GNEIMO Method: Revolutionizing Protein Folding and Refinement with Torsional Dynamics
This article explores the Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method, an advanced internal coordinate molecular dynamics (ICMD) technique transforming the study of protein folding and structure refinement.
GNEIMO Method: Revolutionizing Protein Folding and Refinement with Torsional Dynamics
Abstract
This article explores the Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method, an advanced internal coordinate molecular dynamics (ICMD) technique transforming the study of protein folding and structure refinement. Aimed at researchers, scientists, and drug development professionals, we detail GNEIMO's foundational principles that overcome traditional MD limitations by constraining high-frequency motions to focus sampling on essential torsional degrees of freedom. The content covers methodological protocols for applications like homology model refinement and folding studies, alongside optimization strategies such as the 'freeze and thaw' clustering and Replica Exchange MD. Finally, we present rigorous validation against experimental data and comparative analyses demonstrating GNEIMO's ability to consistently refine protein models by 1.3-2.0 Å, offering powerful implications for computational biology and structure-based drug design.
Beyond Traditional MD: The Foundational Principles of GNEIMO Torsional Dynamics
The Protein Dynamics Sampling Bottleneck in All-Atom Cartesian MD
Molecular dynamics (MD) simulations are an indispensable tool in computational chemistry and drug discovery, providing crucial insights into the dynamic behavior of biomolecular systems. However, the utility of traditional all-atom Cartesian MD is significantly limited by substantial computational costs that restrict accessible timescales. The core bottleneck lies in the intensive calculation of non-bonded forces, which scales quadratically with the number of atoms. Furthermore, accurately resolving high-frequency atomic vibrations necessitates extremely small time steps (on the order of femtoseconds), severely limiting the simulation of biologically relevant processes that often span microseconds to milliseconds [1]. This sampling bottleneck represents a fundamental challenge in studying protein folding, ligand unbinding, and other critical biomolecular processes. Within this context, the GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method emerges as a powerful constrained dynamics approach that addresses these limitations through torsional angle dynamics and hierarchical clustering schemes, enabling enhanced conformational sampling for protein folding research.
Understanding the Fundamental Bottlenecks
Limitations of All-Atom Cartesian Molecular Dynamics
All-atom Cartesian MD simulations face several inherent limitations that create the sampling bottleneck:
- Computational Cost Scaling: The calculation of non-bonded forces, particularly van der Waals and electrostatic interactions, scales quadratically with the number of atoms [1]
- Time Step Restrictions: The need to resolve high-frequency atomic vibrations requires extremely small time steps (typically 1-2 femtoseconds), severely limiting the total simulation time that can be practically achieved [1] [2]
- Timescale Gap: Biologically relevant processes such as protein folding and ligand unbinding occur on timescales of microseconds to milliseconds, which remains computationally intensive for traditional MD [1]
Comparative Analysis of MD Approaches
Table 1: Comparison of All-Atom Cartesian MD and Constrained MD Approaches
| Feature | All-Atom Cartesian MD | Constrained MD (GNEIMO) |
|---|---|---|
| Degrees of Freedom | 3N (where N = number of atoms) | Approximately N/10 (significantly reduced) |
| Time Step Size | 1-2 femtoseconds | 3-5 femtoseconds (2-5x larger) |
| Computational Scaling | Quadratic for force calculations | Linear with NEIMO algorithm |
| Conformational Sampling | Limited by high-frequency vibrations | Enhanced through torsional space exploration |
| Applicable Timescales | Nanoseconds to microseconds | Microseconds to milliseconds effectively |
The GNEIMO Method: A Constrained Dynamics Framework
Fundamental Principles and Architecture
The GNEIMO method provides a generalized framework for constrained molecular dynamics that addresses the sampling bottleneck through several key innovations:
- Reduced Degrees of Freedom: By replacing high-frequency bond stretching and angle bending motions with hard holonomic constraints, GNEIMO reduces the number of degrees of freedom by approximately an order of magnitude compared to all-atom models [2] [3]
- Rigid Body Clustering: Molecules are modeled as collections of rigid bodies (clusters) connected by flexible torsional hinges, with cluster sizes ranging from a few atoms to entire protein domains [3]
- Internal Coordinate Formulation: The equations of motion are formulated in internal coordinates (primarily torsional angles) rather than Cartesian coordinates, eliminating high-frequency vibrations [2]
- Computational Efficiency: The NEIMO algorithm solves the coupled equations of motion with O(N) computational cost using Spatial Operator Algebra, compared to conventional O(N³) methods [2]
Hierarchical "Freeze and Thaw" Clustering
A distinctive feature of the GNEIMO framework is its hierarchical clustering capability, which allows researchers to strategically "freeze and thaw" different parts of a protein during simulations:
GNEIMO Hierarchical Clustering Workflow
This hierarchical approach enables targeted sampling where stable secondary structure elements (like α-helices) can be treated as rigid bodies while sampling only the torsional degrees of freedom connecting these clusters, leading to faster convergence in sampling the native state of proteins [2].
Application Notes and Protocols
Protein Structure Refinement Protocol
The following protocol details the application of GNEIMO constrained MD for refining low-resolution homology models:
Table 2: GNEIMO Structure Refinement Protocol Components
| Component | Specification | Purpose |
|---|---|---|
| Force Field | AMBER99 | Energy calculations and atomic interactions |
| Solvation Model | GB/SA OBC implicit solvent | Efficient solvation effects |
| Integrator | Lobatto integrator | Numerical integration of equations of motion |
| Time Step | 5 fs | Enabled by constrained dynamics |
| Sampling Method | Replica Exchange MD (REXMD) | Enhanced conformational sampling |
| Temperature Range | 310K to 415K (8 replicas) | Thermodynamic sampling |
| Cluster Definition | User-defined rigid bodies | Focused sampling of flexible regions |
Step-by-Step Protocol:
Initial Structure Preparation:
- Begin with low-resolution decoy structures generated from homology modeling (e.g., using MODELLER)
- Perform energy minimization using 1000 steps of steepest descent followed by 1000 steps of conjugate gradient method
- Use AMBER force field with Generalized Born (GB) solvent model with non-bond cutoff of 20Å [3]
System Setup:
- Define rigid body clusters based on secondary structure elements or functional domains
- Select clustering scheme: all-torsion, hierarchical, or mixed based on protein architecture
- Set up replica exchange parameters with 8 replicas across temperature range 310K-415K [3]
Constrained MD Simulation:
- Perform GNEIMO dynamics using Lobatto integrator with 5 fs time step
- Exchange temperatures between replicas every 2 ps (400 time steps)
- Run each replica for 5-15 ns (total 40-120 ns aggregate simulation time) [3]
Analysis and Validation:
- Calculate RMSD to experimental structures
- Analyze population density of native-like conformations
- Compare with all-atom Cartesian MD results for benchmarking
Protein Folding Application Protocol
For protein folding studies, GNEIMO employs a specialized approach:
Experimental Setup:
- Start from extended conformations of the peptide/protein sequence
- Apply conjugate gradient minimization with convergence factor of 10⁻² Kcal/mol/Å
- Use GB/SA OBC implicit solvation model with interior dielectric 1.75 and exterior dielectric 78.3 [2]
Folding Simulation Parameters:
- Apply all-torsion constrained MD or hierarchical clustering based on secondary structure prediction
- Implement replica exchange with 6-8 replicas in temperature range 325K-500K
- Exchange temperatures every 2ps with total simulation duration up to 20ns per replica [2]
Hierarchical Strategy for Mixed-Motif Proteins:
- For proteins with both α-helical and β-sheet regions, treat either motif as rigid bodies
- Freeze backbone atoms of secondary structure elements while sampling side chains as all-torsion
- This approach aligns with the zipping-and-assembly folding model and enhances native structure sampling [2]
Performance and Validation
Quantitative Assessment of Sampling Enhancement
Table 3: Performance Metrics of GNEIMO Constrained MD
| Metric | All-Atom Cartesian MD | GNEIMO Constrained MD | Improvement |
|---|---|---|---|
| Structure Refinement RMSD | Limited improvement or worsening | ~2 Å improvement | Significant enhancement [3] |
| Native Conformation Enrichment | Sparse sampling | Increased population density | Better thermodynamic sampling [3] |
| Replica Count Requirement | Proportional to sqrt(3N dofs) | Proportional to sqrt(N/10 dofs) | ~3x reduction in replicas [2] |
| Simulation Time Scale | Nanoseconds to microseconds | Effective millisecond processes | 2-3 order magnitude enhancement [1] [4] |
Case Study: Trp-Cage Protein Folding
In folding studies of the Trp-cage miniprotein, hierarchical constrained MD simulations demonstrated superior performance:
- Wider Conformational Search: Compared to all-atom MD, GNEIMO exhibited broader exploration of conformational space [2]
- Native Structure Enrichment: Increased sampling of near-native structures was observed with hierarchical clustering [2]
- Principal Component Analysis: Projection of trajectories onto the first two principal components showed more extensive coverage of essential conformational space [2]
- Cluster Analysis: K-means clustering of simulation trajectories revealed better representation of native-like folds in hierarchical GNEIMO simulations [2]
Integration with Advanced Sampling Methods
Synergy with Replica Exchange Molecular Dynamics
The GNEIMO method demonstrates particular effectiveness when combined with replica exchange MD (REXMD):
- Reduced Replica Requirements: Due to fewer degrees of freedom, constrained MD requires approximately one-third the number of replicas compared to all-atom MD [2]
- Enhanced Sampling Efficiency: The combination of torsional dynamics with temperature exchange enables more thorough exploration of conformational landscapes [3]
- Practical Implementation: Temperature exchanges every 2ps with 5fs time steps provides optimal balance between sampling and computational efficiency [2]
Complementary Machine Learning Approaches
Recent machine learning methods offer complementary approaches to the sampling bottleneck:
- BioMD Framework: Uses a hierarchical framework of forecasting and interpolation to generate long-timescale protein-ligand dynamics [1]
- Predictive Information Bottleneck (PIB): Employs deep neural networks to identify predictive reaction coordinates that capture essential dynamics [4]
- Flow Matching Models: Continuous normalizing flows provide efficient, simulation-free training of generative models for molecular trajectories [1]
Solutions to the Sampling Bottleneck
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Tools for Protein Dynamics Studies
| Tool/Resource | Type | Function | Application Context |
|---|---|---|---|
| GNEIMO Software | Computational Method | Constrained MD simulations | Protein folding, structure refinement |
| AMBER Force Fields | Parameter Set | Molecular mechanical energies | Biomolecular simulations |
| GB/SA Solvation Models | Implicit Solvent | Efficient solvation effects | MD simulations without explicit water |
| MODELLER | Software Tool | Homology model generation | Initial structure preparation |
| Replica Exchange MD | Sampling Algorithm | Enhanced thermodynamic sampling | Overcoming energy barriers |
| Principal Component Analysis | Analysis Method | Dimensionality reduction | Identifying essential dynamics |
| Spatial Operator Algebra | Mathematical Framework | Efficient equation solving | O(N) solution of constrained dynamics |
The protein dynamics sampling bottleneck in all-atom Cartesian MD presents a significant challenge in computational biology and drug discovery. The GNEIMO constrained dynamics method effectively addresses this limitation through its innovative approach of reducing degrees of freedom, enabling larger time steps, and implementing hierarchical "freeze and thaw" clustering schemes. When combined with replica exchange methods and modern machine learning approaches, GNEIMO provides a powerful framework for studying protein folding, structure refinement, and biomolecular dynamics across biologically relevant timescales. The continued development and application of these advanced sampling methods will be crucial for accelerating drug discovery and deepening our understanding of protein function and dynamics.
Constraining High-Frequency Degrees of Freedom with Holonomic Constraints
The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method is a constrained molecular dynamics (MD) simulation approach designed to enhance conformational sampling in protein folding and structure refinement. This method addresses a fundamental bottleneck in all-atom Cartesian MD simulations: the computational intractability of simulating biologically relevant timescales due to the large number of degrees of freedom and limitations imposed by high-frequency atomic vibrations [2] [3].
GNEIMO replaces high-frequency degrees of freedom (such as bond stretching and angle bending) with hard holonomic constraints, modeling a protein as a collection of rigid bodies ("clusters") connected by flexible torsional hinges [5] [3]. This formulation reduces the number of degrees of freedom by approximately an order of magnitude, allowing for larger integration time steps (typically 5 fs compared to 1-2 fs in Cartesian MD) and focusing computational resources on sampling the functionally relevant low-frequency torsional space [2] [6]. The method employs an efficient O(N) algorithm to solve the coupled equations of motion in internal coordinates, making it computationally feasible for protein systems [2] [3].
Performance and Quantitative Assessment
The GNEIMO method demonstrates significant advantages in conformational sampling efficiency and refinement capability over traditional Cartesian MD. The following table summarizes key quantitative improvements observed in protein structure refinement applications.
Table 1: Performance of GNEIMO in Protein Structure Refinement [5] [3]
| Metric | All-Atom Cartesian MD | GNEIMO Constrained MD | Improvement |
|---|---|---|---|
| Integration Time Step | 1-2 fs | 5 fs | 2.5-5x increase |
| Degrees of Freedom | ~3N (Cartesian) | ~N (Torsional) | ~3x reduction |
| RMSD Refinement | Limited improvement, often requires restraints | Up to 1.3-2.0 Å improvement | Significant, without experimental restraints |
| Replicas in REMD | Proportional to √(3N) | Proportional to √(N) | ~√3 reduction (fewer replicas needed) |
| Sampling Enhancement | Limited conformational search | Wider search, increased enrichment of near-native structures | Enhanced "native-like" conformation population |
Application Notes and Experimental Protocols
Protocol 1: All-Torsion Protein Folding and Refinement
This protocol is designed for de novo folding of small proteins or refinement of low-resolution homology models [2] [3].
- Initial Structure Preparation: Start from an extended polypeptide conformation or a low-resolution decoy structure (e.g., from homology modeling with MODELLER).
- Energy Minimization: Perform conjugate gradient minimization on the initial structure using an AMBER force field (e.g., parm99 or AMBER99SB) until a convergence gradient of 10⁻² kcal/mol/Å is reached [2].
- Simulation Parameter Setup:
- Force Field: AMBER99/AMBER99SB [2] [5].
- Solvation Model: Implicit solvent Generalized-Born/Surface Area (GB/SA) OBC model [2] [5].
- Dielectric Constants: Interior = 1.5-1.75; Exterior (solvent) = 78.3 [2] [5].
- Non-Bonded Cutoff: 20 Å, with forces smoothly switched off [2].
- Integrator: Lobatto integrator for constrained dynamics [2] [5].
- Time Step: 5 fs [2] [5].
- Temperature Control: Nose-Hoover thermostat [5].
- Enhanced Sampling: Employ the Temperature Replica Exchange MD (REXMD) method [2] [5].
- Simulation Duration: Conduct simulations for 5-100 ns per replica, depending on system size and research goal [5] [3].
- Analysis: Monitor Root-Mean-Square Deviation (RMSD), fraction of native contacts, radius of gyration, and population densities in principal component space [2].
Protocol 2: Hierarchical "Freeze and Thaw" Clustering
This protocol uses a multi-scale strategy for more efficient sampling, particularly effective for proteins with pre-formed secondary structural elements or mixed motifs [2] [3].
- Preliminary Analysis: Perform a short all-torsion GNEIMO simulation (as in Protocol 1) to identify stable secondary structural regions (e.g., α-helices, β-sheets).
- Cluster Definition: Define "rigid clusters" based on the stable regions identified. For example, the backbone atoms of a stable helix can be frozen into a single rigid body [2].
- Dynamic Model Setup: Configure the GNEIMO simulation to treat the defined clusters as rigid bodies. Only the torsional degrees of freedom connecting these rigid bodies, along with all side-chain torsions, are sampled [2] [3].
- Simulation Execution: Run the GNEIMO-REXMD simulation using the parameters from Protocol 1, but with the hierarchical clustering model active.
- Iterative Refinement (Optional): For complex folding pathways, the "freeze and thaw" process can be iterative, dynamically adjusting which clusters are rigid based on simulation progress [2].
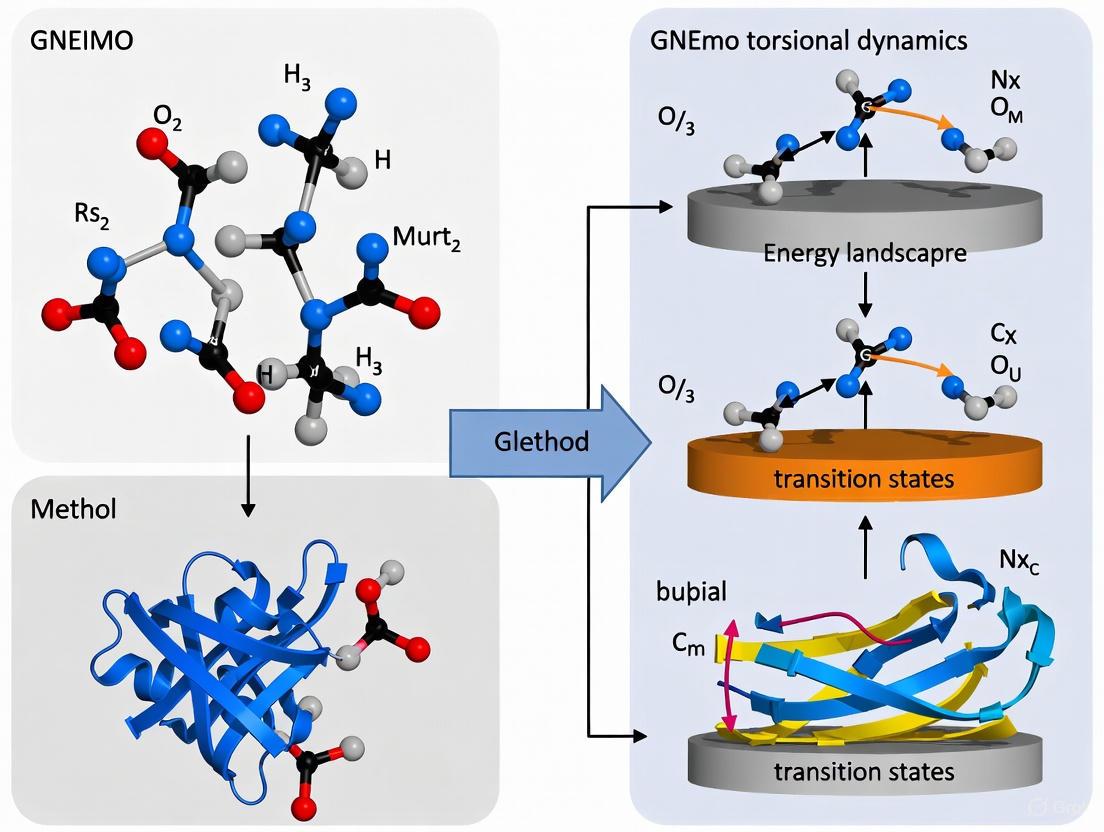
Diagram 1: GNEIMO simulation workflow for protein folding and refinement.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Computational Tools for GNEIMO Simulations
| Reagent/Software | Function/Description | Application Note |
|---|---|---|
| GNEIMO Code | Software package implementing the constrained MD algorithm. | Core engine for performing torsional dynamics simulations [2] [3]. |
| AMBER99SB Force Field | Empirical potential energy function for proteins. | Provides accurate energy terms for bonded and non-bonded interactions; compatible with GNEIMO [5]. |
| GB/SA OBC Implicit Solvent | Generalized Born/Surface Area solvation model with Onufriev-Bashford-Case parameters. | Models solvent effects efficiently without explicit water molecules, reducing computational cost [2] [5]. |
| Lobatto Integrator | Numerical integrator for differential equations. | Specially suited for constrained dynamics, enables stable 5 fs time steps [2] [5]. |
| REXMD Algorithm | Temperature Replica Exchange Molecular Dynamics protocol. | Enhances conformational sampling by allowing replicas at different temperatures to exchange [2] [5]. |
| Homology Modeling Tool (e.g., MODELLER) | Software for generating low-resolution initial models from related structures. | Used to create starting decoy structures for refinement studies [5] [3]. |
Practical Applications and Case Studies
The GNEIMO method has been rigorously tested in various challenging scenarios relevant to structural biology and drug development.
- Folding of Small Proteins: GNEIMO-REXMD successfully folded peptides like polyalanine, WALP16, and the mixed-motif Trp-cage protein from extended states to native-like structures. The hierarchical clustering scheme accelerated convergence by stabilizing partially formed native secondary structures [2].
- Refinement of CASP Targets: In blind tests on targets from the Critical Assessment of Protein Structure Prediction (CASP), GNEIMO refined low-resolution models, improving the Global Distance Test (GDT_TS) scores by up to 14.2 points and reducing Cα-RMSD by up to 1.3 Å without using experimental restraints, a significant achievement over unrestrained Cartesian MD [5].
- Sampling Conformational Dynamics: GNEIMO has efficiently sampled long-timescale conformational changes, such as the transition of calmodulin from the Ca²⁺-bound (holo) to the Ca²⁺-free (apo) state, and populated known conformational substates of fasciculin, transitions that are challenging for conventional MD [6].
Diagram 2: Conceptual comparison of degrees of freedom sampled in different MD methods.
The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method is an advanced computational framework for simulating protein dynamics that addresses a fundamental challenge in molecular dynamics (MD): the computationally expensive nature of all-atom Cartesian simulations. GNEIMO utilizes a constrained molecular dynamics approach, where a protein is modeled as a collection of rigid bodies (clusters) connected by flexible torsional hinges. This physical representation dramatically reduces the number of degrees of freedom in the system by approximately an order of magnitude compared to all-atom models. By replacing high-frequency bond vibrations with hard holonomic constraints and focusing sampling on the slower, more biologically relevant torsional degrees of freedom, GNEIMO enables significantly larger integration time steps (typically 5 femtoseconds) and enhanced conformational sampling, making it particularly valuable for studying protein folding and large-scale conformational changes that occur on biologically relevant timescales [2] [3].
Physical Model and Theoretical Foundation
The Rigid Cluster and Torsional Hinge Architecture
At the core of the GNEIMO physical model is the treatment of proteins as multibody systems composed of interconnected rigid clusters. These clusters are collections of atoms within which all bond lengths and bond angles are fixed using hard holonomic constraints. The clusters are connected to each other by flexible hinges that allow torsional rotation, effectively making torsional angle coordinates the primary degrees of freedom instead of atomic Cartesian coordinates. The size and composition of these rigid clusters can be varied according to the specific research needs, ranging from small clusters containing just a few atoms to large clusters encompassing entire protein domains or secondary structure elements. This flexibility in modeling is referred to as the "freeze and thaw" capability, allowing researchers to selectively rigidify certain protein regions while maintaining flexibility in others [2] [3].
Mathematical Framework and Computational Advantages
The GNEIMO method adapts algorithms from the Spatial Operator Algebra (SOA) mathematical framework for multibody dynamics to efficiently solve the coupled equations of motion in internal coordinates. Unlike conventional O(N³) algorithms for solving internal coordinate equations of motion (where N is the number of degrees of freedom), the GNEIMO implementation of the Newton-Euler Inverse Mass Operator (NEIMO) algorithm solves these equations with O(N) computational cost, making it practical for studying large protein systems. This computational efficiency, combined with the reduced degrees of freedom and elimination of high-frequency vibrations, enables GNEIMO to achieve stable dynamics with larger time steps and access longer simulation timescales than conventional all-atom MD [2] [6].
Table: Comparison of GNEIMO Constrained MD vs. All-Atom Cartesian MD
| Parameter | GNEIMO Constrained MD | All-Atom Cartesian MD |
|---|---|---|
| Degrees of Freedom | ~10% of all-atom models | All atomic Cartesian coordinates |
| Integration Time Step | 5 fs (typical) | 1-2 fs (typical with SHAKE/RATTLE) |
| Computational Scaling | O(N) with NEIMO algorithm | O(N) to O(NlogN) for optimized MD |
| High-Frequency Vibrations | Eliminated via constraints | Explicitly simulated |
| Conformational Sampling | Enhanced in torsional space | Limited by timescale barriers |
Application Notes and Protocols
Protein Folding Studies
GNEIMO has been successfully applied to study the folding mechanisms of various small proteins with different secondary structural motifs. The method is particularly effective when combined with replica exchange molecular dynamics (REXMD) and implicit solvation models to enhance conformational sampling.
Protocol: Protein Folding Using All-Torsion GNEIMO with Replica Exchange
Initial System Preparation: Begin with an extended conformation of the peptide or protein sequence. Perform conjugate gradient minimization with a convergence criterion of 10⁻² Kcal/mol/Å in force gradient [2].
Force Field and Solvation: Utilize the AMBER parm99 forcefield with the GB/SA OBC implicit solvation model. Set the GB/SA interior dielectric value to 1.75 for the solute and exterior dielectric constant to 78.3 for water (adjust to 40.0 for membrane environments). Use a solvent probe radius of 1.4Å for the nonpolar solvation energy component [2].
Simulation Parameters: Employ the Lobatto integrator with an integration step size of 5 fs. Apply a non-bonded force cutoff of 20Å, with forces smoothly switched off at this distance [2].
Replica Exchange Setup: Configure 6-8 replicas in the temperature range of 325K to 500K (in steps of 25K for small peptides). Attempt temperature exchanges between replicas every 2ps. The total simulation time typically ranges up to 20ns per replica [2].
Analysis: Monitor folding progress using metrics such as fraction of residues in native secondary structure, root mean square deviation (RMSD) from native structures, and population density of near-native conformations [2].
Hierarchical Clustering for Mixed Motif Proteins
For proteins with mixed secondary structures like Trp-cage, a hierarchical "freeze and thaw" approach can be implemented:
Initial All-Torsion Simulation: Perform an initial all-torsion GNEIMO simulation to identify partially formed secondary structure regions [2].
Cluster Identification: Analyze trajectories to identify regions with persistent secondary structure formation, particularly helical elements [2].
Freeze Structured Regions: Treat the identified structured regions as rigid clusters, freezing their backbone atoms while maintaining side-chain flexibility [2].
Sampling of Connecting Regions: Sample primarily the torsional degrees of freedom connecting these rigid clusters, significantly reducing the conformational search space [2].
This hierarchical approach has been shown to better sample near-native structures and aligns with the zipping-and-assembly folding model proposed for many proteins [2].
Table: GNEIMO Folding Performance for Various Protein Systems
| Protein System | Structural Motif | Simulation Approach | Key Results |
|---|---|---|---|
| Polyalanine (20-mer) | α-helix | All-torsion REMD (6 replicas) | Achieved helical content comparable to native state at 300K [2] |
| WALP16 | Transmembrane α-helix | All-torsion REMD with membrane dielectric | Successfully folded in membrane-mimetic environment [2] |
| 1E0Q | β-turn | All-torsion REMD (8 replicas) | Sampled near-native structures with proper β-turn formation [2] |
| Trp-cage | Mixed motif | Hierarchical clustering REMD | Enhanced sampling of native states; agreement with zipping-assembly model [2] |
| Fasciculin | Conformational substates | All-torsion REMD | Sampled two experimentally established conformational substates [6] |
| Calmodulin | Domain motion | All-torsion REMD | Captured Ca²⁺-bound to Ca²⁺-free conformational transition [6] |
Protein Structure Refinement
GNEIMO has demonstrated significant promise in addressing the challenge of refining low-resolution homology models towards native-like structures.
Protocol: Structure Refinement of Homology Models
Decoy Generation: Generate low-resolution decoy structures using homology modeling tools such as MODELLER. Select templates with 60-70% sequence identity to the target. Cluster the resulting 100 homology models by structural diversity into 5 clusters and select representative structures with the most secondary structure content [3].
Simulated Annealing: Perform simulated annealing using all-torsion GNEIMO dynamics with temperatures ranging from 310K to 1200K in 50K increments to "swell" the homology models to lower resolution structures (2-5Å backbone RMSD from native) [3].
Energy Minimization: Conduct unconstrained Cartesian MD energy minimization using 1000 steps of steepest descent followed by 1000 steps of conjugate gradient method [3].
GNEIMO Replica Exchange Refinement: Perform all-torsion GNEIMO REXMD simulations with 8 replicas in the temperature range of 310K to 415K with 15K intervals. Exchange temperatures every 2ps. Run each replica for 5-15ns, totaling 40-120ns of simulation time [3].
Analysis and Validation: Calculate RMSD to experimental structures and analyze population density of native-like conformations. Typically, refinement improvements of approximately 2Å RMSD have been observed across various protein systems [3].
The following workflow diagram illustrates the hierarchical GNEIMO protocol for structure refinement:
Diagram: Hierarchical GNEIMO Refinement Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Computational Tools for GNEIMO Simulations
| Tool/Reagent | Function/Description | Application Context |
|---|---|---|
| GNEIMO Software | Implements constrained MD algorithm with O(N) scaling | All GNEIMO simulation protocols |
| AMBER parm99 Forcefield | Provides potential energy functions | Protein energy calculation in implicit solvent |
| GB/SA OBC Solvation Model | Implicit solvent model for biomolecules | Solvation effects without explicit water |
| Lobatto Integrator | Numerical integration method for equations of motion | Molecular dynamics trajectory propagation |
| Replica Exchange Algorithm | Enhanced sampling technique | Overcoming energy barriers in folding/refinement |
| Spatial Operator Algebra | Mathematical framework for multibody dynamics | Efficient solution of constrained equations of motion |
| Principal Component Analysis | Dimensionality reduction for trajectory analysis | Identifying essential motions in protein dynamics |
| K-means Clustering | Machine learning for conformation classification | Grouping structurally similar protein conformations |
Concluding Remarks
The GNEIMO physical model represents a powerful approach to computational protein studies that strategically reduces computational complexity while maintaining physical accuracy where it matters most. By focusing sampling on torsional degrees of freedom and enabling flexible "freeze and thaw" clustering schemes, GNEIMO addresses critical challenges in protein folding and structure refinement that have proven difficult for conventional all-atom MD. The method's ability to enhance conformational sampling of near-native states, coupled with its computational efficiency, makes it particularly valuable for researchers investigating protein dynamics, folding mechanisms, and structure prediction. As computational capabilities continue to advance, GNEIMO's unique physical model offers a promising framework for tackling increasingly complex problems in structural biology and drug development.
Spatial Operator Algebra (SOA) represents a sophisticated mathematical framework adapted from multibody dynamics to overcome one of the most significant bottlenecks in molecular dynamics (MD) simulations: the computational expense of simulating biological macromolecules such as proteins. Conventional all-atom Cartesian MD simulations become computationally prohibitive for studying processes like protein folding that occur on microsecond to millisecond timescales. The SOA framework provides the mathematical foundation for the Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method, a constrained MD approach that enables longer timescale simulations by dramatically reducing the number of degrees of freedom in the system [2].
In the GNEIMO method, proteins are modeled as collections of rigid bodies (clusters) connected by flexible torsional hinges, with fixed bond lengths and bond angles serving as holonomic constraints. This representation reduces the number of degrees of freedom by approximately an order of magnitude compared to all-atom Cartesian models. While conventional algorithms for solving the resulting coupled equations of motion in internal coordinates scale with the cubic power of the number of degrees of freedom (O(N³)), the SOA-based NEIMO algorithm achieves linear scaling (O(N)) through efficient recursive formulations [2] [3]. This mathematical advancement enables stable dynamics with larger integration time steps (typically 5 fs), leading to a significant decrease in computational cost while maintaining physical accuracy [2].
Table 1: Key Computational Advantages of SOA-Based Constrained MD
| Parameter | All-Atom Cartesian MD | Constrained MD (GNEIMO) | Improvement Factor |
|---|---|---|---|
| Degrees of Freedom | ~3N (atomic coordinates) | ~N (torsional angles) | ~10x reduction [2] |
| Integration Time Step | 1-2 fs | 5 fs | 2.5-5x increase [2] |
| Computational Scaling | O(N) to O(N²) | O(N) with SOA | Dramatic improvement for large systems [3] |
| Replica Exchange Requirements | Proportional to √(3N) | Proportional to √(N) | ~3x reduction in replicas [2] |
Application Note: Protein Folding Studies Using SOA-Enhanced Sampling
The GNEIMO method, powered by the SOA mathematical engine, has demonstrated remarkable success in protein folding studies of small proteins with various secondary structural motifs. Research has shown that constrained MD replica exchange methods exhibit wider conformational search capabilities than all-atom MD with increased enrichment of near-native structures [2]. This enhanced sampling capability stems from the more efficient exploration of conformational space when high-frequency bond vibrations are constrained, allowing the simulation to focus on the functionally relevant torsional degrees of freedom that drive protein folding.
In studies of polyalanine (α-helix), WALP16 (transmembrane peptide), β-turn peptides (1E0Q), and mixed motif proteins (Trp-cage), the GNEIMO method with replica exchange successfully folded these systems using only 6-8 replicas in the temperature range of 325K to 500K [2]. The simulations were initiated from extended conformations and utilized the AMBER forcefield with GB/SA implicit solvation. The "hierarchical" constrained MD approach, where partially formed helical regions were frozen while sampling other torsional degrees of freedom, demonstrated superior sampling of near-native structures compared to all-torsion constrained MD simulations [2]. This finding aligns with the zipping-and-assembly folding model and highlights how SOA-enabled flexible clustering schemes can strategically guide conformational sampling toward biologically relevant regions of the energy landscape.
Table 2: Performance of SOA-Based Methods in Protein Structure Applications
| Application | System Studied | Key Performance Metrics | Experimental Validation |
|---|---|---|---|
| Protein Folding | Poly-alanine, WALP16, β-turn, Trp-cage | Enhanced enrichment of near-native structures; wider conformational sampling [2] | Comparison with known native structures; principal component analysis [2] |
| Structure Refinement | 8 proteins with various motifs (all-α, α/β, all-β) | ~2 Å improvement in RMSD to experimental structures [3] | X-ray crystal structures and NMR structures as reference [3] |
| Hierarchical Clustering | Mixed α-helix/β-sheet proteins | Faster convergence to native state; reduced computational cost [2] [3] | Population density analysis of native-like conformations [2] |
Experimental Protocol: All-Torsion Folding Simulation with GNEIMO
System Preparation and Minimization
Begin with an extended conformation of the peptide or protein sequence. Perform conjugate gradient minimization with a convergence criterion of 10⁻² kcal/mol/Å in force gradient using the AMBER forcefield (parm99) with GB/SA implicit solvation. For the solvation model, set the GB/SA interior dielectric constant to 1.75 for the solute and the exterior dielectric constant to 78.3 for the solvent, using a solvent probe radius of 1.4Å for the nonpolar solvation energy component [2]. Apply a non-bonded force cutoff of 20Å with a smooth switching function.
Replica Exchange Configuration
Configure the replica exchange molecular dynamics (REXMD) simulation with 8 replicas for most systems (6 replicas for simple systems like polyalanine) distributed across a temperature range from 325K to 500K in increments of 25-35K [2]. The number of replicas is determined by the square root of the number of degrees of freedom, which is significantly reduced in constrained MD, thereby requiring fewer replicas than comparable all-atom simulations [2].
Constrained Dynamics Production Run
Execute the GNEIMO constrained MD simulation using the Lobatto integrator with a 5 fs time step. Perform temperature exchanges between replicas every 2 ps (400 time steps). Continue the production run for up to 20 ns per replica, though shorter durations may suffice for smaller systems [2]. For the all-torsion model, treat all torsional degrees of freedom as flexible while maintaining rigid bond lengths and bond angles through holonomic constraints.
Trajectory Analysis and Clustering
After simulation completion, analyze the trajectories using principal component analysis (PCA) by constructing covariance matrices of the Cα atom coordinates from simulation snapshots [2]. Project the trajectories onto the first two principal components to visualize conformation population density distributions. Employ K-means clustering algorithm to partition structures into structurally similar subsets, with representative structures generated by averaging 1000 snapshots from each cluster. Calculate population percentages as the fraction of conformations belonging to each cluster group.
Experimental Protocol: Hierarchical "Freeze and Thaw" Clustering for Structure Refinement
Decoy Set Generation and Preparation
Generate low-resolution decoy structures through homology modeling using software such as MODELLER, selecting templates with 60-70% sequence identity to the target [3]. Cluster the resulting top 100 homology models by structural diversity into 5 clusters and select representative structures with the most secondary structure content. Perform simulated annealing with all-torsion GNEIMO dynamics, sweeping temperatures from 310K to 1200K in 50K increments to swell the homology models to lower resolution structures (2-5 Å backbone RMSD from native) [3]. Finally, conduct energy minimization using unconstrained Cartesian MD with 1000 steps of steepest descent followed by 1000 steps of conjugate gradient method.
Hierarchical Clustering Configuration
Implement the "freeze and thaw" strategy by identifying stable secondary structure elements (α-helices or β-sheets) in the decoy structures. Freeze the backbone atoms of these stable regions as rigid clusters while allowing side-chain flexibility [3]. For the remaining protein regions, maintain all-torsion flexibility. This hybrid approach reduces the conformational search space while maintaining flexibility in structurally ambiguous regions.
Replica Exchange Refinement Simulation
Configure the GNEIMO REXMD simulation with 8 replicas across a temperature range of 310K to 415K with 15K intervals [3]. Run each replica for 5-15 ns, totaling 40-120 ns of aggregate simulation time. Perform temperature exchanges every 2 ps (400 time steps) to enhance conformational sampling. Utilize the AMBER forcefield with GB/SA implicit solvation, maintaining the same dielectric and non-bonded cutoffs as in the folding protocol.
Refinement Validation and Analysis
Evaluate refinement success by calculating backbone RMSD to the known experimental structure across the simulation trajectory. Identify the lowest-energy structures and assess improvement in native-like character through population density analysis of near-native conformations [3]. Compare the performance of hierarchical clustering against all-torsion constrained MD and unconstrained Cartesian MD to quantify the enhancement in conformational sampling efficiency.
Table 3: Key Research Reagent Solutions for SOA-Based Protein Simulations
| Resource Category | Specific Implementation | Function and Purpose |
|---|---|---|
| Force Fields | AMBER parm99/AMBER99 [2] | Defines potential energy function for protein interactions |
| Solvation Models | GB/SA OBC implicit solvent [2] | Represents solvent effects without explicit water molecules |
| Constrained MD Software | GNEIMO package [3] | Implements SOA mathematics for efficient constrained dynamics |
| Replica Exchange Framework | Custom implementation in GNEIMO [2] | Enhances conformational sampling through parallel tempering |
| Structure Analysis Tools | Principal Component Analysis, K-means clustering [2] | Identifies and characterizes conformational populations |
| Homology Modeling | MODELLER software [3] | Generates initial decoy structures for refinement protocols |
| Rigid Body Clustering | "Freeze and Thaw" hierarchical scheme [3] | Strategically reduces conformational search space |
Spatial Operator Algebra has proven to be a transformative mathematical framework that addresses fundamental limitations in molecular dynamics simulations of proteins. By enabling efficient O(N) scaling for constrained dynamics, the SOA-based GNEIMO method has opened new avenues for studying protein folding and structure refinement that were previously computationally prohibitive. The hierarchical "freeze and thaw" approaches made possible by this framework align with physical folding models and provide researchers with strategic tools for enhancing conformational sampling. As computational methods continue to play an increasingly vital role in structural biology and drug discovery, the mathematical efficiency provided by Spatial Operator Algebra will remain essential for bridging the gap between simulation timescales and biologically relevant phenomena.
The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method represents a significant advancement in molecular dynamics simulations for protein folding and structure refinement. This constrained molecular dynamics approach enhances conformational sampling by focusing on low-frequency torsional motions while constraining high-frequency bond vibrations. The methodology enables larger integration time steps and provides more efficient exploration of protein conformational space compared to traditional Cartesian molecular dynamics. Within protein folding research, GNEIMO has demonstrated particular utility in refining homology models, folding small proteins, and studying conformational transitions, offering researchers a powerful tool for investigating protein dynamics and facilitating drug design efforts.
Proteins are dynamic molecules whose functions are intrinsically linked to their three-dimensional structures and conformational flexibility. Understanding protein folding remains a central challenge in structural biology with significant implications for drug development. Traditional all-atom Cartesian molecular dynamics (MD) simulations face substantial limitations in simulating biologically relevant timescales due to computational constraints. The high-frequency bond vibrations in these simulations necessitate small integration time steps (typically 1-2 fs), severely limiting conformational sampling.
The GNEIMO method addresses these limitations through a constrained dynamics approach that fundamentally transforms the simulation paradigm. By treating proteins as collections of rigid bodies connected by flexible torsional hinges, GNEIMO significantly reduces the number of degrees of freedom and enables enhanced sampling of functionally relevant conformational states. This application note details the theoretical foundations, practical implementations, and research applications of the GNEIMO method, providing researchers with protocols to leverage its advantages in protein folding studies.
Core Methodological Advantages
Reduction of Degrees of Freedom
The GNEIMO method employs holonomic constraints to fix bond lengths and bond angles, effectively modeling proteins as collections of rigid bodies ("clusters") connected by flexible torsional hinges [3] [2]. This approach reduces the number of degrees of freedom by approximately an order of magnitude compared to all-atom Cartesian MD simulations [2]. For example, in a typical protein system with thousands of atoms, Cartesian MD would simulate 3N degrees of freedom (where N is the number of atoms), while GNEIMO focuses primarily on torsional degrees of freedom, drastically reducing the computational complexity of the simulation.
Increased Integration Time Steps
By constraining high-frequency vibrational modes, GNEIMO enables significantly larger integration time steps of 5 fs compared to the 1-2 fs typically used in Cartesian MD [3] [2]. This 2.5-5 fold increase in time step size directly translates to longer effective simulation timescales within the same computational budget. The method employs a Lobatto integrator to maintain numerical stability at these larger time steps while preserving the accuracy of conformational sampling [3] [2].
Enhanced Low-Frequency Conformational Search
GNEIMO enhances sampling of functionally relevant low-frequency collective motions by focusing computational resources on torsional degrees of freedom that dominate large-scale conformational changes in proteins [6]. Research has demonstrated that GNEIMO simulations can capture conformational transitions and substate distributions that remain inaccessible to conventional Cartesian MD within similar simulation timeframes [6]. For example, GNEIMO has successfully simulated the transition of calmodulin from Ca²⁺-bound to Ca²⁺-free states and sampled multiple conformational substates of fasciculin, illustrating its enhanced sampling capabilities for biologically relevant motions [6].
Table 1: Quantitative Comparison Between Traditional Cartesian MD and GNEIMO Method
| Parameter | Traditional Cartesian MD | GNEIMO Constrained MD |
|---|---|---|
| Degrees of Freedom | 3N (all atoms) | Approximately N/10 (primarily torsional) [2] |
| Typical Time Step | 1-2 fs [2] | 5 fs [3] [2] |
| Computational Scaling | O(N) to O(N²) | O(ndof) for solving equations of motion [2] |
| Conformational Sampling | Limited by high-frequency vibrations | Enhanced low-frequency torsional sampling [6] |
| Replicas Required in RE-MD | Proportional to √(3N) | Approximately 1/3 of Cartesian MD [2] |
Performance and Validation Data
Protein Structure Refinement Applications
In protein structure refinement applications, GNEIMO has demonstrated consistent improvement in model quality. Using an all-torsion GNEIMO protocol coupled with replica exchange molecular dynamics (REXMD), researchers achieved RMSD improvements of approximately 2 Å across eight different proteins when refining low-resolution homology models [3]. The method also showed enrichment in native-like conformations in the population density, indicating not just structural improvement but also more effective sampling of biologically relevant states.
Table 2: GNEIMO Performance in Protein Structure Refinement
| Protein Type | Starting RMSD Range (Å) | Refinement Protocol | RMSD Improvement (Å) |
|---|---|---|---|
| All-α | 2-5 | All-torsion GNEIMO REXMD | ~2 [3] |
| All-β | 2-5 | All-torsion GNEIMO REXMD | ~2 [3] |
| α/β Mixed | 2-5 | All-torsion GNEIMO REXMD | ~2 [3] |
| α/β Mixed | 2-5 | Hierarchical "Freeze and Thaw" | Comparable or better than all-torsion [3] |
Protein Folding Applications
GNEIMO has successfully folded various small proteins and peptides starting from extended conformations, including α-helical peptides (polyalanine, WALP16), β-turn structures (1E0Q), and mixed motif proteins (Trp-cage) [2]. The method demonstrated faster convergence to native-like states compared to Cartesian MD, with increased population of near-native conformations in the sampled ensemble. Hierarchical clustering schemes, where partially formed secondary structure elements were treated as rigid bodies, further enhanced sampling efficiency according to the zipping-and-assembly folding model [2].
Experimental Protocols
All-Torsion GNEIMO Protocol for Structure Refinement
The following protocol details the application of GNEIMO for refining protein homology models:
Initial System Preparation
- Generate low-resolution decoy structures using homology modeling tools such as MODELLER [3]
- Select template structures with 60-70% sequence identity to target sequence
- Cluster resulting homology models by structural diversity and select representative structures
Energy Minimization
- Perform unconstrained Cartesian MD energy minimization
- Apply 1000 steps of steepest descent followed by 1000 steps of conjugate gradient method
- Utilize AMBER force field and Generalized Born (GB) solvent model with non-bond cutoff of 20 Å [3]
GNEIMO REXMD Simulation
- Employ all-torsion GNEIMO method coupled with replica exchange molecular dynamics (REXMD)
- Use 8 replicas across temperature range of 310 K to 415 K with 15 K intervals [3]
- Apply Lobatto integrator with 5 fs time step
- Exchange temperatures between replicas every 2 ps (400 time steps)
- Run each replica for 5-15 ns (total simulation time 40-120 ns)
- Utilize AMBER99 force field with GB/SA OBC implicit solvation model [3]
Trajectory Analysis
- Calculate RMSD to known experimental structures
- Analyze population density of native-like conformations
- Identify lowest energy structures from the ensemble
Hierarchical "Freeze and Thaw" Clustering Protocol
For proteins with mixed α-helix and β-sheet motifs, the hierarchical clustering approach enhances refinement:
Cluster Identification
- Identify stable secondary structure elements (α-helices or β-sheets) in initial models
- Select either α-helical or β-sheet regions for rigid body treatment [3]
Dynamics Setup
- Treat selected secondary structure elements as rigid bodies (frozen backbone atoms)
- Allow full side-chain flexibility within rigid regions
- Model rest of protein with all-torsion dynamics
- Define torsional hinges connecting rigid clusters to flexible regions [3]
Simulation Execution
- Implement constrained dynamics with mixed rigid-flexible treatment
- Apply similar temperature REXMD protocol as all-torsion approach
- Utilize same force field and solvation parameters
Comparative Analysis
- Compare structural refinement metrics with all-torsion results
- Evaluate sampling efficiency and native-state enrichment
Protein Folding Protocol
For ab initio folding of small proteins and peptides:
Initial Structure Preparation
- Start from extended conformation of peptide/protein sequence [2]
- Perform conjugate gradient minimization with convergence factor of 10⁻² Kcal/mol/Å
Constrained MD Simulation Setup
- Apply parm99 forcefield within AMBER99 with GB/SA OBC implicit solvation [2]
- Set GB/SA interior dielectric to 1.75 and exterior dielectric to 78.3 (or 40.0 for membrane environments)
- Use solvent probe radius of 1.4 Å for nonpolar solvation energy
- Apply non-bond force cutoff at 20 Å with smooth switching
Replica Exchange Configuration
- Implement 8 replicas across temperature range 325K to 500K (25K intervals) [2]
- Exchange temperatures every 2 ps
- Run simulations for up to 20 ns per replica
Analysis Methods
- Perform principal component analysis (PCA) on Cα atom coordinates [2]
- Apply k-means clustering to identify structurally similar subsets
- Calculate population percentages of each cluster
- Measure helicity by fraction of residues with φ/ψ angles within 20° of ideal α-helical values
Research Reagent Solutions
Table 3: Essential Research Tools for GNEIMO Simulations
| Tool/Resource | Type | Function | Availability |
|---|---|---|---|
| GneimoSim | Software Package | Modular Internal Coordinates MD Simulation | Free academic download [7] |
| AMBER99 | Force Field | Physics-based potential energy functions | Commercial with academic licenses [3] |
| GB/SA OBC | Solvation Model | Implicit solvent for efficient hydration | Included in AMBER [3] |
| MODELLER | Homology Modeling | Generation of initial low-resolution models | Free academic license [3] |
| PHENIX | Experimental Refinement | Integration with X-ray crystallography data | Free for academic use [7] |
Visualization of Conformational Sampling
The GNEIMO method provides a robust framework for protein structure refinement and folding studies through its innovative approach to constrained molecular dynamics. The core advantages of reduced degrees of freedom, larger integration time steps, and enhanced low-frequency conformational sampling collectively address fundamental limitations of traditional Cartesian MD simulations. The protocols outlined in this application note provide researchers with practical methodologies for implementing GNEIMO in various protein studies, from refining homology models to investigating folding pathways. As computational approaches continue to complement experimental structural biology, GNEIMO represents a valuable tool for advancing our understanding of protein dynamics and facilitating structure-based drug design efforts.
Protocols and Applications: Implementing GNEIMO for Protein Refinement and Folding
The GneimoSim software package represents a significant advancement in the field of molecular dynamics (MD) simulations by implementing the Generalized Newton Euler Inverse Mass Operator (GNEIMO) method for internal coordinates molecular dynamics (ICMD). As a modular ICMD platform, GneimoSim addresses longstanding challenges in molecular simulations by enabling researchers to study protein dynamics, refine protein structures, and investigate large-scale conformational changes with enhanced sampling efficiency. This platform is particularly valuable for protein folding research and drug development applications where understanding conformational dynamics is critical [8].
Traditional all-atom Cartesian MD simulations, while widely used, face limitations in simulating biologically relevant timescales due to computational constraints. The GneimoSim approach utilizes internal coordinates (Bond, Angle, Torsion) which are more natural for describing the bonded structure of proteins and other polymers. By constraining high-frequency bond length and bond angle degrees of freedom, GneimoSim focuses computational resources on the functionally relevant low-frequency torsional degrees of freedom, enabling longer time steps and enhanced conformational sampling [8] [2].
Theoretical Foundations of the GNEIMO Method
Internal Coordinates Molecular Dynamics Framework
The GNEIMO method fundamentally differs from Cartesian MD by modeling molecules as collections of rigid bodies (clusters) connected by flexible hinges with one to six degrees of freedom. These clusters can range in scale from single atoms to entire protein domains, allowing researchers to control the granularity of the dynamics model based on their specific research objectives [8]. This modular approach to molecular representation enables multi-scale simulation strategies that can adapt to different research needs, from atomic-level detail to domain-level motions.
A key innovation in GNEIMO is the use of Spatial Operator Algebra (SOA) methodology, originally developed for spacecraft and robot dynamics, which reduces the computational complexity of solving the ICMD equations of motion from O(n³) to O(n) – where n represents the number of degrees of freedom [8]. This algorithmic efficiency enables the application of ICMD to proteins of biologically relevant sizes that were previously computationally prohibitive.
Advanced Statistical Mechanical Foundations
The GNEIMO method incorporates several theoretical advancements that ensure physical accuracy in constrained dynamics simulations:
Generalized Equipartition Principle: GNEIMO includes a novel equipartition principle derived specifically for internal coordinates, enabling thermodynamically correct initialization of velocities in ICMD simulations [8].
Fixman Potential Compensation: The method includes a low-cost, general-purpose algorithm for computing the Fixman potential, which eliminates systematic statistical biases introduced by the use of hard constraints [8]. This potential ensures that the probability density function of conformational states matches that of unconstrained dynamics, making thermodynamic predictions from constrained dynamics reliable [9].
The mathematical formulation accounts for the position-dependent mass metric tensor in internal coordinates, which differs fundamentally from the constant diagonal mass matrix in Cartesian coordinates. The Fixman potential compensates for this discrepancy, ensuring proper sampling of the Boltzmann distribution [9].
GneimoSim Software Architecture and Features
Modular Design and Extensibility
GneimoSim was designed with modularity and extensibility as core principles, allowing researchers to leverage established force fields and sampling algorithms while utilizing the advanced ICMD capabilities of the package. The software features interfaces to several widely used third-party force field packages including LAMMPS, OpenMM, and Rosetta [8]. This design approach enables the molecular modeling community to integrate GneimoSim into existing workflows without requiring complete methodology overhauls.
The package provides a comprehensive Python interface to the underlying C++ classes and their methods, offering users a powerful and versatile mechanism to develop simulation scripts that configure simulations and control simulation flow [8]. This scripting capability enables sophisticated simulation protocols that can adapt based on intermediate results, facilitating complex computational experiments that would be difficult to implement in more rigid MD software architectures.
Advanced Sampling and Dynamics Capabilities
GneimoSim incorporates multiple state-of-the-art sampling algorithms and dynamics methods specifically adapted for internal coordinates:
- Temperature Replica Exchange MD (REMD): Implemented to enhance conformational sampling in torsional space [5]
- Accelerated MD (aMD): Provides an alternative enhanced sampling approach [8]
- Langevin Dynamics: Available for simulating dynamics in implicit solvent [8]
- Nosé-Hoover NVT Method: Extended for ICMD simulations [8]
The software supports multiple numerical integrators including Runge-Kutta, Lobatto, adaptive CVODE, and Verlet integrators, allowing users to select the most appropriate integration method for their specific system and research objectives [8]. The stability of these integrators has been verified for long timescale simulations (up to microseconds) on proteins ranging from 30 to 300 residues [8].
Table 1: Key Simulation Features in GneimoSim
| Feature Category | Specific Methods | Key Applications |
|---|---|---|
| Integration Algorithms | Runge-Kutta, Lobatto, CVODE, Verlet | Stable long-timescale simulations |
| Enhanced Sampling | REMD, Accelerated MD | Protein folding, conformational transitions |
| Thermostat Methods | Nosé-Hoover, Langevin Dynamics | Temperature control, implicit solvent |
| Solvation Models | GBSA, Periodic Boundary Conditions | Implicit and explicit solvation |
Application Notes and Protocols
Protein Structure Refinement Protocol
GneimoSim has been successfully applied to protein structure refinement of homology models, demonstrating improvement of up to 1.3-1.5 Å in root-mean-square deviation (RMSD) from native crystal structures without requiring experimental restraints [8] [5]. The following protocol outlines the standard methodology for protein structure refinement using GneimoSim:
Protocol 1: Protein Structure Refinement Using GNEIMO-REMD
Initial Structure Preparation:
- Obtain starting decoy structures from homology modeling (e.g., using MODELER) or CASP targets
- Perform all-atom conjugate gradient minimization using AMBER's "sander" program with AMBER99SB force field
- Cluster models and select representative structures based on validation scores (e.g., procheck G-factor) [5]
Simulation Parameters:
- Force Field: AMBER99SB
- Solvation: Generalized Born/Surface Area (GB/SA) OBC implicit solvation model
- Interior dielectric: 1.5 for solute
- Exterior dielectric: 78.3 for solvent
- Solvent probe radius: 1.4 Å for nonpolar solvation energy
- Nonbonded force cutoff: 20 Å with switching function [5]
GNEIMO-REMD Configuration:
- Number of replicas: 32
- Temperature range: 310-415 K
- Temperature exchange attempts: Every 5 ps using Metropolis algorithm
- Integrator: Lobatto with 5 fs time step
- Simulation duration: 15-100 ns per replica [5]
Analysis:
- Evaluate refinement using RMSD, Global Distance Test (GDT), and TM-score metrics
- Compare with best CASP submissions for benchmarking
- Identify lowest energy structures from REMD trajectories
Table 2: Representative Refinement Results for CASP Targets Using GNEIMO
| Target Protein | Starting GDT_TS | Refined GDT_TS | RMSD Improvement (Å) |
|---|---|---|---|
| TR429 | 31.5 | 45.7 | 1.06 |
| TR435 | 80.2 | 87.9 | 0.49 |
| TR453 | 86.6 | 91.5 | 0.41 |
| TR454 | 58.5 | 71.0 | 1.26 |
Conformational Dynamics Mapping Protocol
For studying large-scale conformational changes in proteins, GneimoSim enables enhanced sampling of functionally relevant transitions that occur on timescales difficult to access with conventional Cartesian MD [6]. The following protocol has been successfully applied to proteins such as calmodulin and fasciculin:
Protocol 2: Mapping Conformational Dynamics
System Setup:
- Start from crystal or NMR structures of different conformational states
- Define clusters appropriate to the system (e.g., domains in multi-domain proteins)
- Select torsional degrees of freedom connecting rigid clusters
Simulation Parameters:
- Force Field: AMBER99SB or compatible
- Solvation: Implicit solvent (GB/SA) or explicit solvent with PBC
- Integrator: Lobatto with 4-5 fs time step
- Temperature control: Nosé-Hoover thermostat
Enhanced Sampling:
- Implement REMD with 16-32 replicas depending on system size
- Temperature range: 300-500 K adjusted for specific protein
- Alternatively, use accelerated MD to lower energy barriers
Analysis of Conformational Transitions:
- Monitor inter-domain distances and angles
- Calculate radius of gyration and other global parameters
- Identify transition pathways using principal component analysis
- Validate against experimental NMR or FRET measurements [6]
Protein Folding Studies Protocol
GneimoSim enables efficient folding simulations of small proteins and peptides through its hierarchical constrained dynamics approach, which can accelerate sampling of native-like structures [2]:
Protocol 3: Protein Folding Using Hierarchical GNEIMO
Initial Conditions:
- Start from extended conformation of peptide/protein sequence
- Perform conjugate gradient minimization (convergence: 10⁻² Kcal/mol/Å)
Simulation Parameters:
- Force Field: AMBER99 (parm99)
- Solvation: GB/SA OBC implicit solvation model
- Interior dielectric: 1.75
- Solvent probe radius: 1.4 Å
- Nonbonded cutoff: 20 Å with switching function
- Integrator: Lobatto with 5 fs time step [2]
Replica Exchange Setup:
- Number of replicas: 6-8 (temperature-dependent)
- Temperature range: 325-500 K (in steps of 25K)
- Exchange attempts: Every 2 ps
- Simulation duration: Up to 20 ns per replica [2]
Hierarchical Clustering Options:
- All-torsion dynamics: All torsional degrees of freedom flexible
- Hierarchical dynamics: Pre-formed secondary structure elements as clusters with flexible connecting torsions
- Adaptive clustering: Change clustering scheme during simulation based on emerging structural elements
Diagram 1: GNEIMO Protein Folding Workflow. The workflow shows the parallel sampling approaches using all-torsion and hierarchical clustering methods.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for GNEIMO Simulations
| Tool/Component | Function | Implementation in GneimoSim |
|---|---|---|
| Force Fields | Defines potential energy terms | Interfaces to AMBER99SB, LAMMPS, OpenMM, Rosetta |
| Solvation Models | Mimics solvent effects | GBSA OBC implicit solvation; PBC for explicit solvent |
| Integrators | Numerical solution of equations of motion | Lobatto, Runge-Kutta, CVODE, Verlet |
| Clustering Schemes | Defines rigid and flexible regions | User-defined clusters from atoms to domains |
| Enhanced Sampling | Accelerates conformational search | REMD, Accelerated MD |
| Analysis Modules | Extracts structural and dynamic information | Python interface for trajectory analysis |
Diagram: GNEIMO Method Workflow
Diagram 2: GNEIMO Method Workflow. The process begins with a PDB structure and progresses through cluster definition, internal coordinate transformation, and efficient solution of equations of motion.
The GneimoSim software package provides a robust, modular platform for internal coordinates molecular dynamics that addresses fundamental challenges in molecular simulations. Through its implementation of the GNEIMO method with advanced features such as the Fixman potential, generalized equipartition principle, and efficient O(n) algorithms, GneimoSim enables researchers to study protein dynamics, refine protein structures, and investigate conformational changes with enhanced sampling efficiency. The protocols outlined in this application note provide practical guidance for leveraging GneimoSim in protein folding research and structure-based drug design, offering the scientific community powerful tools to explore complex biological phenomena at molecular detail.
Standard All-Torsion Protocol for Protein Structure Refinement
Within the broader research on the GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method for torsional dynamics, the standard all-torsion protocol represents a foundational approach for protein structure refinement. This method addresses a critical challenge in computational biology: the refinement of low-resolution protein models derived from homology modeling or other prediction techniques towards more accurate, native-like structures [3]. Traditional all-atom Cartesian molecular dynamics (MD) simulations are often limited in their conformational sampling capabilities for this task due to computational expense and timescale limitations [2] [3]. The GNEIMO-based all-torsion protocol overcomes these constraints by employing a reduced coordinate system that focuses sampling on the most relevant degrees of freedom for protein folding and refinement, enabling more efficient exploration of the conformational landscape and enrichment of native-like structures [2] [3] [6].
Theoretical Foundation and Advantages
The standard all-torsion protocol utilizes a constrained dynamics approach where high-frequency bond stretching and angle bending vibrations are replaced with hard holonomic constraints. In this model, the protein is treated as a collection of rigid bodies connected by flexible torsional hinges, effectively reducing the number of degrees of freedom by approximately an order of magnitude compared to all-atom Cartesian MD [2] [3].
This theoretical framework provides two significant advantages for protein structure refinement. First, the elimination of high-frequency motions allows for larger integration time steps (typically 4-5 fs), extending the accessible simulation timescales [2] [3]. Second, the focus on torsional degrees of freedom naturally enhances sampling of the slow, large-amplitude motions that dominate protein folding and conformational changes [6]. Research has demonstrated that this torsional dynamics approach can capture long-timescale conformational transitions that remain challenging for conventional MD methods, such as the transition between conformational substates in fasciculin and the holo to apo transition in calmodulin [6].
The diagram below illustrates the comprehensive workflow for the standard all-torsion protein structure refinement protocol using the GNEIMO method:
Materials and Reagents
Table 1: Essential Research Reagent Solutions for All-Torsion Refinement Protocol
| Item | Specification/Function | Example/Notes |
|---|---|---|
| Molecular Dynamics Software | Must implement GNEIMO constrained dynamics algorithm | Custom GNEIMO code [2] [3] |
| Force Field | AMBER parm99/AMBER99 for energy calculations | Provides parameters for bonded and non-bonded interactions [2] |
| Solvation Model | Implicit solvation using Generalized-Born/Surface Area (GB/SA) | GB/SA OBC model with εint=1.5-1.75, εext=78.3 [2] [3] |
| Starting Structures | Low-resolution protein models requiring refinement | Typically 2-5 Å RMSD from native structure [3] |
| Computational Resources | High-performance computing cluster | Multiple processors for parallel replica simulations |
Step-by-Step Protocol
System Preparation and Minimization
Begin with an extended conformation or low-resolution model of the target protein. If using homology models, generate decoys through standard homology modeling packages like MODELLER and select representative structures from different clusters [3]. Perform initial energy minimization using a conjugate gradient approach with a convergence criterion of 10⁻² kcal/mol/Å in force gradient to remove any steric clashes and prepare the structure for dynamics [2].
Replica Exchange Molecular Dynamics Parameters
Table 2: Standard All-Torsion GNEIMO Simulation Parameters
| Parameter | Standard Setting | Alternative/Range |
|---|---|---|
| Integration Time Step | 5 fs | 4-5 fs [2] [3] |
| Integrator | Lobatto | Suitable for constrained dynamics |
| Number of Replicas | 8 | Scales with √(number of degrees of freedom) [2] |
| Temperature Range | 310K - 415K | 15K intervals [3] |
| Exchange Frequency | Every 2 ps (400 steps) | 1-4 ps depending on system [2] [3] |
| Simulation Duration | 5-15 ns per replica | 40-120 ns total simulation time [3] |
| Non-bonded Cutoff | 20 Å | With smooth switching function [2] |
| Dielectric Constants | Interior: 1.5-1.75Exterior: 78.3 | Environment-dependent [2] [3] |
Enhanced Sampling Strategy
The replica exchange molecular dynamics (REMD) protocol is integral to the all-torsion refinement method. The reduced number of degrees of freedom in constrained dynamics decreases the number of required replicas compared to Cartesian MD, improving computational efficiency [2]. Temperature exchanges should occur at regular intervals (typically every 2 ps) to facilitate crossing of energy barriers and ensure adequate sampling of the conformational landscape.
Trajectory Analysis and Clustering
Following simulations, analyze trajectories using principal component analysis (PCA) to visualize conformational sampling in the space of the first two principal components [2]. Employ K-means clustering or similar algorithms to group structurally similar conformations and identify representative structures from each cluster. Calculate population percentages as the fraction of conformations belonging to each cluster to quantify sampling efficiency.
Expected Outcomes and Validation
Successful implementation of the standard all-torsion protocol typically achieves refinement improvements of approximately 2 Å in RMSD towards the known experimental structures [3]. The method demonstrates enhanced enrichment of near-native structures compared to all-atom MD, with a wider conformational search space [2]. Validation should include assessment of both global metrics (RMSD to native, radius of gyration) and local structure quality (favored rotamers, steric clashes, hydrogen bonding patterns).
Methodological Variations
Hierarchical Clustering Approach
For proteins with mixed structural motifs (α-helix and β-sheet), a "freeze and thaw" hierarchical clustering strategy can be employed where stable secondary structure elements are treated as rigid bodies while sampling torsional degrees of freedom in connecting regions [3]. This approach has shown improved sampling of near-native structures for the Trp-cage protein and aligns with zipping-and-assembly folding models [2].
Application to Different Structural Motifs
The diagram below illustrates the methodological relationships and applications of the all-torsion protocol across different protein systems:
Troubleshooting and Optimization
- Limited Structural Improvement: If refinement shows minimal RMSD improvement, extend simulation duration or adjust temperature spacing in the replica exchange ladder to enhance sampling efficiency.
- Secondary Structure Loss: For proteins experiencing disruption of native secondary structure, consider applying the hierarchical clustering approach to preserve stable structural elements during refinement.
- Sampling Barriers: Implement additional biasing potentials or targeted sampling if specific conformational transitions remain inaccessible within simulation timescales.
- Force Field Selection: Evaluate alternative force fields if structural quality metrics indicate systematic deviations from expected protein geometry.
The standard all-torsion protocol for protein structure refinement using the GNEIMO method provides a robust framework for enhancing the accuracy of protein structural models, with particular value for refining low-resolution homology models and enriching native-like conformational ensembles.
The accuracy of three-dimensional protein models is a critical factor for detailed mechanistic studies, including structure-based drug discovery, protein docking, and function prediction [10]. Pharmaceutical applications, in particular, often require structures with near-experimental accuracy [10]. While template-based modelling (TBM) methods can generate reliable initial models, these predicted 3D structures are often flawed with local and global errors such as irregular contacts, steric clashes, and unusual bond angles [10]. The refinement of these low-resolution homology models serves as the crucial final step in the structure prediction pipeline to bridge the gap towards experimental-level accuracy [10].
The challenge in protein structure prediction using homology modeling has historically been the lack of reliable methods to refine these initial models [3]. Traditional unconstrained all-atom molecular dynamics (MD) simulations often prove inadequate for structure refinement due to their limited conformational sampling capabilities and the risk of deviating from the native structural basin due to force-field inaccuracies [3] [10]. Within this context, the GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method emerges as an advanced constrained dynamics approach that addresses these limitations through enhanced conformational sampling in internal coordinates [3].
Theoretical Foundation of the GNEIMO Method
The GNEIMO method is a generalized constrained MD method that operates in internal coordinates, specifically designed for multibody dynamics of macromolecules [3]. Its fundamental innovation lies in replacing high-frequency degrees of freedom with hard holonomic constraints, modeling proteins as collections of rigid body clusters connected by flexible torsional hinges [3]. This theoretical framework offers several advantages over conventional Cartesian MD simulations:
- Reduced Degrees of Freedom: By constraining high-frequency vibrations, the method substantially decreases the number of degrees of freedom that need to be sampled [3].
- Enhanced Computational Efficiency: The constraint formulation enables larger integration time steps (typically 5 fs), significantly extending the effective simulation timescale [3].
- Expanded Conformational Search: The rigid body cluster representation enhances exploration of the conformational space relevant to protein folding and refinement [3].
The method's name derives from its mathematical foundation—the Generalized Newton-Euler Inverse Mass Operator algorithm—which efficiently solves the coupled equations of motion in internal coordinates with computational cost that scales linearly with the number of degrees of freedom, unlike conventional algorithms that scale cubically [3].
Experimental Protocols and Methodologies
GNEIMO Refinement Workflow
A robust protocol for protein structure refinement using the GNEIMO method involves the following key stages:
Initial Model Preparation
Generate low-resolution decoy sets through homology modeling using tools like MODELLER, selecting templates with 60-70% sequence identity to the target [3]. Cluster the resulting top 100 homology models by structural diversity and select representative structures with the most secondary structure content [3].
Model Swelling via Simulated Annealing
Perform simulated annealing with temperature ranging from 310 K to 1200 K in 50 K increments using all-torsion GNEIMO dynamics to expand the homology model to a lower resolution structure (target backbone RMSD range of 2-5 Å with respect to the native structure) [3]. Select multiple swollen snapshots from the trajectory for refinement.
Energy Minimization
Conduct unconstrained Cartesian MD energy minimization using 1000 steps of steepest descent method followed by 1000 steps of conjugate gradient method [3]. Utilize appropriate force fields (e.g., AMBER) with implicit solvent models (e.g., Generalized Born) and non-bond cutoffs of 20 Å [3].
GNEIMO Replica Exchange Molecular Dynamics (REXMD)
Execute the core refinement using all-torsion GNEIMO method coupled with REXMD with 8 replicas across a temperature range of 310 K to 415 K with 15 K intervals [3]. Run each replica for 5-15 ns (totaling 40-120 ns simulation time per decoy) with temperature exchanges occurring every 400 time steps (2 ps) [3]. Employ the Lobatto integrator with a 5 fs time step [3].
Hierarchical "Freeze and Thaw" Clustering Strategy
The GNEIMO method enables a unique hierarchical clustering approach where specific protein regions can be treated as rigid bodies while others remain fully flexible [3]. For mixed α/β motif proteins, either the α-helix or β-sheet motifs can be frozen as rigid bodies (backbone atoms only) while side chains and connecting regions sample torsional space [3]. This strategy reduces computational complexity while maintaining focused refinement on potentially problematic regions.
Performance Metrics and Quantitative Results
Refinement Efficacy Across Protein Classes
The GNEIMO constrained MD method has demonstrated significant improvement in structural accuracy across diverse protein architectures. In a systematic evaluation using eight proteins with different secondary structural motifs (all-α, α/β, and all-β), the method consistently enhanced model quality [3].
Table 1: GNEIMO Refinement Performance Across Protein Structural Classes
| Protein Structural Class | Number of Proteins Tested | Starting Decoy RMSD Range (Å) | Average RMSD Improvement (Å) | Key Observations |
|---|---|---|---|---|
| All-α | 3 | 2.0-5.0 | ~2.0 | Consistent improvement in helical packing and side-chain positioning |
| α/β | 3 | 2.0-5.0 | ~2.0 | Enhanced positioning of loop regions between secondary structural elements |
| All-β | 2 | 2.0-5.0 | ~2.0 | Improved β-sheet formation and strand alignment |
Comparative Sampling Efficiency
The conformational sampling efficiency of GNEIMO was quantitatively compared against traditional Cartesian MD simulations, revealing significant advantages in native-like conformation enrichment [3].
Table 2: Sampling Efficiency Comparison: GNEIMO vs. Cartesian MD
| Parameter | GNEIMO Constrained MD | Traditional Cartesian MD |
|---|---|---|
| Integration Time Step | 5 fs | Typically 1-2 fs |
| Degrees of Freedom | Reduced (torsional only) | Full Cartesian |
| Conformational Search | Enhanced | Limited |
| Native-like Enrichment | Significant | Minimal |
| Applicability to Refinement | Effective | Often worsens starting model [3] |
Research Reagent Solutions and Computational Tools
Successful implementation of the GNEIMO refinement protocol requires integration of specialized software tools and computational resources.
Table 3: Essential Research Reagents and Computational Tools for GNEIMO Refinement
| Tool/Resource | Type | Function in Refinement Protocol |
|---|---|---|
| GNEIMO Code | Constrained MD Engine | Core computational method for performing constrained dynamics in internal coordinates; enables all-torsion and hierarchical clustering simulations [3]. |
| AMBER Force Field | Molecular Mechanics Potential | Provides physics-based energy terms for bonded and non-bonded interactions; AMBER99 with GB/SA OBC implicit solvation recommended [3]. |
| MODELLER | Homology Modeling | Generates initial low-resolution decoy sets from templates with 60-70% sequence identity to target [3]. |
| REXMD Algorithm | Enhanced Sampling Method | Implements temperature replica exchange molecular dynamics to overcome energy barriers and enhance conformational sampling [3]. |
| GB/SA OBC Solvent Model | Implicit Solvation | Models solvent effects without explicit water molecules; uses interior dielectric 1.5, exterior dielectric 78.3, solvent probe radius 1.4 Å [3]. |
| Lobatto Integrator | Numerical Integration | Solves equations of motion for constrained dynamics; enables 5 fs time steps [3]. |
Applications in Drug Discovery and Structural Biology
The refinement of low-resolution homology models using GNEIMO has significant implications for structure-based drug discovery, particularly for challenging target classes:
- GPCR Drug Discovery: Accurate refined models enable structure-based hit identification and lead optimization for G protein-coupled receptors, a prominent drug target class [11].
- Binding Site Characterization: Refined models provide more accurate binding pocket geometries for virtual screening and ligand docking [12].
- SAR Rationalization: High-resolution refined structures facilitate understanding of structure-activity relationships in compound series [11].
The integration of AI-predicted structures from tools like AlphaFold2 with GNEIMO refinement presents a particularly powerful approach, as AI-generated models often require refinement of binding site geometries and side-chain conformations for effective drug discovery applications [11] [12].
Protocol Optimization and Troubleshooting
Critical Parameters for Success
- Temperature Selection: The 310-415 K range with 15 K intervals between replicas provides optimal balance between enhanced sampling and structural integrity [3].
- Simulation Duration: 5-15 ns per replica (40-120 ns total) typically suffices for observable refinement; excessively long simulations may not provide additional benefits [3] [10].
- Clustering Strategy Selection: Use all-torsion dynamics for global refinement and hierarchical "freeze and thaw" for localized improvements in specific secondary structure elements [3].
Common Challenges and Solutions
- Force Field Inaccuracies: Implement distance restraints from predicted contacts or experimental data to guide sampling toward native basin [10].
- Model Selection Difficulty: Combine energy-based scoring with model quality assessment programs (MQAPs) to identify most native-like conformations [10].
- Limited Improvement: Ensure starting decoys have sufficient structural diversity (RMSD 2-5 Å from native) to enable meaningful refinement [3].
The GNEIMO method for refining low-resolution homology models represents a significant advancement in computational structural biology, consistently delivering approximately 2 Å improvement in model accuracy toward experimental resolution and enabling more reliable protein structures for drug discovery and mechanistic studies [3].
The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method provides a transformative approach for ab initio folding simulations by employing constrained dynamics in internal coordinates. Unlike conventional all-atom Cartesian molecular dynamics (MD), which suffers from limited conformational sampling due to numerous high-frequency degrees of freedom, GNEIMO treats proteins as collections of rigid body clusters connected by flexible torsional hinges. This strategic reduction of degrees of freedom enables significantly enhanced conformational sampling and permits larger integration time steps, making it particularly suitable for studying protein folding from extended states [3] [2].
The method addresses a critical bottleneck in physics-based protein structure prediction: the inefficient sampling of the vast conformational space. Traditional unconstrained all-atom MD simulations often fail to achieve sufficient sampling for folding events within practical computational timeframes. GNEIMO overcomes this limitation through its internal coordinate framework, which focuses sampling on the essential low-frequency torsional degrees of freedom that primarily govern protein backbone rearrangements during folding [6]. This approach has demonstrated remarkable efficiency in folding diverse structural motifs, including α-helices, β-turns, and mixed-motif proteins, starting from completely extended conformations [2].
Experimental Protocols
All-Torsion Folding Protocol for Small Proteins
The all-torsion protocol employs the complete set of torsional degrees of freedom within the GNEIMO framework and is particularly effective for small proteins and peptides [2].
Initial System Preparation: Simulations commence from an extended conformation of the protein sequence. The initial structure undergoes conjugate gradient minimization with a convergence criterion of 10⁻² kcal/mol/Å in force gradient to eliminate steric clashes [2].
Force Field and Solvation: The AMBER parm99 force field is employed for energy calculations. An implicit solvation model (GB/SA OBC) is used to account for solvent effects, with an interior dielectric value of 1.75 for the solute and an exterior dielectric constant of 78.3 for the solvent. A solvent probe radius of 1.4 Å is used for non-polar solvation energy calculations [2].
Constrained Dynamics Parameters: Simulations utilize a Lobatto integrator with an integration time step of 5 fs—significantly larger than the 1-2 fs steps typical of Cartesian MD due to the constraint of high-frequency bond vibrations. Non-bonded forces are smoothly switched off at a cutoff radius of 20 Å [3] [2].
Enhanced Sampling with Replica Exchange: The replica exchange molecular dynamics (REXMD) method is integrated with GNEIMO to further enhance conformational sampling. Typically, 6-8 replicas are distributed across a temperature range of 325-500 K, with exchange attempts occurring every 2 ps. The reduced number of degrees of freedom in constrained MD models reduces the number of replicas required compared to Cartesian MD [2].
Hierarchical "Freeze and Thaw" Clustering Protocol
For more complex folding scenarios, GNEIMO enables hierarchical clustering schemes where specific protein regions can be "frozen" or "thawed" to guide the conformational search [3] [2].
Cluster Identification: Secondary structure elements (e.g., α-helices or β-sheets) identified from preliminary folding trajectories or sequence-based predictions are defined as rigid clusters. Only the backbone atoms within these motifs are frozen, while side chains remain fully flexible [3].
Dynamic Model Specification: The frozen clusters are connected to the rest of the protein via movable torsional degrees of freedom. The remaining protein regions continue to be sampled with all-torsion dynamics [3] [2].
Iterative Refinement: The hierarchical protocol may involve multiple cycles where different regions are frozen and thawed to systematically explore the folding landscape. This approach aligns with the "zipping-and-assembly" folding model and has demonstrated faster convergence to native-like structures [2].
Simulation Workflow
The following diagram illustrates the typical workflow for an ab initio folding simulation using the GNEIMO method:
Key Research Findings and Data
Quantitative Folding Performance
Extensive validation studies have demonstrated GNEIMO's effectiveness in ab initio folding simulations across diverse protein structural classes [2].
Table 1: GNEIMO Folding Performance Across Different Structural Motifs
| Protein/Peptide | Structural Motif | Simulation Time (per replica) | Key Results |
|---|---|---|---|
| Polyalanine (Ala₂₀) | α-helix | Up to 20 ns | Achieved high helicity content at 300K without elevated temperatures [2] |
| WALP16 | α-helix (membrane) | Up to 20 ns | Correct folding in membrane-mimetic environment (dielectric constant=40) [2] |
| Trp-cage | Mixed (α+β) | Up to 20 ns | Successful folding to near-native structures; hierarchical clustering showed superior sampling [2] |
| β-hairpin (1E0Q) | β-turn | Up to 20 ns | Formation of native-like β-turn conformations [2] |
Enhanced Sampling Efficiency
Comparative studies reveal significant advantages of GNEIMO over conventional Cartesian MD in sampling efficiency and native-state enrichment [3] [6].
Table 2: GNEIMO vs. Cartesian Molecular Dynamics for Protein Folding
| Parameter | GNEIMO Constrained MD | Cartesian All-Atom MD |
|---|---|---|
| Degrees of Freedom | ~10% of Cartesian MD [2] | All atomic coordinates |
| Integration Time Step | 5 fs [3] [2] | 1-2 fs |
| Sampling Enhancement | Enhanced torsional sampling; "Freeze and Thaw" capability [3] | Limited by high-frequency motions |
| REXMD Replicas | Fewer required due to reduced DOFs [2] | More replicas required |
| Native-State Enrichment | Higher population of near-native conformations [2] | Limited native-state sampling |
Table 3: Key Computational Tools and Parameters for GNEIMO Folding Simulations
| Resource | Specification/Version | Function in Protocol |
|---|---|---|
| GNEIMO Code | Custom implementation [3] | Core constrained MD simulation engine |
| Force Field | AMBER parm99/AMBER99 [3] [2] | Energy calculation and atomic interactions |
| Solvation Model | GB/SA OBC [3] [2] | Implicit solvent representation |
| REXMD Module | Integrated with GNEIMO [3] | Enhanced conformational sampling |
| Lobatto Integrator | 5 fs time step [2] | Numerical integration of equations of motion |
| Analysis Tools | Principal Component Analysis, K-means clustering [2] | Trajectory analysis and conformation classification |
Concluding Remarks
The GNEIMO method represents a significant advancement in physics-based protein folding simulations by addressing the fundamental challenge of conformational sampling. Its constrained dynamics framework enables efficient exploration of the protein folding landscape from extended states, making ab initio structure prediction more computationally tractable. The unique hierarchical clustering capabilities further enhance its utility for studying complex folding pathways. Validation across diverse structural motifs confirms GNEIMO's ability to enrich native-like conformations, providing researchers with a powerful tool for investigating protein folding mechanisms and energetics. As force fields continue to improve in balancing protein-water interactions and torsional parameters [13], the integration of these advancements with the GNEIMO methodology promises even more accurate and efficient protein folding simulations in the future.
Within the framework of the GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method for protein folding research, integrating enhanced sampling techniques is paramount for studying large-scale conformational changes. Torsional space molecular dynamics provides a powerful alternative to traditional Cartesian methods by constraining high-frequency bond vibrations and focusing computational resources on the low-frequency torsional degrees of freedom that dominate large-scale protein motions [8]. This approach enables longer integration time steps (e.g., 5 fs) and more efficient exploration of the conformational landscape [5]. The GNEIMO method implements internal coordinate molecular dynamics (ICMD) by modeling proteins as collections of rigid clusters connected by flexible torsional hinges, effectively reducing the system's dimensionality while maintaining physical accuracy [8]. This application note details protocols and case studies for integrating Replica Exchange MD (REMD) and Accelerated MD (aMD) within the GNEIMO framework to address the critical sampling challenges in protein folding, structure refinement, and conformational analysis.
Theoretical Framework and GNEIMO Advantages
Torsional Dynamics with the GNEIMO Method
The GNEIMO method represents a significant advancement in internal coordinate molecular dynamics by addressing longstanding challenges in computational efficiency and thermodynamic accuracy. Key innovations include:
- Spatial Operator Algebra (SOA): Provides low-cost algorithms that scale linearly with the number of degrees of freedom, compared to conventional cubic scaling [8]
- Fixman Potential: Compensates for systematic statistical biases introduced by hard constraints, ensuring correct equilibrium probability distributions [8]
- Modified Equipartition Principle: Enables thermodynamically correct velocity initialization in internal coordinates [8]
- Hierarchical Clustering: Allows users to define rigid bodies ranging from single atoms to entire protein domains, providing control over model granularity [8]
Enhanced Sampling Rationale in Torsional Space
Traditional all-atom MD simulations face limitations in crossing energy barriers, leading to inadequate sampling of functionally relevant states. The integration of REMD and aMD within the torsional space addresses these limitations through complementary mechanisms:
- REMD: Overcomes kinetic trapping by running parallel simulations at different temperatures and allowing configuration exchanges [14]
- aMD: Accelerates barrier crossing by adding a non-negative bias potential to energy minima, enhancing transitions between low-energy states [8]
- Torsional Focus: Both methods benefit from application in torsional space where essential conformational changes occur, reducing the ineffective sampling of high-frequency vibrations [6]
Table 1: Comparison of Enhanced Sampling Methods in GNEIMO
| Feature | REMD | aMD |
|---|---|---|
| Sampling Mechanism | Temperature-based configuration exchange | Bias potential applied to potential energy |
| Barrier Crossing | High-temperature replicas surmount barriers | Boost potential reduces effective barrier heights |
| Computational Cost | Higher (multiple replicas) | Lower (single replica) |
| Temperature Control | Multiple thermostats (e.g., Nosé-Hoover) | Single thermostat |
| Implementation in GNEIMO | Full integration with replica exchange manager | Available as bias potential module |
| Optimal Use Cases | Global folding, thermodynamic properties | Local transitions, conformational dynamics |
Integrated REMD-aMD Protocol in GNEIMO
System Setup and Preparation
Initial Structure Preparation:
- Obtain starting protein structure from homology modeling (e.g., MODELLER [5]) or experimental data
- Perform energy minimization using AMBER tools with AMBER99SB force field [5]
- Generate cluster definitions based on structural motifs:
- All-torsion model: All dihedral angles free
- Hierarchical model: Secondary structure elements as rigid clusters with flexible connecting regions [3]
- Define solvation parameters:
REMD-specific Parameters:
- Number of replicas: 16-64 (typically 32 for proteins up to 300 residues) [5]
- Temperature range: 310-415K with exponential distribution [5]
- Exchange attempt frequency: Every 2-5 ps [3]
- Replica sorting: Metropolis criterion based on potential energy [14]
aMD-specific Parameters:
- Boost potential definition:
- Total energy boost: Applied when E < Eₜₕᵣₑₛₕₒₗd
- Dihedral energy boost: Applied when Vₜₒᵣ < Vₜₕᵣₑₛₕₒₗd [8]
- Threshold estimation: Based on short conventional MD equilibration
Integrated Workflow Implementation
The following diagram illustrates the integrated REMD-aMD workflow within the GNEIMO framework:
Simulation Execution and Monitoring
Dynamics Configuration:
- Integrator: Lobatto integrator with 5 fs time step [5]
- Temperature control: Nosé-Hoover thermostat for each replica [8]
- Constraint handling: Hard holonomic constraints on rigid clusters
- Non-bonded interactions: Cutoff at 20Å with smooth switching [3]
- Simulation duration: 15-100 ns per replica (total 40-120 ns aggregate time) [3]
Convergence Monitoring:
- Track RMSD to native structure (if known)
- Monitor replica exchange acceptance rates (target: 20-30%)
- Calculate potential energy and radius of gyration time series
- Assess convergence via block analysis of thermodynamic quantities
Case Studies and Validation
Protein Structure Refinement of CASP Targets
The GNEIMO-REMD method has been extensively validated through the refinement of CASP (Critical Assessment of Structure Prediction) targets. In a comprehensive study of 30 CASP target proteins:
Table 2: GNEIMO-REMD Refinement Performance on CASP Targets [5]
| Target Protein | Starting RMSD (Å) | Refined RMSD (Å) | Refinement (Å) | Simulation Time (ns) |
|---|---|---|---|---|
| TR429 | 6.82 | 5.76 | 1.06 | 15-100 |
| TR435 | 2.14 | 1.65 | 0.49 | 15-100 |
| TR453 | 1.51 | 1.10 | 0.41 | 15-100 |
| TR454 | 3.26 | 2.00 | 1.26 | 15-100 |
| Average | 3.43 | 2.63 | 0.80 | 15-100 |
The protocol demonstrated consistent refinement of up to 1.3Å RMSD across diverse protein folds without using experimental restraints [5]. This represents significant improvement over traditional Cartesian MD, which often requires restraints to prevent structural deviation from the native state.
Conformational Dynamics of Flexible Proteins
Application of GNEIMO with enhanced sampling to conformationally flexible proteins reveals its capability to capture large-scale structural transitions:
Fasciculin Dynamics:
- GNEIMO-REMD simulations sampled two experimentally established conformational substates [6]
- Transitions between substates occurred within simulation timescales
- Cartesian MD simulations failed to sample these transitions without enhanced sampling [6]
Calmodulin Conformational Transition:
- GNEIMO successfully simulated the transition from Ca²⁺-bound to Ca²⁺-free conformation [6]
- Generated ensembles satisfied approximately 50% of both short- and long-range interresidue distances from NMR structures [6]
- Demonstrated efficient sampling of domain motions relevant to biological function
Performance Comparison with Cartesian MD
Table 3: GNEIMO-REMD vs. Cartesian MD for Structure Refinement
| Parameter | GNEIMO-REMD | Cartesian MD |
|---|---|---|
| Time Step | 5 fs | 1-2 fs |
| Refinement Without Restraints | Yes (up to 1.3Å improvement) | Limited (often requires restraints) |
| Sampling Efficiency | Enhanced in torsional space | Limited by high-frequency vibrations |
| Computational Cost | Lower per nanosecond due to reduced DOF | Higher due to more degrees of freedom |
| Application Size Range | Tested on 40-300 residue proteins | Limited by system size and timescale |
| Native-like Conformation Enrichment | Significant enrichment observed | Limited enrichment |
Research Reagent Solutions
Table 4: Essential Research Reagents and Computational Tools
| Reagent/Tool | Function | Implementation in GNEIMO |
|---|---|---|
| GneimoSim Software | ICMD simulation engine | Primary simulation platform with Python API [8] |
| AMBER99SB Force Field | Energy calculation | Primary force field for protein interactions [5] |
| GB/SA OBC Solvation Model | Implicit solvent treatment | Default solvation model for efficiency [5] |
| Lobatto Integrator | Numerical integration of equations of motion | Provides stable long-timescale integration [8] |
| LAMMPS/OpenMM/Rosetta | External force field packages | Interfaced for specialized energy calculations [8] |
| Temperature REPLICA | Replica exchange management | Manages configuration exchanges between replicas [5] |
| Fixman Potential | Statistical correction | Compensates for constraint-induced bias [8] |
Technical Implementation Details
Temperature Replica Exchange Mechanism
The REMD algorithm in GNEIMO employs a temperature-based exchange process illustrated below:
The acceptance probability for exchange between replicas i and j is given by:
[ P{\text{accept}} = \min\left(1, \exp\left[(\betai - \betaj)(Ei - E_j)\right]\right) ]
where β = 1/kBT and E is the potential energy of the configuration [14].
aMD Boost Potential Formulation
In the accelerated MD implementation, the modified potential energy is defined as:
[ V^\ast(r) = V(r) + \Delta V(r) ]
where the boost potential ΔV(r) is applied when the potential energy falls below a threshold:
[ \Delta V(r) = \frac{(E - V(r))^2}{\alpha + (E - V(r))} ]
Parameters E and α determine the strength and shape of the boost potential [8].
The integration of REMD and aMD within the GNEIMO torsional dynamics framework provides a powerful platform for addressing challenging problems in protein folding and conformational dynamics. The method leverages the inherent advantages of internal coordinates—reduced dimensionality, larger time steps, and focused sampling on functionally relevant degrees of freedom—while overcoming sampling limitations through sophisticated enhanced sampling techniques. Validation on CASP targets and flexible protein systems demonstrates the robustness of this approach for protein structure refinement and dynamics studies. The modular architecture of GneimoSim and its interfaces with widely used force field packages make this integrated approach accessible to researchers studying biomolecular systems across a range of scientific and therapeutic applications.
Hierarchical 'Freeze and Thaw' Clustering for Targeted Domain Motions
Understanding the conformational dynamics of proteins is crucial for elucidating their biological function and for rational drug design. The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method is an advanced torsional dynamics simulation technique that enhances the sampling of protein conformational dynamics by focusing on low-frequency torsional degrees of freedom. This approach enables the simulation of long-timescale domain motions that are challenging to capture with conventional Cartesian molecular dynamics [15].
This application note details a protocol that integrates the GNEIMO method with hierarchical clustering—specifically a "Freeze and Thaw" approach—to systematically identify, classify, and analyze functionally relevant domain motions in proteins. This methodology provides a powerful framework for researchers and drug development professionals to decode complex protein dynamics, which can inform the identification of allosteric sites and the design of targeted therapeutics.
Theoretical Foundation: GNEIMO Torsional Dynamics
The GNEIMO method is a multi-body dynamics method formulated in the space of internal torsion angles of proteins. Unlike Cartesian molecular dynamics, which can be computationally limited for studying millisecond-timescale events, GNEIMO enhances conformational sampling by restricting the exploration to the essential torsional degrees of freedom. This allows for more efficient simulation of large-scale conformational changes [15].
Key features of the GNEIMO method include:
- It performs conformational search in the low-frequency torsional degrees of freedom, which are the primary drivers of large-scale protein motion.
- It has been successfully applied to study conformationally flexible proteins like fasciculin and calmodulin, capturing transitions between experimentally known conformational substates that are difficult to sample with unconstrained Cartesian simulations [15].
- The method can be combined with replica exchange protocols to further improve the sampling of complex energy landscapes.
Hierarchical 'Freeze and Thaw' Clustering Protocol
The "Freeze and Thaw" clustering protocol is an agglomerative hierarchical approach designed to build a tree of conformational states from GNEIMO simulation trajectories. This reveals the relationships between different metastable states and the pathways connecting them.
The following diagram illustrates the complete experimental workflow, from simulation to analysis:
Step-by-Step Experimental Protocol
Phase 1: Trajectory Generation and Preprocessing
- System Setup: Prepare the protein structure file (e.g., PDB format). Define the torsional degrees of freedom to be active during the simulation.
- GNEIMO Simulation: Perform torsional dynamics simulations using the GNEIMO method. The simulation should be long enough to sample the conformational transition of interest. Using replica exchange GNEIMO is recommended for enhanced sampling [15].
- Trajectory Alignment: Superimpose all saved trajectory frames onto a reference structure (e.g., the initial crystal structure) using a stable domain as a reference to remove global rotation and translation.
- Feature Extraction: Calculate a set of features that describe the conformational state for each frame. Recommended features include:
- The values of key dihedral angles (backbone and sidechain) in domains of interest.
- Inter-residue distances or domain-level distances between centers of mass.
Phase 2: The "Freeze" Phase - Defining Microstates
- Initial Clustering: Perform a fine-grained clustering (e.g., using K-Means with a high k value) on the entire set of preprocessed trajectory frames. The goal is to group structurally similar conformations into a large number of microstates.
- Representative Structure: For each resulting microstate cluster, calculate its centroid (the frame closest to the geometric center of the cluster). This "freezes" the continuous trajectory into a discrete set of representative conformations.
Phase 3: The "Thaw" Phase - Hierarchical Agglomeration
This phase uses agglomerative hierarchical clustering, a bottom-up approach that starts by treating each microstate as a singleton cluster and successively merges the most similar pairs until only one cluster remains [16] [17].
- Compute Distance Matrix: Calculate an N×N distance matrix, where N is the number of microstate centroids. The distance between two centroids should be a Root Mean Square Deviation (RMSD) calculated over the Cα atoms of the flexible domains.
- Merge with Linkage Criterion: Identify the two closest clusters and merge them. The distance between clusters is defined by the linkage criterion. Ward's method is recommended as it minimizes the increase in total within-cluster variance, tending to create compact, spherical clusters [16].
- Update and Iterate: Update the distance matrix to reflect the new cluster and repeat the merging process until all microstates belong to a single cluster.
- Construct Dendrogram: Record the entire merging process and distances at which merges occur to build a dendrogram—a tree diagram visualizing the hierarchical relationships and dissimilarity between clusters [17].
Phase 4: Analysis and State Identification
- Cut the Dendrogram: Analyze the dendrogram to determine the optimal number of major conformational states. This is done by drawing a horizontal line across the longest vertical line(s) that do not intersect any clusters; the number of vertical lines intersected indicates the number of significant clusters [16].
- Characterize States: For each major cluster (macrostate), analyze the representative structures to characterize the domain motions. This includes quantifying the range of motion, hinge points, and population of each state.
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Essential computational tools and resources for implementing the protocol.
| Item Name | Function/Description | Example/Note |
|---|---|---|
| GNEIMO MD Package | Software to perform torsional dynamics simulations. Enhances sampling of conformational changes by focusing on torsional degrees of freedom [15]. | Custom or academic software. |
| Trajectory Analysis Suite | Software library for aligning trajectories, calculating RMSD, and extracting features. | MDAnalysis (Python), CPPTraj (Amber). |
| Clustering Library | Software providing implementations of K-Means and hierarchical clustering algorithms, plus dendrogram visualization. | Scikit-learn, SciPy (Python). |
| Molecular Viewer | Visualization software to inspect and analyze 3D protein structures and conformational states. | PyMOL, UCSF Chimera, VMD. |
| High-Per Computing (HPC) | Computer clusters are essential for running long GNEIMO simulations and processing large trajectory datasets. | Cloud-based or local institutional HPC resources. |
Data Presentation and Analysis
Quantitative Analysis of Clustering
The following table summarizes key quantitative parameters and results that should be extracted from the protocol for a typical study on a two-domain protein.
Table 2: Summary of quantitative data from a hierarchical clustering analysis of protein domain motions.
| Parameter | Description | Example Value for Calmodulin-like System |
|---|---|---|
| Simulation Length | Total time of the GNEIMO simulation. | 100 ns |
| Number of Microstates | Initial fine-grained clusters from the "Freeze" phase. | 250 |
| Optimal Macrostates | Number of major conformational states identified from the dendrogram. | 4 |
| Major State Population | Percentage of simulation frames assigned to each major state. | State 1: 45%, State 2: 30%, State 3: 15%, State 4: 10% |
| Inter-Domain RMSD Range | The range of RMSD values observed between the open and closed states. | 5.0 Å – 12.5 Å |
| Key Hinge Residues | Residues identified as the center of rotational domain motion. | 75, 78, 82 |
Visualizing the Clustering Logic
The logic of the "Freeze and Thaw" clustering process, from microstates to a hierarchy of macrostates, can be visualized as follows:
Application in Drug Development
For drug development professionals, this protocol offers a strategic advantage. By mapping the hierarchy of conformational states, one can identify cryptic allosteric pockets that are absent in static crystal structures but present in low-population, dynamically sampled states. The quantitative data on state populations and transition pathways can guide the design of stabilizers or inhibitors that trap a specific conformational state, enabling highly targeted therapeutic strategies. Integrating these computational insights with experimental validation creates a powerful pipeline for accelerating structure-based drug discovery.
Optimizing Simulations: Overcoming Challenges and Enhancing GNEIMO Performance
Internal Coordinate Molecular Dynamics (ICMD) represents a powerful alternative to traditional Cartesian coordinate simulations for studying biomolecular systems. By using bond lengths, bond angles, and torsion angles (BAT) as natural coordinates for describing molecular structure, ICMD offers significant advantages for conformational sampling. The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method is an advanced ICMD approach that enables efficient simulation of protein dynamics by focusing computational resources on the low-frequency torsional degrees of freedom most relevant to large-scale conformational changes [18] [8].
A longstanding challenge in constrained dynamics methods, including torsional MD where all bond lengths and bond angles are held rigid, has been the introduction of systematic statistical biases into simulations. These biases adversely affect the calculated thermodynamic and kinetic properties, potentially leading to inaccurate predictions of protein behavior [19]. The Fixman potential provides a rigorous mathematical framework for compensating these constraint-induced biases, thereby restoring the correct statistical mechanical behavior in ICMD simulations [18] [19].
This application note details the theoretical foundation, practical implementation, and experimental validation of the Fixman potential within the GNEIMO ICMD framework, providing researchers with protocols for addressing statistical bias in protein folding and dynamics studies.
Theoretical Foundation
Statistical Bias in Constrained Dynamics
In unconstrained molecular dynamics simulations using BAT coordinates, the probability density function ρ(α,q) for the configuration coordinates is proportional to the square root of the determinant of the mass matrix multiplied by the Boltzmann factor:
ρ(α,q) ∝ [det M_B (α,q)]^1/2 e^(-U(α,q)/kT [19]
When rigid constraints are applied to freeze the high-frequency bond length and bond angle degrees of freedom (denoted as q), the configuration space partition function for the constrained model becomes:
Z(T) = c3 ∫ dα det M^1/2 (α) e^(-U(α,q0)/kT [19]
The critical issue arises because the mass matrix determinant det M(α) for the constrained system differs from its counterpart in the flexible system, leading to systematic biases in the probability distribution of the remaining torsional degrees of freedom (α) [19]. This bias manifests as altered probability density functions for conformational states, incorrect transition barrier crossing rates, and distorted free energy surfaces [18].
The Fixman Compensation Potential
Fixman proposed a compensating potential to correct for these statistical biases introduced by rigid constraints. The Fixman potential (U_F) is defined as:
U_F = (1/2) kT ln[det M(α)] [19]
When this potential is included in the dynamics, the partition function for the constrained system becomes:
Z(T) = c3 ∫ dα e^[-(U(α,q0) + U_F(α))/kT]
The inclusion of U_F effectively compensates for the bias introduced by the constraints, restoring the correct statistical mechanical behavior [19]. For torsional MD simulations, this means that the probability distribution functions of conformational states, transition barrier crossing rates, and free energy surfaces align more closely with those obtained from unconstrained all-atom Cartesian simulations [18].
Computational Implementation in GNEIMO
Spatial Operator Algebra Framework
The GNEIMO method implements the Fixman potential using spatial operator algebra (SOA), a mathematical framework originally developed for spacecraft and robot dynamics [18] [19]. This approach overcomes the historical computational bottleneck associated with calculating the Fixman potential for large, branched molecules.
Key innovations of the GNEIMO-Fixman implementation include:
- Recursive Algorithms: The computational cost scales linearly with the number of degrees of freedom, instead of the cubic scaling of prior methods [19]
- General Topology Support: Capability to handle both serial chains and complex branched molecules of arbitrary size [19]
- Gradient Calculation: Efficient computation of Fixman potential partial derivatives (Fixman torque) for dynamics simulations [19]
The SOA-based implementation makes the inclusion of the Fixman potential computationally tractable for protein systems, with only a modest increase in computational cost compared to standard ICMD simulations [19].
GneimoSim Software Architecture
The GneimoSim software package provides a comprehensive implementation of the GNEIMO-Fixman method with the following capabilities:
Table 1: GneimoSim Software Features and Capabilities
| Feature Category | Specific Capabilities | Supported Molecular Systems |
|---|---|---|
| Dynamics Methods | Torsional MD, Hybrid ICMD, Langevin dynamics | Proteins, polymeric materials |
| Enhanced Sampling | Temperature replica exchange (REMD), Accelerated MD (aMD) | Proteins of 40-300 residues |
| Thermostats | Nosé-Hoover NVT method | All supported systems |
| Integrators | Runge-Kutta, Lobatto, adaptive CVODE, Verlet | Long timescale (microseconds) |
| Solvation Models | Generalized Born (GB/SA), Periodic boundary conditions | Implicit and explicit solvent |
| Force Field Interfaces | LAMMPS, OpenMM, Rosetta | Custom force field support |
GneimoSim's modular architecture allows researchers to leverage established force field packages while utilizing the advanced ICMD capabilities of the GNEIMO method [8]. The software includes a comprehensive Python interface to the underlying C++ classes, enabling flexible configuration of simulation parameters and control of simulation flow [8].
Experimental Protocols
Protocol 1: ICMD with Fixman Potential for Protein Structure Refinement
Application: Refinement of protein homology models to higher accuracy without experimental restraints [5]
Step-by-Step Procedure:
System Setup
- Obtain starting decoy structure from homology modeling (e.g., using MODELER)
- Perform all-atom conjugate gradient minimization using AMBER99SB force field
- Define cluster topology based on desired granularity (default: rigid bodies connected by torsional hinges)
Simulation Parameters
- Force Field: AMBER99SB with GB/SA OBC implicit solvation model
- Interior dielectric: 1.5, Exterior dielectric: 78.3
- Solvent probe radius: 1.4 Å for nonpolar solvation energy
- Nonbonded forces cutoff: 20 Å with switching function
- Integrator: Lobatto with 5 fs time step
- Temperature control: Nosé-Hoover method
Enhanced Sampling Configuration
- Replica Exchange MD (REXMD) with 32 replicas
- Temperature range: 310-415 K
- Exchange attempts every 5 ps using Metropolis criterion
- Total simulation time: 15-100 ns per replica
Fixman Potential Activation
- Enable Fixman compensation in GneimoSim input parameters
- Set Fixman potential update frequency (typically every dynamics step)
- Verify Fixman torque application in simulation log output
Trajectory Analysis
- Extract lowest energy structure from ensemble
- Calculate RMSD and GDT_TS scores against reference structure
- Compare probability distributions of torsion angles with and without Fixman potential
Validation Metrics: Successful refinement demonstrates improvement in GDT_TS scores and reduction in RMSD compared to starting models, with typical refinement of up to 1.3 Å RMSD reported for CASP target proteins [5].
Protocol 2: Bias Validation in Model Systems
Application: Quantitative evaluation of Fixman potential effectiveness in removing statistical bias [19]
Step-by-Step Procedure:
System Preparation
- Select model systems of increasing complexity (butane → pentane → alanine dipeptide)
- Generate initial configurations with varied torsion angles
- Define constrained model with rigid bonds and angles
Simulation Setup
- Force Field: Bond angle and bond length potentials only (no torsion potential)
- Dynamics: Langevin dynamics with low friction coefficient
- Temperature: 300 K
- Simulation time: Sufficient to achieve convergence in torsion distributions
Comparative Simulations
- Case A: Constrained dynamics without Fixman potential
- Case B: Constrained dynamics with Fixman potential
- Case C: Unconstrained dynamics as reference
Data Collection
- Record torsion angle values throughout trajectory
- Compute probability distribution functions for each torsion
- Calculate potential of mean force from distributions
Analysis
- Compare distributions from Cases A and B against reference (Case C)
- Quantify deviation from uniform distribution for systems without torsion potential
- Assess recovery of correct statistical behavior with Fixman potential
Expected Outcome: With Fixman potential, torsion angle distributions should approach the uniform distribution expected for systems without torsion potentials, demonstrating effective bias removal [19].
Visualization of Method Integration
The following workflow diagram illustrates how the Fixman potential integrates within the GNEIMO ICMD method:
GNEIMO-Fixman ICMD Workflow: Integration of the Fixman potential (red) within the constrained dynamics simulation loop.
Research Reagent Solutions
Table 2: Essential Research Reagents and Computational Tools for GNEIMO-Fixman Studies
| Reagent/Tool | Function/Purpose | Implementation Notes |
|---|---|---|
| GneimoSim Software | Primary ICMD simulation platform with Fixman potential support | Modular architecture with Python API; interfaces with external force fields [8] |
| Spatial Operator Algebra (SOA) | Mathematical framework for efficient mass matrix operations | Enables linear scaling of Fixman potential computation [18] [19] |
| AMBER99SB Force Field | Protein force field for potential energy calculations | Compatible with GNEIMO method; parameterized for BAT coordinates [5] |
| GB/SA Solvation Model | Implicit solvent treatment for biomolecular simulations | OBC variant used with interior dielectric 1.5, exterior 78.3 [5] |
| REMD Framework | Enhanced sampling for conformational exploration | 32 replicas across 310-415 K temperature range; exchange every 5 ps [5] |
| Lobatto Integrator | Numerical integration of equations of motion | Supports 5 fs time steps in constrained dynamics [5] |
Applications and Validation
Protein Structure Refinement
The GNEIMO-Fixman method has been successfully applied to refine protein homology models for 30 CASP target proteins, demonstrating refinement of up to 1.3 Å in RMSD without using experimental data as restraints [5]. This represents significant improvement over unrestrained all-atom Cartesian MD simulations, which typically require restraints to achieve similar refinement.
Table 3: Representative Refinement Results for CASP Targets Using GNEIMO-REXMD
| Target Protein | Starting GDT_TS | Refined GDT_TS | RMSD Improvement (Å) |
|---|---|---|---|
| TR429 | 31.5 | 45.7 | 1.06 |
| TR435 | 80.2 | 87.9 | 0.49 |
| TR453 | 86.6 | 91.5 | 0.41 |
| TR454 | 58.5 | 71.0 | 1.26 |
Recovery of Correct Statistics
Validation studies on molecules of increasing complexity demonstrate that the Fixman potential effectively recovers the expected probability distribution functions for torsion angles [19]. In systems with only bond angle and bond length potentials, the inclusion of the Fixman potential restores the uniform distribution of torsion angles that is characteristic of unconstrained systems, thereby annulling the biases caused by constraining bond lengths and angles [19].
The GNEIMO-Fixman method represents a significant advancement in constrained dynamics, enabling researchers to leverage the sampling efficiency of ICMD while maintaining the statistical accuracy required for reliable thermodynamic and kinetic predictions in protein folding research and drug development.
The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) constrained molecular dynamics method addresses a significant challenge in protein structure prediction by enhancing the refinement of low-resolution homology models. A critical component of its success is the "Freeze and Thaw" clustering strategy, which involves the strategic selection of rigid bodies within a protein to enhance conformational sampling. This application note provides a detailed protocol for identifying and selecting these rigid bodies, framed within the broader context of using torsional dynamics for protein folding research. We summarize quantitative performance data, provide step-by-step methodologies for implementing hierarchical clustering, and visualize the underlying logic and workflows to aid researchers in effectively applying this technique for protein structure refinement and drug development.
Conventional all-atom molecular dynamics (MD) in Cartesian coordinates is often ineffective for refining low-resolution protein structural models due to its limited conformational search capabilities [3]. The GNEIMO method overcomes this by employing an internal coordinates molecular dynamics (ICMD) approach, where a protein is modeled as a collection of rigid bodies (clusters) connected by flexible torsional hinges [3] [8]. This formulation allows for the replacement of high-frequency degrees of freedom with hard holonomic constraints, enabling larger integration time steps and a more efficient exploration of the conformational landscape [3].
The 'Freeze and Thaw' dynamics is an advanced strategy within the GNEIMO framework that allows the user to guide the dynamics by controlling the granularity of the protein model [3]. Specifically, parts of the protein can be "frozen" into rigid bodies, reducing the number of active degrees of freedom, while other parts are "thawed" and sampled with full torsional flexibility. This hierarchical clustering is particularly valuable for refining low-resolution decoys derived from homology modeling, where it has been shown to achieve improvements of approximately 2 Å in RMSD to known experimental structures [3] [20]. The ability to selectively freeze stable structural motifs enables a more targeted and computationally efficient conformational search, making it a powerful tool for researchers and drug development professionals focused on obtaining high-quality protein models.
Core Principles of Rigid Body Selection
The process of selecting which parts of a protein to freeze is central to the effectiveness of the protocol. The following principles, derived from application studies, guide this selection to enhance structural refinement.
- Focus on Secondary Structural Elements (SSEs): The most suitable candidates for rigid clusters are well-defined, stable secondary structures such as α-helices and β-sheets [3]. These elements typically maintain their structural integrity during dynamics and can be treated as single, cohesive units. This approach reduces the number of degrees of freedom without sacrificing the accuracy of the conformational search.
- Target Mixed-Motif Proteins: The 'Freeze and Thaw' strategy is particularly advantageous for proteins with mixed α-helix and β-sheet motifs [3]. For such proteins, one can freeze either the α-helical or β-sheet motifs as rigid bodies while leaving the rest of the protein, including connecting loops and side chains, fully flexible. This allows the simulation to focus sampling on the more dynamic and often less-predicted regions of the protein.
- Utilize Experimental and Predictive Data: Whenever available, use high-resolution experimental structures (e.g., from X-ray crystallography or NMR) or high-confidence predictive models to identify stable regions [3]. The initial clustering can be based on the secondary structure assignment from these reference models.
- Balance Rigidity and Flexibility: The goal is not to freeze the entire protein, but to find an optimal balance. Over-constraining the system by freezing too many clusters can hinder necessary conformational adjustments. Conversely, freezing too little may not provide sufficient sampling enhancement. A thoughtful, hierarchical approach is required.
Quantitative Performance of Clustering Strategies
The table below summarizes the performance of different GNEIMO dynamics protocols in protein structure refinement studies, demonstrating the effectiveness of the method.
Table 1: Performance of GNEIMO Dynamics in Protein Structure Refinement
| Protein Motif Type | Refinement Protocol | Starting RMSD (Å) | Final RMSD (Å) | Improvement (Å) | Key Observation |
|---|---|---|---|---|---|
| Various (All-α, α/β, All-β) | All-Torsion GNEIMO REXMD [3] | 2-5 | ~2 | ~2 | Enrichment of native-like conformations [3]. |
| α/β Mixed Motif | Hierarchical 'Freeze and Thaw' Clustering [3] | Information Not Specified | Information Not Specified | Information Not Specified | Enhanced localized conformational search; fewer degrees of freedom than all-torsion dynamics [3]. |
| 30 CASP Targets | GNEIMO-REMD with Fixman Potential [8] | Information Not Specified | ≤ 1.5 | Information Not Specified | Refinement achieved without experimental restraints [8]. |
The data shows that the GNEIMO method is a robust tool for structure refinement across different protein motifs. The hierarchical 'Freeze and Thaw' approach provides a specialized strategy for mixed-motif proteins, offering a pathway to high-resolution models.
Experimental Protocol: Implementing Hierarchical Clustering
This section provides a detailed, step-by-step protocol for implementing a 'Freeze and Thaw' simulation for protein structure refinement using the GneimoSim software package [8].
Pre-processing and System Setup
- Initial Structure Preparation: Obtain the low-resolution protein decoy to be refined. Decoys can be generated through homology modeling with tools like MODELLER [3].
- Force Field and Solvation: Set up the system using a compatible force field (e.g., AMBER99 [3]) and an implicit solvation model, such as the Generalized Born/Surface Area (GB/SA) model [3] [8].
- Energy Minimization: Perform initial energy minimization of the starting decoy using a method like steepest descent followed by conjugate gradient to remove any steric clashes [3].
Defining Rigid Clusters (Freezing)
- Secondary Structure Analysis: Analyze the initial protein model to identify regions of stable secondary structure (α-helices and β-strands/sheets).
- Cluster Definition:
- For α-helices: Define each continuous α-helix as a single rigid cluster. The backbone atoms within the helix are frozen relative to each other.
- For β-sheets: Define a cluster comprising all β-strands that form a single, continuous β-sheet. This treats the entire sheet as a single rigid unit.
- Side Chain Treatment: The side chains of amino acids within a frozen cluster can be treated as flexible (all-torsion) to allow for local packing adjustments [3].
- Hinge Definition: The torsional degrees of freedom connecting these large, rigid clusters are defined as the flexible hinges. The rest of the protein (loops, termini, etc.) is treated as fully flexible.
Diagram: Logical decision process for defining rigid clusters in a protein structure.
Running 'Freeze and Thaw' Dynamics
- Simulation Configuration: In GneimoSim, configure the simulation to use the defined cluster model. Employ the Lobatto integrator with a time step of 5 fs [3].
- Enhanced Sampling: To improve conformational sampling, use the Replica Exchange MD (REXMD) algorithm. A typical setup uses 8 replicas spanning a temperature range of 310 K to 415 K [3].
- Simulation Execution: Run the simulation, allowing exchanges between replicas periodically (e.g., every 2 ps). The total simulation time per replica may range from 5 ns to 15 ns, amounting to 40-120 ns of aggregate sampling [3].
- Trajectory Analysis: After completion, analyze the trajectories from all replicas. Calculate the backbone Root-Mean-Square Deviation (RMSD) of the sampled structures relative to a known experimental reference structure to assess refinement. Monitor the population of native-like conformations.
Diagram: Workflow for executing a GNEIMO 'Freeze and Thaw' simulation with replica exchange.
The Scientist's Toolkit: Essential Research Reagents and Software
The following table details key software tools and computational resources required to implement the described protocols.
Table 2: Essential Research Reagent Solutions for GNEIMO Simulations
| Item Name | Function/Brief Explanation | Example/Reference |
|---|---|---|
| GneimoSim Software Package | The primary software for performing Internal Coordinates MD (ICMD) simulations using the GNEIMO method. | [8] |
| Homology Modeling Tool | Generates initial low-resolution protein decoy structures from a target sequence and template. | MODELLER [3] |
| Force Field | Provides the potential energy functions and parameters for the simulation. | AMBER99 [3] |
| Implicit Solvation Model | Efficiently models the effect of solvent (water) on the protein without explicit water molecules. | Generalized Born/Surface Area (GB/SA) [3] [8] |
| Analysis and Visualization Software | Used for visualizing protein structures, defining clusters, and analyzing simulation trajectories (e.g., RMSD calculation). | VMD, PyMOL, MDTraj |
The strategic selection of rigid bodies is a cornerstone of applying the 'Freeze and Thaw' dynamics within the GNEIMO framework. By focusing on stable secondary structural elements as rigid clusters, researchers can significantly enhance the efficiency and effectiveness of conformational sampling for protein structure refinement. The detailed protocols, performance data, and visual workflows provided in this application note offer a practical guide for scientists to implement this powerful technique, thereby advancing research in protein folding, structure prediction, and rational drug design.
The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method addresses a fundamental challenge in internal coordinate molecular dynamics (ICMD): the thermodynamically correct initialization of velocities. Traditional Cartesian molecular dynamics simulations benefit from a straightforward relationship between velocity initialization and temperature. In contrast, ICMD models, where high-frequency degrees of freedom are constrained, require a specialized approach to avoid statistical biases in sampling conformational states. The GNEIMO method introduces a new equipartition principle that generalizes the classical concept to internal coordinate models, forming the foundation for rigorous velocity initialization in torsional dynamics simulations of proteins [8].
This principle is particularly crucial for protein folding research and refinement, as proper thermalization ensures accurate exploration of the free energy landscape. The equipartition principle enables the definition of "modal velocity coordinates" that provide a mathematically sound method for initializing velocities in ICMD simulations, ensuring that the resulting conformational sampling adheres to correct thermodynamic distributions [8]. This theoretical advancement eliminates systematic errors that could otherwise propagate through long-timescale simulations of protein dynamics and folding pathways.
Performance Data for GNEIMO in Protein Structure Refinement
The GNEIMO method with proper velocity initialization has demonstrated significant success in protein structure refinement applications. The following table summarizes key quantitative results from studies on CASP target proteins:
Table 1: GNEIMO Refinement Performance on CASP Target Proteins
| Metric | Performance Range | Experimental Context |
|---|---|---|
| RMSD Improvement | Up to 1.3-2.0 Å reduction [5] [3] | 30 CASP8 & CASP9 targets; 8 protein test set [5] [3] |
| Simulation Time | 15-100 ns per replica [5] | 32 replicas in REXMD [5] |
| Temperature Range | 310-415 K [5] | Temperature replica exchange MD [5] |
| Time Step | 5 fs [5] [3] | Enabled by rigid cluster constraints [5] [3] |
Table 2: Refinement of CASP Target Structures Using GNEIMO-REXMD
| CASP Target | Starting GDT_TS | Best GNEIMO GDT_TS | Best CASP GDT_TS | RMSD Improvement (Å) |
|---|---|---|---|---|
| TR429 | 31.5 | 45.7 | 39.8 | 1.06 [5] |
| TR435 | 80.2 | 87.9 | 83.4 | 0.49 [5] |
| TR453 | 86.6 | 91.5 | 86.6 | 0.41 [5] |
| TR454 | 58.5 | 71.0 | 60.2 | ~1.26 [5] |
The data demonstrates that GNEIMO consistently refines protein models beyond the best CASP submissions, achieving substantial improvements in both global distance test (GDT_TS) scores and root-mean-square deviation (RMSD) values. This performance highlights the effectiveness of the torsional dynamics approach with proper thermodynamic initialization.
Experimental Protocol: GNEIMO-REXMD for Protein Structure Refinement
System Preparation and Equilibration
- Starting Structure Selection: For structure refinement targets, obtain decoy structures from the CASP repository (
predictioncenter.org). For de novo structure prediction, generate homology models using MODELLER with templates of 30-80% sequence identity, excluding the target protein and close homologues [5]. - Initial Clustering: Generate 100 models using MODELLER and cluster them into 5 groups based on structural diversity. Select the representative structure with the best procheck G-factor from each cluster [5].
- Energy Minimization: Perform all-atom conjugate gradient minimization using the AMBER
sanderprogram with the AMBER99SB force field to remove steric clashes and prepare the structure for dynamics [5].
GNEIMO-REXMD Simulation Parameters
Force Field and Solvation:
- Employ the AMBER99SB force field [5] [3].
- Use the Generalized Born/Surface Area (GB/SA) OBC implicit solvation model [5] [3].
- Set interior dielectric to 1.5 and exterior dielectric to 78.3 [5] [3].
- Apply a solvent probe radius of 1.4 Å for nonpolar solvation energy [5] [3].
- Implement a nonbonded force cutoff of 20 Å with a smooth switching function [5] [3].
Dynamics and Sampling:
- Apply the GNEIMO torsional MD method with all torsional degrees of freedom [5].
- Apply the Nosé-Hoover thermostat for constant temperature dynamics [5] [8].
- Use the Lobatto integrator with a 5 fs time step [5] [8].
- Implement Temperature Replica Exchange MD (REXMD) with 32 replicas [5].
- Set temperature range from 310 K to 415 K [5].
- Perform temperature exchange attempts every 2-5 ps (400-1000 steps) using the Metropolis criterion [5] [3].
Simulation Duration:
Trajectory Analysis and Structure Selection
- Conformation Sampling: Collect structures from the lowest temperature replica at regular intervals throughout the simulation.
- Ensemble Analysis: Cluster the collected structures and select representative conformations from the largest clusters.
- Validation: Evaluate refined structures using RMSD, GDT_TS, TM-score, and MolProbity validation metrics.
Research Workflow and Logical Relationships
The following diagram illustrates the complete GNEIMO protein refinement protocol, from system preparation to final structure selection:
Table 3: Essential Research Reagents and Computational Resources for GNEIMO Simulations
| Resource | Type | Function/Purpose |
|---|---|---|
| GneimoSim Package | Software | Primary ICMD simulation platform implementing GNEIMO method [8] |
| AMBER99SB Force Field | Parameter Set | Physics-based energy function for protein interactions [5] [3] |
| GB/SA OBC Solvent Model | Solvation Method | Implicit solvation for efficient aqueous environment simulation [5] [3] |
| MODELER | Software | Homology modeling for generating initial protein structures [5] [3] |
| LAMMPS/OpenMM/Rosetta | Software | Optional external force field interfaces [8] |
| Temperature Replica Exchange | Algorithm | Enhanced conformational sampling across energy barriers [5] [8] |
The GneimoSim package provides the core infrastructure for implementing the equipartition principle and conducting torsional dynamics simulations. Its modular architecture allows integration with established force field packages while maintaining the theoretical rigor of the internal coordinates approach. The combination of these tools enables researchers to apply the GNEIMO method to challenging problems in protein structure prediction, refinement, and folding pathway characterization.
Force Field and Solvation Model Selection for Stable Long-Timescale Simulations
Long-timescale molecular dynamics (MD) simulations are indispensable for studying critical biological processes such as protein folding and conformational changes. However, the accuracy and stability of these simulations are profoundly influenced by the choice of force field and solvation model. Inaccurate potentials can lead to significant artifacts, such as the formation of overly compact unfolded states or a bias towards non-native secondary structures, ultimately compromising the biological relevance of the simulation data [21] [22]. This application note, framed within the broader research on the GNEIMO torsional dynamics method, provides detailed protocols and comparisons to guide researchers in selecting and optimizing these critical parameters for stable and accurate long-timescale simulations.
Challenges in Long-Timescale Simulations
Selecting an appropriate molecular mechanics force field and an accompanying solvation model is a foundational step in MD simulation setup. A poor choice can lead to simulation instability, thermodynamic inaccuracies, and failure to reproduce experimentally observed properties.
A primary challenge is force field bias, where the underlying energy functions incorrectly stabilize non-native conformations. A seminal study on the Fip35 mutant of the human Pin1 WW domain demonstrated this problem vividly. In 10 µs simulations, the protein failed to fold into its native three-strand β-sheet structure, instead forming an array of non-native helical structures. Subsequent free energy calculations revealed that the force field used (CHARMM22 with CMAP corrections) favored these misfolded helical states by 4.4–8.1 kcal/mol over the native state, explaining the folding failure [21].
Another common artifact is over-compaction, a tendency observed in many implicit solvent models and some explicit solvent force fields to produce overly compact protein structures and denatured states. This is particularly problematic when simulating intrinsically disordered proteins (IDPs) or unfolded states, as it misrepresents their true conformational ensemble [22].
Finally, the computational expense of explicit solvent simulations can prohibit access to biologically relevant timescales. While explicit water models provide a detailed physical description, the number of solvent molecules often constitutes 80-90% of the particles in a simulation, creating a massive computational burden [23].
Comparison of Force Fields and Solvation Models
The selection of a solvation model involves a trade-off between computational efficiency and physical accuracy. The table below summarizes the key characteristics, advantages, and limitations of the predominant approaches.
Table 1: Comparison of Solvation Models for Protein Dynamics Simulations
| Solvation Model | Resolution | Computational Speed | Key Advantages | Key Limitations / Artifacts |
|---|---|---|---|---|
| Explicit Solvent (e.g., TIP4P) | Atomistic | Baseline (1x) | Physically detailed water structure and dynamics [23] | High computational cost; slow conformational sampling [23] |
| Coarse-Grained Solvent (e.g., ELBA) | Coarse-Grained | ~6x faster than atomistic [23] | Good balance of speed and accuracy for backbone dynamics [23] | Larger deviations in side-chain dynamics [23] |
| Implicit Solvent (GBSW, GBMV2) | Continuum Dielectric | Varies; can be much faster | Dramatically accelerated sampling; no explicit solvent viscosity [22] | Over-compaction bias; tendency to stabilize helical structures [21] [22] |
The performance of these models can be evaluated by comparing computed observables against experimental data, such as NMR order parameters ((S^2)). The following table summarizes a comparative study on the proteins BPTI and Galectin-3.
Table 2: Performance of Solvent Models in Reproducing NMR Order Parameters ((S^2)) [23]
| Solvent Model | Backbone NH (S^2) Deviation | Side-Chain (S^2) Deviation | Interpretation |
|---|---|---|---|
| All-Atom (TIP4P) | 0.03 - 0.06 | 0.13 - 0.17 | Reproduces backbone dynamics well; larger errors for side-chains. |
| Coarse-Grained (ELBA) | 0.03 - 0.06 | 0.13 - 0.17 | Comparable to all-atom for backbone; similar side-chain deviations. |
| Implicit (Generalized Born) | 0.03 - 0.06 | 0.13 - 0.17 | All models perform equally for backbone; no clear "winner" overall. |
Optimized Implicit Solvation with GBMV2
Given the computational advantages of implicit solvation for long-timescale simulations, significant effort has been dedicated to optimizing these models. The Generalized Born using Molecular Volume (GBMV2) model is a leading implicit solvent that closely reproduces the molecular surface definition, which helps eliminate unphysical high-dielectric pockets inside the protein [22].
A recent re-optimization of the GBMV2 model with the CHARMM36 protein force field leveraged a multi-scale enhanced sampling (MSES) technique to overcome the slow convergence that had previously hampered its parameterization. The key optimized parameters included [22]:
- Atomic Input Radii: Adjusted to achieve a better balance of solvation forces.
- Surface Tension Coefficient (γ): For the non-polar solvation term.
- Peptide Backbone Torsion Energetics (CMAP): Corrected to compensate for residual biases.
This optimized force field has demonstrated a marked reduction in over-compaction bias and can successfully recapitulate the structural ensembles of both folded model peptides (α-helical and β-hairpin) and intrinsically disordered proteins (IDPs) [22].
The GNEIMO Torsional Dynamics Method
The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method is an internal-coordinate torsional dynamics approach designed for enhanced conformational sampling [24]. Its application is highly relevant for studying long-timescale processes like protein folding and large-scale conformational changes.
The GNEIMO method enhances sampling efficiency by freezing high-frequency degrees of freedom (bond lengths and angles) and performing the simulation in the space of low-frequency torsional degrees of freedom. The protein is partitioned into rigid clusters (which can be as large as an entire domain) connected by torsional hinges. This reduces the number of active degrees of freedom and allows for a larger integration time step (e.g., 5 fs) [24]. The method is often combined with the replica-exchange (REXMD) technique for further sampling enhancement. The following diagram illustrates a typical GNEIMO simulation workflow.
Key Applications and Case Studies
GNEIMO has proven effective in simulating complex conformational changes that are difficult to observe with standard Cartesian MD within practical computational timescales [24].
- Fasciculin Neurotoxin: GNEIMO simulations, starting from the closed state (apo form), spontaneously sampled transitions to the known open state (holo form) without the application of any biasing potential. This transition, characterized by significant flexibility in loop I (residues 6–12), had previously only been observed using steered MD with an external bias [24].
- Calmodulin (CAM) Dynamics: Simulations initiated from the extended, Ca2+-bound state of CAM successfully sampled the transition towards the Ca2+-free state. This involved two major conformational changes: the collapse of the central helix linking the N- and C-terminal domains, and the dynamics of the relative orientations of the two domains. The generated ensemble of conformations satisfied about half of the short- and long-range inter-residue distances from NMR structures of the holo-to-apo transition [24].
Experimental Protocols
Protocol: Free Energy Comparison of Folded vs. Misfolded States
This protocol is based on the method used to identify the force field bias in the Pin1 WW domain study [21].
1. Objective: To calculate the free energy difference between the native fold and a stable misfolded state observed in simulation. 2. System Preparation: - Software: Use a package like NAMD. - Force Field: CHARMM22 with CMAP corrections. - Solvation: Solvate the protein in a cubic box of TIP3P water molecules. Neutralize the system with ions (e.g., 30 mM NaCl). 3. Simulation Steps: - Equilibration: Minimize the system for 3,000 steps. Perform a 100 ps NVT equilibration. - Production Trajectories: Run multiple microsecond-scale simulations (≥ 3 μs) starting from different initial conditions (e.g., fully extended and thermally denatured structures) at the target temperature (e.g., 337 K). 4. Analysis: - Cluster Analysis: Use a tool like the GROMOS clustering method in GROMACS to identify dominant conformational states from the trajectories. - Free Energy Calculation: Employ the Deactivated Morphing (DM) method to compute the free energy difference between reference structures for the native state and the misfolded state(s). This method restrains the system to each reference state and morphs between them via a "dummy" state to calculate the free energy difference.
Protocol: GNEIMO Torsional Dynamics Simulation
This protocol outlines the steps for setting up and running a protein simulation using the GNEIMO method [24].
1. Objective: To enhance conformational sampling of a protein using torsional dynamics. 2. System Preparation: - Initial Structure: Obtain a PDB file of the protein. - Solvation and Equilibration: Solvate the protein in an explicit solvent box (e.g., TIP3P water), neutralize, and add ions to 0.15 M ionic strength. Perform energy minimization and equilibration (NPT ensemble, 310 K, 1 atm, 5 ns) using a standard Cartesian MD package (e.g., AMBER). 3. GNEIMO Simulation Setup: - Force Field and Solvent: Use the AMBER ff99sb force field with a Generalized Born (GB) implicit solvation model (interior dielectric=4.0, exterior=78.3). A nonpolar solvation term based on solvent-accessible surface area (SA) is included. - Simulation Parameters: Perform simulations in the NVT ensemble using a Nosé-Hoover thermostat. Use a cutoff of 20 Å for nonbonded interactions. Set the integration time step to 5 fs using the Lobatto integrator. - Enhanced Sampling: For complex transitions, employ the Replica Exchange (REXMD) method with GNEIMO. 4. Analysis: - Analyze the trajectory for root-mean-square deviation (RMSD), radius of gyration, and other relevant metrics. - Compare sampled conformations against known experimental structures (e.g., from NMR ensembles or different crystal forms).
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Item Name | Function / Application | Specifications / Notes |
|---|---|---|
| CHARMM36 Force Field | A widely used, all-atom force field for proteins. | Often paired with CMAP cross-terms to correct backbone torsion profiles [21] [22]. |
| AMBER ff99SB Force Field | Another high-quality all-atom force field for biomolecules. | Commonly used with the GNEIMO method and for explicit solvent benchmarks [24] [23]. |
| GBMV2 Implicit Solvent | A Generalized Born model using molecular volume. | Accurately reproduces a molecular surface; requires parameter optimization to avoid over-compaction [22]. |
| GBSW Implicit Solvent | A Generalized Born model with a switching function. | An alternative GB model that has also been successfully optimized for protein folding [22]. |
| TIP3P Water Model | A standard 3-site explicit water model. | Commonly used as a benchmark for comparing solvation models [21] [23]. |
| Deactivated Morphing (DM) | A free energy calculation method. | Used to compute free energy differences between distinct protein conformations [21]. |
| Multi-Scale Enhanced Sampling (MSES) | An enhanced sampling technique. | Couples coarse-grained and all-atom models to accelerate sampling for force field optimization [22]. |
The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method is an advanced internal coordinate molecular dynamics (ICMD) technique that has emerged as a powerful tool for studying protein folding and large-scale conformational changes. By constraining high-frequency bond and angle vibrations and modeling a protein as a collection of rigid clusters connected by torsional hinges, GNEIMO enables enhanced conformational sampling in the low-frequency torsional space. This approach allows for larger integration time steps and a more efficient exploration of the protein energy landscape compared to traditional Cartesian molecular dynamics. [3] [8]
However, like any sophisticated simulation methodology, GNEIMO presents unique challenges related to energy conservation, sampling efficiency, and convergence monitoring. This article addresses these common pitfalls within the context of protein folding research, providing application notes and protocols to help researchers, scientists, and drug development professionals optimize their simulations. We frame these solutions within the broader thesis that GNEIMO's torsional dynamics approach offers distinct advantages for mapping complex protein folding pathways and energy landscapes, particularly for systems with rugged energy surfaces such as intrinsically disordered proteins. [25]
Understanding the GNEIMO Framework and Its Application to Protein Folding
Theoretical Foundations of GNEIMO
The GNEIMO method represents a paradigm shift from conventional Cartesian molecular dynamics. Its fundamental innovation lies in treating proteins as multibody systems with internal coordinates, where high-frequency degrees of freedom are replaced with hard holonomic constraints. This formulation reduces the system's dimensionality from 3N coordinates (where N is the number of atoms) to primarily torsional degrees of freedom, significantly enhancing computational efficiency for exploring slow conformational transitions relevant to protein folding. [8] [26]
A key advancement in GNEIMO is the application of Spatial Operator Algebra (SOA) from multibody dynamics, which enables O(N) computational scaling compared to the O(N³) scaling of conventional constrained dynamics algorithms. This efficiency gain is crucial for simulating biologically relevant timescales in protein folding studies. Additionally, GNEIMO incorporates the Fixman potential to correct for systematic statistical biases introduced by hard constraints, ensuring proper thermodynamic sampling—a critical consideration for accurately mapping folding energy landscapes. [8]
Relevance to Protein Folding and Design
The torsional focus of GNEIMO makes it particularly suited for protein folding research. Studies on de novo designed proteins have revealed that local backbone structures, governed by torsional preferences, play a crucial role in determining folding ability and exceptional thermal stability. GNEIMO's enhanced sampling in torsional space directly addresses these determinants, enabling more efficient exploration of the folding landscape. [27]
For intrinsically disordered proteins (IDPs), which possess rugged energy landscapes with multiple states separated by shallow energy barriers, GNEIMO's ability to facilitate transitions between conformational states is particularly valuable. The method has demonstrated success in simulating conformational transitions in flexible proteins like fasciculin and calmodulin that challenge conventional MD approaches. [6] [25]
Common Pitfalls and Troubleshooting Strategies
Energy Conservation Issues
Problem: Non-physical energy drift or poor temperature control in GNEIMO simulations.
Root Causes and Solutions:
Incorrect Velocity Initialization: The equipartition theorem for internal coordinates differs from Cartesian formulations. GNEIMO implements a specialized equipartition principle with "modal velocity coordinates" for thermodynamically correct velocity initialization. [8]
Improper Fixman Potential Application: The use of hard constraints distorts the effective potential energy surface. The Fixman potential compensates for this bias but has been historically challenging to compute. GNEIMO includes a low-cost, general-purpose SOA-based algorithm for including the Fixman correction, which is essential for recovering proper equilibrium probability distributions. [8]
Incorrect Thermostat Implementation: GNEIMO extends the Nosé-Hoover NVT method for internal coordinates, and improper application can cause energy drift. The software includes properly adapted thermostat implementations for ICMD. [8]
Diagnostic Protocol:
- Monitor total energy, kinetic energy, and temperature over time in equilibrium simulations
- Compare potential energy distributions with reference data
- Validate conformational distributions against known experimental or theoretical results
Sampling Efficiency Challenges
Problem: Inadequate exploration of conformational space in protein folding simulations.
Optimization Strategies:
Enhanced Sampling Integration: GNEIMO has been successfully combined with replica exchange molecular dynamics (REMD) and accelerated MD (aMD). The temperature replica exchange method is particularly effective, with standard protocols using 32 replicas across 310-415 K with exchanges attempted every 5 ps. [5] [8]
Hierarchical "Freeze and Thaw" Clustering: This GNEIMO-specific feature allows selective rigidification of protein domains (e.g., α-helices or β-sheets) while maintaining torsional flexibility in connecting regions. This reduces computational cost while maintaining essential flexibility for studying domain motions in folding. [3]
Adaptive Clustering Schemes: Adjust cluster definitions during simulations based on emerging structural features—initially smaller clusters for local folding events, transitioning to larger clusters for domain rearrangement.
Table 1: GNEIMO Enhanced Sampling Parameters for Protein Folding Applications
| Parameter | Recommended Setting | Alternative Options | Application Context |
|---|---|---|---|
| REMD Temperatures | 32 replicas, 310-415 K [5] | 8 replicas, 310-415 K (small proteins) [3] | General protein folding |
| Exchange Frequency | Every 5 ps [5] | Every 2 ps [3] | Rapidly folding systems |
| Integration Time Step | 5 fs [3] [5] | 4-6 fs depending on system | All-torsion dynamics |
| Simulation Duration | 15-100 ns/replica [5] | 5-15 ns/replica (small systems) [3] | Target-dependent |
Convergence Monitoring and Validation
Problem: Determining when protein folding simulations have adequately sampled the relevant conformational space.
Monitoring Framework:
Torsion-Based Metrics: Conventional Cartesian metrics like RMSD may miss important torsional transitions. Implement:
- Cα Torsion Angle Analysis: Torsion angles between four consecutive Cα atoms provide alignment-independent measures of conformational change. Create heat maps to visualize spatial and temporal domains of structural changes. [28]
- Side Chain Rotamer Distributions: Monitor convergence of χ-angle distributions for key residues.
Energy Landscape Analysis:
- Track the smoothness of energy gradients toward native-like states
- Monitor frustration indicators—regions with conflicting structural preferences
- For IDPs, validate the ruggedness of the landscape against experimental observations [25]
Experimental Validation:
- Compare with NMR chemical shifts and J-couplings
- Validate against SAXS profiles for overall dimensions
- Check consistency with FRET measurements for distance distributions [25]
Figure 1: GNEIMO Troubleshooting Workflow for Protein Folding Simulations
Application Notes for Specific Protein Systems
Structured Protein Folding
For well-folded proteins with funnel-like energy landscapes, GNEIMO protocols should focus on efficiently navigating toward the native state while avoiding kinetic traps.
Protocol:
- Initialization: Use all-torsion model with AMBER99SB force field and GB/SA implicit solvent (interior dielectric=1.5, exterior=78.3) [5]
- Enhanced Sampling: Implement REMD with 32 replicas across 310-415 K temperature range
- Cluster Analysis: Employ spatial clustering in torsion space to identify folding intermediates
- Validation: Compare with experimental melting temperatures and native structure metrics
Intrinsically Disordered Proteins
IDPs present unique challenges with their rugged energy landscapes and heterogeneous structural ensembles.
Protocol:
- Extended Sampling: Combine GNEIMO with accelerated MD to enhance transitions between metastable states
- Ensemble Validation: Use ensemble-averaged experimental observables (SAXS, NMR) for validation
- Multi-Scale Approaches: Integrate with knowledge-based methods like TraDES or flexible-meccano for initial ensemble generation [25]
- Analysis Focus: Monitor formation and dissolution of transient secondary structure elements
Table 2: Research Reagent Solutions for GNEIMO Protein Folding Studies
| Reagent/Resource | Type | Function in GNEIMO Protocol | Implementation Notes |
|---|---|---|---|
| AMBER99SB Force Field [5] | Force Field | Provides energy parameters | Standard with GB/SA implicit solvent |
| GB/SA OBC Solvent Model [3] [5] | Solvation Model | Implicit solvation for efficiency | Dielectric constants: 1.5 (int), 78.3 (ext) |
| GneimoSim Software [8] | ICMD Package | Main simulation engine | Interfaces with LAMMPS, OpenMM, Rosetta |
| Lobatto Integrator [3] [8] | Numerical Method | Integration of equations of motion | 5 fs time step for all-torsion dynamics |
| Fixman Potential Algorithm [8] | Correction Method | Eliminates constraint-induced bias | Essential for proper thermodynamics |
| Cα Torsion Analysis [28] | Analysis Method | Monitor conformational changes | Alignment-independent metric |
The GNEIMO method represents a significant advancement in molecular dynamics for protein folding research, particularly through its focus on torsional degrees of freedom and efficient conformational sampling. By addressing common challenges in energy conservation through proper application of the Fixman potential and thermostating, enhancing sampling via replica exchange methods and adaptive clustering, and implementing robust convergence monitoring using torsion-based metrics, researchers can overcome key pitfalls in protein folding simulations. The protocols and application notes provided here offer a framework for leveraging GNEIMO's unique capabilities to advance our understanding of protein folding mechanisms, with particular relevance for both structured proteins and intrinsically disordered systems that play crucial roles in cellular function and drug development.
Proof of Performance: Validating GNEIMO Against Experimental and Computational Benchmarks
The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method is an internal coordinate molecular dynamics (MD) technique designed for long-time scale simulation of biomolecular dynamics. Developed by applying JPL's Spatial Operator Algebra computational framework, GNEIMO enables efficient conformational sampling by focusing computational resources on low-frequency torsional degrees of freedom [29]. As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with, the contents by NLM or the National Institutes of Health. Learn more: PMC Disclaimer | PMC Copyright Notice.
Protein structure refinement remains a critical challenge in computational structural biology. While comparative modeling methods can generate initial structural models, these often contain significant deviations from experimental structures that limit their utility for detailed functional analysis and drug design [5]. The Critical Assessment of protein Structure Prediction (CASP) experiments have established rigorous blind testing grounds for evaluating refinement methodologies, highlighting both the pressing need and significant difficulty in consistently improving model accuracy [30] [31].
This application note details a specific implementation of the GNEIMO method that achieved refinement of up to 1.3 Å RMSD for 30 CASP target proteins, demonstrating its potential as a powerful tool for researchers and drug development professionals seeking to enhance the accuracy of protein structural models [5].
Experimental Design and Quantitative Results
Protein Target Selection and Classification
The refinement protocol was validated using 30 target proteins from the CASP8 and CASP9 experiments, carefully selected to represent different prediction scenarios [5]:
- 23 proteins from the structure refinement category (TR), where participants refine provided decoy structures.
- 7 proteins from the structure prediction category (T0), where homology models were generated using MODELER with templates identified via PDB sequence query search (30-80% sequence identity).
This dual approach tested GNEIMO's capability to improve existing models and refine newly generated homology models, addressing the most common use cases in computational structural biology.
Key Refinement Metrics and Performance
The GNEIMO method demonstrated significant improvement across multiple assessment metrics when applied to the 30 CASP targets. The results are summarized in Table 1.
Table 1: Refinement Performance Metrics for Selected CASP Targets Using GNEIMO Torsional MD
| Target | Starting GDT_TS | Refined GDT_TS | Starting RMSD (Å) | Refined RMSD (Å) | RMSD Improvement (Å) |
|---|---|---|---|---|---|
| TR429 | 31.5 | 45.7 | 6.82 | 5.76 | 1.06 |
| TR435 | 80.2 | 87.9 | 2.14 | 1.65 | 0.49 |
| TR453 | 86.6 | 91.5 | 1.51 | 1.10 | 0.41 |
| TR454 | 58.5 | 71.0 | 3.26 | 2.36 | 0.90 |
The GNEIMO torsional MD method achieved refinement of up to 1.3 Å in root-mean-square deviation (RMSD) without using any experimental data as restraints during simulations [5]. This performance contrasted with unconstrained all-atom Cartesian MD methods conducted under identical conditions, which required restraints during simulations to achieve refinement. The improvement was observed consistently across diverse protein targets, with the most significant gains occurring for models that started with lower accuracy.
Table 2: Comparative Analysis of Refinement Methods in Protein Modeling
| Method | Sampling Approach | Constraints | Restraints Required | Typical RMSD Improvement |
|---|---|---|---|---|
| GNEIMO-REXMD | Torsional dynamics with replica exchange | Holonomic (rigid clusters) | No | Up to 1.3 Å |
| All-Atom Cartesian MD | Cartesian dynamics | Soft (SHAKE/RATTLE) | Yes | Limited without restraints |
| Galaxy-Refine-Complex | Restrained MD with side-chain perturbation | Backbone and positional restraints | Yes | Varies by target |
| Rosetta FastRelax | Monte Carlo with minimization | Backbone fixed (side-chain only) | No | Moderate for side-chains |
Detailed GNEIMO Refinement Protocol
System Preparation and Minimization
The refinement protocol begins with comprehensive structure preparation:
- Decoy Acquisition: Starting decoys for structure refinement targets were downloaded from the CASP website (www.predictioncenter.org).
- Homology Modeling: For structure prediction targets, generate 100 models using MODELER and cluster into five groups. Select the best representative structure based on Procheck G-factor.
- Energy Minimization: Perform all-atom conjugate gradient minimization using the "sander" program with the AMBER99SB force field to remove steric clashes and prepare structures for dynamics.
GNEIMO-REXMD Simulation Parameters
The core refinement protocol employs specific parameters optimized for protein structure refinement:
- Force Field: AMBER99SB [5]
- Solvation Model: Generalized Born/Surface Area (GB/SA) OBC implicit solvation with interior dielectric of 1.5 and exterior dielectric of 78.3 [5]
- Nonbonded Cutoff: 20 Å with switched forces [5]
- Integrator: Lobatto integrator with 5 fs time step [5]
- Replica Exchange: 32 replicas across 310-415 K temperature range [5]
- Exchange Attempts: Every 5 ps using Metropolis criterion [5]
- Simulation Duration: 15-100 ns per replica [5]
GNEIMO Methodological Framework
The GNEIMO method implements a constrained dynamics approach with several distinctive features:
- Constrained Model: The protein is modeled as a collection of rigid clusters connected by flexible torsional hinges, with high-frequency bond length and bond angle vibrations eliminated using holonomic constraints.
- Internal Coordinates: Dynamics occurs in torsional space rather than Cartesian coordinates, reducing the number of degrees of freedom by approximately an order of magnitude.
- Computational Efficiency: The Spatial Operator Algebra framework enables O(N) computational cost for solving equations of motion, compared to conventional O(N³) methods.
- Hierarchical Clustering: Users can define rigid clusters at different scales - from individual atoms to secondary structure elements - enabling "freeze and thaw" strategies for enhanced sampling.
Figure 1: GNEIMO Refinement Workflow. The protocol begins with structure preparation, proceeds through constrained dynamics simulation with replica exchange, and concludes with trajectory analysis to select refined models.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for GNEIMO Refinement
| Item | Type | Function in Protocol | Implementation Notes |
|---|---|---|---|
| GNEIMO Software | Computational Method | Internal coordinate MD with rigid clusters | JPL-developed; uses Spatial Operator Algebra for O(N) computation [29] |
| AMBER99SB Force Field | Molecular Mechanics | Energy calculation and conformational sampling | Includes corrections for protein backbone representation [5] |
| GB/SA OBC Solvation Model | Implicit Solvent | Approximates aqueous solvation effects | OBC model for Generalized Born solvation [5] |
| Temperature Replica Exchange | Sampling Enhancement | Accelerates conformational sampling | 32 replicas across 310-415 K range [5] |
| MODELER | Homology Modeling | Generate starting models for T0 targets | Used for initial model generation [5] |
| AMBER "sander" | Energy Minimization | Structure preparation before dynamics | Conjugate gradient minimization [5] |
Technical Advantages of the GNEIMO Approach
Enhanced Conformational Sampling
The GNEIMO method provides significant advantages for conformational sampling compared to traditional Cartesian MD:
- Larger Time Steps: Elimination of high-frequency vibrations enables 5 fs time steps, compared to 1-2 fs typical in Cartesian MD [2].
- Reduced Degrees of Freedom: Focusing on torsional space decreases the conformational search space by approximately an order of magnitude.
- Barrier Crossing: The force-driven nature of MD enables overcoming energy barriers that may trap Monte Carlo methods [5].
- Replica Exchange Efficiency: Fewer degrees of freedom reduce the number of replicas needed for effective temperature exchange [2].
Applications Beyond Single-Domain Proteins
While this protocol focused on monomeric protein refinement, the GNEIMO method has demonstrated success in broader applications:
- Domain Motion Mapping: GNEIMO has been used to study large-scale domain motions in proteins like calmodulin and phosphoglycerate kinase [6].
- Protein Folding: The method has successfully folded small proteins and peptides from extended conformations, including α-helical and β-sheet motifs [2].
- Protein Complex Refinement: Though more challenging, refinement methods can be extended to protein-protein interfaces with appropriate protocols [32].
The GNEIMO torsional dynamics method provides an effective protocol for protein structure refinement, consistently improving model accuracy by up to 1.3 Å RMSD across diverse protein targets. Its constrained dynamics approach, combined with replica exchange sampling, enables efficient exploration of conformational space in the biologically relevant torsional degrees of freedom. For researchers in structural biology and drug development, this methodology offers a powerful approach to enhance the quality of protein structural models, potentially reducing reliance on extensive experimental structure determination while providing more accurate models for functional analysis and rational drug design.
The integration of physical molecular dynamics with enhanced sampling techniques positions GNEIMO as a valuable tool in the computational structural biology toolkit, particularly as the field addresses increasingly challenging targets including multi-domain proteins and molecular complexes.
Within the broader investigation of the GNEIMO method for torsional dynamics in protein folding research, this case study examines its specific application in enriching native-like conformations from protein folding trajectories. The longstanding challenge in computational protein structure prediction has been the refinement of low-resolution models into highly accurate atomistic structures useful for detailed structural and drug discovery studies [5]. Traditional all-atom Cartesian molecular dynamics (MD) simulations have shown limited success in this refinement without the application of restraints [5]. The GNEIMO (Generalized Newton-Euler Inverse Mass Operator) method addresses this limitation through an internal coordinate MD technique that enhances conformational sampling of biologically relevant states [5] [6]. This study quantitatively evaluates the GNEIMO approach applied to 30 CASP target proteins, demonstrating significant refinement toward native-like conformations through specialized torsional dynamics protocols.
Methodological Framework
GNEIMO Torsional Dynamics
The GNEIMO method is a constrained MD simulation technique based on internal coordinates that enhances sampling efficiency [5] [6]:
- Rigid Body Modeling: High-frequency degrees of freedom are frozen by modeling the protein as a collection of user-defined rigid bodies ("clusters") connected by flexible torsional hinges [5].
- Computational Efficiency: This approach allows larger integration time steps (5 fs) and focuses conformational search exclusively on low-frequency torsional degrees of freedom [5].
- Enhanced Sampling: The method reduces computational cost while enabling more effective crossing of energy barriers compared to traditional MD methods [5].
Replica Exchange Implementation
The GNEIMO method was combined with temperature replica exchange MD (REXMD) to further enhance conformational sampling [5]:
- Temperature Range: 32 replicas spanning 310-415 K [5]
- Exchange Attempts: Temperature sorting based on Metropolis algorithm every 5 ps [5]
- Simulation Duration: 15-100 ns per replica for each target protein [5]
Experimental Protocol
System Preparation
Homology Model Generation (for structure prediction targets):
- Identify template structures using PDB sequence query search (30-80% sequence identity) [5]
- Generate 100 models using MODELER [5]
- Cluster models into five groups and select best representative structure based on Procheck G-factor [5]
Initial Structure Minimization:
- Perform all-atom conjugate gradient minimization using "sander" program [5]
- Utilize AMBER FF99SB force field [5]
- Apply GB/SA OBC implicit solvation model with interior dielectric of 1.5 and exterior dielectric of 78.3 [5]
GNEIMO-REXMD Simulation Parameters
| Parameter Category | Specification |
|---|---|
| Force Field | AMBER99SB [5] |
| Solvation Model | Generalized Born/Surface Area (GB/SA) OBC implicit solvent [5] |
| Nonbonded Cutoff | 20 Å with switch-off [5] |
| Integration Method | Lobatto integrator with 5 fs time step [5] |
| Temperature Control | Nose-Hoover thermostat [5] |
| Replica Configuration | 32 replicas across 310-415 K [5] |
| Exchange Frequency | Every 5 ps using Metropolis criterion [5] |
Analysis Methods
- Structure Quality Assessment: RMSD, GDT_TS, and TM-Score calculations relative to native structures [5]
- Conformational Clustering: Identification of dominant conformational substates [6]
- Free Energy Analysis: Examination of local motions through virtual-bond angles θ and dihedral angles γ [33]
Results and Discussion
Quantitative Refinement of CASP Targets
Application of GNEIMO-REXMD to 30 CASP target proteins demonstrated substantial improvement in model quality across multiple metrics:
Table 1: Representative Refinement Results for CASP Targets [5]
| Target ID | Category | Starting GDT_TS | Refined GDT_TS | Starting RMSD (Å) | Refined RMSD (Å) | Refinement (Å) |
|---|---|---|---|---|---|---|
| TR429 | Refinement | 31.5 | 45.7 | 6.82 | 5.76 | 1.06 |
| TR435 | Refinement | 80.2 | 87.9 | 2.14 | 1.65 | 0.49 |
| TR453 | Refinement | 86.6 | 91.5 | 1.51 | 1.10 | 0.41 |
| TR454 | Refinement | 58.5 | 71.0 | 3.26 | 2.36 | 0.90 |
| T0435 | Prediction | 47.3 | 62.5 | 4.92 | 3.86 | 1.06 |
The GNEIMO method achieved refinement of up to 1.3 Å RMSD without using experimental restraints, outperforming traditional Cartesian MD simulations which typically require restraints to prevent structural collapse [5].
Conformational Substates Sampling
In studies of conformationally flexible proteins, GNEIMO demonstrated exceptional capability in sampling biologically relevant states:
Table 2: Conformational Sampling Performance [6]
| Protein System | Structural Feature | GNEIMO Performance | Cartesian MD Performance |
|---|---|---|---|
| Fasciculin | Two conformational substates | Sampled both known experimental substates [6] | Failed to sample transitions [6] |
| Calmodulin | Holo to apo transition | Sampled transition pathway; 50% satisfaction of NMR distances [6] | Failed to sample transitions [6] |
| Crambin | Structural fluctuations | Reproduced experimental B-factors [6] | Comparable to explicit solvent [6] |
| BPTI | Structural fluctuations | Reproduced experimental B-factors [6] | Comparable to explicit solvent [6] |
The method's ability to sample functionally relevant conformational transitions in fasciculin and calmodulin demonstrates its particular value for drug development applications where understanding conformational dynamics is crucial [6].
Mirror-image Conformation Resolution
Analysis of folding trajectories revealed GNEIMO's capability in resolving subtle conformational states, including mirror-image topologies observed in symmetrical proteins like the B domain of protein A [33]. Through local free-energy profile analysis along amino acid sequences, the method identified key residues responsible for mirror-image formation, particularly in the second loop and third helix region (Asp29–Asn35) [33]. This resolution of energetically competitive native-like conformations provides critical insights for understanding protein misfolding phenomena relevant to neurodegenerative diseases [33].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Reagent/Solution | Function/Application | Specifications |
|---|---|---|
| AMBER99SB Force Field | Physics-based energy function for MD simulations [5] | Includes corrections for protein backbone parameters [5] |
| GB/SA OBC Implicit Solvent | Efficient solvation model without explicit water molecules [5] | Solvent probe radius: 1.4 Å; dielectric constants: 1.5 (interior), 78.3 (exterior) [5] |
| MODELER Software | Comparative protein structure modeling [5] | Generates homology models from templates [5] |
| GNEIMO Algorithm | Torsional dynamics simulation engine [5] [6] | Internal coordinate MD with rigid body clusters [5] |
| Replica Exchange Framework | Enhanced sampling methodology [5] | 32 replicas; 310-415 K temperature range; exchanges every 5 ps [5] |
Workflow Visualization
GNEIMO Refinement Workflow
Cotranslational Folding Pathway
{start of main content}
Comparative Analysis: GNEIMO vs. Unconstrained Cartesian MD in Structure Refinement
The refinement of low-resolution protein models into structures that closely resemble experimental atomic coordinates remains a significant challenge in computational biology. This application note provides a comparative analysis of two molecular dynamics (MD) methodologies for protein structure refinement: the constrained internal coordinate method GNEIMO (Generalized Newton-Euler Inverse Mass Operator) and traditional unconstrained Cartesian MD. Within the broader thesis of GNEIMO's application to torsional dynamics in protein folding research, we demonstrate that GNEIMO's enhanced conformational sampling leads to superior refinement efficacy, achieving approximately 2 Å improvement in RMSD over unconstrained methods. We detail explicit protocols for decoy generation, refinement simulations, and analysis, providing researchers and drug development professionals with practical frameworks for implementing these techniques.
Proteins are dynamic molecular machines whose functions are intimately linked to their three-dimensional structures and conformational flexibility [7]. Accurately determining and refining protein structures is therefore crucial for understanding biological mechanisms and for structure-based drug design. While experimental techniques like X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy provide high-resolution structural information, they are often resource-intensive and may yield incomplete data. Computational structure prediction and refinement methods serve as vital complements to these experimental approaches.
A persistent challenge in the field of protein structure prediction, particularly in homology modeling, is the reliable refinement of low-resolution models toward native-like structures [3]. Traditional unconstrained all-atom MD simulations in Cartesian coordinates have demonstrated limited effectiveness for this refinement task, primarily due to inadequate conformational sampling resulting from the large number of degrees of freedom and the high-frequency bond vibrations that restrict integration time steps [3] [2].
The GNEIMO method addresses these limitations through a constrained dynamics approach rooted in internal coordinates [3] [2]. By replacing high-frequency degrees of freedom with hard holonomic constraints, GNEIMO models proteins as collections of rigid bodies connected by flexible torsional hinges. This formulation reduces the number of degrees of freedom by approximately an order of magnitude, enables larger integration time steps (5 fs versus typically 1-2 fs in Cartesian MD), and enhances exploration of conformational space [3] [2] [7]. This application note presents a structured comparison of these methodologies, providing quantitative performance assessments and detailed implementation protocols to guide researchers in selecting and applying these techniques for protein structure refinement.
Theoretical Foundations and Methodological Comparison
GNEIMO (Constrained Internal Coordinate MD)
The GNEIMO method employs a mathematical framework based on Spatial Operator Algebra to efficiently solve the equations of motion in internal coordinates [3] [2]. In this approach:
- Molecular Model: The protein is represented as a series of rigid clusters (from single atoms to entire domains) connected by flexible torsional hinges.
- Degree of Freedom Reduction: Bond lengths and angles are constrained, leaving only torsional degrees of freedom, reducing the total degrees of freedom by approximately 90% compared to Cartesian MD.
- Computational Efficiency: The NEIMO algorithm solves the coupled equations of motion with O(N) computational cost, where N is the number of torsional degrees of freedom, compared to conventional O(N³) methods [2].
- Enhanced Sampling: The combination of reduced degrees of freedom and larger integration time steps (typically 5 fs) enables more extensive exploration of conformational space within equivalent simulation time [3].
- Hierarchical Clustering: A unique feature of GNEIMO is the "freeze and thaw" capability, allowing specific protein regions (e.g., secondary structure elements) to be treated as rigid bodies while sampling torsions in connecting regions [3] [2].
Unconstrained Cartesian MD
Traditional Cartesian MD simulations model all atoms without constraints:
- Complete Coordinate System: All atomic positions (x, y, z coordinates) are independent variables, resulting in approximately 3N degrees of freedom for an N-atom system.
- High-Frequency Vibrations: The inclusion of bond stretching and angle bending vibrations (with periods of ~10-100 fs) necessitates small integration time steps (typically 1-2 fs) for numerical stability.
- Sampling Limitations: The combination of numerous degrees of freedom and small time steps severely limits the conformational space that can be sampled in practical simulation timescales [3].
- Energy Landscape Navigation: Cartesian MD must navigate rough energy landscapes with numerous local minima, often requiring enhanced sampling techniques to overcome energy barriers.
Quantitative Performance Comparison
Table 1: Comparative Performance Metrics for Structure Refinement
| Performance Metric | GNEIMO Constrained MD | Unconstrained Cartesian MD |
|---|---|---|
| RMSD Improvement | ~2.0 Å improvement from starting decoys [3] | Limited or no improvement; often worsens starting models [3] |
| Sampling Efficiency | Enhanced conformational search; enrichment of native-like conformations [3] | Limited conformational sampling; poor enrichment of native states [3] |
| Degrees of Freedom | ~10% of Cartesian MD (torsional DOFs only) [2] | 100% (all atomic coordinates) |
| Integration Time Step | 5 fs [3] [2] | 1-2 fs [2] |
| Replica Exchange Requirements | Fewer replicas (8 sufficient for systems tested) [3] | More replicas needed due to higher dimensionality |
| Applicable Systems | All-α, α/β, and all-β proteins [3]; small protein folding [2] | Limited effectiveness for refinement [3] |
| Special Features | "Freeze and Thaw" hierarchical clustering; all-torsion dynamics [3] [2] | Standard all-atom simulation |
Table 2: Test System Details from Referenced Studies
| System Characteristic | GNEIMO Study Details |
|---|---|
| Proteins Tested | Eight proteins with varying secondary structures: all-α, α/β, and all-β motifs [3] |
| Starting Structures | Low-resolution decoys (2-5 Å RMSD from native) generated via homology modeling [3] |
| Simulation Duration | 5-15 ns per replica (40-120 ns total with 8 replicas) [3] |
| Solvation Model | Generalized Born/Surface Area (GB/SA) implicit solvent [3] [2] |
| Force Field | AMBER99 [3] [2] |
| Temperature Scheme | Replica Exchange MD with 8 replicas (310-415 K, 15 K intervals) [3] |
Experimental Protocols
Protocol 1: Decoy Set Generation for Refinement Studies
Purpose: To generate low-resolution structural decoys for refinement simulations.
Steps:
- Homology Modeling: Use MODELLER [3] or similar software with templates having 60-70% sequence identity to the target.
- Initial Model Selection: Generate 100 homology models and cluster by structural diversity into 5 clusters. Select representative structures from each cluster with the most secondary structure content.
- Simulated Annealing: Perform simulated annealing using all-torsion GNEIMO dynamics with temperature ranging from 310 K to 1200 K in 50 K increments to "swell" homology models to lower resolution.
- Decoy Selection: Choose three swollen snapshots from the trajectory with backbone RMSD in the range of 2-5 Å with respect to the native experimental structure.
- Energy Minimization: Perform unconstrained Cartesian MD energy minimization using 1000 steps of steepest descent followed by 1000 steps of conjugate gradient method with AMBER force field and Generalized Born solvent model (non-bond cutoff of 20 Å) [3].
Applications: This protocol generates structurally diverse starting points for refinement studies, essential for evaluating the robustness of refinement methods.
Protocol 2: GNEIMO All-Torsion Refinement Protocol
Purpose: To refine low-resolution protein models using GNEIMO all-torsion dynamics.
Steps:
- System Preparation: Initialize the decoy structure with AMBER99 force field parameters.
- Solvation Model Setup: Employ Generalized Born/Surface Area (GB/SA) OBC implicit solvation model with interior dielectric of 1.5 for solute and exterior dielectric of 78.3 for solvent. Use solvent probe radius of 1.4 Å for non-polar solvation energy component.
- Simulation Parameters: Set non-bond forces cutoff at 20 Å with smooth switching. Use Lobatto integrator with 5 fs time step.
- Replica Exchange Setup: Configure 8 replicas in temperature range 310-415 K with 15 K intervals.
- Production Simulation: Run each replica for 5-15 ns, with temperature exchange attempts every 2 ps (400 steps). Total simulation time: 40-120 ns.
- Trajectory Analysis: Monitor RMSD to native structure, radius of gyration, secondary structure evolution, and energy trends.
Applications: Refinement of homology models, generating native-like conformational ensembles, preparatory sampling for drug docking studies.
Protocol 3: Hierarchical "Freeze and Thaw" Clustering Strategy
Purpose: To enhance localized conformational sampling using GNEIMO's flexible clustering capability.
Steps:
- Secondary Structure Identification: Analyze the protein structure to identify well-defined α-helical and β-sheet regions.
- Clustering Scheme Design: For mixed α/β motif proteins, treat either α-helix or β-sheet motifs as rigid bodies (freezing backbone atoms) while leaving side chains and connecting regions available for all-torsion sampling.
- Simulation Setup: Implement the defined clustering scheme in GNEIMO with similar parameters as Protocol 2 (GB/SA solvation, 5 fs time step, AMBER99 force field).
- Comparative Analysis: Run parallel simulations with different clustering schemes (e.g., helical regions frozen vs. sheet regions frozen) and compare sampling efficiency and refinement outcomes.
Applications: Targeted refinement of specific domains or structural motifs, studying allosteric mechanisms, efficient sampling of localized conformational changes relevant to function.
Protocol 4: Unconstrained Cartesian MD for Refinement (Reference Protocol)
Purpose: To provide a reference refinement protocol using traditional Cartesian MD for comparative studies.
Steps:
- System Setup: Prepare the decoy structure with the same force field (AMBER99) and solvation model (GB/SA) as GNEIMO protocols for direct comparison.
- Simulation Parameters: Use Langevin dynamics for temperature control, 2 fs time step, and SHAKE/RATTLE algorithms for bond constraints.
- Replica Exchange: Implement temperature replica exchange with more replicas (typically 12-16) to compensate for higher dimensionality, covering similar temperature range (310-415 K).
- Production Run: Execute simulations for durations comparable to GNEIMO runs (5-15 ns per replica).
- Analysis: Compute identical metrics as GNEIMO simulations for direct comparison: RMSD to native, secondary structure content, and energy landscapes.
Applications: Baseline comparison for constrained methods, studies requiring full atomic flexibility, systems with significant bond angle or length variations.
Workflow Visualization
GNEIMO vs Cartesian MD Refinement
Table 3: Essential Computational Tools for Structure Refinement Studies
| Tool/Resource | Type | Function in Research | Implementation Examples |
|---|---|---|---|
| GneimoSim | Software Package | Primary simulation engine for GNEIMO constrained MD simulations | Structure refinement, protein folding, conformational dynamics [7] |
| AMBER | MD Software Suite | Reference simulations with unconstrained Cartesian MD; force field parameters | Comparative studies, energy minimization, force field implementation [3] |
| AMBER99 Force Field | Force Field | Potential energy function for protein interactions | Primary force field for both GNEIMO and Cartesian MD simulations [3] [2] |
| GB/SA OBC Model | Solvation Model | Implicit solvent treatment for efficient hydration effects | Standard solvation model for refinement protocols [3] [2] |
| MODELLER | Homology Modeling | Generation of initial low-resolution decoy structures | Creating starting models for refinement studies [3] |
| Replica Exchange MD | Sampling Algorithm | Enhanced conformational sampling through temperature cycling | Implementation in both GNEIMO and Cartesian protocols [3] |
| Principal Component Analysis | Analysis Method | Dimensionality reduction for trajectory analysis | Identifying essential dynamics and collective motions [2] |
Discussion and Strategic Implementation Guidelines
Performance Interpretation and Method Selection
The quantitative data demonstrates GNEIMO's superior performance in protein structure refinement applications, particularly for improving low-resolution homology models. The observed ~2 Å RMSD improvement represents a significant advancement toward experimental accuracy. Several factors contribute to this enhanced performance:
- Efficient Phase Space Exploration: By focusing sampling on torsional degrees of freedom most relevant to protein conformational changes, GNEIMO more efficiently explores functionally relevant regions of conformational space [3].
- Native-State Enrichment: GNEIMO simulations show increased population of native-like conformations compared to Cartesian MD, suggesting better navigation of the protein energy landscape [3].
- Hierarchical Advantage: The "freeze and thaw" capability enables targeted sampling strategies that align with theoretical folding models like the "zipping-and-assembly" mechanism [2].
Applications in Drug Development and Structural Biology
The implementation of GNEIMO for structure refinement has several practical implications for drug development professionals:
- High-Throughput Refinement: The method's efficiency enables refinement of multiple protein models or mutants, supporting structural genomics initiatives and mutant characterization studies.
- Allosteric Site Identification: Refined conformational ensembles provide insights into allosteric communication pipelines and potential allosteric drug targets [7].
- GPCR Drug Design: GNEIMO has been specifically applied to G-protein coupled receptors (GPCRs), important drug targets where conformational dynamics profoundly influence function [7].
- Integration with Experimental Data: GNEIMO can complement experimental techniques like NMR and cryo-EM by providing atomic-level interpretation of low-resolution data [7].
Limitations and Future Directions
While GNEIMO demonstrates superior refinement capabilities, researchers should consider certain limitations:
- Force Field Dependence: Like all MD methods, GNEIMO's performance is contingent on the accuracy of the underlying force field [34].
- Implicit Solvent Limitations: The use of GB/SA implicit solvent, while efficient, may not capture specific solvent effects crucial for certain protein interactions.
- Rigid Body Assumptions: The constraint of bond lengths and angles, while generally valid, may limit applicability to systems where these parameters undergo significant changes.
Future developments may integrate machine learning approaches [35] with constrained dynamics methods, potentially combining the sampling advantages of GNEIMO with the pattern recognition capabilities of deep learning for further improvements in refinement accuracy and efficiency.
This comparative analysis demonstrates that the GNEIMO constrained dynamics method provides significant advantages over unconstrained Cartesian MD for protein structure refinement applications. Through its reduced degrees of freedom, larger integration time steps, and flexible hierarchical clustering capabilities, GNEIMO achieves approximately 2 Å improvement in RMSD from starting decoys and enhanced sampling of native-like conformations. The detailed protocols provided herein offer researchers practical frameworks for implementing these methods in structural biology and drug discovery pipelines. As molecular simulation continues to play an increasingly important role in complementing experimental structural biology, methods like GNEIMO that enhance conformational sampling efficiency will prove invaluable for advancing our understanding of protein dynamics and function.
{end of main content}
Understanding protein conformational dynamics is crucial for elucidating biological function and guiding drug discovery efforts. The study of conformational transitions in proteins like fasciculin and calmodulin presents significant challenges due to the long timescales over which these dynamics occur, often reaching into the millisecond range and beyond [15]. Conventional all-atom molecular dynamics simulations have historically struggled to sample these rare but biologically critical events within practical computational timeframes [15] [6].
The Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method addresses this fundamental limitation through an internal coordinate molecular dynamics approach that enhances sampling efficiency [5] [29]. By treating proteins as collections of rigid clusters connected by flexible torsional hinges, GNEIMO enables the simulation of long-timescale conformational changes that are essential for understanding protein function [5]. This application note details protocols for applying GNEIMO to map the conformational landscapes of two biologically significant proteins: fasciculin, a picomolar inhibitor of acetylcholinesterase, and calmodulin, a calcium-dependent signaling protein [36] [15].
Biological Significance and Quantitative Dynamics
Fasciculin: A Model for Synaptic Inhibition
Fasciculin-2 (FAS2) is a three-fingered neurotoxin isolated from snake venoms that acts as a potent inhibitor of synaptic acetylcholinesterase (AChE) with picomolar affinity [36]. This inhibition occurs through binding to the peripheral anionic site of AChE, effectively prolonging the action of acetylcholine in synapses [37]. The primary interactions between FAS2 and AChE occur at the finger tip residues of loops I and II, with conformational flexibility playing a critical role in the binding mechanism [36].
Crystallographic studies have identified two major conformational substates in fasciculin-2 [36]:
- FAS2a substate: Thr-9 near the tip of loop I packs against a hydrophobic pocket formed by residues Tyr-4, Ala-12, Tyr-61, and Arg-37
- FAS2b substate: Thr-9 extends into solution, with the hydrophobic pocket occupied by either detergent molecules (in apo forms) or by Val-73 of AChE (in the complexed form)
Molecular dynamics trajectories of 0.15-0.3 μs have revealed that the high energy barrier between these substates leads to transitions that are slow on the timescale of diffusional encounter, suggesting that conformational readjustments may occur after the initial binding event [36].
Calmodulin: Calcium-Mediated Signaling
Calmodulin serves as a primary calcium sensor in eukaryotic cells, undergoing substantial conformational changes between calcium-bound (holo) and calcium-free (apo) states [15] [38]. This transition enables calmodulin to regulate numerous target proteins involved in diverse cellular processes including muscle contraction, neurotransmitter release, and metabolic regulation [38].
The conformational transition of calmodulin involves large-scale domain rearrangements that have proven challenging to capture with conventional simulation methods. GNEIMO simulations have successfully sampled the holo to apo transition, generating ensembles that satisfy approximately half of both short- and long-range interresidue distances obtained from NMR structures [15].
Table 1: Key Characteristics of Fasciculin and Calmodulin
| Parameter | Fasciculin-2 | Calmodulin |
|---|---|---|
| Protein Size | 61 residues, ~7 kDa | 148 residues, ~16.7 kDa |
| Biological Function | AChE inhibition, synaptic modulation | Calcium sensing, signal transduction |
| Conformational States | FAS2a (closed) and FAS2b (open) | Apo (Ca²⁺-free) and Holo (Ca²⁺-bound) |
| Key Structural Features | Three-fingered toxin with flexible loops | Two globular domains connected by flexible linker |
| Transition Timescale | Submicrosecond to microsecond [36] | Microsecond to millisecond [15] |
| Primary Experimental Validation | X-ray crystallography, MD simulations [36] | NMR, X-ray crystallography [15] |
GNEIMO Methodology and Experimental Protocols
GNEIMO Theoretical Framework
The GNEIMO method employs a constrained dynamics approach in internal coordinates, where high-frequency degrees of freedom are frozen and the protein is modeled as a collection of rigid clusters connected by torsional hinges [5]. This physical model allows larger integration time steps (typically 5 fs) and focuses conformational search in the low-frequency torsional degrees of freedom that dominate large-scale protein motions [5].
The computational implementation uses the Spatial Operator Algebra framework to efficiently solve the equations of motion, with computational cost scaling linearly with the number of degrees of freedom [29]. This represents a significant advantage over conventional Cartesian molecular dynamics where computational cost scales cubically with system size [5].
Protocol for Fasciculin Conformational Sampling
System Preparation:
- Obtain starting structures from Protein Data Bank entries 1FAS (FAS2a substate) and 1FSC (FAS2b substate) [36]
- Solvate the systems with TIP3 water molecules and add counterions to neutralize charge
- For torsional dynamics, define rigid clusters based on secondary structural elements with flexible hinges at loop regions
Simulation Parameters:
- Force Field: AMBER99SB [5]
- Solvation Model: Generalized Born/Surface Area (GB/SA) OBC implicit solvent [5]
- Nonbonded Cutoff: 20 Å [5]
- Integration Time Step: 5 fs [5]
- Temperature Control: Nose-Hoover thermostat [5]
Enhanced Sampling Protocol:
- Perform temperature replica exchange MD (REXMD) with 32 replicas across 310-415 K temperature range [5]
- Exchange replicas every 5 ps using Metropolis criterion [5]
- Collect aggregate simulation time of 15-100 ns per replica [5]
- Use cluster analysis with k-means algorithm to identify conformational families in essential configuration space [36]
Analysis Methods:
- Calculate root mean-square deviation (RMSD) of loop I residues to monitor transitions
- Compute free energy profiles using molecular potential energy landscape: ΔGₘ = -RTln(ρ), where ρ is distribution density in essential configuration space [36]
- Perform normal mode analysis to estimate configurational entropy of each energy basin [36]
Protocol for Calmodulin Holo-to-Apo Transition
System Setup:
- Begin with calcium-bound crystal structure (e.g., PDB 1CLL)
- Define rigid clusters corresponding to individual α-helices with flexible hinges in connecting loops
- Remove calcium ions from EF-hand motifs to initiate transition
Simulation Parameters:
- Force Field: AMBER99SB with GB/SA implicit solvent [5]
- Temperature: 32 replicas from 310-415 K for REXMD [5]
- Integration: 5 fs time step using Lobatto integrator [5]
- Simulation Duration: 20-50 ns per replica
Transition Analysis:
- Monitor interdomain distances and angles to quantify conformational change
- Calculate satisfaction of NMR-derived distance constraints [15]
- Identify intermediate states along transition pathway
Table 2: GNEIMO Simulation Performance Metrics
| Performance Measure | Fasciculin Simulations | Calmodulin Simulations |
|---|---|---|
| Simulation Time per Replica | 15-100 ns [5] | 20-50 ns [15] |
| Number of Replicas | 32 [5] | 32 [5] |
| Temperature Range | 310-415 K [5] | 310-415 K [5] |
| Sampling Enhancement | 10-100x over Cartesian MD [15] | 10-100x over Cartesian MD [15] |
| Transition Events Captured | FAS2a FAS2b transitions [15] | Holo Apo transitions [15] |
| Key Validation Metrics | Comparison to crystal structures [36] | NMR distance constraints [15] |
Workflow Visualization
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Reagent/Software | Specifications | Application in Protocol |
|---|---|---|
| GNEIMO Software | JPL-developed ICMD package [29] | Core torsional dynamics simulation engine |
| AMBER99SB Force Field | Optimized for protein simulations [5] | Potential energy calculations |
| GB/SA OBC Solvent Model | Implicit solvation with 1.4 Å probe radius [5] | Solvation energy approximation |
| TIP3 Water Model | Transferable Intermolecular Potential [36] | Explicit solvation (optional) |
| Fasciculin Structures | PDB: 1FAS (FAS2a), 1FSC (FAS2b) [36] | Initial coordinates for simulations |
| Calmodulin Structures | PDB: 1CLL (Holo), 1CFD (Apo) | Reference structures for validation |
| Temperature Replica Exchange | 32 replicas, 310-415 K range [5] | Enhanced conformational sampling |
| Cluster Analysis | k-means algorithm in essential space [36] | Identification of conformational states |
Applications in Drug Discovery
The conformational dynamics of fasciculin and calmodulin have significant implications for pharmaceutical development. Fasciculin's picomolar affinity for acetylcholinesterase makes it a valuable template for designing novel inhibitors targeting the cholinergic system, with potential applications in neurodegenerative disorders like Alzheimer's disease [37]. Understanding its conformational transitions provides insights for optimizing therapeutic compounds that modulate acetylcholinesterase activity.
Calmodulin's role as a calcium sensor implicated in numerous signaling pathways makes it an attractive target for pharmacological intervention in cardiovascular diseases, neurological disorders, and cancer [38]. The ability to simulate its calcium-dependent conformational transitions using GNEIMO enables structure-based drug design approaches targeting specific calmodulin states or transition pathways.
The GNEIMO methodology has demonstrated particular value in protein structure refinement, achieving improvements of up to 1.3 Å RMSD for CASP target proteins without experimental restraints [5]. This capability directly enhances the accuracy of homology models used in drug discovery when experimental structures are unavailable.
The GNEIMO torsional dynamics method provides a powerful framework for capturing long-timescale conformational transitions in biologically essential proteins like fasciculin and calmodulin. Through its innovative use of internal coordinates and enhanced sampling techniques, GNEIMO enables the simulation of dynamic processes that remain inaccessible to conventional molecular dynamics approaches. The protocols detailed in this application note offer researchers practical guidance for implementing these methods to advance understanding of protein dynamics and facilitate structure-based drug design. As computational capabilities continue to evolve, GNEIMO represents a promising approach for bridging the gap between theoretical models and experimental observations in structural biology.
The accuracy of computational protein structure prediction is paramount for advancing structural biology and drug development. Reliable validation metrics are essential to assess the quality of predicted models. This application note details three core sets of validation metrics—Root-Mean-Square Deviation (RMSD), population density of native states, and stereochemical quality—within the context of the GNEIMO (Generalized Newton-Euler Inverse Mass Operator) torsional dynamics method. GNEIMO enhances conformational sampling by focusing on low-frequency torsional degrees of freedom, making it particularly useful for protein structure refinement and the study of folding dynamics [5] [6]. We provide structured protocols and data presentation standards to help researchers rigorously validate their protein structures, ensuring model reliability for downstream applications.
Quantitative Validation Metrics
Root-Mean-Square Deviation (RMSD)
RMSD measures the average distance between atoms of superimposed protein structures, quantifying global structural similarity to a native or reference structure. It is a fundamental metric for assessing refinement accuracy.
Table 1: RMSD Refinement for CASP Targets using GNEIMO-REXMD
| CASP Target | Starting RMSD (Å) | Refined RMSD (Å) | Refinement (ΔÅ) |
|---|---|---|---|
| TR429 | 6.82 | 5.76 | 1.06 |
| TR435 | 2.14 | 1.65 | 0.49 |
| TR453 | 1.51 | 1.10 | 0.41 |
| TR454 | 3.26 | 2.33 | 0.93 |
Data derived from CASP refinement category targets shows GNEIMO-REXMD can improve model quality by over 1.0 Å RMSD [5]. The protocol successfully refined 30 CASP targets without experimental restraints, outperforming unrestrained all-atom Cartesian molecular dynamics [5].
Population Density of Native States
This metric describes the distribution and occupancy of conformational substates sampled during simulation, reflecting the ensemble nature of protein folding and flexibility. GNEIMO enhances sampling of these native-like states.
Table 2: Conformational Substates Sampled by GNEIMO
| Protein (PDB ID) | Type/Transition | Key Sampled Feature | Experimental Validation |
|---|---|---|---|
| Fasciculin (1FAS, 1FSC) | Closed (Apo) to Open (Holo) | Loop I (residues 6-12) flexibility | X-ray crystallography [24] |
| Calmodulin (1CLL, 1DMO) | Ca²⁺-bound (Holo) to Ca²⁺-free (Apo) | Central helix collapse & domain reorientation | NMR ensemble [6] [24] |
GNEIMO simulations sampled transitions between experimentally known substates for fasciculin and calmodulin. The method generated an ensemble satisfying approximately 50% of short- and long-range interresidue distances from NMR structures for the calmodulin transition [24].
Stereochemical Quality
Stereochemical quality assessment evaluates local atomistic geometry, including bond lengths, angles, and torsional angles, to ensure the model is physically plausible.
Table 3: Stereochemical Quality Z-scores for Predicted Structures
| Protein | Prediction Method | Overall Z-score | Assessment |
|---|---|---|---|
| Gαi1 | Homology Modeling | 0.67 | Optimal |
| Gαi1 | AlphaFold | 0.74 | Optimal |
| Gαs | Homology Modeling | 0.52 | Optimal |
| Gαs | AlphaFold | 0.41 | Optimal |
| Hx | Homology Modeling | -1.07 | Satisfactory |
| Hx | AlphaFold | -1.16 | Satisfactory |
The Z-score indicates deviation from average high-resolution crystal structure quality; values ≥0 are optimal, while negative values indicate declining quality [39]. The predicted Local Distance Difference Test (pLDDT) in AlphaFold provides residue-level confidence scores, with functional motifs like heme-binding sites and switch regions often modeled at moderate to high confidence [39].
Experimental Protocols
Protocol 1: GNEIMO-REXMD for Protein Structure Refinement
This protocol refines protein homology models using GNEIMO Torsional Replica Exchange Molecular Dynamics (REXMD) [5].
Workflow: GNEIMO-REXMD Refinement
Step-by-Step Procedure:
- Structure Preparation: Obtain initial decoy structure from homology modeling (e.g., MODELLER [5]) or CASP refinement target.
- Energy Minimization: Perform all-atom conjugate gradient minimization using the
sanderprogram in AMBER suite with the AMBER FF99SB force field to remove steric clashes [5]. - GNEIMO-REXMD Setup:
- Solvation Model: Use Generalized Born/Surface Area (GB/SA) OBC implicit solvation model [5].
- Dielectric Constants: Set interior dielectric to 1.5 and exterior dielectric to 78.3 [5].
- Replicas: Set up 32 replicas spanning 310–415 K temperature range [5].
- Non-bonded interactions: Apply a 20 Å cutoff with smooth switching [5].
- Simulation Execution:
- Integrator: Use the Lobatto integrator with a 5 fs time step [5].
- Thermostat: Employ the Nose-Hoover method for temperature control [5].
- Simulation Time: Run for 15–100 ns per replica [5].
- Replica Exchange: Attempt exchanges between neighboring replicas every 5 ps based on the Metropolis criterion [5].
- Trajectory Analysis:
- Superpose trajectory frames onto the experimental (native) structure.
- Calculate Ca RMSD for all frames to identify the conformation with the lowest RMSD to the native structure.
Protocol 2: Validation of Stereochemical Quality and Native State Sampling
This protocol provides a standardized workflow for comprehensive model validation using multiple metrics [39] [24].
Workflow: Protein Model Validation
Step-by-Step Procedure:
- Stereochemical Quality Checks:
- Use validation servers like WHAT IF in YASARA to calculate an overall Z-score, which averages Ramachandran plot, backbone conformation, and 3D packing quality [39].
- A Z-score ≥ 0 indicates optimal quality comparable to high-resolution crystal structures; negative values suggest declining quality [39].
- pLDDT Analysis for AlphaFold Models:
- Extract per-residue pLDDT scores from AlphaFold predictions.
- Residues with scores > 90 indicate high confidence, 70-90 indicate good confidence, 50-70 indicate low confidence, and <50 indicate very low confidence [39].
- Pay special attention to functional regions like binding sites.
- Native State and Conformational Analysis:
- For simulation trajectories, cluster conformations based on Ca RMSD to identify highly populated native-like states.
- Calculate the population density of clusters; a high population in the cluster closest to the native state indicates good sampling.
- Functional Site Validation:
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents and Computational Tools
| Item Name | Function/Application | Specification Notes |
|---|---|---|
| AMBER99SB Force Field | Provides potential energy functions for MD simulations. | Used for energy minimization and in GNEIMO-REXMD simulations [5]. |
| Generalized Born (GB/SA) Implicit Solvent | Approximates solvent effects without explicit water molecules. | OBC model used in GNEIMO; dielectric constants: interior=1.5-4.0, exterior=78.3 [5] [24]. |
| GNEIMO Software | Torsional MD package for enhanced conformational sampling. | Enables rigid body definitions and all-torsion MD with 5 fs time steps [5] [6]. |
| MODELLER | Homology modeling software to generate initial protein decoys. | Used to create starting models for refinement when experimental structures are unavailable [5]. |
| YASARA Structure | Software suite for validation and analysis. | Calculates overall Z-score for stereochemical quality assessment [39]. |
| AlphaFold2 | AI-based protein structure prediction server. | Provides models with per-residue pLDDT confidence metrics [39]. |
Conclusion
The GNEIMO torsional dynamics method represents a significant paradigm shift in computational structural biology, effectively addressing the critical challenge of conformational sampling. By focusing on low-frequency torsional degrees of freedom, it enables enhanced exploration of the protein energy landscape, leading to consistent and reliable refinement of protein models and insightful folding studies. The integration of advanced features like the Fixman potential, hierarchical clustering, and replica exchange protocols ensures both thermodynamic rigor and computational efficiency. As the field moves forward, GNEIMO's ability to generate high-accuracy, near-experimental structural models holds profound implications. For biomedical and clinical research, this translates into more reliable structures for rational drug design, a deeper understanding of protein function and malfunction in diseases, and the potential to model large-scale conformational changes critical for drug targeting. Future developments will likely focus on integrating GNEIMO with AI-based prediction tools like AlphaFold for multi-scale modeling, further expanding its impact on biology and medicine.
References
- 1. BioMD: All-atom Generative Model for Biomolecular ... [https://arxiv.org/html/2509.02642v1]
- 2. Folding of Small Proteins Using Constrained Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3124276/]
- 3. Structure Refinement of Protein Low Resolution Models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3377353/]
- 4. Past–future information bottleneck for sampling molecular ... [https://www.nature.com/articles/s41467-019-11405-4]
- 5. Protein Structure Refinement of CASP Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3985798/]
- 6. Mapping Conformational Dynamics of Proteins Using ... [https://www.sciencedirect.com/science/article/pii/S0006349513001835]
- 7. Vaidehi Lab Research | Computational and Quantitative ... [https://www.cityofhope.org/research/beckman-research-institute/computational-and-quantitative-medicine/vaidehi-lab/vaidehi-lab-research]
- 8. GneimoSim: A Modular Internal Coordinates Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4211970/]
- 9. Hamiltonian Monte Carlo with Constrained Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5634954/]
- 10. Methods for the Refinement of Protein Structure 3D Models [https://www.mdpi.com/1422-0067/20/9/2301]
- 11. AI meets physics in computational structure-based drug ... [https://www.nature.com/articles/s44386-025-00019-0]
- 12. Structure Prediction & Target Enablement - Schrödinger [https://www.schrodinger.com/life-science/solutions/structure-prediction-and-target-enablement/]
- 13. Optimized protein-water interactions and torsional ... [https://www.nature.com/articles/s41467-025-65603-4]
- 14. Replica exchange molecular dynamics method for protein ... [https://pubmed.ncbi.nlm.nih.gov/16957325/]
- 15. Mapping conformational dynamics of proteins using torsional ... [https://pubmed.ncbi.nlm.nih.gov/23663843/]
- 16. Mastering Hierarchical Clustering : From Basic to Advanced [https://medium.com/@sachinsoni600517/mastering-hierarchical-clustering-from-basic-to-advanced-5e770260bf93]
- 17. Hierarchical Clustering in Machine Learning [https://www.geeksforgeeks.org/machine-learning/hierarchical-clustering/]
- 18. Internal Coordinate Molecular Dynamics: A Foundation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4315417/]
- 19. Fixman compensating potential for general branched ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3888462/]
- 20. Structure refinement of protein low resolution models using ... [https://pubmed.ncbi.nlm.nih.gov/22260550/]
- 21. Force Field Bias in Protein Folding Simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2711430/]
- 22. Optimization of the GBMV2 Implicit Solvent Force Field for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5407932/]
- 23. Effect of solvent model when probing protein dynamics with ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326316303552]
- 24. Mapping Conformational Dynamics of Proteins Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3647154/]
- 25. Recent Advances in Computational Protocols Addressing ... [https://www.mdpi.com/2218-273X/9/4/146]
- 26. NASA Jet Propulsion Laboratory California Institute of ... [https://dartslab.jpl.nasa.gov/GNEIMO/index.php]
- 27. Robust folding of a de novo designed ideal protein even ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7739874/]
- 28. Analysis and visual summarization of molecular dynamics ... [https://jcheminf.biomedcentral.com/articles/10.1186/1758-2946-6-S1-O16]
- 29. GNEIMO - JPL Robotics [https://www-robotics.jpl.nasa.gov/what-we-do/research-tasks/gneimo/]
- 30. Home - Prediction Center [https://predictioncenter.org/]
- 31. Critical Assessment of Methods of Protein Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5394799/]
- 32. Benchmarking of Structure Refinement Methods for Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8671191/]
- 33. Accounting for a mirror-image conformation as a subtle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4060689/]
- 34. S-EPMC6420231 - Validating Molecular Dynamics Simulations ... [https://www.omicsdi.org/dataset/biostudies-literature/S-EPMC6420231]
- 35. A beginner's approach to deep learning applied to VS and MD ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-00985-7]
- 36. Binding of Fasciculin-2 to Acetylcholinesterase - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1432132/]
- 37. Mambalgins, Fasciculins, and Dendrotoxins [https://www.mdpi.com/1422-0067/26/20/9895]
- 38. Differences in Calmodulin and Calmodulin-Binding Proteins in ... [https://pubmed.ncbi.nlm.nih.gov/11742802/]
- 39. Quality Assessment of Selected Protein Structures Derived ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10747200/]