter set (e.g., GBOBC)
- Surface Area Model: SASA with appropriate atomic radii (e.g., LCPO algorithm)
Step-by-Step Procedure:
System Preparation:
- Obtain initial protein coordinates from experimental structure or prediction tools (e.g., AlphaFold) [13]
- Process structure using appropriate tools (e.g., PDB2PQR, tleap) to add missing atoms, hydrogens, and determine protonation states
- Parameterize the system using the selected force field
Solvation Model Configuration:
- Select GB model variant appropriate for the biological system
- Configure dielectric constants: internal (εint = 1-4), external (εext = 78.5 for water)
- Set SASA method with appropriate surface tension coefficient (typically 0.005-0.007 kcal/mol/Ų)
- Configure ionic strength if simulating physiological conditions (typically 0.15M NaCl)
Simulation Protocol:
- Energy minimization using steepest descent (5,000 steps) followed by conjugate gradient (5,000 steps)
- Gradual heating from 0K to target temperature (300K) over 100ps with weak restraints on protein heavy atoms
- Equilibration at constant temperature (300K) and pressure (1atm) for 1ns
- Production MD simulation using Langevin dynamics with appropriate collision frequency (γ = 1-5 ps-1)
- Extended sampling (100ns-1µs) depending on system size and research question
Analysis Methods:
- Monitor root-mean-square deviation (RMSD) to assess structural stability
- Calculate radius of gyration to track compaction
- Employ clustering algorithms to identify dominant conformations
- Compute free energy landscapes using essential dynamics
Troubleshooting Notes:
- Over-stabilization of salt bridges may indicate insufficient electrostatic screening; consider adjusting ionic strength or GB parameters
- Excessive helix formation may require verification against experimental data and potential force field adjustments
- Unphysical conformational changes may necessitate longer equilibration or alternative initial conditions
Performance Considerations and Optimization
The computational efficiency of implicit solvent models provides significant advantages for specific applications. Speedup factors relative to explicit solvent simulations are highly system-dependent but can be substantial [12]:
Table 2: Comparative Performance of Implicit vs. Explicit Solvent Models
| System Type | Conformational Change Scale | Sampling Speedup* | Key Advantages | Limitations |
|---|---|---|---|---|
| Small Proteins | Dihedral angle flips, sidechain rearrangements | ~1x | Reduced setup complexity | Limited accuracy for specific interactions |
| Miniprotein Folding | Secondary structure formation, collapse | ~7x | Accelerated folding events | Potential bias in native state stability |
| Nucleic Acid Systems | DNA unwrapping, tail collapse | ~1-100x | Efficient sampling of polyelectrolyte behavior | Challenges with ion effects |
| Protein-Protein Association | Binding interface formation, docking | ~1-60x | Rapid exploration of binding modes | Accuracy dependent on interface polarity |
Speedup factors represent conformational sampling efficiency, not computational cost [12]
The performance advantages stem from two factors: reduction in solvent viscosity allowing faster molecular motion, and elimination of explicit solvent degrees of freedom reducing computational overhead [12]. For small systems, the algorithmic efficiency of implicit solvent can provide additional speedup, though this advantage diminishes for larger systems where the cost of surface area and Born radius calculations becomes significant [12].
Table 3: Key Computational Tools for Continuum Solvent Simulations
| Tool/Resource | Function | Application Context |
|---|---|---|
| APBS | Solves Poisson-Boltzmann equation numerically | Electrostatic analysis, binding energy calculations |
| DelPhi | Finite-difference PB solver with focus on biomolecules | Detailed electrostatic mapping, pKa calculations |
| GBOBC | Generalized Born model with Onufriev-Bashford-Case parameters | MD simulations of proteins, efficient conformational sampling |
| SASA/LCPO | Linear Combination of Pairwise Overlaps algorithm | Surface area calculation for nonpolar contributions |
| MM/PBSA | Molecular Mechanics/Poisson-Boltzmann Surface Area | End-state binding free energy calculations |
| MM/GBSA | Molecular Mechanics/Generalized Born Surface Area | Binding affinity ranking, virtual screening |
| COSMO | Conductor-like Screening Model | Quantum chemical calculations with solvation effects |
| PCM | Polarizable Continuum Model | Spectroscopy, reaction modeling in solution |
Applications in Protein Research and Drug Development
Continuum solvent approaches have enabled significant advances in protein science and pharmaceutical development. In protein-ligand binding studies, implicit solvation methods are routinely employed to estimate binding free energies, rank inhibitor potency, and guide lead optimization [9] [10]. The MM/GBSA and MM/PBSA methodologies, which combine molecular mechanics with implicit solvation energies, have become particularly valuable for virtual screening campaigns where computational efficiency is essential for evaluating large compound libraries [7] [9].
For intrinsically disordered proteins (IDPs) that lack stable tertiary structure, implicit solvents enable efficient exploration of vast conformational landscapes that would be prohibitively expensive with explicit solvent [9]. This capability has proven crucial for connecting sequence features with structural ensembles and comparing computational results with experimental data from techniques such as FRET or SAXS [9].
Recent advances in hybrid quantum-continuum approaches allow researchers to incorporate solvation effects directly into electronic structure calculations, with implications for studying enzyme catalysis and photochemical processes [9] [10]. These methods combine quantum mechanical treatment of the solute with continuum descriptions of the solvent, providing a balanced approach to modeling chemically demanding systems where electronic polarization effects are significant [14].
The integration of machine learning techniques with traditional continuum models represents a promising frontier, where ML approaches serve as PB-accurate surrogates, learn solvent-averaged potentials for MD, or supply residual corrections to GB/PB baselines [9]. These hybrid approaches aim to preserve the computational efficiency of implicit solvent while addressing specific limitations through data-driven corrections.
Limitations and Strategic Considerations
Despite their utility, continuum solvent models present several important limitations that researchers must consider when designing simulation studies. A significant challenge is the accurate representation of the hydrophobic effect, which is mostly entropic in nature at physiological temperatures and occurs primarily on the solvent side [7]. While often approximated using solvent-accessible surface area terms with coefficients between 5 and 45 cal/(Ų mol), this treatment remains incomplete [7].
Implicit models also lack the viscosity that water molecules impart through random collisions, which can lead to artificially accelerated dynamics [7]. While this accelerated sampling is often desirable, it produces misleading results when kinetics are of interest [7] [12]. Viscosity may be partially reintroduced using Langevin dynamics with appropriate damping constants, though careful parameterization is required [7].
Specific solute-solvent interactions, particularly hydrogen bonds in the first solvation shell, pose another challenge for continuum representations [7] [14]. While their average energetic contribution can be reproduced with parameterized implicit models, directional and specific interactions often require hybrid approaches that include explicit water molecules in critical locations [7].
The parameter sensitivity of implicit models presents additional challenges, as accuracy depends strongly on the choice of atomic radii, dielectric constants, and empirical coefficients [9]. This sensitivity is particularly pronounced for heterogeneous environments like biological membranes or mixed solvents, where assigning appropriate dielectric properties becomes non-trivial [7] [8].
For protein-protein interaction studies, recent evaluations suggest caution when using predicted structures from AI tools like AlphaFold3 as starting points for thermodynamic analysis, as subtle structural inaccuracies can significantly impact binding affinity predictions despite high overall structural quality metrics [13]. These findings highlight the importance of validating implicit solvent simulations against experimental data when available.
When applied strategically with awareness of their limitations, continuum solvent approaches provide powerful tools for exploring biomolecular structure, dynamics, and interactions across temporal and spatial scales that remain challenging for explicit solvent representations.
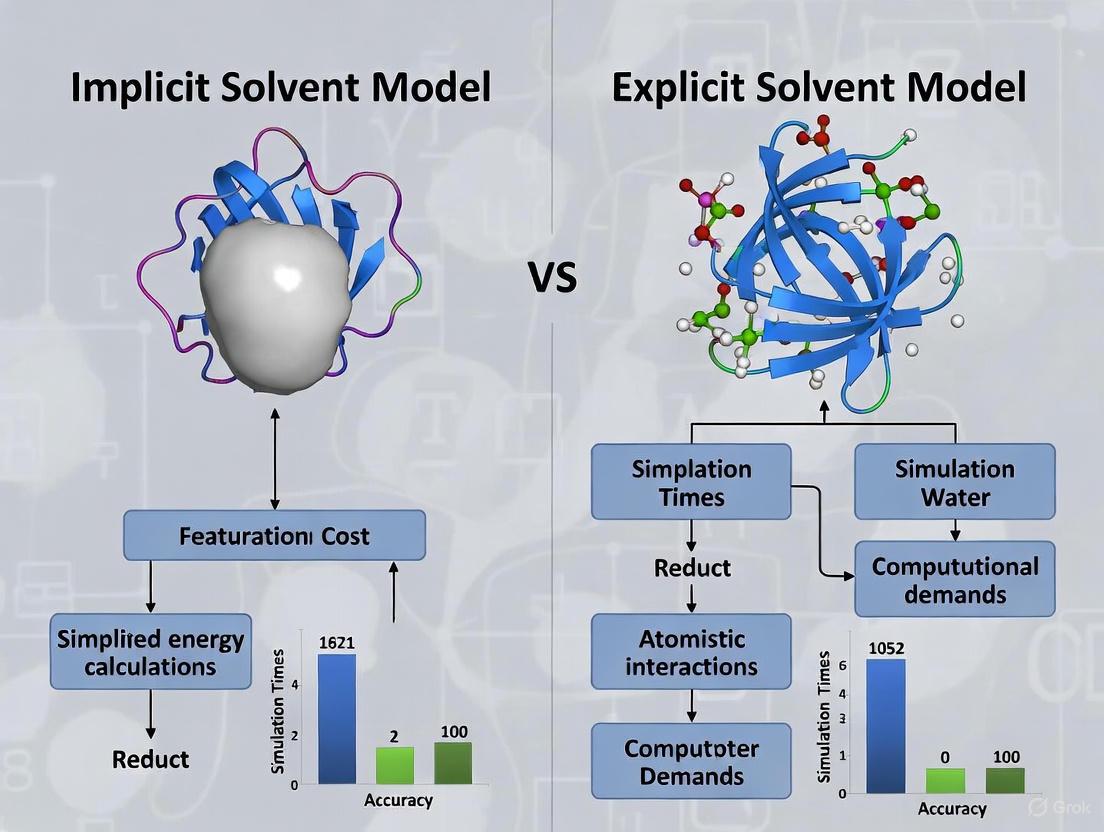
The treatment of the solvent environment is a pivotal consideration in molecular dynamics (MD) simulations of proteins, presenting a fundamental trade-off between computational efficiency and physical accuracy. Researchers must navigate a spectrum of approaches, from explicit solvent models, which treat each solvent molecule as a discrete entity, to implicit solvent models, which replace the solvent with a continuous, polarizable medium [1]. For simulations of small proteins, where resources may be limited or high-throughput screening is desired, this choice critically influences the scope, scale, and reliability of the scientific insights that can be obtained. This application note delineates the core differences between these approaches, provides quantitative comparisons of their performance, and outlines detailed protocols for their application in the study of small protein systems, all within the context of rational drug design.
Core Model Comparison: Explicit vs. Implicit Solvation
At the heart of the accuracy-efficiency trade-off are the distinct underlying physical approximations of explicit and implicit solvent models. The following table summarizes their fundamental characteristics.
Table 1: Fundamental Characteristics of Explicit and Implicit Solvent Models
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Fundamental Representation | Discrete solvent molecules (e.g., TIP3P, SPC water) [1] [15] | Continuous dielectric medium [1] [9] |
| Key Interactions Captured | Specific, atomic-level interactions (e.g., hydrogen bonds, water bridging) [16] [8] | Mean-field electrostatic and non-polar effects [1] [9] |
| Typical Computational Cost | High (70-90% of computation spent on solvent) [3] | Low (dramatically reduced degrees of freedom) [8] [3] |
| Solvent Viscosity & Dynamics | Preserved, can slow conformational sampling [3] | Can be effectively turned off, accelerating sampling [3] |
| Suitability for Sampling | Realistic dynamics, but limited by time scales [16] | Efficient for conformational search and large-scale transitions [3] |
Implicit solvents, also known as continuum models, embed the solute in a cavity surrounded by a homogeneously polarizable medium characterized primarily by its dielectric constant (ε) [1]. The solvation free energy (ΔGsolv) is typically partitioned into polar (electrostatic) and non-polar components. The polar component is calculated by solving the Poisson-Boltzmann (PB) equation or its Generalized Born (GB) approximation, while the non-polar component is often modeled as being proportional to the Solvent-Accessible Surface Area (SASA) [8] [9]. In contrast, explicit models utilize classical force fields, which include terms for bond stretching, angle bending, torsions, and non-bonded interactions (Lennard-Jones and Coulomb potentials), to simulate the interactions of every solvent molecule with the solute and with each other [1] [15].
The following diagram illustrates the core structural difference between these two modeling approaches.
Diagram 1: Representation of Explicit vs. Implicit Solvent Models
Quantitative Performance Benchmarks
The theoretical trade-offs between solvent models manifest in concrete performance differences in practical applications. The table below benchmarks various solvent treatment methods based on recent studies for tasks like binding free energy estimation and conformational sampling.
Table 2: Performance Benchmarks of Different Solvent Treatment Methods
| Method | Application Context | Reported Performance | Computational Cost & Notes |
|---|---|---|---|
| Explicit Solvent (TIP3P-FB) [17] | Conformational sampling of 9 proteins (10-224 residues) | Ground truth for benchmarking; uses extensive sampling (4 ns/starting point) | Very high; requires explicit water, ions, PME for electrostatics [17] |
| QM/MM-GBSA (Single Trajectory) [18] | Binding affinity for 16 benzimidazole inhibitors vs. FabI | R² = 0.88 (vs. experiment) with 6 ns MD trajectories [18] | High; QM calculations add significant cost over pure MM |
| QM/MM on M2 Conformers (QCharge-MC-FEPr) [19] | Binding free energy for 203 ligands across 9 targets | MAE = 0.60 kcal/mol, R = 0.81 (vs. experiment) [19] | Moderate; more efficient than FEP, uses implicit solvent for sampling |
| MM/GBSA (Multiple Sampling) [18] | Binding affinity for 16 benzimidazole inhibitors vs. FabI | R² = 0.84 (vs. experiment) with six 0.25 ns MD simulations [18] | Lower; "Multiple independent sampling" improves robustness/cost |
| Alchemical FEP (FEP+) [19] | Binding free energy for 199 ligands across 8 targets | MAE = 0.8-1.2 kcal/mol, R = 0.5-0.9 (vs. experiment) [19] | Very High; considered a high-accuracy but costly benchmark |
These benchmarks highlight a consistent theme: higher accuracy often comes with a steep computational price. Methods like alchemical Free Energy Perturbation (FEP) in explicit solvent are considered gold standards but are often computationally prohibitive for large-scale virtual screening [19]. Hybrid strategies, such as combining implicit solvent conformational sampling with subsequent QM/MM refinement, offer a promising middle ground, achieving accuracy comparable to expensive methods at a fraction of the cost [19] [18].
Detailed Application Protocols
Protocol for MM/GBSA Binding Affinity Estimation
This protocol is ideal for the intermediate-throughput ranking of lead compounds during a drug discovery campaign [18].
System Preparation:
- Obtain the protein structure from the PDB. Prepare it using a tool like
pdbfixerto add missing atoms, residues, and protons, setting protonation states appropriate for pH 7.0 [17]. - Prepare the ligand structure, assigning partial charges with the AM1-BCC method or via RESP fitting after quantum chemical optimization [18].
- Parameterize the protein and ligand using a force field like AMBER FF14SB/GAFF or CHARMM36 [17] [18].
- Obtain the protein structure from the PDB. Prepare it using a tool like
Explicit Solvent MD Simulation (for sampling):
- Solvate the protein-ligand complex in an orthorhombic water box (e.g., TIP3P) with a 10 Å buffer.
- Add ions to neutralize the system's charge and bring the ionic concentration to a physiological level (e.g., 0.15 M NaCl).
- Perform a standard minimization, heating, and equilibration protocol using PME for long-range electrostatics.
- Run a production MD simulation (e.g., 1-10 ns) under NPT conditions (300 K, 1 bar) to sample the complex's conformational space. Save hundreds to thousands of snapshots for subsequent analysis [18].
MM/GBSA Calculation:
- Extract snapshots evenly from the stable portion of the MD trajectory.
- For each snapshot, calculate the binding free energy using the MM/GBSA formula:
ΔGbind = Gcomplex - (Gprotein + Gligand) ≈ ΔEMM + ΔGGB + ΔGSA - TΔS
- ΔEMM: Gas-phase interaction energy (electrostatic + van der Waals) from the force field.
- ΔGGB: Polar solvation energy calculated via the Generalized Born equation.
- ΔGSA: Non-polar solvation energy, often estimated from SASA (γ × SASA + β).
- TΔS: Conformational entropy change, often omitted for relative ranking due to high cost and noise [18].
- The final binding affinity is the average ΔG_bind across all snapshots.
Protocol for Hybrid QM/MM-Based Free Energy Estimation
This protocol leverages the sampling efficiency of implicit solvent with the electronic accuracy of QM for high-accuracy, absolute binding free energy prediction [19].
Initial Implicit Solvent Sampling (Mining Minima):
- Perform a conformational search for the protein-ligand complex using an implicit solvent model (e.g., GB/SA) to identify multiple low-energy binding poses (conformers). This step is performed with classical force field atomic charges [19].
QM/MM Charge Refinement:
- Select the most probable conformers (e.g., those representing >80% of the Boltzmann population).
- For each selected conformer, set up a QM/MM calculation where the ligand is treated with a QM method (e.g., DFT) and the protein environment is treated with an MM force field.
- Compute a new, polarized set of atomic charges for the ligand (e.g., ESP charges) derived from the QM/MM electron density. This captures charge transfer and polarization effects from the binding site [19].
Free Energy Processing (FEPr):
- Replace the original force field charges on the ligand in the selected conformers with the new QM/MM-derived ESP charges.
- Recalculate the free energy of the complex, protein, and ligand using the implicit solvent model, but now with the refined charges. This step does not typically involve a new conformational search.
- Compute the final absolute binding free energy as a Boltzmann-weighted average over the free energies of the selected conformers. A universal scaling factor (e.g., 0.2) may be applied to the calculated ΔG to align with experimental absolute values [19].
The workflow for this hybrid protocol is summarized below.
Diagram 2: Hybrid QM/MM Free Energy Estimation Workflow
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Software Tools and Parameters for Solvent Model Implementation
| Category | Item / Software | Key Function / Application Note |
|---|---|---|
| Simulation Engines | AMBER [18], GROMACS, OpenMM [17], CHARMM | Core software for running MD simulations with both explicit and implicit solvent capabilities. OpenMM allows for GPU acceleration. |
| Implicit Solvent Models | Generalized Born (GB) [8] [3] (e.g., GBOBC, GBneck2) | Efficient approximation for polar solvation; different radii sets (mbondi, mbondi2) impact accuracy [18]. |
| Poisson-Boltzmann (PB) [8] [9] Solvers (e.g., APBS, DelPhi) | More rigorous, but computationally heavier, solution for electrostatic solvation. | |
| SASA Models [8] [9] | Computes non-polar solvation energy; often used in conjunction with GB/PB (i.e., GB/SA). | |
| Force Fields | AMBER (FF14SB, FF19SB) [17], CHARMM36, OPLS-AA | Provide parameters for protein, nucleic acids, and lipids. AMBER FF14SB combined with GAFF for small molecules is a common choice [18]. |
| Explicit Water Models | TIP3P [17], TIP4P [15], SPC [1] | 3-site or 4-site rigid water models for explicit solvation. TIP3P is one of the most widely used. |
| Analysis & Workflows | MMPBSA.py (AMBER Tools) [18] | Automated tool for performing MM/PBSA and MM/GBSA calculations from MD trajectories. |
| WESTPA [17] | Weighted Ensemble Simulation Toolkit for enhanced sampling of rare events. | |
| Quantum Mechanics | Gaussian, ORCA | Software for QM calculations to derive polarized charges for QM/MM protocols [19] [18]. |
The choice between implicit and explicit solvent models is not a matter of identifying a single superior option, but rather of selecting the right tool for the specific research question and computational budget.
For large-scale conformational sampling, rapid screening of ligands, or simulations of large systems, implicit solvent models (MM/GBSA) offer an unparalleled balance of speed and reasonable accuracy [3] [18]. Their ability to mitigate solvent viscosity allows for more efficient exploration of conformational space.
When high accuracy for binding affinity prediction is paramount and sufficient resources are available, hybrid QM/MM protocols that leverage implicit solvent for sampling and QM for charge refinement provide an excellent compromise, achieving accuracy rivaling explicit solvent FEP at lower cost [19].
Finally, for studying processes that rely on specific, atomic-level solvent interactions (e.g., hydrogen bonding networks, water-mediated binding, or ion transport), explicit solvent simulations remain the indispensable gold standard, despite their high computational cost [1] [16].
By understanding these trade-offs and applying the structured protocols outlined herein, researchers can make informed, strategic decisions to maximize the impact of their computational investigations in small protein research and drug development.
The choice between implicit and explicit solvent models is a fundamental consideration in molecular dynamics (MD) simulations of small protein systems, directly influencing the accuracy, computational cost, and biological relevance of the research. Implicit solvent models treat the solvent as a continuous dielectric medium, replacing discrete solvent molecules with a mathematical representation of their average influence [9] [20]. In contrast, explicit solvent models represent each solvent molecule, typically water, as an individual interacting particle, often using models like TIP3P, TIP4P, or OPC [21]. For researchers and drug development professionals, selecting the appropriate model requires balancing physical fidelity against computational demand. This application note provides a structured framework for this decision-making process, detailing specific use cases, providing executable protocols, and comparing performance characteristics for small protein systems.
Comparative Analysis of Solvent Approaches
The table below summarizes the core characteristics, performance, and optimal application spaces for implicit and explicit solvent models in small protein simulations.
Table 1: Comparative Analysis of Implicit vs. Explicit Solvent Models for Small Protein Systems
| Feature | Implicit Solvent Models | Explicit Solvent Models |
|---|---|---|
| Fundamental Approach | Continuum dielectric medium [9] [20] | Discrete solvent molecules (e.g., TIP3P, TIP4P, OPC) [21] |
| Computational Speed | High (Orders of magnitude faster than explicit) [9] [22] | Low (Computationally expensive) [22] |
| Sampling Efficiency | Excellent for rapid conformational exploration [9] | Limited by solvation dynamics and slower timescales |
| Key Strengths | - Binding free energy estimates (MM-PBSA/GBSA) [9]- Rapid folding/unfolding landscapes [22]- Sampling of intrinsically disordered proteins (IDPs) [9] | - Accurate specific solvent interactions (H-bonds, water bridges) [21]- Realistic dielectric relaxation- Detailed solvation structure |
| Key Limitations | - Poor capture of specific solvent effects [9]- Inaccurate ion-specific effects [9]- Simplified treatment of solvent entropy [9] | - High computational cost limits sampling [22]- Sensitivity to water model parameterization [21] |
| Ideal Application Spaces | - High-throughput ligand screening [9]- Long-timescale conformational changes- Initial stages of drug discovery | - Detailed binding mechanism studies- Systems with crucial water-mediated interactions- Final validation of high-priority candidates |
Experimental Protocols and Workflows
Protocol for Implicit Solvent MD Simulation
This protocol is adapted for running simulations using implicit solvent models, which is often the default in many MD packages for certain types of calculations [9] [23].
System Preparation:
- Obtain the initial protein structure from a PDB file.
- Protonation: Assign protonation states to residues using a tool like
PDB2PQRor the protein ionization tools in BIOVIA Discovery Studio, ensuring correct states for histidines and other titratable groups at the target pH [23]. - Force Field Selection: Choose an appropriate force field (e.g.,
charmm36,amber/ff14SB) [23] [24].
Energy Minimization:
- Objective: Remove bad steric clashes and prepare the system for dynamics.
- Method: Perform energy minimization using a steepest descent algorithm. Typically, 5,000 steps are sufficient for a small protein system [24].
- Restraints: Light positional restraints may be applied to the protein backbone during initial steps.
System Equilibration:
- Solvent Model: Set the implicit solvent model, typically a Generalized Born (GB) model variant [9].
- Thermalization: Gradually heat the system to the target temperature (e.g., 298.15 K) using a Langevin integrator with a low collision rate (e.g., 1.0/ps) [24].
- Equilibration Run: Run a short equilibration simulation (e.g., 10-100 ps) in the NVT ensemble to stabilize the temperature.
Production MD:
- Production Run: Execute the production simulation. Due to the speed of implicit solvent, longer timescales (hundreds of nanoseconds to microseconds) can be achieved more readily [9].
- Analysis: Analyze trajectories for properties like root-mean-square deviation (RMSD), radius of gyration, and conformational clustering.
Protocol for Explicit Solvent MD Simulation
This protocol outlines the key steps for setting up and running a simulation with explicit solvent, which provides a more detailed physical model [21] [24].
System Preparation:
- Protein Preparation: Follow the same initial steps as the implicit solvent protocol (PDB loading, protonation).
- Solvation: Place the protein in an explicit solvent box (e.g., cubic or octahedral) with a sufficient padding distance (e.g., 1.2 nm) from the protein to its nearest box edge [24].
- Solvent Model: Choose an explicit water model. TIP3P is a common default, but OPC or TIP4P/2005 may offer improved accuracy for specific properties [21].
- Neutralization: Add ions (e.g., Na⁺, Cl⁻) to neutralize the system's net charge and to achieve a desired physiological concentration (e.g., 0.15 M NaCl) [24].
Energy Minimization:
System Equilibration:
- NVT Equilibration: Equilibrate the system with restraints on the protein at the target temperature (e.g., 300 K) for 100-125 ps. This allows the solvent and ions to relax around the protein [21].
- NPT Equilibration: Further equilibrate the system without restraints in the isothermal-isobaric ensemble (NPT) for 100 ps - 1 ns to stabilize the density (pressure ~1 atm) [24].
Production MD:
- Production Run: Run the production simulation. The length will be constrained by computational resources, but for small proteins, 100 ns to 1 µs is often targeted.
- Trajectory Output: Save frames at an interval appropriate for the dynamics of interest (e.g., every 100 ps) [24].
The Scientist's Toolkit: Essential Research Reagents and Software
Successful execution of MD simulations relies on a suite of software tools and theoretical models. The table below catalogs key resources referenced in the protocols and literature.
Table 2: Essential Research Reagents and Software Solutions
| Tool/Model Name | Type | Primary Function | Application Context |
|---|---|---|---|
| CHARMM36m [23] [21] | Force Field | Defines potential energy functions for proteins, nucleic acids, and lipids. | Accurate simulation of biomolecules; particularly noted for glycosaminoglycans like heparin [21]. |
| AMBER/ff14SB [24] | Force Field | A standard force field for proteins and DNA. | Often used in protein-ligand simulations with explicit solvent [24]. |
| TIP3P [21] [24] | Explicit Water Model | A 3-site model for water molecules. | Common default; offers a balance of efficiency and reliability [21]. |
| OPC [21] | Explicit Water Model | A 4-site model optimized for accurate water properties. | Improved accuracy for global features of biomolecules like heparin [21]. |
| Generalized Born (GB) [9] [23] | Implicit Solvent Model | Approximates Poisson-Boltzmann electrostatics for rapid calculation of solvation energies. | Core of many implicit solvent simulations; used in MD and docking [9] [23]. |
| OpenMM [25] [24] | MD Simulation Engine | A high-performance toolkit for molecular simulation. | Used in pipelines like drMD and OpenFE for running simulations on GPUs [25] [24]. |
| BIOVIA Discovery Studio [23] | Software Suite | Integrated environment for simulation and analysis, utilizing CHARMm and NAMD. | Provides tools for simulation, protein preparation, and binding energy calculations [23]. |
| LSNN (λ-Solvation Neural Network) [26] | Machine Learning Model | A graph neural network for predicting solvation free energies. | Emerging tool for achieving explicit-solvent accuracy with implicit-solvent speed [26]. |
Application Selection Guide and Decision Framework
Choosing between implicit and explicit solvents is not a binary decision but a strategic one based on the specific scientific question. The following guide provides actionable recommendations.
Choose IMPLICIT Solvent for These Applications:
- High-Throughput Virtual Screening: When you need to rapidly rank hundreds or thousands of small molecules for binding affinity to a protein target. The speed of implicit models enables the use of methods like MM-PBSA or MM-GBSA [9].
- Exploring Large Conformational Changes: For studies of protein folding, unfolding, or the dynamics of intrinsically disordered proteins (IDPs), where the computational efficiency of implicit solvent allows for enhanced sampling of the conformational landscape [9] [22].
- Rapid Hypothesis Testing: In the early stages of a project, to quickly test the stability of a protein mutant or a proposed protein-ligand complex before committing resources to more expensive explicit solvent simulations [9].
Choose EXPLICIT Solvent for These Applications:
- Studying Detailed Binding Mechanisms: When the specific role of water molecules is critical, such as in characterizing water bridges mediating ligand-protein interactions, or probing the stability of hydrogen-bonding networks [21].
- Validation and Final Analysis: For producing high-quality, publication-ready results for a select number of key systems, providing a more physically realistic and defensible representation of the solvated system [21].
- Systems with Dense Electrostatic Interactions: For simulating molecules like highly sulfated glycosaminoglycans (e.g., heparin), where the explicit treatment of water and ions is crucial for accurate conformational dynamics [21].
Consider HYBRID or ADVANCED Models for Future Work:
- Machine Learning-Augmented Models: Emerging approaches like LSNN aim to bridge the accuracy-speed gap by using neural networks to learn highly accurate implicit solvent potentials from explicit solvent data, showing promise for direct free energy calculations [26].
- Coarse-Grained (CG) Models: For very large systems or extremely long timescales, CG models can offer a compromise, with some recent machine-learned models showing impressive transferability across protein sequences [22].
Practical Implementation: Setting Up and Running Simulations with Different Solvent Models
Implicit solvent models are a critical class of coarse-grained models that significantly reduce the computational expense of molecular dynamics (MD) simulations by representing the solvent as a continuous medium rather than explicit molecules [27]. Among these, Generalized Born (GB) models stand out for their favorable balance between computational efficiency and accuracy, making them particularly valuable for studies involving small proteins and drug discovery applications where high-throughput screening or extensive conformational sampling is required [28]. The fundamental approximation of the GB model is expressed in the equation developed by Still et al. [29]:
where q_i and q_j are partial atomic charges, r_ij is the distance between atoms i and j, R_i and R_j are the effective Born radii of atoms i and j, and ε is the dielectric constant of the solvent [29]. The accuracy of a GB model heavily depends on the method used to calculate these effective Born radii, which has led to the development of various "flavors" including GB-HCT, GB-OBC, and GB-Neck series [29].
Generalized Born Model Variants: Theoretical Foundations and Evolution
GB-HCT (Hawkins, Cramer, Truhlar)
The GB-HCT model, developed by Hawkins, Cramer, and Truhlar, employs a pairwise descreening approximation to calculate effective Born radii [29]. This model uses the van der Waals surface to define the solute-solvent boundary, making it computationally efficient but prone to underestimating the effective Born radii of buried atoms within proteins [30]. This limitation arises from the creation of false high-dielectric regions in the protein interior [30]. Despite this drawback, GB-HCT remains implemented in major MD packages like AMBER and CHARMM due to its computational speed [28].
GB-OBC (Onufriev, Bashford, Case)
The GB-OBC model introduced empirical correction parameters (α, β, γ) to address the limitations of GB-HCT, specifically applying a scaling function to increase the effective Born radii of buried atoms [29]. The correction takes the form:
where $\tilde{\rho}_i$ represents the modified intrinsic radius, and ψ is a parameter related to the degree of burial [29]. This correction significantly improved accuracy, with GB-OBC demonstrating smaller average errors compared to GB-HCT in folding simulations of mini-proteins like chignolin [30]. The model exists in two variants (OBC I and OBC II) with different parameter sets [30].
GB-Neck and GB-Neck2
The GB-Neck model introduced a physically motivated "neck" correction to better approximate the molecular surface, particularly addressing gaps between atoms at short distances where explicit water molecules would typically be excluded [29]. The correction modifies the Coulomb integral:
where I_MS and I_vdw represent the integrals using molecular surface and van der Waals volume, respectively [29]. While theoretically promising, the original GB-Neck model showed limitations in maintaining stable protein and nucleic acid structures [31].
GB-Neck2 addressed these limitations by expanding the parameter set from 8 to 18 parameters, making the correction factors atom-type dependent [29]. This refinement significantly improved agreement with Poisson-Boltzmann calculations for both solvation energies and effective radii across diverse test systems [29]. GB-Neck2 also demonstrated enhanced capability in reproducing experimental structures and thermal stability profiles for various peptide systems [29].
Table 1: Key Features and Applications of Generalized Born Models
| Model | Theoretical Basis | Key Improvements | Primary Limitations |
|---|---|---|---|
| GB-HCT | Pairwise descreening approximation with VdW surface [29] | Computational efficiency; Simple implementation [28] | Underestimates Born radii of buried atoms; Structural bias in proteins [30] [31] |
| GB-OBC | Empirical scaling function for buried atoms [29] | Improved accuracy for buried atoms; Better balance of speed/accuracy [30] | Limited transferability to nucleic acids; Helical bias in proteins [31] |
| GB-Neck | Neck correction between atom pairs [29] | Better approximation of molecular surface [29] | Structural instability in MD simulations [31] |
| GB-Neck2 | Atom-type dependent parameters with neck correction [29] | Improved accuracy for proteins and nucleic acids; Better stability [29] [31] | Parameterization complexity; Salt bridge strength requires adjustment [29] |
Quantitative Performance Comparison
A comprehensive comparative study evaluated eight GB models across 60 biomolecular complexes from four classes: protein-protein, protein-drug, RNA-peptide, and small complexes [32]. The performance was assessed by comparing electrostatic binding free energies ($ΔΔG_{el}$) to reference Poisson-Boltzmann calculations [32].
Table 2: Performance of GB Models Across Biomolecular Complex Types (Correlation R² with PB reference) [32]
| GB Model | Small Complexes | Protein-Protein | Protein-Drug | RNA-Peptide | Overall |
|---|---|---|---|---|---|
| GB-HCT | 0.9854 | 0.9591 | 0.8992 | 0.8247 | 0.9171 |
| GB-OBC | 0.9912 | 0.9785 | 0.9528 | 0.9016 | 0.9560 |
| GB-Neck2 | 0.9982 | 0.9953 | 0.9874 | 0.9741 | 0.9888 |
| GBNSR6 | 0.9986 | 0.9971 | 0.9928 | 0.9825 | 0.9949 |
| GBMV | 0.9741 | 0.9458 | 0.8837 | 0.8129 | 0.9041 |
The data reveals several important trends. First, small neutral complexes present the least challenge for most GB models, with all major variants achieving R² > 0.98 [32]. Second, RNA-peptide and protein-drug complexes prove most challenging, with GB-HCT showing particularly poor performance (R² = 0.8247 and 0.8992, respectively) [32]. Third, newer models like GBNSR6 and GB-Neck2 demonstrate significant improvement across all complex types, with GB-Neck2 showing notable enhancement over its predecessors [32].
In folding simulations of chignolin, populations of the native structure differed significantly between GB models: GB-HCT, OBC I, and OBC II all achieved native structure populations exceeding 30%, while GBn yielded only about 15% [30]. The OBC methods demonstrated smaller average errors compared to GB-HCT, making them preferable for obtaining reliable free-energy landscapes [30].
Experimental Protocols and Methodologies
Protocol 1: Calculation of Electrostatic Binding Free Energies
Objective: Calculate and compare electrostatic binding free energies ($ΔΔG_{el}$) for biomolecular complexes using different GB models [32].
System Preparation:
- Obtain PDB structures of the biomolecular complexes and separate into receptor and ligand components [28].
- Prepare the structures using standard molecular preparation workflows: add hydrogens, assign partial charges using an appropriate force field (e.g., ff99SB), and ensure missing heavy atoms are completed [28].
- Generate multiple conformational snapshots for each complex through molecular dynamics sampling or crystal structure variations [32].
Electrostatic Binding Free Energy Calculation:
- For each snapshot, calculate the electrostatic solvation free energy (
$ΔG_{el}$) for the complex, receptor, and ligand using the target GB model [32]. - Compute the electrostatic binding free energy as:
$ΔΔG_{el} = ΔG_{el}(complex) - ΔG_{el}(receptor) - ΔG_{el}(ligand)$[32]. - Repeat calculations for all GB models being evaluated and the reference Poisson-Boltzmann method [32].
Analysis:
- Compare
$ΔΔG_{el}$values from GB models to PB reference using correlation analysis (R²) and root-mean-square deviation [32]. - Evaluate performance across different complex types (small molecules, protein-protein, protein-drug, RNA-peptide) [32].
Protocol 2: Folding Simulations of Mini-Proteins
Objective: Assess GB model accuracy in reproducing native structure populations and free-energy landscapes [30].
System Setup:
- Select a mini-protein system (e.g., chignolin, a 10-residue mini-protein) [30].
- Use extended polypeptide chain as starting conformation [30].
- Employ appropriate force field parameters (e.g., modified ff99 parameters with generalized Born implicit solvation) [30].
Simulation Procedure:
- Perform multicanonical molecular dynamics simulations to enhance conformational sampling [30].
- Conduct simulations of sufficient length (e.g., 300 ns) to ensure convergence of conformational distributions [30].
- Repeat simulations with each GB model (HCT, OBC I, OBC II, GBn) using identical simulation parameters [30].
Analysis:
- Convert conformational ensembles to canonical distributions at 300 K [30].
- Cluster structures and identify native cluster based on experimental reference [30].
- Calculate population of native structure and compare with experimental value (~60% for chignolin at 300 K) [30].
- Compare free-energy landscapes and identify lowest free-energy minima [30].
Diagram 1: Workflow for comprehensive assessment of GB model performance, integrating binding energy calculations and folding simulations.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for GB Model Implementation
| Tool/Resource | Function | Implementation Examples |
|---|---|---|
| AMBER | Molecular dynamics package with multiple GB implementations [28] | GB-HCT, GB-OBC, GB-Neck, GB-Neck2 [29] [31] |
| CHARMM | Molecular simulation program with GB capabilities [28] | GBMV, GBMV2, GBSW [31] |
| Poisson-Boltzmann Solver | Reference method for solvation energy calculations [32] | Accuracy benchmark for GB models [32] [29] |
| Test Molecular Systems | Diverse structures for validation [32] | Small complexes, protein-drug, RNA-peptide, protein-protein complexes [32] |
| Stability Validation | Assessment of long-term simulation behavior [31] | DNA/RNA duplexes, quadruplexes, protein-nucleic acid complexes [31] |
The evolution of Generalized Born models from GB-HCT to GB-Neck2 represents significant progress in implicit solvent methodology. While GB-HCT offers computational efficiency and GB-OBC provides improved accuracy for buried atoms, the GB-Neck2 model currently represents the most balanced option for simulations involving both proteins and nucleic acids. The quantitative performance data and standardized protocols provided here enable researchers to select appropriate GB models based on their specific system requirements, balancing accuracy and computational efficiency for drug discovery applications and beyond. Future developments in machine learning-based implicit solvent models show promise for further bridging the accuracy gap between implicit and explicit solvent simulations [26].
| Category | Item/Model | Function/Description |
|---|---|---|
| Web-Based Platform | CHARMM-GUI Implicit Solvent Modeler (ISM) | Automated workflow for building molecular systems and preparing input files for GB implicit solvent simulations with various MD packages [33]. |
| Generalized Born (GB) Models | GB-OBC, GB-Neck, GB-HCT, GBSW, GBMV | Calculate the polar component of solvation energy; different models offer varying accuracy in estimating effective Born radii and defining the solute-solvent boundary [33]. |
| Molecular Dynamics (MD) Packages | AMBER, CHARMM, GENESIS, NAMD, OpenMM, Tinker | Simulation engines for which CHARMM-GUI ISM can generate complete input files, enabling cross-package consistency [33] [34]. |
| Force Fields (FF) | CHARMM36(m), AMBER (ff14SB, ff19SB), GAFF, OpenFF | Sets of parameters defining potential energy for proteins, nucleic acids, glycans, and ligands; selection in ISM automatically adapts the system [33]. |
| System Preparation | PDB Reader & Manipulator | Integrated module to read, modify, and protonate input structures, including handling covalent ligands [35]. |
{## Introduction}
Implicit solvent models are a cornerstone of computational biophysics, dramatically accelerating molecular dynamics (MD) simulations by replacing explicit solvent molecules with a continuum representation. This approach reduces computational cost and system viscosity, enabling longer timescales and enhanced sampling for studying protein folding, large-scale motions, and ligand binding [33]. The CHARMM-GUI Implicit Solvent Modeler (ISM) provides a standardized, automated platform for setting up Generalized Born (GB) implicit solvent simulations across multiple popular MD programs [33]. This Application Note details the use of CHARMM-GUI ISM, providing a structured protocol and validation data to empower researchers in leveraging this tool for efficient and reliable small protein simulations.
{## CHARMM-GUI ISM Overview and Capabilities}
CHARMM-GUI ISM is a specialized web-based module designed to streamline the setup of biomolecular simulations within a continuum solvent environment. Its core function is to generate simulation systems and input files that are consistent and reproducible across different computational environments [33]. The module supports a wide array of technical combinations, summarized in Table 1.
Table 1: Supported GB Models, Force Fields, and MD Packages in CHARMM-GUI ISM. This table synthesizes information from the ISM documentation, highlighting key compatible components. Note: Support varies by package; GBSW and GBMV are specific to CHARMM, while GB-OBC and GB-Neck are prevalent in AMBER and OpenMM [33].
| MD Package | CHARMM FF | AMBER FF | GB-HCT | GB-OBC | GB-Neck | GBMV | GBSW |
|---|---|---|---|---|---|---|---|
| AMBER | - | ✓ | ✓ | ✓ | ✓ | - | - |
| CHARMM | ✓ | - | - | - | - | ✓ | ✓ |
| GENESIS | ✓ | ✓ | - | - | - | ✓ | - |
| NAMD | ✓ | ✓ | - | - | - | ✓ | - |
| OpenMM | - | ✓ | ✓ | ✓ | ✓ | - | - |
| Tinker | - | ✓ | - | ✓ | ✓ | - | - |
The solvation free energy (ΔGsolv) in these GB models is a sum of polar (ΔGpol) and non-polar (ΔGnp) contributions. The polar term is calculated by the GB equation, while the non-polar term is typically approximated using a solvent-accessible surface area (SASA) term and a surface tension parameter [33]. ISM handles this complex parameterization automatically based on the user's selection.
{## Step-by-Step Protocol for System Setup}
This protocol outlines the process for setting up a simulation for a small protein-ligand complex using CHARMM-GUI ISM. The workflow is visualized in Diagram 1.
Access Module and Input Structure: Navigate to the CHARMM-GUI website and select the Implicit Solvent Modeler (ISM). On the initial page, select "Implicit Solvent" for a standard solution simulation. Provide your protein structure by either uploading a PDB/mmCIF file or by entering a PDB ID for direct download from the RCSB PDB [33].
Force Field Selection and Parameterization: After the structure is processed, the PDB Reader & Manipulator provides options for structure modification. Here, users can perform tasks like protonation state adjustment and introducing mutations. A critical step is ligand parameterization. ISM supports several force fields, including CGenFF, GAFF2, and OpenFF. Users can select the desired force field, and the interface will display the parameterization status for all ligands in the system [36] [35].
Implicit Solvent Model Selection: The next step is dedicated to selecting the GB implicit solvent model. Choose from the available models based on your target MD package and accuracy requirements. For instance, GB-Neck2 is known for its improved accuracy over earlier models like GB-OBC [33]. This step configures the dielectric constants for the solute and solvent, defining the continuum environment.
MD Program Selection and Input Generation: Finally, select the molecular dynamics program for the simulation. ISM supports AMBER, CHARMM, GENESIS, NAMD, OpenMM, and Tinker. After selection, configure final simulation parameters, such as the number of simulation steps and hydrogen mass repartitioning (HMR) options. HMR allows for the use of larger integration time steps, thereby accelerating the simulation [33]. The system will then generate a downloadable archive containing all necessary input files for energy minimization, equilibration, and production MD.
{## Experimental Validation and Performance}
The systems and input files generated by CHARMM-GUI ISM have been rigorously validated to ensure reliability and reproducibility across different simulation packages.
Table 2: Validation of ISM-Generated Systems through Protein Folding Simulations. This table summarizes data from the ISM validation study, which simulated 20 different protein structures and compared the results to experimental references [33].
| Protein (Example) | Simulation Type (GB Model) | Cα RMSD from Experimental Structure (Å) | Key Observation |
|---|---|---|---|
| Villin Headpiece | Implicit (GB-OBC) | ~2.0 - 3.0 | Maintained native fold with reasonable structural deviation over multi-microsecond simulations [33]. |
| BBA5 | Implicit (GB-OBC) | ~2.0 - 3.0 | Simulated structures showed good agreement with experimental NMR data [33]. |
| Trp-Cage | Implicit (GB-OBC) | ~2.0 - 3.0 | Stable simulation of the folded mini-protein was achieved [33]. |
A key application of implicit solvent simulations is in the early-stage screening of drug candidates. ISM facilitates this through high-throughput preparation of protein-ligand complexes. In a validation study, 88 protein-ligand docking structures were simulated using ISM-generated inputs. The results demonstrated that implicit solvent simulations, assessed by metrics like ligand root-mean-square deviation (RMSD) and Molecular Mechanics Generalized Born Surface Area (MM/GBSA) calculations, provided better discrimination between correct and incorrect ligand-binding modes than docking scores alone [33]. While explicit solvent simulations may offer higher accuracy, the performance of implicit solvent simulations presents a favorable trade-off between computational cost and reliability for screening applications [36] [33].
{## Conclusion}
CHARMM-GUI ISM offers a robust, automated, and standardized solution for setting up Generalized Born implicit solvent simulations. By simplifying the complex process of system preparation and input generation for a wide range of MD packages, it enhances reproducibility and accessibility. As demonstrated by validation studies, it enables efficient and reliable simulations for studying protein dynamics and ligand binding, making it an invaluable tool for researchers in computational biophysics and drug development.
Implicit solvent models, which treat the solvent as a continuous polarizable medium rather than individual molecules, provide a foundational strategy for accelerating molecular dynamics (MD) simulations. By eliminating the need to simulate thousands of explicit solvent molecules, these models significantly reduce computational complexity. This efficiency gain is particularly valuable for simulating biological processes like protein folding and conformational changes, which often occur on timescales that are prohibitively long for standard explicit-solvent MD [37] [1]. This Application Note details the integration of implicit solvent models with advanced sampling algorithms to achieve enhanced conformational exploration of small proteins, providing both theoretical context and practical protocols for researchers in computational biophysics and drug discovery.
Background and Theoretical Foundation
Implicit Solvent Models in Biomolecular Simulations
Implicit solvents, or continuum solvents, replace explicit solvent molecules with a homogeneously polarizable medium characterized primarily by its dielectric constant (ε). The solvation free energy (ΔG) in these models is typically decomposed into polar and non-polar contributions [37] [1]:
- Polar Component (ΔGpolar): Describes electrostatic interactions, often calculated using models based on the Poisson-Boltzmann (PB) equation or the Generalized Born (GB) approximation.
- Non-Polar Component (ΔGnon-polar): Accounts for cavity formation, dispersion, and repulsion energies, frequently estimated using solvent-accessible surface area (SASA) terms.
Common implicit models include the Polarizable Continuum Model (PCM), the Solvation Model based on Density (SMD), and Generalized Born models (e.g., GBSA) [1]. The central advantage is a dramatic reduction in the number of particles in a simulation system, which decreases computational cost and facilitates the application of enhanced sampling techniques.
The Need for Accelerated Sampling
Proteins sample a complex energy landscape with multiple metastable states separated by high energy barriers. Conventional MD simulations often remain trapped in local energy minima, making it difficult to observe functionally relevant conformational transitions or folding events within practical simulation timescales [38] [39]. Accelerated sampling methods overcome this limitation by applying a bias to the system, encouraging exploration of conformational space that would otherwise be inaccessible.
Accelerated Sampling Methods with Implicit Solvent
The computational efficiency of implicit solvent models makes them particularly suitable for use with a variety of enhanced sampling algorithms. The table below summarizes key methods and their performance characteristics.
Table 1: Accelerated Sampling Methods Compatible with Implicit Solvent Models
| Method | Key Principle | Reported Speedup | Key Applications |
|---|---|---|---|
| Discard-and-Restart MD [39] | Iteratively runs short MD bursts, discarding unproductive trajectories and restarting with new velocities. | Up to 2000x for sketching folding pathways | Protein folding, partial unfolding events (e.g., α-tubulin) |
| Steered MD (SMD) [38] | Applies a biasing force along a predefined Collective Variable (CV) to drive conformational change. | Highly system/CV dependent | Inducing transitions with low-resolution data (e.g., T4 Lysozyme) |
| Variational Explicit-Solute Implicit-Solvent (VESIS) [27] | Combines a force field for solute atoms with a variational implicit-solvent model for efficient free-energy minimization. | Significant improvement over CPU implementation | Protein-protein binding (e.g., p53-MDM2), molecular conformation |
| AI2BMD [40] | Uses an AI-based ab initio potential with a polarizable AMOEBA solvent model for highly accurate dynamics. | >1,000,000x faster than DFT for large proteins | Protein folding with ab initio accuracy, free-energy calculations |
| Multiple Time Step (MTS) with GB [41] | Computes computationally expensive solvation forces less frequently than fast bonded forces. | Stable with 4x longer steps for GB forces | General protein dynamics |
Machine Learning-Enhanced Implicit Solvation
Recent advances are closing the accuracy gap between implicit and explicit solvent models. Machine learning (ML) is being used to develop more precise implicit solvent potentials. For instance, the LSNN (Solvation Neural Network) model uses a graph neural network (GNN) that is trained not only on forces but also on derivatives with respect to alchemical variables (λelec and λsteric). This approach ensures that the model can accurately compute absolute solvation free energies, a task where traditional ML potentials trained only on force-matching fail [26]. Such models promise near-explicit solvent accuracy at a fraction of the computational cost.
Furthermore, large-scale neural network potentials (NNPs) like those trained on Meta's Open Molecules 2025 (OMol25) dataset are achieving unprecedented accuracy in energy and force calculations for biomolecules. These models can be integrated with implicit solvent frameworks to perform highly efficient and accurate simulations of large systems [42].
Application Notes and Protocols
Protocol 1: Discard-and-Restart MD for Folding Pathway Sketching
This protocol is adapted from recent work demonstrating up to 2000-fold speedups in mapping protein folding pathways [39].
Workflow Overview:
Detailed Steps:
System Preparation:
- Obtain the initial unfolded or partially folded protein structure.
- Solvation: Employ an implicit solvent model such as GBSA (Generalized Born with Surface Area term). The OBC (Onufriev-Bashford-Case) model is a widely used and stable variant for proteins [41].
- Energy Minimization: Perform steepest descent minimization for 5,000 steps or until the maximum force is below 5 kJ/mol/nm.
Define a Collective Variable (CV) Loss Function:
- The CV should quantitatively describe progress toward the target folded state. Effective CVs for folding include [39]:
- Root Mean Square Deviation (RMSD) of the protein backbone relative to the native structure.
- Radius of Gyration (Rg), which measures the compactness of the structure.
- Number of Native Contacts (Q), the fraction of residue contacts in the native state that are formed.
- Solvent Accessible Surface Area (SASA), particularly the burial of hydrophobic residues.
- The CV should quantitatively describe progress toward the target folded state. Effective CVs for folding include [39]:
Iterative Discard-and-Restart Cycle:
- Run Short MD Burst: Perform a short, unbiased MD simulation (e.g., 10-20 ps) starting from the current conformation.
- Calculate Loss: After the burst, compute the value of the CV loss function for the final conformation.
- Decision Logic:
- IF the trajectory has moved closer to the target (loss decreased), ACCEPT the final conformation and use it as the starting point for the next MD burst.
- IF the trajectory has moved away from the target (loss increased), DISCARD the entire trajectory. Generate a new Maxwell-Boltzmann velocity distribution at the simulation temperature and restart a new short MD simulation from the previous accepted conformation.
- Termination: The cycle continues until the system reaches the target state (e.g., RMSD < 2.0 Å from native) or a maximum number of iterations.
Protocol 2: Steered MD (SMD) with Low-Resolution Experimental Constraints
This protocol is designed for inducing conformational changes when only low-resolution structural data (e.g., from FRET or XL-MS) is available for the target state, as demonstrated on T4 Lysozyme [38].
Workflow Overview:
Detailed Steps:
Define a Minimal Set of Collective Variables (CVs):
- Convert low-resolution experimental data into a small set of CVs. For example:
- Distances: Between specific atom pairs measured by FRET or cross-linking.
- Angles: Hinge-bending angles between protein domains.
- A minimal, physically meaningful set of CVs (e.g., 1-2 key distances and 1 angle) is more effective than a large, uncurated set [38].
- Convert low-resolution experimental data into a small set of CVs. For example:
System Setup and Equilibration:
- Solvate the protein in an implicit solvent box using a GB model.
- Equilibrate the system with the protein's coordinates restrained to allow solvent relaxation, followed by a brief period of unrestrained equilibration.
SMD Simulation:
- Apply a harmonic biasing potential to each of the selected CVs. The potential is defined as ( U{bias} = \frac{1}{2} k (ξ - ξ{target})^2 ), where ( k ) is the force constant, ( ξ ) is the current CV value, and ( ξ_{target} ) is the value from the target state.
- Steering: Gradually "pull" the CVs toward their target values over the course of the simulation. The steering speed should be slow enough to prevent unrealistic protein deformation (e.g., 0.0003 - 0.0005 Å/fs for distances) [38].
- Force Constant: Use a moderate spring constant (e.g., 0.02 - 0.025 Ha/Bohr²) to guide the system without over-constraining it.
Analysis and Validation:
- Pathway Analysis: Cluster the resulting conformations and analyze the pathway of the transition. Identify key breaking and forming interactions (e.g., salt bridges, hydrophobic contacts).
- Validation with TAMD: Use Temperature-Accelerated MD (TAMD) as a validation tool. Run TAMD simulations using the CVs identified for SMD. If the SMD-selected CVs are meaningful, TAMD should also successfully drive the conformational transition without a target bias, confirming their importance [38].
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Tool/Solution | Type | Function in Research | Example/Reference |
|---|---|---|---|
| Generalized Born (GB) Models | Implicit Solvent Model | Computes electrostatic solvation energy efficiently; core of many accelerated protocols. | OBC, HCT, STILL models [41] |
| GROMACS | MD Software | High-performance MD engine; supports GB/SA implicit solvent and enhanced sampling methods. | [38] [39] |
| CP2K | MD/Quantum Chemistry Software | Performs QM/MM and force field MD simulations; used for explicit-solvent validation. | [43] |
| Collective Variable (CV) Module | Software Library | Defines and biases CVs (e.g., distances, angles, RMSD) in enhanced sampling simulations. | PLUMED, Colvars |
| AI2BMD Potential | ML Force Field | Provides ab initio accuracy for energy/forces; can be paired with implicit solvent. | [40] |
| OMol25-trained NNPs | Neural Network Potential | Pre-trained models (e.g., eSEN, UMA) for highly accurate and fast energy calculations. | [42] |
| LSNN Model | ML Implicit Solvent | Graph Neural Network that accurately predicts solvation free energies, not just forces. | [26] |
The strategic integration of implicit solvent models with advanced sampling algorithms represents a powerful paradigm for accelerating conformational exploration in proteins. Methods like Discard-and-Restart MD and Steered MD leverage the computational efficiency of continuum solvents to achieve speedups of several orders of magnitude, enabling the study of folding, unfolding, and large-scale conformational transitions. The emerging integration of machine learning, through both accurate force fields like AI2BMD and corrective models for implicit solvation like LSNN, is poised to further bridge the gap between computational efficiency and physical accuracy. These tools collectively empower researchers to use simulations not just for interpretation, but for the predictive discovery of protein dynamics and function.
Molecular dynamics (MD) simulations serve as a powerful tool for elucidating the folding mechanisms of proteins at atomic resolution. The choice of solvent model—implicit or explicit—profoundly impacts the balance between computational efficiency and biological realism. This application note provides a detailed comparative analysis of these approaches through case studies of two well-characterized fast-folding proteins: the 35-residue Villin Headpiece (HP-35) and the 20-residue Trp-cage miniprotein. We summarize critical quantitative data, outline reproducible experimental protocols, and visualize complex workflows to guide researchers in selecting appropriate simulation strategies for small protein systems.
Protein Systems and Structural Properties
Table 1: Characterized Protein Systems for Folding Studies
| Protein | Residues | PDB ID | Native Fold | Experimental Folding Rate | Key Structural Features |
|---|---|---|---|---|---|
| Villin Headpiece (HP-35) | 35 | 1YRF | Three α-helices packed into a hydrophobic core [44] | ~0.5–5 μs⁻¹ (folding); ~800 μs⁻¹ (unfolding at 300 K) [44] | Hydrophobic core; folding nucleus between helices II and III [44] |
| Trp-cage (TC5b) | 20 | 1L2Y | Structured hydrophobic core with buried Trp side chain [45] | ~4.1 μs [45] | Buried indole ring of Trp residue between Pro rings; stable folded state [45] |
Quantitative Simulation Performance Data
Table 2: Comparative Performance of Implicit vs. Explicit Solvent Simulations
| Performance Metric | Villin Headpiece (Implicit Solvent) | Villin Headpiece (Explicit Solvent) | Trp-cage (Implicit Solvent) |
|---|---|---|---|
| Sampling Method | SRMSTIS Path Sampling [44] | Conventional MD [46] | Replica-Exchange MD (REMD) [45] |
| Total Simulation Time | Not Specified (Equilibrium Kinetic Network) [44] | >50 μs [46] | 2 ns per replica × 23 replicas (46 ns total production) [45] |
| Reported Folding Time | Agreement with experiment [44] | Similar to experimental timescales [46] | 1.5–8.7 μs (kinetics study cited) [45] |
| Thermodynamic Stability | Nine (meta)stable states identified [44] | Folds to native conformation reliably [46] | Stable folded state with melting temp ≈400 K [45] |
| Structural Accuracy (Cα RMSD) | Native state defined as RMSD < 0.13 nm [44] | Folds to native conformation [46] | <1.0 Å from NMR structure [45] |
Detailed Experimental Protocols
Replica-Exchange MD (REMD) of Trp-cage with Implicit Solvent
This protocol is adapted from simulations that successfully folded Trp-cage to within 1.0 Å of its NMR structure [45].
1. System Setup:
- Initial Structure: Build the TC5b sequence (NLYIQWLKDGGPSSGRPPPS) in an extended conformation [45].
- Termini & Protonation: Set both N- and C-termini as charged, and all ionizable side chains to their pH 7 protonation state [45].
- Force Field: Use the Cornell et al. all-atom force field (parm94/amber94) [45].
- Solvent Model: Employ the Generalized Born/Solvent-Accessible Surface Area (GB/SA) model. Key parameters: Bondi radii, solvent dielectric constant ε=78.5, and surface tension 0.005 cal·mol⁻¹·Å⁻² [45].
2. Energy Minimization & Equilibration:
- Perform 5,000 steps of steepest descent minimization on the extended structure.
- Equilibrate for 50 ps at 298 K using a Berendsen thermostat (coupling constant 1 ps⁻¹). Use the final state as the initial conformation for all replicas [45].
3. Replica-Exchange Simulation:
- Replica Configuration: Simulate 23 replicas across a temperature range from 273 K to 630 K [45].
- Dynamics Parameters: Use a 1-fs time step (to ensure energy conservation). Apply the SHAKE algorithm to constrain all bond lengths. Maintain temperature via velocity reassignment and a Berendsen thermostat [45].
- Exchange Attempts: Attempt replica swaps every 5 ps of simulation. The acceptance probability is given by the Monte Carlo criterion:
P(accept) = min[1, e^(+(β_lo–β_hi)(U_hi–U_lo))], where β is the inverse temperature and U is the potential energy [45]. - Sampling: After an initial 2 ns equilibration period, save conformations every 0.25 ps during a 2 ns production simulation per replica.
4. Data Analysis:
- Calculate the Cα root-mean-square deviation (RMSD) relative to the experimental NMR ensemble (PDB ID 1L2Y) to identify folded states (<1.0 Å RMSD) [45].
- Analyze the fraction of folded conformations as a function of temperature to determine thermodynamic stability and estimate the melting temperature [45].
Kinetic Network Sampling of Villin Headpiece with Implicit Solvent
This protocol uses the single-replica multiple-state transition-interface sampling (SRMSTIS) method to elucidate the equilibrium kinetic network, capable of overcoming high unfolding barriers [44].
1. System Setup:
- Initial Structure: Use the experimental NMR structure (PDB: 1YRF) [44].
- Force Field: Employ the CHARMM27 force field with the multibody CMAP correction [44].
- Solvent Model: Use the GBSA (Generalized Born Surface Area) implicit solvent model [44].
- Software: Perform all simulations with GROMACS-4.5.4 [44].
2. System Preparation:
- Place the protein in a 6-nm cubic box.
- Conduct energy minimization via the steepest-descent method for 30 ps.
- Dynamics Parameters: Use a 2-fs time step. Maintain temperature at 300 K using the v-rescale thermostat. Set the nonbonded van der Waals cutoff to 1.4 nm and the Coulomb cutoff to 4.8 nm [44].
3. State and Interface Definition:
- Order Parameter: Use the Cα RMSD of residues 2–32 relative to the native state, denoted
rmsd2-32[44]. - State Definition: Define the native state (S01) by
rmsd2-32 < 0.13 nm[44]. - Interface Definition: Define a series of 10 non-intersecting interfaces around the native state, starting at 0.17 nm with 0.04 nm intervals [44].
- Order Parameter: Use the Cα RMSD of residues 2–32 relative to the native state, denoted
4. Path Sampling with SRMSTIS:
- Sample the path ensemble for each interface using a one-way shooting algorithm [44].
- Randomly select a shooting point from an existing path, reverse momenta if shooting backward, and integrate a new trajectory until it reaches any stable state.
- Accept or reject the new trial path based on a Metropolis Monte Carlo rule.
- Iteratively identify additional metastable states and include them in the kinetic network.
5. Network and Rate Analysis:
- Apply Transition Path Theory (TPT) to the sampled network to compute folding and unfolding rates, committor probabilities, and flux pathways [44].
Explicit Solvent MD of Villin Headpiece
This protocol is based on simulations that achieved reliable folding to the native state on timescales similar to experiments [46].
1. System Setup:
- Initial Structure: Obtain the structure from the PDB.
- Solvation: Solvate the protein in an explicit solvent box (e.g., using TIP3P water) with a minimum padding distance (e.g., 1.0 nm). Add ions to neutralize the system.
- Force Field: Select an appropriate all-atom force field (e.g., AMBER or CHARMM).
2. Simulation Run:
- Software: Use a MD package supporting explicit solvent, such as GROMACS or OpenMM. For reference, a benchmark study used OpenMM 8.2.0 with the AMBER14 force field and TIP3P-FB water [17].
- Parameters: Use a 2-4 fs time step. Maintain temperature (e.g., 300 K) with a Langevin thermostat and pressure (1 atm) with a Monte Carlo barostat. Treat long-range electrostatics with Particle Mesh Ewald (PME). Constrain bonds involving hydrogen [17].
- Execute multiple long-timescale simulations (totaling tens of microseconds) to observe spontaneous folding and unfolding events [46].
Workflow Visualization
Simulation Strategy Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Resources for Protein Folding Simulations
| Category | Item / Software / Model | Specific Function / Application |
|---|---|---|
| Simulation Software | AMBER [45], GROMACS [44], OpenMM [17] | Molecular dynamics engines for running simulations with various force fields and solvent models. |
| Analysis & Visualization | VMD [44], WESTPA 2.0 [17], PLUMED [21] | Trajectory visualization, weighted ensemble sampling, and analysis of collective variables and free energies. |
| Implicit Solvent Models | GB/SA [45], GBSA [44] | Continuum solvent models that significantly reduce computational cost for enhanced conformational sampling. |
| Explicit Solvent Models | TIP3P [21], OPC [21], SPC/E [21], TIP4P, TIP5P [21] | Atomistic water models differing in geometry/charge distribution; critical for capturing specific solvent effects. |
| Sampling Algorithms | Replica-Exchange MD (REMD) [45], Weighted Ensemble (WE) [17], Transition-Interface Sampling (TIS) [44] | Enhanced sampling methods to overcome free energy barriers and simulate rare events like folding. |
| Benchmark Datasets | Standardized Protein Benchmark [17] | Curated datasets of diverse proteins (e.g., Trp-cage, Villin, BBA) for rigorous method validation and comparison. |
This application note demonstrates that both implicit and explicit solvent models can successfully simulate the folding of small, fast-folding proteins like Villin Headpiece and Trp-cage. The choice between them involves a fundamental trade-off: implicit solvent models like GB/SA offer superior computational efficiency and facilitate advanced sampling techniques for probing thermodynamics and complex kinetic networks. In contrast, explicit solvent models provide greater physical realism by capturing specific water-mediated interactions at a significantly higher computational cost. Researchers should select their approach based on the specific scientific question—whether it demands high-throughput exploration of conformational space or atomic-level detail of the folding pathway. The provided protocols, data, and toolkits offer a foundation for rigorous and reproducible folding simulations.
Protein structure refinement, the process of improving the accuracy of preliminary protein models towards native-like conformation, represents a critical final step in computational structure prediction. The Critical Assessment of protein Structure Prediction (CASP) experiments provide a rigorous blind testing ground for evaluating refinement methodologies against experimentally determined structures [47] [48]. Despite the revolutionary advances in protein structure prediction through deep learning approaches like AlphaFold2, consistent and reliable refinement remains challenging [48] [49]. This application note examines successful refinement strategies demonstrated in CASP challenges, focusing on their methodologies, quantitative performance, and practical implementation for researchers pursuing structural accuracy at atomic resolution.
Quantitative Assessment of Refinement Success in CASP
The evaluation of refinement methodologies in CASP relies on robust metrics that quantify the degree of improvement over starting models. Key metrics include the Global Distance Test (GDT_TS), Root Mean Square Deviation (RMSD), and TM-Score, which assess global fold accuracy, while molecular mechanics energy and stereochemical quality evaluate physical realism [50] [51]. Successful refinement is defined as a statistically significant improvement in these metrics.
Table 1: Notable Refinement Improvements in CASP Experiments
| CASP Target | Starting GDT_TS | Refined GDT_TS | Improvement | Method Category | Key Methodology |
|---|---|---|---|---|---|
| TR435 [50] | 80.2 | 87.9 | +7.7 | Torsional MD | GNEIMO-REXMD with implicit solvent |
| TR453 [50] | 86.6 | 91.5 | +4.9 | Torsional MD | GNEIMO-REXMD with implicit solvent |
| TR454 [50] | 58.5 | 71.0 | +12.5 | Torsional MD | GNEIMO-REXMD with implicit solvent |
| TR884 [47] | 66 | 76 | +10 | Not Specified | CASP12 Example |
| TR894 [47] | 75 | 96 | +21 | Not Specified | CASP12 Example |
| TR896 [47] | 61 | 77 | +16 | Not Specified | CASP12 Example |
Analysis of CASP results reveals two predominant refinement philosophies. Conservative approaches typically make minimal structural adjustments, often preserving or slightly improving starting model quality with high consistency. In contrast, adventurous approaches attempt more significant conformational changes, achieving dramatic improvements on some targets while risking degradation on others [51] [49]. The GNEIMO-REXMD method exemplifies a balanced approach, demonstrating refinement of up to 1.3 Å RMSD for 30 CASP target proteins without experimental restraints [50].
Methodologies and Sampling Strategies
Successful refinement relies on sophisticated sampling strategies to explore conformational space near the starting model while maintaining native-like features.
Molecular Dynamics-Based Approaches
Molecular dynamics simulations form the backbone of many successful refinement protocols, employing both all-atom and constrained methodologies:
All-Atom Cartesian MD: Traditional MD simulations with all atomic degrees of freedom often require restraints to prevent structural degradation during refinement. Unrestrained simulations frequently drive models away from native conformation due to force field inaccuracies [50] [49].
Constrained Dynamics Methods: The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method implements internal coordinate MD by treating proteins as collections of rigid clusters connected by torsional hinges. This approach freezes high-frequency degrees of freedom, enabling larger integration time steps (5 fs) and focused sampling in torsional space [50].
Replica Exchange Enhancement: Temperature replica exchange MD (REXMD) runs multiple simulations at different temperatures (e.g., 310–415 K range with 32 replicas), enhancing conformational sampling by allowing periodic swapping between replicas based on the Metropolis criterion [50].
Solvent Models in Refinement Protocols
The choice between implicit and explicit solvent models represents a critical computational trade-off in refinement simulations:
Table 2: Solvent Models for Protein Structure Refinement
| Solvent Model | Computational Efficiency | Key Strengths | Limitations | Representative Applications |
|---|---|---|---|---|
| Implicit (GB/SA) | High (4-5x faster than explicit) [52] | Efficient sampling of electrostatic interactions; Good for globular proteins [8] | Misses specific water-mediated interactions; May overestimate H-bond strengths [8] [52] | GNEIMO-REXMD protocol with OBC model [50] |
| Explicit (TIP3P) | Low (High computational cost) | Models specific water interactions accurately; More physically realistic dynamics | Limited sampling due to computational demands; Water viscosity slows conformational search | Reference simulations for validation [52] |
| SASA-Based | Very High | Fast estimation of nonpolar solvation energies | Limited electrostatic treatment; Often paired with GB/PB [8] | Nonpolar component in composite models |
The GB/SA OBC implicit solvation model has demonstrated particular success in refinement protocols, offering a favorable balance between computational efficiency and physical accuracy. This model employs an interior dielectric of 1.5 for the solute and 78.3 for the solvent, with a nonpolar solvation term based on solvent-accessible surface area [50] [8].
Experimental Protocols for Structure Refinement
GNEIMO-REXMD Refinement Protocol
The following protocol outlines the successful GNEIMO-REXMD methodology applied to CASP target refinement:
Initial Structure Preparation
- Obtain starting model from homology modeling (e.g., MODELLER) or CASP decoys [50]
- Perform all-atom conjugate gradient minimization using AMBER99SB force field [50]
- Remove close contacts and optimize stereochemistry while maintaining global fold
Simulation Parameters
- Force Field: AMBER99SB for protein energetics [50]
- Solvation: Generalized Born/Surface Area (GB/SA) OBC implicit solvent model [50]
- Dielectric Settings: Interior dielectric = 1.5, exterior dielectric = 78.3 [50]
- Nonbonded Cutoff: 20 Å with switching function [50]
- Integration: Lobatto integrator with 5 fs time step (enabled by rigid cluster constraints) [50]
Replica Exchange Configuration
- Number of Replicas: 32 across temperature range 310-415 K [50]
- Exchange Attempt Frequency: Every 5 ps of simulation time [50]
- Sorting Algorithm: Metropolis criterion for replica swapping [50]
- Simulation Duration: 15-100 ns per replica (target-dependent) [50]
Conformational Sampling and Analysis
- Perform constrained dynamics in torsional space using rigid clusters
- Collect snapshots from lowest temperature replica for analysis
- Cluster trajectories to identify dominant conformational states
- Select refined models based on lowest energy and best stereochemical quality
Post-Refinement Model Selection
Identifying successfully refined structures remains challenging due to subtle differences between improved and degraded models:
Energy-Based Selection: Prioritize models with favorable molecular mechanics energy terms, particularly improved solvation energy and reduced atomic clashes [49]
Quality Assessment: Apply Model Quality Assessment Programs (MQAPs) to identify native-like structural features [49]
Ensemble Analysis: Select representatives from largest conformational clusters to ensure structural stability [50]
Validation Tools: Verify stereochemical quality using PROCHECK G-factor and physics-based validation scores [50] [49]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools for Protein Structure Refinement
| Tool/Category | Specific Implementation | Function in Refinement | Availability |
|---|---|---|---|
| Molecular Dynamics Engines | GNEIMO [50], AMBER [51], CHARMM [52] | Conformational sampling using physical force fields | Academic licensing, Open source |
| Implicit Solvent Models | GBSW [52], EEF1.1 [52], GB/SA OBC [50] | Efficient solvation energy calculation | Bundled with MD packages |
| Force Fields | AMBER99SB [50], CHARMM19/22 [52] | Potential energy functions for proteins | MD package dependencies |
| Quality Assessment | PROCHECK [50], MolProbity, MQAPs [49] | Stereochemical and model quality validation | Standalone tools, Servers |
| Replica Exchange | Temperature REX [50] | Enhanced conformational sampling | Custom implementation |
Workflow Visualization
Diagram 1: Protein Structure Refinement Workflow. The iterative process begins with an initial model, proceeds through preparation and sampling stages guided by solvent model selection, and culminates in analysis and validation of the refined structure.
Structure refinement methodologies have demonstrated measurable success in CASP challenges, particularly through MD-based approaches enhanced by replica exchange and optimized implicit solvent models. The GNEIMO-REXMD protocol exemplifies how targeted sampling in torsional space with efficient solvation treatment can achieve consistent refinement improvements of 1-3 Å RMSD across diverse protein targets. Future advancements will likely integrate deep learning approaches with physical force fields, improved implicit solvent parametrization, and enhanced model selection techniques to push refinement accuracy closer to experimental resolution. As these methods mature, they will increasingly support critical applications in drug design and functional analysis where atomic-level accuracy is paramount.
Optimization Strategies: Addressing Limitations and Improving Simulation Outcomes
Molecular dynamics (MD) simulation is a powerful computational microscope, enabling the study of biological processes at atomistic resolution. For simulations of small proteins, particularly those containing intrinsically disordered regions (IDRs), the choice of solvent model—implicit or explicit—is foundational. However, this choice is inextricably linked to two major sources of error: force field inconsistencies, which affect the accuracy of the interatomic potentials, and sampling artifacts, which arise from an inability to adequately explore the conformational landscape. This application note details these common pitfalls, provides protocols for their mitigation, and integrates these concepts into a practical research framework.
Force Field Inconsistencies
Force fields (FFs) are mathematical models that calculate the potential energy of a system of atoms. Traditional FFs, parameterized using data from folded, globular proteins, often fail to accurately describe the conformational ensembles of IDPs or the delicate balance of interactions in solution.
The Problem with Standard Force Fields
Simulations using standard force fields like AMBERff99SB or CHARMM36 with traditional water models (e.g., TIP3P) often produce IDP ensembles that are overly collapsed and structured, contradicting experimental observations [53] [54]. For instance, long MD simulations of the Aβ40 peptide with Amber99ffsb-ildn and TIP3P water resulted in close to 90% β-sheet content, whereas experiments show it is largely a random coil [53]. This bias occurs because these FFs were optimized for stable secondary and tertiary structures, leading to excessive protein-protein interactions at the expense of protein-solvent interactions.
Advances in Force Field Development
Recognizing these shortcomings, the field has developed modified FFs. These improvements often involve refining backbone torsion potentials, adjusting protein-water dispersion interactions, and using more accurate water models [53] [54]. The performance of a FF is highly system-dependent, and no single FF is universally best for all proteins.
Table 1: Benchmarking of Select Force Fields and Water Models for Disordered Protein Regions (e.g., R2-FUS-LC)
| Force Field | Water Model | Key Characteristics | Reported Performance |
|---|---|---|---|
| CHARMM36m [54] | TIP3P (modified) | Modified non-bonded terms for folded/disordered proteins. | Balanced description of structured and disordered regions; good for FUS protein [54]. |
| a99SB-disp [54] | Modified TIP4P-D | Derived from ff99SB; uses a water model with increased dispersion. | Designed for both structured and disordered regions; accurate for various proteins [54]. |
| ff99SB*-ILDN [54] | TIP4P-D | Includes backbone corrections and ILDN side-chain modifications. | Produces expanded conformations for IDPs in agreement with NMR/SAXS [54]. |
| DES-Amber [54] | Optimized TIP4P-D | Derived from ff99SB by D.E. Shaw Research. | High accuracy for structured and disordered regions in benchmark tests [54]. |
| AMBERff19SB [54] | OPC | More recent AMBER FF paired with a 4-point water model. | Improved description of IDPs compared to TIP3P simulations [54]. |
Table 2: Implicit vs. Explicit Solvent Models: A Comparative Overview
| Characteristic | Implicit Solvent | Explicit Solvent |
|---|---|---|
| Computational Cost | Lower; faster conformational search [1] [3]. | Higher; requires simulating many solvent molecules [1] [3]. |
| Sampling Speed | Faster due to lack of viscous solvent drag [3]. | Slower; solvent viscosity slows conformational transitions [3]. |
| Treatment of Solvent | Continuum dielectric medium (mean-field) [1]. | Individual, discrete molecules with explicit coordinates [1]. |
| Key Limitations | Lacks specific, atomic-level solvent-solute interactions (e.g., individual H-bonds) [1]. | Computationally demanding; potential force field inaccuracies in water-water/solute interactions [1]. |
| Common Models | Generalized Born (GB), Poisson-Boltzmann (PB) [1] [3]. | TIP3P, TIP4P, SPC, OPC, TIP4P-D [54]. |
The following workflow diagram outlines a systematic approach for selecting and validating a force field and solvent model for a given protein system:
Sampling Artifacts
Even with a perfect force field, the utility of an MD simulation depends on achieving adequate sampling—the collection of a statistically representative set of conformations. Biomolecular energy landscapes are rough, with many local minima separated by high barriers, making sufficient sampling a major challenge [55].
The Sampling Problem in IDPs
Intrinsically disordered proteins (IDPs) exemplify the sampling problem due to their flat energy landscapes and numerous conformational substates [53]. Standard MD simulations can easily become trapped in local energy minima, leading to artifacts such as non-convergent structural properties (e.g., radius of gyration) and an incorrect weighting of sub-populations [53] [55]. For example, a simulation might over-populate collapsed states and miss extended, functionally relevant conformations.
Enhanced Sampling Techniques
Enhanced sampling methods are algorithms designed to accelerate the exploration of configuration space and overcome energy barriers.
- Temperature Replica Exchange (TREx): Also known as Parallel Tempering, this method runs multiple replicas of the system at different temperatures. Periodically, exchanges between replicas are attempted, allowing conformations to escape deep energy wells at high temperatures and propagate to lower temperatures [55]. While widely used, TREx can be inefficient for large systems with entropic barriers, as the number of required replicas grows significantly [53].
- Metadynamics: This method discourages the system from revisiting previously sampled states by adding a history-dependent repulsive bias potential to the Hamiltonian [55]. This "fills up" the free energy wells, forcing the system to explore new regions. Its efficiency depends critically on a priori identification of a small number of collective variables that describe the process of interest.
- Temperature Cool Walking (TCW): A non-equilibrium alternative to TREx, TCW uses only one high-temperature replica to generate trial moves for the target temperature replica. It has been shown to converge more quickly and at a lower computational cost for systems like the Aβ peptide, producing ensembles in better agreement with experimental data compared to TREx with the same force field [53].
The diagram below illustrates the process of setting up and validating an enhanced sampling simulation:
Integrated Experimental-Computational Protocols
A robust strategy to mitigate force field and sampling pitfalls is the close integration of simulations with experimental data.
Experiment-Directed Ensemble Refinement
Experimental data can be used to refine computational ensembles. This can be done a posteriori by reweighting the snapshots from a simulation so that the averaged back-calculated experimental observables (e.g., J-couplings from NMR, chemical shifts, or FRET efficiencies) match the experimental values [56]. Alternatively, data can be incorporated a priori through experiment-biased simulations, where an empirical energy term is added to the force field to bias the simulation toward conformations that are consistent with the data [56].
Force Field Optimization
Beyond refining a single ensemble, experimental data can be used to improve the force field itself, creating a transferable and generally better physical model. This involves adjusting force field parameters to minimize the discrepancy between simulated and experimental observables across a range of different proteins and states [56].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool / Reagent | Type | Primary Function | Example Use Case |
|---|---|---|---|
| CHARMM36m [54] | Force Field | Models both structured proteins and IDPs. | Simulating systems with ordered domains and disordered linkers. |
| a99SB-disp [54] | Force Field | Balanced description for folded/unfolded states. | High-accuracy IDP simulations with excellent agreement to SAXS data. |
| OPC / TIP4P-D [54] | Water Model (Explicit) | Four-point models with improved accuracy. | Providing a more realistic solvation environment than TIP3P. |
| Generalized Born (GB) [1] [3] | Solvent Model (Implicit) | Approximates electrostatic solvation effects. | Rapid conformational sampling and docking studies. |
| PLUMED [57] | Software Library | Analyzes MD trajectories and performs enhanced sampling. | Implementing metadynamics and defining complex collective variables. |
| PySAGES [57] | Software Library | GPU-accelerated enhanced sampling methods. | Running efficient adaptive biasing force calculations on GPUs. |
| OpenMM [53] [57] | MD Simulation Package | High-performance toolkit for molecular simulation. | Running customizable MD simulations with hardware acceleration. |
| AMOEBA [1] [40] | Force Field | Polarizable force field for higher accuracy. | Studying systems where electronic polarization effects are critical. |
Emerging Methods and Future Outlook
The field is rapidly evolving with new methods that aim to circumvent traditional limitations.
- Machine Learning Force Fields (MLFFs): Models like AI2BMD are trained on quantum mechanical (DFT) data and can achieve ab initio accuracy at a fraction of the computational cost, showing promise for accurately simulating protein folding and dynamics [40].
- AI-Enhanced Conformation Sampling: Generative AI models, particularly diffusion models, are now being used to predict and sample diverse protein conformations directly, moving beyond the static structures provided by tools like AlphaFold2 [58].
- Advanced Sampling Frameworks: Libraries like PySAGES are integrating machine learning with enhanced sampling, for instance, using artificial neural networks to approximate free energy surfaces and their gradients, thereby improving the efficiency of sampling complex biomolecular transitions [57].
Success in molecular simulations of small proteins hinges on a careful and critical approach to force field selection and sampling adequacy. The choice between implicit and explicit solvents involves a trade-off between computational efficiency and physical detail, a decision that must be made in the context of the specific biological question. By leveraging modern, validated force fields, employing robust enhanced sampling techniques, and consistently integrating experimental data for validation and refinement, researchers can avoid common pitfalls and generate reliable, insightful models of protein dynamics.
The biological function of a protein is governed not by a single, static three-dimensional structure but by its dynamic existence as a population of interconverting conformations, known as a structural ensemble [59]. Accurately characterizing these ensembles is therefore crucial for a mechanistic understanding of protein activity, allostery, and molecular recognition, with significant implications for biomedical sciences and drug discovery [60] [59]. Molecular dynamics (MD) simulations have long been the standard computational tool for exploring these conformational landscapes [17]. However, the rugged energy landscapes of proteins, with their high kinetic barriers, often render functional transitions rare events on practical MD simulation timescales, leading to inadequate sampling and trapping in local minima [60].
The rapidly evolving field of deep generative modeling presents a promising alternative, offering a computationally efficient path to generating conformational diversity [60]. These machine learning (ML) models, trained on simulation data, can directly produce physically realistic conformational ensembles at a negligible fraction of the cost of traditional MD [59] [61]. Despite their promise, a central challenge persists: different ensemble generation methods, including various ML approaches and MD using different solvent models, can produce structurally divergent ensembles [21] [61]. These deviations arise from fundamental differences in how methods approximate physical reality, from the treatment of solvent to the underlying sampling algorithm. This Application Note details the sources of these structural deviations and provides protocols for their diagnosis and mitigation, specifically within the context of implicit versus explicit solvent models for small protein simulations.
Current Methodological Landscape
The landscape of ensemble generation methods has diversified significantly, broadly splitting into physics-based simulations and machine learning generators, each with distinct strengths and weaknesses that contribute to structural deviations.
Table 1: Categories of Ensemble Generation Methods
| Method Category | Key Principles | Causes of Structural Deviation |
|---|---|---|
| Explicit Solvent MD | Solvent molecules (e.g., water) are modeled as discrete, interacting particles; uses force fields like CHARMM36m and water models like TIP3P, OPC [21]. | Choice of water model (TIP3P vs. TIP4P vs. OPC) significantly impacts conformational dynamics, especially for charged systems like glycosaminoglycans [21]. Computationally expensive, limiting sampling timescale. |
| Implicit Solvent MD | Solvent is a continuous dielectric medium; approximates solvation effects via models like Poisson-Boltzmann (PB) or Generalized Born (GB) [10]. | Fails to capture specific solvent-mediated interactions (e.g., water bridges, hydrogen bonds); poor representation of entropic effects and ion specificity can distort ensemble [10]. |
| ML Generators (Trained on MD) | Learns the probability distribution of conformations from MD data; uses architectures like GANs (idpGAN), Diffusion Models (aSAM), or Flow Matching (EPO) for fast sampling [59] [61] [60]. | Inherits biases and sampling inadequacies of the training MD data. May struggle to generalize to states far from the initial structure or to unseen environmental conditions [61]. |
| Online-Refined ML Generators | Refines a pre-trained generator using physical energy functions without additional MD data (e.g., Energy Preference Optimization - EPO) [60]. | Aims to correct misalignment from the Boltzmann distribution but depends on the accuracy of the energy function used for refinement. |
A critical differentiator, and a primary source of divergence, is the treatment of solvent. Explicit solvent models provide a more detailed physical representation but at a high computational cost. Studies on systems like heparin dodecamers show that the choice of explicit water model (TIP3P, TIP4P, TIP5P, SPC/E, OPC) can introduce substantial variation in global structural properties such as the radius of gyration and end-to-end distance [21]. In contrast, implicit solvent models offer speed, enabling rapid hypothesis testing and large-scale design sweeps, but at the cost of simplifying away explicit solvent structure, which can lead to inaccuracies in capturing specific interactions critical for function [10]. Machine learning generators like idpGAN, aSAM, and AlphaFlow attempt to bypass the sampling limitations of MD but are ultimately constrained by the data on which they were trained [59] [61]. A nascent trend involves hybrid approaches, such as ML-augmented implicit solvent models that use neural networks to provide residual corrections to continuum model baselines, aiming to bridge the accuracy-efficiency gap [10].
Quantitative Comparison of Method-Dependent Deviations
Systematic benchmarking reveals that methodological choices significantly impact key structural metrics. The deviations are particularly pronounced when comparing implicit and explicit solvents, and when assessing the fidelity of ML generators against long MD simulations.
Table 2: Benchmarking Structural and Sampling Metrics Across Methods
| Metric | Explicit vs. Implicit Solvent MD | ML Generator vs. Reference MD |
|---|---|---|
| Radius of Gyration (Rg) | Explicit solvents (OPC, TIP5P) can show greater structural variability compared to TIP3P/SPC/E or implicit models [21]. | ML models (e.g., AlphaFlow, aSAM) often yield lower mean RMSD to the initial structure, indicating undersampling of distant states [61]. |
| Local Flexibility (RMSF) | Not directly compared in results, but implicit solvents may dampen fluctuations due to lack of explicit solvent friction and stochastic collisions. | High Pearson correlation (PCC ~0.9) for Cα RMSF is achievable, though some models (e.g., AlphaFlow PCC 0.904) may statistically outperform others (e.g., aSAMc PCC 0.886) [61]. |
| Torsion Angle Distributions | Implicit models may misrepresent ring puckering or dihedral distributions due to inadequate treatment of specific solvation [21]. | ML models with all-atom resolution (e.g., aSAM) better capture backbone (φ/ψ) and side-chain (χ) torsion distributions than Cβ-only models [61]. |
| Energy Landscape Coverage | Implicit solvents can alter the relative stability of conformational basins. | Methods like EPO and aSAMt (trained at high T) show improved coverage of conformational space and energy landscapes [60] [61]. |
| Computational Cost | Explicit solvent MD is vastly more expensive, limiting sampling. Implicit solvents allow for faster exploration [10]. | ML generators offer massive speed-ups (seconds vs. months of MD) for ensemble generation after training [59]. |
The divergence between explicit and implicit solvent simulations is starkly illustrated in studies of highly charged systems like heparin. For instance, TIP3P and SPC/E explicit water models yield stable heparin conformations, whereas TIP4P, TIP5P, and OPC introduce greater structural variability, and implicit solvent models can poorly reproduce experimental ring puckering conformations [21]. This highlights that for systems dominated by electrostatics, the choice of solvent model is not merely a technical detail but a determinant of conformational outcomes.
For ML generators, benchmarks on datasets like ATLAS show that while they excel at reproducing local flexibility, they often systematically undersample conformations that are far from the input starting structure [61]. This suggests a failure to fully capture complex multi-state transitions, a key limitation that can lead to significant deviations from the true biological ensemble. Furthermore, the choice of architectural paradigm influences the types of deviations: models using latent diffusion (aSAM) demonstrate superior performance in learning realistic distributions of backbone and side-chain torsion angles compared to models that predict only Cβ positions (AlphaFlow) [61].
Experimental Protocols for Diagnosis and Mitigation
To ensure reliable results, researchers must implement standardized protocols for diagnosing structural deviations and mitigating their impact. The following workflows provide a structured approach for method comparison and validation.
Protocol 1: Diagnosing Solvent Model-Induced Deviations
Objective: To systematically quantify the structural impact of choosing an implicit versus an explicit solvent model, or different explicit water models, for a small protein system.
Materials:
- Protein Structure: A small protein or peptide (e.g., from the provided benchmark set [17]: Chignolin, Trp-cage, BBA).
- Software: A molecular dynamics engine (e.g., GROMACS, OpenMM, AMBER).
- Force Field: A consistent protein force field (e.g., CHARMM36m, AMBER14).
- Solvent Models:
- Explicit: TIP3P, OPC, TIP4P/2005.
- Implicit: Generalized Born (GB) model, e.g., as implemented in the MD software.
- Analysis Tools: GROMACS (
gmx rms,gmx gyrate,gmx rmsf), PLUMED, VMD/MDAnalysis.
Procedure:
- System Setup:
- Prepare the protein structure in a folded or unfolded state.
- For each solvent model, create a corresponding simulation system:
- Explicit: Solvate in a periodic water box with ~1.0 nm padding, add ions to neutralize.
- Implicit: Set the dielectric constant for the solvent (e.g., 78.5 for water).
- Equilibration & Production:
- For all systems, perform energy minimization and a standardized equilibration protocol (NVT and NPT for explicit solvent; NVT for implicit).
- Run production simulations for a fixed, comparable duration (e.g., 100-500 ns). Use a common temperature (300 K) and pressure (1 atm, if applicable).
- Trajectory Analysis:
- Global Structure: Calculate the Radius of Gyration (Rg) and End-to-End Distance over time. Compare distributions.
- Structural Drift: Compute the Root-Mean-Square Deviation (RMSD) of the protein backbone relative to the starting structure.
- Local Dynamics: Calculate the Root-Mean-Square Fluctuation (RMSF) of Cα atoms.
- Conformational Clustering: Perform clustering (e.g., using GROMACS) to identify and visualize the dominant conformational states sampled by each solvent model.
Mitigation Strategies:
- If discrepancies are large, particularly for charged or flexible regions, prioritize explicit solvent models with proven accuracy for electrostatic interactions (e.g., OPC) [21].
- For implicit solvents, consider hybrid approaches that incorporate machine-learned corrections to improve accuracy [10].
Protocol 2: Validating ML Generators Against Reference Data
Objective: To assess the physical realism and diversity of ensembles generated by an ML model (e.g., idpGAN, aSAM, EPO) by comparing them against a reference MD ensemble.
Materials:
- Reference Data: A long, well-converged explicit solvent MD trajectory of the target protein, or a standardized benchmark dataset (e.g., ATLAS [61], mdCATH [61], or the 9-protein benchmark [17]).
- ML Generator: A pre-trained generative model (e.g., aSAM, EPO, BioEmu).
- Analysis Suite: A benchmarking framework capable of computing multiple metrics, such as the one described in [17].
Procedure:
- Ensemble Generation:
- Use the ML generator to produce an ensemble of at least 10,000 conformations for the target protein.
- Comparative Analysis:
- Distributional Fidelity: Use the Wasserstein-1 distance or Kullback-Leibler divergence to compare distributions of Rg, end-to-end distance, and bond lengths/angles against the reference [17].
- Landscape Coverage: Perform Principal Component Analysis (PCA) on the combined (ML + reference) ensemble. Plot the free energy landscape projected onto the first two PCs to visually inspect overlap.
- Local Metrics:
- Calculate the Pearson correlation of per-residue Cα RMSF.
- Use scores like WASCO-global and WASCO-local to compare inter-atomic contact maps and joint distributions of backbone dihedral angles (φ/ψ), respectively [61].
- State Discovery: Check if the ML generator samples known rare or functionally relevant states present in the reference data but absent in the training set.
- Online Refinement (if applicable):
- If the generator supports it (e.g., an EPO-like method), apply energy preference optimization using a physical energy function to steer the ensemble toward a more thermodynamically consistent distribution without requiring additional MD data [60].
Mitigation Strategies:
- If the model undersamples diverse states, use temperature-conditioned models (e.g., aSAMt) or generators explicitly trained on high-temperature MD data to enhance exploration [61].
- If local geometry is poor, employ a brief energy minimization step post-generation to relieve atomic clashes and improve stereochemistry [61].
Workflow for Method Comparison and Validation
The Scientist's Toolkit: Essential Research Reagents
Successful ensemble generation and validation rely on a suite of computational tools, datasets, and software. The table below catalogs key resources cited in this note.
Table 3: Key Research Reagents and Resources
| Resource Name | Type | Primary Function | Relevance to Ensemble Generation |
|---|---|---|---|
| CHARMM36m [21] | Force Field | Defines potential energy terms for atoms in MD. | A modern force field for proteins; essential for running physics-based simulations. |
| TIP3P, OPC [21] | Explicit Solvent Model | Represents water molecules with 3 or 4 interaction sites. | OPC often provides higher accuracy for electrostatic systems; TIP3P is a common default. |
| Generalized Born (GB) [10] | Implicit Solvent Model | Approximates solvent as a dielectric continuum. | Enables faster sampling and is useful for large-scale screening. |
| idpGAN [59] | ML Generator (GAN) | Generates coarse-grained conformational ensembles. | Demonstrates conditional generation for IDPs; fast sampling from MD-trained models. |
| aSAM / aSAMt [61] | ML Generator (Latent Diffusion) | Generates all-atom ensembles, conditioned on temperature. | Provides high-resolution torsion angles and models environmental effects. |
| EPO [60] | ML Refinement Algorithm | Aligns a pre-trained generator with the Boltzmann distribution using energy preferences. | Corrects model misalignment without needing additional MD trajectories. |
| WESTPA [17] | Enhanced Sampling Software | Implements weighted ensemble sampling for rare events. | Generates efficient, reference-quality ground truth data for benchmarking. |
| ATLAS / mdCATH [61] | MD Dataset | Collections of MD trajectories for diverse protein domains. | Used for training and benchmarking ML generative models. |
Method Selection and Workflow Integration
Navigating the trade-offs between different ensemble generation methods requires a strategic approach tailored to the specific research question. The following diagram and guidance outline a logical decision process for method selection.
Decision Logic for Ensemble Generation Method Selection
Guiding Principles for Selection:
- Prioritize Explicit Solvent MD when studying processes where atomic-level details of water-mediated interactions, ion specificity, or precise side-chain packing are paramount (e.g., enzyme catalysis, tight binding affinity calculations) [21] [10].
- Opt for Implicit Solvent MD or ML Generators for high-throughput applications, initial exploratory scans of conformational landscapes, or when simulating large systems over long timescales where explicit solvent is computationally prohibitive [10] [59].
- Leverage ML Generators when needing to generate large, diverse ensembles rapidly from a single input structure, especially if the model has been trained on high-quality, diverse MD data relevant to your system [61] [60].
- Employ a Hybrid Validation Strategy irrespective of the primary method chosen. Use the protocols in Section 4 to compare a subset of your results against a higher-fidelity method (e.g., explicit solvent MD) or experimental data to quantify and control for structural deviations.
Addressing structural deviations between ensemble generation methods is not about identifying a single "best" method, but rather about understanding the inherent trade-offs and applying the right tool for the biological question at hand. The divergence between implicit and explicit solvent models underscores the fundamental role of solvation physics, while the limitations of ML generators highlight the irreplaceable value of comprehensive physical sampling for creating accurate training datasets. By adopting the standardized benchmarking protocols, diagnostic workflows, and strategic selection criteria outlined in this Application Note, researchers can systematically quantify, diagnose, and mitigate these deviations. This rigorous approach ensures that computational explorations of protein conformational ensembles yield robust, reliable, and biologically insightful results, thereby strengthening their impact in fields like structural biology and drug discovery.
Implicit solvent models are a cornerstone of modern computational biophysics, enabling feasible simulations of biological processes by representing the solvent as a continuous medium rather than explicit molecules. The core concept involves partitioning the solvation free energy (ΔGsolv) into physically distinct components, most commonly a polar (electrostatic) term and a nonpolar term [10] [9]. The accuracy of these models is highly dependent on their parameterization. Proper optimization of key physical parameters is therefore not merely a technical exercise but a fundamental requirement to achieve a balanced force field that can accurately recapitulate the conformational equilibria of biomolecular systems, particularly for challenging applications like simulating intrinsically disordered proteins (IDPs) [62]. This application note provides a detailed protocol for the systematic optimization of implicit solvent models, with a specific focus on the Generalized Born with Molecular Volume (GBMV2) model.
Key Parameters for Optimization
The performance of an implicit solvent model hinges on a small set of physical parameters. The optimization strategy involves a recursive adjustment of these parameters to reproduce reference data, which can include solvation free energies, potentials of mean force (PMFs), and most critically, the known conformational equilibria of model peptides [62].
Table 1: Key Parameters for Implicit Solvent Model Optimization
| Parameter | Description | Physical Effect | Optimization Target |
|---|---|---|---|
| Atomic Input Radii | Defines the location of the solute-solvent dielectric boundary [62]. | Directly influences the Born radii and electrostatic solvation energy (ΔGele) [62]. | Reproduction of PB-derived Born radii and experimental hydration free energies of small molecules [62]. |
| Surface Tension Coefficient (γ) | Empirical coefficient scaling the nonpolar solvation energy to the solvent-accessible surface area (SASA) [33] [62]. | Controls the balance between solute-solute and solute-solvent van der Waals interactions; critical for preventing over-compaction [62]. | Agreement with explicit solvent PMFs and realistic radii of gyration for peptides and IDPs [62]. |
| Backbone Torsion Energetics (CMAP) | A correction map for the dihedral angles of the protein backbone [62]. | Corrects inherent biases in the underlying force field's secondary structure propensity (e.g., over-stabilization of α-helices) [62]. | Accurate stabilization of both helical and β-sheet structures in model peptides [62]. |
| Effective Born Radius Algorithm | The method for calculating the degree of burial of an atom (e.g., HCT, OBC, Neck) [33]. | Determines accuracy of ΔGele approximation versus PB; models with "neck" corrections better mimic molecular surfaces [33]. | Close reproduction of solvation energies and forces from reference Poisson-Boltzmann calculations [33]. |
Experimental Protocols
Protocol 1: Recursive Optimization of the GBMV2 Force Field
This protocol outlines the recursive parameter optimization strategy successfully employed for the GBMV2 model with the CHARMM36 force field [62].
- Initial Parameter Set. Begin with the default GBMV2 parameters and the CHARMM36 protein force field.
- Optimize Atomic Input Radii.
- Reference Data: Calculate the electrostatic solvation free energies (ΔGele) for a set of peptide backbone and side chain analogs using a high-resolution Poisson-Boltzmann (PB) solver.
- Iteration: Adjust the atomic input radii for key atom types to minimize the difference between the GBMV2-calculated and PB-calculated ΔGele values.
- Optimize Surface Tension Coefficient (γ).
- Reference Data: Calculate the potentials of mean force (PMFs) for pairwise interactions of small molecules in explicit solvent (e.g., TIP3P water).
- Iteration: Adjust the γ value in the SASA term so that the PMFs generated with the implicit solvent model match the reference explicit solvent PMFs.
- Optimize Backbone Torsions (CMAP).
- Reference Data: Perform converged conformational sampling on a set of model helical (e.g., Ala5) and β-hairpin (e.g., GB1p-derived peptides) peptides. This requires enhanced sampling methods (see Protocol 2).
- Iteration: Adjust the CMAP torsion cross terms so that the simulated conformational ensembles show a balanced representation of helical and β-sheet content, in agreement with experimental data.
- Validation. The optimized force field must be validated on a separate set of peptides and proteins not used in the training. A critical test is the simulation of intrinsically disordered proteins (IDPs) to ensure the model does not produce artificially compact structures and generates ensembles consistent with experimental observables (e.g., SAXS data) [62].
Protocol 2: Enhanced Sampling for Parameter Validation
Converged sampling is essential for evaluating conformational equilibria during parameter optimization. Traditional temperature replica exchange (T-REMD) can be prohibitively expensive for models with sharp solvent boundaries. This protocol leverages the Multi-Scale Enhanced Sampling (MSES) method [62].
- System Setup. Prepare the atomistic system (e.g., a β-hairpin peptide) with the current implicit solvent parameters.
- Coarse-Grained Coupling. Concurrently, create a coarse-grained (CG) representation of the same system using a simplified model that allows for faster conformational transitions.
- Hamiltonian Replica Exchange. Set up a Hamiltonian replica exchange simulation where the different replicas are coupled to the CG system with varying coupling strengths.
- Sampling. Run the MSES simulation. The replica with no coupling provides an unbiased atomistic ensemble, while the strong coupling to the CG model drives rapid folding and unfolding events in other replicas, enhancing the overall sampling efficiency.
- Analysis. Analyze the unbiased ensemble to determine the population of native-like states, the radius of gyration, and other relevant metrics for comparison with experimental data.
The following workflow diagram illustrates the recursive optimization and validation process.
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Item / Resource | Function / Description | Application Note |
|---|---|---|
| CHARMM-GUI Implicit Solvent Modeler (ISM) [33] | A web-based platform for setting up implicit solvent simulations and generating input files for various MD packages (AMBER, CHARMM, OpenMM, etc.). | Supports major GB models (GBMV, GBSW, GB-OBC, GB-Neck) and force fields, ensuring reproducible system preparation. |
| GBMV2/GBSW Implicit Solvent | Implicit solvent models that use a molecular surface (MS) definition for the dielectric boundary, providing a more physical representation than van der Waals surfaces [62]. | More accurate but computationally demanding; may require 1-1.5 fs time steps due to sharper forces. |
| GB-Neck2 Implicit Solvent | A GB model incorporating a "neck" correction to approximate the molecular surface, offering high accuracy and speed [62]. | An excellent alternative to GBMV/GBSW, optimized for use with GPU-accelerated MD codes. |
| Multi-Scale Enhanced Sampling (MSES) [62] | An enhanced sampling method that couples all-atom and coarse-grained models via Hamiltonian replica exchange. | Drastically accelerates conformational sampling, making force field optimization feasible. |
| CHARMM36 & Amber ff19SB | All-atom protein force fields, commonly used as the base for implicit solvent parameter optimization [33] [62]. | Underlying force field parameters (especially CMAP) must be co-optimized with solvation parameters. |
| Model Peptide Set | A curated set of small peptides with well-characterized structures (e.g., helices, β-hairpins, IDPs) [62]. | Serves as the primary benchmark for evaluating and optimizing conformational equilibria. |
Performance and Application
Successful parameter optimization leads to a balanced implicit solvent force field capable of accurately modeling diverse biological systems. The primary performance gain is not just computational speed but correct conformational sampling.
- Sampling Speedup: Implicit solvent simulations can provide a ∼1 to over ∼100-fold speedup in conformational sampling compared to explicit solvent (PME), depending on the system and the conformational change being studied [12]. This is largely due to reduced solvent viscosity.
- Structural Accuracy: An optimized GBMV2 force field can recapitulate the native structures of folded proteins and generate structurally accurate ensembles for intrinsically disordered proteins (IDPs) that align with experimental data [62].
- Practical Application - Ligand Screening: Implicit solvent simulations, particularly when used with MM/GBSA calculations, provide a reliable tool for early-stage ligand screening. They demonstrate better performance than docking alone for discriminating correct binding modes [33].
Fine-tuning implicit solvent models is a critical and non-trivial process essential for achieving physical accuracy in biomolecular simulations. The recursive optimization of atomic radii, the surface tension coefficient, and backbone torsion profiles—validated against experimental data and reference calculations using enhanced sampling techniques—is a proven strategy. The resulting parameter sets enable realistic simulations of a wide range of systems, from stable folded proteins to flexible IDPs and protein-ligand complexes, making optimized implicit solvent models an indispensable tool in computational structural biology and drug discovery.
The accurate modeling of solvent effects is fundamental to molecular dynamics (MD) simulations of biological systems, particularly in drug development where binding affinities and protein-ligand interactions are central to the research. Two predominant philosophies have emerged: explicit solvent models, which simulate individual solvent molecules and offer high accuracy at a steep computational cost, and implicit solvent models (also known as continuum models), which represent the solvent as a dielectric medium, offering computational efficiency but often at the expense of accuracy, especially for specific solute-solvent interactions [26] [63]. Hybrid approaches seek to combine the strengths of both methods by using an explicit solvent representation in critical regions and an implicit representation in others. This strategy aims to achieve a favorable balance between computational tractability and physicochemical accuracy, making it highly suitable for the study of small proteins and their interactions with ligands over biologically relevant timescales.
The table below summarizes the core characteristics of the different solvent modeling approaches.
Table 1: Comparison of Solvent Modeling Methodologies
| Feature | Explicit Solvent | Implicit Solvent | Hybrid Approaches |
|---|---|---|---|
| Solvent Representation | Individual molecules (e.g., water, ions) | Continuum dielectric medium | Explicit in key regions, implicit elsewhere |
| Computational Cost | Very High | Low | Moderate |
| Treatment of Specific Solute-Solvent Interactions | Atomistically detailed | Averaged/Approximated | Explicit in defined zones |
| Sampling Speed | Slower (due to solvent viscosity) | Faster (no solvent friction) | Intermediate |
| Handling of Long-Range Electrostatics | Complex (e.g., PME) with periodic artifacts | Straightforward (infinite volume) | Varies by implementation |
| Best Suited For | Detailed mechanistic studies, validation | High-throughput screening, conformational sampling | Balancing accuracy & efficiency in drug discovery |
Key Hybrid Methodologies and Protocols
The Hybridizing Ions Treatment (HIT) Method
The HIT method is specifically designed to address the challenge of simulating highly charged biomolecules like DNA, RNA, and highly charged proteins, where traditional implicit solvent models struggle to balance the system's net charge effectively [64].
- Objective: To realistically calculate electrostatic potentials for highly charged biomolecules by integrating explicit ion data into an implicit solvent framework.
- Principle: The method hybridizes the implicit solvent model with explicit ion information. It first uses an explicit solvent simulation to predict the distribution of "bound" ions. These pre-calculated ion positions are then incorporated as a fixed component into subsequent implicit solvent (e.g., DelPhi) calculations for electrostatic analysis [64].
- Protocol:
- Explicit Simulation for Ion Distribution: Perform a molecular dynamics simulation of the biomolecule in an explicit solvent containing ions.
- Identification of Bound Ions: Analyze the trajectory to identify ions that remain persistently associated with the biomolecule, classifying them as "bound."
- Parameter Optimization: Use dedicated training sets to optimize the parameters for integrating these bound ions into the implicit model.
- Implicit Calculation with Fixed Ions: Run the final electrostatic potential calculation using the implicit solvent method, with the bound ions included as part of the solute molecule.
- Application Note: This method significantly improves the accuracy of electrostatic calculations for systems like molecular motors (myosin, kinesin) and nucleic acids, providing mechanistic insights that align with experimental findings [64].
Brownian Dynamics/Molecular Dynamics (BD/MD) Multiscale Pipelines
This hybrid approach leverages the strengths of both Brownian Dynamics and all-atom Molecular Dynamics to efficiently compute bimolecular association rate constants ((k_{on})), which are critical indicators of drug efficacy [65].
- Objective: To accurately calculate protein-ligand association rates ((k_{on})) while accounting for long-range diffusion and short-range molecular interactions.
- Principle: The method divides the binding process into two stages managed by different physics-based models. BD simulations, which use an implicit solvent model and often rigid-body approximations, efficiently handle the long-range diffusional encounter of the ligand and protein to form an encounter complex. Subsequently, these BD-generated complexes serve as initial structures for more detailed, but computationally expensive, MD simulations with explicit solvent, which capture the final short-range interactions, solvent displacement, and induced fit [65].
- Protocol:
- BD Simulation Setup: Define the protein's position and create two spherical surfaces: an outer "q-surface" and an inner "b-surface" near the binding site.
- Trajectory Propagation: Launch numerous BD trajectories with the ligand starting on the b-surface. Trajectories end when the ligand either meets the reaction criteria (forms an encounter complex) or escapes to the q-surface.
- Encounter Complex Harvesting: Collect the structures from BD trajectories where the ligand comes very close to the binding site.
- MD Simulation Initiation: Use the harvested encounter complexes as starting points for multiple, shorter all-atom MD simulations with explicit solvent.
- Analysis: Compute the association rate constant ((k_{on})) by combining the probability of encounter complex formation from BD with the probability of forming the final bound state from MD [65].
The following diagram illustrates this multi-stage workflow:
Workflow of a BD/MD Hybrid Approach
Machine Learning-Augmented Implicit Solvent Models
Recent advancements introduce machine learning (ML) to correct the limitations of traditional implicit solvent models, creating a different form of hybrid methodology that leverages data from explicit solvent simulations [26].
- Objective: To develop implicit solvent models with near-explicit solvent accuracy for solvation free energies and forces, which are suitable for absolute free energy comparisons.
- Principle: A Graph Neural Network (GNN) is trained not only on forces from explicit solvent simulations (force-matching) but also on the derivatives of the solvation energy with respect to alchemical variables (e.g., electrostatic ((\lambda{elec})) and steric ((\lambda{steric})) coupling factors). This ensures the model learns a scalar potential that accurately represents the Potential of Mean Force (PMF), overcoming the arbitrary constant problem in energy predictions [26].
- Protocol:
- Data Generation: Generate a large dataset (e.g., ~300,000 small molecules) from explicit-solvent alchemical simulations.
- Network Architecture: Employ a graph neural network (e.g., λ-Solvation Neural Network, LSNN) to represent the molecular system.
- Multi-Term Loss Function: Train the network using a modified loss function that combines:
- Force-matching loss ((wF))
- Electrostatic coupling derivative loss ((w{elec}))
- Steric coupling derivative loss ((w_{steric}))
- Simulation & Prediction: Use the trained LSNN model as the potential for MD simulations, enabling the prediction of solvation free energies with accuracy comparable to explicit solvent but at a computational speed comparable to implicit solvent [26].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 2: Key Research Reagents and Computational Tools for Hybrid Solvent Studies
| Tool/Solution | Type | Primary Function |
|---|---|---|
| HIT Package [64] | Software Suite | Implements the Hybridizing Ions Treatment method for electrostatic calculations of highly charged biomolecules. |
| DelPhi [64] | Software Module | A Poisson-Boltzmann solver for implicit solvent electrostatics; used within the HIT protocol. |
| λ-Solvation Neural Network (LSNN) [26] | Machine Learning Potential | A GNN-based implicit solvent model trained for accurate solvation free energy and force prediction. |
| SEEKR [65] | Software Plugin | A tool (e.g., for OpenMM) that implements the BD/MD milestoning approach to calculate binding kinetics. |
| GBSA/PBSA Models [26] [63] | Implicit Solvation Model | Provides a fast, approximate solvation energy (polar + non-polar) and serves as a baseline for ML model improvement. |
| Active Learning (AL) Loop [66] | Computational Strategy | An iterative protocol for efficiently building training sets for ML potentials by identifying under-sampled regions of chemical space. |
Hybrid explicit-implicit solvent methodologies represent a powerful and evolving paradigm in molecular simulation. By strategically combining the accuracy of explicit solvent in critical regions with the speed of implicit solvent elsewhere, methods like HIT, BD/MD pipelines, and ML-augmented models effectively bridge the gap between computational feasibility and biological realism. These protocols enable researchers to tackle challenging problems, such as simulating highly charged biomolecules, predicting drug-protein association rates, and calculating absolute solvation free energies—tasks that are often prohibitively expensive for purely explicit solvent models. As machine learning continues to be integrated into these frameworks, hybrid approaches are poised to become even more accurate and automated, solidifying their role as an indispensable tool in computational drug discovery and the study of small protein dynamics.
Molecular dynamics (MD) simulations are indispensable for studying biological processes, yet a central challenge persists: accurately and efficiently modeling the aqueous environment. The explicit solvent model, which treats each water molecule as an individual entity, is considered the gold standard for accuracy. It captures specific solute-solvent interactions, such as hydrogen bonding and bridging water molecules, which are critical for processes like protein-ligand recognition. However, this accuracy comes at a prohibitive computational cost, limiting the accessible timescales and system sizes [8]. Implicit solvent (or continuum) models offer a computationally efficient alternative by replacing the explicit water molecules with a dielectric continuum, thereby drastically reducing the number of degrees of freedom in the system. While orders of magnitude faster, traditional implicit models can lack accuracy, particularly for processes where local solvent structure is important [1] [8]. This application note provides a structured framework for researchers to navigate this trade-off, presenting tuned protocols and modern tools that enable highly efficient simulations of small proteins without compromising the fidelity of results.
Quantitative Comparison of Solvation Models
Selecting an appropriate solvation model requires a clear understanding of their performance characteristics. The data in the table below summarizes key metrics from published studies, providing a benchmark for expectations regarding accuracy and computational demand.
Table 1: Performance and Accuracy Comparison of Common Implicit Solvent Models vs. Explicit Solvent
| Solvent Model | Correlation with Explicit Solvent (Ligands) | Correlation with Explicit Solvent (Proteins) | Typical Computational Speed vs. Explicit Solvent | Key Limitations |
|---|---|---|---|---|
| Explicit (TIP3P) | 1.00 (Reference) | 1.00 (Reference) | 1x (Base) | High computational cost; limits sampling speed [4] [26] |
| Poisson-Boltzmann (APBS) | 0.953 - 0.966 | 0.65 - 0.99 | 10-100x faster | Sensitive to cavity definition; higher computational cost than other implicit models [4] |
| Generalized Born (GBNSR6) | 0.953 - 0.966 | 0.65 - 0.99 | 10-100x faster | Approximates PB; accuracy can vary with parameterization [4] |
| PCM | 0.87 - 0.93 | 0.65 - 0.99 | 10-100x faster | High numerical accuracy but computationally intensive among implicit models [4] |
| COSMO | 0.87 - 0.93 | 0.65 - 0.99 | 10-100x faster | Conductor-like approximation; performance depends on implementation [4] |
| SASA-Only | Moderate | Low | >100x faster | Misses crucial electrostatic polarization effects; often used for non-polar contributions [8] |
The data reveals that while all tested implicit models show high correlation with explicit solvent for small molecules, their performance for proteins and protein-ligand complexes is more variable, with correlations ranging from 0.65 to 0.99. This underscores the critical importance of model parameterization for biomolecular systems [4]. Furthermore, the choice of underlying force field and partial charge assignment significantly impacts the results, highlighting the need for a consistent and well-validated parameter set [4].
Tuned Protocols for Specific Research Objectives
The following section provides detailed, step-by-step protocols optimized for different research scenarios commonly encountered in the study of small proteins.
Protocol 1: Rapid Binding Affinity Estimation for Ligand Screening
This protocol is designed for high-throughput screening of small molecules against a protein target, where relative ranking is more critical than absolute binding free energy values.
Table 2: Key Research Reagents and Computational Tools
| Reagent/Solution | Function/Description | Application Note |
|---|---|---|
| GPU-Accelerated MD Engine (e.g., apoCHARMM, OpenMM) | Performs the molecular dynamics calculations with high computational efficiency. | Essential for achieving the required speed for high-throughput sampling [67] [23]. |
| Implicit Solvent Model (e.g., GBNSR6, PBSA) | Models solvation effects without explicit water molecules, drastically speeding up calculations. | GBNSR6 has shown high accuracy in estimating desolvation penalties in binding [4]. |
| Force Field (e.g., Amber12, CHARMM36) | Provides the functional form and parameters for potential energy. | Consistent parameterization for both protein and ligand is critical [4]. |
| Ligand Parametrization Tool (e.g., MATCH) | Generates force field parameters for small molecule inhibitors. | Ensures compatibility between the ligand and the chosen force field [23]. |
Step-by-Step Workflow:
System Preparation:
- Obtain the protein structure from a database (e.g., PDB) and prepare it using standard tools (e.g., BIOVIA Discovery Studio). This includes adding missing hydrogen atoms, assigning protonation states, and optimizing side-chain orientations.
- For each ligand, generate 3D coordinates and assign partial charges and force field parameters using a tool like MATCH [23].
Initial Docking:
- Perform flexible ligand docking into the protein's active site using a fast docking engine like CDOCKER [23] to generate an ensemble of plausible initial binding poses.
Implicit Solvent Equilibration:
- For each docked pose, solvate the protein-ligand complex in an implicit solvent model (e.g., GBSA). A recommended starting point is the GBNSR6 model [4].
- Carry out energy minimization followed by a brief MD simulation (e.g., 1-5 ns) to equilibrate the complex at the target temperature (e.g., 300 K).
Binding Free Energy Calculation:
- Use the equilibrated trajectories to compute the binding free energy via a method such as Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) or its Generalized Born equivalent (MM/GBSA) [23].
- The calculation decomposes the free energy into gas-phase interaction energy and solvation free energy components.
Analysis and Ranking:
- Rank all ligands based on their calculated binding free energies. This protocol is highly effective for identifying the most promising candidates from a large library for further experimental validation [23].
Protocol 2: Enhanced Conformational Sampling of Protein States
This protocol leverages the reduced friction of implicit solvent to achieve broader conformational sampling, which is useful for studying folding or large-scale conformational changes.
Step-by-Step Workflow:
System Setup:
- Prepare the protein system as in Protocol 1, but without a ligand.
Solvation and Minimization:
- Place the protein in a continuum solvent with a high dielectric constant (e.g., ε=80 for water). Models like PCM or a well-parameterized GB model are suitable [4].
- Perform extensive energy minimization to remove any steric clashes.
Extended Sampling Simulation:
- Run a long, unbiased MD simulation (hundreds of nanoseconds to microseconds) using the implicit solvent model. The reduced viscosity allows the protein to transition between states more rapidly than in explicit solvent.
Advanced Sampling (Optional):
- To further accelerate the sampling of rare events, integrate the implicit solvent with enhanced sampling techniques. Gaussian accelerated MD (GaMD) is an excellent choice as it is implemented in major software suites like BIOVIA Discovery Studio and does not require predefined reaction coordinates [23].
- Configure and run a GaMD simulation, allowing it to automatically parametrize and apply boost potentials to the system's potential energy.
Trajectory and Free Energy Analysis:
- Analyze the resulting trajectory to identify metastable states and conformational pathways.
- If GaMD was used, estimate the free energy landscape from the simulation data, which may require statistical reweighting [23].
Protocol 3: Hybrid Explicit/Implicit Solvent for Balanced Performance
For processes where first-shell water molecules are critical, but bulk solvent effects are less specific, a hybrid QM/MM-style approach can be optimal.
Step-by-Step Workflow:
Define System Layers:
- Layer 1 (QM/Explicit): Define the core region (e.g., the protein active site with a bound ligand and a few critical water molecules) to be treated with explicit solvent.
- Layer 2 (MM/Continuum): Treat the remainder of the protein and the bulk solvent using an implicit model.
Simulation Execution:
- Use a simulation package that supports multi-layer calculations, such as a QM/MM-enabled engine.
- Run the MD simulation, ensuring self-consistency between the different layers of the system.
Validation:
- Critically, validate the results of this hybrid approach against a shorter, fully explicit solvent simulation or experimental data to ensure key interactions are preserved [43]. Studies have shown that for some systems, like certain catalytic reactions, implicit models can suffice if no specific solvent participation occurs [43].
Workflow Visualization and Decision Logic
The following diagram illustrates the logical process for selecting the most appropriate performance-tuning strategy based on the research goal.
Diagram 1: Solvent Model Selection Workflow
Advanced Methods and Implementation Tools
To achieve maximal performance, leveraging modern hardware and algorithms is essential.
- GPU Acceleration: Implement simulations on Graphics Processing Units (GPUs). Modern MD engines like apoCHARMM and OpenMM are optimized for GPU architectures, providing massive performance gains over CPU-based calculations. apoCHARMM, for instance, is designed for single-GPU support of multiple Hamiltonians, enabling highly efficient free energy methods and minimizing data transfer bottlenecks [67] [27].
- Machine Learning Potentials: Emerging machine learning (ML) approaches offer a path to near-explicit solvent accuracy at implicit solvent speeds. These models, such as the λ-Solvation Neural Network (LSNN), are trained on large datasets from explicit solvent simulations. They learn to predict solvation forces and free energies, addressing a key limitation of traditional implicit models [26].
- Variational Implicit Solvent Models: For studying large-scale processes like protein-protein interactions, advanced continuum models like the Variational Explicit-Solute Implicit-Solvent (VESIS) model are highly efficient. VESIS uses a binary level-set method to capture the solute-solvent interface and is implemented on GPUs, leading to significant speedups for determining equilibrium conformations [27].
- High-Throughput Simulation Datasets: For formulation or mixture design, leveraging large-scale MD datasets can bypass the need for exhaustive custom simulations. Machine learning models trained on such datasets (e.g., 30,000 solvent mixtures) can predict properties like enthalpy of mixing, providing instant estimates for novel systems [68].
The trade-off between computational efficiency and accuracy in molecular simulations is no longer a rigid compromise. By strategically selecting and applying the protocols outlined in this document—ranging from tuned implicit solvent setups for specific tasks to the adoption of GPU-accelerated engines and machine-learning potentials—researchers can dramatically increase the scale and scope of their simulations. The key to success lies in matching the tool to the problem: use fast implicit models for high-throughput screening and enhanced sampling, and reserve hybrid or explicit solvent models for systems where atomic-level solvent detail is non-negotiable. As the field progresses, the integration of machine learning and advanced continuum models will continue to push the boundaries, making accurate, microsecond-to-millisecond simulations of small proteins a routine endeavor in drug discovery and structural biology.
Benchmarking Performance: Quantitative Comparison of Solvent Model Accuracy
The choice of solvent model is a critical determinant of efficiency in molecular dynamics (MD) simulations of biological macromolecules. This Application Note provides a structured framework for quantifying the acceleration of conformational sampling achieved when using implicit solvent models compared to explicit solvent treatments. For researchers investigating small protein dynamics and drug development professionals, understanding these speedups is essential for allocating computational resources effectively and designing feasible simulation projects. The protocols outlined herein are framed within a broader thesis on implicit versus explicit solvent models, focusing on practical assessment methodologies rather than philosophical debate about model superiority. We summarize quantitative findings from controlled comparisons and provide detailed protocols for reproducing these assessments, enabling researchers to make informed decisions based on their specific system requirements and accuracy tolerances.
Quantitative Speedup Analysis
The acceleration of conformational sampling varies significantly depending on the size of the molecular system and the type of conformational change being studied. The following table synthesizes quantitative speedup factors from comparative studies.
Table 1: Conformational Sampling Speedup of Generalized Born (GB) Implicit Solvent vs. PME Explicit Solvent
| Type of Conformational Change | System Description | Nominal Simulation Time | Sampling Speedup (GB vs. PME) | Combined Speedup (Algorithm + Sampling) |
|---|---|---|---|---|
| Small (Dihedral angle flips) | Phospholipase C (4,812 atoms) | Nanoseconds to microseconds | Approximately 1-fold [69] [70] | Approximately 2-fold [69] [70] |
| Large (Nucleosome tail collapse, DNA unwrapping) | Nucleosome complex (25,100 atoms) | Nanoseconds to microseconds | ~1-fold to ~100-fold [69] [70] | ~1-fold to 60-fold [69] [70] |
| Mixed (Miniprotein folding) | CLN025 protein (166 atoms) | Nanoseconds to microseconds | Approximately 7-fold [69] [70] | Approximately 50-fold [69] [70] |
The primary factor driving these speedups is the reduction of solvent viscosity in implicit solvent models, which lowers the friction opposing conformational transitions [69] [70]. This effect is modulated by the Langevin collision frequency parameter in implicit solvent simulations; lower collision frequencies (effectively lower viscosity) yield greater sampling speedups [69] [70]. The table also distinguishes between "Sampling Speedup" (the rate of conformational transitions per nominal simulation time unit) and "Combined Speedup" (which additionally accounts for algorithmic efficiency gains measured by simulation time per processor hour) [69] [70]. For small systems, the purely computational advantage of implicit solvent is minimal or non-existent, but it becomes substantial for larger systems where explicit solvent calculations become prohibitively expensive due to the large number of solvent atoms [69] [70].
Experimental Protocols
Protocol: Comparative Sampling Speed Assessment
This protocol describes how to quantitatively compare the speed of conformational sampling between implicit and explicit solvent models for a protein system of interest.
Step 1: System Preparation
- Obtain the initial protein structure from a database such as the Protein Data Bank (PDB) or through computational modeling.
- For explicit solvent simulations, solvate the protein in a pre-equilibrated water box (e.g., using TIP3P water model) with a minimum distance of 10-12 Å between the protein and box edges. Add ions to neutralize system charge and achieve desired physiological concentration [70].
- For implicit solvent simulations, prepare the protein structure in vacuum, as the solvent effects will be modeled continuously. No explicit water molecules or ions are added, though ionic strength can be parameterized within the implicit model [70] [8].
Step 2: Simulation Parameters
- Software: Use a package that supports both solvent models, such as AMBER [69] [70].
- Explicit Solvent Model: Use the Particle Mesh Ewald (PME) method for long-range electrostatics [69] [70].
- Implicit Solvent Model: Employ a Generalized Born (GB) model, such as the popular GB model implemented in AMBER [69] [70]. Set the Langevin collision frequency to a low value (e.g., 1.0 ps⁻¹) to maximize the viscosity reduction effect [69] [70].
- Common Parameters: Use identical force fields (e.g., AMBER ff99SB), temperature (300 K), and time step (e.g., 2 fs) for both simulations to ensure a fair comparison. Apply similar positional restraints, if any, during equilibration phases.
Step 3: Production Simulation and Analysis
- Run multiple independent production simulations for both setups for identical nominal time periods (e.g., 100 ns - 1 µs, depending on system size).
- Quantify Sampling Speed: For the conformational event of interest (e.g., dihedral flip, folding/unfolding):
- Define a collective variable (CV) or order parameter that characterizes the transition (e.g., dihedral angle, radius of gyration, fraction of native contacts).
- Count the number of complete transitions between states along the simulation trajectory.
- Calculate the transition rate as the number of transitions divided by the total simulation time.
- Calculate Speedup: Compute the ratio of the transition rate in the implicit solvent simulation to the rate in the explicit solvent simulation.
Validation: Where possible, compare the free-energy landscapes along relevant CVs generated from both simulations to assess whether the implicit solvent model alters the thermodynamics in addition to the kinetics [69] [52].
Workflow Visualization
The following diagram illustrates the logical workflow for the comparative assessment protocol, highlighting the parallel paths for explicit and implicit solvent simulations.
The Scientist's Toolkit
This section details the essential computational reagents and their functions for performing the comparative speed assessments described in this protocol.
Table 2: Research Reagent Solutions for Solvent Model Comparison
| Tool/Reagent | Function in Protocol | Example Implementations / Notes |
|---|---|---|
| MD Simulation Package | Core engine for performing calculations; must support both solvent models. | AMBER [69] [70], CHARMM [52], GROMACS, NAMD. |
| Explicit Solvent Model | Represents water as discrete molecules; provides the benchmark for comparison. | TIP3P water model with Particle Mesh Ewald (PME) for long-range electrostatics [69] [70] [52]. |
| Implicit Solvent Model | Approximates solvent as a continuum; the model whose efficiency is being assessed. | Generalized Born (GB) models, such as the GB model in AMBER or GBSW [69] [70] [52]. |
| Protein Force Field | Defines the potential energy function and parameters for the solute (protein). | AMBER ff99SB, CHARMM19, CHARMM22 with CMAP. Use the same force field for both solvent simulations [70] [52]. |
| Visualization & Analysis Tool | Used to visualize trajectories and quantify conformational changes and transition rates. | VMD, Chimera, MDTraj, in-house scripts for transition rate calculation. |
| Langevin Thermostat | Controls temperature in simulations; collision frequency parameter controls effective viscosity in implicit solvent. | Set to a low value (e.g., 1.0 ps⁻¹) for GB to maximize speedup [69] [70]. |
This Application Note establishes that the acceleration of conformational sampling with implicit solvent models is not a fixed value but is highly system- and problem-dependent. The provided quantitative data and detailed protocol offer researchers a framework for anticipating and measuring this speedup for their specific systems of interest. The primary mechanism of acceleration is the reduction of solvent friction, which can be strategically tuned via the Langevin collision frequency parameter. When selecting a solvent model, researchers must weigh the significant speed advantages of implicit solvent, especially for larger systems and large-scale conformational changes, against potential alterations to the free-energy landscape, which were beyond the scope of this assessment but remain a critical consideration for achieving biologically accurate results.
Accurate calculation of binding free energy is a central goal in structure-based drug design, playing a key role in ranking potential drug compounds according to their binding affinities [71]. Among computational methods, Molecular Mechanics with Generalized Born and Surface Area solvation (MM/GBSA) has emerged as a popular endpoint technique that offers a balance between computational efficiency and accuracy [72] [73]. This application note examines the consistency and limitations of MM/GBSA within the broader context of implicit versus explicit solvent models for small protein molecular dynamics (MD) simulations. While explicit solvent models like TIP3P, TIP4P, and OPC provide detailed atomic representation of solvent interactions, they remain computationally expensive for large-scale binding affinity studies [21] [26]. Implicit solvent models, including GB and Poisson-Boltzmann (PB) approaches, significantly reduce computational cost by treating the solvent as a continuous dielectric medium, but this simplification introduces approximations that affect accuracy [27] [21]. Understanding these trade-offs is essential for researchers applying these methods in drug discovery workflows.
Theoretical Framework of MM/GBSA
Energy Components
The MM/GBSA method estimates the absolute binding free energy (ΔGbind) of a protein-ligand complex through the thermodynamic relationship shown in Equation 1 [73] [74]:
Equation 1: ΔGbind = ΔH - TΔS
Where ΔH represents the enthalpy change and TΔS represents the entropic contribution at absolute temperature T. In practical implementation, the binding free energy is calculated using a thermodynamic cycle that separates vacuum and solvation contributions [73], yielding the component decomposition in Equation 2:
Equation 2: ΔGbind = ΔEMM + ΔGsolv - TΔS
The molecular mechanics energy (ΔEMM) includes internal (bond, angle, dihedral), electrostatic (ΔEele), and van der Waals (ΔEvdW) components [75] [74]. The solvation free energy (ΔGsolv) is further decomposed into polar (ΔGGB) and non-polar (ΔGSA) contributions [73] [74]. The complete decomposition is summarized in Table 1.
Table 1: Components of MM/GBSA Binding Free Energy Calculation
| Energy Component | Description | Calculation Method |
|---|---|---|
| ΔEMM | Gas-phase molecular mechanics energy | Molecular mechanics force field |
| ΔEinternal | Bond, angle, and dihedral energy | Force field |
| ΔEele | Electrostatic interaction energy | Coulomb's law |
| ΔEvdW | Van der Waals interaction energy | Lennard-Jones potential |
| ΔGsolv | Solvation free energy | ΔGGB + ΔGSA |
| ΔGGB | Polar solvation contribution | Generalized Born equation |
| ΔGSA | Non-polar solvation contribution | Solvent-accessible surface area |
| -TΔS | Entropic contribution | Normal mode or quasi-harmonic analysis |
The typical MM/GBSA workflow involves multiple stages from system preparation through energy analysis. Figure 1 illustrates this process, highlighting the key steps where methodological decisions significantly impact results.
Figure 1. MM/GBSA calculation workflow showing key stages from system preparation through binding free energy estimation.
Critical Assessment of Limitations
Solvation Model Approximations
A fundamental limitation of MM/GBSA stems from its simplified treatment of solvation. While explicit solvent models like TIP3P, TIP4P, and OPC individually represent water molecules, the Generalized Born model approximates water as a continuous dielectric, which affects the accuracy of electrostatic interactions [21]. This is particularly problematic for systems where specific water molecules mediate binding interactions. Comparative studies reveal that implicit solvent models often poorly reproduce experimental ring puckering conformations in glycosaminoglycans like heparin, whereas explicit solvents offer improved accuracy [21]. The non-polar solvation term (ΔGSA) represents another significant simplification, as it is typically estimated using a simple linear relationship with solvent-accessible surface area (SASA), which may not adequately capture complex hydrophobic effects [26].
Conformational Sampling and Entropy Estimation
The neglect of adequate conformational sampling and entropic contributions represents a major source of inconsistency in MM/GBSA calculations. Configurational entropy change (-TΔS) is computationally expensive to calculate, leading many studies to omit it entirely, which results in overestimation of binding affinity [73] [76]. Normal mode analysis (NMA) provides a theoretically sound approach but becomes prohibitively expensive for large systems [73] [76]. As shown in Table 2, truncated NMA and interaction entropy (IE) methods have emerged as practical alternatives, with IE demonstrating particular promise as it does not incur additional computational cost and provides comparable or better results than truncated NMA in many cases [76].
Table 2: Performance Comparison of Entropy Calculation Methods in MM/GBSA
| Method | Description | Advantages | Limitations | Recommended Use |
|---|---|---|---|---|
| Normal Mode Analysis (NMA) | Calculates vibrational modes from Hessian matrix | Theoretically rigorous | Computationally expensive for large systems | Small systems (<100 atoms) |
| Truncated NMA | NMA on reduced system around binding site | Reduces computational cost | May miss long-range interactions | Protein-ligand complexes with εin = 4 |
| Interaction Entropy (IE) | Estimates entropy from fluctuation of interaction energy | No additional computational cost | Requires sufficient sampling | Diverse datasets with εin = 1-2 |
Ensemble sampling approaches significantly improve reliability. Studies on DNA-intercalator complexes demonstrated that binding energies averaged over multiple simulation replicas show much better agreement with experimental values than single trajectories [77]. Notably, 6 replicas of 100 ns or 8 replicas of 10 ns provided an optimal balance between computational cost and accuracy within 1.0 kcal/mol of experimental values [77].
Dielectric Constant Selection and Force Field Dependence
The choice of internal dielectric constant (εin) significantly impacts MM/GBSA predictions. Higher values (εin = 4) generally work better with truncated NMA entropy calculations, while lower values (εin = 1-2) pair better with the interaction entropy method [76]. While MM/GBSA predictions show some sensitivity to force field choice, comparative studies indicate that differences between modern force fields are relatively modest, with the ff03 force field performing slightly better in benchmark studies [76].
Recommended Protocols
Standard Binding Free Energy Calculation
This protocol describes the MM/GBSA procedure for estimating protein-ligand binding affinities, optimized based on recent benchmark studies [73] [76].
System Preparation
- Obtain the protein-ligand complex structure from crystallography, docking, or MD simulation.
- Process structures using standard tools (e.g., CHARMM-GUI, tleap) to add hydrogen atoms and missing residues.
- Assign partial charges to the ligand using appropriate methods (e.g., AM1-BCC for GAFF ligands).
Molecular Dynamics Simulation
- Solvate the system in an explicit solvent box (e.g., TIP3P water) with 6-10 Å padding from the solute.
- Neutralize the system with counterions (e.g., Na⁺/Cl⁻).
- Perform energy minimization using steepest descent algorithm until convergence (<10 kJ/mol/nm).
- Equilibrate the system first in NVT ensemble (125 ps) followed by NPT ensemble (125 ps) at 300 K with positional restraints on protein heavy atoms (400 kJ/mol/nm²).
- Run production MD simulation for 50-100 ns with a 2 fs time step, saving frames every 100 ps for analysis.
MM/GBSA Calculation
- Extract 500-1000 evenly spaced snapshots from the stabilized MD trajectory (discard initial 10-20% as equilibration).
- For each snapshot, calculate gas-phase energy components (ΔEMM) using molecular mechanics force fields.
- Compute polar solvation energy (ΔGGB) using the GBNSR6 model with Bondi or OPT1 radii.
- Calculate non-polar solvation energy (ΔGSA) from SASA using LCPO algorithm with γ = 0.0072 kcal/mol/Ų.
- Estimate entropy using interaction entropy method based on fluctuation of interaction energies from the MD trajectory.
Analysis and Validation
- Calculate average and standard error of binding free energy across all snapshots.
- Compare with experimental data if available.
- Perform energy decomposition to identify key residues contributing to binding.
Truncated System Approach for Large Complexes
For large systems like supramolecular assemblages, a truncated approach significantly reduces computational cost while maintaining accuracy [71] [73]:
Mixed-Resolution Modeling
- Model the binding site at atomistic resolution while coarse-graining the remaining structure.
- Generate plausible conformers using Anisotropic Network Model (ANM) to sample functional motions.
System Truncation
- Identify key binding site residues (typically within 9-12 Å of ligand center of mass).
- Create a truncated system containing only these residues plus the ligand.
- Apply harmonic restraints (25-50 kcal/mol·Å²) to backbone atoms in buffer regions to prevent disintegration.
MD Simulation of Truncated System
- Perform multiple independent MD simulations (3-5 replicates) of 100 ns each.
- Confirm that 100 ns simulations provide sufficient convergence for binding affinity estimates [71].
Binding Free Energy Calculation
- Use MM/GBSA on truncated system trajectories.
- Employ interaction entropy method for efficient entropy calculation.
Emerging Methodologies
Machine Learning-Enhanced Implicit Solvation
Recent advances integrate machine learning with implicit solvation models to address accuracy limitations. Graph Neural Networks (GNNs) trained on explicit solvent simulations can capture local solvation effects more accurately than traditional GB models [26]. The λ-Solvation Neural Network (LSNN) extends beyond force-matching by incorporating derivatives of alchemical variables (λsterics and λelec), enabling accurate absolute free energy predictions with computational speed comparable to implicit solvent models [26].
Variational Implicit Solvent Models
Variational explicit-solute implicit-solvent (VESIS) models combine explicit solute representation with implicit solvent description, enabling efficient determination of equilibrium conformations [27]. These methods employ binary level-set methods for capturing solute-solvent interfaces and adaptive-mobility gradient descent optimization for solute atoms, significantly improving efficiency for large systems like protein-protein interactions [27].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for MM/GBSA Studies
| Category | Item | Function/Application |
|---|---|---|
| Software Packages | AMBER | MD simulation and MM/GBSA implementation |
| GROMACS | MD simulation with MM/PBSA extension | |
| CHARMM | All-atom force field for biomolecular systems | |
| Schrödinger Suite | Commercial platform for drug discovery | |
| Force Fields | ff03 | Recommended for MM/GBSA with proteins |
| GAFF | General AMBER force field for small molecules | |
| CHARMM36m | Latest version for proteins, lipids, carbohydrates | |
| Solvent Models | GBNSR6 | Generalized Born model for polar solvation |
| TIP3P | Explicit water model for reference MD simulations | |
| OPC | Optimized 4-point water model for improved accuracy | |
| Analysis Tools | MDAnalysis | Python toolkit for trajectory analysis |
| VMD | Molecular visualization and trajectory analysis | |
| PyMol | Molecular visualization system | |
| Specialized Methods | ANM (Anisotropic Network Model) | Efficient conformational sampling of proteins |
| LSNN (λ-Solvation Neural Network) | ML-based implicit solvation for free energy |
MM/GBSA represents a valuable tool for binding free energy estimation in drug discovery, offering a reasonable balance between computational efficiency and predictive accuracy. Its consistency is highly dependent on multiple factors including conformational sampling, entropy treatment, solvation model selection, and system preparation. When applied with careful attention to these factors and appropriate validation against experimental data, MM/GBSA provides meaningful insights into protein-ligand interactions. For large systems and high-throughput applications, truncated approaches and emerging machine learning methods offer promising avenues for maintaining accuracy while reducing computational cost. Researchers should select protocols based on their specific system characteristics and accuracy requirements, recognizing that methodological choices significantly impact results.
Molecular dynamics (MD) simulation serves as a "virtual molecular microscope," providing atomistic details into protein dynamics that often complement traditional biophysical techniques [78]. The usefulness of this fundamental technology for life sciences research depends critically on its accuracy and efficiency [40]. For simulations of small proteins, a central methodological choice concerns solvent representation: explicit models that simulate individual water molecules offer high accuracy at substantial computational cost, while implicit models approximate solvent effects mathematically for improved speed [8] [26]. This Application Note establishes structured protocols for validating simulation outcomes against experimental data within the context of this implicit versus explicit solvent framework, providing researchers with clear methodologies for assessing computational results.
Theoretical Framework: Implicit vs. Explicit Solvation
Implicit Solvent Models
Implicit solvent models replace explicit solvent molecules with a potential of mean force (PMF), dramatically reducing computational cost by eliminating solvent degrees of freedom [8]. These models approximate the free energy of solvation (ΔGsol) through various mathematical formulations:
- Continuum Electrostatic Methods: Generalized Born (GB) and Poisson-Boltzmann (PB) models calculate the polar contribution to solvation energy (ΔGele) by treating the solvent as a dielectric continuum [8].
- Non-Polar Contributions: Solvent-accessible surface area (SASA) models approximate the non-polar components of cavity formation and van der Waals interactions (ΔGcav + ΔGvdW) [8].
- Machine Learning Enhancements: Recent graph neural network approaches, such as the λ-Solvation Neural Network (LSNN), extend beyond traditional force-matching to enable accurate free energy predictions [26].
Explicit Solvent Models
Explicit models simulate solvent molecules individually using classical force fields, providing highly accurate sampling of specific solvent-solute interactions, including hydrogen bonding and water dipole reorientation [8] [26]. These models are considered the gold standard for calculating solvation energies but come with significantly higher computational demands, limiting their use in large-scale screening applications [26].
Table 1: Fundamental Characteristics of Solvent Models in MD Simulations
| Characteristic | Implicit Solvent Models | Explicit Solvent Models |
|---|---|---|
| Solvent Representation | Continuum dielectric medium [8] | Discrete molecules (e.g., TIP3P, TIP4P-EW) [78] [26] |
| Computational Cost | Lower; fewer degrees of freedom [8] [26] | Higher; many solvent molecules to simulate [26] |
| Key Strengths | Computational efficiency; suitable for enhanced sampling [8] | Accurate specific interactions (H-bonds, bridging waters) [8] |
| Key Limitations | Approximates isotropic, bulk solvent behavior [8] | Lengthy simulations needed for solvent relaxation [26] |
| Free Energy Components | ΔGele (GB/PB) + ΔGcav + ΔGvdW (SASA) [8] | Naturally emerges from molecular interactions |
Figure 1: Decision Framework for Solvent Model Selection in Small Protein Simulations
Experimental Validation Methodologies
Nuclear Magnetic Resonance (NMR) Validation
NMR provides powerful experimental data for validating protein conformational ensembles from MD simulations.
Protocol 1: Validating Against Scalar Coupling Constants (³J-Couplings)
- Objective: Quantify accuracy of protein backbone dihedral angle sampling [40].
- Procedure:
- Run MD simulations using both implicit and explicit solvent models.
- Extract backbone dihedral angles (φ and ψ) from simulation trajectories at regular intervals.
- Calculate theoretical ³J-coupling constants using Karplus relationships.
- Compare computed ³J-values with experimental NMR measurements.
- Validation Metric: Mean absolute error between computed and experimental values; correlation coefficients.
Thermodynamic Validation
Accurate reproduction of thermodynamic properties represents a stringent test for simulation methodologies.
Protocol 2: Protein Folding Free Energy Calculations
- Objective: Validate simulation predictions against experimentally determined protein stability [40].
- Procedure:
- Perform alchemical free energy calculations or equilibrium unfolding simulations using both solvent models.
- Compute free energy differences (ΔG) between folded and unfolded states.
- Compare with experimental ΔG values from thermal or chemical denaturation experiments.
- Validation Metric: Error in ΔG prediction (kcal/mol); ability to reproduce melting temperatures [40].
Structural Validation
Protocol 3: Root Mean Square Deviation (RMSD) and Fluctuation (RMSF) Analysis
- Objective: Assess structural integrity and flexibility predictions.
- Procedure:
- Align simulation structures to experimental reference (NMR ensemble or crystal structure).
- Calculate RMSD for backbone atoms to measure overall structural drift.
- Compute RMSF for individual residues to quantify local flexibility.
- Compare RMSF patterns with experimental B-factors from crystallography or NMR order parameters.
- Validation Metric: RMSD stability over time; correlation between predicted and experimental flexibility profiles.
Table 2: Key Experimental Observables for MD Simulation Validation
| Experimental Technique | Observable | Validation Metric | Information Gained |
|---|---|---|---|
| NMR Spectroscopy | ³J-coupling constants [40] | Mean absolute error | Backbone dihedral angle distributions |
| Residual dipolar couplings | Correlation coefficient | Overall structural alignment | |
| Relaxation parameters | Order parameters (S²) | Side-chain dynamics | |
| X-ray Crystallography | B-factors | RMSF correlation | Residue flexibility |
| Electron density | Real-space correlation | Atomic-level structural accuracy | |
| Thermal Denaturation | Melting temperature (Tm) [40] | ΔTm prediction | Protein stability |
| Free energy of folding (ΔG) | ΔG error (kcal/mol) [40] | Thermodynamic stability |
Case Study: Engrailed Homeodomain and RNase H
A comprehensive validation study compared four MD simulation packages (AMBER, GROMACS, NAMD, and ilmm) using three force fields for two globular proteins with distinct topologies: Engrailed homeodomain (EnHD, 54 residues) and RNase H (155 residues) [78].
Experimental Protocol:
- System Preparation: Initial coordinates from PDB entries 1ENH (EnHD) and 2RN2 (RNase H) [78].
- Simulation Conditions: Multiple 200 ns replicates at 298 K with explicit solvent (TIP4P-EW for AMBER/GROMACS, CHARMM-modified TIP3P for NAMD) [78].
- Validation Data: NMR chemical shifts, scalar couplings, and NOEs for EnHD; various biophysical measurements for RNase H.
- Key Findings: While all packages reproduced experimental observables reasonably well at room temperature, subtle differences emerged in conformational distributions and sampling extent. Greater divergence occurred during thermal unfolding, with some packages failing to properly unfold proteins at high temperature or producing results inconsistent with experiment [78].
Emerging AI-Enhanced Approaches
Recent advances in artificial intelligence are creating new paradigms for biomolecular simulation that transcend traditional implicit/explicit distinctions:
AI-Driven Ab Initio Accuracy: AI2BMD uses protein fragmentation and machine learning force fields to achieve DFT-level accuracy for proteins exceeding 10,000 atoms, with a speedup of several orders of magnitude compared to conventional DFT [40]. The system demonstrated remarkable agreement with experimental ³J-couplings from NMR and accurately reproduced protein folding/unfolding processes [40].
Generative Equilibrium Ensembles: BioEmu implements a diffusion model-based generative AI system that simulates protein equilibrium ensembles with 1 kcal/mol accuracy using a single GPU, achieving a 10,000-100,000x speedup for equilibrium distributions [79]. This approach enables high-throughput prediction of conformational changes, free energy distributions, and cryptic pocket formation for drug targeting [79].
Figure 2: AI-Enhanced Simulation Workflow Combining Multiple Data Sources
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Simulation and Validation
| Tool Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| MD Simulation Packages | AMBER, GROMACS, NAMD [78] | Integration of equations of motion | Core simulation engine for both implicit and explicit solvent |
| Implicit Solvent Models | SASA, GB, PBSA [8] | Continuum solvation approximation | Accelerated sampling; free energy calculations |
| Explicit Water Models | TIP3P, TIP4P-EW [78] | Atomic-level solvent representation | High-accuracy sampling of specific solvent interactions |
| Force Fields | AMBER ff99SB-ILDN, CHARMM36 [78] | Empirical potential energy functions | Determines accuracy of physical interactions |
| AI/ML Tools | AI2BMD, BioEmu, LSNN [40] [79] [26] | Machine learning force fields | Ab initio accuracy with reduced computational cost |
| Validation Software | VMD, MDAnalysis, SHIFTX2 | Analysis of simulation trajectories | Calculation of experimental observables for comparison |
Robust validation against experimental data remains essential for establishing the predictive capability of molecular dynamics simulations, particularly when choosing between implicit and explicit solvent models for small protein systems. The structured protocols presented here provide researchers with comprehensive methodologies for assessing simulation accuracy across multiple experimental domains. Emerging AI-enhanced approaches promise to bridge the accuracy-efficiency gap, potentially transforming how we simulate and validate biomolecular systems. As these technologies mature, rigorous validation against experimental data will become increasingly critical for ensuring their reliable application in drug discovery and basic research.
Within the context of molecular dynamics (MD) simulations of small proteins, the choice of force field and its compatibility with the solvent model is a critical determinant of success. This application note examines the performance of various biomolecular force fields across different protein secondary structure classes, providing a structured comparison of their strengths and limitations. The content is framed within a broader research thesis comparing implicit and explicit solvent models, offering practical guidance to researchers and drug development professionals on selecting appropriate simulation parameters for their specific systems. We summarize quantitative benchmarking data, provide detailed experimental protocols from key studies, and visualize the logical workflows for force field selection. The performance of a force field is not absolute but is intrinsically linked to the protein system under investigation, necessitating a careful, system-informed selection process to ensure reliable and predictive simulations.
Performance Benchmarking Across Protein Systems
The accuracy of a force field is highly dependent on the class of protein being simulated. A benchmark study assessing accelerated MD (aMD) protocols on eight peptides—comprising two helical, three β-hairpin, and three intrinsically disordered (ID) peptides—revealed significant performance variations across different force field and solvent model combinations [80].
Table 1: Force Field Performance Across Protein Secondary Structure Types (from [80])
| Protein Type | Recommended Force Field/Solvent Combination | Key Performance Findings |
|---|---|---|
| Helical Peptides | ff99SB, ff14SB, or ff19SB with TIP3P or OPC | All three tested force field combinations performed well on helical peptides. |
| β-Hairpin Peptides | ff19SB with TIP3P | ff19SB performed well, with a slight preference for the TIP3P solvation model. Performance was sequence- and conformation-dependent. |
| Intrinsically Disordered Peptides | ff19SB with OPC | The ff19SB/OPC combination demonstrated the best performance. |
| General Note | --- | The ff14SB/TIP3P combination suffered from a strong helical bias. |
This data underscores that no single force field is universally superior. Researchers must prioritize force fields based on the dominant secondary structure of their target system to avoid known biases, such as the helical bias of ff14SB/TIP3P.
Key Research Reagents and Computational Tools
Selecting the appropriate computational "reagents" is as crucial as selecting laboratory reagents for experimental work. The table below details essential components for setting up and running protein MD simulations.
Table 2: Essential Research Reagent Solutions for Protein MD Simulations
| Item Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Protein Force Fields | AMBER ff19SB, ff14SB, ff15ipq, CHARMM C36 [80] [81] [82] | Empirical potential energy functions describing atomistic interactions. Choice is critical and system-dependent (see Table 1). |
| Explicit Water Models | TIP3P, OPC, TIP4P-EW, SPC/E [80] [82] | Explicitly represent water molecules. OPC shows promise for IDPs and β-hairpins; TIP3P is widely used. |
| Implicit Solvent Models | GBNeck2, EEF1, SASA/VOL models [8] [83] | Replace explicit solvent with a continuous dielectric medium, offering significant speedup (up to 100x) but with potential accuracy trade-offs, especially for disordered proteins [69] [83]. |
| Ligand Force Fields | GAFF2.2, OpenFF [82] [84] | Provide parameters for small organic molecules in protein-ligand binding studies. |
| Simulation & Analysis Software | AMBER, OpenMM, CHARMM-GUI [82] [83] [84] | Software packages used to run simulations, set up systems, and perform subsequent trajectory analysis. |
| Validation Benchmark Sets | JACS Set (BACE, CDK2, etc.), Peptide Folding Set (e.g., Trp-cage, BBA) [82] [83] [22] | Standardized sets of proteins and transformations for validating simulation protocols and force field accuracy. |
Experimental Protocols for Key Applications
Protocol 1: Benchmarking Force Fields for Peptide Folding using aMD
This protocol is adapted from a study benchmarking force fields for peptide secondary structure prediction using accelerated Molecular Dynamics (aMD) [80].
System Setup:
- Peptide Preparation: Select peptides with known native structures (e.g., helical: H1, H2; β-hairpin: B1, B2, B3; intrinsically disordered: ID1, ID2, ID3). Cap termini with acetyl (ACE) and N-methyl amide (NHE) groups.
- Initial Conformations: Generate two independent starting structures for each peptide: (1) an extended conformation (ψ = φ = ω = 180°), and (2) a misfolded conformation (e.g., α-helix for a β-hairpin peptide, and vice-versa).
- Force Field/Solvent Selection: Prepare topologies and coordinates using a tool like
tLEaP, testing combinations of force fields (e.g., ff99SB, ff14SB, ff19SB) and explicit solvation models (e.g., TIP3P, OPC).
Simulation Parameters:
- Boost Parameters: Run a preliminary 20 ns classical MD (cMD) simulation in the NPT ensemble to calculate average dihedral and total potential energies, which are used to set the aMD boost parameters.
- aMD Production Run: Perform aMD simulations in the NPT ensemble (e.g., Langevin thermostat, Berendsen barostat) for 1500 ns. Use a timestep of 2 fs, a non-bonded cutoff of 8 Å, and the Particle Mesh Ewald method for long-range electrostatics. Apply the SHAKE algorithm to constrain bonds involving hydrogen.
Analysis and Convergence:
- Convergence Check: Monitor convergence using time-dependent Root Mean Square Deviation (RMSD) and Dictionary of Secondary Structure of Proteins (DSSP) analysis throughout the trajectory.
- Trajectory Analysis: Once converged (typically the last 500 ns), analyze the data. Perform cluster analysis on Cα atoms to identify dominant conformers. Calculate the fraction of native secondary structure present and compare against experimental reference data.
Protocol 2: Relative Binding Free Energy (RBFE) Calculations with AMBER-TI
This protocol outlines the steps for performing Relative Binding Free Energy (RBFE) calculations using Thermodynamic Integration (TI) within the AMBER framework, as validated in large-scale benchmark studies [82] [84].
System Preparation:
- Protein-Ligand Complexes: Obtain structures from a benchmark set (e.g., the JACS set: BACE1, TYK2, CDK2, etc.). Prepare protein structures by adding standard termini (protonated amine for N-terminus, charged carboxylate for C-terminus).
- Force Field Selection: Assign parameters. A recommended starting combination is ff14SB for the protein, GAFF2.2 for the ligand, and TIP3P for water, with AM1-BCC partial charges for the ligand [82] [84].
- System Generation: Use tools like CHARMM-GUI Free Energy Calculator to generate inputs for both the complex and the ligand in solution for each alchemical transformation.
Simulation Procedure:
- Alchemical Transformation: Define the non-physical pathway connecting ligands L0 and L1 using a coupling parameter λ. A set of 12 or 21 λ windows is typically sufficient.
- Equilibration & Production: For each λ window, minimize the system and equilibrate for 5 ps in the NPT ensemble. Follow with a production run of 5 ns per window using a 4 fs timestep with hydrogen mass repartitioning. Employ a softcore potential (e.g., SSC(2)) for van der Waals interactions.
- Sampling Enhancement: Use Hamiltonian replica exchange between adjacent λ windows to improve sampling.
- Independent Runs: Conduct 4 independent runs for each transformation to ensure statistical robustness.
Free Energy Analysis:
- TI Calculation: Calculate the free energy change (ΔG) for the transformation in both the complex and the ligand alone using the trapezoidal rule for numerical integration over λ. Use the last 4 ns of each window for analysis.
- ΔΔGbind Calculation: Compute the relative binding free energy using the equation: ΔΔGbind = ΔGcomplex(L0→L1) - ΔGligand(L0→L1).
- Validation: Compare the calculated ΔΔGbind values against experimental data to determine the mean unsigned error (MUE) and correlation coefficients, which validate the force field combination's performance.
Workflow Visualization for Force Field Selection
The following diagram illustrates a logical decision pathway for selecting and validating a force field for a new protein simulation project, based on insights from the benchmark data.
The diagram emphasizes an iterative validation process. The "Force Field & Solvent Preselection" node should be informed by the benchmark data in Table 1; for example, selecting ff19SB/OPC for intrinsically disordered proteins or ff19SB/TIP3P for β-hairpin systems.
Advanced Methods and Future Directions
Addressing the Intrinsically Disordered Protein (IDP) Challenge with Implicit Solvent
A primary challenge in MD simulations is the accurate modeling of IDPs. Traditional implicit solvent models often cause IDPs to adopt over-compacted, overly rigid, and incorrectly helical conformations [83]. To address this, advanced parameterization techniques are being employed. For instance, Differentiable Molecular Simulation (DMS) has been used to jointly optimize 108 parameters of the a99SB-disp force field and the GBNeck2 implicit solvent model [83]. This method involves:
- Target Data: Running short (5 ns) differentiable simulations and using the KL divergence to match residue-residue distance distributions from long, accurate explicit solvent reference simulations.
- Outcome: The resulting force field, GB99dms, shows improved reproduction of the radius of gyration and secondary structure content for disordered proteins compared to its predecessor, while maintaining reasonable performance on folded systems [83].
Machine-Learned Coarse-Grained (CG) Models
For accessing longer timescales and larger systems, machine-learned coarse-grained (CG) models represent a transformative advance. These models are parameterized using deep learning and variational force-matching on large datasets of all-atom simulations [22]. A key development is the creation of transferable CG force fields that can be applied to protein sequences not included in the training data [22].
- Performance: Such models can successfully predict metastable states of folded and unfolded structures, fluctuations of IDPs, and relative folding free energies of mutants.
- Computational Gain: These CG models achieve performance comparable to all-atom MD for many applications while being several orders of magnitude faster, showcasing their potential as a universal and efficient simulation tool [22].
This application note synthesizes current best practices for assessing force field compatibility across diverse protein systems. The core principle is that force field performance is not universal; it is context-dependent. Success in molecular dynamics simulations requires a strategic selection process, informed by benchmark data and validated through iterative testing. The emergence of methodologies like differentiable programming for implicit solvent refinement and machine-learned coarse-grained models points toward a future of more automated, accurate, and accessible biomolecular simulations. By adhering to the structured protocols and selection logic outlined herein, researchers can make more informed decisions, thereby enhancing the reliability and predictive power of their computational studies.
The accurate prediction of protein-ligand binding affinity is a cornerstone of computational drug design. Achieving accuracy, however, hinges on a critical methodological choice: the treatment of the solvent environment in molecular dynamics (MD) simulations. Explicit solvent models, which represent each water molecule as a discrete entity, are considered the gold standard for accuracy but come at an extreme computational cost, limiting their use in high-throughput screening. Implicit solvent models, which treat the solvent as a continuous dielectric medium, offer a faster alternative but have historically struggled with accuracy, particularly in capturing specific solvent-mediated effects crucial for reliable affinity prediction. This application note examines the current state of binding affinity prediction methodologies, focusing on the trade-offs between implicit and explicit solvent models, and provides detailed protocols for their application in protein-ligand interaction studies.
Theoretical Background: Solvent Models in MD Simulations
Fundamental Principles
Solvation profoundly influences the structure, dynamics, and function of biomolecules. In biological systems, water mediates essential processes including protein folding, ligand binding, and molecular recognition. Implicit solvent models emerged from early dielectric theories of solvation established by Onsager and Debye, who treated solvents as dielectric continua for estimating solvation energies based on bulk properties like dielectric constant and molecular polarizability [10].
The solvation free energy (ΔGsolv) is typically partitioned into physically distinct components. The most common partition includes a polar (electrostatic) term and a nonpolar term [10]:
ΔGsolv = ΔGele + ΔGnp
The polar component (ΔGele) accounts for the interaction of the solute's charge distribution with the dielectric environment, while the nonpolar component (ΔGnp) describes contributions from cavity formation, solvent-accessible surface area (SASA), and van der Waals interactions. In traditional implicit solvent frameworks like MM/GBSA and MM/PBSA, this is often approximated as [26]:
ΔG ≈ ΔGGB + ΔGSASA
Here, ΔGGB represents the polar contribution calculated using Generalized Born models, while ΔGSASA accounts for the nonpolar component. However, this simplification, particularly the treatment of the nonpolar term using a simple SASA approximation, is prone to significant errors [26].
Comparative Analysis of Solvent Models
Table 1: Characteristics of Explicit and Implicit Solvent Models
| Feature | Explicit Solvent Models | Implicit Solvent Models |
|---|---|---|
| Solvent Representation | Discrete water molecules (TIP3P, TIP4P, TIP5P, OPC, SPC/E) | Continuum dielectric medium |
| Computational Cost | High (computationally demanding) | Low (computationally efficient) |
| Sampling Speed | Slower due to viscous damping | Faster due to low solvent viscosity |
| Accuracy for Specific Solvent Effects | High (captures water bridges, hydrogen bonds) | Low (misses specific solvent interactions) |
| Treatment of Long-Range Electrostatics | Requires sophisticated algorithms (PME) | Naturally incorporates dielectric response |
| Common Applications | Gold-standard MD, precise thermodynamics | High-throughput screening, enhanced sampling |
Explicit solvent models provide highly accurate sampling but at a substantial computational cost that often prevents comprehensive conformational sampling. The most popular explicit water models include TIP3P, TIP4P, TIP5P, SPC/E, and OPC, which differ in their geometry, charge distribution, and performance in reproducing experimental observables [21]. For instance, studies on heparin dodecamer simulations revealed that TIP3P and SPC/E yield stable conformations, whereas TIP4P, TIP5P, and OPC introduce greater structural variability, highlighting the critical impact of water model selection on simulation outcomes [21].
Implicit solvent models eliminate the need to simulate numerous solvent molecules, significantly reducing computational overhead and enabling faster exploration of biomolecular conformations. However, the continuum approximation introduces inherent limitations, including inaccuracies in capturing specific solvent-mediated interactions (e.g., water bridges, hydrogen bonds) and representing entropic contributions of solvent molecules [10].
Current Methodologies in Binding Affinity Prediction
Physics-Based Approaches
Physics-based methods for binding affinity prediction include absolute free energy calculations, free energy perturbation (FEP), and MM/PBSA or MM/GBSA approaches. These methods can yield high accuracy but typically require substantial computational resources [85]. The MM/GBSA approach, for instance, decomposes binding free energy into gas-phase enthalpy, solvent correction, and entropy penalty:
ΔG ≈ ΔHgas + ΔGsolvent - TΔS
In practice, the first two terms have large magnitudes (on the order of 100 kcal/mol) with opposite signs, making the entropic term relatively small but notoriously difficult to calculate accurately [86].
Machine Learning-Enhanced Approaches
Recent machine learning (ML) advancements have created opportunities to overcome traditional limitations in implicit solvent modeling. Neural network potentials now allow accurate calculations of solvation forces for diverse molecular conformations [26]. However, a significant drawback of many ML-based methods has been their reliance solely on force-matching, which predicts energies only up to an arbitrary constant, rendering them unsuitable for absolute free energy comparisons [26].
The novel λ-Solvation Neural Network (LSNN) addresses this limitation by extending training procedures to incorporate derivatives of alchemical variables (electrostatic and steric coupling factors, λelec and λsteric). This approach ensures that solvation free energies can be meaningfully compared across chemical species. The modified loss function for LSNN training incorporates multiple terms [26]:
ℒ = wF(〈∂Usolv/∂ri〉 - ∂f/∂ri)2 + welec(〈∂Usolv/∂λelec〉 - ∂f/∂λelec)2 + wsteric(〈∂Usolv/∂λsteric〉 - ∂f/∂λsteric)2
Trained on approximately 300,000 small molecules, LSNN achieves free energy predictions with accuracy comparable to explicit-solvent alchemical simulations while offering significant computational speedup [26].
Trajectory-Based Affinity Prediction
Alternative strategies evaluate binding affinity by comparing protein dynamic behavior upon ligand binding using MD simulations. Yasuda et al. proposed a method where MD simulations are performed for both apo and ligand-bound protein forms, quantifying differences in binding site residue dynamics using distance metrics [85]. This approach was improved by replacing computationally intensive deep learning-based Wasserstein distances with Jensen-Shannon (JS) divergence, significantly reducing computation time while maintaining comparable accuracy [85].
The JS divergence between probability density functions of two ligand systems i and j is calculated as:
Wij = (1/Na) Σa=1Na JSa(i∥j)
JS(i∥j) = (1/2)(KL(i∥M) + KL(j∥M))
M(x) = (1/2)(i(x) + j(x))
where KL(·∥·) denotes the Kullback-Leibler divergence [85]. This method successfully correlates the first principal component from trajectory analysis with experimental ΔG values.
Performance Benchmarking
Table 2: Performance Comparison of Binding Affinity Prediction Methods
| Method | Accuracy (RMSE) | Speed | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Explicit FEP/TI | ~1 kcal/mol [86] | Very Slow (12+ GPU hours) [86] | Considered gold standard for accuracy | Extremely computationally intensive |
| Docking | 2-4 kcal/mol [86] | Fast (<1 minute CPU) | Very high throughput | Low accuracy, limited correlation (R≈0.3) |
| MM/GBSA | Variable | Medium | Better than docking, faster than FEP | Noisy results, sensitive to parameters |
| ML/GBSA | Unreliable | Medium | Potential improvement over forcefields | Fails due to error propagation [86] |
| LSNN (ML implicit) | Near-explicit accuracy [26] | Fast (vs explicit) | Meaningful absolute free energies | Requires extensive training data |
| Trajectory JS Divergence | Comparable to original method [85] | Medium (vs original) | Reduced computational cost | Requires MD simulations |
Recent research has revealed significant challenges in properly validating ML-based scoring functions. Studies show that train-test data leakage between the PDBbind database and CASF benchmarks has severely inflated performance metrics of many deep-learning-based binding affinity prediction models [87]. When trained on a properly filtered dataset (PDBbind CleanSplit) that eliminates this leakage, the performance of many state-of-the-art models drops substantially, indicating their previously reported high performance was largely driven by data memorization rather than genuine understanding of protein-ligand interactions [87].
Experimental Protocols
LSNN-Based Free Energy Calculation Protocol
The LSNN approach provides a methodology for accurate solvation free energy calculations using machine-learned implicit solvent models. Below is a detailed protocol for implementing this method:
System Preparation
- Molecular Structure Input: Prepare solute molecular structures in a format compatible with your MD simulation package (e.g., PDB, MOL2).
- Parameterization: Assign partial atomic charges and force field parameters using tools like antechamber or similar utilities.
- Alchemical Parameters: Define the electrostatic (λelec) and steric (λsteric) coupling parameters that will scale the interaction energy during the free energy calculation.
Training Data Generation (if training new models)
- Reference Simulations: Perform explicit solvent MD simulations (e.g., using TIP3P water model) for a diverse set of small molecules.
- Force Matching Data: Extract mean applied forces (MAFs) on solute atoms from explicit solvent trajectories.
- Alchemical Derivatives: Calculate derivatives of the solvation potential with respect to λelec and λsteric coupling parameters.
- Dataset Curation: Assemble a dataset of approximately 300,000 diverse small molecules for comprehensive training [26].
Network Architecture and Training
- Graph Neural Network Setup: Implement a graph neural network where atoms represent nodes and chemical bonds represent edges.
- Multi-Term Loss Function: Use the combined loss function that incorporates force-matching and alchemical derivative terms with empirically tuned weights (wF, welec, wsteric).
- Training Protocol: Train the network using reference data until convergence of all loss terms.
Free Energy Calculation
- Solvation Force Prediction: Use the trained LSNN to predict solvation forces for new molecular conformations.
- Conservative Vector Field: Ensure the model constructs a conservative vector field through the inclusion of alchemical derivatives.
- Potential of Mean Force: Calculate the scalar potential f that approximates the true Potential of Mean Force (PMF).
- Free Energy Estimation: Compute solvation free energies by appropriate thermodynamic integration of the predicted potentials.
Validation
- Explicit Solvent Comparison: Validate LSNN predictions against explicit-solvent alchemical simulations for benchmark systems.
- Experimental Comparison: Compare predicted solvation free energies with experimental values where available.
- Transferability Testing: Evaluate performance on molecular structures not represented in the training set.
JS Divergence Trajectory Analysis Protocol
This protocol describes an approach for predicting binding affinity differences by comparing protein dynamics in apo and ligand-bound states using Jensen-Shannon divergence.
System Preparation and MD Simulation
- Initial Structures: Prepare protein-ligand complex structures using crystal structures from the PDB or docking poses.
- Apo Protein Preparation: Generate the apo protein structure by removing the ligand from the complex.
- Structure Optimization: Add hydrogen atoms using tools like H++ at physiological pH (7.0) to complete missing atoms.
- Solvation and Neutralization: Solvate systems in a cubic box with a 10.0 Å buffer region using TIP3P water model, then neutralize system charge by adding sodium or chloride ions.
- MD Simulation Setup:
- Apply appropriate force fields (e.g., ff14SB for proteins, GAFF for ligands)
- Perform energy minimization (5,000 steps steepest descent)
- Conduct restrained NVT simulation (100 ps, 300 K, 10 kcal/mol·Å² restraint on heavy atoms)
- Perform restrained NPT simulation (100 ps, 300 K, 1 bar, same restraints)
- Execute production run (400 ns, NPT ensemble, save trajectories every 2 ps)
- Run three independent trials for each system with random initial velocities
Binding Site Residue Identification
- Activity Ratio Calculation: For each amino acid residue, calculate n/N, where n is the number of frames where the minimum distance between any heavy atom of the residue and any heavy atom of the ligand is <5Å, and N is the total number of frames.
- Binding Site Definition: Identify residues with activity ratio >0.5 during the first 100 ns of simulation.
- Residue Union Set: Extract the union of such residues across all ligand complexes for consistent analysis.
Trajectory Analysis and JS Divergence Calculation
- Trajectory Alignment: Remove rotation and translation by fitting trajectories to an identical structure in the backbone atoms of the binding site residues.
- Probability Density Estimation: For each protein or protein-ligand complex, estimate probability density functions for the trajectories of binding site residues using kernel density estimation with a Gaussian kernel.
- Bandwidth Selection: Determine bandwidth using Silverman's rule-of-thumb for optimal smoothing.
JS Divergence Calculation: Compute the Jensen-Shannon divergence between probability density functions from different ligand systems using the formula:
Wij = (1/Na) Σa=1Na JSa(i∥j)
Dimensionality Reduction and Affinity Correlation
- Distance Matrix Construction: Build a matrix of all pairwise JS divergences between systems.
- Nonlinear Dimensionality Reduction: Embed the distance matrix into three-dimensional space using techniques like multidimensional scaling.
- Principal Component Analysis: Perform PCA on the embedded space and extract the first principal component (PC1).
- Correlation with Binding Affinity: Correlate PC1 values with experimental ΔG values.
- Sign Determination: Use coarse ΔG estimations from AutoDock Vina to determine the sign of correlation between PC1 and ΔG in the absence of experimental data.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Binding Affinity Studies
| Tool/Resource | Type | Primary Function | Application Notes |
|---|---|---|---|
| AMBER | MD Software Suite | Biomolecular simulation with explicit/implicit solvent | Supports multiple force fields; includes GB models |
| CHARMM-GUI | Web-based platform | Input file generation for MD simulations | Prepares systems for various solvent models [21] |
| AutoDock Vina | Docking software | Protein-ligand docking and scoring | Provides coarse ΔG estimates for correlation sign determination [85] |
| OpenMM | MD Library | High-performance MD simulations | Supports custom forces and ML potentials |
| PDBbind CleanSplit | Curated Dataset | Training and validation of affinity prediction models | Reduces data leakage for proper model evaluation [87] |
| OMol25 Dataset | Quantum Chemistry Dataset | Training neural network potentials | 100M+ calculations at ωB97M-V/def2-TZVPD level [42] |
| GB-neck2 | Implicit Solvent Model | Generalized Born solvation calculations | Optimized for proteins and nucleic acids [88] |
| LSNN Framework | ML Solvation Model | Graph neural network for solvation free energies | Incorporates alchemical variables for absolute free energies [26] |
The field of binding affinity prediction continues to evolve rapidly, with implicit solvent models increasingly closing the accuracy gap with explicit solvent approaches through advanced machine learning techniques. Methods like LSNN that incorporate physical constraints and alchemical variables demonstrate that ML-based implicit solvent models can achieve near-explicit accuracy while maintaining computational efficiency. Similarly, trajectory-based approaches using JS divergence offer practical compromises between computational cost and predictive accuracy.
Future advancements will likely focus on several key areas: (1) improved dataset curation to eliminate biases and ensure proper model evaluation, (2) development of universal models trained on expansive, chemically diverse datasets like OMol25, (3) hybrid approaches that combine physical principles with data-driven corrections, and (4) increased integration of quantum mechanical accuracy into classical simulations through neural network potentials. As these methodologies mature, robust binding affinity prediction will become increasingly accessible, accelerating drug discovery and expanding our understanding of biomolecular interactions.
Conclusion
The choice between implicit and explicit solvent models represents a fundamental trade-off between computational efficiency and physical accuracy in small protein simulations. Implicit solvents offer significant speed advantages for conformational sampling and are increasingly reliable for structure refinement and early-stage drug screening, while explicit solvents remain essential for studying detailed solvation effects and achieving highest accuracy. Future directions include developing more accurate implicit solvent parametrizations, creating robust hybrid approaches, and leveraging these methods for challenging biomedical applications such as GPCR signaling and protein design. As computational power increases and methods refine, the strategic selection and optimization of solvent models will continue to enhance our ability to simulate complex biological processes and accelerate therapeutic development.
References
- 1. Solvent model [https://en.wikipedia.org/wiki/Solvent_model]
- 2. Modelling chemical processes in explicit solvents with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11271496/]
- 3. Implicit Solvent Models in Molecular Dynamics Simulations [https://www.sciencedirect.com/science/article/abs/pii/S1574140008000078]
- 4. Accuracy comparison of several common implicit solvent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5313374/]
- 5. Molecular dynamics simulations as a guide for modulating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10933209/]
- 6. Introducing Titratable Water to All-Atom Molecular ... [https://www.sciencedirect.com/science/article/pii/S0006349513007455]
- 7. Implicit solvation [https://en.wikipedia.org/wiki/Implicit_solvation]
- 8. Design and application of implicit solvent models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4045398/]
- 9. Implicit Solvent Models and Their Applications in Biophysics [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468003/]
- 10. Implicit Solvent Models and Their Applications in Biophysics [https://www.mdpi.com/2218-273X/15/9/1218]
- 11. Accurate coarse grained models for protein association ... [https://pubmed.ncbi.nlm.nih.gov/40324844/]
- 12. comparing explicit and implicit solvent molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/25762327/]
- 13. AlphaFold3 prediction of protein-protein complex: Is it ... [https://www.sciencedirect.com/science/article/pii/S2001037025004180]
- 14. Continuum vs. atomistic approaches to computational ... [https://pubs.rsc.org/en/content/articlehtml/2023/cc/d2cc07079k]
- 15. An Introduction to Molecular Dynamics Simulations [https://portal.valencelabs.com/blogs/post/an-introduction-to-molecular-dynamics-simulations-C9nXeGJ8hbNYghL]
- 16. Insights from molecular dynamics simulations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5321087/]
- 17. A Standardized Benchmark for Machine-Learned ... [https://arxiv.org/html/2510.17187v1]
- 18. Comparison of Radii Sets, Entropy, QM Methods, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4688044/]
- 19. Accurate protein-ligand binding free energy estimation ... [https://www.nature.com/articles/s42004-024-01328-7]
- 20. Implicit Solvent Models and Their Applications in Biophysics [https://www.preprints.org/manuscript/202507.2627]
- 21. Benchmark of Available Explicit Solvent Models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12498386/]
- 22. Navigating protein landscapes with a machine-learned ... [https://www.nature.com/articles/s41557-025-01874-0]
- 23. Molecular Dynamics Simulations | BIOVIA [https://www.3ds.com/products/biovia/discovery-studio/simulations]
- 24. Running a Molecular Dynamics (MD) simulation of ... [https://docs.openfree.energy/en/stable/tutorials/md_tutorial.html]
- 25. drMD: Molecular Dynamics for Experimentalists [https://www.sciencedirect.com/science/article/pii/S0022283624005485]
- 26. Extending machine learning model for implicit solvation to ... [https://arxiv.org/html/2510.20103v1]
- 27. Explicit-solute implicit-solvent molecular simulation with ... [https://www.sciencedirect.com/science/article/abs/pii/S0021999122007367]
- 28. Accuracy Comparison of Generalized Born Models in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12199658/]
- 29. Improved Generalized Born Solvent Model Parameters for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4361090/]
- 30. A comparison of generalized Born methods in folding ... [https://www.sciencedirect.com/science/article/abs/pii/S000926140800746X]
- 31. Refinement of Generalized Born Implicit Solvation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4805114/]
- 32. Accuracy Comparison of Generalized in the Calculation... Born Models [https://pubmed.ncbi.nlm.nih.gov/29378399/]
- 33. CHARMM-GUI Implicit Solvent Modeler for Various ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9551160/]
- 34. Version 3.8 (2022. July) [https://charmm-gui.org/?doc=log&version=3.8]
- 35. CHARMM-GUI PDB Reader and Manipulator: Covalent ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283624001499]
- 36. High-Throughput Simulator: Solution Systems [https://www.charmm-gui.org/demo/hts]
- 37. Implicit Solvent Models and Their Applications in Biophysics [https://pubmed.ncbi.nlm.nih.gov/41008525/]
- 38. Minimal Collective Variables for Conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12128028/]
- 39. A discard-and-restart MD algorithm for the sampling of ... [https://www.sciencedirect.com/science/article/abs/pii/S0006349525001985]
- 40. Ab initio characterization of protein molecular dynamics ... [https://www.nature.com/articles/s41586-024-08127-z]
- 41. Integrating Multiple Time Step Method and Implicit Solvent ... [https://cjcp.ustc.edu.cn/hxwlxb/article/doi/10.1063/1674-0068/cjcp2505069]
- 42. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 43. Assessment of reactivities with explicit and implicit solvent ... [https://pubs.rsc.org/en/content/articlehtml/2019/nj/c9nj04003j]
- 44. Equilibrium Kinetic Network of the Villin Headpiece in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4302211/]
- 45. Replica-exchange simulations of ``Trp-cage'' miniproteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC164630/]
- 46. Article Common Structural Transitions in Explicit-Solvent ... [https://www.sciencedirect.com/science/article/pii/S0006349509013599]
- 47. Protein Structure Prediction Center [https://predictioncenter.org/]
- 48. Home - CASP15 [https://predictioncenter.org/casp15/]
- 49. Methods for the Refinement of Protein Structure 3D Models [https://www.mdpi.com/1422-0067/20/9/2301]
- 50. Protein Structure Refinement of CASP Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3985798/]
- 51. Assessment of protein structure refinement in CASP9 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3238793/]
- 52. Comparison of implicit solvent models and force fields in ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261411011353]
- 53. The combined force field-sampling problem in simulations of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6417907/]
- 54. Benchmarking Molecular Dynamics Force Fields for All- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11169342/]
- 55. Enhanced Sampling Techniques in Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4339458/]
- 56. Integrating molecular simulations with experimental data [https://www.sciencedirect.com/science/article/pii/S1877117319302121]
- 57. PySAGES: flexible, advanced sampling methods ... [https://www.nature.com/articles/s41524-023-01189-z]
- 58. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 59. Direct generation of protein conformational ensembles via ... [https://www.nature.com/articles/s41467-023-36443-x]
- 60. EPO: Diverse and Realistic Protein Ensemble Generation ... [https://arxiv.org/html/2511.10165v1]
- 61. Deep generative modeling of temperature-dependent ... [https://www.nature.com/articles/s42004-025-01737-2]
- 62. Optimization of the GBMV2 Implicit Solvent Force Field for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5407932/]
- 63. Implicit Solvation - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/implicit-solvation]
- 64. Hybrid method for representing ions in implicit solvation ... [https://www.sciencedirect.com/science/article/pii/S2001037021000246]
- 65. A Multiscale Simulation Approach to Compute Protein–Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570144/]
- 66. Modelling chemical processes in explicit solvents with ... [https://www.nature.com/articles/s41467-024-50418-6]
- 67. apoCHARMM: High-performance molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/40341929/]
- 68. Leveraging high-throughput molecular simulations and ... [https://www.nature.com/articles/s41524-025-01552-2]
- 69. Speed of Conformational Change: Comparing Explicit and ... [https://www.sciencedirect.com/science/article/pii/S000634951500003X]
- 70. Speed of Conformational Change: Comparing Explicit and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375717/]
- 71. A Computationally Efficient Method to Generate Plausible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12344705/]
- 72. The MM/PBSA and MM/GBSA methods to estimate ligand- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4487606/]
- 73. An Effective MM/GBSA Protocol for Absolute Binding Free ... [https://www.mdpi.com/1420-3049/26/8/2383]
- 74. 3.8. Free Energy Calculation Using MM/GBSA Approach [https://bio-protocol.org/exchange/minidetail?id=7502613&type=30]
- 75. In-silico molecular modelling, MM/GBSA binding free ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.991369/full]
- 76. Assessing the performance of MM/PBSA and MM/GBSA ... [https://pubs.rsc.org/en/content/articlelanding/2018/cp/c7cp07623a]
- 77. Accurate prediction of DNA-Intercalator binding energies [https://www.sciencedirect.com/science/article/pii/S0141813025019592]
- 78. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 79. BioEmu: AI‐Powered Revolution in Scalable Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12643044/]
- 80. Accelerated Molecular Dynamics for Peptide Folding: Benchmarking... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10207344/]
- 81. Current Status of Protein Force Fields for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4554537/]
- 82. Assessing the effect of forcefield parameter sets on the ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9549959/]
- 83. Differentiable simulation to develop molecular dynamics force ... fields [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d3sc05230c]
- 84. Practical Guidance for Consensus Scoring and Force Field ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9772090/]
- 85. Protein–ligand affinity prediction via Jensen-Shannon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12399510/]
- 86. Exploring Protein–Ligand Binding-Affinity Prediction | Rowan [https://rowansci.com/blog/exploring-protein-ligand-binding-affinity-prediction]
- 87. Resolving data bias improves generalization in binding ... [https://www.nature.com/articles/s42256-025-01124-5]
- 88. Molecular Dynamics Simulations of RNA Stem-Loop Folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12593105/]