Correcting Bonded Parameter Imbalance in Biomolecular Force Fields: From Foundations to Next-Generation Solutions
Accurate biomolecular force fields are fundamental to reliable molecular dynamics simulations in drug discovery and structural biology.
Correcting Bonded Parameter Imbalance in Biomolecular Force Fields: From Foundations to Next-Generation Solutions
Abstract
Accurate biomolecular force fields are fundamental to reliable molecular dynamics simulations in drug discovery and structural biology. However, traditional force fields often suffer from imbalances between bonded parameters (bonds, angles, dihedrals) and non-bonded interactions, leading to artifacts like artificial aggregation and incorrect protein dynamics. This article explores the foundational principles of bonded parameter imbalance, reviews methodological advances for its correction—including automated parameterization, NBFIX, and Bayesian learning—and provides a troubleshooting guide for common pitfalls. Finally, it establishes a robust framework for force field validation, comparing the performance of contemporary solutions and their implications for simulating complex biological processes.
The Root of the Problem: Understanding Bonded Parameter Imbalance in Molecular Mechanics
FAQs and Troubleshooting Guide
This guide addresses common challenges researchers face when working with the bonded terms of Class I potential energy functions in biomolecular force fields, providing targeted solutions based on established methodologies.
FAQ 1: My molecular dynamics simulations are producing unrealistic conformational distributions in drug-like molecules. How can I determine if the issue is with the dihedral parameters?
- Problem Identification: Unrealistic conformational distributions often stem from inaccurate torsional energy profiles. This is a known challenge, as the ability to correctly reproduce conformational energetics is an essential criterion for a force field's usefulness [1].
- Diagnostic Protocol:
- Isolate the Dihedral: Identify the specific rotatable bond(s) associated with the unrealistic conformation.
- Perform a Dihedral Scan: Conduct a series of single-point energy calculations, rotating the dihedral angle in small increments (e.g., 10-15°) while keeping the rest of the molecule fixed, using both the force field and a higher-level quantum mechanical (QM) method like MP2 [2] [3].
- Compare Profiles: Plot the energy profiles from the force field and QM calculations against the dihedral angle. Significant deviations indicate a potential problem with the dihedral parameters (
Kϕ,n,n,δnin the potential function) [2].
- Solution: Reparameterize the problematic dihedral term. This involves fitting new parameters for the torsional energy term (
Kϕ,n,n,δn) to more closely match the QM-derived potential energy surface (PES) [2]. The table below summarizes the core components of the Class I potential energy function that require calibration.
Table 1: Key Bonded Interaction Terms in Class I Potential Energy Functions [1]
| Interaction Type | Functional Form | Key Parameters | Physical Significance |
|---|---|---|---|
| Bond Stretching | $E = \sum Kb(b - b0)^2$ | $Kb$ (force constant), $b0$ (eq. length) | Vibrational energy of covalent bonds. |
| Angle Bending | $E = \sum K\theta(\theta - \theta0)^2$ | $K\theta$ (force constant), $\theta0$ (eq. angle) | Energy of valence angle deformation. |
| Dihedral Torsion | $E = \sum \sum{n} K{\phi,n}(1 + \cos(n\phi - \delta_n))$ | $K{\phi,n}$ (amplitude), $n$ (multiplicity), $\deltan$ (phase) | Energy barrier for rotation around bonds. |
| Improper Dihedral | $E = \sum K\varphi(\varphi - \varphi0)^2$ | $K\varphi$ (force constant), $\varphi0$ (eq. angle) | Maintains planarity (e.g., in aromatic rings). |
FAQ 2: During force field parametrization for a novel platinum-based anticancer drug, how do I handle the lack of pre-existing bonded parameters for the platinum atom and its ligands?
- Problem Identification: Transition metals like platinum are often poorly covered in standard force fields, requiring novel parametrization [3]. The challenge is to derive accurate bond, angle, and dihedral parameters that reflect the metal's unique coordination chemistry.
- Methodology Based on Recent Research: A 2025 study on Pt derivatives outlines a robust protocol [3]:
- Quantum Mechanical Benchmarking: Perform a comprehensive benchmark using density functional theory (DFT) methods on small model systems (e.g.,
PtH,PtCl,[PtCl4]2-) to identify the best level of theory for predicting structural parameters and vibrational frequencies [3]. - Parameter Derivation:
- Bond & Angles: The equilibrium bond lengths (
b0) and angles (θ0) can be taken directly from the QM-optimized geometry. Force constants (Kb,Kθ) can be fitted to reproduce QM-calculated vibrational frequencies [3]. - Charges: Calculate partial atomic charges using a method like CHELPG (Charges from Electrostatic Potentials using a Grid-based method), which was shown to effectively describe the chemical environment of platinum [3].
- Bond & Angles: The equilibrium bond lengths (
- Validation in Solid State: Validate the derived parameters by simulating known crystal structures (e.g., cisplatin polymorphs) and comparing the results against experimental structural data [3].
- Quantum Mechanical Benchmarking: Perform a comprehensive benchmark using density functional theory (DFT) methods on small model systems (e.g.,
FAQ 3: When using a Class I force field, my simulations fail to maintain planarity in aromatic rings and amide groups. What is the most common cause and how can it be fixed?
- Problem Identification: The failure to maintain molecular planarity typically points to an issue with the out-of-plane bending terms. In Class I force fields, this is often handled by an improper dihedral term [1].
- Diagnosis and Solution:
- Verify Parameter Assignment: First, confirm that improper dihedral parameters are correctly assigned to all atoms requiring planarity enforcement (e.g., all atoms in an aromatic ring, or the C, O, N, and H atoms in an amide group) [4].
- Check Force Constants: The primary culprit is often an insufficient force constant (
Kφ). If the energy penalty for moving out of plane is too low, the structure will distort easily during dynamics. Consult the documentation for your specific force field (e.g., CHARMM, AMBER) for the recommended improper dihedral parameters for these chemical groups [1] [4]. - Alternative Approach: Some force fields or molecular builders may use alternative methods, such as defining an "out-of-plane" angle bending term, but the improper dihedral is the most common implementation in Class I functions [1].
Featured Experimental Protocol: Calibrating Bonded Parameters via Splitting-Tensile Strength
The following protocol, adapted from a 2024 study on asphalt mixtures, exemplifies a rigorous empirical approach to calibrating the parameters of a bonded contact model (a conceptual analog to molecular "bonds") in a discrete element simulation, ensuring macroscopic experimental data matches simulation outcomes [5].
Objective: To calibrate the bonding parameters (normal stiffness, shear stiffness, critical normal stress, critical shear stress) of an EDEM simulation model by using the splitting tensile strength of laboratory specimens as a benchmark [5].
Workflow Overview:
Key Materials and Reagents:
Table 2: Research Reagent Solutions for Parameter Calibration [5]
| Material / Tool | Function / Specification | Role in the Protocol |
|---|---|---|
| SBS-Modified Asphalt | Styrene-butadiene-styrene polymer modified binder. | Provides the cohesive bonding material in the mixture. Its properties are represented by the bonding parameters in the simulation. |
| Coarse & Fine Aggregate | Technical indicators conforming to JTG 3432-2024 standards. | Forms the granular skeleton of the mixture. Modeled as discrete particles in the simulation. |
| SYD-0730A Automatic Tester | Multi-functional asphalt pressure tester. | Used to conduct the laboratory splitting tensile strength test to obtain the benchmark macroscopic property (R_T). |
| EDEM 2023 Software | Discrete Element Method (DEM) simulation platform. | Environment for building the virtual specimen and conducting the virtual splitting tests. |
| Hertz-Mindlin with Bonding | A specific contact model in EDEM. | The mathematical model that defines how particles interact and "bond," containing the parameters to be calibrated. |
| SolidWorks 2018 | 3D CAD modeling software. | Used to create realistic 3D shapes of the coarse aggregate particles for import into EDEM. |
| Response Surface Methodology (RSM) | A statistical and mathematical technique. | Designs an efficient set of simulation runs to understand the relationship between multiple input parameters and the output (tensile strength). |
Detailed Methodology:
Laboratory Testing:
- Prepare Marshall specimens (e.g., φ101.6 mm × 63.5 mm) with the specified aggregate gradation (e.g., AC-13) and binder content (e.g., 5% oil-stone ratio) [5].
- Conduct splitting tests at a controlled temperature (e.g., 15°C) and loading rate (e.g., 50 mm/min). Record the maximum load (
PT) at failure for each specimen [5]. - Calculate the actual splitting tensile strength (
RT) in MPa using the standard formula:RT = 0.006287 * PT / h, wherehis the specimen height in mm. This provides the target value for calibration [5].
Simulation Model Construction:
- Model three representative coarse aggregate particles in 3D using CAD software and import them into the DEM environment [5].
- Select the "Hertz-Mindlin with Bonding" contact model. This model introduces "bonds" between particles that can break when critical stress thresholds are exceeded, simulating the failure of the asphalt binder [5].
- Define plausible value ranges for the four key bonding parameters to be calibrated: Normal Stiffness Per Unit Area, Shear Stiffness Per Unit Area, Critical Normal Stress, and Critical Shear Stress [5].
Parameter Calibration via RSM:
- Design a Response Surface Methodology experiment (e.g., a Central Composite Design) to structure a limited number of simulation runs that efficiently explore the defined parameter space [5].
- For each combination of parameters in the experimental design, run a virtual splitting test in the DEM software and record the simulated tensile strength.
- Fit a quadratic regression model that describes the splitting tensile strength as a function of the four bonding parameters. Analyze the model to determine the significance of each parameter and identify their optimal values that yield a simulated tensile strength matching the laboratory-measured
RT[5].
Validation:
- Use the final calibrated parameter set in a new, independent simulation of the splitting test.
- Calculate the relative error between the simulation and actual laboratory results. The feasibility of the method is demonstrated by a low relative error (e.g., -2.48% as reported in the source study) [5].
Table 3: Key Software and Data Resources for Force Field Parameterization
| Tool / Resource | Type | Primary Function in Parameter Workflow |
|---|---|---|
| QM Software (e.g., ORCA, Gaussian) | Computational Chemistry | Provides target data (geometries, vibrational frequencies, conformational energies) for parameter derivation and validation at the electronic structure level [2] [3]. |
| Force Field Parametrization Tools (e.g., CHARMMgui, AmberTools, Moltemplate) | Utility Software | Assist in the assignment of atom types, bonded parameters, and partial atomic charges for molecules within specific force fields like CHARMM, AMBER, or OPLSAA [6] [4]. |
| Platinum Diverse Dataset | Benchmarking Dataset | A high-quality set of protein-bound ligand conformations used to benchmark a force field's ability to reproduce bioactive conformations [2]. |
| MMFF94 Bond-Charge-Increment (BCI) Solver | Specialized Algorithm | A tool for deriving partial atomic charges based on the MMFF94 force field's methodology, which considers chemical polarization through bond-charge increments [3]. |
Advanced Visualization: The Parameter Interrelationship
The accuracy of a Class I force field depends on the careful balance of its bonded terms. The following diagram conceptualizes how these terms work together to define a molecule's energy landscape, and where imbalances often occur, leading to the need for troubleshooting.
In biomolecular molecular dynamics (MD) simulations, the force field is the underlying model that determines the accuracy and reliability of the results. A fundamental challenge in force field development is the critical, and often problematic, interdependence between different parameter types. Specifically, the assignment of atomic partial charges is inextricably linked to the parameterization of bonded terms, such as bond stretching, angle bending, and dihedral torsions. When this link is not carefully managed, it creates a parameter imbalance that can lead to systematic errors in simulations, including unrealistic protein aggregation, collapsed denatured states, and distorted nucleic acid structures [7]. This technical support guide addresses the specific issues arising from this interdependence and provides methodologies for their identification and correction, forming a crucial component for researchers aiming to correct bonded parameter imbalances.
Troubleshooting Guides
Problem 1: Artificial Aggregation in Multi-Component Systems
Observed Issue: During simulations of proteins, nucleic acids, or lipid assemblies, biomolecules exhibit unrealistic clumping or aggregation that is not experimentally observed.
Underlying Cause: This is a classic symptom of an imbalance between non-bonded and bonded parameters. Overly attractive interactions between charged or hydrophobic groups, often stemming from inaccurately high partial charge magnitudes, are not sufficiently counterbalanced by the repulsive forces or torsional barriers defined in the bonded parameters [7]. The force field is effectively "out of balance."
Diagnosis and Solution:
- Diagnostic Check: Calculate the radial distribution function (RDF) between charged groups (e.g., amine-phosphate) or between hydrophobic residues. Compare against known experimental data or well-behaved reference systems.
- Recommended Solution: Implement pair-specific corrections to the Lennard-Jones parameters using the NBFIX method [7]. This approach surgically corrects the overestimated attractive interactions between specific atom pairs (e.g., ions-carboxylates) without perturbing the carefully parameterized bonded terms or hydration free energies.
- Methodology:
- Obtain Reference Data: Use experimental osmotic pressure data for binary solutions (e.g., electrolyte or amino acid solutions) as a calibration target [7].
- Setup Simulation: Employ a two-compartment MD setup to compute osmotic pressure computationally [7].
- Calibrate: Systematically adjust the LJ parameters for the specific atom pair in question (e.g., Na+-Cl-) until the simulated osmotic pressure matches the experimental data.
Problem 2: Distorted Crystal Lattice Parameters
Observed Issue: When simulating a carbohydrate or small molecule crystal, the unit cell dimensions deviate significantly from the experimental crystallographic data during an MD run.
Underlying Cause: The set of partial atomic charges is unsuitable for the condensed phase. Crystal geometries are highly sensitive to intermolecular non-bonded forces. If the partial charges overestimate polarity, they can distort the delicate balance of intermolecular hydrogen bonds and van der Waals contacts that maintain the crystal lattice [8].
Diagnosis and Solution:
- Diagnostic Check: Perform an MD simulation of the crystal structure under periodic boundary conditions and monitor the evolution of the unit cell vectors (a, b, c) over time.
- Recommended Solution: Re-derive the partial charges using a Restrained Electrostatic Potential (RESP) fitting procedure with an appropriate restraint weight [8].
- Methodology:
- Quantum Calculation: Compute the molecular electrostatic potential at the HF/6-31G* level for a representative conformation [8].
- Charge Fitting: Fit the atomic partial charges to the electrostatic potential using a restraint function (Eq. 4) [8]:
χ_resp² = χ_esp² + k_rstr * Σ [ √(q_j² + b²) - b ]. - Validation: Test the new charge set in a crystal simulation. A restraint weight (
k_rstr) of 0.01 was found to yield the best agreement with the neutron diffraction structure of α-d-glucopyranose [8].
Problem 3: Inaccurate Torsional Energy Barriers and Conformational Sampling
Observed Issue: The simulated molecule does not sample its conformational landscape correctly, getting stuck in incorrect torsional minima or exhibiting unrealistic torsional energy barriers compared to quantum mechanical (QM) data.
Underlying Cause: The traditional treatment of 1-4 interactions—atoms separated by three bonds—creates a problematic interdependence. These interactions are modeled using a hybrid of bonded dihedral terms and scaled non-bonded (electrostatic and Lennard-Jones) interactions [9]. Standard non-bonded functions do not account for charge penetration effects at short distances, forcing an arbitrary scaling of 1-4 interactions. This inaccurate physics then requires the dihedral terms to be over-fitted to compensate, reducing transferability and potentially yielding inaccurate forces [9].
Diagnosis and Solution:
- Diagnostic Check: Compare the torsional energy profile of a model compound (e.g., alanine dipeptide) from your MD simulation against a high-level QM calculation.
- Recommended Solution: Adopt a bonded-only treatment for 1-4 interactions [9]. This approach uses bonded coupling terms (torsion-bond, torsion-angle) to accurately capture the energetics, thereby decoupling the parameterization of torsions from non-bonded interactions.
- Methodology:
- Automated Parameterization: Utilize an automated parameterization toolkit like Q-Force to derive the necessary bonded coupling terms [9].
- QM Reference Data: Generate a high-quality QM dataset for the target molecule, including energy and force information.
- Fitting: The toolkit fits the bonded coupling terms (e.g., Morse potentials for bonds, etc.) to the QM reference data, eliminating the need for any 1-4 non-bonded interactions [9].
Problem 4: Transferability Failure in Chemically Modified Residues
Observed Issue: Simulations of post-translationally modified proteins or non-standard amino acids produce unrealistic geometries or dynamics.
Underlying Cause: The partial charges and bonded parameters for the modified residue were likely derived in isolation without considering the consistency with the existing force field for standard residues. The charges may overpolarize the new functional group, disrupting the existing balance of bonded and non-bonded terms.
Diagnosis and Solution:
- Diagnostic Check: Compare the hydration free energy and conformational preferences of the modified residue model compound against QM calculations or available experimental data.
- Recommended Solution: Ensure charge derivation for new residues uses the exact same protocol (e.g., RESP with HF/6-31G* and a consistent restraint weight) as the parent force field. Avoid mixing charge sets from different derivation methods.
- Methodology:
- Consistent Protocol: Derive charges for the new molecule using the standard RESP procedure and restraint weight established for the force field (e.g.,
k_rstr = 0.01for carbohydrates in GLYCAM) [8]. - Experimental Validation: Where possible, leverage new experimental methods like ionic Scattering Factors (iSFAC) modelling with electron diffraction to obtain experimental partial charges for validation [10]. This technique can refine a charge parameter for each atom against diffraction data.
- Consistent Protocol: Derive charges for the new molecule using the standard RESP procedure and restraint weight established for the force field (e.g.,
Frequently Asked Questions
How can I experimentally validate my partial charge set?
Traditionally, validating partial charges has been challenging. However, a recent breakthrough method called ionic Scattering Factors (iSFAC) modelling now allows for the experimental determination of partial charges for any crystalline compound using electron diffraction [10]. This method refines a charge parameter for each atom against diffraction data, providing an absolute scale for partial charges. It has been successfully demonstrated on amino acids like histidine and tyrosine, showing a strong correlation with quantum chemical computations [10].
My water model is known to be problematic. Should I change it or use NBFIX?
Modifying the water model is a global change that affects all solute-solute and solute-solvent interactions. While new water models can improve the behavior of denatured proteins, they may also destabilize folded conformations or disrupt other fine-tuned interactions [7]. The NBFIX approach is a surgical correction that targets only specific, problematic atom pairs. It is often preferable when the core issue is traceable to a few over-strong interactions, as it minimizes unintended side-effects on the rest of the system [7].
What is the most common mistake in deriving partial charges for bonded parameter fitting?
The most common mistake is the over-polarization of bonds when using unrestrained electrostatic potential (ESP) charges derived from quantum mechanical calculations. ESP charges computed with a 6-31G* basis set are known to overestimate bond polarities, which can artificially strengthen hydrogen bonds and disrupt the conformational balance [8]. Always using a restrained ESP (RESP) fit with an appropriate weight (e.g., 0.001-0.01) to attenuate charge magnitudes is critical for condensed-phase simulations [8].
The table below summarizes key experimental and computational methods for diagnosing and correcting parameter imbalance.
Table 1: Protocols for Parameter Troubleshooting
| Method Name | Primary Application | Key Measurable | Technical Summary |
|---|---|---|---|
| Osmotic Pressure Calibration (NBFIX) [7] | Correcting artificial ion & solute aggregation. | Osmotic pressure of binary solutions. | MD simulation in a two-compartment setup; adjust LJ parameters for specific atom pairs to match experimental pressure. |
| Crystal Lattice Validation [8] | Validating partial charges for condensed phase. | Unit cell parameters (a, b, c). | MD simulation of a crystal structure; compare simulated unit cell dimensions to experimental X-ray/neutron diffraction data. |
| Bonded-Only 1-4 Treatment [9] | Fixing inaccurate torsional barriers & forces. | Torsional energy profile (QM vs. MM). | Use automated parameterization (Q-Force) to replace scaled 1-4 non-bonded interactions with bonded coupling terms. |
| iSFAC Modelling [10] | Experimental charge determination. | Partial charge per atom (in e). | Refine atomic scattering factors against 3D electron diffraction data, introducing a charge parameter for each atom. |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Data Resources for Force Field Correction
| Item Name | Function / Purpose | Relevance to Parameter Balance |
|---|---|---|
| CHARMM/AMBER | Molecular dynamics simulation software. | Platforms for implementing NBFIX corrections and performing validation simulations (e.g., crystal, osmotic pressure). |
| Q-Force Toolkit [9] | Automated force field parameterization. | Derives bonded coupling terms for a bonded-only treatment of 1-4 interactions, decoupling torsions from non-bonded terms. |
| RESP Fit Restraint Weight [8] | A hyperparameter in charge fitting. | Critical for attenuating bond polarity; a value of ~0.01 is often optimal for carbohydrates and other biomolecules. |
| HF/6-31G* ESP | Quantum mechanical reference data. | The standard level of theory for generating electrostatic potentials for RESP charge fitting in AMBER/CHARMM. |
| Experimental Osmotic Pressure Data [7] | Reference data for calibration. | Used as a benchmark to calibrate and correct over-strength attractive interactions via the NBFIX method. |
Workflow Visualization
The following diagram illustrates the logical relationship between partial charge models and the subsequent parameterization of bonded terms, highlighting key troubleshooting points.
Troubleshooting Guides
Guide 1: Diagnosing and Correcting Artifacts from Bonded Parameter Imbalance
Q1: My simulation of an intrinsically disordered protein (IDP) shows an unnatural structural collapse into a globular state, unlike experimental data. What is the cause and how can I fix it?
A: This artifact is a classic symptom of bonded parameter imbalance, specifically an overestimation of protein backbone compactness or inaccurate interactions with the solvent model [11].
- Primary Cause: The imbalance often stems from force field parameters that were primarily optimized for stable, folded proteins, and certain water models (like TIP3P) that can promote this artificial collapse [11].
- Diagnosis Steps:
- Compare the radius of gyration (Rg) from your simulation trajectory against experimental Small-Angle X-Ray Scattering (SAXS) data.
- Check if backbone chemical shifts predicted from your trajectory deviate from experimental NMR chemical shifts.
- Analyze if transient secondary structural elements, observed in NMR, are retained or lost in your simulation [11].
- Solution:
- Switch Force Fields and Water Models: Re-run your simulation using a modern force field and water model combination designed for disordered proteins. Benchmarks have shown that combinations like CHARMM36m with the TIP4P-D water model significantly improve performance for IDPs and hybrid proteins [11].
- Validate with NMR Relaxation: Use NMR relaxation parameters (R1, R2, hetNOE) as a sensitive benchmark, as they are highly sensitive to force-field imperfections and the artificial collapse [11].
Q2: How can I identify if a bonded parameter imbalance is affecting the conformational landscape of a drug-like small molecule in my protein-ligand simulation?
A: Inaccurate torsion parameters for the ligand can lead to incorrect populations of conformational states, which directly impacts the calculation of binding affinities [12].
- Primary Cause: Traditional look-up table-based force fields may have poor coverage or inaccurate parameters for specific chemical moieties present in your ligand [12].
- Diagnosis Steps:
- Perform a conformational search for the isolated ligand using quantum mechanics (QM) methods (e.g., at the B3LYP-D3(BJ)/DZVP level).
- Compare the QM-derived torsional energy profiles and low-energy conformers against those generated by your molecular mechanics (MM) force field. Significant deviations indicate poor parameterization [12].
- Solution:
- Adopt a Data-Driven Force Field: Use a modern, data-driven force field like ByteFF or Espaloma that employs graph neural networks (GNNs) to predict parameters across a broad chemical space, ensuring more accurate torsion profiles [12].
- Parameterize Specifically: If a widespread force field is insufficient, consider generating specific torsion parameters for the problematic dihedral in your ligand using QM calculations.
Q3: I am setting up a simulation with a novel small molecule. What is the most reliable way to assign force field parameters to avoid inherent imbalances from the start?
A: The key is to start with a complete and correct chemical representation of your molecule [13].
- Primary Cause: Starting from file formats that lack explicit bond order and formal charge information (like many PDB or legacy topology files) forces parameter assignment tools to guess the chemical identity, which is a primary source of initial error and parameter imbalance [13].
- Diagnosis: Check the source of your initial molecular structure. If it was generated by a simulation package without access to bond order information, the parameters assigned are likely based on inference and may be incorrect.
- Solution:
- Use Chemoinformatic Inputs: Always begin with file formats that explicitly define chemical identity. The most reliable sources are [13]:
- Isomeric SMILES strings
.mol2files from a chemoinformatics toolkit (e.g., RDKit, OpenEye)- InChI strings or IUPAC names
- Apply a Direct Chemical Perception Force Field: Use a force field with direct chemical perception like the SMIRNOFF format, which assigns parameters based on SMIRKS patterns applied to the molecule's full chemical identity, avoiding the inaccuracies of indirect atom-typing [13].
- Use Chemoinformatic Inputs: Always begin with file formats that explicitly define chemical identity. The most reliable sources are [13]:
Experimental Validation Protocols
Protocol 1: Validating Simulated Conformational Ensembles Against Experimental NMR Data
This protocol is essential for diagnosing artifacts in protein or peptide dynamics [11].
- Sample Preparation: Prepare a ~0.3-1.0 mM sample of the protein/peptide in a suitable aqueous buffer. For proteins with IDRs, ensure the buffer conditions (pH, salt) match the physiological state of interest.
- NMR Data Collection:
- Backbone Assignments: Acquire standard triple-resonance experiments (HNCA, HNCOCA, etc.) for backbone chemical shift assignment.
- Relaxation Measurements: Acquire ¹⁵N R1 (longitudinal) and R2 (transverse) relaxation rates and ¹⁵N-{¹H} steady-state heteronuclear Overhauser effect (hetNOE) data at least one magnetic field strength.
- Residual Dipolar Couplings (RDCs): Acquire in-phase/anti-phase (IPAP) spectra in isotropic and aligned media (e.g., stretched polyacrylamide gel) to measure RDCs [11].
- Data Prediction from Simulation:
- From your MD trajectory, calculate the following for direct comparison:
- Backbone Chemical Shifts: Use predictors like SPARTA+ or SHIFTX2.
- R1, R2, and hetNOE: Compute from the simulation trajectory using the relaxation theory formalism.
- RDCs: Calculate from the average molecular alignment tensor and the trajectory [11].
- From your MD trajectory, calculate the following for direct comparison:
- Analysis: Quantitatively compare the predicted parameters from your simulation with the experimental data. Poor agreement, especially in Rg and relaxation parameters, strongly indicates force field artifacts [11].
Protocol 2: Benchmarking Force Field Performance Using SAXS and Multi-Parametric NMR
A robust benchmarking protocol to select the optimal force field for your system, particularly for proteins with both structured and disordered regions [11].
- Experimental Data Collection:
- SAXS: Collect SAXS data to determine the experimental radius of gyration (Rg) and molecular form factor.
- NMR: Collect chemical shifts, RDCs, and paramagnetic relaxation enhancement (PRE) data if possible. PREs are especially sensitive to transient long-range contacts.
- Molecular Dynamics Simulations:
- Set up multiple simulation systems of the same protein using different force field and water model combinations (e.g., Amber99SB-ILDN/TIP3P, CHARMM36m/TIP4P-D).
- Run multiple replicas of microsecond-length simulations to ensure adequate sampling.
- Prediction and Comparison:
- From each trajectory, predict the Rg (for SAXS comparison), chemical shifts, RDCs, and PREs.
- Systematically compare the accuracy of each force field combination against the full suite of experimental data.
- Selection Criterion: The force field that most accurately reproduces the entire set of experimental observables, particularly the NMR relaxation parameters and Rg, should be selected for production runs [11].
Frequently Asked Questions (FAQs)
Q: What are the most common artifacts caused by bonded parameter imbalance, and what are their experimental signatures?
A: The table below summarizes key artifacts and how to spot them.
| Artifact | Description | Experimental Signature |
|---|---|---|
| Artificial Collapse | IDPs or disordered regions unnaturally collapse into overly compact states [11]. | - Rg (from SAXS) is significantly lower than experiment.- NMR-derived R2 rates are too high, indicating restricted motion. |
| Loss of Transient Helicity | Disordered regions with a propensity for helical structure lose these transient elements [11]. | - Deviations in backbone chemical shifts (Cα, Cβ) from random coil values.- Disagreement between simulated and experimental RDCs. |
| Inaccurate Torsional Landscapes | Small molecules or side chains populate incorrect rotameric states [12]. | - Torsional energy profiles from simulation deviate from QM calculations.- Predicted conformational populations do not match NMR-derived populations. |
Q: Which modern force fields and water models are recommended to mitigate these imbalance issues?
A: Benchmarks suggest the following combinations are effective, especially for systems containing disorder:
| Force Field | Recommended Water Model | Best For / Key Feature |
|---|---|---|
| CHARMM36m [11] | TIP4P-D [11] | Hybrid proteins (with both structured and disordered regions); prevents artificial collapse. |
| ByteFF [12] | TIP3P, SPCE, etc. | Drug-like small molecules; expansive chemical space coverage via machine learning. |
| SMIRNOFF (OpenFF) [13] | Compatible with multiple | Direct chemical perception for accurate parameter assignment based on chemical identity. |
Q: My research focuses on drug discovery. How can bonded parameter imbalance affect my results?
A: The consequences are direct and critical [12]:
- Inaccurate Binding Affinity Predictions: If the ligand's torsional energy landscape is wrong, the free energy of binding will be incorrect.
- Misidentification of Binding Poses: An imbalanced force field may stabilize non-native ligand conformations or protein-ligand interactions, leading to false positives in virtual screening.
- Poor Reliability in Lead Optimization: Predictions of how chemical modifications affect potency (SAR) will be unreliable if the underlying conformational energetics are flawed.
Diagnostic Workflow and Signaling Pathways
The Scientist's Toolkit: Research Reagent Solutions
| Research Reagent | Function / Application |
|---|---|
| CHARMM36m Force Field [11] | A widely benchmarked force field for simulating proteins, particularly effective for systems with intrinsically disordered regions when combined with TIP4P-D water. |
| TIP4P-D Water Model [11] | A modified water model designed to correct for the over-stabilization of protein-protein interactions, crucial for preventing artificial collapse in IDP simulations. |
| ByteFF [12] | A data-driven, Amber-compatible force field for drug-like molecules. It uses a graph neural network trained on a massive QM dataset to accurately predict parameters across a broad chemical space. |
| SMIRNOFF Force Field Format [13] | A force field specification that uses direct chemical perception (SMIRKS patterns) for parameter assignment, avoiding the ambiguities of atom-typing and improving transferability. |
| B3LYP-D3(BJ)/DZVP QM Method [12] | A quantum mechanics method that provides a good balance of accuracy and computational cost for generating reference data (geometries, Hessians, torsion profiles) for force field training and validation. |
| geomeTRIC Optimizer [12] | An optimizer used for geometry optimization at the QM level during the generation of training data for force field development. |
Frequently Asked Questions (FAQs)
Q1: What are anharmonicity and cross-terms in the context of biomolecular force fields, and why are they important? Anharmonicity refers to deviations from a simple harmonic (quadratic) potential energy surface. In force fields, this means that the energy of bond stretching or angle bending is not perfectly described by a single parabolic curve, especially for larger displacements [1]. Cross-terms are potential energy components that describe the coupling between different internal coordinates, such as how the stretching of one bond affects the stretching of an adjacent bond or an angle [1] [14].
Their importance lies in achieving higher accuracy. Class I force fields (like AMBER, CHARMM) use only harmonic terms for bonds and angles, which is often sufficient for simulations at room temperature [1] [14]. However, for accurately reproducing quantum mechanical potential energy surfaces, vibrational spectra, and properties of systems under stress or at high temperatures, Class II and III force fields introduce anharmonic and cross-terms [1] [14]. This is particularly critical for modeling functional materials and systems with light nuclei, like hydrogen-bonded networks, where quantum fluctuations and large-amplatomic motions are significant [15].
Q2: My simulations of a protein backbone are showing unrealistic flexibility in certain motifs. Could this be related to a lack of cross-terms? Yes, this is a plausible explanation. The local conformational energetics of proteins are dominated by torsional potentials. While Class I force fields rely primarily on proper dihedral parametrization, they can sometimes fail to capture coupled motions. A specific and advanced solution to this issue is the introduction of a torsion-torsion cross-term, such as the CMAP (Correction Map) term used in the CHARMM protein force field [1]. CMAP provides a grid-based energy correction as a function of two dihedral angles (e.g., Φ and Ψ in the protein backbone), which directly addresses correlated conformational changes and helps to better stabilize secondary structures like alpha-helices and beta-sheets [1].
Q3: I am parameterizing a novel small molecule drug candidate. When should I consider using a force field that includes anharmonicity? For most routine applications in computational structure-based drug discovery (CSBDD), such as estimating binding affinities or conducting molecular dynamics of a protein-ligand complex, Class I additive force fields are the standard and are sufficient [1] [16]. The computational cost of using more complex force fields is often not justified for these purposes.
You should consider anharmonic force fields (Class II or III) or machine learning potentials when your study specifically focuses on:
- Vibrational spectroscopy: If you need to directly compare computed vibrational frequencies to experimental IR or Raman spectra with high accuracy [1].
- Properties at extreme conditions: When simulating systems at very high temperatures or pressures where bonds and angles are driven far from their equilibrium values [15] [17].
- Systems with strong quantum nuclear effects: This is crucial for materials containing light elements like hydrogen, where quantum fluctuations are significant and can even determine dynamical stability [15].
Q4: What are the main practical challenges and trade-offs when implementing force fields with anharmonicity and cross-terms? The primary trade-off is between accuracy and complexity.
- Parametrization Complexity: Introducing anharmonic and cross-terms multiplies the number of parameters that need to be optimized [1]. This requires a much larger set of high-quality target data from quantum mechanics (QM) calculations and/or experiments for meaningful parameter optimization [1].
- Computational Cost: While the evaluation of these additional terms adds some computational overhead, the more significant cost often lies in the parameter optimization process itself [1].
- Transferability: Ensuring that the more complex parameters work well across a wide range of chemical environments and molecules is a major challenge [1] [18]. Highly specialized parameters might work perfectly for a small training set but fail to generalize.
Troubleshooting Guides
Issue: Inaccurate Vibrational Spectra from Simulation
Problem: The vibrational frequencies (e.g., from a Fourier transform of the velocity autocorrelation function) of my molecule do not match experimental infrared or Raman spectra.
Diagnosis: This is a classic symptom of the limitations of harmonic force fields. Harmonic oscillators cannot capture frequency shifts due to bond dissociation limits or the broadening of peaks at elevated temperatures.
Solution: Consider moving to a force field that includes anharmonic corrections.
- Identify Critical Motions: Determine which vibrational modes (e.g., O-H stretch, C=O stretch) are showing the largest discrepancies.
- Select a Force Field: Choose a Class II force field (e.g., MMFF94) or a machine learning potential (MLP) that has been trained on QM data including these anharmonic motions [14] [18].
- Protocol for MLP-assisted Anharmonicity: For the highest accuracy, particularly with quantum effects, a protocol pairing MLPs with the Stochastic Self-Consistent Harmonic Approximation (SSCHA) can be used [15]. The workflow for this advanced method is outlined below.
Diagram: Workflow for incorporating quantum anharmonicity using MLPs and SSCHA [15].
Issue: Parameter Transferability and Imbalance in Novel Molecules
Problem: After manually adding parameters for a novel residue or small molecule, the simulation becomes unstable, or the molecule adopts incorrect conformations, indicating a parameter imbalance.
Diagnosis: Manually derived parameters, especially for one part of a molecule, can lack consistency with the existing force field. This disrupts the careful balance of bonded and non-bonded terms, leading to instabilities.
Solution: Use a systematic, software-assisted parameterization workflow that fits against QM and experimental data.
- Initial Parameter Generation: Use a tool like CHARMM General Force Field (CGenFF) or the Open Force Field Toolkit to get initial parameters for your molecule [16].
- Target Data Calculation: Perform QM calculations on the molecule or its fragments to generate target data. This includes:
- conformational energies
- torsional energy profiles
- electrostatic potential (for deriving partial charges)
- vibrational frequencies
- Parameter Optimization: Use a framework like ForceBalance to automatically optimize the force field parameters against the QM and experimental target data [19]. This process minimizes an objective function that quantifies the difference between the force field's predictions and the target data.
Diagram: Automated parameter optimization workflow using ForceBalance [19].
The following table summarizes the key differences between force field classes relevant to this troubleshooting issue.
Table: Comparison of Force Field Classes Regarding Anharmonicity
| Feature | Class I (e.g., AMBER, CHARMM) | Class II (e.g., MMFF94) | Class III (Polarizable, e.g., AMOEBA) |
|---|---|---|---|
| Bond/Angle Potential | Harmonic only [14] | Adds cubic/quartic anharmonic terms [1] [14] | May include anharmonicity; key feature is polarization [14] [16] |
| Cross-Terms | Typically absent (exception: CMAP in CHARMM) [1] | Present (e.g., bond-bond, bond-angle) [1] [14] | Present and more complex due to polarization [16] |
| Primary Application | Routine biomolecular MD simulations [1] [16] | Higher accuracy for small molecules & vibrational spectra [14] | Systems where electronic polarization is critical [16] |
| Computational Cost | Low | Moderate | High |
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Tools for Advanced Force Field Development and Testing
| Tool / Reagent | Function | Relevant Context |
|---|---|---|
| ForceBalance | A software package for the systematic optimization of force field parameters against quantum mechanical (QM) and experimental target data [19]. | Essential for refining bonded parameters and correcting imbalances; can handle anharmonic terms [19]. |
| Open Force Field Toolkit | A tool for parameterizing small molecules using the SMIRNOFF format, enabling direct chemical perception and automation [19] [16]. | Used to generate initial parameters for novel drug-like molecules in a consistent manner [16]. |
| Machine Learning Interatomic Potentials (MLIPs) | Machine-learned potentials (e.g., MTPs, GAP) that approximate QM-level accuracy at near-MM cost [15] [18]. | Used to capture complex anharmonic potential energy surfaces and quantum effects, as in the SSCHA workflow [15]. |
| Stochastic Self-Consistent Harmonic Approximation (SSCHA) | An advanced computational method to treat quantum nuclear effects and anharmonicity beyond perturbation theory [15]. | Crucial for studying materials like metal hydrides where quantum fluctuations determine stability [15]. |
| CMAP (CHARMM) | An empirical correction map that acts as a dihedral cross-term to better model protein backbone conformational energetics [1]. | A specific solution to correct for correlated torsional motions in proteins, addressing backbone flexibility issues [1]. |
Experimental Protocols
Protocol 1: Optimizing Force Field Parameters with ForceBalance
This protocol outlines the steps to optimize force field parameters, such as those for anharmonic terms, using ForceBalance in conjunction with the OpenFF Evaluator [19].
- Define the Training Set: Curate a set of experimental physical property data (e.g., density, enthalpy of vaporization) for molecules relevant to your system. This data set is saved in a JSON file [19].
- Prepare the Initial Force Field: Load a starting force field (e.g., OpenFF Parsley) and add cosmetic "parameterize" tags to the specific parameters you wish to optimize (e.g., Van der Waals
epsilonandrmin_half) [19]. - Set Up the Directory Structure: Create directories named
forcefield/(for the tagged force field file) andtargets/pure_data/(for the training set and options) [19]. - Configure the Optimization: Create a ForceBalance input file (
optimize.in) and anoptions.jsonfile. In the JSON file, define:calculation_layers: Set to "SimulationLayer" to use MD for property evaluation.weightsanddenominators: Define the relative weight and scaling for each property type in the objective function [19].
- Launch the Optimization Server: Start an
EvaluatorServer(e.g., using aDaskLocalCluster) to handle the computational workload. Execute ForceBalance, which will iteratively adjust the tagged parameters to minimize the difference between simulated and experimental data [19].
Protocol 2: Modeling Anharmonicity and Quantum Effects with MLPs and SSCHA
This protocol describes the advanced workflow for efficiently modeling strong anharmonicity and quantum effects, as applied to materials like PdCuH2 [15].
- Initial Harmonic Calculation: Start with a small supercell of your material. Perform a DFT calculation to obtain the harmonic force constants as an initial guess for the dynamical matrices [15].
- Active Learning and MTP Training:
- The SSCHA algorithm generates a population of displaced atomic configurations.
- For the first populations, compute the Energies, Forces, and Stresses (EFS) for all configurations using DFT.
- Use these structures and EFS data to train a Moment Tensor Potential (MTP) [15].
- SSCHA Optimization with MLP: In subsequent populations, use the trained MTP to predict the EFS for the majority of configurations. Only configurations where the MTP prediction is uncertain (high "extrapolation grade," γ) are calculated with DFT and added to the training set. The combined EFS data is fed into the SSCHA to optimize the force constants and atomic centroids [15].
- Iterate and Upscale: Repeat step 3 until the SSCHA converges for the current supercell size. Then, upscale to a larger supercell. Use the converged dynamical matrix from the smaller cell to initialize the larger one, and repeat the SSCHA+MLP cycle. This drastically reduces the number of required DFT calculations in the larger cell (e.g., from 100% to under 3%) [15].
Modern Strategies for Balanced Parameter Development and Implementation
Biomolecular force fields are the foundation of molecular dynamics (MD) simulations, but a known limitation in standard fixed-charge models is their tendency to overestimate attractive interactions between charged and hydrophobic groups. This can promote artificial aggregation in simulations of multi-component protein, nucleic acid, and lipid systems, compromising the accuracy of the results [7]. This overbinding arises because the standard Lennard-Jones (LJ) parameters, typically combined using the Lorentz-Berthelot rules, are not perfectly balanced for all atom pairs.
The NBFIX (Non-Bonded FIX) correction method provides a surgical solution to this problem. Instead of globally modifying parameters, NBFIX allows for pair-specific adjustments to the LJ interactions between specific atom types. This method corrects the balance of intermolecular forces without affecting other well-calibrated properties, such as hydration free energy or solution density, making it an essential tool for force field refinement [7].
Core NBFIX Concepts and Key Applications
The NBFIX approach directly adjusts the LJ parameters for a specific pair of atom types, overriding the standard combination rules. The standard LJ potential is given by:
[ V_{LJ}(r) = 4\varepsilon \left[ \left(\frac{\sigma}{r}\right)^{12} - \left(\frac{\sigma}{r}\right)^6 \right] ]
In practice, the parameters ( \sigma ) (distance where potential is zero) and ( \varepsilon ) (depth of the potential well) are optimized for problematic atom pairs [20] [21]. The force derived from this potential is:
[ F(r) = -\frac{\partial U(r)}{\partial r} = 48 \varepsilon \left[ \frac{\sigma^{12}}{r^{13}} - \frac{\sigma^6}{r^7} \right] ]
In MD software, this radial force must be decomposed into its x, y, and z components for the simulation. If calculating the force on particle i due to particle j, the force component in the x-direction is:
[ Fx = F(r) \frac{(xi - x_j)}{r} ]
The same logic applies to the y and z components [22].
NBFIX corrections have been successfully developed and applied to numerous specific interaction types. The table below summarizes key NBFIX applications documented in the literature.
Table 1: Documented NBFIX Corrections for Specific Atom Pairs
| Type of NBFIX Correction | Key Targeted Atom Pairs | References |
|---|---|---|
| Ion Pairs | Li/Na/K/Mg–Cl/COO/PO4; Ca–Cl/COO/PO4; Cs/Rb/Br/I | [7] |
| Amine-Containing Pairs | Amine–COO/PO4/SO4 | [7] |
| Biological Molecule Pairs | Urea–urea; Hydrocarbon–hydrocarbon; Guanidinium–COO | [7] |
| Sugar & Alcohol Pairs | Carbohydrate–carbohydrate; Hydroxyl–hydroxyl | [7] |
Implementation Protocol: A Step-by-Step Guide
This section provides a detailed methodology for developing and implementing NBFIX corrections, from identifying problematic interactions to validating the final parameters.
The following diagram illustrates the logical workflow for implementing NBFIX corrections, from problem identification to final validation.
Identifying Problematic Interactions
The first step is to identify which specific atomic interactions are causing inaccuracies in your simulations. Key indicators include:
- Artificial Aggregation: Urealistic clustering of proteins, nucleic acids, or lipids that is not observed experimentally [7].
- Osmotic Pressure Discrepancies: A common and quantitatively measurable symptom. Simulations of binary salt or amino acid solutions often show significant deviation from experimental osmotic pressure measurements if ion-ion or ion-solute interactions are too attractive [7].
- Over-stabilized Salt Bridges: Protein simulations showing artificially rigid and long-lived salt bridges between amine (e.g., in Lysine) and carboxylate (e.g., in Aspartate or Glutamate) groups [21].
Parameter Optimization Procedure
Once a target atom pair is identified, follow this protocol to optimize its LJ parameters:
- Obtain Reference Data: Gather high-quality experimental data for calibration. Osmotic pressure data for binary solutions is highly effective because it is sensitive to solute-solute interactions and can be accurately obtained from both experiment and simulation [7].
- Set Up Simulation for Calibration: Use a two-compartment MD setup to compute osmotic pressure directly, or run simulations of the target system (e.g., a salt solution) and calculate the pair distribution function or potential of mean force.
- Optimize Parameters Systematically:
- Start with the standard LJ parameters for the atom pair, derived from the force field's combination rules.
- Adjust the well-depth (ε) and radius (σ) parameters iteratively. A common surgical correction is to increase the ( R{min} ) (or σ) value to reduce overly attractive interactions. For example, in CHARMM 36m, the amine-phosphate N-O=P cross-term ( R{min} ) was increased by 0.16 Å [21].
- The optimization goal is to minimize the difference between the simulated property (e.g., osmotic pressure) and the experimental reference data.
Table 2: Example NBFIX Parameter Adjustments in CHARMM36m
| Target Atom Pair | Description | Δ R_min Adjustment | Final R_min Value |
|---|---|---|---|
| Amine–Carboxylate (N–O=C) | Corrects over-stabilized salt bridges in proteins. | +0.08 Å | 3.63 Å |
| Amine–Phosphate (N–O=P) | Reduces artificial DNA-protein and DNA-DNA aggregation. | +0.16 Å | 3.71 Å |
Troubleshooting FAQs and Common Issues
Q1: After applying NBFIX corrections, my simulation shows unrealistic behavior in parts of the system not directly targeted by the fix. What could be the cause?
This "side effect" usually indicates that the NBFIX parameters are too drastic or that other force field terms have become unbalanced. To resolve this:
- Verify System Thermodynamics: Check that the corrected simulation still reproduces correct densities and enthalpies of vaporization for neat liquids of the involved components [23] [24].
- Check Hydration Free Energies: Ensure the NBFIX correction has not adversely affected the solvation thermodynamics of the involved atoms. A key advantage of a well-tuned NBFIX is that it should not significantly alter hydration free energies [7].
- Re-calibrate Iteratively: Slightly scale back the magnitude of the NBFIX correction and re-run validation. The optimal correction is the smallest one that resolves the original overbinding issue.
Q2: How many different NBFIX types are actually necessary to achieve an accurate force field?
Recent research suggests that highly simplified LJ typing schemes can achieve accuracy competitive with complex ones. One study found that a model with just five LJ types (one each for C, O, and N, and two for H - polar and apolar) performed nearly as well as models with over 15 types [24]. This implies that the number of unique NBFIX corrections needed might be smaller than anticipated. Start by correcting the most egregious, well-documented pairs (like those in Table 1) before attempting to develop a large library of custom corrections.
Q3: My NBFIX-corrected simulation fails to reproduce experimental osmotic pressure. What is the next step?
- Validate the Reference Data: Double-check the experimental osmotic pressure data and the conditions (temperature, concentration) under which it was measured.
- Inspect the Simulation Setup: Ensure your two-compartment simulation setup is technically correct and has reached equilibrium. Use a sufficient number of particles and long enough simulation time to gather adequate statistics.
- Check for Parameter Conflicts: Ensure that no other force field parameters (e.g., torsional angles, bond strengths) have been recently changed that could conflict with the NBFIX optimization. Parameter optimization should be a sequential and systematic process.
Table 3: Key Software and Computational Resources for NBFIX Development
| Tool / Resource | Function | Availability / Notes |
|---|---|---|
| CHARMM | MD software that natively supports NBFIX commands. | Commercial & Academic Licenses |
| AMBER | MD software that implements similar corrections via the LJEDIT command. |
AmberTools (includes utilities) |
| GROMACS | MD software that uses nonbond_param entries for pair-specific LJ modifications. |
Open Source |
| ForceBalance | A systematic tool for optimizing force field parameters against experimental and QM data. | Open Source [24] |
| GAAMP | A web server and script for automated atomic model parameterization, useful for initial parameter generation. | http://gaamp.lcrc.anl.gov/ [23] |
| Quantum Chemical Software (e.g., Gaussian) | Used for high-level ab initio calculations to generate target data for charge fitting and torsional parameterization. | Commercial Licenses [23] |
Frequently Asked Questions (FAQs)
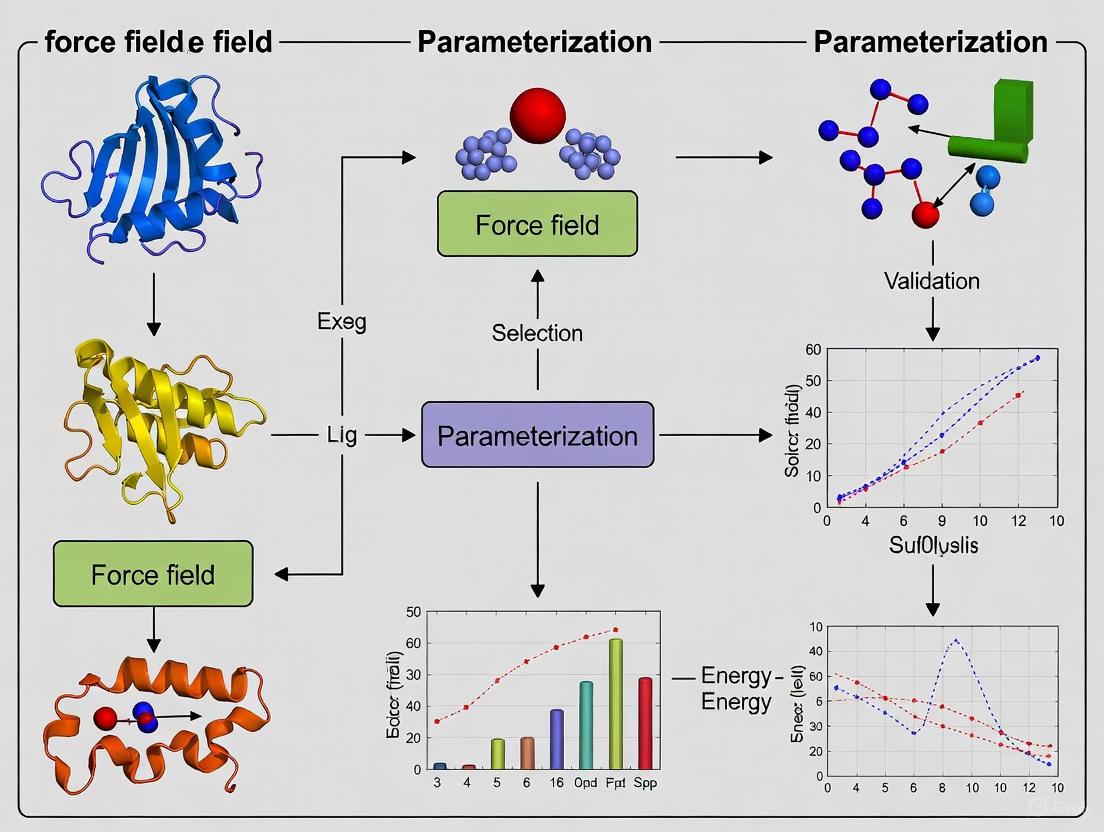
1. What is automated force field parameterization and why is it important? Automated force field parameterization uses software tools to systematically derive the parameters (e.g., bonds, angles, dihedrals, charges) required for molecular dynamics simulations. This process is crucial because manual parameterization is time-consuming, error-prone, and can introduce inconsistencies, especially for unusual molecules like nucleotide analogues, functionalized carbohydrates, and modified amino acids that are poorly described by standard force fields [25]. Automation enhances reproducibility, reduces human error, and accelerates the creation of large parameter sets for machine learning and high-throughput screening in drug discovery [25] [26].
2. My simulations show artificial aggregation of biomolecules. What is the likely cause and how can I fix it? Artificial aggregation in simulations of proteins, DNA, or lipids is a common artifact indicating an imbalance in non-bonded interactions, often from an overestimation of attractive forces between charged and hydrophobic groups [7]. This is a known shortcoming of several standard force fields.
- Solution: Apply pair-specific Lennard-Jones parameter corrections, known as NBFIX (in CHARMM) or CUFIX (for AMBER/CHARMM). These are surgically calibrated against experimental data like osmotic pressures to reduce unrealistic attraction without introducing new artifacts [7] [27]. For example, the CUFIX corrections for amine-carboxylate and amine-phosphate interactions have been shown to correct DNA-DNA interactions and prevent unrealistic protein compaction [27].
3. How can I validate newly generated parameters to ensure they are physically realistic? A robust validation protocol should compare simulation results with experimental and quantum mechanical (QM) target data. Key properties to check include:
- Pure-solvent properties: Density, heat of vaporization (target <15% error from experiment) [26].
- Solvation free energies: Should be within ±0.5 kcal/mol of experimental values [26].
- Osmotic pressure: For electrolyte and amino acid solutions, simulated osmotic pressure should match experimental measurements [7].
- Quantum mechanical targets: Compare conformational energies and dihedral scans from molecular mechanics (MM) calculations against high-level QM reference data [26].
4. I am parameterizing a small molecule for drug discovery. What is a reliable automated workflow? A reliable automated workflow integrates several steps to ensure parameter quality:
- Input Preparation: Provide a 3D molecular structure.
- Atom Typing: Use tools like the ParamChem webserver to assign initial CHARMM-compatible atom types [26].
- Parameter Generation: Use an automated tool like AutoParams [25] [28] or the Force Field Toolkit (ffTK) [26] to generate bond, angle, dihedral, and charge parameters.
- Charge Fitting: Employ QM-based methods (e.g., water-interaction profiles for CHARMM, RESP for AMBER) to derive partial atomic charges [26].
- Validation: Run short simulations of the molecule in solution and pure solvent to compute density, enthalpy of vaporization, and free energy of solvation, comparing against known experimental data [26].
5. What are the best practices for training a Machine-Learned Force Field (MLFF)? When using on-the-fly learning with tools like VASP's MLFF, follow these guidelines to ensure a robust model [29]:
- Exploration: Train in the NpT ensemble (ISIF=3) if possible, as cell fluctuations improve force field robustness. Avoid the NVE ensemble.
- Sampling: Gradually heat the system during training to explore a larger region of phase space.
- System Focus: If your system has different components (e.g., a crystal surface and an adsorbing molecule), train them separately before combining them.
- Stress Handling: For systems with surfaces or isolated molecules, set the stress weight (
ML_WTSIF) to a very small value to avoid training on unphysical stresses from vacuum layers. - Convergence: Do not set
MAXMIX > 0, as this can lead to non-converged electronic structures between ab-initio calculations.
6. How do I choose between all-atom and coarse-grained automated parameterization? The choice depends on your research question and the scale of the system.
- All-atom (AA): Provides atomic-resolution detail. Use for studying specific molecular interactions, ligand binding, or protein folding. Tools include ffTK [26] and AutoParams [25].
- Coarse-grained (CG): Allows simulation of larger systems and longer timescales by grouping multiple atoms into single interaction sites. Use for studying membrane partitioning, large assemblies, or long-timescale dynamics. Tools like CGCompiler can automate Martini 3 force field parametrization, using experimental log P values and atomistic density profiles as targets [30].
Troubleshooting Guides
Issue 1: Parameterization Yields Poor Solvation Free Energies or Densities
Problem: After parameterizing a small molecule, simulations in water yield a solvation free energy or pure-solvent density that deviates significantly from experimental values.
Diagnosis and Solutions:
- Check Partial Atomic Charges: Inaccurate partial charges are a primary source of error in solvation properties.
- Action: If using the CHARMM force field, ensure charges are derived from QM water-interaction profiles, not just electrostatic potential fitting [26]. For AMBER, verify that the RESP fitting procedure was performed correctly.
- Tool: Use ffTK to re-optimize charges against QM water-interaction energies [26].
- Verify Lennard-Jones (LJ) Parameters: LJ parameters significantly influence density and solvation.
- Action: Compare your LJ parameters against those for similar atom types in the established force field (GAFF, CGenFF). Tools like ParamChem provide penalty scores for transferred parameters; a high penalty indicates a poor match and a need for optimization [26].
- Tool: The ForceBalance method can automatically optimize LJ parameters against experimental density and enthalpy of vaporization data [31].
Validation Workflow:
Issue 2: Unrealistic Dihedral Energy Profiles
Problem: A rotational energy profile generated with your new parameters does not match the target quantum mechanical (QM) scan.
Diagnosis and Solutions:
- Insufficient Target Data: The dihedral parameter was fit to a narrow range of conformations.
- Action: Ensure the QM dihedral scan covers the full 360-degree rotation with adequate resolution (e.g., 15-degree increments).
- Overfitting: The parameter is too strong and fits noise in the QM data.
- Action: Use a regularization penalty during optimization. The ForceBalance tool includes Tikhonov regularization to prevent overfitting [31].
- Lack of Phase Terms: A single dihedral term may be insufficient to capture the complex QM profile.
- Action: Add multiple Fourier terms (multiple periods) for the same dihedral angle. The ffTK dihedral fitting tool is designed to handle this [26].
Recommended Protocol:
- Perform a QM Scan: Use your preferred QM software (e.g., Gaussian, Psi4) to calculate the single-point energy of the molecule while rotating the dihedral of interest in small increments.
- Fit the Parameters: Input the QM energy profile into a tool like ffTK. The tool will perform a least-squares optimization to find the dihedral force constants (V) and phase angles (δ) that best reproduce the QM profile with the MM energy function [26].
- Cross-Validate: Test the fitted parameters in a different molecular context or check if they reproduce the relative energies of known stable conformers from the literature.
Issue 3: System Crashes or Instabilities During Dynamics
Problem: Simulations crash shortly after energy minimization or during the initial equilibration steps, often with errors related to "bond too long" or excessive force.
Diagnosis and Solutions:
- Incorrect Bonded Parameters: This is the most common cause, specifically bad bond or angle equilibrium values/force constants.
- Action:
- Verify Geometry Optimization: Ensure the initial molecular geometry used for parameterization was fully optimized at a QM level [26].
- Check Bond/Angle Parameters: Use ffTK to plot the potential energy surface for bonds and angles around their equilibrium values. Compare the MM energy curve against QM calculations. Large discrepancies indicate poor parameters that need re-optimization [26].
- Action:
- Charge Imbalance: A non-neutral molecule can cause catastrophic instabilities.
- Action: Ensure the sum of partial atomic charges is exactly an integer (e.g., 0 for a neutral molecule, +1 for a cation). Most automated tools enforce this, but it is critical to check [26].
- Steric Clashes: Overly attractive LJ parameters can cause atoms to come too close.
- Action: Check for very short non-bonded distances in the initial crash. Consider applying NBFIX corrections if the issue involves specific ion pairs or charged groups [7].
Research Reagent Solutions: Essential Tools for Automated Parameterization
Table 1: Key Software Tools for Automated Force Field Development
| Tool Name | Compatible Force Field(s) | Core Function | Key Feature |
|---|---|---|---|
| AutoParams [25] [28] | AMBER | Automated parameter generation for unusual molecules. | Web-based service; minimal user input; integrates charge generators (Psi4, TeraChem). |
| Force Field Toolkit (ffTK) [26] | CHARMM/CGenFF | GUI-driven workflow for ab-initio parameterization. | VMD plugin; provides modular workflow for charges, bonds, angles, and dihedrals. |
| ForceBalance [31] | Multiple (e.g., TIP3P-FB, TIP4P-FB) | Systematic force field optimization. | Fits parameters to flexible combinations of experimental and QM target data. |
| CGCompiler [30] | Martini 3 (Coarse-Grained) | Automated parametrization of small molecules. | Uses particle swarm optimization against experimental log P and density profiles. |
| NBFIX/CUFIX [7] [27] | CHARMM, AMBER | Correction for non-bonded interactions. | Surgical correction of LJ parameters to prevent artificial aggregation. |
| CHAPERONg [32] | GROMACS | Automation of simulation setup and analysis. | Bash/Python tool that automates GROMACS MD pipelines and trajectory analysis. |
Experimental Protocols
Protocol 1: Deriving Bond and Angle Parameters Using the Force Field Toolkit (ffTK)
This protocol outlines the process of deriving bond and angle parameters that reproduce Quantum Mechanical (QM) potential energy surfaces [26].
1. Generate QM Target Data:
- Starting from the QM-optimized geometry, perform a series of single-point QM calculations where specific internal coordinates (bonds or angles) are displaced from their equilibrium values.
- For a bond, typically displace the length by ±0.05 Å in 0.01 Å increments. For an angle, displace by ±15 degrees in 2.5-degree increments.
- This generates a potential energy surface (PES) around the minimum.
2. Set Up Optimization in ffTK:
- In the "Bonds & Angles" module of ffTK, load the generated QM data.
- The tool will automatically set up the objective function, which is the sum of squared differences between the MM and QM energies across all displacement points.
3. Execute Optimization:
- ffTK uses a least-squares optimization algorithm (e.g., Levenberg-Marquardt) to adjust the force constant (k) and equilibrium value (r₀/θ₀) for the bond or angle.
- The optimization iteratively runs MM calculations with trial parameters until the MM PES converges to the QM target.
4. Validate:
- Visually inspect the overlaid QM and MM energy curves provided by ffTK.
- A good fit shows the MM curve closely following the QM curve, especially near the energy minimum.
The following diagram illustrates this iterative workflow:
Protocol 2: Applying NBFIX Corrections to Ameliorate Aggregation Artifacts
This protocol describes how to apply non-bonded fixes (NBFIX) to correct for artificially strong interactions in DNA and protein simulations [7] [27].
1. Identify Problematic Interactions:
- The artifact often manifests as unrealistic clustering of DNA duplexes, overly compact denatured proteins, or incorrect ion binding.
- Common culprits are overestimated attractions between amine-nitrogen and carboxylate-oxygen, or between aliphatic carbon atoms.
2. Obtain Correction Parameters:
- Download the published CUFIX or NBFIX parameter files for your force field (AMBER or CHARMM) from the developer's website [27].
- These files contain the corrected Lennard-Jones ε and Rmin/2 parameters for specific atom pairs (e.g.,
NG2S2 O2F1for amine-carboxylate in CHARMM).
3. Integrate into Simulation Setup:
- For CHARMM: Replace the standard
toppar_water_ions.strfile with the one provided in the CUFIX package. - For AMBER in GROMACS: Add an
#include "cufix.itp"statement to yourforcefield.itpfile and update your DNA/RNA residue topology files (dna.rtp,rna.rtp) to use the new atom types (e.g.,ON2for phosphate oxygens) [27]. - Ensure you are using the correct water model (TIP3P) and ion parameters as specified.
4. Validate the Correction:
- Re-run the simulation that previously showed aggregation.
- The corrected simulation should show proper biomolecular separation and, if available, reproduce experimental data such as osmotic pressure or solution scattering profiles.
Quantitative Data for Force Field Validation
Table 2: Key Target Properties for Validating Biomolecular Force Fields
| Property Category | Specific Target | Acceptable Error | Reference Method |
|---|---|---|---|
| Liquid Properties | Density | < 1.5% from experiment | Experiment [26] |
| Enthalpy of vaporization | < 1.5% from experiment | Experiment [26] | |
| Static dielectric constant | ~1.5% from experiment (e.g., 77.3 vs. 78.4) | Experiment [31] | |
| Solvation | Free energy of solvation | ± 0.5 kcal/mol from experiment | Experiment [26] |
| Osmotic pressure | Match experimental curve | Experiment [7] | |
| Quantum Mechanics | Dihedral energy profile | RMSE < 0.5 kcal/mol | High-level QM calculation [26] |
| Interaction energy with water | RMSE < 1.0 kcal/mol | QM water-interaction profile [26] |
The IPolQ (Implicitly Polarized Charge) scheme is an advanced method for developing biomolecular force fields that aims to account for the electronic polarization of a solute induced by its solvent environment, while remaining compatible with standard, fixed-charge molecular dynamics simulation packages [33]. In classical fixed-charge force fields, atomic partial charges remain constant regardless of their environment, which is a significant physical approximation because real molecules experience charge redistribution when moving from vacuum to aqueous solution or between different protein microenvironments [34] [35]. The IPolQ method addresses this limitation through a dual-charge approach that utilizes two distinct sets of charges for each amino acid: one set appropriate for vacuum conditions and another appropriate for aqueous solution [33].
This methodology represents a philosophically superior compromise for combining well-defined sets of quantum mechanical (QM) information in a thermodynamically defensible way [33]. The fundamental insight driving IPolQ's development is the recognition that deriving bonded parameters (angle terms and torsion potentials) using gas-phase QM calculations—while necessary for practical reasons—creates an inconsistency when those parameters are used with solution-phase charges in molecular dynamics simulations [35] [36]. By maintaining separate charge sets for parameter development and production simulations, IPolQ seeks to provide a more physically consistent treatment of solvent-induced polarization effects within the framework of fixed-charge force fields [33] [35].
Table: Core Components of the IPolQ Scheme
| Component | Description | Purpose |
|---|---|---|
| QVac Charge Set | Gas-phase partial charges | Used for deriving bonded parameters (angle and torsion terms) |
| QIPol Charge Set | Solution-phase partial charges | Used in production molecular dynamics simulations |
| Parameter Fitting | Automated optimization to QM data using gas-phase charges | Ensures bonded parameters match vacuum potential energy surfaces |
| Simulation Phase | Uses polarized charges in solvent environment | Accounts for water-induced electronic polarization |
Theoretical Foundation and Workflow
The IPolQ method is built upon a clear physical assumption: the energetic consequences of solvating a biomolecule are predominantly electrostatic in nature [33]. This foundation justifies the approach of deriving bonded parameters in the context of gas-phase charges and then transferring them to simulations employing solution-phase charges. The protocol recognizes that QM calculations can accurately map potential energy surfaces in vacuum, providing an ideal data source for developing bond, angle, and torsion parameters [33]. Simultaneously, the implicitly polarized charge scheme converges to an optimal fixed charge representation of a molecule in a polarizing medium like water [33].
The workflow begins with quantum mechanical calculations performed at the MP2/cc-pvTZ level of theory on various amino acids and short peptides [33]. These calculations generate single-point energies for approximately 265,000 conformations, providing comprehensive coverage of the conformational space [33]. The key innovation of IPolQ is that these QM calculations are performed in both vacuum and in the presence of a reaction-field potential that models water, yielding two sets of electrostatic parameters [35] [36]. The solution-phase charges are obtained by averaging the charges from these two environments, creating implicitly polarized parameters that account for the dielectric screening effects of water [35].
The IPolQ scheme was first implemented in the ff14ipq force field, where partial charges were positioned halfway between the QM charges of a dipeptide in vacuum and in the presence of a reaction-field potential [35]. This approach allowed the developers to implicitly account for polarization effects when deriving dihedral parameters using gas-phase potential energy surfaces [35]. The method was subsequently refined in the ff15ipq force field, which introduced new atom types for backbone atoms, enabling more specific dihedral refinements [33] [35]. Validation simulations demonstrated that ff15ipq produced good agreement with challenging experimental data, including the temperature-dependent unfolding of peptides and folding events of intrinsically disordered proteins upon binding [35].
Frequently Asked Questions (FAQs)
Q1: What is the fundamental advantage of IPolQ over traditional fixed-charge methods?
The primary advantage of IPolQ is its physically consistent approach to handling the different environments in which parameters are derived versus applied. Traditional force fields often derive bonded parameters from gas-phase quantum mechanical calculations but use them in condensed-phase simulations with the same charge set, creating an inherent inconsistency [33] [35]. IPolQ acknowledges that gas-phase QM calculations provide the most reliable potential energy surfaces for parameter development, while also recognizing that solvated biomolecules experience significant electronic polarization [33]. By separating the charges used for parameter development (QVac) from those used in production simulations (QIPol), IPolQ provides a more rigorous treatment of solvent effects without requiring explicit polarization in the simulation itself [33].
Q2: How does IPolQ compare to explicitly polarizable force fields?
IPolQ occupies a middle ground between traditional fixed-charge force fields and explicitly polarizable models. While polarizable force fields like AMOEBA or those using the Drude oscillator model explicitly calculate electronic polarization in response to the instantaneous electric field during simulations [34], IPolQ incorporates polarization effects implicitly through its dual-charge approach [33]. The advantage of IPolQ is that it maintains the computational efficiency of fixed-charge force fields, as it doesn't require the iterative self-consistent field calculations or extended Lagrangian methods needed by polarizable models [33] [34]. This makes IPolQ suitable for longer timescale simulations that might be prohibitively expensive with fully polarizable force fields, while still offering improved physical accuracy over traditional fixed-charge models.
Q3: What are the known limitations of the IPolQ method?
Despite its theoretical advantages, IPolQ has several important limitations. First, as a mean-field approach, it cannot capture the dynamic polarization response to specific, instantaneous molecular configurations, unlike explicitly polarizable force fields [33] [34]. Second, validation studies have revealed some systematic biases; for instance, the ff15ipq-Qsolv variant (where bonded parameters were optimized with solution-phase charges) was found to overstabilize α-helical structures in certain peptides [33]. Additionally, the method still relies on the accuracy of the quantum mechanical calculations and the transferability of the parameters across different molecular environments [33] [35]. Finally, the IPolQ scheme does not address other limitations of fixed-charge force fields, such as the inability to model anisotropic charge distributions or charge penetration effects [34] [37].
Q4: How does RESP2 differ from the IPolQ approach?
RESP2 is another method that addresses similar challenges as IPolQ but with a different implementation. While IPolQ uses explicit solvent QM calculations followed by charge averaging [33] [35], RESP2 generates partial charges as linear combinations of gas- and aqueous-phase charges, with a mixing parameter δ that can be optimized against experimental condensed-phase data [38]. RESP2 with δ ≈ 0.6 (60% aqueous, 40% gas-phase) has been shown to be an accurate and robust method for generating partial charges [38]. Both methods share the fundamental insight that charges for condensed-phase simulations should reflect an intermediate polarization state between gas and aqueous phases, but they differ in their specific implementation and parameterization strategies.
Troubleshooting Common Issues
Issue 1: Overstabilization of Secondary Structures
Problem: Simulations using force fields derived with the IPolQ method show excessive α-helical content or overstable helices in certain peptide systems [33].
Diagnosis: This issue was particularly observed in the ff15ipq-Qsolv variant, where bonded parameters were optimized in the context of solution-phase charges [33]. The problem likely stems from the bonded parameters counteracting the solution-phase characteristics of the charge set when sampling local structure [33].
Solution:
- Use the standard ff15ipq parameters rather than the Qsolv variant
- Validate against multiple peptide systems with known experimental structural preferences
- Consider adjusting simulation temperature or using enhanced sampling techniques
- Verify that the balance between solute-solvent and solute-solute interactions is appropriate for your specific system
Issue 2: Instability of Globular Proteins
Problem: Simulations result in incorrect unfolding of globular proteins or loss of native structure [33].
Diagnosis: This issue was notably observed in the ff15ipq-Vac variant, where gas-phase charges were used in production simulations instead of the solution-phase charges [33]. The vacuum charges lack the polarization needed to properly stabilize protein structure in aqueous environment.
Solution:
- Ensure you are using the correct charge set (QIPol) for production simulations, not the gas-phase charges (QVac)
- Verify force field file implementation to confirm the proper charge set is loaded
- Check that your water model is compatible with the protein force field
- Monitor simulation metrics such as RMSD and radius of gyration early to detect instability
Issue 3: Inaccurate Peptide Conformational Sampling
Problem: Short peptides do not show correct metastability or conformational preferences compared to experimental data [33].
Diagnosis: This may indicate limitations in how the force field handles the balance between different conformational states, potentially due to incomplete treatment of environment-dependent effects [33].
Solution:
- Extend simulation time to ensure adequate sampling of conformational space
- Replicate specific experimental conditions from literature validation studies
- Compare results across multiple force field variants (standard ff15ipq, Qsolv, Vac) to identify systematic biases
- Consider targeting additional experimental data such as J-coupling constants or NMR observables for validation
Experimental Protocols and Methodologies
Parameter Derivation Protocol
The development of IPolQ-based force fields follows a rigorous protocol for deriving accurate parameters:
Quantum Mechanical Calculations:
- Perform QM calculations at the MP2/cc-pvTZ level on amino acids and short peptides [33]
- Collect single-point energies for approximately 265,000 conformations to ensure comprehensive coverage of conformational space [33]
- Conduct calculations in both gas phase and with a reaction-field potential representing water [35]
Charge Derivation:
Bonded Parameter Optimization:
- Fit angle terms and torsion parameters to reproduce gas-phase quantum energy surfaces using QVac charges [33]
- Utilize automated fitting tools to optimize over 60 angle parameters and 700 torsion parameters [33]
- Employ iterative optimization and force-field guided structure manipulation to ensure parameter convergence [33]
Table: Comparison of Charge Generation Methods
| Method | QM Level | Solvent Treatment | Mixing Ratio | Key Features |
|---|---|---|---|---|
| IPolQ | MP2/cc-pvTZ | Explicit solvent reaction field or implicit hydration model | 50:50 (gas:aqueous) | Dual charge sets; physically rigorous |
| RESP2 | PW6B95/aug-cc-pV(D+d)Z | Implicit solvent model | 40:60 (gas:aqueous, δ=0.6) | Optimized δ parameter; co-optimized with LJ parameters |
| Traditional RESP | HF/6-31G* | Gas phase only | 100:0 (gas:aqueous) | Relies on fortuitous overpolarization of HF/6-31G* |
Force Field Validation Protocol
To ensure the reliability of IPolQ-based force fields, comprehensive validation is essential:
System Preparation:
- Select diverse set of validation systems: globular proteins, short peptides, and intrinsically disordered proteins [33] [35]
- Include proteins with different secondary structure compositions (e.g., α-helical, β-sheet)
- Prepare systems with appropriate solvation and ion concentration for biological relevance
Simulation Parameters:
Validation Metrics:
- Monitor stability of globular protein structures using RMSD and native contact analysis [33]
- Quantify secondary structure content using DSSP or similar algorithms [33]
- Compare with experimental data such as J-coupling constants, NMR order parameters, and conformational distributions [33] [35]
- Assess temperature-dependent unfolding behavior for peptides with known experimental profiles [33]
Research Reagent Solutions
Table: Essential Computational Tools for IPolQ Method Development
| Tool/Resource | Function | Application in IPolQ Development |
|---|---|---|
| Quantum Chemistry Software | High-level QM calculations | Generate reference data for charge derivation and torsional profiles [33] |
| ForceBalance | Automated parameter optimization | Simultaneously fit multiple parameters to experimental and QM target data [35] [36] |
| MD Simulation Packages | Molecular dynamics engine | Conduct validation simulations and production runs [33] |
| IPolQ-Mod | Efficient charge derivation | Implicit solvent version of IPolQ that saves computational time [38] |
| Trajectory Analysis Tools | Simulation output analysis | Calculate validation metrics and compare with experimental data [33] |
Frequently Asked Questions
Q1: My Bayesian-optimized force field fails to reproduce correct densities at high concentrations, even though it performs well at the concentration used for training. What could be wrong? This is a common challenge related to transferability. The parameterization using small molecular fragments in solution is typically performed at concentrations accessible to ab initio molecular dynamics (AIMD). If your production simulations run at a significantly higher concentration, explicit solute-solute interactions missing from the training data can cause inaccuracies.
- Solution: Always validate your optimized force field against a known experimental property, such as aqueous solution density across a range of concentrations, before using it for production runs on your target system [39].
Q2: The Bayesian inference process is computationally prohibitively slow. How can I accelerate it? The computational bottleneck is the need to run countless classical molecular dynamics (FFMD) simulations to evaluate candidate parameter sets during Markov Chain Monte Carlo (MCMC) sampling.
- Solution: Implement a surrogate model, such as a Local Gaussian Process (LGP) regressor, to emulate the quantities of interest (QoIs) from the FFMD simulations. This surrogate predicts structural properties from trial parameters at a fraction of the computational cost, making MCMC sampling efficient. LGP models are often preferred over neural networks for this purpose because they can incorporate interpretable, physics-informed priors [39] [40].
Q3: How can I be sure that the uncertainty estimates from my Gaussian Process model are reliable for active learning? Reliable uncertainty estimates require a well-defined model that separates different types of error.
- Solution: Ensure your GP model provides interpretable uncertainties by distinguishing between epistemic uncertainty (σiα, due to distance from training data) and noise uncertainty (σn, inherent variability in the data). The trustworthiness of these estimates hinges on using a low-dimensional, physically motivated kernel (e.g., based on 2- and 3-body interactions) and rigorously optimizing the GP's hyperparameters by maximizing the log marginal likelihood of the training data [41].
Q4: I need to parameterize a new molecule. What is the minimal reference data required to start the Bayesian learning process? You can begin with a surprisingly small amount of targeted reference data.
- Solution: The process can be initiated with AIMD simulation data of the solvated molecular fragment. An active learning framework can then automate training by using the GP's internal uncertainty to decide when to perform new ab initio calculations, often requiring only on the order of ~100 DFT calculations to build a robust force field for complex systems [41].
Q5: When refining bonded parameters in a coarse-grained system using Bayesian optimization, how do I avoid overfitting to the target properties? Overfitting is a key risk when optimizing against a small number of properties.
- Solution: The Bayesian framework inherently penalizes unnecessary complexity. Furthermore, you should validate the optimized parameters by checking their performance for higher degrees of polymerization than were used in the training set. This ensures the parameters are transferable and not just fitted to a specific chain length [42].
Troubleshooting Guides
Problem: Imbalanced Force Field After Introducing New Parameters
Symptoms: After adding parameters for a new residue or ligand, the simulation shows unrealistic aggregation, distorted solvation structures, or instability.
| Investigation Step | Action | Reference / Tool |
|---|---|---|
| Check Parameter Penalties | If using tools like CGenFF, a high penalty score indicates a large chemical difference from existing parameters, necessitating manual validation. | Force Field Toolkit (ffTK) in VMD [43] |
| Validate Partial Charges | Compare the dipole moment of the new molecule from QM calculations with the dipole moment produced by the assigned partial charges in the force field. | Population analysis (e.g., RESP) [39] |
| Validate Bonded Terms | Perform quantum chemical potential energy scans for dihedrals and compare them with the classical energy profile from the force field. | Quantum Chemistry software (e.g., Gaussian, ORCA) [43] |
| Test in Condensed Phase | Run a short simulation of the solvated molecule and compare radial distribution functions (RDFs) against reference AIMD data. | Bayesian Inference Workflow [39] |
Problem: Inadequate Sampling of Rare Events Despite Accurate Force Field
Symptoms: The force field is known to be accurate, but your molecular dynamics (MD) simulation fails to observe expected conformational changes or binding events.
| Solution Approach | Key Implementation Details |
|---|---|
| Perform Replicate Simulations | Initiate multiple independent simulations from different starting points in phase space (e.g., different random velocities or configurations) [43]. |
| Employ Enhanced Sampling | Use methods like metadynamics or Gaussian-accelerated MD (GaMD) to apply a bias potential that encourages the system to escape local energy minima [43]. |
| Use Active Learning | Implement an on-the-fly method like FLARE. The GP model can identify uncertain configurations during dynamics, triggering new ab initio calculations to expand the training set and improve the model for rare events [41]. |
Problem: Bayesian Optimization Fails to Converge on a Robust Parameter Set
Symptoms: The optimization process is unstable, or the final parameters are highly sensitive to the initial guess.
| Potential Cause | Solution |
|---|---|
| Poorly Informative Data | Ensure your target data (e.g., from AIMD) includes diverse states: the aqueous solute, the solute with a direct contact ion, and the solute with a solvent-shared ion [39]. |
| Incorrect Prior Distributions | Re-evaluate your prior distributions for parameters. Use physically motivated priors (e.g., truncated normal distributions) based on typical force field values [39] [44]. |
| High-Dimensional Search Space | Reduce the dimensionality of the parameter space. For coarse-grained topologies, focus on optimizing a low-dimensional set of bonded parameters (bond lengths, angles, and their constants) [42]. |
Experimental Data & Protocols
Key Quantitative Performance of the Bayesian Framework
The table below summarizes the performance of a Bayesian framework when applied to 18 biologically relevant molecular motifs, demonstrating consistent improvement over a established force field (CHARMM36) [39].
| System Class | Metric | Performance vs. CHARMM36 | Key Improvement |
|---|---|---|---|
| Most Molecular Species | Hydration Structure (RDFs) | < 5% error | Robust reproduction of solvation structure [39] |
| Most Molecular Species | Hydrogen Bond Counts | < 10-20% deviation | Larger variability due to rigid water model limitations [39] |
| Charged Systems (esp. Anions) | Ion-Pair Distance Distributions | < 20% deviation (typically) | Restored balanced electrostatics [39] |
| Charged Species Solutions | Density vs. Experiment | Deviation < 1% | Validated across a range of concentrations [39] |
Core Protocol: Bayesian Force Field Parameterization from AIMD Data
Objective: Refine partial charge distributions for a molecular fragment using reference data from ab initio molecular dynamics (AIMD) simulations [39].
Step-by-Step Workflow:
Generate Reference Data: Run an AIMD simulation of the solvated molecular fragment. From this trajectory, extract condensed-phase Quantities of Interest (QoIs), such as:
- Radial distribution functions (RDFs) between solute and solvent atoms.
- Hydrogen bond counts.
- Ion-pair distance distributions (for charged species) [39].
Define Parameter Prior: Choose a prior distribution for the partial charges. A common choice is a truncated normal distribution, with bounds based on physically meaningful ranges from established force fields [39].
Build a Training Set: Sample thousands of charge distributions from the prior. For each set, run a classical force field MD (FFMD) simulation and compute the same QoIs. This creates a mapping from parameters to observables [39].
Train a Surrogate Model: Use the training set to build a Local Gaussian Process (LGP) surrogate model. This model will learn to instantly predict the QoIs for any new set of partial charges, avoiding the cost of running a full FFMD simulation for every candidate [39].
Sample the Posterior with MCMC: Use a Markov Chain Monte Carlo sampler (e.g., using Pyro or similar tools) to explore the parameter space. At each step, the likelihood of a candidate parameter set is evaluated against the AIMD reference data using the LGP surrogate's prediction [39] [40].
Validate the Optimized Force Field: The output is a posterior distribution of parameters. Draw multiple samples from this posterior and:
- Validate that the QoIs match the AIMD reference.
- Test the force field's transferability by comparing simulated properties (e.g., density) against experimental data not used in training [39].
This workflow is visualized in the following diagram:
The Scientist's Toolkit: Essential Research Reagents & Software
| Item | Function / Application | Key Notes |
|---|---|---|
| Ab Initio MD (AIMD) | Generates reference data incorporating electronic polarization and many-body effects. | Typically based on Density Functional Theory (DFT); provides the "ground truth" for training [39]. |
| Local Gaussian Process (LGP) | Serves as a fast surrogate model to emulate MD simulation outputs. | Accelerates Bayesian inference by predicting properties without running full simulations; offers interpretable uncertainty [39]. |
| MCMC Sampler (e.g., NUTS) | Samples the posterior distribution of force field parameters. | Core engine for Bayesian learning; requires efficient implementation (e.g., via Pyro) [40]. |
| Active Learning Framework (e.g., FLARE) | Automates on-the-fly training and uncertainty quantification for force fields. | Uses GP uncertainty to decide when to run new ab initio calculations, ideal for rare events [41]. |
| Bayesian Optimization (BO) | Optimizes costly black-box functions, like CG force field parameterization. | Efficiently balances exploration and exploitation; useful for refining bonded parameters in coarse-grained models [42]. |
Diagnosing and Correcting Common Force Field Artifacts and Pitfalls
Addressing Overly Compact Denatured States and Artificial Aggregation
Frequently Asked Questions (FAQs)
1. What are the symptoms of an imbalanced biomolecular force field in my simulation? You may be observing artificial aggregation of proteins, DNA, or lipids, where molecules clump together in ways not observed experimentally. Another common symptom is an overly compact denatured state or unfolded state ensemble during protein folding simulations, where the simulated random coil is more collapsed than real disordered proteins would be [27] [45].
2. What is the fundamental "bonded parameter imbalance" causing these issues? The core issue often isn't with the bonded parameters themselves, but with an imbalance in non-bonded interactions. Specifically, most standard force fields overestimate attractive interactions between charged groups (e.g., amine-carboxylate or amine-phosphate) and between hydrophobic aliphatic carbons. This excessive attraction drives unrealistic aggregation and collapse [27] [46].
3. How can I fix artificial aggregation in my simulations without introducing new artifacts? A widely recommended method is to apply pair-specific corrections to non-bonded interactions, known as NBFIX (for CHARMM) or CUFIX (for AMBER). These are "surgical corrections" that refine the Lennard-Jones parameters for specific atom pairs. They are calibrated against experimental data like osmotic pressure and do not alter bonded parameters or solvation free energies, thus minimizing the risk of new artifacts [27] [46].
4. My protein folding simulation looks unrealistic. Could a folding intermediate be affecting the outcome? Yes. The structural features of the denatured state ensemble (DSE) and any intermediate states (ISE) significantly impact aggregation propensity. Simulations have shown that proteins with rapidly formed, native-like folding intermediates that have contacts spread across the protein aggregate more slowly. In contrast, proteins with more localized native structure in the DSE are more vulnerable to domain-swapping and fast aggregation [45].
5. I am incorporating unnatural amino acids. Where can I find reliable parameters? Specialized force field parameters for unnatural amino acids (UAAs) are an active area of research. You may need to derive them yourself using quantum mechanics (QM) calculations followed by RESP charge fitting, or look for published parameter sets. For instance, parameters for 18 phenylalanine and tyrosine derivatives compatible with the AMBER ff14SB force field have been developed and tested [47].
Troubleshooting Guides
Issue 1: Artificial Aggregation of Biomolecules
Problem: During a simulation of multiple proteins, nucleic acids, or a mixture, you observe chains sticking together or forming clusters that are not biologically relevant.
Diagnosis Steps:
- Verify Observation: Confirm that the aggregation is not a transient, rare event but a persistent state.
- Check Concentration: Ensure your simulation box size and number of molecules result in a physiologically realistic concentration. Artificially high concentrations can force aggregation.
- Identify Interacting Groups: Analyze which chemical groups (e.g., charged side chains, hydrophobic patches) are making persistent, non-native contacts.
Solutions:
- Apply NBFIX/CUFIX Corrections: The most direct solution is to incorporate these corrections. For the CHARMM force field, this involves replacing the standard
toppar_water_ions.strfile with a corrected version [27].- For the AMBER force field (ff99/ff14 variants), you can download a complete corrected package or integrate the corrections by:
- Adding a
#include "cufix.itp"statement to yourforcefield.itpfile. - Replacing specific atom types in your nucleic acid topology files (e.g., changing O1P and O2P to ON2).
- Using the ion parameters provided with the correction, which are often optimized with the TIP3P water model [27].
- Adding a
- For the AMBER force field (ff99/ff14 variants), you can download a complete corrected package or integrate the corrections by:
- Use Updated Force Fields: Consider using modern force fields that have incorporated these lessons. For example, the DES-Amber force field includes revisions to nonbonded and torsion parameters for DNA and RNA to prevent over-stabilization of interactions, making it more suitable for simulating protein-nucleic acid complexes [48].
Issue 2: Overly Compact Denatured or Unfolded States
Problem: In protein folding simulations or simulations of intrinsically disordered proteins, the unfolded chain appears too tightly packed, failing to match the expected dimensions from experimental techniques like SAXS or FRET.
Diagnosis Steps:
- Compare with Experiment: Quantify the radius of gyration (Rg) or end-to-end distance of your simulated denatured state and compare it to known experimental values, if available.
- Analyze Intra-chain Contacts: Calculate the number of native versus non-native intra-chain contacts in the denatured state. An imbalance may indicate over-stable non-native interactions.
Solutions:
- Refine Specific Non-bonded Interactions: As with aggregation, applying NBFIX corrections for amine-carboxylate and aliphatic carbon-carbon pairs has been shown to significantly increase the size of denatured protein conformations without destabilizing the native state [27].
- Validate with a Test System: If developing your own parameters, test them on a simple system like amino acid solutions. The corrected parameters should reproduce the experimental osmotic pressure of these solutions [27].
Experimental Protocols & Data
Protocol 1: Implementing NBFIX/CUFIX Corrections in AMBER Simulations
This protocol outlines the steps to integrate CUFIX corrections into an AMBER force field for use in GROMACS [27].
- Acquire the Correction Package: Download the
ff99sb-ildn-phi-bsc0-cufixpackage from the Aksimentiev Group website. - Integrate Files: Copy the files
cufix.itp,mg-sol6.itp,mg-sol6.pdb,ca-sol7.itp, andca-sol7.pdbinto your force field directory. - Modify Nucleic Acid Topology: In the
dna.rtpandrna.rtpfiles, change the atom type for all O1P and O2P atoms fromO2toON2. TheON2atom type is defined withincufix.itp. - Include the Correction File: Edit your
forcefield.itpfile. Add the line#include "cufix.itp"between the lines#include "ffnonbonded.itp"and#include "ffbonded.itp". - Update Ion Parameters: Delete the existing definitions for ions (Li, Na, K, Cl, MG, Rb, Cs, F, Br, I) from your
ffnonbonded.itpfile. The CUFIX package uses updated ion parameters from the Cheatham group. - Simulate: Run your simulation, ensuring you use the compatible TIP3P water model (
tip3p.itp).
Protocol 2: Deriving Force Field Parameters for Unnatural Amino Acids
This workflow, adapted from Wang and Li, describes parametrizing an UAA compatible with AMBER ff14SB [47].
Key Reference Data for Parametrization
Table 1: Target Data for UAA Parameter Validation
| System Type | Target Benchmark | Acceptable Threshold |
|---|---|---|
| Dipeptide (α- and β-backbone) | QM Relative Energies (MP2/cc-pVTZ) | Reproduced within force field error |
| Optimized Dipeptide Structure | Root-Mean-Square Deviation (RMSD) from QM structure | ~0.1 Å [47] |
| Protein with incorporated UAA | RMSD from crystal structure | ~1.0 Å [47] |
Protocol 3: A Bayesian Framework for Robust Parameter Learning
For the highest accuracy, consider a modern data-driven approach. This method learns parameters directly from ab initio molecular dynamics (AIMD) data, which inherently includes environmental effects like electronic polarization [49].
- Define Molecular Fragments: Break down the system of interest into smaller, chemically meaningful fragments.
- Generate Reference Data: Run AIMD simulations of each fragment in explicit solvent to obtain reference structural data (e.g., radial distribution functions, RDFs).
- Learn the Posterior: Use a Bayesian inference framework with Markov chain Monte Carlo (MCMC) sampling to learn the posterior distribution of force field parameters (like partial charges) that best reproduce the AIMD data.
- Validate Transferability: Test the resulting parameters on properties not included in the training, such as the density of aqueous solutions at high concentration [49].
Table 2: Performance of a Bayesian-Learned Force Field on Biomolecular Fragments
| Quantity of Interest (QoI) | Typical Error vs. AIMD | Improvement over CHARMM36 |
|---|---|---|
| Hydration Structure (RDFs) | < 5% for most species | Systematic improvement, especially for anions [49] |
| Hydrogen Bond Counts | < 10-20% deviation | Consistent improvement [49] |
| Ion-Pair Distance Distributions | < 20% deviation | Notable improvement for charged species [49] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Correcting Force Field Imbalances
| Resource / Reagent | Function / Purpose | Key Features / Notes |
|---|---|---|
| CUFIX/NBFIX Parameters [27] | Corrects over-attraction in amine-carboxylate, amine-phosphate, and carbon-carbon interactions. | Optimized for CHARMM36/27/22 and AMBER ff99/ff14; uses TIP3P water. |
| DES-Amber Force Field [48] | Integrated force field for proteins and nucleic acids with improved nonbonded and torsion parameters. | Designed for accuracy in single-/double-stranded DNA/RNA and compatibility with DES-Amber proteins. |
| RESP Charge Model [47] [50] | Derives partial atomic charges from QM electrostatic potentials. | Higher accuracy for solvation free energies; preferred over AM1-BCC for GAFF/GAFF2. |
| Bayesian Force Field Framework [49] | Learns parameters and their uncertainties directly from AIMD data. | Provides confidence intervals; naturally incorporates solvation effects. |
| Artificial Chaperone Assays [51] | Experimental method to assess and assist protein refolding, benchmarking against aggregation. | Used to validate simulations by comparing refolding yields and pathways. |
Frequently Asked Questions (FAQs)
Q1: What are 1-4 interactions, and why are they a "challenge" in force field development? 1-4 interactions are the non-bonded interactions (electrostatic and van der Waals) between atoms separated by exactly three covalent bonds. The challenge arises because the standard practice of applying arbitrary scaling factors to these interactions often creates an imbalance. This imbalance can lead to inaccurate molecular geometries, conformational energies, and thermodynamic properties, ultimately reducing the predictive power of simulations for drug design [7].
Q2: My MD simulations show overly compact denatured proteins or artificial aggregation. Could this be linked to 1-4 parameters? Yes, this is a recognized symptom. Commonly used biomolecular force fields (like AMBER and CHARMM) can be out of balance, sometimes overestimating attractive interactions between charged and hydrophobic groups. This promotes unnatural aggregation in simulations of proteins, nucleic acids, and lipid systems. Improving the description of 1-4 and other non-bonded interactions is a key step to resolving this [7].
Q3: What are the main alternatives to using scaled potentials for 1-4 interactions? The primary alternative is a surgical approach that directly optimizes the underlying Lennard-Jones parameters for specific atom pairs. This is often implemented via corrections such as:
- NBFIX (Non-Bonded FIX): Overrides the standard combination rules for specific atom pairs, calibrating them against experimental data like osmotic pressure [7].
- Machine Learning (ML)-driven optimization: Uses ML surrogate models or evolutionary algorithms to efficiently navigate high-dimensional parameter spaces and optimize Lennard-Jones parameters against quantum chemical or experimental target data [52] [53].
Q4: I am optimizing force field parameters and the process is extremely slow. How can I improve efficiency? A highly effective modern approach is to substitute the most time-consuming molecular dynamics calculations within the optimization loop with a machine learning surrogate model. Research has demonstrated that this can reduce the required optimization time by a factor of approximately 20 while still producing force fields of similar quality [52].
Q5: During geometry optimization with a reactive force field (ReaxFF), I encounter convergence issues due to discontinuities. What can I do? Discontinuities in the energy derivative can often be traced to the bond order cutoff. To improve stability, you can:
- Switch to the 2013 formula for torsion angles (set
Engine ReaxFF%Torsionsto2013) for smoother behavior at lower bond orders. - Decrease the bond order cutoff (
Engine ReaxFF%BondOrderCutoff), which reduces the discontinuity's magnitude at a minor computational cost. - Use tapered bond orders (
Engine ReaxFF%TaperBO) as proposed by Furman and Wales [54].
Troubleshooting Guides
Issue 1: Artificial Aggregation in Biomolecular Simulations
Problem: Your simulation of multiple proteins, nucleic acids, or lipids shows unrealistic clumping or aggregation that is not observed experimentally.
Diagnosis: This is a classic sign of an imbalance in the force field, where attractive non-bonded interactions (including 1-4 interactions) are overestimated [7].
Solution Pathway:
- Step 1: Implement NBFIX Corrections. Adopt a force field that includes NBFIX corrections, which are surgically calibrated to specific atom pairs (e.g., ion-phosphate, amine-carboxylate) to reproduce experimental data like solution osmotic pressure [7].
- Step 2: Validate System Properties. After applying NBFIX, ensure the corrections do not adversely affect other properties. A well-designed NBFIX correction should not degrade the accuracy of hydration free energies, solution densities, or backbone torsion profiles [7].
Issue 2: Inefficient Force Field Parameter Optimization
Problem: The computational cost of iteratively running MD simulations to optimize new parameters (like 1-4 LJ terms) is prohibitively high.
Diagnosis: Traditional parameter optimization that relies on full MD simulations at every step is inherently slow and limits exploration of the parameter space.
Solution Pathway:
- Step 1: Employ a Machine Learning Surrogate Model. Replace the inner-loop MD calculations with a trained ML model that predicts the key target properties (e.g., density, conformational energies) from the parameter set. This dramatically speeds up the optimization [52].
- Step 2: Use Advanced Optimization Algorithms. Leverage sophisticated frameworks like the Alexandria Chemistry Toolkit (ACT), which uses genetic algorithms and Monte Carlo methods for "force field science," or hybrid methods like simulated annealing combined with particle swarm optimization (PSO) for ReaxFF parameterization [53] [55].
The diagram below illustrates a modern, ML-accelerated workflow for force field parameter optimization.
Table 1: Key Experimental Data for Validating 1-4 and Non-Bonded Interactions
| Experimental Property | Role in Force Field Validation | Reference in Methodology |
|---|---|---|
| Osmotic Pressure | Used to calibrate ion-ion and ion-solute LJ parameters (NBFIX) via MD simulation in a two-compartment setup to prevent artificial aggregation [7]. | Yoo & Aksimentiev, 2012/2016 [7] |
| Quantum Chemical Energies | SAPT (Symmetry-Adapted Perturbation Theory) energy components serve as targets for training intermolecular force field parameters from scratch, ensuring a physically grounded balance [53]. | van der Spoel et al., 2025 [53] |
| Liquid Density & Enthalpy of Vaporization | Critical condensed-phase properties used for final validation of a newly parameterized force field, testing its performance beyond gas-phase training data [53]. | van der Spoel et al., 2025 [53] |
| Relative Conformational Energies | Target property for optimizing LJ parameters (e.g., for carbon and hydrogen) to ensure the force field correctly reproduces different molecular conformations [52]. | Strickstrock et al., 2025 [52] |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Algorithms for Force Field Correction
| Tool / Algorithm | Function | Application Context |
|---|---|---|
| NBFIX (CHARMM) / LJEDIT (AMBER) | Overrides the standard Lorentz-Berthelot combination rules to apply custom LJ parameters for specific atom pairs, correcting systematic attraction errors [7]. | Correcting artificial aggregation in biomolecular simulations; calibrating ion parameters. |
| Alexandria Chemistry Toolkit (ACT) | An open-source software for machine learning of physics-based force fields using genetic algorithms and Monte Carlo methods to optimize parameters against large quantum chemical datasets [53]. | Developing new force fields or refining existing parameters with high transferability. |
| Machine Learning Surrogate Model | A neural network or other ML model trained to predict target properties from parameters, substituting for slow MD simulations during optimization [52]. | Drastically speeding up (by ~20x) force field parameter optimization workflows. |
| Hybrid SA/PSO with CAM | A multi-objective optimizer combining Simulated Annealing (SA) and Particle Swarm Optimization (PSO), with a Concentrated Attention Mechanism (CAM) that weights key data more heavily [55]. | Efficient and accurate optimization of complex force fields like ReaxFF. |
| P-LINCS | A parallel constraint algorithm essential for MD simulations with domain decomposition, allowing constraints to cross the boundaries between computational domains [56]. | Enabling stable, large-scale simulations on parallel computing architectures. |
Experimental Protocols
Protocol 1: Calibrating NBFIX Corrections Using Osmotic Pressure
This protocol is based on methodologies established in [7].
1. Objective: To surgically correct overestimated attractive interactions between specific atom pairs (e.g., Na⁺-Cl⁻, amine-COO⁻) by calibrating their Lennard-Jones parameters to reproduce experimental osmotic pressure.
2. Materials and Software:
- MD Simulation Engine: Software supporting NBFIX corrections (e.g., CHARMM, AMBER, GROMACS).
- Force Field: A base biomolecular force field (e.g., CHARMM36, AMBER).
- System Setup: A two-compartment simulation box separated by a semi-permeable membrane.
3. Procedure:
- Step 1: Obtain Reference Data. Acquire experimental osmotic pressure data for binary solutions of the molecules/ions of interest (e.g., NaCl, amino acids) [7].
- Step 2: Simulate Osmotic Pressure. Set up an MD simulation with the two-compartment geometry. One compartment contains the solute at a specific concentration, while the other contains pure solvent. The force exerted on the membrane is related to the osmotic pressure.
- Step 3: Iterative Calibration. Run simulations and adjust the LJ parameters (σ and ε) for the target atom pair (e.g., Na⁺-Cl⁻) until the simulated osmotic pressure matches the experimental data across a range of concentrations.
- Step 4: Validation. Incorporate the new NBFIX parameters and validate them on systems not included in the training, such as the stability of DNA Holliday junctions or the correct folding of proteins, to ensure no side effects were introduced.
Protocol 2: ML-Accelerated Optimization of Lennard-Jones Parameters
This protocol synthesizes approaches from [52] and [53].
1. Objective: To efficiently optimize the Lennard-Jones parameters for carbon and hydrogen atoms to reproduce target properties like n-octane's relative conformational energies and bulk-phase density.
2. Materials and Software:
- Optimization Framework: Custom scripting or toolkit like the Alexandria Chemistry Toolkit (ACT) [53].
- ML Library: Libraries for building neural networks (e.g., TensorFlow, PyTorch).
- Target Data: Quantum chemical calculations (for conformational energies) and experimental data (for liquid density).
3. Procedure:
- Step 1: Generate Training Data. Perform a sampling of the parameter space. For selected parameter sets, run full MD simulations to compute the target properties (density, energies). This creates a dataset of
{parameters -> properties}. - Step 2: Train the Surrogate Model. Train a machine learning model (e.g., a neural network) on the generated dataset. The model learns to instantly predict the target properties from any given parameter set, bypassing the need for MD.
- Step 3: Perform Optimization. Run the chosen optimization algorithm (e.g., genetic algorithm, particle swarm optimization). In the inner loop, the surrogate model—not an MD simulation—is used to evaluate the fitness of parameter sets.
- Step 4: Final Validation. Once the optimization converges, run a full MD simulation using the final optimized parameters to confirm they accurately reproduce the target properties.
Frequently Asked Questions
Q1: What are the most common pitfalls when assigning atom types for biomolecular force fields? A common and critical pitfall is the inconsistent treatment of 1-4 interactions—interactions between atoms separated by three bonds. Traditional force fields often use a hybrid approach that combines bonded torsional terms with empirically scaled non-bonded interactions. This can lead to inaccurate forces and geometries, creates interdependence between dihedral and non-bonded terms, and complicates parameterization, reducing its transferability [9]. Another frequent issue is the failure to account for chemical symmetry. Atoms that are chemically equivalent (e.g., the two oxygen atoms in a carboxyl group) must be assigned identical force field parameters; otherwise, the energy surface will be incorrectly described [12].
Q2: How can I identify if my simulation errors are caused by poor atom typing or parameter assignment? Errors often manifest in several ways. Key indicators include:
- Erroneous geometries and an inability to reproduce quantum mechanical (QM) reference structures, even for small molecules or dipeptides [9] [57].
- Inaccurate torsional energy profiles, which directly impact conformational sampling [9] [12].
- Poor reproduction of relative energies between different backbone conformers (e.g., α-helix vs. β-strand) when compared to benchmark QM data [57].
- SHAKE algorithm convergence failures can sometimes indicate underlying force field issues, such as atomic clashes from problematic initial structures or parameters [58].
Q3: My research involves unnatural amino acids (UAAs). What is the best practice for deriving missing parameters? A robust protocol for deriving parameters for UAAs or other novel molecules involves a multi-step QM-driven process [57]:
- Model System Construction: Construct dipeptides of the UAA in both α- and β-backbone conformations, blocked with N-methyl and acetyl groups (Ace-XXX-NMe).
- QM Optimization and Calculation: Perform structural optimization at a level like B3LYP/6-31G*, followed by single-point energy calculations at a higher level (e.g., MP2/cc-pVTZ).
- Electrostatic Potential (ESP) Fitting: Derive RESP charges by evaluating the ESP at, for instance, the HF/6-31G* level.
- Parameter Assignment and Validation: Generate initial bonded and non-bonded parameters using a tool like GAFF. Crucially, test and refine these parameters by ensuring they reproduce the QM relative energies and optimized structures of the dipeptide conformers.
Q4: Are machine-learned force fields a solution to parameter assignment challenges? Machine-learned force fields (MLFFs) offer a promising, data-driven alternative. They can predict parameters across a broad chemical space, potentially overcoming the limitations of look-up table approaches. However, they require extensive, high-quality QM datasets for training and come with their own set of best practices. For instance, when training an MLFF, it is sometimes necessary to treat atoms of the same element in different chemical environments as separate species to improve accuracy, though this increases computational cost [29]. While MLFFs like ByteFF show excellent performance in predicting geometries and torsional profiles, their computational efficiency is still lower than conventional molecular mechanics force fields [12].
Q5: What tools and resources can help automate and validate parameter assignment? Several modern toolkits facilitate systematic parameterization:
- Q-Force: An automated framework for force field parameterization that can systematically derive bonded coupling terms, essential for a bonded-only treatment of 1-4 interactions [9].
- BioExcel Building Blocks (BioBB): A library of interoperable Python wrappers that can help build and orchestrate biomolecular simulation workflows, including those involving parameterization [59].
- Data-Driven Platforms: Tools like ByteFF use graph neural networks (GNNs) trained on massive QM datasets to predict force field parameters for drug-like molecules, ensuring coverage of an expansive chemical space [12].
Troubleshooting Guides
Issue 1: Inconsistent Torsional Energy Barriers and Molecular Geometries
Problem: Simulations fail to replicate QM energy barriers or stable conformations, often due to the flawed hybrid treatment of 1-4 interactions.
Diagnosis and Solution: Consider moving to a bonded-only model for 1-4 interactions. This approach eliminates the need for arbitrarily scaled non-bonded interactions altogether.
- Theory: The method uses bonded coupling terms (like torsion-bond and torsion-angle couplings) to accurately capture the 1-4 energy surface. This decouples the parameterization of torsional and non-bonded terms, simplifying development and improving transferability [9].
- Protocol: Implement this using an automated parameterization toolkit like Q-Force [9]:
- Generate QM Reference Data: Perform ab initio calculations on target molecules to create a reference potential energy surface (PES).
- Implement Coupling Terms: Extend the force field to include bond-angle and torsion-bond coupling terms.
- Automated Parameter Fitting: Use the toolkit to efficiently determine the necessary coupling terms against the QM data without manual adjustment.
- Validation: Validate the new parameters by comparing the force field PES against QM data for small molecules and key systems like alanine dipeptide in gas and implicit solvent.
The workflow for this methodology is outlined below:
Issue 2: Missing Parameters for Novel Molecules (e.g., Unnatural Amino Acids)
Problem: Standard force fields lack parameters for non-canonical residues or drug-like molecules, halting simulations.
Diagnosis and Solution: Follow a rigorous, QM-validated parameter derivation workflow, as summarized in the table below [57]:
Table 1: Key Research Reagent Solutions for Parameter Derivation
| Item/Reagent | Function in Parameterization |
|---|---|
| Ace-XXX-NMe Dipeptide | Model system representing the novel molecule (XXX) in a protein-like environment for QM calculations. |
| Quantum Mechanics (QM) Software (e.g., Gaussian) | Performs molecular optimization and single-point energy calculations to generate benchmark data. |
| RESP Charge Fitting | Derives electrostatic potential (ESP)-fitted partial charges for the molecule, crucial for non-bonded interactions. |
| General Amber Force Field (GAFF) | Provides a baseline set of bonded and non-bonded parameters for organic, drug-like molecules. |
| Validation Suite (MD, MM/PBSA) | Tests the new parameters in realistic environments like protein-ligand complexes to validate against experimental data. |
The detailed workflow for parameter development is as follows:
Issue 3: Force Field Errors Propagating from Incorrect Local Environments
Problem: Force field inaccuracies arise because parameters assigned from look-up tables fail to capture specific local chemical environments.
Diagnosis and Solution: Adopt a data-driven, chemically aware parameterization strategy.
- Modern Approach: Use a graph neural network (GNN) model trained on a large, diverse dataset of molecular fragments. This model predicts parameters based on the local molecular graph, ensuring that chemically equivalent atoms receive identical parameters [12].
- Protocol for Utilizing Data-Driven Force Fields:
- Input Preparation: Provide a standardized representation of your molecule (e.g., SMILES string or 3D structure).
- Parameter Prediction: The GNN model (e.g., ByteFF, Espaloma) predicts all bonded and non-bonded parameters simultaneously, respecting permutational and chemical symmetry.
- Focus on Torsional Accuracy: Ensure the chosen method prioritizes accurate torsion profiles, as these are critical for conformational sampling in drug discovery [12].
Table 2: Comparison of Traditional vs. Data-Driven Parameter Assignment
| Feature | Traditional Look-up Table | Data-Driven GNN Approach |
|---|---|---|
| Basis | Pre-defined, discrete chemical environments (SMIRKS) | Continuous, learned representation of molecular graph |
| Transferability | Limited by the pre-determined list of types | High, generalizes to new chemical space within training domain |
| Symmetry Handling | Manual and error-prone | Automatic and inherent (permutationally invariant) |
| Coverage | Requires constant expansion for new molecules | Broad coverage from initial training on large, diverse datasets |
| Primary Challenge | Scalability and manual curation | Requires massive, high-quality QM dataset for training |
In biomolecular force field research, the accurate parameterization of non-bonded interactions is paramount for reliable molecular dynamics (MD) simulations. Atomic partial charges are among the most critical parameters, directly influencing electrostatic interactions that govern molecular recognition, binding affinities, and solvation properties. Within the AMBER ecosystem, two predominant methods have emerged for assigning these charges: the Restrained Electrostatic Potential (RESP) method and the Austin Model 1 with Bond Charge Correction (AM1-BCC) approach. While both aim to generate chemically reasonable charge distributions, they are founded on different philosophical and technical principles, making them non-interchangeable without potential consequences for simulation outcomes.
The integrity of empirical force fields depends on maintaining consistency between the parameterization methodology and its application. Using charge models interchangeably can introduce systematic errors that compromise the accuracy of physical property predictions, particularly in structure-based drug design where precise binding affinity calculations are essential [50]. This technical guide examines the fundamental differences between these charge derivation methods, provides protocols for their proper application, and offers troubleshooting advice for researchers navigating charge assignment challenges in biomolecular simulations.
Key Differences Between RESP and AM1-BCC Methods
Fundamental Philosophical and Technical Divergences
RESP and AM1-BCC embody different approaches to balancing computational efficiency with electrostatic accuracy. RESP performs a fitting procedure to reproduce the quantum mechanical (QM) electrostatic potential (ESP) calculated at the Hartree-Fock (HF) level with the 6-31G* basis set [60]. This method captures molecular specificity through explicit QM calculations but at significantly greater computational expense, especially for larger molecules [61]. In contrast, AM1-BCC utilizes a semi-empirical approach that applies parameterized bond charge corrections (BCCs) to AM1 Mulliken charges to approximate HF/6-31G* ESP charges without performing ab initio calculations [62].
A crucial philosophical difference lies in their treatment of molecular context. RESP charges are explicitly derived for each molecular conformation, potentially capturing context-specific electronic effects, while AM1-BCC applies transferable parameters based on local bonding environments [60]. This distinction becomes particularly important for molecules with significant conformational flexibility or unusual bonding arrangements not well-represented in the AM1-BCC training set.
Table 1: Fundamental Comparison of RESP and AM1-BCC Charge Methods
| Feature | RESP | AM1-BCC |
|---|---|---|
| QM Method | HF/6-31G* (typically) | Semi-empirical AM1 |
| Computational Cost | High (hours to days) | Low (seconds to minutes) |
| Molecular Specificity | High (conformation-dependent) | Medium (transferable parameters) |
| Backbone Restraints | Supported [60] | Not supported [60] |
| Primary Application | Force field development [63] | High-throughput ligand parameterization |
Performance Comparison and Limitations
Extensive validation studies reveal that both methods can produce chemically reasonable charge distributions, but with important distinctions in their performance characteristics. Research indicates that RESP and AM1-BCC charges typically agree on the overall charge distribution pattern, though individual atoms may show significant deviations [60]. For instance, a study on natural amino acids found that both methods reproduced literature values with sufficient accuracy, though AM1-BCC could not restrain backbone charges to predetermined values—a standard practice in biomolecular force field development [60].
The performance gap becomes more pronounced in specific chemical contexts. A comparison on isoprene demonstrated significant charge deviations, particularly for carbons engaged in double bonds, with differences exceeding 0.3 charge units for some atoms [61]. For larger molecules such as peptides exceeding 100 atoms, users have reported "too much charge deviation" between the methods, with individual atomic differences ranging from ±0.5 to 1.0 charge units [61].
Notably, the AM1-BCC method has demonstrated systematic errors exceeding 1.0 kcal/mol in solvation free energy calculations for specific functional groups when used with the GAFF2 force field, which was originally parameterized using RESP charges [50]. This highlights the importance of charge model compatibility with the underlying force field parameterization.
Experimental Protocols and Validation Methodologies
RESP Charge Derivation Protocol
The standard RESP protocol involves multiple steps to ensure charge quality and conformational robustness:
Conformational Sampling: Generate multiple conformations for the target molecule, differing in backbone dihedrals (φ, ψ) and side-chain rotamers (χ1, χ2). For natural amino acids, literature protocols use statistical analysis of crystallized proteins to identify relevant conformations [60].
Quantum Mechanical Calculations:
ESP Fitting with Restraints:
- Implement two-stage RESP fitting with hyperbolic restraint on heavy atoms
- Apply symmetry constraints to chemically equivalent atoms
- Restrain backbone atoms (N, H, C, O) to predetermined values when extending existing force fields [60]
Validation: Compare derived charges with reference values and assess molecular dipole moment.
Diagram 1: RESP Charge Derivation Workflow
AM1-BCC Charge Derivation Protocol
The AM1-BCC method offers a significantly streamlined approach:
Input Preparation: Prepare a single reasonable molecular geometry (typically energy-minimized)
Charge Calculation:
- Execute AM1 calculation to obtain Mulliken charges
- Apply bond charge corrections (BCCs) from parameterized library
- No capacity for restraining backbone atoms to predetermined values [60]
Validation: Assess charge reasonableness through visual inspection and comparison with similar chemical motifs.
Advanced implementations may incorporate the newer ABCG2 parameter set, specifically optimized for GAFF2, which significantly reduces errors in hydration free energy predictions (MUE reduced from 1.03 kcal/mol to 0.37 kcal/mol) [63].
Next-Generation Methods: RESP2
Recent advances have led to the development of RESP2, which addresses a key limitation of traditional RESP by incorporating both gas-phase and aqueous-phase ESPs in charge determination [38]. This approach uses a mixing parameter δ (typically 0.6, representing 60% aqueous and 40% gas-phase) to better represent the polarized state of molecules in condensed phases [38]. RESP2 has demonstrated improved accuracy in predicting liquid properties and hydration free energies when combined with re-optimized Lennard-Jones parameters [38].
Table 2: Quantitative Performance Comparison of Charge Methods
| Charge Method | HFE MUE (kcal/mol) | Computational Time | Recommended Use |
|---|---|---|---|
| RESP (HF/6-31G*) | ~1.0 [50] | Hours to days | Force field development; publication-quality simulations |
| AM1-BCC (Original) | ~1.03 [63] | Seconds to minutes | High-throughput screening; initial studies |
| AM1-BCC (ABCG2) | 0.37 [63] | Seconds to minutes | GAFF2-compatible production simulations |
| RESP2 (δ=0.6) | Improved vs RESP [38] | Hours to days | Highest accuracy for condensed phase |
Troubleshooting Common Charge Assignment Issues
Frequently Asked Questions
Q1: I've noticed significant charge differences between RESP and AM1-BCC for my molecule, particularly for buried atoms. Which should I trust?
This is a common observation, especially for carbon atoms in hydrophobic regions. RESP charges are known to exhibit numerical instabilities for buried atoms, where charges can vary widely with minimal effect on the ESP fit RMSD [62]. In such cases, AM1-BCC may provide more chemically reasonable values due to its parameterized approach. However, for functional groups directly involved in molecular recognition, RESP charges derived from multiple conformations typically provide superior performance [60]. We recommend comparing both charge sets against experimental data (e.g., solvation free energies, dipole moments) when possible.
Q2: Can I use AM1-BCC charges with force fields parameterized using RESP?
While AM1-BCC was designed to approximate RESP charges, they are not equivalent. Using AM1-BCC charges with force fields like GAFF2 (which was parameterized with RESP charges) can introduce systematic errors, particularly for specific functional groups [50]. For the most accurate results with GAFF2, either use RESP charges or the newer ABCG2 parameter set specifically optimized for GAFF2 [63].
Q3: Why do I get different charges when I use different conformations with RESP?
Charge distributions exhibit conformational dependence due to changes in molecular polarization, steric effects, and intramolecular interactions [60]. This is not a methodological flaw but rather a reflection of physical reality. The standard protocol addresses this by using multiple conformations to derive robust, conformationally-averaged charges [60]. If using a single conformation, select one representative of the molecular state in your system of interest.
Q4: How should I handle charged groups like ammonium (-NH3+) where AM1-BCC and RESP differ significantly?
For charged groups, RESP typically provides more accurate charge distributions as it explicitly captures the polarization response of the entire molecule to the formal charge. The significant differences observed for ammonium groups [64] reflect the limitations of transferable corrections in capturing context-dependent polarization. For critical applications involving charged species, we recommend RESP charges derived using multiple conformations.
Diagnostic Guide: Charge Assignment Problems and Solutions
Table 3: Troubleshooting Guide for Charge Assignment Issues
| Problem | Possible Causes | Solutions |
|---|---|---|
| Large charge fluctuations between similar molecules | Inconsistent conformation selection; RESP fitting instability | Use multiple conformations; apply stronger RESP restraints; verify chemical equivalence |
| Systematic errors in hydration free energy | Charge model/force field incompatibility; inadequate parameterization | Use ABCG2 with GAFF2; validate with experimental data; consider RESP2 |
| Unphysical charges on buried atoms | RESP fitting instability; inadequate ESP sampling | Use AM1-BCC; apply distance-dependent RESP restraints; inspect ESP fit quality |
| Charges incompatible with existing force field | Different charge derivation protocol; missing backbone restraints | Derive new charges with proper restraints; maintain force field integrity |
Research Reagent Solutions: Essential Tools for Charge Derivation
Table 4: Essential Software Tools for Charge Derivation
| Tool Name | Function | Compatibility |
|---|---|---|
| ANTECHAMBER | Automated parameterization including AM1-BCC | AMBER, GAFF/GAFF2 |
| RESP | Two-stage RESP fitting with restraints | AMBER force fields |
| PyRESP | Python implementation of RESP | AMBER with additional flexibility |
| Red Server | Web-based multi-conformation RESP | Various force fields |
| NWChem | QM calculations for ESP generation | Multi-platform |
| Gaussian | QM calculations for geometry optimization and ESP | Professional license |
| Psi4 | Open-source QM calculations for RESP | RESP2 development |
Based on the documented differences between RESP and AM1-BCC charge models, we recommend the following strategic approach:
For force field development and publication-quality simulations where accuracy is paramount, use the RESP method with multiple conformations and proper restraints to maintain compatibility with existing biomolecular force fields.
For high-throughput screening and initial investigations where computational efficiency is prioritized, use AM1-BCC with the ABCG2 parameter set specifically optimized for your target force field (e.g., GAFF2).
For the highest accuracy in condensed-phase simulations, consider the RESP2 method with optimized δ parameter (approximately 0.6), which systematically balances gas-phase and aqueous-phase polarization effects.
Always maintain consistency between the charge derivation method and the original force field parameterization philosophy to preserve force field integrity. Document your charge derivation methodology thoroughly to enable reproducibility and proper interpretation of simulation results.
Benchmarking and Validation Frameworks for Robust Force Field Assessment
For decades, the root mean square deviation (RMSD) has been a cornerstone metric for validating biomolecular force fields in molecular dynamics (MD) simulations. While useful for measuring average structural drift, RMSD alone provides an incomplete picture, often failing to capture critical failures like artificial aggregation, distorted energy landscapes, and unstable trajectories. This technical support center provides troubleshooting guides and FAQs to help researchers identify and correct these issues, with a special focus on the challenge of bonded parameter imbalance that can undermine simulation accuracy.
FAQs: Addressing Common Force Field Challenges
Q1: My simulations of denatured proteins or multi-component systems show unrealistic aggregation. What is the likely cause and how can I fix it?
A: A common cause is overestimation of attractive interactions between charged and hydrophobic groups in standard force fields [7]. This is often a non-bonded parameter issue. To address it:
- Apply NBFIX Corrections: Utilize the NBFIX (Non-Bonded FIX) method to surgically calibrate pair-specific Lennard-Jones parameters against experimental data like osmotic pressure [7]. This corrects overly attractive interactions without destabilizing other force field components.
- Validate with Osmotic Pressure: Set up a two-compartment MD simulation to compute osmotic pressure and calibrate your force field parameters until they match experimental measurements for relevant solutions (e.g., electrolytes, amino acids) [7].
Q2: My force field has excellent RMSD on a folded protein but fails to reproduce correct unfolded state ensembles or conformational dynamics. What is wrong?
A: This indicates a potential imbalance in your force field's energy function. Many force fields are calibrated against folded structures and may be out of balance for denatured or disordered states [7] [65]. The problem may lie in an incorrect balance between bonded and non-bonded terms.
- Multi-State Validation: Do not validate only on a single, static structure. Use a multi-metric approach that includes data from multiple conformational states.
- Check Radius of Gyration: Compare the radius of gyration (Rg) of simulated disordered states against experimental data from techniques like SAXS. Overly compact denatured states are a known symptom of force field imbalance [7].
- Incorporate Ensemble Data: Leverage databases of dynamic conformations (e.g., ATLAS, GPCRmd) for validation against a broader conformational landscape [65].
Q3: My machine-learning force field (MLFF) achieves low force mean absolute error (MAE) during training, but the MD simulation becomes unstable and diverges. Why?
A: A low force MAE does not guarantee simulation stability [66]. This often occurs when the model encounters atomic configurations outside its training domain, leading to unphysical force predictions.
- Prioritize Pre-Training: Pre-train your MLFF on large, diverse datasets (e.g., the OC20 dataset) before fine-tuning on your specific system. This significantly improves generalizability and simulation stability [66].
- Monitor Stability Metrics: Track simulation longevity and structural fidelity. A stable model should sustain long trajectories without catastrophic bond breakage or unrealistic configurations [66].
- Validate Structurally: Use the pair-distance distribution function, ( h(r) ), to check whether the MLFF preserves the correct molecular structure throughout the simulation [66].
Troubleshooting Guides
Guide 1: Diagnosing and Correcting Artificial Aggregation
Symptoms: Unrealistic clustering of proteins, nucleic acids, or lipids in simulation; failure to reproduce experimental scattering data for multi-component systems.
Diagnostic Table:
| Metric to Measure | What it Diagnoses | Expected Outcome (Non-Aggregating System) |
|---|---|---|
| Inter-molecular distance distribution | Strength of solute-solute attraction | A distribution matching experimental references or theory. |
| Osmotic pressure of a binary solution | Balance of solute-solute vs. solute-solvent interactions | Computed osmotic pressure matches experimental data [7]. |
| Radial distribution function (RGF) between solute molecules | Short-range ordering and clustering | No unnatural, sharp peaks at short distances. |
Experimental Protocol: Osmotic Pressure Calibration for NBFIX
- System Setup: Create a simulation box with two compartments separated by a semi-permeable membrane. One compartment contains a solution (e.g., NaCl), and the other contains pure solvent [7].
- Run Simulation: Perform an MD simulation and measure the pressure difference (the osmotic pressure, ( \pi )) between the two compartments.
- Compare to Experiment: Compare the simulated ( \pi ) to known experimental values for your solution at the same concentration [7].
- Iterate and Correct: Adjust the Lennard-Jones parameters for specific atom pairs (e.g., Na-Cl) using the NBFIX procedure in your MD package (e.g.,
NBFIXin CHARMM,LJEDITin AMBER). - Re-run and Validate: Repeat the osmotic pressure simulation until the results agree with experiment. This calibrated force field should now show reduced artificial aggregation in your biomolecular simulations.
Logical Workflow for Diagnosing Aggregation
Guide 2: Validating the Balance of Bonded Parameters
Symptoms: Incorrect protein secondary structure propensities; distorted backbone dihedral distributions; inability to fold proteins or reproduce known conformational transitions.
Diagnostic Table:
| Metric to Measure | What it Diagnoses | Expected Outcome (Balanced Force Field) |
|---|---|---|
| Backbone dihedral angle (Φ/Ψ) distribution | Accuracy of torsional potentials | Free energy landscape matches high-quality quantum mechanics/data-derived maps. |
| J-coupling constants from NMR | Local backbone conformation | Computed J-couplings from simulation match experimental NMR data [7]. |
| Chemical shift predictions | Local electronic environment | Predicted shifts (e.g., from SHIFTX2) correlate with experimental NMR shifts. |
Experimental Protocol: Benchmarking on Multiple Conformational States
- Select Benchmark Set: Choose a set of proteins or peptides with known and diverse conformational states (e.g., folded, unfolded, intrinsically disordered regions, multiple conformations from PDBFlex [65]).
- Run Ensemble Simulations: Perform simulations starting from different conformations (folded and unfolded) to sample the conformational landscape.
- Compute Multi-Metric Profile: For each state, calculate a suite of metrics: RMSD (for folded state), Rg (for unfolded state), dihedral distributions, and NMR observables.
- Compare to Ensemble Data: Validate your results not against a single structure, but against conformational ensembles from databases like CoDNaS 2.0 or MD databases like ATLAS [65]. A balanced force field should reproduce the distribution of states.
Guide 3: Ensuring MLFF Stability and Transferability
Symptoms: Simulation "blows up" (energy/forces become extreme); unphysical bond breaking; unrealistic geometries despite good training metrics.
Diagnostic Table:
| Metric to Measure | What it Diagnoses | Expected Outcome (Stable MLFF) |
|---|---|---|
| Simulation longevity (ps before failure) | General stability and extrapolation behavior | Sustains nanosecond-to-microsecond trajectories without failure [66]. |
| Pair-distance distribution function, ( h(r) ) | Structural fidelity over time | Distribution matches the reference method (e.g., DFT) throughout the trajectory [66]. |
| Energy/Force distribution over time | Presence of non-physical outliers | Forces and energies remain within a physically plausible range. |
Experimental Protocol: Stress-Testing an MLFF with Extended MD
- Train/Finetune: Train your MLFF from scratch or, preferably, fine-tune a pre-trained model (e.g., GemNet-T pre-trained on OC20) on your target dataset (e.g., MD17) [66].
- Run Long Simulations: Initiate multiple, independent MD simulations at the target temperature (e.g., 500 K) using a velocity Verlet integrator and a thermostat (e.g., Nosé–Hoover) [66].
- Monitor for Instability: Run for hundreds of picoseconds, monitoring for catastrophic failures like extreme atomic displacements or energy drift.
- Analyze Structural Integrity: Compute the pair-distance distribution function ( h(r) ) from the trajectory and compare it to the expected distribution. A stable MLFF will preserve the molecule's fundamental structure [66].
MLFF Stability Validation Pathway
Table 1: Key Computational Tools for Force Field Validation
| Tool / Resource | Function | Relevance to Validation |
|---|---|---|
| CHARMM [7], AMBER [7], GROMACS [65], OpenMM [65] | MD Simulation Engines | The core platforms for running simulations and implementing parameter corrections (e.g., NBFIX). |
| NBFIX / LJEDIT [7] | Non-Bonded Parameter Correction | Directly corrects overestimated attractive interactions between specific atom pairs. |
| OC20 Dataset [66] | Large-Scale Pre-training Data | A diverse dataset of catalysts for pre-training MLFFs to improve stability and generalizability. |
| ATLAS [65], GPCRmd [65] | Protein Dynamics Databases | Provide reference MD trajectories and conformational ensembles for validating dynamic behavior. |
| CoDNaS 2.0 [65], PDBFlex [65] | Conformational Ensemble Databases | Collections of alternative protein conformations from the PDB, useful for multi-state validation. |
| Pair-Distance Distribution ( h(r) ) [66] | Structural Metric for MLFFs | Validates the structural integrity of molecules during simulations with MLFFs, especially for non-periodic systems. |
Molecular dynamics (MD) simulations are an indispensable tool in academic and industrial research, providing atomic-level insight into biomolecular processes. The accuracy of these simulations is fundamentally dependent on the empirical force fields used to describe interatomic interactions. Among the most widely used are AMBER, CHARMM, and GROMOS, which have been refined over decades but employ different parametrization strategies and exhibit distinct performance characteristics. This technical resource is framed within a broader thesis on correcting bonded parameter imbalances in biomolecular force fields, where the interplay between torsion potentials, angle terms, and charge models is critical for physical accuracy. Below, we provide a comparative analysis, troubleshooting guides, and FAQs to assist researchers in selecting and implementing these force fields effectively.
Performance Comparison Tables
| Force Field | Year/Version | Parametrization Philosophy | Key Strengths | Key Limitations |
|---|---|---|---|---|
| AMBER | ff15ipq (2017) [33] | IPolQ scheme with dual gas/solution phase charges; automated fitting tools [33]. | Accurate for globular proteins and metastable peptides; good with cyclic β-amino acids [67] [33]. | Lacks some neutral termini; may over-stabilize helices in some variants (ff15ipq-Qsolv) [67] [33]. |
| CHARMM | CHARMM36m (2017) [67] | Torsional path matching to QM calculations; optimization to water interaction energies [67] [33]. | Best overall for β-peptides; accurate experimental structure reproduction; good for oligomer stability [67]. | Requires specific GROMACS mdp settings (e.g., force-switch) [68]. |
| GROMOS | 54A7, 54A8 (2013-2015) [67] | United-atom; parametrized with twin-range cut-off; physically incorrect with modern integrators [67] [68]. | Supports β-peptides "out of the box" [67]. | Lowest performance for β-peptide secondary structures; unstable with single-range cut-off; missing termini [67] [68]. |
Table 2: Performance on Specific Systems
| Biomolecular System | AMBER | CHARMM | GROMOS | Key Findings |
|---|---|---|---|---|
| β-Peptides (Monomeric) | Accurate for 4/7 sequences, especially with cyclic residues [67]. | Accurate reproduction of experimental structures for all 7 sequences tested [67]. | Struggled to reproduce experimental secondary structures [67]. | CHARMM's QM-derived backbone dihedrals show superior performance [67]. |
| β-Peptides (Oligomeric) | Could not yield spontaneous oligomer formation [67]. | Correctly described all oligomeric examples and stability [67]. | Not reported for oligomer formation [67]. | CHARMM is preferred for self-assembly studies [67]. |
| Globular Proteins | Stable fold depiction [33] [69]. | Stable fold depiction [69]. | Performance depends on version and electrostatic treatment [69]. | All major force fields can be adequate but require validation [69]. |
| Short Peptides/IDPs | Mixed success; some versions show helical bias [33]. | Varying degrees of success [33] [69]. | Challenging to distinguish statistically from others [69]. | Reproducing metastable states remains challenging [33] [69]. |
| Liquid Densities | Reasonable agreement (RMS error ~0.04 g/ml) [70]. | Second only to TraPPE in accuracy [70]. | Not the top performer [70]. | CHARMM is a strong choice for condensed-phase properties [70]. |
Experimental Protocols
Workflow for Force Field Comparison on a Curated Protein Set
The following diagram outlines a general protocol for comparing force field performance, based on methodologies used in recent studies [67] [69].
Detailed Methodology
The protocol below is adapted from a 2023 study comparing force fields for β-peptides, which provides a robust framework for a curated protein set [67].
System Preparation (Sequence Curation):
- Select a diverse set of protein sequences encompassing various secondary structures (e.g., helices, sheets, hairpins) and states (monomeric, oligomeric).
- Build molecular models using software like PyMOL. Critical Step: Pay meticulous attention to assigning the correct protonation states and terminal groups (e.g., neutral amine, N-methylamide) as reported in the experimental literature. Missing termini in some force fields (e.g., Amber, GROMOS) can prevent simulation of certain peptides [67].
Topology Generation:
- Use force-field-specific tools to generate topologies:
- AMBER: Tools like
antechamberandACPYPEcan be used to generate GAFF parameters for small molecules, which can be combined with protein force fields like ff19SB [68] [71]. - CHARMM: The
CHARMM-GUIweb server or the MacKerell lab website provide topology files in GROMACS format [68]. - GROMOS: The GROMOS software suite (e.g.,
make_top) can be used, though compatibility with modern MD engines requires caution [67] [68].
- AMBER: Tools like
- Solvation and Ionization: Place the protein in the center of a simulation box (e.g., cubic) with a minimum 1.4 nm distance to the box wall. Add pre-equilibrated solvent (water, methanol, etc.). Add ions to neutralize the system and, for aqueous systems, include additional salt to a physiological concentration (e.g., 50 mM) [67].
- Use force-field-specific tools to generate topologies:
Simulation Parameters in GROMACS:
- Energy Minimization: Perform steepest descent energy minimization, first in vacuo and then on the solvated system with position restraints on protein heavy atoms, to remove steric clashes [67].
- Equilibration:
- NVT Ensemble: Run a short MD simulation (e.g., 100 ps) at 300 K using a weak coupling algorithm (e.g.,
Berendsen) with position restraints on heavy atoms [67]. - NPT Ensemble: Run a simulation (e.g., 100 ps) to equilibrate pressure at 1 bar using a Parrinello-Rahman barostat, again with position restraints [67]. For unstable systems (e.g., containing heme groups), running two NPT equilibriations—first with and then without restraints—may be necessary [72].
- NVT Ensemble: Run a short MD simulation (e.g., 100 ps) at 300 K using a weak coupling algorithm (e.g.,
- Production MD: Run multiple, long production simulations (now typically hundreds of ns to μs) for each force field and each starting conformation without restraints. Use a modern integrator like
velocity Verletand a timestep of 2 fs. Employ PME for long-range electrostatics. The choice of van der Waals cutoff and modifier is force-field-dependent [67] [69] [71].
Analysis and Validation:
- Analyze trajectories using tools within GROMACS or packages like
MDAnalysis. Calculate a range of structural properties [69]:- Root-mean-square deviation (RMSD) from the experimental structure.
- Radius of gyration (Rg) and Solvent-accessible surface area (SASA).
- Hydrogen bond counts and patterns.
- Secondary structure prevalence (e.g., via DSSP).
- J-coupling constants and NOE intensities if experimental NMR data is available.
- Crucial: Use a curated test set of high-resolution structures (X-ray/NMR) and compare multiple metrics simultaneously, as improvements in one property can be offset by losses in another [69].
- Analyze trajectories using tools within GROMACS or packages like
Troubleshooting Guides and FAQs
FAQ 1: My simulation becomes unstable and crashes with "bonds rotated more than 30 degrees" or "cannot settle water molecules." What should I do?
This indicates a severe instability, often due to bad contacts, incorrect topology, or inadequate equilibration.
- Solution A (General): Extend your equilibration protocol. Run separate NPT equilibration steps: first with strong position restraints on all protein heavy atoms, and then a second step with restraints only on the protein backbone. This allows side chains to relax gradually [72].
- Solution B (Specific to Heme Groups): If simulating a complex like hemoglobin, ensure the metal center (e.g., Fe²⁺) is properly restrained during equilibration. The topology for the heme group must be correctly parameterized, which is non-trivial. The GROMOS 54a7 force field includes some heme parameters, but for other force fields, you may need to use specialized servers (e.g., CHARMM-GUI for CHARMM) or derive parameters via quantum mechanical calculations [72] [73].
- Solution C (Check Topology): Verify the topology of all molecules, especially non-standard residues or cofactors, for missing atoms, incorrect bonds, or bad atom types.
FAQ 2: How do I choose the correct non-bonded interaction settings (cut-offs, modifiers) for my force field in GROMACS?
The mdp parameters are critical for consistency with the force field's parametrization.
- For CHARMM36: Use the following settings, as recommended by the developers [68]:
- For AMBER (e.g., ff14SB, ff19SB): The parameters have evolved. While older versions like ff14SB used 0.8 nm cutoffs, newer ones like ff19SB use 1.0 nm. When using the Verlet cut-off scheme in GROMACS, the
Potential-shiftmodifier is applied to both Coulomb and van der Waals potentials by default, which is consistent with AMBER's approach of applying a continuum model correction [71]. The key is to consult the specific publication for the force field version you are using. - For GROMOS: A major warning: The GROMOS force fields were parametrized with a physically incorrect twin-range cut-off scheme. Using them with a single-range cut-off (as is standard in modern GROMACS) can lead to incorrect densities and other properties. You should check the latest literature to see if validated parameters for modern integrators are available [68].
FAQ 3: My force field over-stabilizes a particular secondary structure (e.g., alpha-helices). Is this a known issue and how can I address it?
Yes, this is a recognized form of bonded parameter imbalance. For example, the ff15ipq-Qsolv variant was shown to overestimate helical content in peptides like AAQAA3 and even induce helicity in disordered peptides [33]. This can occur when bonded parameters are optimized in a way that inadvertently biases the backbone dihedrals.
- Solution A (Force Field Selection): If your system is known to be flexible or disordered, research which force fields have been validated for such systems. The standard CHARMM36m and AMBER ff19SB have been developed with efforts to correct such imbalances, but testing multiple force fields is recommended.
- Solution B (Informed Analysis): Be cautious in interpreting results related to secondary structure propensity. Cross-validate your findings with experimental data (e.g., NMR chemical shifts, CD spectroscopy) whenever possible [69].
While force fields have improved significantly, inaccuracies persist. The primary challenge is that force field parametrization is a poorly constrained problem with highly correlated parameters [69]. Key sources of inaccuracies include:
- Bonded Parameter Imbalance: The delicate balance between torsion potentials, angle terms, and the charge model. Incorrect balance can lead to systematic biases in secondary structure preference [33] [74].
- Fixed Charge Models: Standard force fields cannot account for electronic polarization changes in different environments, which can affect binding energies and conformational dynamics [33].
- Validation Limitations: It is difficult to determine if a force field is fundamentally better, as improvements in one property (e.g., J-couplings) may worsen another (e.g., structural compactness) [69].
Ongoing research focuses on developing more systematic parametrization methods, such as the IPolQ charge scheme [33] and using extensive QM calculations to derive bonded terms [67] [73], to combat these inaccuracies.
Table 3: Essential Software Tools for Force Field Implementation and Testing
| Tool Name | Function/Brief Explanation | Relevant Force Fields |
|---|---|---|
| GROMACS | High-performance MD engine capable of running simulations with AMBER, CHARMM, and GROMOS force fields, allowing for impartial comparison [67]. | All |
| CHARMM-GUI | Web-based portal for building complex systems and generating input files and topologies for various MD engines, including GROMACS [68]. | CHARMM, AMBER, others |
| AmberTools | A suite of programs, including antechamber and tleap, for preparing systems and generating topologies within the AMBER ecosystem. |
AMBER, GAFF |
| ACPYPE | A tool for converting AMBER topologies (including those from GAFF) to GROMACS format, helpful for simulating small molecules [68]. | AMBER, GAFF |
| PyMOL | Molecular graphics system used for visualizing structures, building initial models, and analyzing simulation trajectories. | All |
| Automated Force Field Fitting Tools | Software (e.g., used for ff15ipq) that leverages cluster computation to derive parameters with less human intervention, improving reproducibility [33]. | All (development) |
| Multiwfn | A multifunctional program for wavefunction analysis, used for deriving RESP charges in quantum mechanics-based parameterization [73]. | All (development) |
| ATB (Automated Topology Builder) | Online server for generating molecular topologies and parameters, primarily for the GROMOS force field [72]. | GROMOS |
Confronting Sampling and Statistical Significance in Validation Studies
Technical Support Center
Troubleshooting Guide: Sampling and Significance in Force Field Validation
How do I diagnose insufficient sampling in my molecular dynamics trajectories?
Problem: Simulation results are not reproducible, and calculated properties show high variance between different simulation runs. Solution:
- Convergence Analysis: Monitor key observables (e.g., RMSD, radius of gyration, potential energy) over time. Ensure they plateau and fluctuate around a stable average. If trends are still observed, sampling is likely insufficient [33].
- Block Averaging: Divide your trajectory into consecutive blocks. Calculate the property of interest within each block. If the average changes significantly with additional blocks, longer sampling is required.
- Statistical Uncertainty: Quantify the standard error of the mean for your key metrics. A large uncertainty relative to the mean value indicates more sampling is needed.
Why is my bonded parameter validation showing statistically insignificant results despite long simulations?
Problem: Simulations are long, but differences between force fields or parameter sets are not statistically significant. Solution:
- Power Analysis: Conduct an a priori power analysis. For this, you need to define:
- Independent Replicas: Do not rely on a single long simulation. Perform multiple independent simulations starting from different initial conditions (e.g., different velocities). This provides a better estimate of variance and allows for robust statistical testing [33].
How can I choose the right sampling method for my validation study?
Problem: Uncertainty in whether the chosen sampling strategy adequately represents the configurational space. Solution: Select a method based on your system and research goal. The table below summarizes the primary approaches.
Table 1: Sampling Techniques for Simulation Validation
| Sampling Method | Description | Best Use Case in Force Field Validation |
|---|---|---|
| Simple Random | Every possible simulation starting point has an equal chance of being selected [77] [76]. | Testing a force field on a large, well-defined set of small peptides or folds. |
| Stratified Random | The population is divided into homogeneous subgroups (strata), and random samples are drawn from each [77] [76]. | Ensuring adequate representation of different secondary structure types (e.g., alpha-helix, beta-sheet, coil) in your test set. |
| Cluster | Population is divided into clusters; a random sample of clusters is chosen [77] [76]. | When validating on very large protein families; clusters could be different protein folds. |
| Convenience | Selection based on availability and accessibility [77] [76]. | Not recommended for final validation. Can be useful for initial, rapid testing of parameters. |
| Purposive | Selection based on the researcher's judgment and specific needs of the study [76]. | Creating a targeted benchmark set of proteins known to be challenging for force fields. |
The following workflow outlines a systematic approach to diagnosing and resolving these common sampling and significance issues:
Frequently Asked Questions (FAQs)
Q1: What is the difference between a "population" and a "sample" in the context of force field validation?
In force field validation, the population is the complete set of all possible conformational states and dynamic behaviors of the biomolecular system you are studying. The sample is the finite set of conformations and trajectories you actually generate and analyze through your molecular dynamics simulations. Conclusions about the force field's accuracy are drawn from the sample and inferred to the population [76].
Q2: My simulation time is limited. Should I run one very long simulation or multiple shorter replicas?
For assessing statistical significance, running multiple independent replicas is generally more robust than a single long simulation. Replicas allow you to estimate the variance and uncertainty in your results. A single long simulation might appear stable but could be trapped in a local energy minimum, giving a false sense of convergence. Multiple starts help ensure broader coverage of conformational space [33].
Q3: How many independent simulations are sufficient for my validation study?
There is no universal number. The required number of replicas depends on the inherent variability of the system you are studying and the effect size you want to detect. A disordered peptide may require more replicas than a stable, globular protein to achieve the same statistical confidence. You should determine this through a power analysis before beginning your production runs [75].
Q4: What are the consequences of using convenience sampling in validation?
Convenience sampling (e.g., only testing your new parameters on a few well-behaved, easy-to-simulate proteins) introduces a high risk of sampling bias [77] [76]. Your force field may perform well on this biased sample but fail dramatically when applied to other protein folds or systems it was not tuned for. This severely limits the generalizability and predictive power of your force field.
Q5: How can I design a validation set that is representative of the broader biomolecular space?
Use a stratified sampling approach. Divide the "population" of protein structures into meaningful strata based on criteria relevant to force field performance, such as:
- Secondary structure content (e.g., all-alpha, all-beta, alpha/beta)
- Presence of specific ligands or cofactors
- Membrane-bound vs. soluble Then, select a representative sample from each stratum to ensure your validation set covers a wide range of challenging scenarios [77] [76].
Experimental Protocols
Protocol 1: Standard Workflow for Validating Bonded Parameter Corrections
This protocol is designed to systematically test new bonded parameters, such as those derived for the ff15ipq force field, against a reference [33].
System Preparation:
- Select a diverse validation set of proteins and peptides using a stratified random sampling method (see Table 1).
- Prepare the simulation systems using standard tools (e.g.,
solvatein GROMACS [78] orCHARMM-GUI). Ensure systems are neutralized with ions and solvated in a sufficient water box.
Equilibrium Molecular Dynamics:
- Perform energy minimization until the maximum force is below a chosen threshold (e.g., 1000 kJ/mol/nm).
- Gradually heat the system to the target temperature (e.g., 300 K) over 100-200 ps in the NVT ensemble.
- Equilibrate the density by running a short simulation (100-200 ps) in the NPT ensemble.
- Run production simulations in the NPT ensemble. The length will depend on the system, but for peptide validation, >1 µs per replica is often necessary [33].
Data Analysis:
- Convergence: Calculate properties like RMSD and radius of gyration as a function of time to ensure equilibrium has been reached.
- Property Calculation: Compute the experimental observables of interest (e.g., J-coupling constants, chemical shifts, helical content, native structure stability).
- Statistical Comparison: Use statistical tests (e.g., t-tests) to compare the simulation-derived observables with experimental data or with results from other force fields. The unit of analysis for these tests should be the independent replica, not time points from a single trajectory.
The logical flow and key decision points for this protocol are summarized in the following diagram:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for Force Field Validation Studies
| Item / Reagent | Function / Purpose |
|---|---|
| Stratified Test Set | A carefully selected ensemble of proteins/peptides that represent diverse structural classes, ensuring validation is not biased towards specific folds [77] [76]. |
| Molecular Dynamics Software | Software like GENESIS [58] or GROMACS [78] to run the simulations. These provide the computational engine for sampling conformational space. |
| Enhanced Sampling Algorithms | Methods like replica-exchange MD (REMD) available in GENESIS [58] or Gaussian accelerated MD (GaMD) to improve sampling efficiency, especially for overcoming high energy barriers. |
| Trajectory Analysis Tools | Programs like rmsd_analysis, hb_analysis, and wham_analysis in GENESIS [58], or equivalent tools in other packages, to compute observables from raw simulation trajectories. |
| Statistical Power Analysis Software | Tools (e.g., in R or G*Power) to calculate the required sample size (number of replicas) before running costly simulations, ensuring the study is designed to detect meaningful effects [75]. |
| Reference Quantum Mechanical Data | High-level QM calculations (e.g., MP2/cc-pvTZ [33]) on small model systems used for initial parameter derivation and validation of intrinsic energetics. |
| Experimental Data Repository | Publicly available experimental data (NMR J-couplings, chemical shifts, stability data) used as the ultimate benchmark for validating simulation outcomes [33] [74]. |
Frequently Asked Questions (FAQs)
Q1: Why are THz spectroscopy and J-couplings considered together for force field validation? These techniques probe complementary aspects of molecular structure and dynamics. Terahertz (THz) spectroscopy captures low-frequency collective motions, weak intermolecular interactions (like hydrogen bonds and van der Waals forces), and lattice vibrations, making it ideal for assessing bulk condensed-phase properties and hydration shells [79] [80]. J-couplings (or scalar couplings) are measured through NMR and provide detailed information on torsion angles and conformational preferences in molecules like peptides [81]. Using them together offers a multi-scale validation strategy: J-couplings help refine torsional parameters (a key bonded term), while THz spectroscopy validates the resulting condensed-phase behavior and non-bonded interactions.
Q2: My simulations show good agreement with J-coupling data but poor THz spectra. What does this indicate? This is a classic symptom of a bonded parameter imbalance. Your force field's torsional parameters (which strongly influence dihedral angles probed by J-couplings) may be well-tuned, but the imbalance likely lies in the non-bonded parameters or the water model. Poor THz spectra agreement suggests inaccuracies in how the model handles:
- Intermolecular forces: The balance between solute-solute, solute-water, and water-water interactions may be off [79].
- Polarization: The lack of explicit polarization in standard additive force fields can lead to an incorrect representation of the dipole moment fluctuations and the collective dynamics of the water network, which are critical for THz spectra [79] [16].
- Water model compatibility: The partial charges and van der Waals parameters of the solute must be developed consistently with the chosen water model [79].
Q3: What are the key advantages of polarizable force fields like AMOEBA in this context? Polarizable force fields explicitly model the change in a molecule's electron distribution in response to its environment. This directly addresses a major limitation of additive force fields. For validation:
- AMOEBA incorporates polarization via induced dipoles, uses permanent atomic multipoles (not just point charges), and employs a softer buffered 14-7 van der Waals potential. This has been shown to improve the prediction of hydration shell structure, hydrogen bonding, and diffusion kinetics, all of which contribute to more accurate THz spectroscopic predictions [79].
- A more physically realistic treatment of electrostatics provides a better foundation for simultaneously reproducing J-coupling (conformational) and THz (collective, environmental) observables [16].
Q4: How can Machine Learning Force Fields (MLFFs) help overcome these challenges? MLFFs like MACE-OFF are trained on high-level quantum mechanical data and can capture complex electronic effects without relying on fixed functional forms. They offer a path to improved validation by:
- Intrinsic Accuracy: They demonstrate high accuracy for a wide range of properties, including dihedral torsion scans (relevant for J-couplings) and condensed-phase properties of molecular liquids and crystals (relevant for THz spectroscopy) [81].
- Transferability: They are designed to generalize across chemical space, potentially providing accurate parameters for diverse molecules, including post-translationally modified proteins and drug-like molecules, which are challenging for classical force fields [81] [16].
Troubleshooting Guides
Issue 1: Poor Agreement with Experimental THz Absorption Spectra
Problem: The simulated THz spectrum does not match the position or intensity of peaks in the experimental data.
Potential Causes and Solutions:
- Cause 1: Inaccurate Water Model or Solute-Water Interactions.
- Solution A: Test different water models (e.g., TIP3P, TIP4P, SPC/E) with your force field. Note that TIP3P has been associated with less structured hydration shells and erroneous diffusion coefficients for carbohydrates [79].
- Solution B: Consider switching to a polarizable force field like AMOEBA, which includes a water model developed consistently with its solute parameter set and has shown improvements in modeling hydration dynamics [79].
- Cause 2: Incorrect Balance of Non-bonded Forces.
- Solution: Refine the Lennard-Jones parameters, particularly the repulsive term. The AMOEBA force field uses a "softer" buffered 14-7 potential instead of the standard 12-6 Lennard-Jones, which can improve the description of short-range interactions [79].
- Cause 3: Lack of Polarization Effects.
Issue 2: Discrepancy Between Calculated and Experimental J-Couplings
Problem: J-couplings back-calculated from an MD simulation trajectory do not agree with NMR measurements.
Potential Causes and Solutions:
- Cause 1: Inaccurate Torsional Energy Profiles.
- Solution: Perform torsion scans of the relevant dihedral angles (e.g., in a model compound) using high-level QM calculations and compare them to the force field's profile. If a systematic deviation is found, re-parameterize the torsional terms. MLFFs like MACE-OFF are specifically noted for producing accurate dihedral torsion scans for unseen molecules [81].
- Cause 2: Inadequate Conformational Sampling.
- Solution: Extend the simulation time or use enhanced sampling techniques (e.g., replica-exchange molecular dynamics) to ensure all relevant conformational states are visited [16].
- Cause 3: Improparametrized Partial Atomic Charges.
- Solution: Re-derive charges using a QM method that accurately reproduces the molecular electrostatic potential, ensuring compatibility with the force field's philosophy and the water model.
Experimental Protocols for Validation
Protocol 1: Validating Force Fields with Terahertz Time-Domain Spectroscopy (THz-TDS)
1. Principle: THz-TDS measures the electric field of a THz pulse transmitted through (or reflected from) a sample. By comparing the pulse with and without the sample, the absorption coefficient and refractive index in the 0.1-10 THz range can be extracted. This "fingerprint" region is sensitive to weak intermolecular interactions and collective motions [80].
2. Key Steps:
- Sample Preparation:
- For solid samples (e.g., crystalline drugs or carbohydrates), the powder is often mixed with a non-absorbing matrix like polyethylene and pressed into a pellet.
- For liquid samples, a transmission cell with suitable windows (e.g., quartz) is used. Attenuated Total Reflection (ATR) THz-TDS is particularly effective for strongly absorbing materials like polar liquids [80].
- Data Collection:
- Acquire the time-domain waveform of the THz pulse through the sample and a reference (e.g., an empty pellet or cell).
- Use a Fourier transform to convert the time-domain signals into frequency-domain spectra.
- Calculate the complex optical parameters (absorption, refractive index) from the amplitude and phase differences between the sample and reference pulses [80].
- Simulation and Comparison:
- Run a classical MD simulation of the system (e.g., a solute in explicit solvent).
- Calculate the THz spectrum from the MD trajectory, typically by Fourier transformation of the dipole moment time-correlation function of the simulation box.
- Compare the simulated and experimental absorption spectra.
3. Workflow Diagram:
Protocol 2: Validating Force Fields with J-Couplings
1. Principle: J-couplings (^3J) are measured via NMR and depend on the dihedral angle between atoms (e.g., the backbone φ and ψ angles in peptides). They provide a quantitative measure of conformational populations.
2. Key Steps:
- NMR Measurement:
- Acquire NMR spectra (e.g., COSY, HSQC) of the molecule (e.g., a peptide) in solution to extract experimental ^3J-coupling values.
- Calculation from MD Trajectory:
- Run an MD simulation of the molecule under the same conditions (solvent, temperature, pH).
- From the trajectory, extract the time series of the relevant dihedral angles.
- Use a Karplus equation (e.g.,
^3J(θ) = A cos²(θ) + B cos(θ) + C) to convert each dihedral angle in the trajectory into an instantaneous J-coupling value. The parameters (A, B, C) are typically taken from the literature and are derived from QM calculations or NMR data. - Average the instantaneous J-values over the entire trajectory to get the calculated ensemble-average J-coupling.
- Comparison and Refinement:
- Compare the calculated and experimental J-couplings.
- If discrepancies are found, the torsional parameters governing those dihedral angles in the force field are prime candidates for re-parameterization.
3. Workflow Diagram:
Quantitative Data for Comparison
The following table summarizes key benchmarks for assessing force field performance against experimental data.
Table 1: Expected Agreement Ranges for Key Experimental Observables
| Observable | Experimental Technique | Target Agreement for a Validated Force Field | Notes and Tolerances |
|---|---|---|---|
| Peak Position in THz Spectrum | THz-TDS [80] | ± 0.1 THz (~3.3 cm⁻¹) | Critical for identifying specific vibrational modes. Larger shifts may indicate incorrect potential energy surfaces. |
| Radial Distribution Function (RDF), first peak position | X-ray/Neutron Scattering (validated by MD) | ± 0.1 Å | Essential for validating solute-water and water-water structure [79]. |
| J-Coupling Value (^3J) | NMR Spectroscopy [81] | ± 0.5 Hz | Direct measure of torsional populations. Discrepancies point to imbalanced dihedral parameters. |
| * Density (ρ) of Molecular Liquid* | Experimental Measurement | ± 0.01 g/cm³ | A key test for overall condensed-phase performance and non-bonded interaction balance [81]. |
Research Reagent Solutions
This table outlines essential computational and experimental "reagents" for conducting these validation studies.
Table 2: Essential Research Reagents and Tools for Force Field Validation
| Item | Function/Brief Explanation | Examples / Notes |
|---|---|---|
| Polarizable Force Field | A force field that explicitly includes electronic polarization effects, crucial for accurate THz spectra and interaction energies. | AMOEBA [79], Drude [16] |
| Machine Learning Force Field (MLFF) | A high-accuracy potential trained on QM data, offering improved transferability for bonded and non-bonded parameters. | MACE-OFF [81], FENNIX [81] |
| Validated Water Model | A water model parameterized specifically for use with a given force field to ensure balanced solute-solvent and solvent-solvent interactions. | TIP3P, TIP4P, SPC/E (Note: Choice is force field dependent) [79] |
| THz-TDS Spectrometer | The core instrument for acquiring experimental THz absorption spectra of solid and liquid samples. | Transmission, Reflection, or ATR systems [80] |
| Karplus Relation Parameters | The empirical equation (J = A cos²θ + B cosθ + C) that translates MD dihedral angles into calculated J-couplings for NMR validation. | Parameters (A, B, C) are specific to the atom types involved (e.g., H-N-C-H) [81]. |
Conclusion
Correcting bonded parameter imbalance is not a singular fix but a continuous process integral to force field development. The integration of advanced methodologies—from automated parameterization and surgical NBFIX corrections to Bayesian learning and ML-driven force fields—signals a shift towards more systematic, data-driven, and physically robust models. A key lesson is the necessity of multi-faceted validation against diverse experimental and quantum mechanical data to prevent overfitting and ensure transferability. For biomedical research, these advancements promise more accurate predictions of drug-target binding, protein folding pathways, and the behavior of intrinsically disordered proteins, ultimately enhancing the reliability of computational insights in drug development. Future efforts must focus on standardized benchmarking, the development of universally accepted validation protocols, and the seamless integration of electronic polarization effects to achieve the next level of predictive power in biomolecular simulation.
References
- 1. Molecular Mechanics - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4026342/]
- 2. Accuracy evaluation and addition of improved dihedral ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-019-0371-6]
- 3. Methodological Approach Based on Structural Parameters ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11886664/]
- 4. Bond , angle, dihedral and improper coefficients in large ... [https://matsci.org/t/bond-angle-dihedral-and-improper-coefficients-in-large-molecules/32221]
- 5. Research on the Calibration Method of the Bonding ... [https://www.mdpi.com/2079-6412/14/12/1553]
- 6. Merck Molecular Force Field (MMFF94) [https://www.charmm-gui.org/charmmdoc/mmff.html]
- 7. Improving the accuracy of biomolecular force fields by pair ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5874203/]
- 8. Restrained electrostatic potential atomic partial charges for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4191892/]
- 9. Improved Treatment of 1-4 interactions in Force Fields for ... [https://arxiv.org/html/2504.14398v2]
- 10. Experimental determination of partial charges with electron ... [https://www.nature.com/articles/s41586-025-09405-0]
- 11. Choice of Force Field for Proteins Containing Structured and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7136338/]
- 12. Data-driven parametrization of molecular mechanics force ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06640e]
- 13. Frequently asked questions (FAQ) — openforcefield 0.7.1 ... [https://open-forcefield-toolkit.readthedocs.io/en/0.7.1/faq.html]
- 14. Force Fields and Interactions - GitHub Pages [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/01-Force_Fields_and_Interactions/index.html]
- 15. Efficient modelling of anharmonicity and quantum effects in ... [https://www.nature.com/articles/s41524-025-01553-1]
- 16. Review The continuous evolution of biomolecular force fields [https://www.sciencedirect.com/science/article/abs/pii/S0969212625001911]
- 17. The molecular dynamic study of anharmonic effects at Cu ... [https://www.sciencedirect.com/science/article/pii/S0375960114003661]
- 18. Recent advances in artificial intelligence-driven ... [https://pubmed.ncbi.nlm.nih.gov/41273961/]
- 19. Tutorial 04 - Optimizing Force Fields — OpenFF Evaluator ... [https://docs.openforcefield.org/projects/evaluator/en/v0.1.1/tutorials/tutorial04.html]
- 20. - Lennard Potential Jones [https://lab.concord.org/doc/models/md2d/lennard-jones-potential/lennard-jones-potential.html]
- 21. CHARMM 36m Additive Force Field [https://www.emergentmind.com/topics/charmm-36m-additive-force-field]
- 22. derivatives - Numerical force due to Lennard potential... Jones [https://math.stackexchange.com/questions/1742524/numerical-force-due-to-lennard-jones-potential]
- 23. Optimized Lennard-Jones Parameters for Drug-Like Small ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5997559/]
- 24. Data-driven analysis of the number of Lennard–Jones ... [https://www.nature.com/articles/s42004-020-00395-w]
- 25. An Automated Web-Based Tool To Generate Force Field ... [https://pubmed.ncbi.nlm.nih.gov/37773638/]
- 26. Rapid parameterization of small molecules using the Force ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3874408/]
- 27. CUFIX: Non-bonded Fix (NBFIX) parameters for ... [https://bionano.physics.illinois.edu/CUFIX]
- 28. An Automated Web-Based Tool To Generate Force Field ... [https://chemrxiv.org/engage/chemrxiv/article-details/64b0a880ae3d1a7b0da82eb6]
- 29. Best practices for machine-learned force fields [https://www.vasp.at/wiki/index.php/Best_practices_for_machine-learned_force_fields]
- 30. Automated parametrization of small molecules within the ... [https://www.nature.com/articles/s41598-025-24757-3]
- 31. Building Force Fields - an Automatic, Systematic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9649520/]
- 32. CHAPERONg: A tool for automated GROMACS-based ... [https://www.sciencedirect.com/science/article/pii/S2001037023003367]
- 33. Links between the charge model and bonded parameter force ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5669220/]
- 34. Polarizable force fields for biomolecular simulations [https://pmc.ncbi.nlm.nih.gov/articles/PMC6520134/]
- 35. Integration of experimental data and use of automated ... [https://www.nature.com/articles/s42004-022-00653-z]
- 36. Integration of experimental data and use of automated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8979544/]
- 37. Supplemental: Overview of the Common Force Fields [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/S01-Force_Fields_Overview/index.html]
- 38. Non-bonded force field model with advanced restrained ... [https://www.nature.com/articles/s42004-020-0291-4]
- 39. Bayesian learning for accurate and robust biomolecular ... [https://arxiv.org/html/2511.05398v1]
- 40. Surrogate-enabled Bayesian sampling of force ... [https://openforcefield.org/community/news/science-updates/bayesian-surrogate-modeling-2021-09-03/]
- 41. On-the-fly active learning of interpretable Bayesian force ... [https://www.nature.com/articles/s41524-020-0283-z]
- 42. Refining coarse-grained molecular topologies: a Bayesian ... [https://www.nature.com/articles/s41524-025-01729-9]
- 43. Robustness in biomolecular simulations [https://pmc.ncbi.nlm.nih.gov/articles/PMC12165335/]
- 44. Bayesian inference-driven model parameterization and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9217127/]
- 45. Influence of denatured and intermediate states of folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2253448/]
- 46. New tricks for old dogs: improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions - PubMed [https://pubmed.ncbi.nlm.nih.gov/29547221/]
- 47. Development and Testing of Force Field Parameters for ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2020.608931/full]
- 48. Development of Force Field Parameters for the Simulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9234960/]
- 49. Bayesian learning for accurate and robust biomolecular force fields [https://arxiv.org/html/2511.05398]
- 50. Preserving the Integrity of Empirical Force Fields - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9399073/]
- 51. Artificial Chaperone-assisted Refolding of Citrate Synthase [https://www.sciencedirect.com/science/article/pii/S0021925819884090]
- 52. Speed up Multi-Scale Force-Field Parameter Optimization ... [https://pubmed.ncbi.nlm.nih.gov/40911030/]
- 53. Evolutionary machine learning of physics-based force in... fields [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00178a]
- 54. Troubleshooting and warnings — ReaxFF 2025.1 ... - SCM [https://www.scm.com/doc/ReaxFF/Troubleshooting.html]
- 55. An improved reactive force field parameter optimization ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025625001193]
- 56. Parallelization - GROMACS 2025-rc documentation [https://manual.gromacs.org/documentation/2025-rc/reference-manual/algorithms/parallelization-domain-decomp.html]
- 57. Development and Testing of Force Field Parameters ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7770134/]
- 58. Frequently Asked Questions - GENESIS [http://mdgenesis.org/docs/FAQ/]
- 59. BioExcel Building Blocks Frequently Asked Questions [https://mmb.irbbarcelona.org/biobb/documentation/faqs]
- 60. Comparison of Charge Derivation Methods Applied to Amino ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6641686/]
- 61. Re: AMBER: RESP vs. AM1-BCC charges for isoprene [https://structbio.vanderbilt.edu/archives/amber-archive/2004/2489.php]
- 62. Re: AMBER: RESP vs. AM1-BCC charges for isoprene [http://archive.ambermd.org/200407/0340.html]
- 63. A fast and high-quality charge model for the next generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7728379/]
- 64. [AMBER] Partial charge discrepancy between AM1-BCC small ... [http://archive.ambermd.org/202207/0162.html]
- 65. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 66. Beyond Force Metrics: Pre-Training MLFFs for Stable MD ... [https://arxiv.org/html/2506.14850v1]
- 67. Comparative Study of Molecular Mechanics Force Fields for β ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10302482/]
- 68. Force fields in GROMACS [https://manual.gromacs.org/current/user-guide/force-fields.html]
- 69. On the Validation of Protein Force Fields Based on Structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11103706/]
- 70. Comparison of the AMBER, CHARMM, COMPASS ... [https://www.sciencedirect.com/science/article/abs/pii/S0378381206003323]
- 71. Mdp parameters for non-bonded interaction using the AMBER ... [https://gromacs.bioexcel.eu/t/mdp-parameters-for-non-bonded-interaction-using-the-amber-force-field/6458]
- 72. Issues with MD Run and Fe Displacement in Hemoglobin ... [https://gromacs.bioexcel.eu/t/issues-with-md-run-and-fe-displacement-in-hemoglobin-simulation/10616]
- 73. Development of force field parameters for the atomic ... [https://www.nature.com/articles/s42004-025-01722-9]
- 74. Systematic design of biomolecular force fields [https://www.sciencedirect.com/science/article/pii/S0959440X2030141X]
- 75. How to choose a sampling technique and determine ... [https://www.sciencedirect.com/science/article/pii/S2772906024005089]
- 76. Sampling Methods | Types, Techniques & Examples [https://www.scribbr.com/methodology/sampling-methods/]
- 77. Sampling methods in Clinical Research; an Educational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5325924/]
- 78. Answers to frequently asked questions (FAQs) [https://manual.gromacs.org/current/user-guide/faq.html]
- 79. Is AMOEBA a Good Force Field for Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12152942/]
- 80. Progress in application of terahertz time-domain spectroscopy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10393043/]
- 81. Short Range Transferable Machine Learning Force Fields ... [https://arxiv.org/html/2312.15211v4]