Explicit vs. Implicit Solvent Models in MD Simulations: A Strategic Guide for Biomedical Researchers
Molecular dynamics (MD) simulations are a cornerstone of modern computational biophysics and drug discovery, where accurately modeling the solvent environment is critical.
Explicit vs. Implicit Solvent Models in MD Simulations: A Strategic Guide for Biomedical Researchers
Abstract
Molecular dynamics (MD) simulations are a cornerstone of modern computational biophysics and drug discovery, where accurately modeling the solvent environment is critical. This article provides a comprehensive guide for researchers and drug development professionals on navigating the critical choice between explicit and implicit solvent models. We first establish the foundational principles of both approaches, explaining their theoretical underpinnings and inherent trade-offs between computational cost and physical detail. The guide then delves into practical methodologies and application-specific considerations, from simulating protein-ligand binding to predicting drug solubility. Furthermore, we address common troubleshooting scenarios and optimization techniques, including the integration of machine learning to enhance accuracy and speed. Finally, we present a rigorous framework for the validation and comparative analysis of simulation results, empowering scientists to select the optimal solvent model for their research objectives and efficiently leverage MD simulations to advance biomedical science.
Understanding the Core Physics: From Discrete Waters to a Dielectric Continuum
Frequently Asked Questions
1. What is an explicit solvent model? An explicit solvent model is a computational approach in molecular modeling where each solvent molecule, such as a water molecule, is represented as an individual entity within the simulation [1]. This allows for a detailed, atomistic treatment of the interactions between the solute (the molecule being studied) and the solvent environment [2] [1].
2. How does an explicit solvent model differ from an implicit one? The core difference lies in how the solvent is represented. Explicit models treat solvent as discrete molecules, whereas implicit models treat it as a continuous, polarizable medium defined by properties like the dielectric constant [2]. Explicit models can capture specific, directional interactions like hydrogen bonding, while implicit models are computationally faster but miss these atomic-level details [2] [1] [3].
3. When should I use an explicit solvent model? Explicit solvents are particularly important when specific solute-solvent interactions are critical to the process being studied. Key use cases include:
- Studying biochemical interactions like molecular recognition and drug binding [1].
- Modeling reactions where solvent participates directly, such as acting as a proton bridge [4].
- Simulating systems with significant hydrophobic effects or other solvent structuring [2] [5].
- Investigating highly charged biomolecules like RNA and DNA, where the ionic atmosphere is complex [6] [7].
- When implicit solvent models have been shown to fail for your specific chemical system [5] [4].
4. What are the main computational challenges of using explicit solvents? The primary challenge is the high computational cost. Including thousands of explicit solvent molecules dramatically increases the number of particles in a simulation, which in turn requires significant computational resources and limits the system size and simulation timescale that can be feasibly studied [1] [8] [5]. Adequate sampling also becomes a major concern due to the many degrees of freedom introduced by the solvent [5].
5. Can explicit and implicit solvent models be combined? Yes, hybrid approaches are common and can offer a balance between computational efficiency and accuracy. For example, a small number of explicit solvent molecules can be placed in the first solvation shell of a solute to capture key interactions, while the bulk solvent is treated as an implicit continuum [2] [9] [4]. Quantum Mechanics/Molecular Mechanics (QM/MM) methods are another powerful hybrid technique, where the reactive core is treated with accurate QM, the surrounding solvent is modeled with explicit MM molecules, and the distant bulk solvent is handled implicitly [2] [3].
Troubleshooting Guides
Challenge: Simulation Instability After Solvation
Problem: Your molecular dynamics simulation "blows up" shortly after starting, often due to high initial forces caused by atomic overlaps or improper system preparation [10].
Solution: Follow a structured equilibration protocol to gradually relax the system. The following ten-step protocol is designed to stabilize an explicitly solvated biomolecule for production simulations [10].
Table: Ten-Step System Preparation Protocol for Stable MD Simulations [10]
| Step | Description | Key Actions & Parameters |
|---|---|---|
| 1 | Initial minimization of mobile molecules | 1000 steps Steepest Descent; positional restraints (5.0 kcal/mol/Ų) on large molecules. |
| 2 | Initial relaxation of mobile molecules | 15 ps NVT MD (1 fs timestep); positional restraints (5.0 kcal/mol/Ų) on large molecules. |
| 3 | Initial minimization of large molecules | 1000 steps Steepest Descent; medium positional restraints (2.0 kcal/mol/Ų) on large molecules. |
| 4 | Continued minimization of large molecules | 1000 steps Steepest Descent; weak positional restraints (0.1 kcal/mol/Ų) on large molecules. |
| 5 | Initial relaxation of large molecule substituents | 15 ps NVT MD; positional restraints (2.0 kcal/mol/Ų) on backbone atoms only. |
| 6 | Minimization of entire system | 500 steps Steepest Descent; no positional restraints. |
| 7 | Short relaxation of entire system | 5 ps NVT MD; no positional restraints. |
| 8 | Second minimization of entire system | 500 steps Steepest Descent; no positional restraints. |
| 9 | Second relaxation of entire system | 10 ps NPT MD; no positional restraints. |
| 10 | Final equilibration | NPT MD until system density stabilizes (monitor for a plateau). |
Challenge: Prohibitively High Computational Cost
Problem: The system size with explicit solvent is too large, making the simulation too slow or impossible to run with available resources.
Solution: Consider the following strategies to improve computational efficiency while retaining accuracy:
- Leverage Machine Learning Potentials (MLPs): Train a machine learning model to act as a fast surrogate for the quantum mechanical potential energy surface. This can make ab initio level simulations of reactions in explicit solvent feasible [3] [5]. Active learning strategies can help build efficient training sets [3].
- Adopt a Hybrid (QM/MM) Approach: Model only the chemically active region (e.g., a reaction site) with high-level quantum mechanics (QM), and treat the surrounding solvent with a faster molecular mechanics (MM) force field. A third, implicit solvent layer can be added to represent the bulk solvent [2] [3].
- Use a Cluster-Continuum Model: For certain properties like pKa or reaction barriers, include a small, well-chosen cluster of explicit solvent molecules (e.g., 3-5 water molecules) around the solute to capture key interactions, and embed this cluster in an implicit solvent model to represent the bulk [9] [4]. The number of explicit molecules needed should be determined through convergence testing [4].
- Benchmark Sampling Speed: Be aware that conformational sampling in explicit solvent is slower. One study found speedups for implicit solvent ranged from approximately 1-fold to 100-fold depending on the system, largely due to reduced solvent viscosity [8].
Table: Comparative Sampling Speed: Explicit vs. Implicit Solvent [8]
| Type of Conformational Change | Approximate Sampling Speedup (Implicit vs. Explicit) |
|---|---|
| Small (e.g., dihedral angle flips) | ~1-fold |
| Large (e.g., nucleosome tail collapse) | ~1 to 100-fold |
| Mixed (e.g., miniprotein folding) | ~7-fold (in low viscosity regime) |
Challenge: Determining the Optimal Number of Explicit Solvent Molecules
Problem: It is unclear how many explicit solvent molecules to include in a simulation to get accurate results without making the system unnecessarily large.
Solution:
- For cluster-continuum models: Start with a small number of solvent molecules placed at key interaction sites (e.g., where hydrogen bonds can form, especially for anions) [4]. Systematically increase the number and monitor the convergence of the property of interest (e.g., solvation free energy, reaction barrier) [9] [4].
- For full solvation in MD: Use a simulation box where the solute is separated from its periodic images by a sufficient distance (e.g., 1.0-1.5 nm). Tools in MD packages like CHARMM, GROMACS, or AMBER can automatically create a solvent box with a specified size [9] [6] [10].
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Computational Tools for Explicit Solvent Simulations
| Tool / Reagent | Function / Description | Example Use Case |
|---|---|---|
| Molecular Dynamics Engines | Software to perform energy minimization and molecular dynamics simulations. | GROMACS [6], AMBER [8] [10], CHARMM [9], NAMD [10], OpenMM. |
| Explicit Water Models | Parametrized, simplified representations of water molecules for MD simulations. | TIP3P [8], TIP4P [2], SPC [2], OPC (4-point model, improved accuracy) [6]. |
| Polarizable Force Fields | Advanced force fields that account for changes in a molecule's charge distribution in response to its environment. | AMOEBA [2], SIBFA [2], QCTFF [2]. Crucial for accurate ion and solvent dynamics. |
| Machine Learning Potential (MLP) Methods | Surrogate models trained on QM data to simulate complex potential energy surfaces at lower cost. | Atomic Cluster Expansion (ACE) [3] [5], Gaussian Approximation Potential (GAP) [3], NequIP [3]. For reactive explicit solvent MD [3]. |
| System Building & Automation Tools | Programs and scripts to set up simulation systems, including placing solvent boxes and adding ions. | CHARMM-GUI [10], PACKMOL, Tinker [9], internal tools in MD suites. |
| Enhanced Sampling Libraries | Software plugins that implement advanced sampling algorithms to improve exploration of conformational space. | PLUMED [6]. Used for techniques like metadynamics or Hamiltonian replica exchange (HREX) [6]. |
Frequently Asked Questions (FAQs)
Q1: What is the fundamental principle behind an implicit solvent model? An implicit solvent model, also known as a continuum solvent, replaces explicit solvent molecules with a homogeneously polarizable medium, characterized primarily by its dielectric constant (ε). The solute is embedded in a cavity within this continuum. The model calculates the solvation energy based on the solute's interaction with this polarizable environment, significantly reducing computational cost compared to simulating individual solvent molecules [2].
Q2: What are the main energy components that contribute to the solvation free energy in implicit models? The solvation free energy (ΔGsolv) is typically partitioned into distinct physical components [11] [2]:
- Polar/Polar Electrostatic (ΔGele): Accounts for the solute's charge distribution polarizing the surrounding dielectric medium.
- Non-Polar (ΔGnp): This can be further broken down into:
- Cavity Formation (ΔGcav): The energy required to create a cavity in the solvent for the solute.
- van der Waals Interactions (ΔGvdW): Includes attractive dispersion and repulsive interactions between the solute and solvent.
A common approximation is ΔGsolv = ΔGGB + ΔGSASA, where the polar term is calculated via a Generalized Born (GB) model and the non-polar term is estimated using the Solvent-Accessible Surface Area (SASA) [12].
Q3: My implicit solvent simulation of an intrinsically disordered protein (IDP) is over-compacting and forming unrealistic rigid structures. What could be wrong? This is a known major limitation of many implicit solvent models, which tend to over-stabilize secondary structures like α-helices in disordered proteins [13]. The inherent lack of explicit, atomistic solvent-solute friction and specific solvent-solute interactions can lead to this issue.
- Troubleshooting Step: Consider using a modernized implicit solvent force field that has been specifically optimized for disordered systems. For example, the GB99dms force field was developed by using differentiable molecular simulations to retrain parameters of the a99SB-disp force field and GBNeck2 implicit solvent model against explicit solvent data for disordered proteins, leading to improved radius of gyration and secondary structure content [13].
Q4: Why are my calculated absolute solvation free energies from a machine learning (ML) implicit model unreliable, even when forces appear correct? Many ML-based implicit solvent models are trained solely using a force-matching approach. This method determines the potential energy only up to an arbitrary constant, making the prediction of absolute free energies, which are thermodynamic state functions, inherently inaccurate [12].
- Troubleshooting Step: Look for or develop ML models that extend beyond simple force-matching. Newer approaches, like the λ-Solvation Neural Network (LSNN), incorporate training on derivatives with respect to alchemical variables (λsteric, λelec) to ensure that solvation free energies can be meaningfully compared across different molecules [12].
Q5: How does the choice of an implicit solvent model's specific implementation (e.g., PCM, GB, COSMO) affect its performance? While the underlying principles are similar, different algorithms and their software implementations can yield varying levels of accuracy and computational speed [14].
- Performance Insight: A comparative study found that for small molecules, various implicit models (PCM, GB, COSMO, Poisson-Boltzmann) showed high correlation with experimental hydration energies. However, for larger systems like protein solvation energies and protein-ligand binding desolvation energies, substantial discrepancies (up to 10 kcal/mol) with explicit solvent references can occur. The study noted that the Poisson-Boltzmann equation (APBS) and Generalized Born method (GBNSR6) were among the most accurate for calculating complex desolvation energies [14].
Troubleshooting Guides
Issue 1: Handling IDP Over-compaction with Differentiable Simulations
Problem: Implicit solvent simulations cause intrinsically disordered proteins (IDPs) to collapse into overly compact, non-physical conformations.
Solution: Employ a parameter optimization workflow using differentiable molecular simulation (DMS) to refine the force field and implicit solvent model parameters against explicit solvent reference data.
Experimental Protocol (Based on GB99dms Development [13]):
- System Preparation: Select a training set of proteins, including both folded (e.g., Trp-cage, BBA) and disordered (e.g., Htt-1-19, histatin-5) systems.
- Reference Data Generation: Run long-timescale (e.g., 2 μs) explicit solvent molecular dynamics simulations for the training systems to obtain reference structural data, such as residue-residue distance distributions.
- Differentiable Simulation Setup: Configure differentiable MD simulations (e.g., using Molly.jl) for the training systems. Initialize the simulations with the force field and implicit solvent model you wish to improve (e.g., a99SB-disp and GBNeck2).
- Parameter Selection: Identify a set of parameters to optimize. The GB99dms study tuned 108 parameters, including:
- Atom-specific partial charge scalings, Lennard-Jones σ and ε parameters.
- Torsion force constants for common proper torsions.
- GBNeck2-specific parameters like atom radii, screening parameters, and the neck cutoff.
- Training Loop: For each training epoch: a. Run a short (e.g., 5 ns) implicit solvent simulation for each protein in the training set. b. Calculate the loss by comparing the simulation's residue-residue distance distributions to the reference explicit solvent data using a metric like the Kullback-Leibler (KL) divergence. c. Use automatic differentiation to compute the gradient of the loss with respect to all tunable parameters. d. Update the parameters to minimize the loss.
- Validation: Validate the final optimized force field (e.g., GB99dms) on a separate set of test proteins and against experimental observables like radius of gyration.
The following diagram illustrates this iterative optimization workflow:
Issue 2: Correcting Systematic Errors in Solvation Free Energy Calculations
Problem: Standard implicit solvent models or ML potentials trained only on forces show systematic errors and poor accuracy in predicting absolute solvation free energies.
Solution: Utilize a machine learning framework that integrates experimental solvation free energy data directly into the training process to correct for systematic biases.
Experimental Protocol (Based on the ReSolv Framework [15]):
Stage 1: Bottom-Up Vacuum Potential Training
- Objective: Learn a potential energy surface for molecules in a vacuum.
- Data: Use a large dataset of small organic molecules with ab initio (e.g., DFT) calculated energies and forces in vacuum (from databases like QM9 or ANI).
- Training: Train a machine learning potential (Uvac) to predict the ab initio energies and forces via force-matching.
Stage 2: Top-Down Implicit Solvent Potential Training
- Objective: Learn a potential for molecules in an implicit solvent (Usol) that reproduces experimental solvation free energies.
- Data: Use a curated database of experimental hydration free energies (e.g., FreeSolv).
- Method: The key is to avoid direct backpropagation through the entire MD simulation. The ReSolv framework uses a path reweighting technique (Differential Trajectory Reweighting - DiffTRe): a. For a given molecule, run simulations to compute the free energy difference (ΔA) between Uvac and the current Usol. b. Calculate the loss as the difference between this computed ΔA and the experimental value. c. Use the DiffTRe method to estimate the gradient of this loss with respect to the parameters of Usol. d. Update the parameters of Usol to minimize the loss.
This two-stage protocol ensures the model is both physically grounded (from ab initio data) and thermodynamically accurate (from experimental data).
Quantitative Data Comparison
This table summarizes the performance of various implicit solvent models in calculating solvation energies, compared against experimental data and explicit solvent calculations.
| Implicit Solvent Model | Correlation with Exp. Data (Small Molecules) | Correlation with Explicit Solvent (Small Molecules) | Correlation with Explicit Solvent (Protein Solvation) | Correlation with Explicit Solvent (Protein-Ligand Desolvation) | Typical RMSE for Small Molecules (kcal/mol) |
|---|---|---|---|---|---|
| Poisson-Boltzmann (APBS) | 0.87 - 0.93 | 0.82 - 0.97 | 0.65 - 0.99 | 0.76 - 0.96 | ~3.6 [15] |
| Generalized Born (GBNSR6) | 0.87 - 0.93 | 0.82 - 0.97 | 0.65 - 0.99 | 0.76 - 0.96 | ~3.6 [15] |
| Polarized Continuum (PCM) | 0.87 - 0.93 | 0.82 - 0.97 | 0.65 - 0.99 | 0.76 - 0.96 | - |
| COSMO | 0.87 - 0.93 | 0.82 - 0.97 | 0.65 - 0.99 | 0.76 - 0.96 | - |
| GBSA (Optimized SASA) | - | - | - | - | ~1.68 [15] |
| Machine Learning (ReSolv) | - | - | - | - | < 1.0 (Close to exp. uncertainty) [15] |
Table 2: Key Research Reagents and Computational Tools
A selection of essential software, models, and datasets used in modern implicit solvent research.
| Item Name | Type | Primary Function / Application |
|---|---|---|
| APBS [14] | Software | Solves the Poisson-Boltzmann equation for electrostatic solvation energy calculations. |
| OpenMM [13] | Software Toolkit | A high-performance toolkit for molecular simulation that supports various implicit solvent models. |
| GBNeck2 [13] | Implicit Solvent Model | A Generalized Born model with a "neck" correction for improved molecular surface prediction. |
| a99SB-disp [13] | Force Field | A force field developed for both folded and disordered proteins in explicit solvent. |
| GB99dms [13] | Optimized Force Field | An a99SB-disp/GBNeck2 derivative optimized via DMS for better IDP performance in implicit solvent. |
| LSNN [12] | ML Solvation Model | A graph neural network model trained to provide accurate solvation free energies. |
| ReSolv [15] | ML Framework | A framework to parametrize ML implicit solvent potentials using experimental free energy data. |
| FreeSolv [15] | Database | A curated database of experimental and calculated hydration free energies for small molecules. |
Troubleshooting Common Computational Issues
Frequently Asked Questions
Q1: My calculated hydration free energies (HFEs) for small molecules are consistently overestimated. What could be the source of this error? Systematic overestimation of HFEs is a known issue in some implicit solvent models, particularly 3D-RISM. This error often stems from an artifactual overestimation of pressure within the model [16]. A reliable diagnostic is to apply a Partial Molar Volume Correction (PMVC) or an Element Count Correction (ECC). If these corrections significantly improve agreement with experimental data, it confirms the error originates from the solvation model itself rather than your solute's force field parameters [16].
Q2: How can I determine if errors in my solvation energy calculations are due to the force field or the solvent model? To isolate the error source, you can use a two-step diagnostic approach [16]:
- Apply a post-calculation correction (like PMVECC) to your results. If the correction resolves the discrepancy, the error likely lies with the solvation model.
- Apply the same correction to results from explicit solvent calculations. If the explicit solvent results also improve, this indicates a systematic error in the solute's force field parameters, particularly the Lennard-Jones parameters for specific elements like Cl, Br, I, or P [16].
Q3: When simulating a large, flexible glycan, my implicit solvent simulation yields different global conformations compared to an explicit solvent simulation. Is this expected? Yes, some divergence is possible. While local conformational properties like dihedral angles and sugar ring puckering are often similar between implicit and explicit solvent models, global conformational sampling can differ [17]. Implicit solvent models lack the specific, directional friction and hydrogen bonding of explicit water molecules, which can alter the sampling of extended versus compact states. For studies focused on global conformation, validating key results with shorter explicit solvent simulations is recommended.
Q4: The desolvation penalty I calculated for a protein-ligand complex is significantly off compared to explicit solvent benchmarks. How can I improve this? The accuracy of desolvation energy calculations is highly sensitive to the chosen methodology and its parameterization [14]. For protein-ligand complexes, the Poisson-Boltzmann (PB) equation and the Generalized Born (GBNSR6) method have been shown to be more accurate than simpler models [14]. Ensure that the partial charges and force field parameters for your protein and ligand are consistent with the parameterization of the implicit solvent model you are using, as this is a major source of error.
Troubleshooting Guide: Solvation Energy Calculations
| Problem | Possible Causes | Diagnostic Steps | Recommended Solutions |
|---|---|---|---|
| Systematic HFE Overestimation [16] | 3D-RISM pressure artifact; Incorrect force field parameters. | Apply PMVC or ECC; Check for element-specific trends (Cl, Br, I, P). | Apply a combined PMVECC; Consider refining Lennard-Jones parameters for problematic elements. |
| High Variance in HFE for Flexible Molecules [16] | Inadequate conformational sampling; Use of a single, rigid conformer. | Run MD in a fast solvent (e.g., GB) to check for HFE standard deviation. | Perform conformational sampling before HFE calculation; Use a flexible subset of structures. |
| Poor Correlation with Explicit Solvent Results [14] | Inaccurate partial charges; Mismatch between force field and solvent model parameterization. | Compare multiple charge models (e.g., AM1-BCC, HF/6-31G*); Check literature for best practices. | Recalculate charges with a higher-level method; Use a solvent model parameterized for your force field. |
| Inaccurate Protein-Ligand Desolvation [14] | Use of an oversimplified implicit model (e.g., S-GB, COSMO). | Compare results against a PB or GBNSR6 reference calculation. | Switch to a more accurate model like PB or GBNSR6 for the final calculation. |
Experimental Protocols & Workflows
Protocol 1: Correcting Systematic Errors in Hydration Free Energy Calculations
This protocol uses the Partial Molar Volume with Element Count Correction (PMVECC) to identify and correct errors in 3D-RISM calculations [16].
- Calculation Setup: Perform 3D-RISM calculations to obtain the initial hydration free energy,
ΔG_RISM, and the partial molar volume,v, for each molecule in your dataset. - Element Counting: For each molecule, count the number of atoms,
N_i, for each chemical element (e.g., C, N, O, Cl, P). - Parameter Fitting: Using a training set with experimental HFE data, perform a multi-variable linear regression to fit the parameters in the following equation:
ΔG_Corrected = ΔG_RISM + a*v + b + Σ(c_i * N_i)Here,aandbare the PMV correction parameters, andc_iare the element-specific correction coefficients [16]. - Error Diagnosis: Analyze the fitted
c_icoefficients. Large values for specific elements (e.g., Cl, Br, I, P) indicate systematic errors in the force field's Lennard-Jones parameters for those elements [16]. - Application: Apply the fitted PMVECC equation to all subsequent calculations to obtain corrected, more accurate HFEs.
Protocol 2: Assessing Conformational Flexibility for Solvation Calculations
This protocol helps determine if a molecule is rigid or flexible, which is crucial for deciding whether conformational sampling is needed before a single-conformer solvation calculation [16].
- System Preparation: Prepare the solute molecule in a gas-phase state using your chosen force field.
- Molecular Dynamics Simulation: Run a short (nanosecond-scale) MD simulation of the solute using a fast implicit solvent model like Generalized Born (GB).
- Energy Analysis: Throughout the simulation, calculate the solvation free energy (
ΔG_GB) for each frame. - Rigidity Assessment:
- Calculate the standard deviation of
ΔG_GBover the simulation trajectory. - Compare the
ΔG_GBfrom the full simulation (ΔG_GB,MD) with the value from the first frame only (ΔG_GB,static).
- Calculate the standard deviation of
- Classification: Molecules with a low standard deviation and a small difference between
ΔG_GB,MDandΔG_GB,staticcan be classified as rigid. Molecules with high variance are flexible and require ensemble averaging for accurate solvation free energies [16].
Workflow: Diagnosing Solvation Free Energy Errors
The diagram below outlines a logical workflow for diagnosing the source of errors in solvation free energy calculations, integrating concepts from the troubleshooting guide and protocols.
Quantitative Data for Method Comparison
Table 1: Accuracy of Implicit Solvent Models for Small Molecules
This table summarizes the performance of various implicit solvent models in calculating hydration free energies (HFEs) for small molecules compared to experimental data and explicit solvent calculations. Correlation coefficients (R) and error metrics are provided where available [14].
| Implicit Solvent Model | Software Implementation | Correlation with Experiment (R) | Correlation with Explicit Solvent (R) | Key Notes |
|---|---|---|---|---|
| 3D-RISM with PMVECC | - | - | - | MUE: 1.01 ± 0.04 kcal/mol, requires <15s CPU time per molecule [16] |
| Generalized Born (GB) | GBNSR6 | 0.87 - 0.93 | 0.82 - 0.97 | Proven high accuracy for small molecules [14] |
| Poisson-Boltzmann (PB) | APBS | 0.87 - 0.93 | 0.82 - 0.97 | High accuracy, computationally more expensive than GB [14] |
| Polarized Continuum (PCM) | DISOLV, MCBHSOLV | 0.87 - 0.93 | 0.82 - 0.97 | High numerical accuracy, slower than S-GB [14] |
| COSMO | DISOLV, MOPAC | 0.87 - 0.93 | 0.82 - 0.97 | Conductor-like screening model [14] |
Table 2: Performance on Proteins and Protein-Ligand Complexes
This table compares the performance of implicit solvent models on larger systems, highlighting the challenge of achieving high accuracy for proteins and desolvation penalties [14].
| Implicit Solvent Model | Protein Solvation Energy | Protein-Ligand Desolvation Energy | Key Notes |
|---|---|---|---|
| All Tested Models | Discrepancy up to 10 kcal/mol with explicit solvent | Discrepancy up to 10 kcal/mol with explicit solvent | Accuracy is highly dependent on parameterization [14] |
| Poisson-Boltzmann (PB) | Correlation: 0.65 - 0.99 | Correlation: 0.76 - 0.96 | One of the most accurate for desolvation energies [14] |
| Generalized Born (GB) | Correlation: 0.65 - 0.99 | Correlation: 0.76 - 0.96 (GBNSR6) | GBNSR6 implementation is particularly accurate [14] |
| PCM / COSMO / S-GB | Correlation: 0.65 - 0.99 | Correlation: 0.76 - 0.96 | Performance similar within the same parameterization [14] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software Tools for Solvation Free Energy Calculations
This table lists key software tools and their functions for conducting and analyzing solvation free energy calculations.
| Tool Name | Type | Primary Function | Relevant Context |
|---|---|---|---|
| FreeSolv Database [16] | Database | Public experimental and calculated hydration free energy benchmark for >600 small molecules. | Essential for validation and force field/solvation model parametrization [16]. |
| 3D-RISM [16] [18] | Solvation Model | An integral equation theory-based implicit solvent model providing full solvent structure and thermodynamics. | Calculates HFEs in a single step; requires corrections for pressure artifacts [16]. |
| APBS [14] | Software | Solves the Poisson-Boltzmann equation for biomolecular solvation. | High-accuracy reference for electrostatic solvation; good for desolvation penalties [14]. |
| GBNSR6 [14] | Software | Implements a fast and accurate Generalized Born model for implicit solvation. | Recommended for accurate and efficient HFE and desolvation calculations [14]. |
| DISOLV / MCBHSOLV [14] | Software | Implements multiple implicit models (PCM, S-GB, COSMO) on a smooth solvent boundary. | Allows direct comparison of different models on the same molecular geometry [14]. |
| CheShift [19] | Validation Tool | Uses QM-calculated chemical shifts to assess the accuracy of MD trajectories and force fields. | Helps diagnose errors in atomic coordinates that affect solvation thermodynamics [19]. |
Troubleshooting Guides and FAQs
Poisson-Boltzmann (PB) Equation
Q: My PB calculation is slow or fails to converge for large biomolecules. What strategies can I try? A: Convergence issues are common with the nonlinear PB equation and complex molecular surfaces.
- Preconditioning: Implement a Calderón preconditioner or scaling strategy to improve the conditioning of the boundary integral system, which can significantly speed up solver convergence for large problems [20].
- Formulation Choice: For small molecules, use a direct boundary integral formulation. For larger systems, switch to more complex second-kind integral equations, which perform better as problem size increases [20].
- Mesh Size: If using finite-difference methods, ensure your grid spacing is appropriate. A very fine mesh (e.g., 0.2 Å) increases accuracy but drastically increases computational cost [21].
Q: How can I accurately handle the dielectric interface and charge singularities in PB calculations? A: The discontinuity at the solute-solvent interface is a major source of error.
- Use Advanced Solvers: Employ a solver that rigorously handles interface conditions. The Matched Interface and Boundary (MIB) method, used in the MIBPB solver, is designed to accurately treat these jumps and achieve second-order accuracy [21].
- Surface Definition: Use a stable molecular surface definition, such as the Eulerian solvent-excluded surface, which provides an analytical representation and improves solver robustness [21].
Generalized Born (GB) Model
Q: My GB model provides fast but inaccurate solvation energies. How can I improve accuracy? A: GB is an approximation to PB, and its accuracy depends heavily on parameterization.
- Parameterization: Ensure you are using an appropriate set of atomic radii and empirical parameters tuned for your specific class of molecules (e.g., proteins, nucleic acids) [22] [11].
- Model Selection: Use modern GB models like GBNSR6, which offer improved accuracy over earlier versions. Consider GB as a starting point for screening, and refine promising results with a more accurate PB model [22].
- Machine Learning Correction: Explore emerging machine learning models that use GB features as input to predict PB-level solvation energies, correcting for the inherent approximation of the GB method [21].
Polarizable Continuum Model (PCM) and Quantum-Chemical Models
Q: When integrating PCM with quantum chemistry calculations, what should I consider for accurate results? A: PCM allows for the inclusion of solvation effects in electronic structure calculations.
- Component Breakdown: Ensure the model accounts for all major contributions to the solvation free energy: electrostatic polarization, cavitation (creating the cavity in the solvent), and dispersion/repulsion interactions [22] [11].
- Model Variant: Select the appropriate variant for your task. IEF-PCM is a standard for many applications, while COSMO-RS is well-regarded for solvation properties in organic solvents [11].
- Quantum Workflows: For studies of reaction mechanisms or spectroscopy, use a workflow that couples the continuum solvation model with conformational sampling to account for solvent-induced structural changes [11].
General Implicit Solvent Issues
Q: My implicit solvent simulation fails to capture specific ion or water effects. What are the limitations? A: This is a fundamental limitation of the continuum approximation.
- Identify the Cause: Implicit models average out solvent molecules and ions. They cannot capture specific, atomic-level interactions like water bridging, explicit hydrogen bonding, or ion-specific (Hofmeister) effects [22] [11].
- Solution - Hybrid Modeling: If these effects are critical, use a hybrid approach. Simulate the solute and a few key solvent molecules or ions explicitly, while treating the bulk solvent implicitly. For ion-specific effects, you may need to use explicit ions or an advanced model that incorporates such corrections [11].
Q: How do I choose the right implicit solvent model for my project? A: The choice involves a trade-off between speed, accuracy, and the specific property of interest.
- For high-speed screening of biomolecular conformations or binding poses: Use a modern Generalized Born (GB) model [22] [11].
- For high-accuracy electrostatic analysis or binding free energies: Use a Poisson-Boltzmann (PB) solver [21].
- For studying chemical reactions, spectroscopy, or electronic properties in solution: Use a quantum-continuum model like PCM, COSMO, or an SMx model [22] [11].
Comparison of Implicit Solvent Models
The table below summarizes the key characteristics of the main implicit solvent frameworks.
| Model | Theoretical Basis | Computational Cost | Typical Applications | Key Strengths | Common Limitations |
|---|---|---|---|---|---|
| Poisson-Boltzmann (PB) | Continuum electrostatics; numerical solution of PB equation [22] [21] | High (especially for large systems or fine grids) [21] | Protein-ligand binding, pKa calculations, electrostatic mapping [21] | Rigorous treatment of electrostatics and ionic effects [22] | Slow for large molecules; sensitive to surface definition and parameters [20] [21] |
| Generalized Born (GB) | Approximate analytical solution to the PB equation [22] [11] | Low [22] [11] | Long-timescale MD simulation, conformational sampling, rapid binding estimates [22] [11] | High computational efficiency; suitable for MD [22] [11] | Less accurate than PB; accuracy depends on parameterization [21] |
| Polarizable Continuum Model (PCM) & COSMO | Continuum dielectric in quantum chemistry [22] [11] | Medium to High (depends on QM method) | Solvation energies, reaction rates in solution, spectroscopy [22] [11] | Directly couples solvation to electronic structure [22] [11] | High cost for large systems; requires QM calculation [22] |
Experimental Protocols
Protocol 1: Calculating Electrostatic Solvation Free Energy using a Poisson-Boltzmann Solver
This protocol outlines the steps to compute the electrostatic solvation free energy, a key quantity in implicit solvent modeling [22] [21].
System Preparation:
- Obtain the atomic coordinates of the biomolecule (e.g., from a PDB file).
- Assign atomic charges and radii using a standard force field (e.g., AMBER, CHARMM).
- Define the internal (solute) and external (solvent) dielectric constants (e.g., ε₁=2-4 for solute, ε₂=80 for water).
- Set the ionic strength by specifying the inverse Debye length (κ).
Surface Generation:
- Generate a molecular surface to define the dielectric boundary (Γ). The solvent-excluded surface (SES) is commonly recommended [21].
Numerical Solution:
Energy Calculation:
- Calculate the electrostatic solvation free energy (ΔG) using Eq. 4:
ΔG = 1/2 * Σ qₖ (φ(rₖ) - φ₀(rₖ))where φ₀ is the potential in a uniform dielectric (e.g., the vacuum state) [21].
- Calculate the electrostatic solvation free energy (ΔG) using Eq. 4:
Protocol 2: Incorporating Solvation Effects in Quantum Chemistry Calculations using PCM
This protocol describes how to perform a quantum chemistry calculation with an implicit solvent [22] [11].
Geometry Selection:
- Obtain a molecular structure (can be a gas-phase optimized geometry).
Model Selection and Parameterization:
- Choose a continuum model (e.g., IEF-PCM, COSMO).
- Select a cavity definition (usually based on atomic radii) and specify the solvent's dielectric constant.
Self-Consistent Field (SCF) Calculation:
- Run a quantum chemistry calculation (e.g., HF, DFT). The PCM will be incorporated directly into the Hamiltonian during the SCF procedure, polarizing the electron density of the solute in response to the continuum solvent.
Free Energy Analysis:
The Scientist's Toolkit: Research Reagent Solutions
The table below lists key software tools and their functions for working with implicit solvent models.
| Tool / Reagent | Function / Application | Relevant Model(s) |
|---|---|---|
| APBS | Solves the Poisson-Boltzmann equation for biomolecular electrostatics [21] | Poisson-Boltzmann |
| DelPhi | Finite-difference PB solver for calculating electrostatic potentials and energies [11] [21] | Poisson-Boltzmann |
| MIBPB | A high-order accurate PB solver using the Matched Interface and Boundary method [21] | Poisson-Boltzmann |
| GBNSR6 | A modern Generalized Born model with improved accuracy for molecular dynamics [22] | Generalized Born |
| AGBNP2 | An analytical Generalized Born model that includes nonpolar solvation terms [22] | Generalized Born |
| PCM & COSMO | Quantum chemistry models for computing solvation effects on electronic structure [22] [11] | PCM, COSMO |
| SMx & SMD | Families of quantum-based solvation models parameterized for a wide range of solvents [22] [11] | PCM/GB variants |
Workflow Visualizations
Frequently Asked Questions
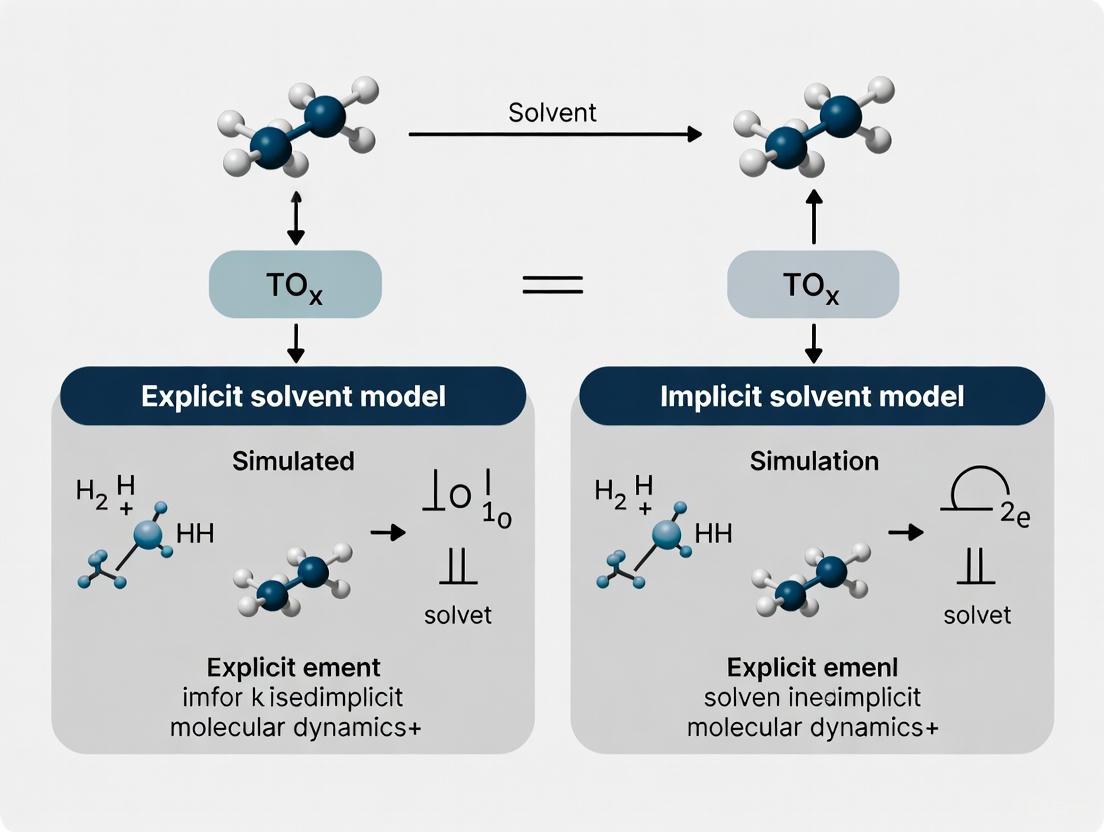
FAQ: What is the core trade-off between explicit and implicit solvent models? The primary trade-off is between computational cost and physical detail. Explicit solvent models individually represent each solvent molecule, offering high physical realism for solute-solvent and solvent-solvent interactions at a high computational cost. Implicit solvent models treat the solvent as a continuous medium, offering significantly faster computational speed but potentially missing specific molecular-level interactions like hydrogen bonding or non-bulk solvent behavior [23] [24].
FAQ: When should I choose an implicit solvent model for my simulation? Implicit solvent models are advantageous when you need to achieve faster conformational sampling, especially for large-scale conformational changes or when computational resources are limited. Studies have shown that for large conformational changes, implicit solvent models can provide a conformational sampling speedup of between approximately 1-fold and 100-fold compared to explicit solvent models [8]. They are also suitable when the phenomenon of interest is primarily influenced by the solvent's electrostatic response rather than specific, individual solvent molecule interactions [24].
FAQ: What are the common pitfalls when switching from an explicit to an implicit solvent model? A common pitfall is the misrepresentation of solvent viscosity, which can lead to an unrealistic speedup in conformational dynamics. Implicit solvent models have reduced friction, which accelerates sampling but may not accurately reflect real-world timescales. Additionally, implicit models might fail to capture specific effects like hydrogen bonding networks, hydrophobic interactions, or the behavior of water molecules in confined protein pockets, which can be critical for biological function [8] [24].
FAQ: How does the Potential of Mean Force (PMF) relate to implicit solvation? The Potential of Mean Force (PMF) is the foundational statistical mechanics concept behind implicit solvent models. It is a free energy quantity that represents the solvent-averaged effective potential governing the solute's behavior. In implicit models, the solvation free energy (ΔGs) is a key component of the PMF, which also includes the solute's internal energy. This decomposition allows implicit models to approximate the thermally averaged effect of the solvent without simulating each solvent molecule explicitly [24].
Troubleshooting Guides
Issue: Inadequate Conformational Sampling in Explicit Solvent
Problem: Your molecular dynamics (MD) simulation in explicit solvent is not sampling the desired conformational space within a practical simulation time.
Solution:
- Consider a Hybrid Approach: Run multiple short simulations with an implicit solvent model to rapidly identify key conformational states. Use these states as starting points for more refined, longer simulations in explicit solvent.
- Validate with Order Parameters: Before relying on implicit solvent results, ensure that the implicit model reproduces key structural properties (e.g., radius of gyration, native contacts) compared to a short explicit solvent control simulation.
- Protocol:
- Set up a simulation using a Generalized Born (GB) implicit solvation model [24].
- Use enhanced sampling techniques (e.g., temperature replica exchange) since the reduced friction in implicit solvent already accelerates sampling [8].
- Cluster the results to identify representative conformations.
- Solvate these representative structures in a box of explicit TIP3P water molecules and run production MD simulations using Particle Mesh Ewald (PME) for electrostatics [8].
Issue: Mismatch Between Calculated and Experimental Reaction Rates or Binding Affinities
Problem: Your implicit solvent simulations yield reaction rates or binding free energies that deviate significantly from experimental values.
Solution:
- Diagnose the Free Energy Components: The solvation free energy in implicit models is typically decomposed into polar and non-polar contributions [24]:
- Polar component (ΔGes): Calculated via Poisson-Boltzmann (PB) or Generalized Born (GB) methods.
- Non-polar component (ΔGnp): Includes cavity formation and van der Waals interactions.
- Check the Non-Polar Term: The non-polar term is often estimated using a simple surface-area relation, which might be insufficient. Consider if more sophisticated approaches are needed for your system.
- Verify Parameter Sets: Ensure that the atomic radii and internal dielectric constant parameters used in the PB/GB calculation are appropriate for your specific solute (e.g., protein, small molecule, nucleic acid). Incorrect parameters are a common source of error [24].
Issue: Unrealistically Fast Dynamics in Implicit Solvent
Problem: The dynamics observed in your implicit solvent simulation appear artificially accelerated compared to experimental data or explicit solvent simulations.
Solution:
- Understand the Cause: The speedup is primarily due to the reduction in solvent viscosity (friction). The conformational sampling speedup increases as the effective viscosity in the model decreases [8].
- Calibrate with a Langevin Thermostat: Use a Langevin thermostat and adjust the collision frequency (γ) to introduce friction back into the system. This parameter can be tuned to match the diffusion coefficients or dynamics observed in explicit solvent simulations [8].
- Interpret Results Carefully: Recognize that the timescales from implicit solvent simulations are often not directly comparable to real-world timescales. Focus on the relative order of events and pathways rather than absolute simulation times.
Quantitative Data Comparison
The table below summarizes a quantitative comparison of conformational sampling speeds between explicit and implicit solvent models, as reported in a systematic study [8].
Table 1: Conformational Sampling Speedup of Implicit vs. Explicit Solvent Models
| Conformational Change Type | Example System | Approximate Sampling Speedup (GB vs. PME) | Primary Cause of Speedup |
|---|---|---|---|
| Small | Dihedral angle flips in a protein | ~1-fold (minimal) | Slight reduction in friction |
| Large | Nucleosome tail collapse, DNA unwrapping | ~1 to 100-fold | Significant reduction in solvent viscosity |
| Mixed | Folding of a miniprotein | ~7-fold (at same T)~50-fold (combined effect) | Reduced viscosity and algorithmic efficiency |
The Scientist's Toolkit: Essential Modeling Components
Table 2: Key Components for Solvation Modeling
| Item | Function in Solvation Modeling |
|---|---|
| Potential of Mean Force (PMF) | The central free energy quantity that serves as the effective potential in implicit solvent models; it averages out the solvent degrees of freedom [24]. |
| Solvation Free Energy (ΔGs) | The energy required to transfer a solute from a vacuum to the solvent. It is a key target for implicit solvent models to predict [24]. |
| Generalized Born (GB) Model | An approximate method for calculating the electrostatic component of the solvation free energy. It is computationally efficient and commonly used in MD simulations [24]. |
| Poisson-Boltzmann (PB) Equation | A more computationally demanding, but often more accurate, approach for calculating the electrostatic solvation free energy compared to GB methods [24]. |
| Continuum Dielectric | Represents the solvent as a medium with a uniform dielectric constant (e.g., ~80 for water), which is a fundamental assumption in most implicit solvent models [24]. |
Decision Workflow for Solvent Model Selection
The following diagram outlines a logical workflow to help researchers choose between explicit and implicit solvent models based on their specific simulation goals and constraints.
Choosing Your Model: Practical Guidance for Key Biomedical Applications
This technical support center provides guidance for researchers grappling with the critical choice between explicit and implicit solvent models in molecular dynamics (MD) simulations. This decision profoundly impacts the physical accuracy, computational cost, and biological relevance of your research, particularly in studies of protein folding, ion-specific effects, and processes where solvent structure is paramount. The following guides and FAQs are designed to help you navigate specific issues and optimize your experimental protocols.
Frequently Asked Questions (FAQs)
Table: Quick Guide to Solvent Model Selection
| Research Context | Recommended Solvent Model | Key Rationale | Primary Trade-off |
|---|---|---|---|
| Protein Folding & Large Conformational Changes | Implicit Solvent (for initial sampling) | Significantly faster conformational sampling (∼1- to 100-fold speedup) [8]. | Potential inaccuracies in free-energy landscapes and solvent-mediated interactions [8]. |
| Ion-Specific Effects & Binding | Explicit Solvent | Captures specific ion-peptide interactions and correct solvent structure [25]. | High computational cost and slower dynamics due to solvent viscosity [25] [8]. |
| Validating/Refining Folding Mechanisms | Explicit Solvent | Provides physically accurate, high-resolution data on the folding process and non-native intermediates [26]. | Computationally challenging; limits system size and simulation time [26]. |
| Calculating Quantitative Thermodynamic Properties | Explicit Solvent | More reliable for rigorous comparison with experiment due to physical treatment of solvent. | Requires extensive sampling to overcome slower conformational dynamics [8]. |
FAQ 1: For studying protein folding, when is it acceptable to use an implicit solvent model to speed up my simulations?
Implicit solvent models, such as Generalized Born (GB), are acceptable for initial studies of protein folding or large conformational changes where sampling speed is the primary concern. Research shows GB can accelerate conformational sampling by approximately 1- to 100-fold compared to explicit solvent models like PME-TIP3P, depending on the size and nature of the conformational change [8]. However, this speedup comes with a significant caveat: the free-energy landscapes may differ substantially from those generated with explicit solvent. Therefore, implicit solvent is best used for exploratory work or when explicit solvent sampling is entirely infeasible, with the understanding that results may require validation [8].
FAQ 2: My research focuses on ion-specific effects. Which solvent model is necessary?
For research into ion specificity, explicit solvent is mandatory. Implicit solvent models treat ions through a mean-field electrostatic approach and cannot capture specific, atomistic interactions between ions and the solute. For example, explicit-solvent MD simulations revealed that sodium ions (Na⁺) can become tightly bound by several carbonyl and carboxylate groups on a peptide, leading to long-lived, compact configurations that dramatically slow α-helical folding kinetics. This highly specific action, which could not be reproduced with an implicit model, creates individual kinetic barriers and reduces the peptide's configurational mobility by an order of magnitude [25].
FAQ 3: What is the primary source of speedup in implicit solvent simulations?
The speedup in implicit solvent simulations is primarily due to two factors:
- Reduction in Solvent Viscosity: The main effect is the removal of the frictional drag exerted by explicit solvent molecules. Conformational sampling speedup increases as the effective viscosity in the simulation decreases [8].
- Computational Efficiency: For small- to medium-sized systems, implicit solvents can be computationally faster because they eliminate thousands of solvent-solute and solvent-solvent non-bonded interactions. However, this algorithmic advantage may diminish or reverse for very large systems [8].
Quantitative Data Comparison
Table: Comparative Performance of Explicit vs. Implicit Solvent Models
| Simulation Type and Metric | Explicit Solvent (PME) | Implicit Solvent (GB) | Observed Speedup (GB vs. PME) |
|---|---|---|---|
| Small Conformational Changes (e.g., dihedral angle flips) | Baseline | Comparable | ~1-fold (minimal speedup) [8] |
| Large Conformational Changes (e.g., nucleosome tail collapse) | Baseline | Faster | ~1- to 100-fold [8] |
| Mixed Changes (e.g., miniprotein folding) | Baseline | Faster | ~7-fold (in conformational sampling) [8] |
| Ion-Specific Effects (e.g., Na⁺ binding kinetics) | Captures specific ion binding and trapping | Cannot capture specific atomistic binding | Not applicable; explicit solvent required [25] |
| Configurational Mobility (Diffusivity) | Baseline | Higher | Increase of ~1 order of magnitude in Na⁺ salts [25] |
Experimental Protocols
Protocol 1: Investigating Ion-Specific Effects on Protein Folding with Explicit Solvent
This protocol details the methodology for studying how specific ions influence protein folding kinetics, as exemplified in studies of α-helical peptides [25].
1. System Setup:
- Peptide Selection: Use a well-defined oligopeptide, such as Ace-AEAAAKEAAAKA-Nme.
- Initial Coordinates: Obtain or generate an initial extended or unfolded structure.
- Solvation: Solvate the peptide in an explicit solvent box (e.g., TIP3P water model) large enough to accommodate the fully extended peptide with a sufficient cutoff distance from periodic images.
- Ion Addition: Add ions (e.g., KCl, NaCl, NaI) to the system to achieve the desired molar concentration and overall charge neutrality. Using different salts allows for the direct comparison of ion-specific effects.
2. Simulation Parameters:
- Force Field: Choose an appropriate force field (e.g., AMBER, CHARMM) with compatible parameters for the ions.
- Electrostatics: Use the Particle Mesh Ewald (PME) method for long-range electrostatic interactions [8].
- Temperature Coupling: Maintain a constant temperature using a thermostat like Nosé-Hoover.
- Pressure Coupling: Use a barostat like Parrinello-Rahman for constant pressure conditions.
- Constraints: Apply constraints to all bond lengths involving hydrogen atoms (e.g., LINCS algorithm) to allow a integration time step of 2 fs [27].
3. Execution & Analysis:
- Equilibration: Conduct a multi-step equilibration to relax the solvent and ions around the peptide.
- Production Run: Perform multiple, long-timescale production simulations to ensure adequate sampling of folding and unfolding events.
- Kinetic Analysis: Calculate the mean first passage times (MFPT) for folding and unfolding.
- Ion Binding Analysis: Monitor residence times of ions in the peptide's first solvation shell and identify long-lived, trapped configurations [25].
- Diffusional Analysis: Model the folding process as diffusion in a one-dimensional free-energy landscape to quantify how ions modify equilibrium properties and introduce kinetic barriers [25].
Protocol 2: Comparing Folding Pathways with Explicit and Implicit Solvent
This protocol provides a framework for benchmarking implicit solvent models against explicit solvent for a specific system, such as a miniprotein or the villin headpiece [8] [26].
1. System Preparation:
- System: Select a well-studied folding system (e.g., villin headpiece).
- Explicit Setup: Prepare the system as in Protocol 1.
- Implicit Setup: For the implicit solvent simulation, use the same initial coordinates and solute parameters, but with a GB solvation model and a corresponding surface-area term for non-polar contributions. No explicit water molecules are added.
2. Simulation Execution:
- Multiple Runs: For both explicit and implicit setups, launch an ensemble of simulations (e.g., 10-100 runs) starting from different unfolded conformations.
- Consistent Parameters: Use identical temperature, integration time steps, and other parameters where possible. Note that implicit solvents may allow for a larger time step.
- Timing: Run simulations for a consistent nominal time (e.g., microseconds) or until folding is observed multiple times.
3. Comparative Analysis:
- Folding Time: Calculate and compare the average folding times between the two solvent models to determine the conformational sampling speedup [8].
- Pathway Analysis: Compare the dominant folding pathways, the presence of intermediates (native and non-native), and the structural properties of the transition state ensembles [26].
- Free-Energy Landscapes: Construct free-energy landscapes as a function of relevant reaction coordinates (e.g., RMSD, radius of gyration) to identify any major differences induced by the solvent model [8].
Troubleshooting Common Issues
Problem: Insufficient Conformational Sampling in Explicit Solvent
- Symptoms: Folding/unfolding events are not observed; simulations get trapped in non-native states; poor statistics on calculated properties.
- Solutions:
- Ensemble Simulations: Run multiple simulations (dozens to hundreds) starting from different initial conditions rather than one long simulation [26].
- Enhanced Sampling: Employ advanced sampling techniques (e.g., metadynamics, replica exchange) to overcome high free-energy barriers.
- Hybrid Approach: Use an implicit solvent model to generate an ensemble of putative folding intermediates, then solvate these structures in explicit solvent for shorter, refinement simulations [8].
Problem: Simulation Instabilities or Crashes with Explicit Solvent
- Symptoms: Simulation terminates with errors related to bond lengths, van der Waals contacts, or particle mesh Ewald (PME).
- Solutions:
- Check Equilibration: Ensure the system is properly equilibrated before starting production runs. Inadequate equilibration can cause sudden pressure or energy spikes.
- Verify Parameters: Double-check all parameters in your molecular dynamics parameter (.mdp) file. Common errors include incorrect cutoffs, thermostat/barostat settings, or PME parameters [28].
- Review Topology: Use tools like
gmx checkto verify the integrity of your run input (.tpr) and trajectory files. Ensure your topology matches your coordinate file and that all necessary molecules are defined [29] [28].
Problem: Unphysical Results with Implicit Solvent
- Symptoms: Over-stabilization of non-native contacts, incorrect protein dynamics, or failure to fold to the native state.
- Solutions:
- Parameter Tuning: The performance of implicit solvent models is highly sensitive to parameters like the effective Born radius and the non-polar solvation term. Experiment with different parameter sets or consider using a more modern variant of the model.
- Temperature Calibration: The effective friction in implicit solvent is controlled by the Langevin collision frequency. Adjusting this parameter and/or the simulation temperature can help match the dynamics observed in explicit solvent or experiment [8].
- Know the Limitations: Accept that some systems, particularly those dependent on specific water-mediated interactions or ion effects, are not suitable for implicit solvent and require an explicit treatment [25].
Visualization of Workflows and Relationships
The Scientist's Toolkit: Essential Research Reagents & Materials
Table: Key Components for Explicit Solvent Protein Folding Studies
| Item | Function / Role | Example / Specification |
|---|---|---|
| Force Field | Defines the potential energy function and parameters for all atoms in the system. | AMBER, CHARMM, OPLS-AA. Must be chosen for compatibility with ions and the protein system [27]. |
| Water Model | Explicitly represents water molecules and their interactions with the solute. | TIP3P, SPC/E, TIP4P. TIP3P is commonly used with the CHARMM force field [8]. |
| Ion Parameters | Define the non-bonded interactions (charge, size) for cations and anions. | Critical for ion specificity studies; parameters must be consistent with the chosen force field [25]. |
| Software Suite | Provides the computational engine to run MD simulations. | GROMACS, AMBER, NAMD. GROMACS is widely used for its performance and analysis tools [29] [28]. |
| Analysis Tools | Used to process simulation trajectories and compute relevant properties. | Built-in GROMACS tools (gmx rms, gmx gyrate), VMD, MDAnalysis, custom scripts [30]. |
| High-Performance Computing (HPC) | Provides the necessary computational power to run explicit solvent simulations. | Computer clusters with multi-core CPUs and GPUs are essential for achieving microsecond-plus timescales [30] [27]. |
Troubleshooting Guides
Common Implicit Solvent Simulation Errors and Solutions
Problem: Inadequate Sampling of Conformational Space
- Symptoms: Failure to observe expected conformational changes (e.g., protein folding, loop movement) within simulation time; high variance in calculated free energies.
- Solutions:
- Utilize the inherent speed advantage of implicit solvent models to run longer simulations [31] [32].
- Combine implicit solvent with enhanced sampling techniques like Replica Exchange Molecular Dynamics (REMD), which is more feasible due to the reduced number of degrees of freedom [33].
- Verify that the reduced solvent viscosity (Langevin collision frequency) is not set too low, as this can be tuned to balance sampling speed and physical realism [31].
Problem: Inaccurate Free Energy Estimates
- Symptoms: Significant deviations from experimental binding affinity or solvation free energy data.
- Solutions:
- For binding affinity calculations, ensure the solvation free energy of the ligand, protein, and complex are calculated consistently. The binding energy is ΔGbind = ΔGs,pl - ΔGs,l - ΔGs,p [24].
- Consider a hybrid explicit/implicit solvent approach. Use implicit solvent to rapidly sample the conformational landscape and calculate free energy differences (ΔG0,A→B), then use targeted explicit solvent simulations to compute corrections (ΔG0→1,A, ΔG0→1,B) for specific basins, as shown in the workflow below [33].
Problem: Force Field and Parameterization Errors
- Symptoms: Systematic errors in hydration free energies for specific classes of molecules (e.g., containing Cl, Br, I, P); instability of native protein structures.
- Solutions:
- Be aware that errors can originate from the solute force field itself, not just the solvent model. Element-specific corrections may be necessary [16].
- Use optimized implicit solvent force fields (e.g., within CHARMM or AMBER) that have been specifically parameterized to improve the balance of solvation and intramolecular interactions, which is crucial for folding simulations [32].
FAQs: Implicit vs. Explicit Solvent Models
Q1: What is the fundamental difference between implicit and explicit solvent models?
- A: In explicit solvent models, water molecules are represented individually, requiring simulation of all solvent degrees of freedom. In implicit models, the solvent is treated as a continuous medium, and its mean influence on the solute is captured by a solvation free energy term (Potential of Mean Force, or PMF), dramatically reducing the number of degrees of freedom [24] [32].
Q2: When should I prioritize using an implicit solvent model?
- A: Implicit solvents are particularly advantageous in the following scenarios [31] [33] [32]:
- Rapid Conformational Sampling: When studying processes like protein folding, large loop movements, or domain rearrangements where the reduction of solvent viscosity accelerates barrier crossing.
- Binding Affinity Estimates: For high-throughput screening of ligand binding where a trade-off between speed and accuracy is acceptable.
- Enhanced Sampling Techniques: When performing REMD, as the smaller system size reduces the number of replicas required.
- System Size Limitations: When simulating large systems where explicit solvent would be computationally prohibitive.
Q3: What are the key limitations of implicit solvent models?
- A: The main limitations stem from the mean-field approximation [24] [32]:
- Loss of Molecular Detail: They cannot capture specific, structured water molecules (e.g., in binding pockets or mediating hydrogen bonds).
- Simplified Non-Polar Interactions: The treatment of hydrophobic effects often relies on simple solvent-accessible surface area (SA) models, which may not capture length-scale dependence accurately.
- Accuracy Trade-off: While faster, they can be less accurate than explicit solvent simulations, especially in environments where water plays a specific chemical role.
Q4: How much faster is conformational sampling with implicit solvents?
- A: The speedup is highly system-dependent. The table below summarizes quantitative findings from a comparative study [31] [8].
Table 1: Conformational Sampling Speedup of Implicit vs. Explicit Solvent
| Conformational Change Type | Example System | Approximate Sampling Speedup (GB vs. PME) |
|---|---|---|
| Small (Dihedral flips) | Protein side chains | 1-fold |
| Large (Macromolecular rearrangements) | Nucleosome tail collapse, DNA unwrapping | Between ~1-fold and ~100-fold |
| Mixed (Folding) | Miniprotein | ~7-fold |
Q5: Can I combine the strengths of both implicit and explicit solvent models?
- A: Yes. Hybrid methods are an active area of research. One approach involves using implicit solvent to quickly map the free energy landscape and identify basins, then performing targeted explicit solvent simulations to calculate accurate correction terms for these specific basins, leveraging a thermodynamic cycle to obtain explicit-solvent quality results at a fraction of the computational cost [33].
Experimental Protocols & Workflows
Protocol: Calculating Conformational Free Energy Difference via a Hybrid Solvent Approach
This protocol is adapted from a method that connects free energy surfaces in implicit and explicit solvent [33].
1. Define the System and Basins: * Choose your solute molecule (e.g., a protein or peptide). * Identify the conformational basins of interest (A and B) using suitable order parameters (e.g., dihedral angles, RMSD).
2. Implicit Solvent Sampling: * Perform extensive molecular dynamics simulation of the solute in an implicit solvent (e.g., a Generalized Born model). * Objective: Achieve sufficient sampling of the transition between basins A and B, which is facilitated by the reduced solvent friction. * Output: The free energy difference between basins A and B in implicit solvent, ΔG0,A→B.
3. Calculate Basin Populations in Implicit Solvent: * From the implicit solvent trajectory, calculate the relative population fractions of a selected cell (a small region within the basin, denoted a1) within basin A, PA0,a1. Do the same for a cell b1 in basin B, PB0,b1.
4. Localized Explicit Solvent Correction Simulations: * For each selected cell (a1 and b1), run a simulation in explicit solvent. * Objective: Calculate the free energy cost of "transferring" the solute conformation from the implicit solvent environment to the explicit solvent environment. This is the localized decoupling free energy, ΔG0→1,a1 and ΔG0→1,b1. * Method: This can be done using free energy perturbation (FEP) or thermodynamic integration (TI) by coupling a lambda parameter to scale the interactions between the solute and the explicit solvent environment.
5. Calculate Cell Populations in Explicit Solvent: * From the (shorter) explicit solvent simulations, calculate the relative population fractions PA1,a1 and PB1,b1 for the same cells.
6. Compute the Total Explicit Solvent Free Energy Difference: * Use the thermodynamic cycle to combine the results. The free energy difference in explicit solvent is given by: * ΔG1,A→B = –ΔG0→1,A + ΔG0,A→B + ΔG0→1,B * Where the basin transfer free energies are calculated as: * ΔG0→1,A = –kBT ln [ PA0,a1 exp(–ΔG0→1,a1/kBT) / PA1,a1 ] * (A similar equation is used for ΔG0→1,B)
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Computational Tools and Models for Solvent Methods
| Item | Function / Description | Example Use Case |
|---|---|---|
| Generalized Born (GB) Model | An approximate implicit solvent model that calculates electrostatic solvation free energy analytically, enabling fast force calculations and MD integration [24] [32]. | Rapid conformational sampling; folding simulations of small peptides and proteins. |
| Poisson-Boltzmann (PB) Equation | A more computationally demanding but often more accurate implicit solvent model for calculating electrostatic solvation by solving a differential equation for the electrostatic potential [24]. | Single-point energy calculations; final accurate solvation energy estimates for static structures. |
| 3D-RISM | An implicit solvent model based on statistical mechanics integral equations that uses an all-atom solvent model, providing complete solvation thermodynamics in a single calculation [16]. | Hydration free energy (HFE) calculations for small molecules; identifying force field errors. |
| Thermodynamic Integration (TI) / Free Energy Perturbation (FEP) | A class of methods used to compute free energy differences by gradually mutating one system into another along a coupling path [24] [33]. | Calculating solvation free energies; performing alchemical transformations for binding affinity estimates. |
| Replica Exchange MD (REMD) | An enhanced sampling technique that runs multiple simulations at different temperatures (or Hamiltonians) and periodically exchanges configurations to improve sampling [33] [32]. | Overcoming free energy barriers in both explicit and implicit solvent simulations, though more efficiently in the latter. |
| Solvent-Accessible Surface Area (SA) | A common method for estimating the non-polar component of the solvation free energy, which includes cavity formation and van der Waals interactions [24] [32]. | Part of the GB/SA or PB/SA implicit solvent model to account for hydrophobic effects. |
Frequently Asked Questions (FAQs)
FAQ: What are the key MD properties for predicting drug solubility with ML? Through rigorous analysis, seven molecular dynamics (MD)-derived properties have been identified as highly effective for predicting aqueous solubility: the octanol-water partition coefficient (logP), Solvent Accessible Surface Area (SASA), Coulombic_t, LJ (Lennard-Jones interaction energy), Estimated Solvation Free Energies (DGSolv), Root Mean Square Deviation (RMSD), and the Average number of solvents in the Solvation Shell (AvgShell) [34] [35] [36]. These properties can be used as input features for machine learning models, with the Gradient Boosting algorithm demonstrating particularly strong performance (R² = 0.87, RMSE = 0.537 on a test set) [34].
FAQ: Should I use an explicit or implicit solvent model for my solubility simulation? The choice depends on your specific goals and the trade-off between computational cost and physical accuracy [37] [8] [2].
- Explicit solvent models treat solvent molecules individually, providing a physically detailed description of specific solute-solvent interactions (like hydrogen bonding) and local solvent structure [5] [2]. However, they are computationally demanding because they add many degrees of freedom [5].
- Implicit solvent models (or continuum models) replace explicit solvent molecules with a polarizable continuum, significantly speeding up calculations and improving conformational sampling speed [37] [8] [2]. Their main drawback is the inability to capture specific, directional interactions between the solute and solvent molecules [5] [2].
For a quick comparison, refer to the table below.
Table 1: Comparison of Explicit vs. Implicit Solvent Models for MD Simulations
| Feature | Explicit Solvent | Implicit Solvent |
|---|---|---|
| Computational Cost | High [5] | Low [37] |
| Sampling Speed | Slower [8] | Faster (1x to 100x speedup reported) [8] |
| Physical Realism | High; captures specific interactions [5] | Lower; models mean-field effects [37] |
| Key Strengths | Accurate hydrogen bonding, local structure, hydrophobic effect [5] | Efficiency, good for bulk electrostatic effects [37] |
| Key Limitations | Expensive, requires extensive sampling [5] | Poor for charged species & specific interactions [5] |
FAQ: My explicit solvent MD simulation is failing or hanging. What should I do? Errors and hangs during explicit solvent MD simulations can stem from various sources. A common example is an OpenMM simulation that runs for a few iterations before failing with an error like "Error downloading array energyBuffer: Invalid error code (700)" or hanging completely [38]. Initial troubleshooting should focus on your computational environment, as such errors can sometimes be related to the hardware or driver stability on the cluster nodes being used [38].
FAQ: Are there new datasets or models that can improve my ML-driven solubility predictions? Yes, the field is advancing rapidly. Meta's Fundamental AI Research (FAIR) team recently released the Open Molecules 2025 (OMol25) dataset, a massive resource of over 100 million high-accuracy quantum chemical calculations [39]. They also released pre-trained neural network potentials (NNPs) like eSEN and the Universal Model for Atoms (UMA), which demonstrate state-of-the-art performance in accurately and quickly computing molecular energies [39]. These tools can be invaluable for generating more accurate training data or serving as foundational models for property prediction in drug discovery.
Troubleshooting Guides
Guide: Implementing an ML Workflow for Solubility Prediction
This guide outlines the methodology from a recent study that successfully predicted drug solubility using MD properties and machine learning [34].
Workflow Overview The following diagram illustrates the key stages of the experimental workflow.
Protocol Details
Data Collection
MD Simulations Setup
- Software: Use an MD software package like GROMACS [34].
- Force Field: Select an appropriate force field, for example, GROMOS 54a7 [34].
- Ensemble: Perform simulations in the isothermal-isobaric (NPT) ensemble to mimic realistic conditions [34].
- Solvation: Solvate the drug molecules in a suitable water model within a simulation box [34].
Feature Extraction
- From the MD trajectories, extract the key physicochemical properties. The study identified the following seven as most impactful [34]:
- logP
- Solvent Accessible Surface Area (SASA)
- Coulombic Interaction Energy (Coulombic_t)
- Lennard-Jones Interaction Energy (LJ)
- Estimated Solvation Free Energy (DGSolv)
- Root Mean Square Deviation (RMSD)
- Average number of solvents in Solvation Shell (AvgShell)
- From the MD trajectories, extract the key physicochemical properties. The study identified the following seven as most impactful [34]:
Feature Selection & ML Model Training
- Use statistical analysis or feature importance methods to select the most relevant properties for your model [34].
- Train ensemble machine learning algorithms. The cited study compared four:
- Random Forest (RF)
- Extra Trees (EXT)
- eXtreme Gradient Boosting (XGB)
- Gradient Boosting Regression (GBR)
- The Gradient Boosting algorithm achieved the best performance in the referenced study [34].
Table 2: Key MD-Derived Properties for Solubility Prediction
| Property | Description | Role in Solubility |
|---|---|---|
| logP | Octanol-water partition coefficient (experimental) | Measures lipophilicity; high logP generally correlates with lower solubility [34]. |
| SASA | Solvent Accessible Surface Area | Represents the surface area a solvent can access; related to solvation energy [34] [37]. |
| DGSolv | Estimated Solvation Free Energy | The free energy change of solvation; more negative values favor solubility [34]. |
| Coulombic_t | Coulombic Interaction Energy | Represents electrostatic solute-solvent interactions [34]. |
| LJ | Lennard-Jones Interaction Energy | Represents van der Waals solute-solvent interactions [34]. |
| AvgShell | Avg. solvents in Solvation Shell | Indicates the local solvation environment and packing [34]. |
Guide: Choosing Between Explicit and Implicit Solvent Models
Use the decision diagram below to help select an appropriate solvent model for your molecular dynamics project.
Key Considerations:
- When Explicit Solvent is Necessary: Choose explicit models when your research question depends on specific, atomistic solute-solvent interactions. This is crucial for processes like detailed hydrogen bonding network analysis, modeling the hydrophobic effect, or studying systems where water acts as a reactant or plays a direct structural role (e.g., ion coordination) [5] [2].
- When Implicit Solvent is Sufficient: Implicit models are a good choice for large-scale conformational sampling, studying folding or large-scale dynamics, or when computational resources are limited and the system is primarily influenced by bulk electrostatic effects rather than specific solvent interactions [37] [8].
- Hybrid Approaches: For a balance, consider hybrid (or "cluster-continuum") approaches. These model a few explicit solvent molecules in the first solvation shell (to capture key interactions) while using an implicit model to represent the bulk solvent, offering a compromise between accuracy and cost [9] [2].
The Scientist's Toolkit
Table 3: Essential Research Reagents & Computational Tools
| Item / Resource | Function / Application |
|---|---|
| GROMACS | A software package for performing MD simulations; used to simulate the dynamics of drug molecules in solution [34]. |
| GROMOS 54a7 Force Field | A force field used to model the molecules' neutral conformation, generating topology and initial coordinate files for simulations [34]. |
| Solvent Accessible Surface Area (SASA) | An MD-derived property that models the non-polar component of solvation free energy; a key descriptor for solubility [34] [37]. |
| ωB97M-V/def2-TZVPD | A high-level of theory in quantum chemistry; used to generate accurate reference data for training advanced models (e.g., in the OMol25 dataset) [39]. |
| Open Molecules 2025 (OMol25) | A massive, high-accuracy dataset of quantum chemical calculations; can be used to train or benchmark models for molecular property prediction [39]. |
| Neural Network Potentials (NNPs) | Machine learning models, such as eSEN or UMA, that provide a fast and accurate way to compute potential energy surfaces, accelerating MD simulations [39]. |
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between explicit and implicit solvent models in molecular dynamics (MD) simulations?
Explicit solvent models treat each solvent molecule as an individual entity, using specific point-charge models like TIP3P, TIP4P, or OPC to represent water. This allows for a detailed, atomistic description of solvent structure and specific solute-solvent interactions, such as hydrogen bonding [40] [11]. In contrast, implicit solvent models replace the explicit solvent with a continuous dielectric medium, approximating its average effect. This is computationally efficient but can struggle to accurately capture specific, local interactions like explicit hydrogen bonds or the entropic contributions of solvent molecules [41] [11].
Q2: When should I choose an explicit solvent model for simulating polymeric nanoparticles (PNPs)?
An explicit solvent model is preferred when your study involves processes where the precise structure and dynamics of the solvent are critical. This includes:
- Analyzing specific molecular recognition events, such as the role of water bridges in stabilizing a drug-polymer complex.
- Studying systems where implicit models are known to fail, particularly for charged species or when explicit inner-sphere solvent-solute interactions (like hydrogen bonding) are important [41].
- Investigating the structure and dynamics at a molecular interface, where the explicit reorganization of solvent molecules can influence the mechanism [41].
Q3: What are the main limitations of implicit solvent models, and how is machine learning (ML) helping to overcome them?
Traditional implicit models have two key limitations: they often lack accuracy for charged species and specific interactions, and they simplify the non-polar solvation free energy using a simple solvent-accessible surface area (SASA) term, which can be a significant source of error [41] [12]. Machine learning is being used to address these issues by developing more accurate neural network potentials. For instance, some novel ML models are trained not only on forces but also on derivatives of alchemical variables, enabling them to predict solvation free energies with an accuracy comparable to explicit solvent calculations, but at a much lower computational cost [12].
Q4: How does the choice of water model (e.g., TIP3P vs. OPC) affect the simulation outcome for a biopolymer like heparin?
The water model can significantly influence conformational dynamics. A recent benchmark study on a heparin dodecamer found that while TIP3P and SPC/E yielded stable conformations, models like TIP4P, TIP5P, and OPC introduced greater structural variability [40]. This highlights that the choice of solvent model is not neutral and can systematically impact simulation results, especially for highly charged and flexible systems like sulfated carbohydrates [40].
Troubleshooting Common Issues
Issue 1: Poor Sampling of Solvent-Driven Conformational Changes
- Problem: Your simulation fails to capture a known solvent-driven conformational change in a polymer or drug-polymer complex, or the sampling is too slow to be practical.
- Diagnosis: This is a classic challenge in MD, where the timescale of the event of interest exceeds the practical simulation time. This is exacerbated in explicit solvent because the system size is larger and the solvent degrees of freedom must also be sampled [41].
- Solution:
- Consider ML-Accelerated Explicit Solvent: A powerful emerging workaround is to use machine learning to create a fast, accurate potential trained on high-level quantum chemical data for your specific system. Once trained, this ML potential can be used to run the MD simulation, enabling the extensive sampling needed to reconstruct free energy surfaces in explicit solvent without prohibitive computational cost [41].
- Utilize Advanced Datasets and Models: Leverage newly released, massive datasets and pre-trained models like Meta's OMol25 and the associated eSEN or Universal Models for Atoms (UMA). These resources provide high-accuracy neural network potentials that are far more efficient than traditional quantum chemistry methods, making them suitable for large systems like polymers [39].
- Hybrid Approach: If using pure explicit solvent is not feasible, consider using an ML-augmented implicit solvent model that is trained to capture local solvation effects more accurately than traditional models, thus providing a better balance between speed and accuracy for conformational sampling [12].
Issue 2: Inaccurate Solvation Free Energy Predictions
- Problem: Your implicit solvent model yields solvation free energies that are consistently off compared to experimental data or explicit solvent benchmarks, undermining the reliability of your predictions for drug loading and release.
- Diagnosis: Traditional implicit solvent models, which use a combination of a polar term (e.g., Generalized Born, Poisson-Boltzmann) and a non-polar SASA term, have inherent accuracy limits. The SASA approximation for the non-polar component is a known source of error [12] [11].
- Solution:
- Adopt a Modern ML-Based Implicit Model: Implement next-generation implicit solvent models like the λ-Solvation Neural Network (LSNN). Unlike models trained only on force-matching, LSNN is also trained on derivatives of alchemical variables, allowing it to predict absolute solvation free energies accurately, bridging the gap between implicit and explicit solvent accuracy [12].
- Verify with High-Accuracy NNPs: Use state-of-the-art neural network potentials (NNPs) trained on large, diverse datasets (e.g., OMol25) as a benchmark to validate your solvation free energy calculations, as these can approach the accuracy of high-level DFT on large systems [39].
Issue 3: High Computational Cost of Explicit Solvent Simulations
- Problem: The computational expense of running explicit solvent simulations for large polymer or PNP systems is prohibitively high, limiting the simulation time or system size.
- Diagnosis: This is the fundamental trade-off of explicit solvent. The number of atoms increases dramatically, and the fast motions of solvent molecules require a small integration time step. Most quantum chemical methods scale poorly (O(N²)–O(N³)) with system size, making explicit solvent QM/MM simulations particularly costly [41].
- Solution:
- ML-Driven Workflow: Follow the workflow outlined in the troubleshooting guide for Issue 1. The core strategy is to replace the expensive quantum mechanical method with a pre-trained or custom-trained ML potential that is both accurate and significantly faster [41] [39].
- Conservative-Force Models: When selecting an ML potential, prefer "conserving" models (e.g., eSEN-cons) over "direct" models. Conserving models ensure energy conservation during dynamics, which is critical for stable and physically meaningful MD simulations [39].
Experimental Protocols & Methodologies
Protocol 1: Active Learning for ML-Potential in Explicit Solvent
This methodology describes how to train a robust machine learning potential for simulating chemical reactions in explicit solvent, as demonstrated in recent research [41].
- 1. Define System and Goals: Identify the specific polymer-solvent system and the chemical process or property you wish to study.
- 2. Generate Diverse Training Structures: Create multiple sets of initial structures to ensure the ML model learns all relevant interactions:
- Substrates only (polymer and drug).
- Substrates with 2 solvent molecules (to capture key specific interactions).
- Substrates with 33 solvent molecules (a small explicit shell).
- Solvent-only clusters (to model bulk solvent behavior) [41].
- 3. Active Learning Loop:
- Use similarity-based descriptors (e.g., geometric or feature-based) to select new molecular configurations for quantum mechanical (QM) calculation, rather than a more expensive energy-based selector.
- Run high-level QM calculations (e.g., ωB97M-V/def2-TZVPD) on these selected configurations to generate reference energies and forces.
- Add this new data to the training set and retrain the ML potential (e.g., an Atomic Cluster Expansion model).
- Iterate until the model's performance on a validation set converges [41].
- 4. Production Simulation: Use the trained ML potential to run molecular dynamics or biased sampling simulations to compute free energy surfaces and analyze reaction mechanisms or conformational dynamics.
Protocol 2: Training an ML Model for Accurate Free Energy Calculations
This protocol is based on the development of the LSNN model, which is designed for accurate solvation free energy predictions within an implicit solvent framework [12].
- 1. Data Collection: Assemble a large dataset (e.g., ~300,000 small molecules) with reference solvation free energies, ideally from explicit-solvent alchemical simulations.
- 2. Model Architecture: Employ a Graph Neural Network (GNN) to represent the molecular structure inherently.
- 3. Loss Function Definition: Move beyond simple force-matching. Use a multi-term loss function that includes:
- Force-matching term: Matches forces on atoms.
- Electrostatic coupling term: Matches derivatives with respect to the alchemical variable λelec.
- Steric coupling term: Matches derivatives with respect to the alchemical variable λsteric [12].
- 4. Model Training: Train the GNN with this expanded loss function, using empirically tuned weights for each term. This ensures the model learns a potential that is suitable for absolute free energy comparisons, not just conformational sampling.
- 5. Validation: Benchmark the predicted solvation free energies against explicit solvent results and experimental data.
Data Presentation
Table 1: Comparison of Common Explicit Water Models
This table summarizes key properties of popular explicit water models to guide selection for simulations of polymer-drug systems [40].
| Water Model | Type (Sites) | Key Features | Best Use Cases | Considerations |
|---|---|---|---|---|
| TIP3P | 3-site | Simple, fast, most widely used [40]. | Standard protein simulations; large systems where speed is critical [40]. | Can introduce structural artifacts; less accurate for thermodynamic properties [40]. |
| TIP4P | 4-site | Extra site improves dielectric properties [40]. | General purpose; improved accuracy over TIP3P [40]. | Slightly more computationally expensive than TIP3P [40]. |
| TIP5P | 5-site | Two lone-pair sites better represent water tetrahedrality [40]. | Studies requiring highly accurate water structure [40]. | Higher computational cost; less common in biomolecular force fields [40]. |
| SPC/E | 3-site | Includes polarization correction for better dynamics [40]. | Simulating dynamic properties and bulk water [40]. | |
| OPC | 4-site | Optimized to reproduce multiple physical properties of water accurately [40]. | High-accuracy studies; parameterizing new systems [40]. | Used in recent benchmark studies showing strong performance [40]. |
Table 2: Explicit vs. Implicit Solvent Models at a Glance
This table provides a high-level comparison to help researchers choose the right approach for their project.
| Feature | Explicit Solvent | Traditional Implicit Solvent | ML-Augmented Implicit Solvent |
|---|---|---|---|
| Computational Cost | Very High [41] | Low [11] | Low to Medium [12] |
| Treatment of Solvent | Individual molecules [40] | Dielectric continuum [11] | Data-driven potential [12] |
| Accuracy for Specific Interactions | High (Gold Standard) [41] | Low (e.g., poor for H-bonds) [41] | Improved [12] |
| Free Energy Calculations | Accurate but expensive [41] | Prone to error, especially non-polar component [12] | Highly accurate (e.g., LSNN) [12] |
| Ideal For | Detailed mechanistic studies; validation [41] | High-throughput screening; large conformational searches [11] | Accurate property prediction (solvation energy); efficient dynamics [12] |
Workflow and Model Architecture Visualizations
Workflow for ML-Accelerated Explicit Solvent Studies
This diagram illustrates the integrated workflow for using machine learning to make explicit solvent simulations computationally feasible for drug delivery applications [41].
Architecture of an ML-Based Implicit Solvent Model
This diagram outlines the key components and training strategy of a modern machine learning-based implicit solvent model, such as LSNN, designed for accurate free energy calculations [12].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Solvent Modeling
This table lists key software, datasets, and models that are instrumental in modern simulations of polymer-solvent systems.
| Tool Name | Type | Function/Benefit |
|---|---|---|
| OMol25 Dataset | Dataset | A massive, high-accuracy dataset of >100M quantum chemical calculations for training and benchmarking NNPs across diverse chemical spaces, including biomolecules and electrolytes [39]. |
| eSEN & UMA Models | Pre-trained ML Model | State-of-the-art neural network potentials trained on OMol25, providing high-accuracy energies and forces for MD simulations of large systems [39]. |
| LSNN (λ-Solvation Neural Network) | ML Model | A GNN-based implicit solvent model trained to predict accurate solvation free energies, overcoming limitations of traditional implicit models [12]. |
| CHARMM-GUI | Web-Based Tool | A widely used platform for setting up input files for MD simulations with various force fields, including solvation in explicit or implicit solvent [40]. |
| Active Learning Frameworks | Methodology | A strategy for efficiently building training sets for ML potentials by iteratively selecting the most informative new data points, reducing the number of expensive QM calculations needed [41]. |
Frequently Asked Questions (FAQs)
Q1: What are the key differences between implicit and explicit solvent models, and when should I choose one over the other? Explicit solvent models simulate individual solvent molecules (e.g., water) around the solute, providing a detailed, atomistic picture of the solvation shell. Implicit solvent models treat the solvent as a continuous medium with an average dielectric constant, which is computationally much faster [42]. You should choose an explicit solvent model when specific interactions between the solute and solvent molecules are critical to the process being studied, such as when solvent molecules participate in the reaction mechanism or when highly specific hydrogen bonding is involved [42]. Implicit models are a good choice for initial screenings, studying systems where the solvent primarily provides a polarizable environment, or when computational resources are limited [42]. For instance, a study on silver-catalyzed furan ring formation found that an implicit model was sufficient to identify the most favorable reaction pathway, as no direct solvent participation occurred [42].
Q2: My QM/MM simulation is producing unrealistic energies at the boundary between the QM and MM regions. What could be wrong? This is a common issue often related to the treatment of covalent bonds that are cut at the QM/MM boundary. The most likely cause is an improperly applied link atom scheme. The link atom scheme, typically using hydrogen atoms, is used to saturate the valencies of the quantum mechanical (QM) region when it is part of a larger molecule [43]. Ensure that your implementation correctly handles the forces on the link atoms and the adjacent MM atoms. Furthermore, verify that the chosen embedding scheme is appropriate for your system. An electrostatic embedding (EE) scheme includes polarization of the QM region by the MM point charges and is generally more accurate than a mechanical embedding (ME) scheme, which treats the QM/MM interaction classically [43].
Q3: How can I improve the transferability and accuracy of a machine-learned (ML) force field? The accuracy and transferability of ML force fields depend heavily on the quality and diversity of the training data [44]. To improve them, consider these strategies:
- Use High-Fidelity Data: Train the ML model on data derived from high-level quantum mechanics (QM) calculations, such as Density Functional Theory (DFT) [44].
- Employ Delta-Learning: Instead of learning the total energy directly, train the model to predict the difference (delta) between a low-level, fast QM method and a high-level, accurate QM method. This embeds physical knowledge and can improve generalization [44].
- Incorporate Active Learning: Use algorithms that dynamically identify and add new, relevant configurations to the training set, ensuring the model is exposed to a diverse and representative set of chemical environments [44].
- Ensure Physical Consistency: The ML model should respect fundamental physical laws, such as energy conservation and correct asymptotic behavior, to produce reliable and stable simulations [44].
Q4: Can I combine implicit and explicit solvent models in a single simulation? Yes, this is known as a cluster-continuum or "semicontinuum" approach [9]. In this method, a few key explicit solvent molecules are included in the QM region to model specific interactions (e.g., hydrogen bonding), while the bulk solvent effect is represented by an implicit model. This balances accuracy and computational cost. Best practices involve ensuring that the property you are studying (e.g., reaction energy) is converged with respect to the number of explicit solvent molecules included [9].
Q5: What are the typical speedups when using an implicit solvent model or an ML-accelerated potential compared to explicit solvent QM/MM? Speedups are highly system- and problem-dependent [8]. The table below summarizes approximate speedups for conformational sampling from a comparative study.
| Conformational Change Type | Approximate Speedup (Implicit vs. Explicit Solvent) | Notes |
|---|---|---|
| Small (e.g., dihedral angle flips) | ~1-fold (minimal speedup) | [8] |
| Large (e.g., nucleosome collapse) | ~1 to 100-fold | Highly variable [8] |
| Mixed (e.g., miniprotein folding) | ~7-fold | [8] |
| ML-Accelerated Force Fields | Near-QM accuracy at MM cost | Several orders of magnitude faster than full QM [44] |
Troubleshooting Guides
QM/MM Simulation Instabilities
Problem: Unstable energy or bonds breaking/forming incorrectly at the QM/MM boundary.
| Possible Cause | Diagnostic Steps | Solution |
|---|---|---|
| Faulty Link-Atom Setup | Check the log files for unusually large forces or energy jumps near the boundary atoms. Visualize the link atom positions. | Implement a robust link atom scheme to cap the QM region. Ensure the forces on the link atom and the classical atom it's connected to are correctly handled [43]. |
| Incorrect Embedding Scheme | Compare energies and charges from a pure QM calculation of the core region with the QM/MM result in a static geometry. | Switch from Mechanical Embedding (ME) to Electrostatic Embedding (EE). EE includes the MM point charges in the QM Hamiltonian, which polarizes the QM electron density and provides a more accurate interaction [43]. |
| Inadequate QM Region Size | Test if key chemical events (e.g., bond breaking) occur near the edge of the QM zone. | Redefine the QM region to ensure the entire chemically active site (e.g., enzyme active site, reacting bonds) is included within the QM zone, with a sufficient buffer [43]. |
Implicit Solvent Model Inaccuracies
Problem: Implicit solvent model yields reaction energies or barrier heights that disagree with experimental data or explicit solvent benchmarks.
| Possible Cause | Diagnostic Steps | Solution |
|---|---|---|
| Missing Specific Solvent Interactions | Analyze the explicit solvent trajectory (if available) for persistent hydrogen bonds or other specific solute-solvent interactions. | Use a cluster-continuum approach. Add explicit solvent molecules to the QM region to model critical interactions, while the bulk solvent is treated implicitly [9]. |
| Incorrect Dielectric Constant | Verify that the dielectric constant used in the implicit model matches the experimental solvent. | Set the dielectric constant to the correct value for your solvent (e.g., ~78.4 for water at 25°C). |
| Limitations of the Model Itself | Benchmark multiple implicit models (e.g., SMD, COSMO, PBSA) against a small set of explicit solvent calculations for your specific system. | Choose a more advanced implicit model or switch to a QM/MM approach with explicit solvent for production runs [42]. |
ML-Potential Failures
Problem: ML-potential produces unphysical geometries, energy drifts, or poor performance on new molecular systems.
| Possible Cause | Diagnostic Steps | Solution |
|---|---|---|
| Insufficient Training Data | Check the model's uncertainty estimation (if available) on the new configuration. Errors will be large in unexplored regions of chemical space. | Retrain the model using an active learning loop. Generate new QM data for configurations where the model is uncertain, and add them to the training set [44]. |
| Lack of Physical Constraints | Monitor the total energy in an NVE simulation; it should be conserved. Check for unrealistic long-range behavior. | Use ML architectures that build in physical invariances (e.g., to rotation, translation) and asymptotic constraints. Consider a delta-learning approach to correct a physics-based baseline method [44]. |
| Out-of-Distribution System | Test the model on a known property (e.g., bond lengths, vibrational frequencies) of the new system before running long simulations. | Develop a new model specifically trained on data relevant to the new chemical domain. Avoid extrapolation with ML potentials [44]. |
Experimental Protocols & Data
Workflow for Benchmarking Solvent Models on a Reaction
This protocol outlines steps to validate an implicit solvent model against a more accurate QM/MM explicit solvent calculation for a chemical reaction [42].
System Setup:
- Solute: Optimize the geometry of the reactant, transition state, and product in the gas phase using a QM method (e.g., DFT).
- Explicit Solvent (QM/MM): Place the solute into a periodic simulation box filled with explicit solvent molecules (e.g., 112 DMF molecules in a 24.4 Å box). Describe the solute with QM and the solvent with an MM force field (e.g., CHARMM) [42].
- Implicit Solvent: Use the gas-phase geometries and apply an implicit solvation model (e.g., SMD) during single-point energy calculations or re-optimization [42].
Simulation & Calculation:
- Explicit Solvent Free Energy: Perform QM/MM molecular dynamics under NVT conditions. Use an enhanced sampling method (e.g., thermodynamic integration or blue moon sampling) along a defined reaction coordinate (e.g., forming bond distance) to calculate the free energy profile [42].
- Implicit Solvent Free Energy: Calculate the electronic energy in the implicit solvent. Add the gas-phase thermal corrections (from a frequency calculation) to obtain the free energy in solution [42].
Benchmarking and Analysis:
- Compare the activation free energies and reaction free energies obtained from both methods.
- Analyze the QM/MM trajectories to check for direct participation of solvent molecules in the reaction. If no specific interactions are found, the implicit model is likely sufficient [42].
Workflow for benchmarking solvent models to guide model selection.
Performance Comparison: Explicit vs. Implicit Solvation
The following table quantifies the performance differences observed in a study comparing explicit (PME) and implicit (GB) solvent models for molecular dynamics simulations [8].
| System / Metric | Explicit Solvent (PME/TIP3P) | Implicit Solvent (GB) | Observed Speedup in\nConformational Sampling |
|---|---|---|---|
| Small Conformational Change | Baseline | Comparable performance | ~1-fold (minimal speedup) [8] |
| Large Conformational Change | Baseline | Significantly faster | ~1 to 100-fold (highly variable) [8] |
| Miniprotein Folding | Baseline | Faster sampling | ~7-fold [8] |
| Primary Cause of Speedup | N/A | N/A | Reduction in solvent viscosity (friction) in the implicit model [8] |
The Scientist's Toolkit: Research Reagent Solutions
| Tool / Resource | Function | Example Use Case |
|---|---|---|
| GROMOS Simulation Package | A molecular simulation software package with enhanced QM/MM functionality, including a link-atom scheme and interfaces to multiple QM programs [43]. | Performing advanced biomolecular simulations, such as enzyme catalysis, where part of the system requires a quantum mechanical description [43]. |
| CHARMM-GUI | A web-based platform that provides input file generators for various simulation packages, including those supporting QM/MM [45]. | Setting up complex simulation systems (e.g., membrane proteins in a lipid bilayer) for programs like GROMACS, NAMD, or AMBER [45]. |
| QM/MM Embedding Schemes | Defines how the QM and MM regions interact energetically. | Mechanical Embedding (MECC/MEDC): Fast but less accurate; suitable for non-polar environments. Electrostatic Embedding (EE): More accurate; includes polarization of the QM region by MM charges [43]. |
| Cluster-Continuum Approach | A hybrid method that combines a few explicit solvent molecules with an implicit solvent model [9]. | Modeling chemical reactions in solution where specific hydrogen bonding with a few water molecules is critical, but simulating a full explicit solvent box is too costly [9]. |
| Delta-Learning ML Models | A machine learning technique where the model learns the difference between a low-level and high-level QM calculation [44]. | Creating highly accurate and transferable force fields at a computational cost much lower than that of the high-level QM method [44]. |
Overcoming Limitations and Enhancing Simulation Performance
Molecular dynamics (MD) simulations are indispensable for understanding biomolecular function, but the explicit treatment of solvent molecules often creates a formidable computational bottleneck. Implicit solvent models address this challenge by replacing explicit water molecules with a continuum representation, dramatically accelerating conformational sampling. This technical resource center provides evidence-based guidance on the performance gains, practical methodologies, and common pitfalls of implicit solvent simulations, equipping researchers with the knowledge to effectively integrate these approaches into their drug discovery and basic research pipelines.
The core acceleration mechanism operates through two primary effects: a significant reduction in computational overhead by eliminating solvent degrees of freedom, and a decrease in effective solvent viscosity that facilitates faster biomolecular dynamics [46]. Quantitative speedups are highly system-dependent, ranging from negligible for small motions to over 100-fold for large-scale conformational changes [31].
Quantitative Speedup Analysis
Table 1: Measured Speedup of Conformational Sampling with Implicit Solvent Models
| Type of Conformational Change | Example System | Nominal Simulation Time Scale | Approximate Sampling Speedup (GB vs. Explicit) | Primary Contributing Factor |
|---|---|---|---|---|
| Small Changes | Dihedral angle flips in a protein | Nanoseconds | ~1-fold | Reduced solvent friction [31] |
| Mixed Changes | Folding of a miniprotein | Microseconds | ~7-fold | Combined effect of reduced viscosity and computational cost [31] |
| Large Changes | Nucleosome tail collapse, DNA unwrapping | Nanoseconds to microseconds | ~1 to 100-fold | Significantly reduced solvent viscosity [31] |
Table 2: Popular Generalized Born (GB) Implicit Solvent Models and Their Attributes
| GB Model | Key Features | Common Implementation(s) | Notable Applications |
|---|---|---|---|
| GB-OBC | Empirical correction for buried atoms; uses vdW surface [46] | AMBER, OpenMM | Protein folding studies [46] |
| GB-Neck | "Neck" correction to better approximate the molecular surface [46] | AMBER, CHARMM, OpenMM | Improved accuracy for salt bridges and dense structures [46] |
| GBSW | Smooth switching function at dielectric boundary; grid-based [47] [46] | CHARMM, NAMD | Membrane simulations; refinement of NMR structures [47] |
| GBMV | Empirical correction to Coulomb Field Approximation; grid-based [46] | CHARMM | |
| GB-HCT | Pair-wise descreening approximation; uses vdW surface [46] | AMBER, OpenMM |
Experimental Protocols & Methodologies
Protocol 1: Quenched Molecular Dynamics for Minimum Energy Conformation Search
This protocol is adapted from studies evaluating implicit solvent models against explicit solvent benchmarks [47].
- System Setup: Construct your solute (e.g., a small peptide) using a molecular modeling package and the desired force field. For explicit solvent control simulations, solvate the solute in a pre-equilibrated water box (e.g., TIP3P).
- High-Temperature MD: Run a production MD simulation at a high temperature (e.g., 1000 K) to facilitate broad conformational exploration. A typical duration is 10 ns.
- Conformation Sampling: Save molecular structures from the trajectory at regular intervals (e.g., every 10 ps).
- Energy Minimization: Perform extensive energy minimization on each saved snapshot. A common protocol is 2500 steps of steepest descent followed by 2500 steps of conjugate gradient minimization, until a root mean-square gradient threshold (e.g., 0.01 kcal/mol/Å) is reached.
- Analysis: Cluster the resulting minimized structures to identify local energy minima. Compare the sets of minima and their relative stability (free energy) obtained from different solvent models.
Protocol 2: Setting Up an Implicit Solvent Simulation via CHARMM-GUI
The CHARMM-GUI Implicit Solvent Modeler (ISM) provides a standardized, error-resistant workflow for preparing simulations across multiple MD packages [46].
- Structure Reading: Upload your initial structure (PDB or similar format). Select "Implicit Solvent" for standard simulations or "Implicit Solvent/Membrane" for membrane systems.
- Force Field Selection: Choose an appropriate force field (e.g., CHARMM36, ff19SB) for your biomolecule (protein, DNA, RNA, glycan, ligand).
- Implicit Solvent Model Selection: Based on your system and the available models in your target MD software, select a specific GB model (e.g., GBSW, GB-OBC, GB-Neck2).
- MD Package and Parameters: Select the MD simulation package (AMBER, CHARMM, OpenMM, etc.) and set up simulation parameters (time step, temperature, etc.). The platform will automatically generate all necessary input files.
The diagram below illustrates the logical workflow for planning and executing an implicit solvent simulation study, from initial setup to analysis.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software and Model Components for Implicit Solvent Simulations
| Tool Category | Specific Item / "Reagent" | Function / Purpose |
|---|---|---|
| MD Software Packages | AMBER, CHARMM/NAMD, OpenMM, GENESIS, Tinker | Execution engines for running implicit solvent MD simulations; each supports different GB models [46]. |
| Web-Based Preparation Platforms | CHARMM-GUI Implicit Solvent Modeler (ISM) | Automated system building and input file generation for various MD packages, reducing manual scripting errors [46]. |
| Generalized Born (GB) Models | GB-OBC, GB-Neck, GBSW, GBMV | Core implicit solvent "reagents" that calculate the polar solvation energy; choice depends on system and accuracy needs [47] [46]. |
| Force Fields (FF) | CHARMM36(m), AMBER (ff14SB, ff19SB) | Define bonded and non-bonded parameters for the solute; must be compatible with the chosen GB model [46]. |
| Solvation Parameters | Surface tension coefficient (γ) for nonpolar term | An empirical parameter, typically between 0.005-0.138 kcal/mol/Ų, used to calculate the nonpolar solvation free energy [46]. |
Frequently Asked Questions (FAQs)
Q1: For which systems are implicit solvents least accurate, and what are the alternatives? Implicit solvents often struggle with nucleic acids, particularly RNA, due to their high charge density and the importance of specific ion effects [48]. They can also be inaccurate in systems where explicit water bridges or specific solvent interactions are critical for stability. Alternatives include using a hybrid explicit/implicit approach [49], or employing a novel implicit model that combines a Langevin-Debye treatment of dielectric saturation with a Poisson-Boltzmann description of counter ions [48].
Q2: Why does my implicit solvent simulation show unrealistic structural distortion, especially for RNA? This is a common problem, often stemming from an inadequate description of electrostatics. Standard GB models may fail to fully capture the strong screening needed around the densely charged RNA backbone. Troubleshooting steps include: 1) Increasing the ionic strength parameter (Debye-Hückel κ) in the GB model to better mimic counter-ion screening [48], 2) For systems with divalent ions like Mg²⁺, consider adding a few explicit ions while keeping the solvent implicit [48], and 3) Exploring newer, more physics-based implicit models designed for nucleic acids [48].
Q3: How do I know if the conformational speedup I'm observing is physically meaningful and not an artifact? Validation is key. Follow this protocol: First, run a short explicit solvent simulation of your system (or a similar, smaller system if possible) to establish a baseline for physically realistic dynamics. Then, compare key observables from your implicit solvent simulation against this baseline. Relevant metrics include: 1) Root-mean-square deviation (RMSD) from a known experimental or starting structure, 2) Stability of known secondary structural elements, and 3) Comparison of free-energy landscapes for well-characterized conformational changes, if data is available [31] [47].
Q4: Can I use implicit solvents for protein-ligand binding studies? Yes, implicit solvents are widely used for early-stage ligand screening and binding mode refinement via MM/GBSA (Molecular Mechanics/Generalized Born Surface Area) calculations. While faster than explicit solvent simulations, their accuracy in ranking binders is generally better than docking but may be lower than more rigorous explicit solvent free-energy methods. They are excellent for rapidly narrowing down large lists of potential candidates [46].
Q5: What is the practical impact of the "Langevin collision frequency" parameter in my implicit solvent simulation? This parameter controls the effective viscosity of the implicit solvent. A lower collision frequency reduces the viscous drag on the solute, leading to faster conformational transitions and a higher observed speedup [31]. However, setting it too low can produce non-physiological, gas-phase-like dynamics. It is recommended to use the value prescribed for your chosen GB model or to perform a sensitivity analysis for your specific system.
In molecular dynamics (MD) simulations, implicit solvent models are invaluable for their computational efficiency, representing the solvent as a continuous, homogeneous medium rather than explicit molecules [2]. However, this simplification comes at a cost: the loss of specific, atomistic solvent-solute interactions. For researchers in drug development, this pitfall can lead to inaccurate predictions of molecular behavior. This guide addresses the most common issues arising from this limitation and provides practical troubleshooting advice.
FAQ: Understanding the Core Issues
What specific solvent effects do implicit models typically miss?
Implicit solvent models, particularly standard continuum models, struggle to capture several specific solvent-solute interactions [50] [2] [37]:
- Specific Hydrogen Bonding: They cannot accurately model the directional, strong interactions between solute atoms and individual solvent molecules, such as the precise geometry of water-mediated hydrogen bonds [50] [37].
- Solvent Structure Fluctuations: These models represent a thermally averaged solvent environment and cannot capture local fluctuations in solvent density or the specific ordering of solvent molecules around a solute (e.g., solvation shells) [51] [2].
- Bridging Solvent Molecules: They fail to represent single solvent molecules that simultaneously interact with multiple parts of the solute, which can be critical in processes like protein-ligand binding or nucleic acid folding [37].
- Hydrophobic Effects: While the thermodynamic consequence of hydrophobicity (cavity formation) is modeled, the detailed, entropically driven process of water rearrangement around non-polar surfaces is not explicitly captured [37].
When is it absolutely necessary to switch to an explicit solvent model?
You should strongly consider explicit solvents or a hybrid approach in the following scenarios [52] [37]:
- When studying processes where specific hydrogen bonds between the solute and solvent are critical to the mechanism.
- When simulating nucleic acids, as their charged backbones and structure are highly sensitive to the precise distribution of ions and water [37].
- When investigating membrane-associated systems, where the heterogeneous environment is poorly described by a uniform dielectric constant [37].
- When calculating properties that rely on atomic-level detail of the solvent, such as residence times of water molecules in a binding pocket.
Can hybrid approaches solve this problem?
Yes, hybrid (or "cluster-continuum") approaches aim to combine the strengths of both methods [2] [9]. A common strategy is to include a limited number of explicit solvent molecules in the quantum mechanical (QM) region or the primary simulation box to capture the most critical specific interactions (e.g., the first solvation shell), while the bulk solvent is treated with an implicit model. This can provide a good balance between accuracy and computational cost, though it requires careful setup to avoid artifacts [9].
Troubleshooting Guide: Identifying and Mitigating Pitfalls
Problem: Inaccurate Reproduction of Folded or Unfolded Structural Distributions
- Symptom: Your simulation of a protein or polymer fails to stabilize the known native state or produces an unrealistic ensemble of unfolded states compared to experimental data [51] [37].
- Root Cause: The implicit model's averaged description of solvation fails to capture the specific, stabilizing interactions of key water molecules within the protein core or at the surface, and may inadequately model the backbone solvation in unfolded states [37].
- Solution:
- Benchmark: Compare the stability of a known native structure in implicit and explicit solvent.
- Hybrid Method: For critical regions, use a QM/MM approach where a few explicit water molecules are included in the QM region to model specific interactions accurately, while the rest is treated with a faster MM force field and implicit solvent [2] [9].
- Model Selection: Consider using a more advanced implicit model that incorporates a more physical description of non-polar contributions or a water-aware statistical potential [37].
Problem: Poor Prediction of Solvation Free Energies for Polar Molecules
- Symptom: Calculated solvation free energies, particularly for polar solutes or those that can form hydrogen bonds, show significant deviations from experimental values [52].
- Root Cause: The continuum approximation inherently smoothes over the strong, specific electrostatic and hydrogen-bonding interactions that dominate the solvation of polar molecules [2] [52].
- Solution:
- Validation: Always validate your implicit model's performance for a test set of molecules similar to your solute of interest [52].
- Explicit Solvent Reference: For critical applications, use free energy calculations (e.g., TI or FEP) in explicit solvent as a benchmark. A 2017 study found explicit solvent models were in better agreement with experiment for organic molecules than several common implicit models [52].
- Machine Learning Potentials: Explore emerging machine learning-based implicit solvent models trained on explicit solvent simulations, which can learn a potential of mean force that more accurately reproduces specific effects [51].
Problem: Failure to Model Solvent-Sensitive Spectral Properties
- Symptom: Calculated spectroscopic properties (e.g., NMR chemical shifts, UV-Vis absorption spectra) do not match experimental measurements in solution.
- Root Cause: Spectral shifts often depend on the instantaneous, specific orientation and electronic polarization of the solvent molecules in the immediate vicinity of the solute, which a continuum model cannot provide [53] [54].
- Solution:
- Explicit Sampling: Perform the property calculation on multiple snapshots taken from an explicit solvent MD simulation to account for solvent configurational averaging.
- Polarizable Continuum Model (PCM) with State-Specific Setup: For some methods like TD-DFT, ensure the implicit model's parameters (e.g., dielectric constant and refractive index) are correctly set for the property being calculated [53] [54].
Protocol: Benchmarking an Implicit Solvent Model for Protein Dynamics
This protocol helps you systematically evaluate whether an implicit solvent model is suitable for your specific system [27] [37].
- System Preparation: Start with a high-resolution structure of your protein.
- Explicit Solvent Control Simulation: Place the protein in a box of explicit water molecules (e.g., TIP3P) and run a multi-nanosecond MD simulation to establish a reference conformational ensemble.
- Implicit Solvent Test Simulation: Run a simulation of the same duration starting from the same structure using your chosen implicit solvent model (e.g., GBMV, OBC).
- Analysis and Comparison:
- Calculate the root-mean-square deviation (RMSD) of the protein backbone relative to the starting structure for both trajectories.
- Compare key observables: radius of gyration (Rg), secondary structure stability (e.g., via DSSP), and solvent-accessible surface area (SASA).
- If available, compare against experimental data such as NMR order parameters.
- Interpretation: Significant deviations in the implicit solvent trajectory from the explicit solvent reference or experimental data indicate a loss of critical solvent effects.
Protocol: Setting Up a Hybrid QM/MM-MD Simulation with Explicit Waters
This methodology is used when specific electronic polarization or chemical reactivity in an aqueous environment is crucial [51] [9].
- Generate Solvated Configurations: Use an MD package (e.g., GROMACS, CHARMM) to solvate your solute in a box of explicit water. Run a short equilibration simulation.
- Configuration Selection: Extract multiple snapshots from the trajectory at regular intervals to ensure conformational sampling of the solvent.
- Subsystem Definition: For each snapshot, define the QM region (the solute + critical explicit water molecules involved in specific interactions). The remainder is the MM region.
- QM/MM Calculation: Run a QM/MM geometry optimization or MD simulation for each selected snapshot. Software like Q-Chem provides interfaces for this [9].
- Averaging: Average the property of interest (e.g., reaction energy, spectral shift) over all snapshots to account for solvent fluctuations.
Quantitative Data on Solvation Free Energy Accuracy
The table below summarizes findings from a study comparing solvation free energy predictions, highlighting the performance gap for certain molecule types [52].
Table 1: Comparison of Solvation Free Energy Calculation Methods
| Model Type | Example | Performance Note | Key Limitation |
|---|---|---|---|
| Implicit | Generalized Born (GB), SMx models | Can struggle with polar molecules and specific H-bonding; accuracy is system-dependent [52]. | Lacks molecular detail of solvent [2]. |
| Explicit | TIP3P water model with GAFF solute parameters | Found to be in better agreement with experiment for a set of organic molecules than implicit models in a 2017 study [52]. | Computationally intensive; requires careful sampling [27] [2]. |
| Machine Learning-Based | DeepPot-SE implicit model [51] | Can reproduce explicit solvent free energy surfaces with high accuracy (e.g., RMSD < 0.9 kcal/mol for alanine dipeptide) [51]. | Requires extensive training data; potential transferability issues [51] [50]. |
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 2: Essential Computational Tools for Solvent Modeling
| Item / Software | Function in Research |
|---|---|
| Continuum Model Software (ORCA, Q-Chem) | Provides implementations of implicit models like C-PCM, SMD, and IEF-PCM for quantum chemical calculations [53] [9] [54]. |
| MD Engines (GROMACS, AMBER, CHARMM) | Enable simulations with both explicit and implicit solvent models, allowing for direct benchmarking [27] [9]. |
| Force Fields (AMOEBA, CHARMM, GAFF) | AMOEBA is a polarizable force field for more accurate explicit solvent interactions. CHARMM and GAFF are widely used for biomolecules and drug-like molecules [2] [52]. |
| Machine Learning Potentials (DeepPot-SE) | Used to develop new implicit solvent models directly from explicit solvent data, capturing more specific effects [51]. |
| Analysis Tools (VMD, MDAnalysis) | Critical for visualizing trajectories, calculating RMSD, Rg, SASA, and identifying specific solute-solvent interactions [37]. |
Workflow Diagram: Solvent Model Selection Strategy
Frequently Asked Questions (FAQs)
FAQ 1: What is the primary computational advantage of using an implicit solvent model over an explicit one? Implicit solvent models offer two main advantages. First, they are often algorithmically faster as they eliminate the need to compute interactions for thousands of explicit solvent molecules, reducing the number of particles in the system [55] [56]. Second, they can speed up conformational sampling by reducing the effective solvent viscosity that impedes molecular motion, allowing the solute to explore its energy landscape more rapidly [56].
FAQ 2: Why is the choice of atomic radii so critical in implicit solvent models like Poisson-Boltzmann (PB)? Atomic radii are a key parameter because they define the solute-solvent boundary, which directly determines the distribution of dielectric constants around the solute [57]. An inaccurate boundary leads to errors in calculating the solvation free energy. Using an optimized set of atomic radii, parameterized for a specific force field, is essential for accurately reproducing solvation free energies from explicit solvent simulations [57].
FAQ 3: Is the internal dielectric constant of a protein a fixed value? No, the protein dielectric constant is not a universal constant [58]. It is a complex function that reflects the protein's structure and sequence. The dielectric properties are inhomogeneous: the hydrophobic core is tightly packed and has low dielectric values (~6-7), while the protein surface, which is loosely packed and rich in charged residues, has a much higher local dielectric constant (20-30) [58].
FAQ 4: My MM/PBSA simulations fail to preserve the native protein structure. What could be wrong? A common reason for this failure is an improper treatment of the dielectric constant [59]. Using a vacuum dielectric constant (ε=1) for the solute in a PB model often does not work. Some studies have found that applying a higher, homogeneous internal dielectric constant (e.g., ε=10-17) is necessary to obtain stable trajectories, although this poses theoretical challenges [59]. A more physically realistic approach is to use a smooth, position-dependent dielectric function [58].
Troubleshooting Guides
Issue 1: Inaccurate Solvation Free Energy Estimates
Problem: Computed solvation free energies deviate significantly from benchmark explicit solvent results or experimental data.
- Potential Cause 1: Non-optimized Atomic Radii. The atomic radii (PB radii) used to define the dielectric boundary are not optimized for your force field or simulation setup [57].
- Solution: Implement a new set of optimized atomic radii. For example, Yamagishi et al. provide a set for the AMBER force field that discriminates between terminal and non-terminal residues for higher accuracy [57].
- Potential Cause 2: Inhomogeneous Dielectric Effects. Using a single, homogeneous dielectric constant for the entire protein ignores its complex internal electrostatic environment [58].
- Solution: Consider using a Gaussian-based smooth dielectric function that automatically assigns higher dielectric values to charged and polar surface regions and lower values to the hydrophobic core [58].
Issue 2: Poor Sampling of Conformational Transitions
Problem: The simulation does not adequately sample relevant biomolecular conformations within a reasonable computation time.
- Potential Cause: High Effective Viscosity. The implicit solvent model may still be parameterized with an effective viscosity that is too high, slowing down conformational changes [56].
- Solution: In implicit solvent MD simulations, which often use Langevin dynamics, reduce the collision frequency (γ) parameter. This lowers the effective solvent viscosity, which has been shown to speed up conformational sampling by up to 100-fold for large-scale changes [56].
Issue 3: Over-stabilization of Salt Bridges and Helical Structures
Problem: The implicit solvent simulation results in an over-population of salt bridges and alpha-helical content compared to explicit solvent benchmarks or experimental knowledge.
- Potential Cause: Limitations of the Generalized Born (GB) Model. Certain Generalized Born models, particularly when used for molecular mechanics (MM/GBSA), are known to have insufficient electrostatic screening, which can over-stabilize salt bridges and alter secondary structure preferences [60].
- Solution: Be aware of this known limitation when interpreting results. Validation against explicit solvent simulations or experimental data is crucial. For critical applications, the more computationally expensive Poisson-Boltzmann model may be a better choice for the final analysis [60].
Table 1: Performance Comparison of Explicit vs. Implicit Solvent Models for Conformational Sampling
| Type of Conformational Change | System Size (atoms) | Sampling Speedup (Implicit vs. Explicit) | Primary Factor for Speedup |
|---|---|---|---|
| Small (dihedral angle flips) [56] | ~5,000 | ~1-fold (minimal) | N/A |
| Large (DNA unwrapping, tail collapse) [56] | ~25,000 | ~1 to 100-fold | Reduced solvent viscosity [56] |
| Mixed (miniprotein folding) [56] | ~200 | ~7-fold | Reduced solvent viscosity [56] |
Table 2: Typical Dielectric Constant Values Used in Continuum Solvent Models
| Region | Typical Dielectric Constant (ε) | Notes and Rationale |
|---|---|---|
| Bulk Water [2] [58] | ~80 | Represents the high polarizability of bulk water. |
| Protein Interior (Homogeneous model) [59] [58] | 1 - 4 | Accounts for electronic polarization only; often used for rigid structures [58]. |
| Protein Interior (Homogeneous model) [59] [58] | 10 - 20 | An effective value attempting to account for limited side-chain and backbone motions [59] [58]. |
| Protein Surface (Gaussian model) [58] | 20 - 30 | Loosely packed, charged, and polar regions with higher ability to reorganize [58]. |
| Protein Hydrophobic Core (Gaussian model) [58] | 6 - 7 | Tightly packed, uncharged atoms with limited response to electrostatic fields [58]. |
Experimental Protocols
Protocol 1: Optimizing Atomic Radii for Poisson-Boltzmann Calculations
This methodology is based on the work by Yamagishi et al. for deriving accurate atomic radii for the AMBER force field [57].
- Explicit Solvent Reference Simulation: Perform molecular dynamics simulations of the target systems (e.g., large molecular fragments) in an explicit solvent model like TIP3P water [57].
- Calculate Reference Solvation Free Energies: Obtain the solvation free energies for these systems from the explicit solvent simulations [57].
- Define the Optimization Goal: The objective is to find a set of atomic radii that, when used in PB calculations, reproduce the explicit solvent solvation free energies as closely as possible [57].
- Iterative Optimization: Systematically adjust the atomic radii parameters in the PB model and recompute the solvation free energies until the difference with the explicit solvent reference is minimized [57].
- Terminal Group Discrimination: Differentiate the optimized radii for N- and C-terminal residues from those used for non-terminal residues to improve accuracy at the molecular fragment level [57].
Protocol 2: Implementing a Smooth Dielectric Function for Proteins
This protocol outlines the approach for assigning a non-homogeneous dielectric constant, as described by Li et al. [58].
- Input Structure: Utilize the 3D atomic structure of the protein macromolecule [58].
- Gaussian-Based Atomic Densities: Construct a smooth dielectric function for the entire space (protein and water) based on Gaussian distributions of atomic densities, rather than simply smoothing the molecular surface [58].
- Assign Local Dielectric Values: The algorithm automatically assigns higher local dielectric constants to space occupied by loosely packed and charged atoms (common at the protein surface) and lower dielectric constants to regions of tightly packed, uncharged atoms (the hydrophobic core) [58].
- Energy Calculations: Implement the resulting smooth dielectric map into a Poisson-Boltzmann solver (e.g., the DelPhi program) to calculate electrostatic potentials and energies [58].
- Validation: Benchmark the model's performance against experimental data, such as pKa values of residues in proteins [58].
Workflow and Relationship Diagrams
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools and Parameters for Implicit Solvent Modeling
| Item Name | Function / Role | Key Considerations |
|---|---|---|
| Optimized Atomic Radii Sets [57] | Defines the solute-solvent boundary for accurate solvation free energy calculation. | Must be parameterized for your specific force field (e.g., AMBER). Terminal vs. non-terminal residues may need different parameters [57]. |
| Poisson-Boltzmann Solver [59] [58] | Computes the electrostatic potential and polar solvation energy by numerically solving the PB equation. | More accurate but computationally slower than GB. Examples include DelPhi, APBS, and UHBD [59] [58]. |
| Generalized Born (GB) Model [60] [56] | Approximates the PB electrostatics using an analytical formula, offering a speed/accuracy trade-off. | Computationally faster, enabling longer simulations. Can over-stabilize salt bridges and alter secondary structure preferences [60] [56]. |
| Smooth Dielectric Function [58] | Replaces a homogeneous protein dielectric with a position-dependent one for a more physical model. | Better reflects the inhomogeneous nature of protein interiors, improving pKa predictions and energy calculations [58]. |
| Surface Area (SA) Term [60] | Accounts for the non-polar (hydrophobic) contribution to solvation free energy. | Typically proportional to the Solvent Accessible Surface Area (SASA). The proportionality constant (surface tension) is a key parameter [60]. |
Molecular dynamics (MD) simulations are a cornerstone of modern computational biology and drug development, providing atomic-level insight into the behavior of proteins, nucleic acids, and their complexes with ligands [37]. A critical choice in setting up these simulations is how to treat the solvent environment. Explicit solvent models simulate individual water molecules surrounding the solute, offering high accuracy by capturing specific solute-solvent interactions, such as hydrogen bonds, at the cost of dramatically increased computational expense [5]. Implicit solvent models (also known as continuum solvation) represent the solvent as a continuous medium, approximating its average effect on the solute. This approach can speed up conformational sampling significantly—anywhere from approximately 2-fold to over 100-fold depending on the system and the conformational change being studied [8]—by reducing the number of interacting particles and removing the viscous drag of explicit water [60] [8].
Integrating Multiple Time-Step (MTS) methods with implicit solvents presents a powerful strategy to further accelerate simulations. MTS algorithms allow different forces in the system to be calculated at different frequencies, reserving the most expensive calculations for longer time intervals. This technical overview provides a structured guide to the successful implementation, optimization, and troubleshooting of this combined approach.
Key Concepts and Research Reagents
Understanding the core components is essential before attempting integration. The following table details the essential "research reagents" – the computational models and methods central to this field.
Table 1: Key Computational Models and Methods ("Research Reagents")
| Reagent/Method | Type | Primary Function & Description |
|---|---|---|
| Generalized Born (GB) [60] [61] [37] | Implicit Solvent Model | Approximates the electrostatic component of solvation free energy. It is computationally efficient and analytically differentiable, making it highly suitable for MD. Variants include GBSA (with a non-polar Surface Area term) and GBMV2. |
| Poisson-Boltzmann (PB) [60] [37] | Implicit Solvent Model | Provides a more numerically accurate, but computationally expensive, solution for the electrostatic solvation energy by solving the PB equation. Often used as a benchmark for GB models. |
| SASA [60] [37] | Implicit Solvent Component | Solvent Accessible Surface Area. Models the non-polar contribution to solvation free energy (cavity formation and van der Waals interactions). Often paired with GB or PB. |
| Multiple Time-Step (MTS) Integrator | Simulation Algorithm | A numerical integration algorithm that calculates "fast" forces (e.g., bonded interactions) every time step, and "slow" forces (e.g., non-bonded, solvation) less frequently (e.g., every 2, 4, or 10 steps), improving computational efficiency. |
| Langevin Dynamics [60] [8] | Thermostat | A stochastic dynamics method that regulates temperature and adds friction to the system. Crucial in implicit solvent simulations to replace the missing viscous drag of explicit water. |
Performance Benchmarks and Optimization Data
The decision to use an implicit solvent model is often driven by the significant acceleration it provides. The speedup, however, is highly system-dependent. The table below summarizes comparative performance data from the literature.
Table 2: Performance Comparison of Explicit vs. Implicit Solvent Simulations
| Simulation System / Process | Explicit Solvent Model | Implicit Solvent Model | Observed Speedup in Conformational Sampling | Key Notes and References |
|---|---|---|---|---|
| Small Conformational Changes (e.g., dihedral angle flips) | TIP3P (PME) | Generalized Born (GB) | ~1-fold (minimal speedup) | Small-scale motions are not heavily damped by solvent viscosity. [8] |
| Large Conformational Changes (e.g., DNA unwrapping, tail collapse) | TIP3P (PME) | Generalized Born (GB) | ~1 to 100-fold | Speedup is highly dependent on the specific system and the reduction in effective viscosity. [8] |
| Mixed Conformational Changes (e.g., miniprotein folding) | TIP3P (PME) | Generalized Born (GB) | ~7 to 50-fold | Combined effect of reduced viscosity and faster computational speed per step. [8] |
| GPU Acceleration | Explicit (e.g., TIP3P) | GBMV2/SA | ~60 to 70-fold (computational speed) | This is a measure of raw computational speedup on a GPU, not conformational sampling speedup. [61] |
Workflow Diagram: MTS and Implicit Solvent Integration
The following diagram illustrates the logical flow and decision points for implementing a multiple time-step method within an implicit solvent MD simulation.
Frequently Asked Questions (FAQs) & Troubleshooting
Q1: My simulation becomes unstable when I increase the outer time-step for the implicit solvation forces. What could be the cause?
- A: This is a common issue. The implicit solvation energy, particularly in Generalized Born models, is a many-body function—the Born radius of each atom depends on the positions of all other atoms. This makes the "slow" solvation forces highly sensitive to positional changes.
- Solution: Start with a conservative MTS scheme. Use a small outer time-step (e.g., 2-4 fs) for the solvation forces. Gradually increase it while monitoring the total energy and temperature of the system for drift. The maximum stable outer time-step is system-dependent. Ensure that the "fast" and "medium" force groups are comprehensive enough to capture the highest frequency motions that couple to the solvation response.
Q2: My implicit solvent simulation samples conformations much faster, but the results don't match experimental data or explicit solvent benchmarks. What might be wrong?
- A: This can stem from several limitations inherent to implicit solvent models [60] [5] [37].
- Cause 1: Poor Treatment of Specific Solvent Effects. Implicit models struggle with explicit solute-solvent hydrogen bonds, bridging water molecules, and the hydrophobic effect (often crudely approximated by a SASA term).
- Troubleshooting: If your process relies on specific, structured water molecules (e.g., in ligand binding or enzyme catalysis), a pure implicit solvent may be inadequate. Consider a hybrid explicit-implicit approach where a few layers of water are treated explicitly, and the bulk is implicit.
- Cause 2: Over-stabilization of Salt Bridges and Charged Groups. The electrostatic screening in some GB models can be insufficient, leading to overly strong salt bridges and altered protein dynamics.
- Troubleshooting: Validate your results against a known experimental observable or a short explicit solvent simulation. You may need to adjust parameters or try a different, more recently parameterized implicit solvent model.
Q3: How do I manage the lack of viscous drag in implicit solvent simulations?
- A: The absence of thousands of colliding water molecules means the simulation lacks physical viscosity, which can lead to unphysically fast dynamics and poor sampling of the kinetic properties [60].
- Solution: Always use a stochastic thermostat, most commonly Langevin Dynamics. The "collision frequency" (γ) parameter in Langevin dynamics controls the effective viscosity. A value of γ = 1-5 ps⁻¹ is often used to mimic the viscosity of water [8]. However, for maximum conformational sampling speed, you can use a lower γ (lower effective viscosity), acknowledging that the kinetics will be further accelerated [60].
Q4: Are there new methods that improve the accuracy of implicit solvent models?
- A: Yes, Machine Learning (ML) approaches are showing significant promise. Traditional implicit models like GB/SASA can have known inaccuracies in their non-polar terms and are parameterized with simplifications [12].
- Emerging Solution: Researchers are now developing graph neural network-based implicit solvent models (e.g., λ-SNN, LSNN) that are trained on large datasets from explicit solvent simulations [12]. These ML-based models aim to achieve near-explicit solvent accuracy while retaining the computational speed of implicit solvents, and they are specifically designed to be suitable for free energy calculations, a traditional weakness of force-matched ML potentials [12].
Leveraging Machine Learning for Physics-Enforced and Accurate Free Energy Calculations
Frequently Asked Questions (FAQs)
Q1: What are the primary advantages of using implicit solvent models in free energy calculations? Implicit solvent models, such as Generalized Born (GB), can significantly speed up conformational sampling compared to explicit solvent models like Particle Mesh Ewald (PME). This speedup is highly system-dependent but has been observed to range from approximately 1-fold for small conformational changes to over 100-fold for large conformational changes [31]. The primary reason is the reduction in simulated degrees of freedom and the effective lower solvent viscosity, which allows the system to explore phase space more rapidly.
Q2: Can I mix parameters from different force fields when parameterizing a new molecule for my simulation? No. You should not take parameters from one force field and apply them inside another. Molecules parametrized for a specific force field will not behave physically when interacting with molecules parametrized under different standards. If a molecule is missing from your chosen force field, you must parametrize it yourself according to that force field's specific methodology [62].
Q3: My machine learning model for free energy surfaces produces physically implausible results. How can I enforce physical correctness? You can integrate physical constraints directly into the model. A primary method is using physics-informed optimization, such as designing a physics-augmented loss function. This function typically combines a standard data-fitting term (e.g., mean squared error) with a term that penalizes violations of known physical laws, such as the violation of a relevant Partial Differential Equation (PDE) or conservation law [63]. This guides the model toward physically realistic solutions.
Q4: What kind of properties can I fit with machine learning potentials for molecular dynamics? When training Machine Learning Potentials (MLPs), such as with the M3GNet architecture, you can typically train the model to reproduce energies and forces obtained from high-level reference calculations, such as Density Functional Theory (DFT) [64]. These energies and forces are then used to drive the molecular dynamics simulations.
Q5: How do I handle the trade-off between computational speed and accuracy when choosing a solvent model? The choice involves evaluating your specific scientific goal. The table below summarizes key performance differences to guide this decision:
| Criterion | Explicit Solvent (PME) | Implicit Solvent (GB) |
|---|---|---|
| * conformational Sampling Speed* | Baseline (slower) | 1x to >100x faster (system-dependent) [31] |
| Treatment of Solvent Effects | More physically detailed, includes explicit water structure | Approximate, based on a continuum model |
| Computational Cost (System Size) | High (many solvent atoms) | Lower for small systems; can be slower for large systems [31] |
| Recommended Use Case | Final production runs where highest accuracy is critical | Rapid conformational sampling, system setup, and initial screening |
Troubleshooting Guide
Problem 1: Inadequate Conformational Sampling
- Symptoms: Results are not converged, high uncertainty in free energy estimates, failure to observe known transitions.
- Solutions:
- Leverage Implicit Solvent Models: For the initial exploration of the free energy landscape, use an implicit solvent model like Generalized Born (GB). This can provide a ~1 to 100-fold speedup in sampling compared to explicit solvent [31], allowing you to rapidly identify important states.
- Combine with Enhanced Sampling: Use the implicit solvent model in conjunction with advanced sampling techniques (e.g., metadynamics, umbrella sampling) to further accelerate the crossing of high energy barriers.
- Validate with Explicit Solvent: Use the insights and pathways discovered with implicit solvent to run shorter, targeted simulations with an explicit solvent model for final, high-accuracy free energy calculations.
Problem 2: ML Model Failure on Unseen Geometries
- Symptoms: Poor prediction accuracy for molecular configurations not present in the training data.
- Solutions:
- Incorporate Physical Priors: Use a Physics-Informed Neural Network (PINN) architecture. This integrates the physical laws governing your system (e.g., molecular mechanics force fields, relevant PDEs) directly into the model's loss function or architecture, improving its generalizability and robustness [63] [65].
- Implement Active Learning: Set up an active learning workflow where the ML potential is used to run simulations until it encounters configurations with high uncertainty. It then automatically performs a reference calculation (e.g., DFT) for that configuration and retrains itself, thereby improving its dataset and performance iteratively [64].
Problem 3: Inaccurate Force Field Parameters for Novel Molecules
- Symptoms: Unphysical molecular geometries, unrealistic interaction energies, or failure to reproduce experimental data.
- Solutions:
- Follow Force Field Conventions: As emphasized in the FAQs, never mix parameters from different force fields. Parametrize all missing parameters consistently within the chosen force field's framework [62].
- Utilize ML for Parameterization: Employ machine learning models to fit the new molecule's parameters against high-level quantum mechanical data, ensuring they align with the philosophy and functional forms of your target force field.
Experimental Protocols
Protocol 1: Benchmarking Solvent Models for Sampling Speed
This protocol quantitatively compares the conformational sampling efficiency of implicit versus explicit solvent models for your specific system.
- System Preparation: Create a simulation system for a well-characterized molecule or a small segment of your research system (e.g., a protein dihedral or a DNA base pair flip).
- Parallel Simulation Setup:
- Set up two identical systems, one using an explicit solvent model (e.g., TIP3P water with PME for long-range electrostatics).
- Set up the other using an implicit solvent model (e.g., the Generalized Born model).
- Molecular Dynamics: Run multiple, independent molecular dynamics simulations from the same starting structure for both systems. Use the same thermodynamic ensemble (e.g., NVT) and temperature.
- Metric Calculation: Track the rate of a specific conformational change (e.g., dihedral angle flip) over time.
- Analysis: Calculate the speedup factor as the ratio of the transition rate in the implicit solvent simulation to the rate in the explicit solvent simulation. For example, a study found speedups of approximately 7-fold for miniprotein folding [31].
Protocol 2: Implementing a Physics-Informed Neural Network (PINN) for Free Energy Estimation
This methodology outlines how to build a PINN to construct a free energy surface, leveraging physical constraints.
- Data Collection: Use molecular dynamics simulations (potentially accelerated with an implicit solvent model) to collect a set of molecular configurations. The collective variables (CVs) for these configurations (e.g., distances, angles) serve as the input features.
- Model Architecture: Design a neural network where the input is the CV space and the output is the estimated free energy.
- Physics-Informed Loss Function: Construct a composite loss function (Ltotal) that enforces physical plausibility:
- Data Loss (Ldata): Mean squared error between predicted and reference free energy values from a subset of data.
- Physics Loss (L_physics): A penalty term for violating the underlying physical theory, such as the forces derived from the free energy surface (F = -∇G) not matching the observed mean forces from the simulation.
- Total Loss:
L_total = L_data + λ * L_physics, where λ is a weighting hyperparameter [63] [66].
- Training and Validation: Train the network and validate the resulting free energy surface on a held-out test set of simulation data.
Workflow Visualization
The following diagram illustrates a robust, iterative workflow that integrates machine learning, active learning, and different solvent models for efficient and accurate free energy calculations.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key software, tools, and methods essential for implementing ML-enhanced free energy calculations.
| Tool / Method | Type | Primary Function |
|---|---|---|
| Generalized Born (GB) [31] | Implicit Solvent Model | Accelerates conformational sampling by modeling solvent as a continuum, providing significant speedups. |
| Particle Mesh Ewald (PME) [31] | Explicit Solvent Method | Provides a highly accurate treatment of long-range electrostatics with explicit water molecules. |
| Physics-Informed Neural Network (PINN) [63] [65] | Machine Learning Model | Integrates physical laws (e.g., PDEs) into ML models to ensure plausible and generalizable predictions. |
| M3GNet [64] | Machine Learning Potential | A graph neural network for developing accurate molecular dynamics potentials from quantum mechanical data. |
| Active Learning Workflow [64] | Computational Procedure | Iteratively improves an ML model by automatically querying reference calculations for high-uncertainty configurations. |
| GROMACS [62] | Molecular Dynamics Engine | A high-performance software package for simulating biomolecular systems, supporting both implicit and explicit solvent models. |
Benchmarking and Validating Your Solvation Model for Reliable Results
Why is establishing a validation workflow for implicit solvent models critical in molecular dynamics research?
Implicit solvent models significantly speed up molecular dynamics (MD) simulations by treating the solvent as a continuum rather than simulating individual molecules [56]. However, this approximation can alter the system's free-energy landscape and动力学, potentially leading to inaccurate results [56] [12]. A rigorous validation workflow is, therefore, essential to ensure that the gains in computational efficiency do not come at the cost of predictive accuracy, especially for applications in drug discovery where reliable free energy calculations are crucial [12].
FAQ & Troubleshooting Guide
Q1: When we run an implicit solvent simulation, the conformational sampling is much faster than with explicit solvent. Does this mean our results are less accurate?
A1: Not necessarily, but the results must be validated. Faster conformational sampling is a known benefit of implicit solvent models, primarily due to the reduction of viscous drag from the explicit solvent atoms [56]. The table below summarizes potential causes for concern and their solutions.
| Potential Cause | Diagnostic Check | Recommended Action |
|---|---|---|
| Altered free-energy landscape | Compare the populations of key conformational states (e.g., helix vs. coil) or the free energy of a known process (e.g., miniprotein folding) between implicit and explicit solvent simulations. [56] | If landscapes differ significantly, consider using a different implicit solvent model or a machine learning (ML)-based potential that has been validated for similar systems. [12] |
| Inaccurate solvation forces | Calculate solvation free energies for a set of small molecules and compare against explicit solvent results or experimental data. [12] | Use a model specifically trained for free energy calculations, such as those incorporating derivatives with respect to alchemical variables. [12] |
| Poor electrostatic treatment | Analyze the stability of salt bridges or polar interactions in your protein; compare the root mean square deviation (RMSD) of key binding site residues against an explicit solvent benchmark. | Ensure your model's electrostatic parameters (e.g., internal dielectric constant) are appropriate for your system. |
Q2: What is a robust step-by-step protocol to validate a new implicit solvent model or a machine learning potential for our system?
A2: A robust validation protocol should benchmark against both explicit solvent simulations and available experimental data. The workflow below outlines this multi-faceted approach.
Workflow Overview:
Define Validation Metrics: Before running simulations, identify the key properties you need to predict accurately. These depend on your research question and should include:
- Structural Stability: Root mean square deviation (RMSD) of the protein backbone, radius of gyration, secondary structure content over time [67].
- Free Energy: Solvation free energy of small molecules, protein-ligand binding affinity [12].
- Kinetic Properties: Rates of conformational change, although this is more challenging [56].
Run Benchmark Simulations:
- Perform MD simulations using both the implicit solvent model (or ML potential) under investigation and a well-regarded explicit solvent model (e.g., TIP3P water with PME for electrostatics) as a reference [56] [67].
- Ensure all simulations are run for a sufficient duration to achieve adequate sampling for the chosen metrics.
- Use identical solute force fields and starting structures for a direct comparison.
Compare Against Explicit Solvent: Quantitatively compare the validation metrics from step 1. The table below provides a template for this comparison.
Metric Implicit Solvent Result Explicit Solvent Result Agreement Notes Protein Backbone RMSD (Å) 1.5 1.4 Good Values under 1-2 Å generally indicate good structural match. [67] Solvation Free Energy (kcal/mol) -5.0 -5.5 Fair Calculate for multiple small molecules to establish a trend. Distance between key residues (Å) 10.0 ± 1.5 9.8 ± 2.0 Good Monitor specific functional interactions. [67] Compare Against Experimental Data: Where possible, compare simulation results directly with experimental observations. High-quality simulation-derived properties should correlate well with experiments [68]. For example, compare calculated densities and enthalpies of vaporization for pure solvents against experimental measurements [68].
Final Assessment: Synthesize the results from the comparisons. A model can be considered validated for a specific application if it shows consistent agreement with both explicit solvent benchmarks and relevant experimental data within an acceptable margin of error for your study.
Q3: We are using a machine learning-based implicit solvent model trained on explicit solvent data. Our forces look good, but our absolute free energies are off. What is the likely cause and how can we fix it?
A3: This is a known limitation of models trained solely using a force-matching approach [12]. Force-matching determines the potential energy only up to an arbitrary constant, making it unsuitable for predicting absolute free energies, which are essential for calculating binding affinities or solvation free energies [12].
Solution: Seek out and use next-generation ML models that are specifically designed for free energy calculations. These models, such as the λ-Solvation Neural Network (LSNN), extend the training procedure beyond force-matching [12]. They are trained to also match the derivatives of the solvation energy with respect to alchemical variables (e.g., λ_elec and λ_steric), which ensures that the scalar potential can meaningfully approximate the true potential of mean force (PMF) [12]. The diagram below illustrates this advanced training concept.
The Scientist's Toolkit
A successful validation study relies on specific computational tools and datasets.
| Tool / Resource | Function in Validation | Example / Note |
|---|---|---|
| Explicit Solvent MD Software | Provides the benchmark simulation data. | GROMACS [69], AMBER [56] [67]. |
| High-Quality Training Data | Used to train and validate new ML potentials. | Open Molecules 2025 (OMol25) [39], a massive dataset of quantum chemical calculations. |
| Neural Network Potentials (NNPs) | Fast, accurate models that can bridge the gap between QM and MM. | eSEN, UMA models [39], LSNN [12]. |
| Active Learning Workflows | Automates on-the-fly training of ML potentials during MD. | Prevents simulation failures and ensures accuracy [70]. |
| Analysis Suites | Calculates key validation metrics from trajectories. | Tools within GROMACS, AMBER, VMD [67], MDAnalysis. |
Technical FAQ: Solvent Models in Molecular Dynamics
Frequently Asked Questions
Q1: What is the fundamental difference between explicit and implicit solvent models?
- A1: Explicit solvent models simulate individual water molecules (e.g., TIP3P, TIP4P) as distinct particles interacting with the solute. In contrast, implicit solvent models replace the explicit water environment with a continuous dielectric medium, representing solvent effects as an averaged energetic response [71] [55].
Q2: When should I choose an implicit solvent model for my simulation?
- A2: Implicit solvent is advantageous when computational speed is a primary concern, particularly for:
- Rapid Conformational Sampling: It significantly speeds up conformational changes by reducing solvent viscosity [8].
- Large Systems: It can offer better computational scaling for very large molecular systems [55].
- Free Energy Calculations: It provides very effective ways to estimate relative free energies of conformational ensembles [55].
- A2: Implicit solvent is advantageous when computational speed is a primary concern, particularly for:
Q3: When is an explicit solvent model necessary?
- A3: Explicit solvent is often essential for:
- Studying Specific Solvent Interactions: When the role of structured water molecules, hydrogen bonds, or water-mediated interactions is critical to the mechanism being studied [71] [72].
- Accuracy in Biomolecular Simulations: It generally provides a more realistic, albeit computationally expensive, representation of the solvent environment [71] [55].
- A3: Explicit solvent is often essential for:
Q4: How does the choice of solvent model affect the observed conformational dynamics?
- A4: The solvent model can directly influence the speed and pathway of conformational changes. The reduction of solvent friction in implicit models can lead to ~1 to 100-fold speedups in conformational sampling for large changes compared to explicit solvent simulations [8]. Furthermore, the different treatment of electrostatic screening and hydrophobic effects can alter the resulting free-energy landscapes [8].
Q5: For simulations of nucleic acids like DNA, what are the key solvent-related considerations?
- A5: Nucleic acids are highly charged, making electrostatic and ion interactions paramount. Simulations often require careful parameterization of ions and the water model itself [72]. The choice can affect the stability of the double helix, the representation of ion distributions in the major and minor grooves, and the behavior of single-stranded DNA [72].
Troubleshooting Guides
Issue 1: Unrealistically Fast Conformational Changes in Implicit Solvent
- Problem: Your protein or DNA is folding or changing conformation much faster than expected from experimental data or previous explicit solvent simulations.
- Diagnosis: This is a known characteristic of many implicit solvent models due to the lack of viscous drag and the simplified representation of solvent friction [8].
- Solution:
- This may not be an "error" but a feature for enhanced sampling. If quantitative kinetics are required, consider using explicit solvent.
- If you must use implicit solvent, you can tune the effective viscosity by adjusting the Langevin collision frequency in your dynamics parameters to slow down the motions [8].
- Validate the resulting thermodynamic ensembles (e.g., populations of states, free energies) against experimental data or explicit solvent benchmarks.
Issue 2: Poor Stability of DNA Duplex or Protein Secondary Structure
- Problem: Your simulated DNA duplex unwinds or your protein's native secondary structure collapses during an implicit solvent simulation.
- Diagnosis: Some implicit solvent models, particularly older Generalized Born (GB) implementations, do not accurately reproduce the balance of interactions that stabilize secondary structures and base-pairing [71] [55].
- Solution:
- Verify you are using a modern, well-parameterized implicit solvent model (e.g., the latest IGB models in AMBER) that has been tested for nucleic acids or proteins [71].
- Consider switching to an explicit solvent model like TIP3P or TIP4P for production runs, as they are generally more reliable for maintaining structural integrity [71] [72].
- For DNA, ensure you are using a force field with corrected parameters for α/γ torsions and glycosidic torsions (e.g.,
bsc0andχOL4) [72].
Issue 3: Inaccurate Representation of Solvent-Mediated Interactions in Complexes
- Problem: You are simulating a protein-DNA or protein-glycosaminoglycan complex, and the binding interface or conformational dynamics do not match experimental evidence.
- Diagnosis: Specific, structured water molecules are often critical at biomolecular interfaces. For instance, in protein-glycosaminoglycan complexes, about half of the residue contacts are mediated by water [71]. Implicit solvent cannot capture these discrete effects.
- Solution:
- For studying binding interfaces, explicit solvent (e.g., TIP3P, OPC) is highly recommended [71].
- If resources are limited, consider a hybrid approach: equilibrate the system with explicit solvent, then use implicit solvent for specific parts of the analysis or further sampling, while being aware of the limitations.
Quantitative Data Comparison
The table below summarizes key performance characteristics of different solvent models as observed in benchmark studies.
Table 1: Benchmarking Solvent Models for Biomolecular Simulations
| Solvent Model | Model Type | Reported Sampling Speed vs. Explicit* | Key Applications & Notes |
|---|---|---|---|
| TIP3P [71] | Explicit | 1x (Baseline) | Most popular explicit model; general-purpose for proteins and nucleic acids [71]. |
| TIP4P/TIP4PEw [71] [72] | Explicit | Slower than TIP3P | Improved description of peptide conformation and bulk water properties [72]. |
| OPC [71] | Explicit | Slower than TIP3P | High-accuracy model; excellent for reproducing experimental water properties [71]. |
| GB (IGB=1,2,5,7,8) [71] | Implicit | ~1 to 100x faster [8] | Speedup is system-dependent. Faster conformational search but may compromise accuracy of specific solvent effects [71] [8] [55]. |
| GB (with low viscosity) [8] | Implicit | ~50x faster (for miniprotein folding) | Maximum speedup achieved by reducing effective solvent viscosity [8]. |
*Speedup refers to the rate of conformational sampling, not computational performance.
Table 2: Impact of Solvent Model on Heparin (GAG) Molecular Descriptors [71]
| Molecular Descriptor | Variation Across 11 Solvent Models | Implicit vs. Explicit Discrepancy |
|---|---|---|
| End-to-End Distance (EED) | Significant | Yes |
| Radius of Gyration | Significant | Yes |
| Ring Puckering | Moderate | Yes |
| Dihedral Angles | Moderate | Yes |
| Intramolecular H-Bonds | Affected | Yes |
Experimental Protocols
Protocol 1: Standard MD Setup for a DNA Oligomer in Explicit Solvent (Based on [72])
This protocol outlines the key steps for setting up a simulation of a double-stranded DNA molecule in an explicit saline environment.
- 1. System Building:
- Obtain or generate an initial B-DNA structure (e.g., using the
make_naserver or other tools). - Place the DNA in the center of a periodic box (e.g., rectangular or octahedral) with a minimum distance of 8-10 Å between the solute and the box edge.
- Obtain or generate an initial B-DNA structure (e.g., using the
- 2. Solvation and Ion Addition:
- Solvate the system with an explicit water model (e.g., TIP3P, TIP4P-EW).
- Add Na⁺ and Cl⁻ ions to a physiological concentration (e.g., 150 mM). First, add enough Na⁺ ions to neutralize the negative charge of the DNA backbone, then add additional salt pairs to reach the desired concentration.
- 3. Energy Minimization:
- Perform a two-step minimization to remove bad contacts.
- Step 1: Minimize with positional restraints on the DNA heavy atoms (e.g., 100 kcal/mol/Ų), focusing on relaxing the solvent and ions.
- Step 2: Perform a full minimization without any restraints.
- Perform a two-step minimization to remove bad contacts.
- 4. System Equilibration:
- Gradually heat the system from a low temperature (e.g., 50 K) to the target temperature (310 K) in the NVT ensemble over 50-100 ps, maintaining restraints on the DNA.
- Equilibrate the density of the system by running a short simulation (50-100 ps) in the NPT ensemble at the target temperature and pressure (1 atm), still with DNA restraints.
- Release the restraints and equilibrate the entire system further in NPT until the density and potential energy stabilize.
- 5. Production MD:
- Run a long, unrestrained simulation in the NPT ensemble to collect data for analysis. Use a 2 fs timestep, applying constraints to bonds involving hydrogen (e.g., SHAKE or LINCS).
Protocol 2: Comparative Study Using Implicit and Explicit Solvent
This protocol describes a framework for directly comparing the effect of solvent models on a molecule's conformational dynamics, as performed in [71].
- 1. System Preparation:
- Start with the same initial molecular structure (e.g., a protein, DNA, or glycosaminoglycan like heparin).
- 2. Parallel Simulation Setup:
- Setup A: Prepare the system for explicit solvent simulation as described in Protocol 1.
- Setup B: Prepare the system for implicit solvent simulation using a Generalized Born (GB) model (e.g., one of the IGB models in AMBER). No periodic box or explicit water molecules are needed, but counterions may still be used.
- 3. Simulation and Analysis:
- Run multiple, independent MD simulations for each setup. The length will depend on the system size and dynamics of interest.
- For both sets of trajectories, calculate the same set of molecular descriptors (e.g., Radius of Gyration, End-to-End Distance, RMSD, dihedral angle distributions, hydrogen bonding).
- Statistically compare the results from the implicit and explicit solvent simulations to identify significant differences in the conformational ensemble and dynamics.
Solvent Model Selection Workflow
The following diagram illustrates a decision-making process for selecting between implicit and explicit solvent models, based on the research goals and constraints.
Solvent Model Selection Guide
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software and Force Fields for Biomolecular MD
| Tool / Reagent | Type | Function / Application | Example Use |
|---|---|---|---|
| AMBER [71] | MD Software Suite | A comprehensive package for simulating biomolecules. Includes tools for simulation (pmemd) and analysis (cpptraj). | Used for benchmarking solvent models for heparin [71] and comparing conformational sampling speeds [8]. |
| GROMACS [72] [34] | MD Software Suite | A high-performance MD engine for simulating Newtonian dynamics. | Used for simulations of amino acid-DNA interactions [72] and drug solubility studies [34]. |
| GLYCAM06 [71] | Force Field | A force field specifically parameterized for carbohydrates and glycosaminoglycans. | Essential for accurate simulation of heparin and other polysaccharides [71]. |
| AMBER ff99SB-ILDN [72] | Force Field | A force field for proteins, with improvements in sidechain torsions. | Used for simulating amino acid sidechain analogs in DNA solutions [72]. |
| parm99/bsc0/χOL4 [72] | Force Field | A combination of parameter sets providing a high-quality description of DNA conformation. | Recommended for DNA simulations to improve α/γ and glycosidic torsions [72]. |
| TIP3P [71] [72] | Explicit Water Model | A standard 3-site water model; most widely used due to balance of speed and accuracy. | Common default in many MD studies of proteins and nucleic acids. |
| TIP4P-EW [72] | Explicit Water Model | A 4-site model that provides improved descriptions of bulk water and peptide properties. | Chosen for its performance in modeling protein-nucleic acid interactions [72]. |
| OPC [71] | Explicit Water Model | A 4-site model optimized for outstanding agreement with experimental water properties. | Used in high-accuracy benchmarking studies [71]. |
| Generalized Born (GB) [71] [8] | Implicit Solvent Model | A fast, approximate method for calculating solvation energies. Various parameterizations exist (IGB1-8). | Used for rapid conformational sampling and free energy estimation [71] [8]. |
Technical Support Center
This support center provides troubleshooting guidance for researchers calculating hydration free energies and binding affinities, key for drug development within molecular dynamics (MD) simulations. The FAQs address common challenges when working with explicit and implicit solvent models.
Frequently Asked Questions (FAQs)
1. My implicit solvent simulation shows over-stabilized salt bridges and incorrect helix populations. What is the cause and how can I fix this? This is a known limitation of certain implicit solvent models, particularly Generalized Born (GB) models used in isolation. The issue arises from insufficient electrostatic screening and inaccurate modeling of the hydrophobic effect, which alters the protein's energy landscape [60].
- Solution: Consider using a hybrid implicit-explicit solvent approach, where a layer of explicit water molecules surrounds the solute while the bulk solvent is treated implicitly [60]. Furthermore, validate your results against explicit solvent simulations or experimental data where possible. For community-accepted best practices, consult benchmarks like those enabled by large datasets such as Meta's OMol25 [39].
2. Why is my binding free energy calculation taking months to complete, and how can I accelerate it? Prolonged computation times in explicit solvent are often due to the costly sampling of explicit water molecules and the slow conformational dynamics caused by solvent viscosity [5] [56].
- Solution: For faster conformational sampling, you can use an implicit solvent model like Generalized Born (GB). GB simulations can provide a conformational sampling speedup of approximately 1 to 100-fold compared to explicit solvent (PME), depending on the system and the conformational change being studied [56]. Be aware that this speedup may come with a trade-off in accuracy for certain electrostatic interactions.
3. My explicit solvent calculation of a reaction mechanism gives different results compared to an implicit solvent model. Which one should I trust? Discrepancies often occur because implicit solvent models (like PCM, COSMO, or SMD) can fail to describe explicit, specific solvent-solute interactions such as hydrogen bonding. A documented case study on the Baylis-Hillman reaction showed that implicit solvent models could produce solvation free energies that are off by ~10 kcal/mol [5].
- Solution: For processes where specific solvent-solute interactions are critical (e.g., reactions involving charged species), explicit solvent models are generally more reliable. Machine Learning Neural Network Potentials (NNPs) trained on high-accuracy quantum chemical data now offer a path to achieving near-quantum chemistry accuracy at a fraction of the computational cost, making them a powerful alternative for such validation [39] [5].
4. How can I account for pH in my MD simulations when calculating pKa values? Traditional MD simulations use fixed protonation states, which is a source of error when pKa values are near the pH of interest. The Continuous Constant pH Molecular Dynamics (CPHMD) method explicitly includes pH as an external parameter.
- Solution: Implement a CPHMD approach. A hybrid method that uses the GB model for efficiency in calculating protonation-state forces, while propagating conformational dynamics in explicit solvent for accuracy, has been shown to yield an average absolute deviation of 0.53 from experimental pKa values [73].
Quantitative Comparison of Solvent Models
The table below summarizes key metrics to help you select the appropriate solvent model for your project.
| Metric | Explicit Solvent (PME) | Implicit Solvent (GB) | Hybrid & Advanced Methods |
|---|---|---|---|
| Conformational Sampling Speedup (Relative to explicit PME) | 1x (Baseline) | ~1x to 100x (System-dependent) [56] | Varies (e.g., ML-NNPs can be far faster than DFT) [39] |
| Sampling Acceleration Factor | N/A | ~2x to 20x common [56] | N/A |
| Electrostatic Treatment | Explicit water dipole reorientation [5] | Approximate continuum dielectric [60] | Explicit near solute, continuum bulk [60] |
| Computational Cost | High (Many solvent atoms) | Lower (No explicit solvent atoms) | Moderate |
| pKa Prediction Accuracy (AAD) | Feasible but slow convergence [73] | Less accurate for buried residues [73] | 0.53 (CPHMD-explicit) [73] |
| Known Artifacts | Slow sampling, high viscosity [56] | Over-stabilized salt bridges, altered secondary structure populations [60] | Potential boundary effects |
Detailed Experimental Protocols
Protocol 1: Absolute Binding Free Energy Calculation using Alchemical Transformation
This method calculates the reversible work for decoupling the ligand from its environment (protein and solvent) through a series of non-physical intermediate states [74].
System Preparation:
- Obtain the 3D structure of the protein-ligand complex.
- Solvate the complex in a pre-equilibrated water box (e.g., TIP3P) and add ions to neutralize the system's charge.
Equilibration:
- Minimize the energy of the system to remove steric clashes.
- Perform a short MD simulation with positional restraints on the protein and ligand backbone atoms to equilibrate the solvent and side chains.
- Run an unrestrained equilibration simulation to ensure system stability.
Restraining the Ligand:
- Apply harmonic restraints to the ligand's translational, rotational, and conformational degrees of freedom within the binding site. This prevents the ligand from drifting and improves sampling convergence [74].
Alchemical Transformation:
- Define a pathway (e.g., using a coupling parameter, λ) that gradually turns off the non-bonded interactions (electrostatic and van der Waals) between the ligand and its environment (protein and solvent).
- Run a series of simulations at different λ values (e.g., λ = 0, 0.1, 0.2, ..., 1). At λ=1, the ligand is fully interacting; at λ=0, it is completely decoupled.
Free Energy Analysis:
- Use techniques like Free Energy Perturbation (FEP) or Thermodynamic Integration (TI) to compute the free energy change (ΔG_bind) for the alchemical process.
- Rigorously calculate and remove the contribution of the restraining potentials applied in Step 3 to obtain the unbiased standard binding free energy [74].
Protocol 2: Hydration Free Energy Calculation using Neural Network Potentials (NNPs)
This protocol leverages modern machine learning potentials to compute hydration free energies with high accuracy and efficiency [39] [5].
Model Selection and System Setup:
- Select a pre-trained NNP, such as Meta's eSEN or Universal Model for Atoms (UMA), which have been trained on vast datasets like OMol25 [39].
- Prepare the solute molecule (e.g., a small organic compound) in a simulation box.
Explicit Solvent Simulation with NNP:
- Instead of using a traditional force field, use the selected NNP to drive an explicit solvent molecular dynamics (MD) simulation. The NNP provides quantum-mechanics-level accuracy for energies and forces at a much lower computational cost [39].
- A user reported that such models provide "much better energies than the DFT level of theory I can afford" for large systems [39].
Free Energy Calculation:
- Employ an alchemical free energy method (similar to Protocol 1, Step 4) to gradually decouple the solute from the explicit solvent water.
- Alternatively, for some properties, the NNP may allow for direct computation from a sufficiently long MD trajectory.
Validation:
- Compare the computed hydration free energy against experimental values or high-level quantum chemical benchmarks to validate the accuracy of the NNP for your specific system.
Workflow Visualization
The following diagram illustrates the logical decision process for selecting and applying a solvent model, integrating the troubleshooting advice and protocols above.
Diagram: Solvent Model Selection Workflow
Research Reagent Solutions
The table below lists key computational tools and datasets essential for research in this field.
| Reagent / Resource | Type | Function / Application |
|---|---|---|
| OMol25 Dataset [39] | Quantum Chemistry Dataset | Provides over 100 million high-accuracy calculations to train and validate machine learning potentials for biomolecules, electrolytes, and metal complexes. |
| Neural Network Potentials (NNPs) [39] | Machine Learning Model | Offers quantum-mechanical accuracy for molecular energies and forces at a fraction of the computational cost, enabling large-scale explicit solvent simulations. |
| Generalized Born (GB) Model [56] [60] | Implicit Solvent Model | Approximates electrostatic solvation effects for faster conformational sampling and free energy calculations compared to explicit solvent. |
| Continuous Constant pH MD (CPHMD) [73] | Simulation Methodology | Allows pKa calculations and studies of pH-dependent phenomena by dynamically updating protonation states during a simulation. |
| Alchemical Free Energy Perturbation (FEP) [74] | Calculation Method | Computes free energy differences (e.g., binding or hydration affinities) by gradually transforming one state into another via non-physical pathways. |
The Role of High-Quality Datasets and Neural Network Potentials for Benchmarking
Frequently Asked Questions (FAQs)
FAQ 1: My neural network potential (NNP) performs well on training data but fails during molecular dynamics (MD) production runs. What is the likely cause and how can I fix it?
This is typically caused by insufficient sampling of the chemical and conformational space during training, leading to poor extrapolation capabilities [3] [75]. The potential encounters geometries not represented in its training set.
- Solution: Implement an active learning workflow. This involves:
- Using a calibrated committee of NNPs to run short, exploratory MD simulations [75].
- Identifying configurations where the committee members disagree significantly, as this indicates regions of high uncertainty [75].
- Sending these select configurations for ab initio calculation and adding them to the training set [3] [75].
- Retraining the NNP on this augmented dataset. This iterative process ensures the potential learns from its own mistakes and becomes robust across a wider range of atomic environments.
FAQ 2: When benchmarking a solvation model, what are the critical factors beyond model architecture that significantly impact accuracy?
The quality and composition of the benchmark dataset itself are as critical as the model. Key factors often overlooked include:
- Molecular Flexibility: For medium-to-large, drug-like molecules, a single gas-phase geometry is insufficient. You must use exhaustive conformational ensembles for each phase (gas and solvent) to account for solvent-induced structural changes [76].
- Data Diversity: The benchmark set should contain structurally and functionally diverse molecules, not just many data points from a few molecules. This tests the model's generalizability [76].
- Phase-Specific Geometries: Using a single geometry (e.g., a gas-phase optimized structure) for solvation energy calculations can introduce systematic bias. Always use separate, phase-specific optimized geometries or Boltzmann-weighted ensembles [76].
FAQ 3: How can I rigorously benchmark the conformational sampling of a machine-learned MD method against a ground truth?
A standardized benchmark should evaluate multiple metrics across a diverse set of proteins [77].
- Procedure:
- Select a Diverse Ground Truth Dataset: Use a dataset of various proteins with different sizes and folds (e.g., from 10 to over 200 residues) that have been extensively sampled with an established, high-quality method [77].
- Employ Enhanced Sampling: Use a method like Weighted Ensemble (WE) sampling to efficiently explore conformational space, including rare events, within a tractable simulation time [77].
- Compare Multiple Observables: Compute a suite of metrics against the ground truth. This should include [77]:
- Global Properties: Time-lagged Independent Component Analysis (TICA) landscapes, radius of gyration (RoG) distributions, and contact map differences.
- Local Properties: Distributions of bond lengths, angles, and dihedrals.
- Quantitative Divergences: Wasserstein-1 and Kullback-Leibler (KL) divergences for the above distributions.
FAQ 4: What is a key advantage of using a pre-trained, general NNP and fine-tuning it for my specific system?
This strategy, known as transfer learning, dramatically reduces the computational cost and data required to develop an accurate potential for your specific application. A general pre-trained model (e.g., for C, H, N, O elements) already contains a foundational understanding of chemical bonding and interactions. By fine-tuning it with a small amount of new, system-specific data from DFT calculations, you can achieve Density Functional Theory (DFT)-level accuracy for your target material without needing thousands of new expensive quantum calculations [78].
FAQ 5: My implicit solvent simulations are computationally efficient but produce erroneous dynamics for my protein. How should I validate the model?
The accuracy of implicit solvent models is highly force-field dependent and must be validated for each specific system [79].
- Validation Protocol:
- Run a Control Explicit Solvent Simulation: Perform a (shorter) explicit solvent MD simulation of the same system using the same force field [79].
- Compare Key Structural and Dynamic Properties: Benchmark your implicit solvent results against the explicit solvent simulation and any available experimental data. Critical properties to compare include [79]:
- Root-mean-square deviation (RMSD) from a known structure.
- Radius of gyration (RoG).
- Solvent-accessible surface area (SASA).
- NMR order parameters (if available).
- Intra-peptide contact maps. Significant discrepancies indicate that the implicit solvent model or force field is unsuitable for your system and may require reparameterization or the use of an explicit solvent model [79].
Benchmarking Data and Model Performance
The tables below summarize key quantitative data from recent studies for easy comparison of datasets and model performance.
Table 1: Overview of Recent Benchmarking Datasets for MD Simulations
| Dataset Name | System Type / Size | Key Metrics | Description & Purpose |
|---|---|---|---|
| FlexiSol [76] | 1551 molecule-solvent pairs; 25000+ conformers | Solvation energy, Partition ratios (logK) | Benchmarks solvation models for flexible, drug-like molecules using exhaustive conformer ensembles. |
| Standardized WE Benchmark [77] | 9 proteins (10-224 residues) | >19 metrics (TICA, RoG, contact maps, KL divergence) | Provides a ground-truth dataset and framework for evaluating protein conformational sampling. |
| EMFF-2025 Training Data [78] | 20 High-Energy Materials (HEMs) | Energy MAE (< 0.1 eV/atom), Force MAE (< 2 eV/Å) | Dataset for developing a general NNP for C,H,N,O-based materials, validated on structure and decomposition. |
Table 2: Performance of Selected Neural Network and Solvation Models
| Model Name | Model Type | Key Performance Results | Applicability & Notes |
|---|---|---|---|
| EMFF-2025 [78] | General NNP (for HEMs) | Predicts HEM structures, mechanical properties, and decomposition pathways at DFT-level accuracy. | Uses transfer learning for data efficiency; applicable to C, H, N, O systems. |
| Cluster-to-PBC MLP [3] | Machine Learning Potential | MLP trained on cluster data transfers well to Periodic Boundary Conditions (PBC) for Diels-Alder reaction in solvent. | Offers a cost-effective strategy for modeling reactions in explicit solvent. |
| AiiDA-TrainsPot [75] | Automated NNP Training | Achieves state-of-the-art accuracy for carbon allotropes via automated active learning and data augmentation. | Democratizes NNP development; uses calibrated committee disagreement for uncertainty. |
| Physics-Informed ML (Starling) [80] | ML pKa Prediction | Predicts macroscopic pKa, isoelectric points, and logD values in minutes. | Part of the Rowan platform; bridges physics-based and data-driven models. |
Experimental Protocols and Workflows
Protocol 1: Active Learning Workflow for Building a Robust NNP for Solutions
This protocol is adapted from methodologies used to model chemical processes in explicit solvents [3] and automated training frameworks [75].
Initial Data Generation:
- Gas Phase Set: Generate configurations for the solute(s) by randomly displacing atomic coordinates, focusing on the reaction path (including transition states) [3].
- Explicit Solvent Cluster Set: Create cluster models with solute surrounded by explicit solvent molecules. The solvent shell radius should be at least the cut-off radius planned for the MLP to avoid boundary artifacts [3].
Initial MLP Training: Train the first version of the MLP on the combined gas-phase and cluster data.
Active Learning Loop:
- Propagation: Run short MD simulations using the current MLP.
- Uncertainty Quantification & Selection: Use a selector (e.g., a committee of models or a descriptor-based method like SOAP) to identify configurations where the MLP is uncertain [3] [75].
- Ab Initio Labeling: Perform reference quantum mechanical (e.g., DFT) calculations on the selected configurations to get accurate energies and forces.
- Dataset Augmentation & Retraining: Add the newly labeled data to the training set and retrain the MLP.
- Convergence Check: Repeat until errors on energy and forces are below a defined threshold and MD simulations are stable.
The following diagram illustrates this iterative workflow:
Protocol 2: Standardized Benchmarking for Protein Conformational Sampling
This protocol is based on a modular framework for evaluating MD methods using weighted ensemble sampling [77].
System Preparation:
- Select a set of diverse proteins from a ground truth dataset (e.g., the provided set of 9 proteins) [77].
- For each protein, obtain multiple starting structures that cover its conformational space.
Simulation Setup:
Propagation and Sampling:
- Run the WE simulation using the method to be benchmarked (e.g., a new MLP or force field).
- Use a flexible propagator interface to run simulations with any engine [77].
Comprehensive Analysis:
- Run the analysis suite to compute over 19 different metrics and visualizations [77].
- Global Metrics: Compare TICA energy landscapes, contact map differences, and distributions for the radius of gyration.
- Local Metrics: Compare distributions of bond lengths, angles, and dihedrals.
- Quantitative Divergence: Calculate Wasserstein-1 and Kullback-Leibler divergences for all relevant analyses to quantify differences from the ground truth.
The logical flow of this benchmark is as follows:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Datasets for Benchmarking NNPs and Solvation Models
| Item / Resource | Function / Purpose | Example Use Case |
|---|---|---|
| Active Learning Platforms (e.g., AiiDA-TrainsPot [75], DP-GEN [78]) | Automates the iterative process of NNP training, data augmentation, and uncertainty quantification. | Efficiently building a robust NNP for a novel material or molecular system from scratch. |
| Pre-trained General NNPs (e.g., EMFF-2025 [78], Egret-1 [80]) | Provides a foundational potential that can be fine-tuned for specific systems, saving computational resources. | Rapidly developing a specialized potential for a new energetic material or organic molecule. |
| Standardized Benchmark Suites (e.g., WE Framework [77], FlexiSol [76]) | Provides ground-truth data and standardized metrics for fair and reproducible comparison of MD methods. | Objectively evaluating the performance of a new MLP against existing force fields. |
| Descriptor-Based Selectors (e.g., SOAP [3]) | Uses molecular descriptors to assess whether a training set adequately represents a chemical space during active learning. | Identifying and filling gaps in the training data for a complex reaction in solution. |
| Enhanced Sampling Tools (e.g., WESTPA [77]) | Enables efficient exploration of conformational space and sampling of rare events through weighted ensemble methods. | Benchmarking a model's ability to reproduce protein folding dynamics or ligand unbinding. |
| Solvation Benchmark Datasets (e.g., FlexiSol [76]) | Provides high-quality, diverse data on solvation energies and partition ratios for flexible molecules. | Testing and validating the accuracy of implicit or explicit solvation models. |
Frequently Asked Questions (FAQs)
1. Why are my simulation results different when I switch from an explicit to an implicit solvent model?
The differences arise because explicit and implicit solvent models represent the solvent environment in fundamentally different ways, each with inherent strengths and weaknesses [55]. An explicit solvent model simulates individual water molecules, capturing specific effects like hydrogen bonding and solvent structure. In contrast, an implicit solvent model replaces the explicit water with a continuous dielectric, approximating the average effect of the solvent [37]. This fundamental difference can lead to variations in the simulated conformational dynamics, solvation forces, and thermodynamic properties of your solute [55] [81].
2. For which systems are implicit solvent models likely to perform poorly?
Implicit solvent models tend to be less reliable for systems where specific, non-bulk solvent behavior is critical [37]. Performance challenges are often seen with:
- Nucleic Acids: The highly charged backbone and specific ion-binding effects are difficult to model with a simple dielectric [37].
- Membranes and Interfaces: The environment is not a homogeneous, isotropic bulk solution, violating a core assumption of most implicit models [37].
- Systems with Bridging Solvent Molecules: When water molecules form essential bridges between parts of the solute, their explicit absence can lead to inaccurate structures or energies [55].
- Processes Involving Large Solvent Dipole Reorientation: The continuum model cannot capture the specific reorientation of water dipoles in response to solute conformational changes [55].
3. My implicit solvent simulation is running much faster, but are the dynamics physically accurate?
Implicit solvent simulations can be significantly faster—often by two orders of magnitude—because they eliminate the thousands of solvent degrees of freedom [81]. However, this speed comes with a trade-off in dynamical accuracy. The absence of explicit solvent atoms removes viscous damping and atomic-level friction. To maintain a constant temperature, a Langevin dynamics integrator with user-defined friction coefficients is often used [82] [81]. This means the dynamics are a good approximation for sampling conformational space but may not perfectly reproduce the real-world diffusive timescales of motion [55].
4. How can I validate the results from an implicit solvent model?
Validation against more accurate methods is crucial. Recommended approaches include:
- Comparison with Explicit Solvent: Run a shorter, parallel simulation with explicit solvent for the same system and compare key observables like root-mean-square deviation (RMSD) or radius of gyration [81].
- Benchmarking against Experimental Data: Compare your results with available experimental data, such as NMR spectroscopy or small-angle X-ray scattering (SAXS) profiles.
- Comparison with Higher-Level Theory: For small molecules or key interactions, compare the energies and geometries with those obtained from higher-level quantum mechanics (QM) calculations [83].
5. What is the role of the dielectric constant (ε) in implicit solvent models, and how should I choose its value?
The dielectric constant (ε) is a critical parameter that represents the polarizability of the environment [37]. In the Generalized Born (GB) model, for instance, it directly influences the electrostatic contribution to the solvation energy.
- For Aqueous Solvent: A value of ε = 80 is typically used to represent bulk water.
- For Solute Interior: An internal dielectric constant (ε_int) must also be set. A value of 1-4 is common to represent the low polarizability of a protein's interior, while a value of 1 represents a vacuum. The choice can significantly impact outcomes, especially for charged systems. It is often recommended to test the sensitivity of your results to small changes in this parameter.
Troubleshooting Guide
| Symptom | Possible Cause | Solution |
|---|---|---|
| Unphysical clustering of solute molecules | Lack of explicit, repulsive water molecules between solutes; underestimated non-polar solvation contribution [55]. | Check and calibrate the non-polar solvation term (e.g., SASA model). Increase the scaling factors for van der Waals radii to create a larger exclusion volume. |
| Over-stabilization of charged groups | The continuum dielectric may be over-screening electrostatic interactions, especially with a high internal dielectric [37]. | Re-evaluate the choice of internal dielectric constant. Validate salt-bridge or ion-pair interactions against explicit solvent or experimental data. |
| Poor sampling of conformational states | The smoothed energy landscape of implicit solvent reduces energy barriers, leading to "faster" but potentially less accurate dynamics [55]. | Use the simulation for enhanced sampling (e.g., to identify metastable states) and then validate the stability of those states with explicit solvent. |
| Unstable protein structure during dynamics | Inaccurate balance between the solvation energy term and the vacuum force field parameters; missing specific stabilizing H-bonds [55]. | Ensure the force field is compatible with the implicit solvent model. Consider using a force field specifically parameterized for implicit solvation. |
| Energy conservation issues in NVE simulation | Inaccuracies in the calculation of solvation forces, particularly with certain Generalized Born implementations [82]. | Switch to a different implicit solvent implementation or use an NVT ensemble with a thermostat, which is more common and robust for implicit solvent MD. |
Quantitative Comparison of Solvent Models
Table 1: Characteristic Comparison of Explicit and Implicit Solvent Models
| Feature | Explicit Solvent | Implicit Solvent |
|---|---|---|
| Computational Cost | High (80-90% of time spent on solvent) [55] | Low (10-100x faster) [81] |
| Solvent Representation | Individual water molecules | Dielectric continuum |
| Sampling Speed | Slower (viscous damping) | Faster (friction can be tuned) [55] |
| Specific Solvent Effects | Captured (e.g., H-bonds, bridging) | Not captured [55] |
| System Setup | More complex (solvation, ion placement) | Simplified |
| Dielectric Response | Explicit, atomic | Pre-defined, uniform constant [37] |
Table 2: Performance of Modern Implicit Solvent and Neural Network Potentials on Molecular Energy Benchmarks
The table below shows performance metrics (lower values are better) on standardized benchmarks, demonstrating the accuracy of next-generation models. The Wiggle150 benchmark tests the ability to reproduce torsional potential energy surfaces, while GMTKN55 is a broad benchmark of general main-group chemistry [83].
| Model Type | Model Name | Wiggle150 (kcal/mol) | GMTKN55 WTMAD-2 (kcal/mol) |
|---|---|---|---|
| Neural Network Potential (NNP) | eSEN (conserving, medium) | ~0.3 | ~1.0 |
| Neural Network Potential (NNP) | UMA (Universal Model for Atoms) | ~0.3 | ~1.0 |
| High-Accuracy DFT | ωB97M-V/def2-TZVPD | Reference | Reference |
Experimental Protocols
Protocol 1: Comparative Dynamics Using Explicit and Implicit Solvent
This protocol outlines a method to compare the behavior of a system using both solvent modeling approaches, allowing for direct validation of implicit solvent results.
Methodology:
- System Preparation: Start with the same solute structure (e.g., a protein or peptide) and force field.
- Explicit Solvent Simulation:
- Solvate the solute in a pre-equilibrated water box (e.g., TIP3P model) using tools like
gmx solvate(GROMACS). - Add ions to neutralize the system and achieve a desired ionic concentration.
- Energy minimize the system.
- Equilibrate with position restraints on solute heavy atoms, first in the NVT ensemble (constant Number of particles, Volume, and Temperature) for 100 ps, then in the NPT ensemble (constant Number of particles, Pressure, and Temperature) for 100 ps.
- Run a production MD simulation for a defined timeframe (e.g., 100 ns) using an integrator like
md(leap-frog) in GROMACS [82].
- Solvate the solute in a pre-equilibrated water box (e.g., TIP3P model) using tools like
- Implicit Solvent Simulation:
- Using the same initial solute structure, set up a simulation with an implicit solvent model (e.g., Generalized Born).
- In the parameter file (e.g.,
.mdpin GROMACS), setintegrator = sd(stochastic dynamics) to use a Langevin thermostat, which provides temperature control and friction [82]. - Define the implicit solvent and its dielectric constant in the parameters.
- Run a production simulation of equivalent length in time (e.g., 100 ns).
- Analysis:
- Compare key observables from both trajectories, such as:
- Root-mean-square deviation (RMSD) and fluctuation (RMSF).
- Radius of gyration.
- Secondary structure content.
- Interatomic distances for specific interactions (e.g., salt bridges).
- Compare key observables from both trajectories, such as:
Protocol 2: Assessing Solvation Free Energy with a Poisson-Boltzmann/SASA Model
This protocol uses a combination of a Poisson-Boltzmann (PB) solver and a Solvent-Accessible Surface Area (SASA) model to calculate the free energy of solvation, a key metric for validating solvent models against experimental data [37].
Methodology:
- Structure Preparation: Obtain a clean 3D structure of the small molecule or solute.
- Parameterization: Assign atomic partial charges and radii using a compatible force field (e.g., AMBER, CHARMM).
- Electrostatic Calculation (ΔGele):
- Use a PB solver (e.g., APBS) to calculate the electrostatic component of the solvation free energy.
- The calculation is performed by solving the Poisson-Boltzmann equation numerically for the solute in a vacuum (ε=1) and in solvent (ε=80), with the difference yielding ΔGele [37].
- Non-Polar Calculation (ΔG_np):
- Calculate the SASA for each atom in the molecule.
- Compute the non-polar contribution using the formula: ΔGnp = Σ σi * SASAi, where σi is an atom-specific surface tension parameter [37].
- Total Solvation Free Energy:
- Sum the components: ΔGsol = ΔGele + ΔGnp.
- Compare the computed ΔGsol with experimental hydration free energy data to assess the model's accuracy.
Research Reagent Solutions
Table 3: Essential Software Tools for Solvent Model Research
| Item | Function | Example Use Case |
|---|---|---|
| GROMACS | A molecular dynamics package for simulating biomolecular systems [82]. | Running production MD simulations with both explicit (TIP3P) and implicit (Generalized Born) solvent models. |
| AMBER | A suite of biomolecular simulation programs with supported force fields [84]. | Parameterizing molecules and running simulations with the GB(OBC) implicit solvent model. |
| Meta's eSEN/UMA Models | Pre-trained neural network potentials (NNPs) for molecular modeling [83]. | Providing highly accurate and fast energy/force calculations that implicitly include solvent effects, trained on massive quantum chemistry datasets (OMol25). |
| APBS | A software for modeling the electrostatics of biomolecules using the Poisson-Boltzmann equation [37]. | Calculating the electrostatic component of solvation free energy for static structures. |
| CHARMM | A versatile program for classical and quantum mechanics simulations with comprehensive force fields [84]. | Running simulations with the polarizable Drude oscillator model to study polarization effects explicitly. |
Conceptual Diagrams
Diagram 1: Fundamental Representations of Solvent Models.
Diagram 2: Solvent Model Selection Workflow.
Conclusion
The choice between explicit and implicit solvent models is not a matter of one being universally superior, but rather hinges on the specific scientific question and available computational resources. Explicit models remain the gold standard for capturing detailed, specific solvent interactions but at a high computational cost. Implicit models offer unparalleled efficiency for rapid sampling and screening, though they may average out crucial local effects. The most powerful modern strategies involve hybridization—using implicit solvents for extensive pre-sampling or combining them with machine learning correctors to bridge the accuracy gap. Future directions point toward the wider adoption of ML-augmented models that offer near-explicit accuracy at implicit-model speeds, the increased use of multi-scale simulation frameworks, and the integration of quantum–continuum modules for simulating complex reaction mechanisms in solution. These advancements will profoundly impact biomedical and clinical research by enabling more accurate prediction of drug solubility, protein-ligand binding affinities, and the dynamics of large biomolecular systems, thereby accelerating the drug discovery pipeline.
References
- 1. Explicit solvent models - (General Chemistry II) [https://fiveable.me/key-terms/general-chemistry-ii/explicit-solvent-models]
- 2. Solvent model [https://en.wikipedia.org/wiki/Solvent_model]
- 3. Modelling chemical processes in explicit solvents with ... [https://www.nature.com/articles/s41467-024-50418-6]
- 4. When should we use explicit solvent molecules in QM ... [https://longkunxuluke.github.io/posts/2020/07/blog-post-2/]
- 5. Machine Learning for Explicit Solvent Molecular Dynamics [https://corinwagen.github.io/public/blog/20230720_ml_aimd.html]
- 6. Reweighting of molecular simulations with explicit-solvent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8373061/]
- 7. Hybrid method for representing ions in implicit solvation ... [https://www.sciencedirect.com/science/article/pii/S2001037021000246]
- 8. comparing explicit and implicit solvent molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/25762327/]
- 9. Explicit Solvation Models in Q-Chem - How To [https://talk.q-chem.com/t/explicit-solvation-models-in-q-chem/1689]
- 10. A protocol for preparing explicitly solvated systems for stable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7413747/]
- 11. Implicit Solvent Models and Their Applications in Biophysics [https://www.mdpi.com/2218-273X/15/9/1218]
- 12. Extending machine learning model for implicit solvation to ... [https://arxiv.org/html/2510.20103v1]
- 13. Differentiable simulation to develop molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10966991/]
- 14. Accuracy comparison of several common implicit solvent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5313374/]
- 15. Predicting solvation free energies with an implicit solvent ... [https://arxiv.org/html/2406.00183v1]
- 16. Identifying Systematic Force Field Errors Using a 3D-RISM ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9921782/]
- 17. Comparison of the conformational dynamics of an N-glycan ... [https://www.sciencedirect.com/science/article/abs/pii/S0008621522002014]
- 18. Implicit Solvation - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/implicit-solvation]
- 19. Error assessment in molecular dynamics trajectories using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5669388/]
- 20. Towards optimal boundary integral formulations of the ... [https://pubmed.ncbi.nlm.nih.gov/35201634/]
- 21. Poisson-Boltzmann-based machine learning model for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11393697/]
- 22. Implicit Solvent Models and Their Applications in Biophysics [https://www.preprints.org/manuscript/202507.2627]
- 23. Machine-Learned Potentials for Solvation Modeling [https://arxiv.org/html/2505.22402v3]
- 24. Implicit solvent methods for free energy estimation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4310817/]
- 25. Ion specificity in α-helical folding kinetics [https://pubmed.ncbi.nlm.nih.gov/20942455/]
- 26. Article Common Structural Transitions in Explicit-Solvent ... [https://www.sciencedirect.com/science/article/pii/S0006349509013599]
- 27. Best Practices for Foundations in Molecular Simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6884151/]
- 28. Md simulation analysis error - GROMACS forums [https://gromacs.bioexcel.eu/t/md-simulation-analysis-error/3590]
- 29. Common errors when using GROMACS [https://manual.gromacs.org/2024.4/user-guide/run-time-errors.html]
- 30. From complex data to clear insights: visualizing molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11043564/]
- 31. Speed of Conformational Change: Comparing Explicit and ... [https://www.sciencedirect.com/science/article/pii/S000634951500003X]
- 32. Recent advances in implicit solvent based methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2386893/]
- 33. Connecting Free Energy Surfaces in Implicit and Explicit : an... Solvent [https://pmc.ncbi.nlm.nih.gov/articles/PMC4521639/]
- 34. Machine learning analysis of molecular dynamics ... [https://www.nature.com/articles/s41598-025-11392-1]
- 35. Machine learning analysis of molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/40707560/]
- 36. Machine Learning Analysis of Molecular Dynamics ... [https://chemrxiv.org/engage/chemrxiv/article-details/686e69f7728bf9025e8b2fa1]
- 37. Design and application of implicit solvent models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4045398/]
- 38. Problems with explicit solvent MD and CUDA platform #926 [https://github.com/openmm/openmm/issues/926]
- 39. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 40. Benchmark of Available Explicit Solvent Models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12498386/]
- 41. Machine Learning for Explicit Solvent Molecular Dynamics [https://cwagen.substack.com/p/machine-learning-for-explicit-solvent]
- 42. Assessment of reactivities with explicit and implicit solvent ... [https://pubs.rsc.org/en/content/articlehtml/2019/nj/c9nj04003j]
- 43. A Robust and Versatile QM/MM Interface for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11804165/]
- 44. Hybrid Models in Computational Chemistry [https://www.kamithinktank.com/computational-chemist/hybrid-models-in-computational-chemistry]
- 45. FAQ [https://charmm-gui.org/faq]
- 46. CHARMM-GUI Implicit Solvent Modeler for Various ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9551160/]
- 47. Conformational Sampling with Implicit Solvent Models [https://pmc.ncbi.nlm.nih.gov/articles/PMC1697846/]
- 48. A Novel Implicit Solvent Model for Simulating the Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3762369/]
- 49. Bridging Implicit and Explicit Solvent Approaches for ... [https://www.sciencedirect.com/science/article/pii/S0006349502739088]
- 50. Advanced Solvent in Computational Chemistry Effects [https://www.numberanalytics.com/blog/advanced-solvent-effects-computational-chemistry]
- 51. Machine learning based implicit for aqueous-solution... solvent model [https://pmc.ncbi.nlm.nih.gov/articles/PMC9900604/]
- 52. Is there any definitive evidence that implicit solvation ... [https://mattermodeling.stackexchange.com/questions/930/is-there-any-definitive-evidence-that-implicit-solvation-methods-are-more-accura]
- 53. 7.50. Implicit Solvation Models - ORCA 6.0 Manual [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/solvationmodels.html]
- 54. 2.13. Implicit Solvation [https://orca-manual.mpi-muelheim.mpg.de/contents/essentialelements/solvationmodels.html]
- 55. Implicit Solvent Models in Molecular Dynamics Simulations: A Brief Overview - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1574140008000078]
- 56. Speed of Conformational Change: Comparing Explicit and Implicit Solvent Molecular Dynamics Simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375717/]
- 57. A new set of atomic radii for accurate estimation ... [https://pubmed.ncbi.nlm.nih.gov/25220475/]
- 58. On the Dielectric “ Constant ” of Proteins: Smooth Dielectric Function... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3622359/]
- 59. Protocol for MM/PBSA Molecular Dynamics Simulations of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1303073/]
- 60. Implicit solvation - Wikipedia [https://en.wikipedia.org/wiki/Implicit_solvation]
- 61. Accelerating the Generalized Born with Molecular Volume ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7076882/]
- 62. Answers to frequently asked questions (FAQs) [https://manual.gromacs.org/2025.0/user-guide/faq.html]
- 63. When physics meets machine learning: a survey of ... [https://link.springer.com/article/10.1007/s44379-025-00016-0]
- 64. Frequently Asked Questions — Workflows 2025.1 ... - SCM [http://www.scm.com/doc/Workflows/SimpleActiveLearning/FAQ.html]
- 65. Physics-informed machine learning meets renewable ... [https://www.sciencedirect.com/science/article/pii/S0306261925016551]
- 66. Physics-informed machine learning-based real-time long ... [https://www.nature.com/articles/s44172-025-00501-7]
- 67. | Stony-Brook - iGEM 2025 [https://2025.igem.wiki/stony-brook/molec-dynam]
- 68. Leveraging high-throughput molecular simulations and ... [https://www.nature.com/articles/s41524-025-01552-2]
- 69. Answers to frequently asked questions (FAQs) [https://manual.gromacs.org/documentation/2025.4/user-guide/faq.html]
- 70. General — Workflows 2025.1 documentation - SCM [https://www.scm.com/doc/Workflows/SimpleActiveLearning/General.html]
- 71. Solvent Model Benchmark for Molecular Dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10091405/]
- 72. Direct comparison of amino acid and salt interactions with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5543776/]
- 73. Continuous Constant pH Molecular Dynamics in Explicit ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6425487/]
- 74. Computations of Standard Binding Free Energies with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3837708/]
- 75. Automated training of neural-network interatomic potentials [https://arxiv.org/html/2509.11703v1]
- 76. A diverse and chemically relevant solvation model benchmark ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc06406f]
- 77. A Standardized Benchmark for Machine-Learned ... [https://arxiv.org/html/2510.17187v1]
- 78. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 79. Benchmarking Implicit Solvent Folding Simulations of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2719849/]
- 80. Rowan | ML-Powered Molecular Design and Simulation [https://www.rowansci.com/]
- 81. Long time dynamics of Met-enkephalin: comparison of explicit and... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1301977/]
- 82. Molecular dynamics parameters (.mdp options) [https://manual.gromacs.org/2025.0-beta/user-guide/mdp-options.html]
- 83. Exploring Meta's Open Molecules 2025 (OMol25) & Universal Models ... [https://rowansci.com/blog/exploring-open-molecules-2025]
- 84. Practical considerations for Molecular Dynamics - GitHub Pages [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/aio/index.html]