Bridging the Gap: A Practical Guide to Validating MD-Predicted Protein Folding Pathways
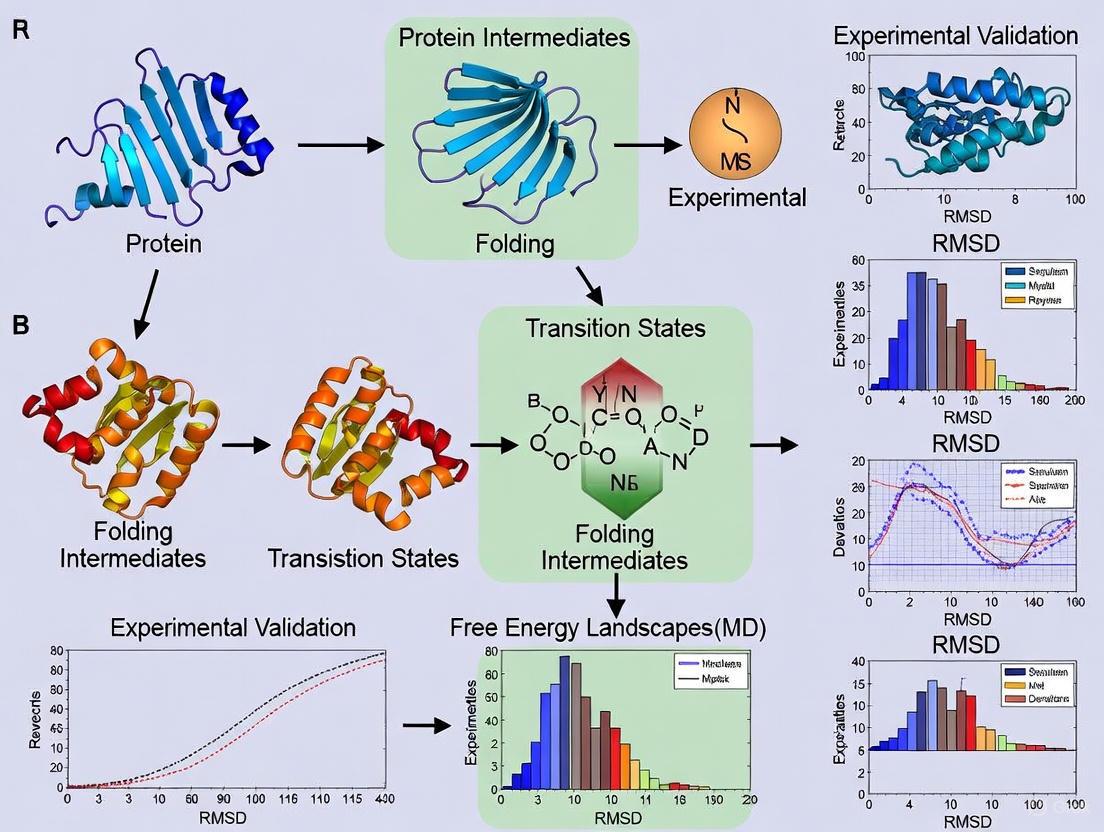
This article provides a comprehensive framework for researchers and drug development professionals seeking to validate molecular dynamics (MD) simulations of protein folding pathways against experimental data.
Bridging the Gap: A Practical Guide to Validating MD-Predicted Protein Folding Pathways
Abstract
This article provides a comprehensive framework for researchers and drug development professionals seeking to validate molecular dynamics (MD) simulations of protein folding pathways against experimental data. It covers foundational concepts, from the fundamental challenges of the protein folding problem revealed by the Levinthal paradox to the revolutionary impact of AI-based structure prediction tools like AlphaFold. The content explores integrated methodological approaches that combine machine learning models with MD simulations to explore conformational ensembles, details common pitfalls in simulation accuracy and sampling, and establishes robust protocols for quantitative comparison with experimental observables. By synthesizing insights across computational and experimental disciplines, this guide aims to enhance confidence in MD-predicted folding mechanisms for critical applications in biomedical research and therapeutic development.
The Protein Folding Landscape: From Anfinsen's Dogma to AI Revolution
In 1969, Cyrus Levinthal posed a fundamental challenge to our understanding of protein folding: if a protein were to fold by randomly sampling all possible conformational states, it would require astronomical timescales far exceeding the age of the universe, yet proteins typically fold in milliseconds to seconds [1]. This discrepancy between theoretical calculation and experimental observation became known as Levinthal's paradox. The resolution of this paradox lies not in faster conformational sampling, but in the nature of the folding process itself—proteins do not fold by exhaustive random search but follow biased, energetically favorable pathways guided by a funnel-shaped energy landscape [2].
The energy landscape theory revolutionized our understanding of protein folding by introducing the concept of a rugged funnel where the folding process is directed toward the native state by decreasing free energy and increasing native-like contacts [2] [3]. This theoretical framework has profound implications for modern structural biology, particularly in validating molecular dynamics (MD)-predicted folding pathways with experimental data. As we move beyond static structure prediction toward dynamic conformational ensembles, integrating computational approaches with experimental validation becomes crucial for understanding protein function and dysfunction in disease states [4].
Theoretical Framework: From Paradox to Funnel
The Levinthal Paradox and Its Implications
Levinthal's paradox highlights a fundamental mathematical contradiction: for a typical protein of 100 amino acids, the number of possible conformations is astronomically large (~3¹⁰⁰), and random sampling would require timescales orders of magnitude longer than observed folding times [2] [1]. This paradox initially suggested that protein folding represented an unsolvable search problem, potentially requiring new physical laws for its explanation [2].
The critical insight for resolving this paradox came from recognizing that proteins are not random heteropolymers. Instead, natural protein sequences have been evolutionarily selected for folding efficiency and minimal frustration [2]. As Bryngelson and Wolynes established, such minimally frustrated sequences exhibit energy landscapes with two key characteristics: a folding transition temperature (TF) and a glass transition temperature (Tg). Easy-to-fold sequences maintain a high TF/Tg ratio, enabling efficient folding without kinetic trapping [2].
Energy Landscape Theory and the Folding Funnel
Energy landscape theory conceptualizes protein folding as navigation on a rugged funnel-shaped landscape [2] [3]. This landscape is "funneled" because its overall slope biases the conformational search toward the native state, while "rugged" due to the presence of metastable intermediates and kinetic barriers [3].
The funnel metaphor captures several essential features of protein folding:
- Global bias: The overall downward slope represents the decreasing free energy as the protein approaches its native state
- Ruggedness: Local minima and barriers correspond to metastable states and folding intermediates
- Progressive narrowing: The funnel narrows toward the native state, reflecting the decrease in conformational entropy
- Multiplicity of pathways: Unlike specific stepwise chemical reactions, folding can proceed through multiple dominant routes [2]
Quantitative studies have confirmed this funneled landscape paradigm. For ordered proteins like HP-35 and WW domain, the landscape slope is approximately -50 kcal/mol, meaning free energy decreases by ~5 kcal/mol upon formation of 10% native contacts. In contrast, intrinsically disordered proteins like pKID exhibit shallower landscapes (slope of -24 kcal/mol), explaining their disorder in isolation. Upon binding to their partners, their landscapes become significantly steeper (slope of -54 kcal/mol), enabling folding [5].
Table 1: Key Characteristics of Protein Folding Energy Landscapes
| Characteristic | Ordered Proteins | Intrinsically Disordered Proteins | Minimally Frustrated Sequences |
|---|---|---|---|
| Landscape Slope | ~ -50 kcal/mol [5] | ~ -24 kcal/mol (free); -54 kcal/mol (bound) [5] | Steeply funneled |
| Frustration Level | Minimal | Varies | Minimal by evolutionary design |
| Metastable States | Limited | Multiple | Few deep traps |
| Folding Timescale | Microseconds to seconds | Context-dependent | Optimized for rapid folding |
| Response to Mutations | Often destabilizing | Can alter binding-induced folding | Sensitive to conservative changes |
Computational Methodologies for Exploring Folding Landscapes
Molecular Dynamics Simulations
Molecular Dynamics (MD) simulations provide an atomistic approach to studying protein folding by numerically solving Newton's equations of motion for all atoms in the system. Conventional all-atom MD with explicit solvent offers high accuracy but comes at extreme computational cost, limiting its application to relatively small proteins and shorter timescales [6].
Advanced sampling techniques have been developed to overcome these limitations:
- Parallel Tempering/Replica Exchange: Simultaneously runs simulations at multiple temperatures, enabling enhanced conformational sampling [6]
- Metadynamics and OPES: Bias simulations along collective variables (CVs) to accelerate rare events like folding/unfolding transitions [3]
- Variational Force-Matching: Uses machine learning to develop coarse-grained force fields that maintain near-atomistic accuracy [6]
Recent work has demonstrated the critical importance of designing effective CVs that capture slow degrees of freedom relevant to folding. Bioinspired CVs that explicitly distinguish protein-protein from protein-water hydrogen bonds and account for side-chain packing can significantly enhance state resolution and reduce degeneracy problems that plague traditional CVs [3].
Machine-Learned Coarse-Grained Models
The development of transferable coarse-grained (CG) models represents a major advancement for simulating folding processes. By combining deep learning with diverse training sets of all-atom simulations, researchers have developed bottom-up CG force fields with chemical transferability that can extrapolate to sequences not used during parameterization [6].
These machine-learned CG models successfully predict metastable states of folded, unfolded, and intermediate structures, fluctuations of intrinsically disordered proteins, and relative folding free energies of protein mutants while being several orders of magnitude faster than all-atom models [6]. For example, CGSchNet demonstrates remarkable transferability, accurately reproducing folding landscapes for proteins with low (<40%) sequence similarity to training examples, indicating that the model learns to represent effective physical interactions rather than merely memorizing structural templates [6].
Table 2: Comparison of Protein Folding Simulation Methods
| Method | Spatial Resolution | Timescale Accessible | Key Applications | Limitations |
|---|---|---|---|---|
| All-Atom MD | Atomic | Nanoseconds to milliseconds | Folding mechanisms, atomistic details | Extreme computational cost |
| Coarse-Grained MD | 3-5 heavy atoms per bead | Microseconds to seconds | Folding thermodynamics, larger proteins | Loss of atomic detail |
| Machine-Learned CG | Coarse-grained (Cα or backbone) | Microseconds to seconds | Metastable states, folding free energies | Training data dependency |
| Enhanced Sampling | Atomic or coarse-grained | Effectively extends accessible times | Free energy landscapes, rare events | Dependent on collective variables |
| Go̅ Models | Cα or backbone | Milliseconds and beyond | Folding principles, large systems | Native-centric, limited for misfolding |
Cotranslational Folding Simulations
The Generalized Protein Cotranslational Folding (GPCTF) simulation framework represents a significant innovation by modeling ribosomal exit tunnels and translation processes. This approach reveals fundamental differences between cotranslational folding in vivo and free folding in vitro, showing that CTF provides more helix-rich initial structures with fewer nonnative long-range contacts compared to FF [7].
GPCTF simulations demonstrate that while subsequent folding follows similar pathways as free folding, the distribution among these pathways is modulated by translation speed. This pathway regulation mechanism helps reconcile discrepancies in previous experimental results and offers significant insights into protein folding processes in physiological contexts [7].
Experimental Validation of Predicted Folding Pathways
Biophysical Techniques for Monitoring Folding
Experimental validation of computationally predicted folding pathways employs multiple biophysical techniques that probe different aspects of protein structure and dynamics:
- Hydrogen-Deuterium Exchange Mass Spectrometry: Measures solvent accessibility and dynamics by tracking deuterium incorporation, providing insights into folding intermediates and protected regions [4]
- Single-Molecule Fluorescence Resonance Energy Transfer (smFRET): Monitors intra-molecular distances and conformational changes in real-time, enabling detection of transient intermediates [4]
- Nuclear Magnetic Resonance (NMR) Spectroscopy: Provides atomic-resolution information on structure and dynamics, particularly powerful for characterizing folding intermediates and residual structure in disordered states [4] [5]
- Circular Dichroism (CD) Spectroscopy: Tracks secondary structure formation during folding by measuring differential absorption of left- and right-circularly polarized light [1]
These techniques generate complementary data that collectively constrain possible folding mechanisms and enable validation of MD-predicted pathways. For example, NMR measurements of protection factors can directly validate predicted hydrogen bonding patterns in folding intermediates, while smFRET time trajectories can confirm predicted folding routes and rates.
Quantitative Landscape Mapping
Advanced experimental approaches now enable quantitative mapping of folding energy landscapes. By combining site-directed mutagenesis with phi-value analysis, researchers can probe the structure of transition states and folding intermediates. Phi-values between 0 and 1 indicate the extent to which a residue's native interactions are formed in the transition state, providing crucial information about folding mechanisms [7].
Recent methodological developments allow explicit construction of free energy landscapes from simulation data. The reduced landscape f(Q) is obtained by averaging the free energy f(r) = Eu(r) + Gsolv(r) over configurations with specific values of an order parameter Q (typically the fraction of native contacts) [5]. This approach distinguishes between the globally funneled landscape f(Q) and the free energy profile F(Q) = -k_BT log P(Q), which includes configurational entropy effects and typically shows unfolded and folded minima separated by a barrier [5].
Case Studies: Successes and Limitations
Mini-Protein Folding Landscapes
Comprehensive studies on mini-proteins like Chignolin and TRP-cage have provided detailed validation of energy landscape principles. Enhanced sampling simulations using specialized collective variables that capture hydrogen bonding and side-chain packing have successfully resolved complex free-energy landscapes and revealed critical intermediates such as the dry molten globule state [3].
These studies demonstrate that convergent folding pathways emerge naturally from the energy landscape, with proteins incrementally forming native contacts through stochastic search. The dry molten globule intermediate, characterized by substantial native-like secondary structure but incomplete side-chain packing and dehydration, appears to be a general feature of the folding process for many small proteins [3].
Multi-Domain Proteins and Prediction Challenges
Despite significant advances, substantial challenges remain, particularly for multi-domain proteins and systems with limited evolutionary information. A case study on the SAML protein revealed severe deviations between experimental structures and AI predictions, with positional divergences exceeding 30 Å and overall RMSD of 7.7 Å [8].
These discrepancies were particularly pronounced in the relative orientation of protein domains, which could not be resolved even with customized searches using low MSA depth, different random seeds, and multiple recycling steps [8]. This highlights current limitations in capturing inter-domain interactions and conformational flexibility, especially when experimental structures represent specific conformations stabilized by crystallization conditions that predictions may not account for [8].
Cotranslational vs. Free Folding
Systematic comparison of cotranslational folding (CTF) and free folding (FF) using the GPCTF framework has revealed fundamental differences in folding mechanisms. Simulations totaling over 8 milliseconds across three proteins with different topologies revealed that CTF produces nascent peptides with more helix-rich structures and fewer long-range contacts upon expulsion from the ribosomal exit tunnel compared to FF [7].
While subsequent folding follows similar pathways, their relative probabilities are modulated by translation speed, demonstrating a pathway regulation mechanism inherent to cotranslational folding. This provides a mechanistic basis for understanding how synonymous codon substitutions that alter translation speed can impact protein structure without changing the amino acid sequence [7].
Research Toolkit: Essential Methods and Reagents
Table 3: Research Reagent Solutions for Protein Folding Studies
| Reagent/Resource | Function/Application | Key Features | Example Uses |
|---|---|---|---|
| GROMACS [4] | Molecular dynamics simulation package | High performance, versatile | Folding/unfolding simulations |
| AMBER [4] | Molecular dynamics software | Specialized for biomolecules | Detailed folding pathway analysis |
| CHARMM [4] | MD simulation program | Comprehensive force fields | Free energy calculations |
| OpenMM [4] | Toolkit for MD simulation | GPU acceleration, customizability | Enhanced sampling methods |
| ATLAS Database [4] | MD simulation database | ~2000 proteins, diverse structural space | Reference dynamics data |
| GPCRmd [4] | Specialized MD database | GPCR-focused, 705 simulations | Membrane protein folding |
| AlphaFold2 [4] | Structure prediction | High accuracy static structures | Initial coordinates for MD |
| CoDNaS 2.0 [4] | Conformational diversity database | Native state ensembles | Conformational variability studies |
Signaling Pathways and Workflows
Protein Folding Pathway Validation Workflow
Folding Funnel with Key Intermediates
The resolution of Levinthal's paradox through energy landscape theory has fundamentally transformed our understanding of protein folding, replacing the concept of random search with guided navigation through funneled landscapes. This theoretical framework provides a robust foundation for integrating computational predictions with experimental validations, enabling increasingly accurate models of folding pathways.
Current research is extending these principles beyond single-domain folding to complex cellular processes. The emerging paradigm recognizes that protein function often depends on dynamic transitions between multiple conformational states rather than static structures [4]. Future advances will require continued development of multi-scale models that connect folding mechanisms to physiological contexts, including cotranslational folding, chaperone-assisted folding, and the impact of cellular environment on energy landscapes.
As computational methods continue to advance, particularly through machine-learned force fields and enhanced sampling techniques, and experimental approaches provide ever more detailed structural and dynamic information, we move closer to a comprehensive understanding of protein folding that bridges from quantum mechanics to biological function. This integrated approach promises not only to solve the fundamental challenge posed by Levinthal but to enable predictive modeling of protein behavior in health and disease.
The remarkable success of AI-based protein structure prediction systems, acknowledged by the 2024 Nobel Prize in Chemistry, has created a paradigm where three-dimensional protein structures can be determined from sequence alone with unprecedented accuracy [9] [10]. However, this triumph of static structure prediction has inadvertently overshadowed a more fundamental biological process: how proteins dynamically navigate their conformational landscape to reach these native states. For researchers in drug discovery and biomedical science, this folding process is not merely academic; misfolded proteins underlie pathologies from Alzheimer's disease to Type II Diabetes, and a protein's folding pathway can determine its functional state, cellular localization, and susceptibility to aggregation [11] [9]. While static snapshots provide crucial architectural blueprints, they cannot reveal the dynamic journey—the multiple routes, transitional intermediates, and kinetic traps—that proteins experience in living systems. This guide examines the critical experimental and computational methodologies bridging this gap, comparing their capabilities in validating and elucidating these essential biological pathways.
Fundamental Concepts: From Energy Landscapes to Multiple Pathways
The conceptual framework for understanding protein folding has evolved significantly from simplistic linear models to a more nuanced energy landscape theory. This theory visualizes folding as a funnel-like multidimensional surface where a protein navigates from an ensemble of unfolded states toward the native conformation with the lowest free energy [12]. A key implication of this landscape is the potential existence of multiple folding pathways, where different molecules of the same protein may reach the identical native state via distinct structural routes [13] [12].
The question of whether proteins with similar architectures fold via conserved pathways remains actively debated. Experimental studies comparing proteins with similar tertiary structures but divergent sequences reveal that some folds display highly conserved transition state structures, while others do not [14]. This suggests that certain topologies may restrict folding to a limited number of pathways, whereas others permit many potential routes to the native state [14]. This principle extends beyond proteins to RNA molecules, where studies have demonstrated that co-transcriptional folding during synthesis in the cellular environment can steer molecules along pathways distinct from those taken during refolding of full-length sequences in vitro [15].
Computational Approaches for Pathway Prediction
Computational methods, particularly Molecular Dynamics (MD) simulations, provide the primary tools for generating atomic-resolution hypotheses about folding pathways. The table below compares the fundamental approaches used to simulate these dynamic processes.
Table 1: Computational Methods for Simulating Folding Pathways
| Method | Fundamental Principle | Key Applications | Notable Limitations |
|---|---|---|---|
| Classical MD [13] | Numerically solves Newton's equations of motion for all atoms under physiological conditions. | Simulating unfolding at high temperature; analyzing denatured state ensembles. | Extremely computationally expensive; limited to microsecond-millisecond timescales. |
| Essential Dynamics Sampling (EDS) [16] | Biases MD simulation to explore configurations along collective motions derived from native state dynamics. | Folding simulations from unfolded states; studying large proteins like cytochrome c. | Relies on predefined collective coordinates; may miss novel pathways. |
| Targeted MD [16] | Applies time-dependent harmonic restraints to steer the system from an initial to a target structure. | Calculating reaction paths between two known conformations. | The chosen path may not be the physiologically relevant one. |
| AI-Based Prediction (AlphaFold) [17] [9] [10] | Uses deep learning on known structures to predict static native conformations from sequence. | Rapid generation of native state models; protein-ligand interaction prediction. | Provides static snapshots; does not model folding kinetics or pathways. |
A significant challenge in comparing MD-generated pathways is developing robust analytical methods. Researchers employ both geometry-based approaches (like root-mean-squared deviation between structures) and property-based analyses (tracking time-dependent changes in parameters like radius of gyration or solvent-accessible surface area) to objectively compare multiple unfolding trajectories and identify convergent and divergent pathways [13].
Experimental Methods for Pathway Validation
Computational predictions require rigorous experimental validation. The following table summarizes key techniques used to probe folding pathways and their specific applications in pathway characterization.
Table 2: Experimental Methods for Validating Folding Pathways
| Method | Measured Parameter | Application to Folding Pathways | Key Innovation for Pathway Analysis |
|---|---|---|---|
| Φ-value (Phi) Analysis [14] | Changes in transition state stability upon mutation. | Inferring transition state structure and key stabilizing residues. | Quantitative comparison of folding pathways for proteins with similar structures. |
| Single-Molecule Spectroscopy [12] | FRET efficiency, force-extension curves of individual molecules. | Direct detection of multiple pathways and transient intermediates. | Observes heterogeneity hidden in bulk measurements; reveals parallel pathways. |
| Bulk Kinetics with Multiple Probes [12] | Fluorescence, circular dichroism, NMR chemical shift. | Detecting sequence of structure formation from different structural perspectives. | Using multiple probes on the same protein can reveal pathway complexity. |
| cDNA Display Proteolysis [18] | Protease resistance of protein variants linked to their cDNA. | Mega-scale measurement of folding stability for hundreds of thousands of variants. | Identifies stability determinants and quantifies thermodynamic couplings. |
A critical advancement is the development of high-throughput experimental methods like cDNA display proteolysis. This method combines cell-free molecular biology with next-generation sequencing to measure thermodynamic folding stability for up to 900,000 protein domains in a single experiment [18]. By comprehensively measuring all single mutants across hundreds of natural and designed domains under identical conditions, this approach provides the quantitative data necessary to test computational predictions of how sequence encodes folding behavior on an unprecedented scale [18].
Detailed Experimental Protocol: cDNA Display Proteolysis
The following workflow outlines the key steps in this high-throughput stability profiling method [18]:
- Library Preparation: A DNA library is created using synthetic oligonucleotide pools, where each oligonucleotide encodes a single test protein variant.
- cDNA Display: The DNA library is transcribed and translated in vitro using a cell-free cDNA display system, resulting in each protein being covalently attached to its own encoding cDNA at the C-terminus.
- Proteolysis Reaction: The protein-cDNA complexes are incubated with a series of increasing concentrations of protease (e.g., trypsin or chymotrypsin).
- Pull-Down and Quantification: Protease-resistant (folded) proteins are isolated via a pull-down step targeting an N-terminal tag. The relative abundance of each variant in the surviving pool is quantified by deep sequencing.
- Stability Inference: A Bayesian kinetic model is applied to the sequencing count data. The model, based on single-turnover protease cleavage kinetics, infers a K50 value (protease concentration for half-maximal cleavage) for each sequence. Thermodynamic folding stability (ΔG) is then calculated by comparing the measured K50 to the sequence's estimated susceptibility in the unfolded state (K50,U) and a universal folded state susceptibility (K50,F).
Diagram 1: cDNA Display Proteolysis Workflow
Integrated Workflow: Validating Predicted Pathways
Bridging computational prediction and experimental validation requires a structured, iterative workflow. The following diagram synthesizes the methodologies from previous sections into a cohesive framework for testing and refining models of protein folding pathways.
Diagram 2: Pathway Validation Workflow
Successful investigation of folding pathways relies on a suite of specialized reagents, databases, and computational tools. The following table details key resources for researchers in this field.
Table 3: Essential Research Reagents and Resources
| Resource Name | Type | Primary Function in Pathway Research |
|---|---|---|
| ACPro Database [11] | Curated Database | Provides verified protein folding kinetics data (lnkf) and experimental conditions for 126 proteins, enabling confident benchmarking of predictive models. |
| AlphaFold Protein Structure Database [17] | Structure Database | Offers open access to over 200 million predicted protein structures, providing initial native state models for simulation and analysis. |
| AMBER ff99bsc0+χOL3 [15] | Force Field | A refined all-atom force field for MD simulations of nucleic acids, critical for studying RNA folding pathways and co-transcriptional folding. |
| GROMACS [16] | MD Software Package | A high-performance molecular dynamics toolkit used to simulate folding/unfolding trajectories with various force fields. |
| cDNA Display Proteolysis Library [18] | Experimental Reagent | A mega-scale library of protein variants enables high-throughput measurement of folding stability for mutational scanning. |
The journey to fully understand protein folding is transitioning from a focus on static endpoints to a dynamic investigation of pathways. While AI-based structure prediction provides an invaluable starting point, the biological imperative lies in deciphering the kinetic and thermodynamic principles that govern the folding process itself [9] [10]. The synergy between sophisticated computational simulations like MD and groundbreaking high-throughput experimental methods is creating an unprecedented opportunity to achieve this goal. For drug discovery professionals and researchers, embracing this shift from static snapshots to dynamic pathways is not merely an academic exercise. It is essential for understanding disease mechanisms, designing stable biologics, and developing therapeutic strategies that target the folding process itself. The future of structural biology lies not just in knowing the destination, but in comprehensively mapping the journey.
The release of AlphaFold represents a watershed moment in structural biology, largely solving the decades-old protein structure prediction problem with unprecedented accuracy. By demonstrating remarkable performance in CASP14 with a global distance test (GDT) score exceeding 90, AlphaFold achieved approximately three times the accuracy of the next best method and a level comparable to experimental methods [19] [20]. This breakthrough has democratized access to protein structural information, with the AlphaFold Protein Structure Database now providing over 200 million predicted structures to the scientific community and supporting research into diseases, antibiotic resistance, and crop resilience [21] [20].
However, beneath this success lies a significant limitation: despite its exceptional ability to predict static structures, AlphaFold provides limited insight into protein dynamics—the conformational changes, folding pathways, and transient states that underlie biological function. This review examines AlphaFold's performance against alternative computational approaches, with a specific focus on its capabilities and limitations in capturing the dynamic nature of proteins, a crucial aspect for understanding cellular mechanisms and advancing rational drug design.
Performance Comparison: AlphaFold Versus the Computational Toolkit
Accuracy in Static Structure Prediction
Independent validations against experimental structures consistently demonstrate AlphaFold's superiority in predicting static protein folds. In comprehensive comparisons with experimental nuclear receptor structures, AlphaFold 2 achieves high accuracy for stable conformations with proper stereochemistry, though systematic limitations emerge in capturing flexible regions and ligand-binding pockets [22].
Table 1: Quantitative Performance Comparison of Protein Structure Prediction Tools
| Method | Primary Strength | Global Structure Accuracy (GDT_TS) | Ligand Binding Pocket Prediction | Dynamic Sampling |
|---|---|---|---|---|
| AlphaFold 2/3 | Static fold accuracy | ~90 [19] | Underestimates volumes by 8.4% on average [22] | Single conformation [23] [24] |
| Molecular Dynamics (mdCATH) | Conformational sampling | N/A (sampling method) | Captues flexibility [25] | Excellent (62 ms accumulated simulation) [25] |
| Boltz 2 | Binding affinity prediction | High (comparable to AF3) [24] | Dual-head affinity prediction [24] | Limited multi-conformation handling [24] |
| Traditional Docking | Pose prediction | N/A | ~60% accuracy (vs AF3's 93%) [26] | Rigid or flexible docking options |
The table reveals a consistent pattern: while AlphaFold excels at global structure prediction, it systematically underestimates ligand-binding pocket volumes by 8.4% on average and misses functionally important conformational diversity, particularly in homodimeric receptors where experimental structures show functionally important asymmetry [22].
Case Studies Highlighting Limitations in Dynamic Prediction
Real-world applications underscore these limitations, particularly for complex multi-domain proteins and flexible systems:
- Marine Sponge Receptor (SAML): A striking case study reveals severe deviations between experimental and AlphaFold-predicted structures for a two-domain protein, with positional divergences beyond 30 Å and an overall RMSD of 7.7 Å. The relative orientation between domains was incorrectly predicted despite moderate confidence metrics (pLDDT) from AlphaFold [27].
- Nuclear Receptor Flexibility: Comprehensive analysis shows that while AlphaFold accurately predicts stable conformations of nuclear receptors, it captures only single conformational states, missing the spectrum of biologically relevant states particularly in flexible ligand-binding domains, which show higher structural variability (CV = 29.3%) compared to more rigid DNA-binding domains (CV = 17.7%) [22].
- Adversarial Testing Reveals Physical Principles Gap: When challenged with binding site mutagenesis that should physically displace ligands, co-folding models like AlphaFold 3 continue to predict biologically implausible binding modes, indicating potential overfitting to training data rather than learning fundamental physical principles [26].
Experimental Protocols for Validation
Experimental Validation Workflows
The accuracy of computational predictions must be validated against experimental data through standardized protocols:
Table 2: Experimental Validation Methods for Computational Predictions
| Experimental Method | Validation Target | Protocol Summary | Key Findings for AlphaFold |
|---|---|---|---|
| X-ray Crystallography | Global fold accuracy | Molecular replacement using predicted structures as search models | AF2 structures work well as search models, closely resembling crystal structures [19] |
| Cryo-EM | Complex architecture | Fitting predicted models into experimental density maps | AF2 structures fit well into cryo-EM maps [19] |
| NMR Spectroscopy | Solution-state conformation | Comparing predicted models with NMR-derived structures | Excellent fit in majority of cases, indicating predictions not overly biased to crystal state [19] |
| Cross-linking Mass Spectrometry | Distance constraints | Validating residue-residue distances in predicted models | Majority of AF2 predictions correct for single chains and complexes [19] |
| Molecular Dynamics | Conformational stability | Running simulations from predicted structures | mdCATH dataset provides 62 ms simulation data for validation [25] |
Diagram 1: Experimental validation workflow for computational predictions
Methodologies for Assessing Dynamic Properties
Specialized experimental and computational protocols are required to evaluate protein dynamics:
Molecular Dynamics Simulation Protocol (based on mdCATH dataset generation):
- System Preparation: Protein domains solvated in TIP3P water with at least 9 Å padding, neutralized and ionized with Na+ and Cl− ions at 0.150 M concentration [25]
- Force Field Parameterization: CHARMM22* forcefield with particle-mesh Ewald summation for long-range electrostatics [25]
- Sampling Strategy: Five replicates at five temperatures (320 K, 348 K, 379 K, 413 K, 450 K) in geometric progression to capture thermal unfolding [25]
- Data Collection: Coordinates and forces recorded every 1 ns, with over 62 ms of accumulated simulation time enabling statistical analysis of unfolding thermodynamics and kinetics [25]
Sequence-Based Dynamics Prediction (based on folding dynamics method):
- Evolutionary Field Extraction: Obtaining precise evolutionary field from observed variations in homologous protein sequences [28]
- Energy Mapping: Mapping energetics to coarse-grained folding model treating protein as string of interacting foldons [28]
- Equilibrium Calculation: Computing equilibrium folding curve and identifying emergence of protein folding sub-domains for any given protein sequence [28]
- Mutation Analysis: Analyzing how mutations perturb both folding stability and cooperativity [28]
Table 3: Key Research Reagents and Computational Resources
| Resource | Type | Primary Function | Access Information |
|---|---|---|---|
| AlphaFold Protein Structure Database | Database | Access to 200+ million predicted structures | https://www.alphafold.ebi.ac.uk/ [20] |
| mdCATH Dataset | Molecular Dynamics Dataset | Proteome-wide dynamic trajectories for 5,398 domains | Available at HuggingFace under CC BY 4.0 license [25] |
| CHARMM22* | Force Field | Empirical energy functions for MD simulations | Standard parameterization in MD packages [25] |
| AlphaFold Server | Prediction Tool | Biomolecular interaction predictions powered by AF3 | Free for non-commercial research [20] |
| Boltz 2 | Prediction Tool | Binding affinity prediction with physics-based steering | Open access to model weights and inference pipeline [24] |
Integrating Static and Dynamic Approaches
The limitations of current AI methods in capturing protein dynamics have prompted the development of integrated approaches that combine deep learning with physics-based simulations:
Diagram 2: Integration of static and dynamic prediction methods
Emerging solutions address AlphaFold's dynamical limitations through several mechanisms:
- Neural Network Potentials: Machine learning approaches that enhance computational protein research by enabling more accurate simulations of dynamic behaviors, potentially trained on MD datasets like mdCATH that include instantaneous forces [25].
- Physics-Informed AI: Models like Boltz 2 that incorporate physics-based steering during inference to improve physical plausibility and overcome limitations like steric clashes and incorrect stereochemistry [24].
- Multi-Temperature Sampling: The mdCATH approach of simulating at multiple temperatures (320-450 K) captures a variety of conformations, including higher energy states encountered during molecular dynamics simulations [25].
AlphaFold has unquestionably revolutionized structural biology by providing rapid, accurate protein structure predictions at an unprecedented scale. However, its limitation in capturing protein dynamics represents a significant frontier for future development. The integration of deep learning with physics-based simulations, enhanced by comprehensive dynamical datasets like mdCATH, points toward a future where computational methods can accurately predict both protein structures and their dynamic behaviors.
For researchers in drug discovery and protein engineering, this evolution is critical. Understanding conformational dynamics, allosteric mechanisms, and folding pathways will enable more sophisticated interventions in biological systems. As the field progresses, the combination of AlphaFold's structural accuracy with the dynamic sampling of molecular dynamics and the emerging class of hybrid AI-physics models promises a more complete computational understanding of protein function, ultimately accelerating therapeutic development and fundamental biological discovery.
Why Validate? Addressing the Sampling and Accuracy Problems in MD Simulations
Molecular Dynamics (MD) simulation has emerged as a fundamental tool for studying protein folding and dynamics at atomic resolution, offering insights that often remain elusive to experimental methods alone [29]. However, MD simulations face two fundamental challenges that necessitate rigorous validation: the sampling problem, where simulations often fail to explore all relevant conformational states due to high-energy barriers and limited timescales, and the accuracy problem, where force field inaccuracies and numerical artifacts can produce unrealistic dynamics [30] [31]. Without proper validation, researchers risk drawing conclusions from incomplete or physically implausible simulations, potentially leading to misleading scientific interpretations [31].
The validation gap becomes particularly critical in the context of protein folding pathway prediction, where the transition between unfolded and native states involves numerous metastable intermediates that are difficult to sample comprehensively [32]. As MD simulations increasingly inform drug discovery and protein engineering, establishing robust validation frameworks ensures that computational predictions align with biophysical reality. This review examines how integrating experimental data with enhanced sampling algorithms and emerging artificial intelligence approaches addresses these fundamental challenges, creating more reliable frameworks for understanding protein folding mechanisms.
Understanding the Core Problems in MD Simulations
The Sampling Problem: Limited Timescales and Energy Barriers
The sampling problem in MD simulations stems from the rough energy landscapes characteristic of biomolecular systems, with many local minima separated by high-energy barriers that govern biomolecular motion [30]. This landscape topography makes it easy for simulations to become trapped in non-functional states for durations exceeding practical simulation timescales. As noted in research on enhanced sampling techniques, "insufficient sampling often limits MD application" due to these inherent energy landscape characteristics [30].
The temporal limitations of MD further exacerbate this sampling challenge. Despite advances in computing power, all-atom MD simulations typically run for tens to hundreds of nanoseconds, up to 1-2 microseconds for state-of-the-art setups [29]. This remains insufficient for many biologically relevant processes, including the folding of many proteins, where folding times can range from microseconds to seconds or longer near physiological conditions. As one analysis of protein folding simulations noted, "refolding from extended states using explicit solvent has been out of reach at these timescales" for many systems of biological interest [29].
The consequences of inadequate sampling are profound for folding pathway studies. A single trajectory rarely captures all relevant conformations, particularly for biological systems with vast conformational spaces that must overcome numerous energy barriers to explore significant states [31]. Without sufficient sampling, simulations may follow pathways that are not statistically representative, potentially missing rare but functionally crucial transition states or intermediates.
The Accuracy Problem: Force Fields and Physical Realism
Beyond sampling limitations, accuracy problems present equally significant challenges for reliable MD simulations. Force field selection profoundly impacts simulation outcomes, as these mathematical models are carefully designed and parameterized for specific molecular classes [31]. Using an inappropriate force field—such as applying a protein-specific model to carbohydrates or nucleic acids—leads to inaccurate energetics, incorrect conformations, or unstable dynamics [31].
Physical realism can be compromised through various other mechanisms as well:
- Incorrect protonation states or missing atoms in starting structures [31]
- Inappropriate simulation parameters, including timestep selection, thermostat/barostat settings, and treatment of periodic boundary conditions [31]
- Mixing incompatible force fields without proper parameterization, disrupting the balance between bonded and non-bonded interactions [31]
- Inadequate equilibration, resulting in systems that don't represent the correct thermodynamic ensemble [31]
These accuracy concerns are particularly problematic because, as noted in common MD mistakes, "MD engines will happily simulate a system even when key components are incorrect" [31]. The simulation may run without crashing while producing physically meaningless results, creating a false sense of security for researchers.
Established Solutions: Enhanced Sampling and Experimental Integration
Enhanced Sampling Algorithms
To address the sampling problem, several enhanced sampling algorithms have been developed that accelerate exploration of conformational space. These methods effectively reduce the energy barriers that limit sampling in conventional MD simulations. The table below compares three major enhanced sampling approaches:
Table 1: Enhanced Sampling Techniques for MD Simulations
| Method | Key Principle | Best For | Limitations |
|---|---|---|---|
| Replica-Exchange MD (REMD) | Parallel simulations at different temperatures exchange configurations [30] | Studying free energy landscapes and folding mechanisms [30] | Computational cost increases with system size; temperature selection critical [30] |
| Metadynamics | "Fills free energy wells" with computational bias potential to discourage revisiting states [30] | Protein folding, molecular docking, conformational changes [30] | Depends on proper selection of a small set of collective coordinates [30] |
| Simulated Annealing | Gradual temperature decrease to reach global energy minimum [30] | Characterizing very flexible systems; large macromolecular complexes [30] | May require multiple runs with different cooling schedules [30] |
These algorithms have demonstrated particular value in studying protein folding pathways. For example, replica-exchange molecular dynamics has been successfully employed to study free energy landscapes and folding mechanisms of various peptides and proteins [30]. Metadynamics has proven effective for exploring protein folding landscapes and conformational changes that would be inaccessible to conventional MD [30].
Experimental Data Integration
Integrating experimental data with MD simulations provides a powerful approach to addressing both sampling and accuracy problems. Biophysical methods including NMR, EPR, HDX-MS, SAXS, and cryo-EM provide valuable but often indirect signals about protein structure and dynamics [33]. Integrative modeling approaches combine these experimental data with physics-based simulations to reveal both stable structures and transient, functionally important intermediates [33].
The workflow below illustrates how experimental data can be integrated with MD simulations to validate and refine folding pathway predictions:
Figure 1: Integrative Framework for Validating Folding Pathways
This integrative approach is particularly valuable for characterizing partially folded states that are "heterogeneous, consisting of many rapidly exchanging conformations" [29]. Ensemble averaging from such states complicates the interpretation of experimental data, while MD provides a molecular framework for interpretation. For example, experimental observables such as B-factors from X-ray crystallography can be compared to root mean square fluctuations (RMSF) from simulations, while NMR measurements like Nuclear Overhauser Effect (NOE) distances and scalar coupling constants can be compared to their simulated counterparts [31].
AI-Generated Ensemble Approaches: A Paradigm Shift
Next-Generation Generative Models for Protein Dynamics
Recent advances in artificial intelligence have introduced a new paradigm for addressing sampling limitations in MD simulations. Generative AI models trained on MD simulation data can now produce structural ensembles at a fraction of the computational cost, effectively learning the underlying physics from expensive simulations and generating diverse conformations without simulating every intermediate step [34].
Several innovative architectures have emerged in this space:
- BioEmu: A diffusion model-based generative AI system that simulates protein equilibrium ensembles with 1 kcal/mol accuracy using a single GPU, achieving a 4-5 orders of magnitude speedup for equilibrium distributions in folding and native-state transitions compared to traditional MD [35].
- AlphaFlow: An AF2-based generative model trained on the ATLAS dataset of protein simulations that accurately reproduces residue fluctuations but struggles with complex multi-state ensembles [34].
- aSAM/aSAMt: A latent diffusion model that generates heavy atom protein ensembles, with a temperature-conditioned version (aSAMt) capable of producing conformational ensembles under varying environmental conditions [34].
These approaches represent a fundamental shift from simulating dynamics to learning and generating physically realistic ensembles. As the BioEmu developers note, their approach "overcomes the sampling bottleneck of traditional MD simulations," sampling thousands of structures per hour on a single GPU compared to months on supercomputing resources [35].
Performance Comparison: AI vs. Traditional Methods
The table below quantitatively compares the performance of AI-based generative approaches with traditional MD simulations for ensemble generation:
Table 2: Performance Comparison of MD and AI-Based Ensemble Generation Methods
| Method | Computational Cost | Sampling Rate | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Conventional MD | Months on supercomputers [35] | Limited by simulation time | Physical realism, explicit solvent [29] | Inadequate for large conformational changes [30] |
| Enhanced Sampling MD | Days to weeks on HPC clusters [30] | Improved for specific coordinates | Accelerates barrier crossing [30] | Requires prior knowledge; may bias sampling [30] |
| BioEmu | Hours on single GPU [35] | Thousands of structures/hour [35] | High thermodynamic accuracy; identifies cryptic pockets [35] | Primarily single-chain proteins; larger complexes require optimization [35] |
| AlphaFlow | Minutes to hours on GPU [34] | Varies by system size | Good local flexibility reproduction [34] | Struggles with multi-state ensembles; poor side chain torsions [34] |
| aSAM/aSAMt | Minutes to hours on GPU [34] | Varies by system size | Accurate backbone/side chain torsions; temperature conditioning [34] | Requires energy minimization; lower MolProbity scores [34] |
The performance advantages of AI approaches are particularly evident in their ability to capture thermodynamic properties. BioEmu demonstrates exceptional thermodynamic accuracy in quantitative prediction tasks, achieving less than 1 kcal/mol accuracy in relative free energy through its Property Prediction Fine-Tuning (PPFT) algorithm, which fine-tunes the model on hundreds of thousands of experimental stability measurements [35].
Implementation Guide: Validation Protocols and Research Tools
Essential Validation Workflow
Implementing a robust validation protocol is essential for ensuring the reliability of MD-predicted protein folding pathways. The following workflow outlines key validation steps:
Figure 2: MD Simulation Validation Workflow
This validation workflow emphasizes several critical aspects often overlooked in MD studies:
- Multiple replicate runs are essential because "a single trajectory rarely captures all relevant conformations" for biological systems with vast conformational spaces [31].
- Verification of equilibration through stabilization of key thermodynamic properties including temperature, pressure, total energy, and system density [31].
- Comparison with experimental observables such as B-factors, NMR measurements, or SAXS profiles to ensure physical realism [31].
Table 3: Essential Tools for MD Simulation and Validation
| Tool Category | Specific Tools | Key Function | Application in Folding Studies |
|---|---|---|---|
| Structure Preparation | PDBFixer, H++ [31] | Fix missing atoms; assign protonation states | Ensures realistic starting structures for folding simulations |
| MD Engines | GROMACS, AMBER, NAMD [30] [31] | Perform molecular dynamics calculations | Provides production MD with various enhanced sampling methods |
| Enhanced Sampling | PLUMED, COCOMO [30] [34] | Implement advanced sampling algorithms | Accelerates exploration of folding energy landscape |
| AI Generators | BioEmu, AlphaFlow, aSAM [35] [34] | Generate structural ensembles from learned distributions | Rapidly explores conformational diversity in folding pathways |
| Validation & Analysis | MDAnalysis, Bio3D, cpptraj [31] | Analyze trajectories and compare to experiments | Quantifies sampling quality and agreement with experimental data |
| Experimental Data Integration | MAXENT, Bayesian Weighing [33] | Incorporate experimental constraints | Refines ensembles using NMR, cryo-EM, SAXS data |
This toolkit provides researchers with essential resources for each stage of folding pathway investigation, from initial structure preparation to final validation against experimental data. Particularly important are tools for experimental data integration, which enable the "integrative approaches that combine experiments with physics-based simulations" needed to reveal both stable structures and transient intermediates [33].
The sampling and accuracy problems in MD simulations present significant but addressable challenges for predicting protein folding pathways. Traditional enhanced sampling algorithms combined with experimental validation provide robust frameworks for improving simulation reliability, while emerging AI-based generative models offer revolutionary advances in computational efficiency. The key insight across all methodologies is that validation against experimental data is not optional—it is essential for transforming computationally convenient narratives into scientifically valid mechanistic models.
As MD simulations continue to inform drug discovery—helping identify cryptic pockets in Fascin for anti-metastatic cancer drugs or revealing binding sites in sialic-acid binding factors for novel antibiotics [35]—the stakes for accurate folding pathway prediction continue to rise. The integration of physical simulations with AI acceleration and experimental constraints represents the most promising path forward, potentially enabling researchers to achieve both the sampling comprehensiveness and physical accuracy needed to fully elucidate protein folding mechanisms.
Integrated Workflows: Combining AI, MD, and Global Optimization for Pathway Prediction
Leveraging AI-Predicted Structures and Distograms as MD Starting Points
The integration of artificial intelligence (AI)-predicted protein structures into molecular dynamics (MD) simulations represents a rapidly evolving paradigm in computational structural biology. AI systems like AlphaFold have demonstrated remarkable accuracy in predicting static protein structures from amino acid sequences alone, even achieving accuracy competitive with experimental methods in many cases [36]. However, proteins are dynamic entities, and understanding their function, folding pathways, and functional mechanisms often requires insight into their conformational dynamics and energy landscapes. Molecular dynamics simulations provide this dynamical perspective by simulating the physical movements of atoms over time, but their success heavily depends on starting from physiologically relevant conformations [37]. This comparison guide objectively examines the performance of using AI-predicted structures—particularly atomic coordinates and distograms (pairwise distance maps)—as initial conditions for MD simulations, evaluating this hybrid approach against traditional MD initialization methods within the critical context of experimental validation.
Performance Comparison: AI-MD Hybrid vs. Traditional Approaches
The table below summarizes key performance metrics when using AI-predicted structures as MD starting points compared to traditional ab initio or homology-modeled starting structures.
Table 1: Performance Comparison of MD Starting Protocols
| Performance Metric | AI-Predicted Starting Structures | Traditional Homology Models | Ab Initio Folding |
|---|---|---|---|
| Time to Reach Converged Ensemble | Significantly reduced for structured regions [37] | Variable; depends on template quality | Prohibitively long for most proteins |
| Sampling of Rare/Transient States | Enhanced through bias-free initialization; improved identification of folding intermediates [37] [38] | Potentially biased by template conformation | Theoretically complete but practically unachievable |
| Accuracy vs. Experimental Data (NMR, SAXS) | Good agreement for ensemble-averaged properties; potential domain orientation errors [37] [27] | Good if correct template is used | Not applicable on relevant timescales |
| Computational Resource Requirements | Lower overall due to faster convergence [37] | Moderate | Extremely high |
| Applicability to IDPs/IDPRs | Emerging methods show promise [37] | Poor due to lack of structured templates | Only practical for very short peptides |
Quantitative Experimental Data
Several studies have provided quantitative data supporting the hybrid AI-MD approach:
Table 2: Experimental Validation Data for AI-MD Hybrid Methods
| Study System | Key Experimental Validation | Result of AI-MD Integration |
|---|---|---|
| ArkA IDP (Yeast) | Circular Dichroism (CD) Spectroscopy [37] | GaMD simulations initiated from AI-generated ensembles better matched experimental CD data, revealing proline isomerization as a conformational switch [37]. |
| SAML (Marine Sponge Receptor) | X-ray Crystallography [27] | Significant deviation (7.7 Å RMSD) in inter-domain orientation between AlphaFold prediction and experimental structure highlighted the need for MD refinement of AI-predicted multi-domain proteins [27]. |
| Ubiquitin | Topological Data Analysis of Folding Landscape [38] | Novel analysis methods on simulation data showed 10x speed improvement in identifying key topological folding features when leveraging efficient representations [38]. |
Experimental Protocols and Methodologies
Protocol 1: Generating AI-Initialized Conformational Ensembles for IDPs
Intrinsically Disordered Proteins (IDPs) lack stable tertiary structures, existing instead as dynamic ensembles. The following protocol leverages AI to generate structurally diverse starting ensembles for MD simulation of IDPs [37].
- Input Preparation: Provide the amino acid sequence of the IDP or IDPR (Intrinsically Disordered Protein Region).
- Deep Learning Sampling: Employ a deep learning model (e.g., trained on large-scale MD data or experimental ensemble data) to generate a diverse set of conformations. These models learn sequence-to-structure relationships without being constrained by physical force fields.
- Experimental Filtering: Filter the generated conformations against available experimental data, such as Small-Angle X-Ray Scattering (SAXS) profiles or NMR chemical shifts, to ensure the ensemble matches known average properties.
- Ensemble Selection: Select a representative subset of conformations spanning the diverse states identified by the AI.
- MD Simulation and Refinement: Use each selected conformation as a starting point for independent, explicit-solvent MD simulations. This step refines the structures and explores the local conformational landscape around each AI-generated state.
- Validation: Compare the resulting simulation trajectories and meta-stable states with experimental observables not used in the filtering step, such as FRET efficiency or hydrodynamic radius.
Protocol 2: Refining AI-Predicted Multi-Domain Proteins with MD
AI models can mispredict the relative orientation of protein domains [27]. This protocol uses MD to refine these structures.
- Initial Structure Prediction: Generate a 3D model of the multi-domain protein using AlphaFold2 or a similar tool.
- Inter-Domain Analysis: Inspect the Predicted Aligned Error (PAE) plot. High PAE between domains suggests low confidence in their relative orientation.
- Targeted MD Setup: If the PAE is high, or if an experimental structure of the full protein is available for validation, place the protein in a solvated simulation box. If the domain orientation is suspected to be incorrect, consider applying weak restraints to the intra-domain atomic coordinates to maintain domain integrity while allowing inter-domain movement.
- Enhanced Sampling: Run an enhanced sampling MD simulation (e.g., Gaussian Accelerated MD (GaMD) or replica-exchange MD). This accelerates the sampling of different domain orientations and identifies the most stable configuration.
- Experimental Cross-Validation: Validate the refined domain arrangement against experimental data. For example, if the crystal structure is available, calculate the RMSD of the MD-refined model. Alternatively, compare with SAXS data or other solution-phase techniques [27].
Diagram 1: AI-MD refinement workflow for multi-domain proteins.
Table 3: Key Resources for AI-MD Integration and Experimental Validation
| Tool / Resource | Type | Primary Function in Workflow |
|---|---|---|
| AlphaFold2/3[citation:2] | AI Structure Prediction | Provides high-accuracy initial atomic coordinates and per-residue/local distance confidence metrics (pLDDT/PAE). |
| RoseTTAFold[ [39]] | AI Structure Prediction | An alternative end-to-end deep learning model for protein structure prediction with capabilities similar to AlphaFold2. |
| GROMACS/AMBER[ [37]] | Molecular Dynamics Engine | Performs the actual MD simulations for refining structures and exploring dynamics using physics-based force fields. |
| Gaussian Accelerated MD (GaMD)[ [37]] | Enhanced Sampling Method | Accelerates the sampling of rare events (e.g., domain reorientation, proline isomerization) in MD simulations. |
| cDNA Display Proteolysis[ [18]] | High-Throughput Experiment | Measures thermodynamic folding stability for hundreds of thousands of protein variants, providing large-scale data for model training and validation. |
| SAXS[ [37]] | Biophysical Technique | Provides low-resolution structural data in solution, used to validate the overall shape and dimensions of AI-generated and MD-refined ensembles. |
| NMR Spectroscopy[ [37]] | Biophysical Technique | Provides atomic-level data on dynamics and chemical environments in solution, a key benchmark for validating MD-predicted conformational states. |
The integration of AI-predicted structures and distograms as starting points for MD simulations presents a powerful hybrid methodology that combines the strengths of deep learning and physics-based simulation. Quantitative data show this approach can significantly accelerate the convergence of MD simulations and enhance the sampling of functionally relevant states, particularly for complex systems like IDPs. However, performance is not universally superior; key limitations remain, especially regarding the prediction of inter-domain orientations in multi-domain proteins and the inherent biases of AI models trained on existing data. The validity of any computational model, including this hybrid approach, must be rigorously assessed against experimental data. The continued development of high-throughput experimental methods, such as cDNA display proteolysis, will provide the essential benchmark data needed to further refine and validate these integrated computational strategies, ultimately enhancing their reliability for drug development and basic biological research.
The accurate prediction of protein folding pathways represents a central challenge in computational biology, with significant implications for understanding cellular function, molecular disease mechanisms, and drug development. This guide objectively compares methodologies for studying these pathways, focusing on their validation against experimental data. While "Action-CSA" is referenced in the title per your requirement, the specific technical details of this particular methodology were not identified in the available literature. This overview instead focuses on well-documented related techniques in the field, framing them within the research context of validating molecular dynamics (MD)-predicted protein folding pathways with experimental data.
The following sections compare the performance, experimental protocols, and applications of these methods, providing researchers with a practical framework for selecting and implementing pathway search methodologies.
Performance Comparison of Pathway Search and Validation Methods
The table below summarizes the primary computational methods used in protein folding studies, highlighting their respective performance characteristics and validation approaches.
Table 1: Comparison of Protein Folding and Stability Analysis Methods
| Method Name | Method Type | Primary Application | Key Performance Metrics | Typical Validation Data |
|---|---|---|---|---|
| EFoldMine [40] [41] | Machine Learning (SVM) | Early Folding Residue (EFR) Prediction | Sensitivity: 73.1%, Specificity: 75.2%, AUC: 80.8% [40] | NMR Pulsed-Labelling HDX [40] |
| QresFEP-2 [42] | Physics-Based (MD/FEP) | Protein Stability & Mutational Effects | High accuracy on 600+ mutations; High computational efficiency [42] | Experimental Protein Stability Data (Thermal Denaturation) [42] |
| Molecular Dynamics (MD) [43] | Physics-Based Simulation | VLP Stability & Self-Assembly | Predicts surface hydrophobicity & structural stability [43] | Experimental Hydrophobicity & Stability Assays [43] |
| AlphaFold Systems [44] | Deep Learning | Protein Structure Prediction | High Reliability in Protein Domain Folding (CASP16) [44] | Experimental Structures (e.g., X-ray, Cryo-EM) [44] |
Detailed Experimental Protocols
EFoldMine: Protocol for Early Folding Residue Prediction
EFoldMine is a sequence-based predictor that identifies residues involved in the initial stages of folding, providing a target for validating MD-predicted pathways [40].
- Input Feature Generation: For a given protein sequence, calculate three sets of features for each residue:
- Model Application: Process the feature vector for each residue using the pre-trained Support Vector Machine (SVM) with a Radial Basis Function (RBF) kernel. The model outputs a continuous "early folding score" for each residue position [40].
- Validation with Experimental Data:
- Data Source: Perform or obtain NMR pulsed-labelling Hydrogen-Deuterium Exchange (HDX) experiments. This technique identifies backbone amide protons protected from exchange due to stable hydrogen bonding within milliseconds of folding initiation [40].
- Correlation: Compare computationally predicted EFRs with experimentally identified protected residues. A successful prediction shows significant overlap, providing residue-level validation for proposed folding nuclei [40].
QresFEP-2: Protocol for Free Energy Calculation
QresFEP-2 is a hybrid-topology Free Energy Perturbation (FEP) protocol used to quantify the effects of point mutations on protein stability, testing specific hypotheses about the energetic contributions of residues along a folding pathway [42].
- System Setup:
- Construct a hybrid topology file for the wild-type (WT) and mutant protein. This topology uses a single representation for the conserved protein backbone and dual representations for the differing side-chain atoms [42].
- Embed the protein in a spherical water droplet with appropriate boundary conditions [42].
- Alchemical Transformation:
- Define a thermodynamic pathway (λ-schedule) that gradually transforms the WT side chain into the mutant side chain.
- Run molecular dynamics simulations at intermediate λ-states to sample the conformational space [42].
- Free Energy Analysis:
- Use the Bennett Acceptance Ratio (BAR) or similar method to calculate the relative free energy change (ΔΔG) between the WT and mutant from the simulation data [42].
- Validation with Experimental Data:
- Data Source: Obtain experimental stability data, such as changes in melting temperature (ΔTm) or free energy of unfolding (ΔΔGunfolding), from techniques like differential scanning calorimetry or circular dichroism for a set of mutations [42].
- Benchmarking: Calculate the correlation coefficient (R²), mean absolute error (MAE), and root mean square error (RMSE) between the computed ΔΔG values and the experimental measurements [42].
MD Simulation: Protocol for Assembly Pathway Validation
Molecular dynamics can simulate the stability and assembly of complex structures like virus-like particles (VLPs), providing insights into supramolecular folding pathways [43].
- Model Construction:
- Build a partial VLP model comprising multiple protein chains (e.g., 17 chains of a modified Hepatitis B core protein) based on a known structural template [43].
- Simulation Execution:
- Solvate the model in an explicit water box with ions.
- Run extensive MD simulations (nanoseconds to microseconds) under physiological temperature and pressure to observe stability and subunit interactions [43].
- Property Calculation:
- Analyze simulation trajectories to calculate metrics such as root mean square deviation (RMSD) for stability, radius of gyration (Rg) for compactness, and solvent-accessible surface area (SASA) for surface hydrophobicity [43].
- Validation with Experimental Data:
- Data Source: Perform biochemical assays on the actual VLP candidates. This can include hydrophobicity probes (e.g., ANS dye binding) and stability assays under various stress conditions (e.g., temperature, pH) [43].
- Correlation: Compare the computationally derived properties (e.g., SASA) with experimental measurements (e.g., fluorescence from hydrophobicity dyes). A strong correlation validates the MD model's predictive power for guiding design [43].
Workflow Visualization
The following diagram illustrates the integrated computational and experimental workflow for validating predicted protein folding pathways, synthesizing the protocols described above.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful validation of folding pathways relies on specific experimental reagents and computational tools.
Table 2: Key Reagents and Materials for Folding Pathway Research
| Item Name | Function / Application | Relevance to Pathway Validation |
|---|---|---|
| Deuterated Solvent (D₂O) [40] | Solvent for NMR-based Hydrogen-Deuterium Exchange (HDX) experiments. | Enables tracking of protein folding kinetics by identifying backbone amides protected from exchange. |
| Stability Dyes (e.g., ANS) [43] | Fluorescent probes that bind hydrophobic surfaces. | Used to experimentally measure surface hydrophobicity of folding intermediates or designed proteins, validating MD predictions. |
| QresFEP-2 Software [42] | Open-source FEP software integrated with the Q molecular dynamics package. | Calculates the change in free energy upon mutation, providing a physics-based measure of residue stability for benchmark. |
| STRIDE or DSSP | Algorithms for assigning secondary structure from 3D atomic coordinates. | Used to analyze MD simulation trajectories, tracking the formation and dissolution of secondary structures during folding. |
| Start2Fold Database [40] | Public database of experimental data on protein early folding. | Provides a critical benchmark dataset for training and validating computational predictors like EFoldMine. |
The classical challenge of simulating protein dynamics is the immense computational cost of achieving sufficient sampling, particularly for complex processes like folding or the exploration of conformational landscapes by intrinsically disordered proteins (IDPs). Traditional all-atom Molecular Dynamics (MD) simulations, while highly accurate, are often prohibitively expensive, requiring supercomputers and months of computation to capture rare events [35]. Machine Learning (ML), particularly deep generative models, has emerged as a powerful alternative, offering speedups of several orders of magnitude. However, purely data-driven ML models can sometimes learn statistical shortcuts from their training data rather than underlying physical principles, potentially limiting their generalizability to unseen systems [45]. This guide examines the current state of hybrid pipelines that integrate ML and MD to overcome these individual limitations. By combining the physical rigor of MD with the scalability of ML, these hybrid approaches are enabling the determination of accurate, experimentally-validated conformational ensembles, thereby providing powerful tools for drug discovery and basic research [46] [37].
Comparative Analysis of ML/MD Hybrid Methodologies
The table below summarizes the core architectural and performance characteristics of several contemporary hybrid pipelines, highlighting their distinct approaches to integrating machine learning with molecular dynamics.
Table 1: Comparison of Modern ML/MD Hybrid Pipelines for Conformational Ensemble Generation
| Pipeline Name | Core ML Methodology | MD Integration & Role | Reported Speedup vs. Traditional MD | Key Validation Metrics | Primary Application Scope |
|---|---|---|---|---|---|
| CGSchNet [6] | Deep neural network force field | Bottom-up learning from all-atom MD training data | Several orders of magnitude | Fraction of native contacts, Cα RMSD, folding free energies | Transferable coarse-grained simulation of folded and disordered proteins |
| BioEmu [35] | Diffusion model | Trained on large-scale MD datasets and experimental data; emulates equilibrium ensembles | 4-5 orders of magnitude (on a single GPU) | ~1 kcal/mol thermodynamic accuracy, success rates (55-90%) on domain motion benchmarks | Single-chain protein equilibrium ensembles, cryptic pocket prediction |
| MaxEnt Reweighting [46] | Maximum entropy principle | Reweights frames from long-timescale unbiased MD simulations | N/A (Post-processing of MD data) | Kish ratio, agreement with NMR chemical shifts, SAXS data | Determining force-field independent atomic-resolution ensembles of IDPs |
| DEERFold [47] | Fine-tuned AlphaFold2 | Guided by experimental distance distributions (e.g., from DEER spectroscopy) | N/A (Structure prediction) | Accuracy in switching conformations of membrane transporters | Modeling conformational selection using sparse experimental restraints |
| Hybrid MD-kMC [48] | Kinetic Monte Carlo (kMC) | MD used for local dynamics; kMC for rare events (e.g., secondary/tertiary structure formation) | Faster folding kinetics achieved | Folding intermediates, agreement with experimental folding rates | Protein folding in explicit solvent, pathway exploration |
A critical analysis of these pipelines reveals a trade-off between computational efficiency and physical granularity. CGSchNet and BioEmu represent a paradigm shift toward pure ML emulation, achieving massive speedups by learning to directly generate statistical ensembles from underlying MD or experimental data [6] [35]. In contrast, the MaxEnt Reweighting approach [46] and the Hybrid MD-kMC algorithm [48] represent a tighter, more iterative integration. MaxEnt uses MD as the foundational sampling engine and applies ML principles a posteriori to bias the ensemble toward experimental reality, effectively correcting for force field inaccuracies. The MD-kMC hybrid uses ML-like concepts (kinetic move sets) to steer the MD simulation itself, enabling efficient exploration of complex folding pathways that would be inaccessible to either method alone.
Experimental Protocols and Workflows in Practice
Protocol 1: ML-Driven Coarse-Graining for Transferable Force Fields
The development of a transferable coarse-grained (CG) model like CGSchNet exemplifies a bottom-up hybrid workflow. The protocol involves several key stages [6]:
- Training Set Generation: A diverse dataset of all-atom explicit solvent MD simulations is generated for a wide array of small proteins and peptides. This dataset must encompass varied folded structures and sequences.
- Model Training: A deep neural network (CGSchNet) is trained using the variational force-matching approach. The model learns to predict the effective forces between CG degrees of freedom (e.g., one bead per amino acid) from the all-atom data.
- Simulation and Validation: The trained CG force field is deployed to run simulations on new protein sequences not present in the training data. Performance is validated by comparing the resulting free energy landscapes, metastable states, and fluctuations against reference all-atom simulations and, for larger proteins, experimental data such as relative folding free energies of mutants.
This workflow successfully predicted folding intermediates and unfolded states for several fast-folding proteins, demonstrating that the ML model learned physically meaningful interactions rather than simply memorizing structures [6].
Protocol 2: Integrative Refinement of IDP Ensembles with Maximum Entropy Reweighting
For intrinsically disordered proteins (IDPs), a major challenge is deriving an atomic-resolution ensemble that is consistent with experimental observations. The following workflow, which uses maximum entropy reweighting, has proven effective [46]:
- Initial Ensemble Generation: Long-timescale (e.g., 30 µs) all-atom MD simulations of the IDP are performed using one or more state-of-the-art force fields (e.g., a99SB-disp, Charmm36m).
- Forward Calculation of Observables: For every frame in the MD ensemble, experimental observables (NMR chemical shifts, J-couplings, SAXS curves) are calculated using established forward models.
- Automated Reweighting: A maximum entropy algorithm is applied to assign new statistical weights to each frame in the simulation. The goal is to find the set of weights that provides the best agreement with the raw experimental data while minimizing the deviation from the original MD ensemble (i.e., maximizing the entropy of the reweighted distribution).
- Convergence and Validation: The similarity of reweighted ensembles, derived from MD simulations started with different force fields or initial conditions, is assessed. Highly similar final ensembles indicate a robust, force-field independent result that can be considered a accurate model of the solution-state ensemble [46].
The diagram below illustrates the workflow for generating accurate conformational ensembles of IDPs by integrating MD simulations with experimental data.
Protocol 3: Augmenting Structure Prediction with Experimental Restraints
While AlphaFold2 excels at predicting static structures, it can be modified to generate conformational ensembles guided by experimental data. DEERFold is a prime example of this approach [47]:
- Fine-Tuning: The AlphaFold2 network (implemented via OpenFold) is fine-tuned on a set of structurally diverse proteins. This process explicitly incorporates distance distributions, such as those obtained from Double Electron-Electron Resonance (DEER) spectroscopy, into the training loss.
- Conformational Selection: For a protein of interest, a set of experimental or simulated DEER distance distributions is used to guide the model during inference. The fine-tuned network, DEERFold, uses these sparse restraints to select and predict relevant conformations from the ensemble.
- Benchmarking: The method's performance is benchmarked on systems like membrane transporters, where it can successfully predict alternative conformational states using only a limited number of distance restraints.
This protocol demonstrates that integrating even sparse experimental data directly into an ML architecture can powerfully constrain the conformational landscape and reveal functionally relevant states [47].
Successful implementation of ML/MD hybrid pipelines relies on a suite of software tools, datasets, and computational resources. The table below catalogues key components of the modern computational scientist's toolkit in this field.
Table 2: Key Research Reagent Solutions for ML/MD Hybrid Pipelines
| Tool/Resource Name | Type | Primary Function | Relevance to Hybrid Pipelines |
|---|---|---|---|
| PROTAC-DB / PROTAC-PEDIA [45] | Database | Curated repository of PROTACs and related data | Provides clean, component-aware data for training ML models on ternary complexes. |
| PROTAC-Splitter [45] | Data Processing Tool | Automates parsing of degrader molecules into warhead, linker, and E3-ligand components | Feeds generative AI models with standardized building blocks for de novo design. |
| HADDOCK [45] | Docking Software | Performs data-driven docking of biomolecular complexes | Used in physics-driven pipelines to generate initial models of ternary complexes for MD refinement. |
| Markov State Models (MSMs) [35] | Analytical Framework | Models the kinetics and thermodynamics of molecular systems from MD data | Used to reweight and extract equilibrium distributions from long MD trajectories for training generative models like BioEmu. |
| MEGAscale Dataset [35] | Experimental Dataset | Contains high-throughput protein stability measurements (e.g., melting temperature) | Used for property prediction fine-tuning (PPFT) of generative models, embedding thermodynamic accuracy. |
| AlphaFold2 (OpenFold) [47] | Structure Prediction Model | Predicts protein structures from sequence; platform for fine-tuning. | Base model for developing specialized tools like DEERFold that incorporate experimental data for ensemble generation. |
The trend is toward the creation of a unified, multi-scale computational stack. This stack begins with automated data curation tools, proceeds to generative AI for candidate design, employs high-fidelity hybrid MD/ML for structural and activity prediction, and finally uses rigorous physics-based scoring for final validation [45]. The integration of these tools into cohesive workflows is what ultimately empowers researchers to move from sequence to functionally insightful conformational ensembles with unprecedented speed and accuracy.
Adenylate kinase (ADK) is a pivotal phosphotransferase enzyme essential for cellular energy homeostasis, catalyzing the reversible transfer of a phosphoryl group between adenosine nucleotides (ATP + AMP ⇌ 2 ADP) [49] [50]. This ubiquitous enzyme undergoes large-scale, multi-domain conformational changes during its catalytic cycle, making it a quintessential model system for studying the relationship between protein dynamics and biological function [49] [51]. A comprehensive understanding of its conformational ensemble is crucial, not only for fundamental enzymology but also for applications in rational drug design and enzyme engineering, where targeting dynamic ensembles proves more effective than focusing on static structures [52].
The central challenge in mapping ADK's conformational landscape lies in capturing these dynamics across multiple spatial and temporal scales. This case study objectively compares the performance of modern computational and experimental methods in elucidating the transition pathways and energy landscapes of ADK. We focus on validating molecular dynamics (MD)-predicted folding pathways with experimental data, a critical step in the broader thesis of benchmarking predictive models against empirical reality [53].
Biological Background: ADK Domain Architecture and Conformational States
Escherichia coli adenylate kinase (ADK), a monomeric enzyme, is structurally composed of three primary domains [49]:
- The CORE Domain (residues 1–29, 68–117, and 161–214): A relatively rigid scaffold that forms the structural foundation of the enzyme.
- The AMP-binding Domain (NMP) (residues 30–67): Also known as the NMP domain, this region undergoes conformational changes to bind AMP.
- The ATP-binding Domain (LID) (residues 118–167): A flexible loop that closes over the bound ATP substrate.
In the absence of substrates, ADK predominantly adopts an open conformation, where the LID and NMP domains are displaced away from the CORE domain, providing access to the active site [49]. Upon substrate binding (e.g., to the inhibitor AP5A), the enzyme transitions to a closed conformation, where the LID and NMP domains move inward, encapsulating the substrates and forming a catalytically competent state [49] [50]. These large-scale conformational transitions, occurring on the microsecond to millisecond timescale, are rate-limiting for the catalytic reaction [49].
Comparison of Methodologies for Mapping Conformational Transitions
Diverse methodologies are employed to capture ADK's dynamics, each with distinct strengths, limitations, and operational scales. The following table provides a high-level comparison of these key techniques.
Table 1: Comparison of Methods for Studying ADK Conformational Dynamics
| Method | Core Principle | Temporal Resolution | Key Advantage | Primary Limitation |
|---|---|---|---|---|
| Long-Timescale MD [49] | All-atom simulation with explicit solvent; models physical forces over time. | Nanoseconds to microseconds | Atomic-level detail of interactions and pathways. | Computationally prohibitive for full transition sampling; force field inaccuracies. |
| Enhanced Sampling MD (BE-META) [51] | Accelerates exploration of free energy landscape using bias potentials. | Effectively reaches beyond millisecond scales | Enables calculation of multidimensional free energy landscapes. | Choice of collective variables can bias the observed pathways. |
| Path Sampling (WE) [54] | Statistically rigorous sampling of transition paths without predefined reaction coordinates. | N/A (ensemble-based) | Identifies multiple pathways and intermediates; model-independent. | Typically requires coarse-grained models, sacrificing atomic detail. |
| Crystallography (Multiple States) [50] | Determines atomic structures from diffraction patterns of crystallized proteins. | Static snapshots | Provides high-resolution, experimental structures of different states. | May not represent true solution-state dynamics; crystal packing artifacts. |
Detailed Experimental Protocols
To ensure reproducibility, below are the detailed methodologies for key experiments cited in this guide.
Protocol 1: Long Time-Scale Molecular Dynamics Simulations [49]
- Initial System Setup: Obtain crystal structures of open (PDB ID: 4AKE) and closed (PDB ID: 1AKE) states. Remove nonpolar hydrogen atoms and add polar hydrogens based on calculated pKa values. Parameterize the enzyme using the Amber force field.
- Solvation and Neutralization: Solvate the protein in a simulation box with explicit TIP3P water molecules. Add counterions (e.g., Na+) to neutralize the system's charge.
- Energy Minimization: Perform ~5,000 steps of steepest descent minimization followed by ~5,000 steps of conjugate gradient minimization to remove steric clashes.
- Equilibration: Conduct a short MD simulation with protein atoms fixed to equilibrate the solvent. Then, perform a subsequent equilibration of the entire system without restraints.
- Production Simulation: Run molecular dynamics simulations under constant temperature (300 K) and pressure (NPT ensemble) using a 2-femtosecond time step. Use the particle mesh Ewald (PME) method for long-range electrostatics and a cut-off for van der Waals interactions. For enhanced sampling, initiate multiple independent simulations (10-200 ns) from different frames of initial trajectories.
Protocol 2: Multi-State Crystallographic Analysis [50]
- Protein Preparation: Clone, express, and purify the target ADK (e.g., from Methanotorris igneus) with a C-terminal His-tag. Purify using heat treatment, immobilized metal affinity chromatography (HisTrap HP column), and size-exclusion chromatography (Superdex75 column).
- Crystallization: Grow crystals of different liganded states using the sitting-drop vapor diffusion method at 20°C.
- Apo Form: Reservoir solution containing 30% (v/v) MPD and 100 mM Tris-HCl pH 8.0.
- AMP-bound: Condition with 4 mM AMP, 8% (w/v) PEG 8000, 100 mM magnesium acetate, and 100 mM sodium acetate pH 4.5.
- Ap5A-bound: Reservoir solution with 4 mM Ap5A, 17% (w/v) PEG 3350, and 250 mM sodium malonate.
- Data Collection and Processing: Flash-cool crystals in liquid nitrogen. Collect X-ray diffraction data at a synchrotron beamline (e.g., 100 K). Process data using XDS.
- Structure Determination and Refinement: Solve the structure by molecular replacement using a known homologous ADK structure as a search model. Iteratively refine the model and build the structure using Coot and PHENIX. Perform ensemble refinement to model conformational heterogeneity within the crystal.
Performance Benchmark: Key Findings and Data Comparison
Different methods have yielded insights into ADK's transition pathways, intermediates, and the underlying energy landscape. The following table synthesizes quantitative findings and key observations from various studies.
Table 2: Comparative Performance of Methods in Revealing ADK Dynamics
| Method | Identified Transition Pathways | Key Intermediates Detected | Energetics (Energy Barrier) | Agreement with Experiment |
|---|---|---|---|---|
| Long-Timescale MD [49] | Sequential (LID closes before/after NMP) | Two novel states: LID-open/NMP-closed & LID-closed/NMP-open | N/A | Strong (over 20 transitions captured) |
| Bias-Exchange Metadynamics [51] | Multiple parallel pathways | Multiple intermediate states in the free energy landscape | Shallow barrier (apo); Large barrier (holo, closed) | Strong (explains conformational selection) |
| Weighted Ensemble Path Sampling [54] | Two distinct pathways | Intermediates consistent with previous findings | N/A | Strong (validated by experimental structures) |
| Replica Exchange MD [55] | Efficient sampling of open/closed transition | N/A | N/A | High (consistent with experimental studies) |
Synthesis of Quantitative Results
- Pathway Heterogeneity: Multiple studies confirm that ADK's conformational transition is not restricted to a single pathway. Weighted ensemble path sampling and other simulations consistently identify two major distinct pathways, with the transition order of the LID and NMP domains not being strictly fixed [54] [51].
- Energy Landscape: The free energy landscape of ligand-free (apo) ADK is characterized by a shallow or non-significant energy barrier between the open and closed states, populated by multiple intermediate conformations. In contrast, the ligand-bound (holo) form shows a strong preference for the closed conformation with a large energy barrier to reopen [51].
- Transition Mechanism: The prevalence of multiple, easily accessible intermediates in the apo state supports a conformational selection mechanism, where the ligand binds to a pre-existing closed-like conformation, shifting the population equilibrium [49] [51].
Visualizing the Conformational Transition Pathway
The large-scale domain motion of ADK during its catalytic cycle can be summarized in the following pathway diagram. This diagram integrates findings from multiple molecular dynamics and path-sampling studies [49] [54] [51].
The diagram illustrates the chronological operation of ADK's functional domains and the existence of multiple pathways via different intermediate states, as revealed by path sampling and MD simulations [49] [54] [51].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful experimental and computational analysis of ADK's conformational ensemble relies on a suite of key reagents and tools. The following table details these essential components.
Table 3: Key Research Reagent Solutions for ADK Conformational Studies
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| Stabilized Enzyme Constructs | Provides a homogeneous, stable protein sample for crystallization and biophysics. | C-terminal His-tagged ADK from M. igneus: Allows purification via IMAC; thermostable enzyme improves crystal quality [50]. |
| Chemical Inhibitors / Substrates | Traps the enzyme in specific conformational states for structural studies. | Ap5A (P1,P5-di(adenosine-5')-pentaphosphate): A bisubstrate analog that locks ADK in a closed conformation [49] [50]. AMPNP: A non-hydrolyzable ATP analog used to study intermediate states [50]. |
| Crystallization Reagents | Facilitates the growth of protein crystals for X-ray diffraction. | MPD, PEG 3350, Sodium Malonate: Precipitants used to crystallize different liganded states of ADK [50]. |
| Molecular Dynamics Force Fields | Defines the potential energy function for all-atom simulations. | AMBER ffamber03: A widely used force field for simulating proteins and nucleic acids; provided parameters for ATP/AMP in ADK simulations [51]. |
| Path Sampling Software | Enables statistically rigorous sampling of transition pathways. | Weighted Ensemble (WE) Method: A path-sampling algorithm that efficiently explores rare transitions without biasing the pathway [54]. |
| Enhanced Sampling Plugins | Accelerates the exploration of free energy landscapes in MD simulations. | Bias-Exchange Metadynamics (BE-META): An advanced sampling technique to compute free energy landscapes along multiple collective variables [51]. |
This comparison guide demonstrates that a multi-faceted approach is paramount for comprehensively mapping the conformational ensemble of adenylate kinase. Long-timescale and enhanced-sampling MD simulations provide atomistic detail of pathways and free energy landscapes, while rigorous path sampling methods like the Weighted Ensemble approach confirm the existence of multiple, heterogeneous transition pathways. These computational predictions are critically validated by experimental crystallographic data, which offers high-resolution snapshots of distinct states and, through multi-conformer and ensemble refinement, reveals intrinsic structural heterogeneity [49] [50] [54].
The collective evidence strongly supports a conformational selection and population-shift mechanism for ADK function, where the enzyme intrinsically samples a broad ensemble of states, including closed-like conformations, even in the absence of substrates [49] [51]. The successful benchmarking of these computational methods against experimental data for ADK establishes a powerful framework for investigating conformational dynamics in other medically relevant enzymatic targets, ultimately accelerating drug development by shifting the focus from static structures to dynamic ensembles.
Navigating Pitfalls: Force Field Selection, Sampling Limits, and AI Model Constraints
Molecular dynamics (MD) simulations serve as a computational microscope, enabling researchers to observe the intricate motions of proteins and other biomolecules at an atomic level. The accuracy of these simulations is fundamentally dependent on the force field—a mathematical model that describes the potential energy of a molecular system as a function of its atomic coordinates. Within the context of validating MD-predicted protein folding pathways with experimental data, selecting an appropriate force field becomes paramount. This guide provides an objective comparison of three dominant force field families—AMBER, CHARMM, and GROMACS—evaluating their performance based on experimental validation data, with a particular focus on their application in protein folding and structural dynamics studies.
The AMBER, CHARMM, and GROMOS force fields are implemented in various MD software packages, including the GROMACS engine, which supports all three natively [56]. A critical methodological consideration is that force field comparison does not require identical simulation parameters (such as cutoffs) between different force fields. Instead, it requires the proper implementation of each force field with its own prescribed settings [57]. For instance, when using the CHARMM36 force field in GROMACS, the recommended parameters include a force-switch modifier for van der Waals interactions with rvdw_switch at 1.0 nm and rvdw at 1.2 nm, and Particle Mesh Ewald for electrostatics with rcoulomb at 1.2 nm [56]. Using non-standard settings can lead to deviations from the intended physical properties.
The conversion of files and parameters between different simulation packages is now highly automated using tools like ParmEd and InterMol, which serve as crucial "Research Reagent Solutions" [58]. These converters allow for the direct comparison of energies from single configurations across different molecular dynamics engines, a necessary step for validation. Studies have shown that with careful parameter choices, energy calculations across different engines (GROMACS, AMBER, LAMMPS, DESMOND, CHARMM) can agree to within 0.1% or better for all energy components [58].
Quantitative Performance Comparison
The most reliable method for evaluating force field accuracy is comparison with experimental data. A robust benchmark is the use of cross-solvation free energies, which provide a systematic matrix for comparing a molecule as both a solute and a solvent. A 2021 study employed a 25x25 matrix of experimental values to compare nine condensed-phase force fields, offering a quantitative measure of their performance in reproducing experimental thermodynamics [59].
Table 1: Force Field Performance Against Experimental Cross-Solvation Free Energies
| Force Field | Family | Correlation Coefficient (R) | RMSE (kJ mol⁻¹) | AVEE (kJ mol⁻¹) |
|---|---|---|---|---|
| GROMOS-2016H66 | GROMOS | 0.88 | 2.9 | -1.5 |
| OPLS-AA | OPLS | 0.88 | 2.9 | +1.0 |
| AMBER-GAFF2 | AMBER | 0.84 | 3.3 | Not Specified |
| AMBER-GAFF | AMBER | 0.82 | 3.6 | Not Specified |
| CHARMM-CGenFF | CHARMM | 0.76 | 4.0 | Not Specified |
Data sourced from Kashefolgheta et al., Phys. Chem. Chem. Phys., 2021, 23, 13055 [59]. RMSE: Root-Mean-Square Error; AVEE: Average Error.
The data reveals that while differences between the top-performing force fields (GROMOS-2016H66 and OPLS-AA) and others like AMBER-GAFF2 and CHARMM-CGenFF are statistically significant, they are "not very pronounced" [59]. Furthermore, performance is "distributed rather heterogeneously over the set of compounds within the different force fields," suggesting that the optimal force field may depend on the specific system under investigation [59].
For protein-specific simulations, the choice is nuanced. The protein force fields from AMBER and CHARMM are generally considered "probably equally good," though AMBER may be "somewhat better for dsDNA" [57]. It is critical to use modern, validated parameter sets; for example, some AMBER force fields distributed with older versions of GROMACS are "antiquated and not appropriate for modern simulations of nucleic acids" [57].
Application in Protein Folding and Structural Validation
Long MD simulations are now capable of folding and unfolding proteins multiple times, providing a direct avenue for testing force fields against experimental folding data. Assessments show that modern physical models can accurately reproduce protein folding rates and free energies, as well as the structure and dynamics of folded proteins [60]. However, these same force fields often struggle to accurately reproduce folding enthalpies and the detailed characteristics of the unfolded state [60], highlighting a key area for future improvement.
The rise of AI-based protein structure prediction tools like AlphaFold 2 (AF2) has created new opportunities and challenges for MD validation. While AF2 achieves high accuracy in predicting stable conformations with proper stereochemistry, it shows limitations in capturing the full spectrum of biologically relevant states, particularly in flexible regions and ligand-binding pockets [9] [61]. For instance, AF2 systematically underestimates ligand-binding pocket volumes and captures only single conformational states in systems where experimental structures show functionally important asymmetry [61]. This makes MD simulations with accurate force fields essential for modeling protein dynamics, conformational diversity, and ligand-induced changes that static AF2 models might miss.
Table 2: Capabilities and Limitations in Protein Structure Modeling
| Aspect | MD Simulations with Physical Force Fields | AlphaFold 2 Predictions |
|---|---|---|
| Dynamic Process (Folding) | Directly models pathways and kinetics [60]. | Predicts a single, static structure. |
| Conformational Ensemble | Can, in principle, sample multiple states. | Often captures a single state; misses functional asymmetry in multimers [61]. |
| Ligand-Binding Pockets | Can model induced-fit and conformational selection. | Systematically underestimates pocket volumes [61]. |
| Unfolded/Disordered States | Struggles with accurate characterization [60]. | Low confidence (pLDDT) scores indicate unstructured regions [61]. |
| Key Strength | Provides thermodynamic and kinetic data. | High speed and accuracy for stable core structures [61]. |
Experimental Protocols for Force Field Validation
Protocol 1: Validation via Cross-Solvation Free Energies
This protocol provides a robust method for assessing force field performance in condensed phases [59].
- System Selection: Curate a set of N small organic molecules (e.g., alkanes, ethers, ketones, alcohols) that are liquids under ambient conditions.
- Experimental Matrix Construction: Compile a full N x N matrix of experimental cross-solvation free energies, where each molecule is considered as both a solute (A) and a solvent (B).
- Simulation Setup: Perform solvation free energy simulations (e.g., using thermodynamic integration or free energy perturbation) for all unique pairs in the matrix across the force fields being tested.
- Analysis: Calculate the correlation coefficient, root-mean-square error (RMSE), and average error (AVEE) between the experimental and simulation results for each force field to quantify accuracy.
Protocol 2: Single-Configuration Energy Comparison
This protocol, used in preparation for the SAMPL5 challenge, verifies the correct translation of a model and its parameters between different MD engines [58].
- Initial Parameterization: Define the molecular system and its force field parameters in a single format (e.g., AMBER format using GAFF/RESP parameters).
- Automated Conversion: Use conversion tools (ParmEd and InterMol) to translate the input files into the formats of other MD programs (GROMACS, LAMMPS, DESMOND, CHARMM).
- Energy Calculation: In each program, calculate the potential energy of the system for an identical, single atomic configuration.
- Validation: Compare the energies from all programs. With correct conversion and equivalent nonbonded treatment, the energies should agree within 0.1% or better for all components.
Protocol 3: Assessment of Folding Thermodynamics and Kinetics
This protocol leverages long, equilibrium MD simulations to directly test a force field's ability to describe protein folding [60].
- System Preparation: Select a small, fast-folding protein with known experimental folding data (e.g., rates, free energy).
- Long-Timescale Simulation: Run multiple, long MD simulations (on the microsecond to millisecond scale) at equilibrium conditions, capturing numerous folding and unfolding events.
- Trajectory Analysis: Calculate the folding free energy (ΔG), folding rates, and the structure of the folded and unfolded states from the simulation trajectory.
- Benchmarking: Compare the simulation-derived properties (folding rates, free energies, and native state structure/dynamics) directly against the available experimental data.
The following diagram illustrates the logical workflow for validating a force field against experimental protein folding data, integrating the protocols above.
Diagram 1: Workflow for experimental validation of molecular dynamics force fields.
Table 3: Key Software Tools and Resources for Force Field Comparison and MD Simulation
| Tool / Resource | Type | Function / Purpose |
|---|---|---|
| ParmEd | Software Library | Program-agnostic tool for manipulating molecular topologies and converting files between AMBER, GROMACS, CHARMM, and OpenMM formats [58]. |
| InterMol | Software Tool | An all-to-all converter between molecular simulation file formats (GROMACS, LAMMPS, DESMOND) [58]. |
| AMBER-GAFF/GAFF2 | Force Field | The Generalized Amber Force Field; provides parameters for small molecules compatible with AMBER biomolecular force fields [56]. |
| CHARMM36 | Force Field | A widely used all-atom force field for proteins, lipids, and nucleic acids; requires specific nonbonded parameters in GROMACS [57] [56]. |
| GROMOS 54A7 | Force Field | A united-atom force field; parametrized with a specific cut-off scheme, requiring caution when used with modern integrators [56]. |
| Cross-Solvation Matrix | Benchmark Dataset | A curated set of experimental solvation free energies for validating force field accuracy in condensed-phase simulations [59]. |
The comparative analysis of AMBER, CHARMM, and GROMOS force fields reveals a landscape where performance is increasingly convergent, yet nuanced. Quantitative benchmarks against experimental solvation free energies show that top-performing force fields from different families (e.g., GROMOS-2016H66, OPLS-AA, and AMBER-GAFF2) achieve remarkably similar accuracy, with RMSE values clustering between 2.9 and 3.6 kJ mol⁻¹ [59]. For the specific task of validating MD-predicted protein folding pathways, current force fields demonstrate a capacity to accurately reproduce folding rates, free energies, and the structure of the native state, though challenges remain in modeling folding enthalpies and the unfolded state ensemble [60]. The choice of force field should therefore be guided by the specific biological question, the molecular system under study, and the availability of well-validated parameters. The integration of static structural models from AI predictors like AlphaFold 2 with the dynamic trajectories from MD simulations, powered by physically validated force fields, represents the most promising path forward for a comprehensive understanding of protein folding and function.
Molecular dynamics (MD) simulation serves as a "computational microscope," providing atomistic details of protein folding that often remain hidden from experimental view [62]. However, a fundamental challenge constrains the reliability of these simulations: the sampling problem. This refers to the difficulty in simulating a trajectory long enough to adequately explore the conformational space and reach thermodynamic equilibrium, ensuring results are reproducible and biologically meaningful [62] [63]. For protein folding, where timescales can range from microseconds to minutes, determining when a simulation is 'long enough' is not straightforward [63]. This guide examines the core aspects of this problem, compares simulation methods, and outlines validation protocols to help researchers assess the convergence and reliability of their folding simulations.
The Convergence Dilemma: Theory vs. Practice
A system in thermodynamic equilibrium fully explores its available conformational space (Ω). In practice, MD studies use a working definition: a property is considered equilibrated if its running average stabilizes with small fluctuations for a significant portion of the trajectory after a convergence time [64].
The critical insight is that a simulation can reach partial equilibrium, where some properties converge while others do not. Average structural properties (e.g., root-mean-square deviation or radius of gyration), which depend mainly on high-probability regions of conformational space, may converge in multi-microsecond trajectories [64]. In contrast, properties like transition rates between rarely visited states, which depend on low-probability regions, often require much longer simulation times that remain impractical for many systems [64]. This dichotomy means a simulation can be 'long enough' for one research question but insufficient for another.
Comparative Performance of Simulation Approaches
The ability to sample conformational space effectively depends heavily on the chosen simulation method and force field. The table below summarizes the performance of different approaches in simulating protein folding and dynamics.
Table 1: Performance Comparison of Molecular Simulation Methods
| Method / Force Field | Spatial Resolution | Key Features / Biases | Reported Performance on Protein Folding |
|---|---|---|---|
| All-Atom MD (Standard Force Fields) [62] | All-Atom | AMBER ff99SB-ILDN, CHARMM36; explicit solvent; best practice parameters | Reproduces experimental observables at 298K for some proteins; performance diverges at higher temperatures (498K) simulating unfolding [62]. |
| Gō/Structure-Based Models (SBMs) [63] | Coarse-Grained (Cα or a few beads/residue) | Potential energy biased toward native contacts; minimizes energetic frustration. | Computationally efficient; successfully predicts folding pathways and intermediates for large proteins (e.g., serpins, adenylate kinase) [63]. |
| Essential Dynamics Sampling (EDS) [16] | All-Atom (biased on backbone) | MD simulation biased to not increase distance from target in a subspace of essential degrees of freedom. | Correctly folded cytochrome c from highly unfolded states using only 106 backbone collective degrees of freedom; pathways agreed with experiment [16]. |
| AI2BMD [65] | All-Atom | Machine learning force field (MLFF) with ab initio accuracy; uses protein fragmentation. | Energy MAE: ~0.045 kcal mol⁻¹ (vs. 3.198 for MM); Force MAE: ~0.078 kcal mol⁻¹ Å⁻¹ (vs. 8.125 for MM); demonstrated folding/unfolding [65]. |
| Neural Network Potentials (e.g., eSEN) [66] | All-Atom | Trained on massive quantum chemical datasets (e.g., OMol25); conservative-force models outperform direct-force. | Matches high-accuracy DFT performance on molecular energy benchmarks; enables large-system simulations previously infeasible [66]. |
Experimental Protocols for Validating Convergence and Pathways
Assessing Convergence in MD Simulations
A simulation should not be deemed converged based on a single metric. A multi-faceted validation protocol is essential [64].
- Standard Protocol:
- Run multiple, independent simulations: Initiate simulations from different starting conditions (e.g., different velocities or slightly different structures) [62].
- Monitor property time series: Track the evolution of key properties, including:
- Energetics: Total potential energy, protein-ligand interaction energy.
- Structural Metrics: Root-mean-square deviation (RMSD) from a reference, radius of gyration (Rg).
- Collective Variables: Distances, angles, or dihedrals relevant to the folding process.
- Check for plateau formation: Visually inspect the time series for the point at which the running average of these properties stabilizes and fluctuates around a steady value [64].
- Compare across replicates: Ensure that the final average values of key properties are consistent across the independent simulation replicates [62].
Validating Folding Pathways Against Experimental Data
Agreement with experimental data is the ultimate test for any predicted folding pathway. The following workflow outlines an integrated computational and experimental validation strategy.
The specific experimental observables used for validation include:
- Time-Resolved Circular Dichroism (CD) and Small-Angle X-Ray Scattering (SAXS): These techniques can track secondary structure formation and global compaction, respectively. For instance, SAXS measurements on cytochrome c suggested an initial collapse followed by concerted secondary structure formation, a pattern that EDS simulations were able to reproduce [16].
- Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS): This method identifies regions of the protein that are protected from exchange, indicating stable hydrogen-bonded structures. Computed pathways can be checked to see if they form these protected regions in the same sequence.
- Single-Molecule FRET (smFRET): This provides distance distributions between specific labeled residues. A simulation predicting a specific intermediate should yield a distance distribution for those residues that matches the experimental FRET efficiency [16].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Resources for Protein Folding Simulations
| Category | Item / Software | Primary Function in Folding Studies |
|---|---|---|
| Simulation Software | GROMACS [16] [62], NAMD [62], AMBER [62], OpenMM | Core engines for performing MD simulations; integration of equations of motion and force calculations. |
| Force Fields | AMBER ff99SB-ILDN [62], CHARMM36 [62], GROMOS87 [16] | Empirical potential energy functions defining atomistic interactions (bonds, angles, dihedrals, non-bonded). |
| Specialized Methods | Gō/Structure-Based Models (SBM) [63], Essential Dynamics Sampling (EDS) [16] | Accelerate sampling by using native structure bias or collective motions from essential dynamics. |
| AI/ML Potentials | AI2BMD [65], eSEN/UMA Models [66] | Machine-learning force fields trained on quantum data for ab initio accuracy at near-classical MD cost. |
| Analysis & Validation | MDTraj, PyEMMA, HOOMD | Analyze trajectories, calculate properties (RMSD, Rg), and build Markov State Models to study kinetics. |
| Experimental Data | PDB Structures, HDX-MS data, smFRET data, Kinetic folding rates | Serve as initial coordinates and as critical benchmarks for validating simulation predictions [16]. |
The question of when an MD simulation is 'long enough' for studying protein folding has no universal answer. The sufficient simulation length is dictated by the specific biological question and the property of interest. Convergence of average structural properties is an achievable goal with modern hardware and advanced sampling methods, while the accurate prediction of transition rates between rare states remains a formidable challenge. The most reliable conclusions are drawn from a combination of multiple independent simulations, the use of enhanced sampling techniques like EDS or SBMs where appropriate, and—most critically—rigorous validation against experimental data. The emergence of AI-driven force fields and simulators promises to dramatically expand the accessible timescales and accuracy of these simulations, bringing us closer to the ultimate goal of a truly predictive computational microscope for protein folding.
The advent of AI-based co-folding models represents a transformative advancement in structural biology, enabling the simultaneous prediction of three-dimensional structures for protein-ligand complexes, protein-protein interactions, and assemblies involving nucleic acids. Models such as AlphaFold3, RoseTTAFold All-Atom, Boltz-2, and OpenFold3 have demonstrated remarkable performance on public benchmarks, achieving unprecedented accuracy in predicting native binding poses [67] [68]. These unified frameworks leverage diffusion-based architectures to model arbitrary chemical structures, ostensibly approaching experimental-level accuracy in specific docking scenarios [67]. Their capability to generate structural hypotheses for diverse biomolecular complexes has positioned them as indispensable tools for accelerating drug discovery and protein engineering.
However, beneath these impressive capabilities lies a fundamental challenge: the machine learning methods powering these models are trained on experimentally determined structures from public databases like the Protein Data Bank (PDB), which may not adequately represent the thermodynamic principles governing molecular interactions in physiological environments [9]. Recent critical investigations have revealed that these models often rely on statistical pattern recognition from their training corpus rather than developing a genuine understanding of the physical chemistry that dictates protein-ligand interactions [67] [69]. This limitation becomes particularly evident when models are applied to novel targets or subjected to biologically plausible perturbations that should fundamentally alter binding behavior according to basic physicochemical principles. The resulting discrepancies question whether these models truly learn the physics of molecular interactions or primarily excel at interpolating within their training data distribution.
Fundamental Limitations: Examining the Evidence
The Overfitting Problem: Interpolation vs. Extrapolation
A central limitation of current co-folding models is their pronounced performance degradation when applied to biomolecular systems that diverge significantly from those present in their training data. These models demonstrate exceptional capability when predicting structures similar to those encountered during training but struggle with extrapolation to novel targets.
Performance Decay on Novel Targets: A comprehensive independent benchmark termed "Runs N' Poses," comprising 2,600 structures published after the models' training cut-offs, revealed an almost linear drop in prediction success rates as training set coverage declined [68]. For targets with the sparsest representation in training data (fewer than 100 examples), success rates plummeted to approximately 20%, compared to much higher performance on well-represented targets [68].
Memorization of Training Data: Studies indicate that co-folding models largely memorize ligands from their training data and demonstrate limited generalization to unseen ligand structures [67] [68]. This memorization tendency explains the stark contrast between impressive performance on standardized benchmarks derived from PDB and reduced accuracy on proprietary drug discovery targets that often feature novel chemotypes or protein folds.
Chemical Space Limitations: The relatively small dataset of approximately 100,000 protein-ligand structures available for training creates a fundamental constraint on the diversity of chemical space these models can effectively represent [69]. This data scarcity forces models to rely on superficial pattern recognition rather than learning underlying principles that would enable robust generalization.
Table 1: Performance Comparison of Co-Folding Models on Novel vs. Familiar Targets
| Model | Performance on Familiar Targets (LDDT-PLI > 0.8 & RMSD < 2Å) | Performance on Novel Targets (LDDT-PLI > 0.8 & RMSD < 2Å) | Training Data Dependency |
|---|---|---|---|
| AlphaFold3 | ~93% (with known binding site) [67] | As low as ~20% for sparse chemotypes [68] | High dependence on PDB data distribution |
| RoseTTAFold All-Atom | Lower baseline performance than AF3 [67] | Significant performance drop on novel targets [68] | Similar PDB dependency |
| Boltz-2 | High accuracy on in-distribution complexes [68] | Performance decays with target novelty [68] | Open-source model with similar constraints |
| OpenFold3 | Reproduction of AF3 architecture [68] | Expected similar limitations [68] | Same underlying data limitations |
Lack of Physical Understanding: Adversarial Testing Reveals Fundamental Flaws
Beyond overfitting concerns, research has systematically investigated whether co-folding models internalize the fundamental physical principles governing molecular interactions. Through carefully designed adversarial examples based on established physical, chemical, and biological principles, studies have revealed notable discrepancies in how these models respond to biologically plausible perturbations.
In one revealing experimental approach, researchers subjected Cyclin-dependent kinase 2 (CDK2) in complex with ATP to a series of binding site perturbations [67] [69]:
Binding Site Removal: All binding site residues were replaced with glycine, effectively removing major side-chain interactions that facilitate ATP binding. Despite this drastic alteration that eliminated positively charged residues essential for anchoring negatively charged ATP, all four tested co-folding models continued to predict the ATP-CDK2 complex with nearly identical binding mode, as if the favorable interactions remained present [67].
Binding Site Occlusion: All binding site residues were mutated to phenylalanine, simultaneously removing favorable native interactions and sterically occluding the original binding pocket with bulky aromatic side chains. While models demonstrated some capacity to adapt, predictions remained heavily biased toward the original binding site, with several models placing ATP entirely within the now-nonexistent pocket and generating structures with unphysical atomic overlaps and steric clashes [67].
Charge Reversal and Chemical Mismatching: Additional challenges involved mutating binding site residues to create chemically dissimilar environments with altered charge distributions and steric properties that should disrupt binding. In these scenarios, models consistently failed to respond appropriately, continuing to place ligands in original binding sites despite the absence of complementary interactions [67] [69].
Table 2: Response of Co-Folding Models to Physical Adversarial Challenges
| Adversarial Challenge | Expected Physical Behavior | Model Response | Physical Plausibility of Output |
|---|---|---|---|
| Binding site removal (Glycine mutation) | Ligand displacement due to loss of specific interactions | Continued placement in original pose | Low: Few/no interactions present |
| Binding site occlusion (Phenylalanine mutation) | Complete ligand displacement due to steric hindrance | Biased placement toward original site; steric clashes | Very Low: Unphysical atomic overlaps |
| Dissimilar residue substitution | Altered binding pose or ligand displacement | Minimal pose alteration | Low: Ignores chemical incompatibility |
| Ligand chemical modification | Disrupted binding interactions | >50% failure to account for perturbations [69] | Very Low: Predicts stable complexes that shouldn't exist |
These adversarial tests collectively demonstrate that co-folding models lack a genuine understanding of physicochemical principles such as hydrogen bonding, electrostatic complementarity, and steric constraints [69]. Instead of reasoning from first principles, they appear to pattern-match to memorized binding motifs from their training data, resulting in physically implausible predictions when confronted with novel scenarios.
Experimental Validation: Methodologies for Assessing Model Limitations
Binding Site Mutagenesis Protocol
The experimental methodology for evaluating co-folding models' physical understanding involves systematic mutagenesis of protein binding sites followed by assessment of prediction quality:
Residue Selection: Identify all binding site residues forming contacts with the ligand in the wild-type experimental structure. For CDK2-ATP complexes, this includes residues coordinating the ATP molecule through hydrogen bonding and hydrophobic interactions [67].
Mutation Strategy: Implement three progressive mutation approaches: (1) Replace all binding site residues with glycine to remove side-chain interactions while maintaining backbone flexibility; (2) Replace all binding site residues with phenylalanine to sterically occlude the binding pocket and eliminate favorable interactions; (3) Replace each binding site residue with a chemically dissimilar residue to dramatically alter the site's shape and chemical properties [67].
Prediction and Analysis: Submit wild-type and mutated sequences to co-folding models. Compare predicted structures using root-mean-square deviation (RMSD) for ligand positioning, analysis of interaction preservation, and identification of steric clashes. The positive control is the unmutated wild-type prediction compared to the experimental structure [67].
Independent Benchmarking on Temporal Data Splits
To evaluate overfitting and generalization capabilities, researchers have developed rigorous benchmarking protocols:
Temporal Validation Set: Curate a benchmark of protein-ligand structures published after the models' training cut-off dates. The "Runs N' Poses" benchmark comprises 2,600 such structures, ensuring no possibility of training data contamination [68].
Structural Clustering: Group structures by similarity to quantify training data representation. Calculate the number of similar structures (<2.0Å RMSD) present in the training set for each benchmark entry [68].
Stratified Performance Analysis: Evaluate model success rates (defined as LDDT-PLI > 0.8 and ligand RMSD < 2.0Å) across different levels of training set representation, from well-represented clusters (>100 similar training examples) to sparse clusters (<100 similar examples) [68].
Ligand Perturbation Experiments
Complementary to binding site modifications, researchers have also developed protocols to test model responses to ligand alterations:
Functional Group Modification: Identify key functional groups on ligands that participate in critical interactions with the protein (e.g., hydrogen bond donors/acceptors, charged groups). Systematically modify or remove these groups to disrupt binding interactions [69].
Binding Affinity Assessment: In cases where models provide affinity predictions (e.g., Boltz-2's explicit affinity head), evaluate whether predicted affinities appropriately decrease following ligand perturbations that should disrupt binding [68].
Pose Conservation Analysis: Quantify whether ligand pose predictions adjust appropriately in response to chemical modifications, or whether they remain rigidly fixed in the original binding mode despite no longer forming favorable interactions [69].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Computational Tools for Co-Folding Validation
| Tool/Reagent | Function | Application Context | Access Considerations |
|---|---|---|---|
| AlphaFold3 | Unified diffusion model for predicting protein-ligand complexes | Benchmarking performance on well-characterized targets | Restricted license; research-only, no commercial use [68] |
| RoseTTAFold All-Atom | Three-track neural network for all-atom structure prediction | Comparative performance assessment | More accessible than AF3 but lower accuracy [67] |
| Boltz-2 | Open-source diffusion model with affinity prediction | Flexible modification and commercial application | Full OSS license; enables proprietary use [68] |
| OpenFold3 | Open-source reproduction of AF3 architecture | Transparent benchmarking and customization | Full OSS; promotes reproducibility [68] |
| ModFOLDdock | Independent model quality assessment | Objective evaluation of prediction quality | Public server available [70] |
| PoseBustersV2 | Benchmark for evaluating structural plausibility | Validation of physical realism in predictions | Open benchmarking framework [67] |
| ApherisFold | Local deployment platform for co-folding models | Proprietary data analysis without sharing | On-premise solution for IP protection [68] |
| PDB (Protein Data Bank) | Repository of experimental protein structures | Training data source and ground truth reference | Public access with limitations on novel targets [71] |
Implications for Drug Discovery and Protein Engineering
The limitations of current co-folding models have profound implications for their application in pharmaceutical research and protein design. In drug discovery, where accurate prediction of protein-ligand interactions is crucial for virtual screening and lead optimization, the models' tendency to generate physically implausible binding poses for novel chemotypes could lead to misleading conclusions about biological activity, binding affinity, or specificity [67] [69]. The performance degradation on underrepresented targets is particularly problematic given that novel drug targets often involve previously uncharacterized proteins or unique binding sites [68].
For protein engineers designing novel enzymes or binders, the lack of genuine physical understanding in co-folding models limits their utility for predicting how mutations affect folding pathways and stability. The models' static snapshots cannot capture the dynamic conformational ensembles that proteins sample in solution, which is particularly important for understanding allosteric mechanisms and designing proteins with novel functions [9]. While models like RFdiffusion and ProteinMPNN have demonstrated impressive capabilities in de novo protein design, their success rates remain variable, with only 15% of designed serine hydrolase variants exhibiting detectable catalytic activity in one case [72].
These limitations underscore the continued importance of experimental validation and complementary computational approaches that incorporate physical principles. Molecular dynamics simulations, though computationally intensive, can provide insights into protein flexibility and binding kinetics that static co-folding models cannot [9]. Similarly, physics-based docking approaches using tools like AutoDock Vina, while less accurate than AI models for familiar systems, may offer more physically plausible predictions for novel targets because they explicitly model energetic constraints [67].
Future Directions: Toward Physically-Grounded Models
Addressing the limitations of current co-folding models requires advances along multiple research fronts:
Integration of Physical Priors: Incorporating explicit physical constraints and energy-based scoring during the prediction process, rather than relying solely on pattern recognition, could enhance model robustness [69]. This might involve hybrid architectures that combine deep learning with molecular mechanics force fields.
Federated Learning and Expanded Data Diversity: Initiatives like the AI Structural Biology network enable collaborative training across distributed proprietary datasets without sharing raw data, potentially expanding the chemical space covered by models while preserving IP protection [68].
Dynamics-Aware Architectures: Moving beyond static structure prediction to model conformational ensembles and folding pathways would better represent biological reality. Simple structure-based statistical mechanical models like WSME-L have shown promise in predicting protein folding mechanisms with low computational complexity [73].
Explainability and Uncertainty Quantification: Developing better methods to interpret model predictions and quantify uncertainty would help researchers identify when to trust AI-generated structures and when to seek experimental validation [70].
Closed-Loop Experimental Validation: Iterative cycles of prediction, experimental testing, and model refinement based on experimental feedback can gradually improve model accuracy and physical realism, as demonstrated in some de novo protein design pipelines [72].
The rapid pace of innovation in this field suggests that these limitations are likely to be addressed in coming years. However, researchers should maintain a critical perspective when applying these tools, understanding their current strengths and weaknesses, and complementing AI predictions with physical reasoning and experimental validation.
Best Practices for System Setup, Solvation Models, and Simulation Parameters
Molecular dynamics (MD) simulations have become an indispensable tool for studying protein folding pathways, offering atomic-level insights that complement experimental data. The reliability of these simulations, however, is profoundly dependent on appropriate system setup, careful selection of solvation models, and optimization of simulation parameters. With the emergence of AI-predicted protein structures from tools like AlphaFold 2, which provide high-accuracy static structures but often miss conformational diversity [61], the need for rigorous validation through properly configured MD simulations has never been greater. This guide establishes best practices for MD system configuration, objectively compares solvation models with supporting experimental data, and provides protocols for generating simulation data that can effectively validate predicted protein folding pathways against experimental observations.
Molecular Dynamics System Setup: Fundamental Concepts
Foundational Principles
Molecular Dynamics simulations operate on a particle-based description of molecular systems, where equations of motion are numerically integrated to generate dynamical trajectories [74]. The foundational setup involves several critical considerations:
Force Field Selection: Molecular mechanics force fields calculate non-bonded and bonded interactions through empirical parameters fitted to experimental or quantum mechanical data [74]. Choices include CHARMM, AMBER, and GROMOS families, each with specific strengths for different biological systems.
Boundary Conditions: Periodic boundary conditions are typically employed to simulate bulk systems without surface artifacts, with the simulation box size carefully selected to ensure proper solvation shell representation.
Integration Algorithms: The choice of numerical integrator (e.g., Verlet, Leap-frog) and timestep (commonly 1-2 fs) must balance computational efficiency with numerical stability, with constraints often applied to high-frequency bonds to enable larger timesteps [74].
Proper system setup requires understanding these fundamental components before advancing to specific model selections and parameter optimization aimed at capturing biologically relevant protein dynamics.
System Preparation Workflow
The process of preparing a protein system for MD simulation follows a logical sequence of decisions, each critical to the eventual quality and biological relevance of the results. The following diagram outlines this workflow from initial structure preparation to production simulation:
Solvation Models: Comparative Analysis and Performance Data
Explicit vs. Implicit Solvent Approaches
The treatment of solvent environment represents one of the most fundamental choices in MD simulation setup, with significant implications for computational cost and accuracy. Explicit solvent models represent individual water molecules (e.g., TIP3P, TIP4P) and provide the most physically realistic representation of solute-solvent interactions but come with substantial computational overhead [75]. Implicit solvent models treat water as a continuous dielectric medium, dramatically reducing computational cost but potentially sacrificing accuracy in specific applications [75].
Quantitative Comparison of Solvent Models
Table 1: Performance Metrics of Solvation Models in MD Simulations
| Solvent Model | Computational Speed* | Structural Stability | Electrostatic Treatment | Recommended Applications |
|---|---|---|---|---|
| Explicit (TIP3P) | 1x (baseline) | High | Physically realistic | Folding validation, Membrane proteins, Binding free energies |
| GBSW | 4-5x faster than explicit | High [75] | Generalized Born approximation [75] | Solvation free energy, Native state dynamics |
| EEF1 | 20x faster than explicit [75] | Moderate (varies by force field) [75] | Solvent exclusion model [75] | Initial folding studies, Large systems |
| ACE | 6x faster than explicit [76] | Variable (parameter-dependent) [76] | Analytical continuum electrostatics [76] | Specific ion channels, Specialized applications |
| DDE | 50x faster than explicit [76] | Low | Distance-dependent dielectric | Crude sampling, Very large systems |
*Relative to explicit solvent simulations
The comparative analysis reveals significant trade-offs between computational efficiency and simulation quality. The GBSW implicit model demonstrates the most favorable balance, achieving 4-5x speedup over explicit solvent while maintaining structural stability comparable to explicit simulations [75]. The EEF1 model offers the greatest computational efficiency (20x faster) but exhibits variable performance depending on the paired force field [75].
Force Field and Solvent Model Interdependence
Research demonstrates that solvation model performance is intrinsically linked to force field selection. Studies of the PB1 domain revealed that EEF1 with the CHARMM19 force field induced significant conformational reorientation, while the same solvent model with CHARMM22 force field maintained native-like dynamics similar to GBSW simulations [75]. This underscores the critical importance of testing force field/solvent model combinations for specific protein systems rather than relying on universal recommendations.
Simulation Parameters and System Equilibration
Equilibration Protocols
Proper system equilibration is essential for generating physiologically relevant simulation data. A phased approach is recommended:
- Energy Minimization: Removes steric clashes and improper geometry using steepest descent or conjugate gradient algorithms.
- Solvent Relaxation: Positional restraints on protein heavy atoms allow solvent molecules to organize around the protein.
- Gradual Heating: System temperature is gradually increased to target value (commonly 310K) while maintaining restraints.
- Pressure Equilibration: Isotropic pressure coupling achieves correct system density.
Studies emphasize that insufficient equilibration represents a common pitfall, particularly for simulations aimed at validating folding pathways [77]. Monitoring equilibrium indicators such as stable potential energy, temperature, pressure, and root-mean-square deviation (RMSD) is essential before proceeding to production simulations.
Enhanced Sampling for Folding Studies
Conventional MD simulations face timescale limitations for studying complete folding processes. Several advanced techniques address this challenge:
- Replica Exchange MD: Parallel simulations at different temperatures enable enhanced conformational sampling.
- Metadynamics: History-dependent bias potentials accelerate exploration of free energy landscapes.
- Targeted MD: Guided simulations can rapidly generate transition pathways between states.
When applying these methods to validate AI-predicted structures, it's crucial to recognize that AlphaFold 2 and similar tools tend to predict single conformational states, potentially missing functionally important asymmetry and conformational diversity observed in experimental structures [61]. Enhanced sampling techniques can help explore the complete conformational landscape around AI-predicted structures.
Experimental Protocols for Method Validation
Standardized Benchmarking Setup
To ensure reproducible comparison of simulation methodologies, researchers should implement standardized benchmarking protocols:
System Preparation Protocol:
- Obtain protein structure from experimental source (PDB) or AI prediction (AlphaFold Database)
- Process structure using pdb4amber or CHARMM's psfgen to add missing atoms/residues
- Select appropriate force field (e.g., CHARMM22/27/36 or AMBER ff14SB/19SB)
- Create simulation box with minimum 10-12 Å padding between protein and box edge
- Add solvent molecules and ions to neutralize system charge
- Implement 8-10 Å cutoff for non-bonded interactions with Particle Mesh Ewald for long-range electrostatics
Simulation Protocol:
- Energy minimization: 5,000 steps steepest descent followed by 5,000 steps conjugate gradient
- Solvent equilibration: 100 ps with positional restraints on protein heavy atoms (force constant 5 kcal/mol/Ų)
- System heating: 100 ps gradual heating from 0K to target temperature using Langevin dynamics
- Pressure equilibration: 1-5 ns with semi-isotropic pressure coupling
- Production simulation: Unrestrained MD with timestep appropriate for constraints (typically 2 fs)
Performance Optimization on Modern Hardware
Recent advances in computational hardware offer significant performance improvements for MD simulations. On AWS Graviton3E processors, optimal performance is achieved using:
- Arm Compiler for Linux (ACfL) version 23.04 or later with SVE-enabled binary compilation
- Arm Performance Libraries (ArmPL) for mathematical optimizations
- Open MPI 4.1.5 or later with Elastic Fabric Adapter support [78]
Benchmarking demonstrates that this configuration delivers 19-28% better performance compared to NEON/ASIMD-enabled binaries, with near-linear scalability across multiple nodes [78].
Table 2: Essential Resources for Molecular Dynamics Simulations
| Resource Category | Specific Tools | Function and Application |
|---|---|---|
| Structure Sources | PDB, AlphaFold Database | Provide initial protein structures for simulation [61] |
| Simulation Software | GROMACS, NAMD, AMBER, CHARMM, LAMMPS | MD simulation engines with various optimization profiles [78] |
| Force Fields | CHARMM36, AMBER ff19SB, GROMOS | Parameter sets defining molecular interactions [75] |
| Solvation Models | TIP3P, GBSW, EEF1 | Explicit and implicit solvent treatments [75] |
| Analysis Tools | MDAnalysis, VMD, PyMol, CPPTRAJ | Trajectory analysis and visualization |
| Computational Resources | AWS Hpc7g, X86 clusters, GPU accelerators | Hardware for simulation execution [78] |
The establishment of rigorous best practices for MD system setup, solvation model selection, and parameter optimization is fundamental to generating reliable simulation data for validating protein folding pathways. As AI-based structure prediction tools increasingly provide initial structural models, their limitations in capturing conformational diversity [61] and environmental dependence [9] make MD simulation validation more crucial than ever. By implementing the comparative frameworks, experimental protocols, and optimization strategies outlined in this guide, researchers can generate more reliable simulation data that effectively bridges the gap between computational prediction and experimental reality in protein folding research.
Benchmarks and Metrics: Rigorously Comparing Predictions with Experimental Reality
Validating molecular dynamics (MD) predictions of protein folding pathways requires integration with robust experimental biophysical techniques. This guide compares three key methods—Förster Resonance Energy Transfer (FRET), Nuclear Magnetic Resonance (NMR), and Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)—for their utility in providing experimental observables that can test and refine computational models. These techniques probe protein structure and dynamics across different spatial and temporal resolutions, creating a multi-faceted framework for validation. FRET provides exquisite temporal resolution for monitoring distance changes during folding events [79], NMR offers atomic-level structural and dynamic information, and HDX-MS reveals conformational dynamics and stability patterns across various protein states [80] [81]. Within the context of MD validation, HDX-MS has emerged as a particularly powerful tool when combined with advanced computational approaches like maximum-entropy reweighting to build ensembles that faithfully reproduce experimental data [82] [83].
Technical Comparison of Key Observables
The following table summarizes the core characteristics, key observables, and utility for MD validation of each technique.
Table 1: Comparison of Key Experimental Techniques for Validating MD Simulations
| Feature | FRET | NMR | HDX-MS |
|---|---|---|---|
| Key Observable | Distance between dye-labeled sites (typically 2-8 nm) [79] | Chemical shift, J-coupling, NOEs, RDC | Deuterium incorporation into backbone amides [81] |
| Spatial Resolution | Low (inter-dye distance) | High (atomic) | Medium (peptide-level, 5-20 residues) [82] |
| Temporal Resolution | Nanoseconds to milliseconds (single molecule) [79] | Picoseconds to seconds | Milliseconds to hours (sampling times) [81] |
| Sample Consumption | Low (single molecule) to moderate | High (mmol concentrations) | Low (pmol to μg) [80] [81] |
| Typical System Size | Small proteins to complexes [79] | Small to medium proteins (< ~100 kDa) | No practical upper limit (complexes, viral capsids) [81] |
| Key Strength for MD Validation | Direct measurement of transition path times and rates [79] | Atomic-level structural and dynamic parameters | Comprehensive profiling of conformational dynamics and populations [82] [80] |
| Primary Limitation | Requires labeling which may perturb system | Size limitations, specialized expertise required | Indirect structural interpretation, peptide-level resolution [82] |
Hydrogen-Deuterium Exchange (HDX-MS)
Principle and Workflow
HDX-MS measures the exchange of backbone amide hydrogens for deuterium from the surrounding solvent, which reports on protein structure and dynamics [81]. The exchange rate is influenced by hydrogen bonding and solvent accessibility, providing insights into local stability and conformational dynamics [80]. The following diagram illustrates the core workflow for a continuous-labeling HDX-MS experiment.
Key Experimental Protocols
For studying protein folding or conformational changes, HDX-MS experiments typically employ either continuous labeling (varying D₂O exposure time) or pulsed labeling (fixed labeling time after perturbation) [81]. The protein is first diluted into D₂O buffer and incubated for varying timepoints (e.g., 10 seconds to several hours). The exchange reaction is then quenched by lowering pH and temperature (typically to pH 2.5 and 0°C), which reduces the intrinsic exchange rate by a factor of approximately 14 [81]. The quenched sample is subjected to proteolytic digestion (usually with pepsin) followed by liquid chromatography separation and mass spectrometry analysis to determine deuterium incorporation for each identified peptide [81].
Integration with MD Simulations: The HDXer Approach
The HDX ensemble reweighting (HDXer) methodology provides a rigorous framework for connecting HDX-MS data with MD simulations [82] [83]. This approach applies a maximum-entropy bias to a candidate structural ensemble from MD simulations such that averaged peptide-deuteration levels, predicted by an empirical model, agree with experimental values [82]. The following diagram illustrates this integrative process.
This approach enables researchers to objectively determine whether a given simulation trajectory reproduces the conformational ensemble reflected in experimental HDX data, and if not, to reweight the ensemble to achieve agreement while introducing minimal bias [83].
Application Example: Distinguishing Substrate and Inhibitor Binding
HDX-MS proved particularly powerful in a study of the proton-coupled transporter XylE, where it captured distinct dynamics upon substrate (xylose) versus inhibitor (glucose) binding [80]. Despite nearly identical static structures, HDX-MS revealed that protonation of a conserved aspartate (D27) triggers conformational transition to an inward-facing state only in the presence of substrate, while glucose locks the transporter in an outward-facing state. This allosteric coupling, corroborated by MD simulations, demonstrated HDX-MS's unique ability to distinguish functionally distinct ligands that appear identical in crystal structures [80].
Förster Resonance Energy Transfer (FRET)
Principle and Key Observables
FRET measures non-radiative energy transfer between a donor and an acceptor fluorophore, with efficiency inversely proportional to the sixth power of the distance between them [79]. This makes it exceptionally sensitive to distance changes in the 2-8 nm range, ideal for monitoring protein folding and conformational changes. A particularly powerful application in folding studies is the measurement of transition path times—the actual time a protein spends crossing the free energy barrier between folded and unfolded states [79]. These timescales are logarithmically dependent on barrier height, unlike folding rates which show exponential dependence, providing a more direct probe of the diffusive properties of the polypeptide chain [79].
Experimental Protocols for Folding Studies
In single-molecule FRET protein folding studies, the protein is typically labeled with donor and acceptor fluorophores at specific positions and immobilized on a surface [79]. Experiments are performed near the denaturation midpoint to observe both folding and unfolding transitions. Photon trajectories are analyzed using maximum likelihood methods, sometimes incorporating a virtual intermediate state to extract finite transition path times [79]. This approach has revealed transition path times of several microseconds for small proteins, providing critical tests for all-atom MD simulations [79].
Essential Research Reagents and Solutions
Table 2: Key Research Reagents and Solutions for HDX-MS and FRET Experiments
| Reagent/Solution | Function/Purpose | Technical Considerations |
|---|---|---|
| D₂O Buffer | Deuterium source for HDX labeling | pD must be adjusted (read pH + 0.4 units) [81] |
| Quench Solution | Stops HDX (low pH, low temperature) | Typically pH 2.5, 0°C [81] |
| Immobilized Pepsin | Proteolytic digestion for HDX-MS | Works at low pH and temperature [81] |
| Donor/Acceptor Dyes | FRET pair for distance measurement | Must site-specifically label protein without perturbing folding [79] |
| Denaturants | Modulate protein stability for folding studies | Used to measure stability and rates near denaturation midpoint [79] |
FRET, NMR, and HDX-MS provide complementary experimental observables for validating MD-predicted protein folding pathways. FRET excels at measuring distance changes and transition path times with high temporal resolution, while HDX-MS offers comprehensive profiling of conformational dynamics and populations, even for large systems. NMR provides atomic-resolution structural and dynamic information, though with more size limitations. The integration of these experimental data with MD simulations through approaches like HDX ensemble reweighting represents the cutting edge in protein folding studies, enabling researchers to build dynamically accurate structural ensembles that bridge the gap between simulation and experiment.
Molecular dynamics (MD) simulations provide atomistic detail of protein folding pathways, but their predictive accuracy must be rigorously validated against experimental data. This process relies on quantitative metrics that can bridge computational and experimental approaches, each offering distinct insights into the folding process. Root-mean-square deviation (RMSD) and global distance test (GDT) serve as primary measures for assessing structural accuracy of the final folded state, while transition path time distributions offer a unique window into the actual barrier-crossing events during folding. The integration of these metrics, complemented by experimental techniques such as hydrogen-deuterium exchange mass spectrometry (HDX-MS) and single-molecule Förster resonance energy transfer (smFRET), creates a powerful framework for validating MD-predicted folding mechanisms. This guide provides a comprehensive comparison of these fundamental metrics, their experimental counterparts, and protocols for their application in benchmarking the accuracy of MD simulations against experimental observations, with particular importance for drug discovery professionals who rely on accurate protein structural information.
Quantitative Metrics for Protein Folding Validation
Structural Comparison Metrics
Table 1: Structural Metrics for Folding Validation
| Metric | Calculation Basis | Structural Feature Assessed | Typical Experimental Reference | Strengths | Limitations |
|---|---|---|---|---|---|
| RMSD | Root-mean-square deviation of atomic positions | Global backbone conformation | X-ray crystallography, Cryo-EM, NMR | Simple calculation, Intuitive interpretation | Sensitive to domain rotations, Global measure misses local accuracy |
| GDT | Percentage of Cα atoms within defined distance cutoffs | Global fold correctness, Native contact formation | High-resolution structures from PDB | More robust to local deviations, Better correlates with model quality | Multiple cutoffs required for full picture, Less familiar to non-specialists |
| pLDDT | Predicted local distance difference test per residue | Local structure confidence, Model quality assessment | Experimental lDDT calculated from PDB | Residue-specific confidence scores, No experimental structure required | AF2 self-confidence measure, not direct accuracy validation [61] |
RMSD remains a fundamental metric for structural comparison, calculated as the root-mean-square deviation of atomic positions after optimal superposition. For protein folding validation, Cα-RMSD values below 1-2 Å typically indicate high accuracy in predicting the native state, as demonstrated in MD simulations of villin headpiece where multiple force fields achieved 0.6-1.3 Å Cα-RMSD from experimental structures [84]. GDT provides a complementary perspective by measuring the percentage of Cα atoms within defined distance cutoffs (typically 1, 2, 4, and 8 Å) after superposition, offering a more robust assessment of global fold correctness that is less sensitive to small domain shifts than RMSD.
Kinetic and Transition Path Metrics
Table 2: Kinetic Metrics for Folding Mechanism Validation
| Metric | Temporal Scale | Folding Information | Experimental Method | Relationship to Free Energy | Force Field Dependence |
|---|---|---|---|---|---|
| Folding Rate (kf) | Milliseconds to seconds | Overall folding speed | Stopped-flow, smFRET | Exponential dependence on barrier height: kf ~ exp(-ΔG‡/kBT) [79] | Highly sensitive; different force fields yield varying rates [84] |
| Transition Path Time (tTP) | Nanoseconds to microseconds | Direct barrier crossing duration | Photon-by-photon smFRET analysis | Logarithmic dependence on barrier height [79] | Less sensitive to force field details |
| Transition Path Time Distribution | Molecular timescale | Barrier shape, internal friction | High-time-resolution smFRET | Reveals free energy surface roughness, traps [85] | Reveals fundamental limitations in force fields |
The transition path represents the critical segment of a protein folding trajectory where the free energy barrier between unfolded and folded states is actually crossed. While traditional folding times (inverse of folding rates) measure the waiting time before a successful barrier crossing event, transition path times directly measure the duration of the barrier crossing itself, typically occurring on nanosecond to microsecond timescales [79]. This distinction is crucial because folding times exhibit exponential dependence on free energy barrier height, while transition path times show only logarithmic dependence, making them more robust metrics for comparing simulation and experiment [79]. Recent advances in analyzing transition path time distributions have revealed long-time tails that may indicate the existence of "traps" or wells in the free energy surface along the folding pathway, providing additional mechanistic insights beyond average timescales [85].
Experimental Methodologies for Metric Validation
Single-Molecule FRET for Transition Path Measurements
Experimental Protocol: Transition Path Time Determination via smFRET
Protein Labeling: Site-specifically label the protein of interest with donor (e.g., Cy3) and acceptor (e.g., Cy5) fluorophores at positions that exhibit distinct distance changes between folded and unfolded states.
Immobilization: Immobilize labeled proteins on a passivated glass surface to enable extended observation of individual molecules without diffusion.
Data Acquisition: Conduct experiments near the denaturation mid-point to observe both folding and unfolding transitions. Use high-intensity lasers and sensitive detectors to achieve maximum photon detection rates (critical for nanosecond timescale resolution).
Photon Trajectory Analysis: Apply maximum likelihood methods developed by Szabo and Gopich to analyze unbinned photon trajectories [79]. This approach is essential when transition path times are too short for conventional binning analysis.
Model Comparison: Compare two-state models (assuming instantaneous transitions) with three-state models incorporating a virtual intermediate state with finite lifetime. The lifetime value that maximizes the likelihood function corresponds to the average transition path time.
Statistical Validation: Establish statistical significance through likelihood ratio tests between competing models. If no significant peak exists, determine an upper bound for the transition path time.
This methodology has successfully measured transition path times for several two-state proteins, including the 35-residue WW domain and 56-residue α/β protein, providing crucial experimental benchmarks for MD simulations [79].
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
Experimental Protocol: HDX-MS for Protein Structural Validation
Deuterium Labeling: Incubate the protein in D₂O buffer for defined time periods (seconds to hours) under physiological conditions to allow backbone amide hydrogens to exchange with deuterium.
Quenching: Rapidly quench the reaction by lowering pH to 2.5 and temperature to 0°C to minimize back-exchange.
Digestion: Digest proteins using immobilized pepsin columns under quenched conditions to generate peptides for analysis.
LC-MS Analysis: Separate peptides using reversed-phase chromatography at 0°C and analyze with high-resolution mass spectrometry. Employ electron transfer dissociation (ETD) fragmentation to minimize deuterium scrambling and achieve single-residue resolution [86].
Data Processing: Identify peptides and calculate deuterium uptake using specialized software (e.g., BioPharma Finder). Express results as relative fractional uptake (RFU) normalized to maximum possible deuterium incorporation.
Protection Factor Calculation: Estimate protection factors from protein structures using algorithms that consider heavy atom contacts and hydrogen bonding within defined distance cutoffs [87].
HDX-MS provides exceptional sensitivity to conformational dynamics and can discriminate between native and non-native protein folds through quantitative comparison of experimental and simulated deuterium uptake patterns [87]. Recent advances include artificial intelligence-based HDX (AI-HDX) prediction from sequence, enabling high-throughput dynamics analysis [88].
Cross-linking Mass Spectrometry (XL-MS)
Experimental Protocol: XL-MS for Spatial Restraint Determination
Cross-linking Reaction: Treat protein with bifunctional cross-linking reagents (e.g., DSSO, BS3) that primarily target lysine residues, using appropriate reagent-to-protein ratios and reaction times.
Digestion and Separation: Digest cross-linked proteins with proteases (typically trypsin) and separate cross-linked peptides using liquid chromatography.
MS/MS Identification: Identify cross-linked peptides using tandem mass spectrometry, employing specialized search algorithms (e.g., XlinkX, plink) to detect cross-linked peptide pairs.
Distance Restraint Derivation: Convert identified cross-links into spatial restraints based on cross-linker spacer arm length, typically setting distance thresholds of 20-30 Å between Cα atoms of cross-linked residues.
Computational Integration: Incorporate distance restraints into structure prediction pipelines, such as Rosetta or Integrative Modeling Platform (IMP), either as scoring function penalties or structural filters [89].
XL-MS has been successfully applied to systems ranging from individual proteins to mega-Dalton complexes, providing crucial distance restraints that improve model accuracy when integrated with computational approaches [89].
Research Reagent Solutions for Folding Studies
Table 3: Essential Research Reagents for Protein Folding Studies
| Category | Specific Products/Systems | Application in Folding Studies | Key Features |
|---|---|---|---|
| Mass Spectrometry Systems | Orbitrap Exploris 480, Orbitrap Eclipse Tribrid | HDX-MS, XL-MS, Native MS | High resolution-accurate mass, Multiple fragmentation techniques, ETD capability |
| Chromatography Systems | TRAJAN CHRONECT HDX, Vanquish Neo UHPLC | Peptide separation for HDX-MS | Temperature control (0°C), Low-pH mobile phases, Immobilized pepsin columns |
| Fluorescence Systems | Custom smFRET setups with immobilized proteins | Transition path time measurements | High photon detection rates, Single-molecule sensitivity, Temperature control |
| Software Platforms | BioPharma Finder, DynamX, Rosetta, IMP | Data analysis, Structure prediction, Model validation | Specialized HDX data processing, Integration of sparse experimental data |
| Cross-linking Reagents | DSSO, BS3, DSG | XL-MS for spatial proximity mapping | MS-cleavable variants, Variable spacer lengths, Membrane-permeable options |
Comparative Analysis of MD Force Field Performance
The accuracy of MD-predicted folding pathways depends critically on the force field employed. Studies comparing Amber ff03, Amber ff99SB-ILDN, CHARMM27, and CHARMM22 force fields revealed that while all could reproduce the experimental native state structure of villin headpiece (Cα-RMSD 0.6-1.3 Å) and approximate folding rates (~1 μs experimental vs. 0.8-3.0 μs simulated), they exhibited significant differences in folding mechanisms and unfolded state properties [84]. For instance, CHARMM27 produced an unfolded state with substantially higher helical content (73%/33%/90% for helices 1-3) compared to CHARMM22* (41%/9%/44%), leading to different predominant folding pathways [84]. This highlights the critical importance of validating not just final structures and overall rates, but the complete folding mechanism against experimental data.
Recent developments in structure-based statistical mechanical models, such as the WSME-L model, show promise for predicting folding mechanisms of multidomain proteins with low computational complexity, providing valuable benchmarks for atomistic simulations [73]. These models successfully reproduce experimentally observed folding behaviors by incorporating nonlocal interactions through virtual linkers, enabling prediction of complex folding pathways that involve discontinuous domains and disulfide bond formation [73].
Comprehensive validation of MD-predicted protein folding pathways requires integration of multiple complementary metrics. Structural metrics like RMSD and GDT provide essential validation of the final folded state, while transition path time distributions offer unique insights into the barrier-crossing process itself. Experimental techniques including smFRET, HDX-MS, and XL-MS generate crucial data for benchmarking simulations across different temporal and spatial resolutions. As force field comparisons demonstrate, accurate prediction of native structure and folding rate does not guarantee correct folding mechanisms, emphasizing the need for multifaceted validation approaches. By strategically applying this toolkit of quantitative metrics and experimental methodologies, researchers can rigorously assess and improve the predictive power of molecular dynamics simulations, ultimately advancing applications in protein engineering and drug discovery where understanding folding pathways is critical.
Molecular dynamics (MD) simulations provide a powerful vehicle for capturing the structures, motions, and interactions of biological macromolecules in full atomic detail, enabling the prediction of protein folding pathways. However, the accuracy of such simulations is critically dependent on the force field—the mathematical model used to approximate the atomic-level forces acting on the simulated molecular system [90]. This guide examines two seminal case studies, the FSD-1 designed protein and the WW domain, where MD-predicted folding pathways have been rigorously validated against experimental data. These systems represent important benchmarks for assessing the performance of various computational force fields and simulation methodologies, providing critical insights for researchers investigating protein folding mechanisms and developing more accurate predictive models.
The validation process involves sophisticated experimental techniques including NMR spectroscopy, circular dichroism, calorimetry, and laser-induced temperature-jump spectroscopy, which provide quantitative data on folding kinetics, thermodynamic stability, and native state structures. By comparing simulation results with these experimental observables, researchers can benchmark the performance of different force fields and identify areas needing improvement. This guide provides a comprehensive comparison of these benchmarking efforts, detailing the specific methodologies, key findings, and implications for the field of computational biophysics.
FSD-1: A Designed ββα Ultrafast Folding Protein
FSD-1 is a 28-residue designed ultrafast folder with a ββα (hairpin/helix) fold, featuring a well-defined hydrophobic core and containing only naturally occurring residues [91]. This system was intentionally designed to serve as a model for studying the folding of mixed α/β proteins, bridging the gap between experimental and in silico studies. Unlike more commonly studied ββα proteins which may contain non-natural residues, FSD-1's composition makes it particularly valuable for testing computational force fields. A close analog, FSD-1ss, displays two folding phases (τ₁∼150 ns and τ₂∼4.5 µs) at 322 K, placing it at the top range of known ultrafast folders [91]. The folding kinetics and structural properties of FSD-1 make it computationally tractable while still providing insights relevant to larger, more complex protein systems.
Experimental Benchmarks and Observed Folding Behavior
The thermal unfolding of FSD-1, as determined from Circular Dichroism (CD) and Differential Scanning Calorimetry (DSC), is reversible but weakly cooperative, with a relatively low melting temperature (Tₘ = 315 K) [91]. Early interpretations attributed this broad transition to the melting of the entire protein, though this was later challenged by researchers who proposed it might reflect only the melting of the α-helical segment (residues 14–26). This controversy highlighted the need for detailed MD simulations to resolve the nature of the transition and establish whether FSD-1 could genuinely serve as a model system for studying α/β protein folding.
Table 1: Key Experimental Benchmarks for FSD-1 Folding
| Parameter | Experimental Value | Measurement Technique | Interpretation |
|---|---|---|---|
| Melting Temperature (Tₘ) | 315 K | Circular Dichroism (CD), Differential Scanning Calorimetry (DSC) | Weakly cooperative folding transition |
| Folding Time (FSD-1ss analog) | τ₁∼150 ns, τ₂∼4.5 µs | Laser-induced temperature-jump spectroscopy | Two-phase folding kinetics |
| Native Structure | ββα fold | NMR | Well-defined hydrophobic core |
MD Force Field Performance and Validation
Early simulations of FSD-1 met with mixed and sometimes conflicting results. Replica exchange molecular dynamics (REMD) simulations in explicit solvent using the Amber ff03 force field predicted a melting temperature of 411.59 K, approximately 100 K higher than the experimental value [91]. Similarly, simulations using the OPLS-AA/L 2001 force field produced a melting temperature 84 K higher than experimental observations. These significant discrepancies highlighted substantial limitations in existing force fields.
More successful results were obtained using the Amber ff96 protein force field combined with the implicit water solvent IGB = 5, which has demonstrated a good balance of α/β propensity for small peptides [91]. This combination revealed that the breadth of FSD-1's folding transition arises from the spread in melting temperatures (from ∼325 K to ∼302 K) of individual transitions: formation of the hydrophobic core, β-hairpin and tertiary fold, with the helix forming earlier. The simulations demonstrated that the melting transition corresponds to the melting of the protein as a whole, rather than solely the helix-coil transition, resolving the earlier experimental controversy.
Table 2: Force Field Performance for FSD-1 Folding Simulations
| Force Field | Solvent Model | Predicted Tₘ | Deviation from Experimental Tₘ | Key Findings |
|---|---|---|---|---|
| Amber ff03 | TIP3P (explicit) | 411.59 K | +96.59 K | Overstabilized native state |
| OPLS-AA/L 2001 | TIP4P (explicit) | ~399 K | ~84 K | Overstabilized native state |
| Amber ff96 | IGB = 5 (implicit) | 315-325 K (component-dependent) | Minimal for individual components | Explained broad transition; identified folding hierarchy |
| param99MOD5 | GBSA (implicit) | ~309 K | -6 K | FSD-1 used in force field optimization |
The exhaustive sampling achieved with ff96/igb5 enabled researchers to assess the quality of this force field combination, revealing that while it can predict the correct native fold, it nonetheless overstabilizes the α-helix portion of the protein (Tₘ = ∼387 K) as well as denatured structures [91]. This case study illustrates the importance of comparing multiple thermodynamic and kinetic parameters between simulation and experiment, rather than focusing on a single observable.
WW Domain: A Minimal β-Sheet Folding Model
The WW domain is one of the smallest protein modules, composed of only 40 amino acids, which folds into a meandering triple-stranded antiparallel β-sheet [92]. Named after the presence of two conserved tryptophans (W) spaced 20-22 amino acids apart, this domain mediates specific protein-protein interactions with short proline-rich or proline-containing motifs [92]. WW domains are present in various signaling and structural proteins, including the human Pin1 protein, where it plays important roles in cell signaling and has been implicated in various diseases [93] [92]. Its small size, well-defined structure, and cooperative folding make it an ideal model system for detailed folding studies.
Experimental Benchmarks and Key Folding Insights
The human Pin1 WW domain has been particularly extensively studied. Experimental studies revealed that the rate-limiting step for its folding is the formation of the loop 1 substructure [93]. This six-residue loop positions side chains that are important for mediating protein-protein interactions through binding of Pro-rich sequences. Interestingly, replacement of the wild-type loop 1 primary structure by shorter sequences with a high propensity to fold into a type-I' beta-turn conformation or the statistically preferred type-I G1 bulge conformation accelerates WW domain folding by almost an order of magnitude and increases thermodynamic stability [93].
However, this loop engineering to optimize folding energetics has a significant functional downside: it effectively eliminates WW domain function according to ligand-binding studies [93]. This demonstrates a classic trade-off between folding efficiency and biological function, suggesting that the energetic contribution of loop 1 to ligand binding appears to have evolved at the expense of fast folding and additional protein stability. Thus, the two-state barrier exhibited by the wild-type human Pin1 WW domain principally results from functional requirements, rather than from physical constraints inherent to the loop formation process itself.
MD Simulations and Force Field Validation
The small size of the WW domain has made it amenable to extremely detailed simulation studies. The Shaw laboratory developed a specialized machine that allowed elucidation of the atomic level behavior of the WW domain on biologically relevant timescales, employing equilibrium simulations that identified seven unfolding and eight folding events [92]. These extensive simulations provided unprecedented atomic-level detail of the folding process.
Additionally, research by Ranganathan's team has shown that a simple statistical energy function, which identifies co-evolution between amino acid residues within the WW domain, is necessary and sufficient to specify sequences that fold into native structure [92]. Using such an algorithm, they synthesized libraries of artificial WW domains that functioned very similarly to their natural counterparts, recognizing class-specific proline-rich ligand peptides. This demonstrates how combining MD simulations with bioinformatic approaches can lead to insights applicable to protein design.
Comparative Analysis of Benchmarking Methodologies
Experimental Techniques for Validating Folding Pathways
Both FSD-1 and WW domain studies employed a range of sophisticated experimental techniques to validate MD predictions. These methods provide complementary information about the folding process across different timescales and structural resolutions.
Table 3: Key Experimental Techniques for Validating Folding Pathways
| Technique | Information Provided | Application to FSD-1 | Application to WW Domain |
|---|---|---|---|
| NMR Spectroscopy | Atomic-level structure and dynamics | Limited data provided | Detailed structure determination; ligand interactions |
| Circular Dichroism (CD) | Secondary structure content | Thermal unfolding curves | Not specifically mentioned |
| Differential Scanning Calorimetry (DSC) | Thermodynamics of folding | Reversible, weakly cooperative transition | Not specifically mentioned |
| Laser-induced T-jump | Fast folding kinetics | Used for FSD-1ss analog | Not specifically mentioned |
| Isothermal Titration Calorimetry (ITC) | Binding affinities and thermodynamics | Not used | Quantitative ligand binding studies |
Assessment of Computational Force Fields
Systematic validation of protein force fields against experimental data has revealed significant differences in their abilities to reproduce the structure and fluctuations of folded proteins [90]. For example, comparisons with experimental NMR data for folded proteins like ubiquitin and GB3 showed that while most force fields maintained stable native states, their accuracy in describing backbone scalar couplings, residual dipolar couplings, and order parameters varied considerably [90].
The performance of different force fields has been shown to depend on the specific structural elements being studied. For instance, the CHARMM27 force field severely overstabilizes helical structures [90], while ff99SB-ILDN underestimates helix stability [90]. More recent "helix coil–balanced" force fields (ff99SB-ILDN, ff03 and CHARMM22*) provide better descriptions for peptides with mixed secondary structure preferences [90].
These findings highlight the importance of testing force fields against multiple systems with diverse structural characteristics rather than relying on a single benchmark system. Both FSD-1 (with its mixed ββα fold) and the WW domain (with its triple-stranded β-sheet) provide distinct structural contexts for evaluating force field performance.
Signaling Pathways and Functional Implications
The folding dynamics of WW domains have direct implications for their biological function in critical signaling pathways. WW domain-containing proteins like YAP and WWOX play key roles in the Hippo signaling pathway, which regulates organ size and tumor suppression [92] [94]. The ability of these proteins to interact with multiple ligands through their WW domains makes them important signaling hubs, and their folding kinetics can influence their signaling capabilities.
Diagram 1: WW Domain Function in Hippo Signaling Pathway. The diagram illustrates how WW domain-mediated interactions regulate YAP activity in the Hippo signaling pathway, influencing cell proliferation and growth suppression.
For the WW domain-containing oxidoreductase (WWOX) tumor suppressor, the relationship between folding and function is particularly complex. The WW domains within the WW1–WW2 tandem module physically associate to adopt a fixed spatial orientation relative to each other, with the WW2 domain acting as a chaperone for the WW1 domain [94]. This interaction stabilizes the WW1 domain and enhances its ligand-binding capability, demonstrating how domain-domain interactions can influence both folding and function.
Research Toolkit: Essential Methods and Reagents
Diagram 2: Computational Toolkit for Folding Pathway Studies. The diagram illustrates the relationship between different software tools, force fields, enhanced sampling methods, and analysis utilities used in protein folding simulations.
Table 4: Essential Research Reagents and Solutions for Folding Studies
| Reagent/Resource | Function/Application | Examples/Specifics |
|---|---|---|
| Protein Force Fields | Mathematical models for atomic-level forces | AMBER (ff96, ff99SB, ff03), CHARMM (22, 27, 22*), OPLS-AA |
| Solvent Models | Simulating aqueous environment | TIP3P, TIP4P (explicit); GBSA, IGB=5 (implicit) |
| Specialized Hardware | Enhanced simulation capabilities | Anton specialized computer [90] |
| NMR Spectroscopy | Atomic-level structure validation | Structure determination, dynamics measurements |
| Circular Dichroism | Secondary structure monitoring | Thermal unfolding experiments |
- Calorimetry - Thermodynamic parameter quantification - Differential Scanning Calorimetry (DSC), Isothermal Titration Calorimetry (ITC)
- Fast Kinetics Instruments - Microsecond-nanosecond timescale folding measurements - Laser-induced temperature-jump spectroscopy
The case studies of FSD-1 and the WW domain demonstrate the critical importance of rigorous benchmarking for validating MD-predicted protein folding pathways against experimental data. These systems have served as important testbeds for evaluating force field performance, revealing both successes and limitations in current computational methodologies. The integration of experimental and computational approaches has provided insights that would be impossible to obtain from either methodology alone.
Future directions in the field include the development of more accurate force fields with better balance between different secondary structure elements, improved sampling algorithms to access longer timescales, and more sophisticated methods for comparing simulation results with experimental observables. The recent advancements in AI-based structure prediction tools like AlphaFold, while revolutionary for static structure prediction, have not eliminated the need for MD simulations in understanding folding dynamics and pathways [95]. As both computational power and experimental techniques continue to advance, the integration of multiple approaches will remain essential for unraveling the complexities of protein folding.
The accurate interpretation of confidence scores is fundamental to validating molecular dynamics (MD)-predicted protein folding pathways with experimental data. AlphaFold2 and related AI-based structure prediction tools output per-residue and global confidence metrics that estimate the reliability of their predictions. The pLDDT (predicted local distance difference test) and PAE (predicted aligned error) are crucial for assessing local structure reliability and inter-domain accuracy, respectively. For researchers comparing AI predictions with experimental structures, these scores help identify well-resolved regions suitable for detailed mechanistic studies and flexible regions that may adopt multiple conformations. Experimental validations consistently reveal that while high-confidence regions often match experimental accuracy, medium-to-low confidence areas frequently correspond to biologically critical flexible regions involved in binding and allostery. This analysis provides a framework for scientists to critically evaluate AI-predicted models against experimental benchmarks.
Core Confidence Metrics: Technical Specifications and Interpretation
pLDDT (Predicted Local Distance Difference Test)
The pLDDT is a per-residue local confidence score scaled from 0 to 100, with higher scores indicating higher confidence and typically more accurate prediction. It estimates how well the prediction would agree with an experimental structure based on the local distance difference test Cα (lDDT-Cα), which assesses the correctness of local distances without relying on structural superposition [96]. The pLDDT score varies significantly along a protein chain, indicating regions where the predicted structure may be reliable versus regions unlikely to be accurate.
Table: pLDDT Score Interpretation Guidelines
| pLDDT Range | Confidence Level | Structural Interpretation | Recommended Usage |
|---|---|---|---|
| > 90 | Very high | High backbone and side-chain accuracy; suitable for binding site characterization | Detailed mechanistic studies, drug docking |
| 70 - 90 | Confident | Correct backbone with possible side-chain displacements | Functional analysis, molecular dynamics starting points |
| 50 - 70 | Low | Low confidence; treat with caution | Limited interpretation; may indicate flexibility |
| < 50 | Very low | Likely disordered or unstructured in physiological conditions | Avoid structural interpretation; may indicate intrinsic disorder |
Low pLDDT regions (<50) generally indicate one of two scenarios: either the region is naturally highly flexible or intrinsically disordered and lacks a well-defined structure, or AlphaFold2 does not have enough information to predict it with confidence [96]. Notably, AlphaFold2 may be very confident in the structure of globular domains but less confident in linkers between domains, as linkers are more likely to be naturally variable, less structured, and more flexible [96].
PAE (Predicted Aligned Error)
The PAE matrix represents a pairwise error prediction that estimates the expected distance error in Ångströms between residues after optimal alignment. Unlike pLDDT, which measures local confidence, PAE assesses the relative positional confidence between different parts of the structure, making it particularly valuable for evaluating multi-domain proteins and complexes.
Table: PAE Score Interpretation and Implications
| PAE Value Range (Å) | Structural Relationship | Confidence Interpretation | Biological Implications |
|---|---|---|---|
| 0 - 5 | High confidence | Strong positional constraint | Stable domain or rigid-body relationship |
| 5 - 10 | Medium confidence | Moderate positional uncertainty | Flexible linkers or dynamic interfaces |
| > 10 | Low confidence | Weak positional constraint | Highly flexible or independent domains |
The PAE plot is visualized as a heatmap where the x and y axes represent residue indices, and the color at any point (i,j) indicates the predicted error in the relative position of residue i when the model is aligned on residue j. Well-defined domains typically appear as dark green squares along the diagonal, indicating high internal confidence, while off-diagonal elements reveal inter-domain confidence [97]. Research demonstrates that PAE maps from AlphaFold2 correlate with distance variation matrices from molecular dynamics simulations, revealing that PAE maps can predict the dynamical nature of protein residues [98].
Experimental Validation: Comparing AlphaFold2 Predictions with Experimental Structures
Comprehensive Analysis of Nuclear Receptors
A 2025 comprehensive analysis comparing AlphaFold2-predicted and experimental nuclear receptor structures provides critical insights for structure-based drug design. The study examined root-mean-square deviations, secondary structure elements, domain organization, and ligand-binding pocket geometry across seven human nuclear receptors with available full-length multi-domain experimental structures [61].
Table: AlphaFold2 Performance Across Nuclear Receptor Domains
| Structural Feature | AlphaFold2 Performance | Experimental Correlation | Limitations |
|---|---|---|---|
| Overall Backbone | High accuracy for stable conformations | RMSD < 1.0Å for high pLDDT regions | Misses full spectrum of biological states |
| Ligand-Binding Domains (LBD) | Moderate accuracy (CV = 29.3%) | Captures general fold | Systematic 8.4% underestimation of pocket volumes |
| DNA-Binding Domains (DBD) | High accuracy (CV = 17.7%) | High structural conservation | Less conformational diversity captured |
| Full-Length Multi-domain | Accurate domain structures | Proper stereochemistry | Misses functional asymmetry in homodimers |
The analysis revealed that while AlphaFold2 achieves high accuracy in predicting stable conformations with proper stereochemistry, it shows limitations in capturing the full spectrum of biologically relevant states, particularly in flexible regions and ligand-binding pockets [61]. Statistical analysis revealed significant domain-specific variations, with ligand-binding domains showing higher structural variability (CV = 29.3%) compared to DNA-binding domains (CV = 17.7%) [61]. Notably, AlphaFold2 systematically underestimates ligand-binding pocket volumes and captures only single conformational states in homodimeric receptors where experimental structures show functionally important asymmetry [61].
Correlation with Molecular Dynamics and Flexibility
Research indicates that AlphaFold2 not only predicts protein 3D structure but also provides clues about protein dynamics through both pLDDT scores and PAE matrices. Studies comparing molecular dynamics simulations with AlphaFold2 predictions found that for most protein models, AF2-scores derived from pLDDT are highly correlated with root mean square fluctuations calculated from MD simulations [98]. This correlation suggests that pLDDT scores convey information about residue flexibility, connecting static structures with dynamic personalities.
However, for intrinsically disordered proteins and randomized proteins with no MSA hits, the AF2-scores do not correlate with RMSF from MD, especially for IDPs [98]. This indicates that in AlphaFold2 modeling, biological information through multisequence alignment may not only be translated to structural information but also contains biophysical information about which residues are mobile.
Methodologies for Accessing and Visualizing Confidence Metrics
Technical Workflow for Metric Extraction
After a successful AlphaFold2 run, confidence metrics are stored in specific output files that require processing for visualization and analysis. The key files containing confidence metrics include:
result_model_{1-5}_pred_0.pkl: Contains dictionaries with 'plddt' and 'predictedalignederror' arraysranking_debug.json: Includes quality scores for each model (0-100 scale)relaxed_model_{1-5}_pred_0.pdb: PDB files with pLDDT scores stored in the B-factor field [97]
The following DOT script visualizes the complete workflow for extracting and interpreting AlphaFold2 confidence metrics:
Python Implementation for Metric Visualization
To programmatically extract and visualize confidence metrics, researchers can use Python scripts to unpickle the result files and generate publication-quality plots:
Essential Research Reagents and Computational Tools
Table: Research Reagent Solutions for Confidence Metric Analysis
| Tool/Resource | Type | Primary Function | Access Method |
|---|---|---|---|
| AlphaFold2 Database | Database | Precomputed structures with confidence metrics | https://alphafold.ebi.ac.uk/ |
| AlphaFold2 Open Source | Software | Local structure prediction with confidence scores | GitHub repository |
| MD Simulation Software | Software | Validate dynamic properties against confidence scores | GROMACS, AMBER, NAMD |
| Nuclear Receptor Structures | Experimental Data | Benchmark AF2 predictions in pharmaceutically relevant system | Protein Data Bank (PDB) |
| Python Visualization Scripts | Custom Code | Generate publication-quality metric plots | Custom development |
The interpretation of pLDDT and PAE confidence scores provides researchers with critical guidance for validating MD-predicted protein folding pathways. High-confidence regions (pLDDT > 70) generally provide reliable structural frameworks for drug docking and mechanistic studies, while low-confidence regions (pLDDT < 50) often correspond to biologically important flexible regions or intrinsic disorder. The systematic underestimation of ligand-binding pocket volumes by AlphaFold2 highlights the necessity of integrating experimental data with computational predictions, particularly for structure-based drug design. By strategically applying these confidence metrics, researchers can prioritize experimental resources, identify potential limitations in AI-predicted models, and develop more accurate representations of protein conformational landscapes for drug discovery applications.
Conclusion
Validating MD-predicted protein folding pathways is not merely a technical exercise but a critical step for ensuring the biological relevance of computational models. The integration of AI-based structure prediction with MD simulations and global optimization methods has created powerful, hybrid workflows capable of sampling complex conformational ensembles. However, this synthesis also highlights inherent limitations, including force field inaccuracies, sampling bottlenecks, and the sometimes 'unphysical' nature of deep learning predictions. Success hinges on a rigorous, multi-faceted validation strategy that leverages quantitative metrics and diverse experimental data. Future progress will depend on developing more physiologically accurate force fields, achieving longer simulation timescales, and creating AI models that more deeply incorporate physical principles. For biomedical research, reliably validated folding pathways will accelerate efforts in drug discovery, protein design, and understanding the molecular basis of misfolding diseases, ultimately bridging the gap between computational prediction and clinical application.
References
- 1. Navigating the landscape of protein folding and proteostasis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550075/]
- 2. Evolution, energy landscapes and the paradoxes of protein ... [https://www.sciencedirect.com/science/article/abs/pii/S0300908414003897]
- 3. The Arch from the Stones: Understanding Protein Folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12451740/]
- 4. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 5. Folding Free Energy Landscape of Ordered and ... [https://www.nature.com/articles/s41598-019-50825-6]
- 6. Navigating protein landscapes with a machine-learned ... [https://www.nature.com/articles/s41557-025-01874-0]
- 7. Pathway regulation mechanism by cotranslational protein ... [https://www.nature.com/articles/s42004-025-01636-6]
- 8. Severe deviation in protein fold prediction by advanced AI [https://www.nature.com/articles/s41598-025-89516-w]
- 9. Critical assessment of AI-based protein structure prediction [https://www.sciencedirect.com/science/article/pii/S2950363925000353]
- 10. How DeepMind's AlphaFold 3 Is Transforming Drug Discovery [https://www.iankhan.com/the-ai-protein-folding-revolution-how-deepminds-alphafold-3-is-transforming-drug-discovery/]
- 11. A comprehensive database of verified experimental data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4253821/]
- 12. The road less traveled in protein folding [https://www.sciencedirect.com/science/article/abs/pii/S0959440X20301809]
- 13. Analysis methods for comparison of multiple molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283699928437]
- 14. do proteins with similar structures fold via the same pathway? [https://pubmed.ncbi.nlm.nih.gov/15718132/]
- 15. Transcription reshapes RNA hairpin folding pathways ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013472]
- 16. Molecular Dynamics Simulation of Protein Folding by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1303567/]
- 17. AlphaFold Protein Structure Database [https://alphafold.ebi.ac.uk/]
- 18. Mega-scale experimental analysis of protein folding ... [https://www.nature.com/articles/s41586-023-06328-6]
- 19. How have AlphaFold 2’s predictions of protein structure been... [https://www.ebi.ac.uk/training/online/courses/alphafold/validation-and-impact/how-have-alphafolds-predictions-of-protein-structure-been-validated/]
- 20. - Google DeepMind AlphaFold [https://deepmind.google/science/alphafold/]
- 21. Reshapes APAC Research AlphaFold Protein Prediction [https://www.startuphub.ai/ai-news/ai-research/2025/alphafold-protein-prediction-reshapes-apac-research/]
- 22. Bridging prediction and reality: Comprehensive analysis of ... [https://www.sciencedirect.com/science/article/pii/S2001037025001734]
- 23. Artificial intelligence methods for protein folding and design [https://www.sciencedirect.com/science/article/abs/pii/S0959440X25000843]
- 24. Protein Engineering with AI: OpenFold3 vs Boltz 2 vs ... [https://blackthorn.ai/blog/protein-engineering-with-ai/]
- 25. mdCATH: A Large-Scale MD Dataset for Data-Driven ... [https://www.nature.com/articles/s41597-024-04140-z]
- 26. Investigating whether deep learning models for co-folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12501370/]
- 27. Severe deviation in protein by advanced AI: a case... fold prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC11807112/]
- 28. Predicting protein folding dynamics using sequence ... [https://arxiv.org/abs/2505.17237]
- 29. Combining Experiment and Simulation in Protein Folding [https://pmc.ncbi.nlm.nih.gov/articles/PMC2291534/]
- 30. Enhanced Sampling Techniques in Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4339458/]
- 31. 10 Common Molecular Dynamics Mistakes New ... [https://insilicosci.com/common-molecular-dynamics-mistakes/]
- 32. Protein folding pathway prediction based on conformational ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1011438]
- 33. Integrating experimental data and dynamics [https://www.sciencedirect.com/science/article/pii/S0959440X25001733]
- 34. Deep generative modeling of temperature-dependent ... [https://www.nature.com/articles/s42004-025-01737-2]
- 35. BioEmu: AI‐Powered Revolution in Scalable Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12643044/]
- 36. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 37. Frontiers | Use of AI-methods over MD simulations in the sampling of... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1542267/full]
- 38. [DOCS] Hyperdimensional Graph Kernel Alignment for Topological... [https://gist.github.com/freederia/1ed7cca89fce80d626abfb2e0d79c711]
- 39. AI Models For Protein Structure Prediction [https://frontlinegenomics.com/ai-models-for-protein-structure-prediction/]
- 40. Exploring the Sequence-based Prediction of Folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5562875/]
- 41. Interpreting a black box predictor to gain insights into early ... [https://www.sciencedirect.com/science/article/pii/S200103702100372X]
- 42. Accurate predictions of protein mutational effects ... [https://www.nature.com/articles/s42004-025-01771-0]
- 43. Experimental and molecular dynamics simulation studies ... [https://www.sciencedirect.com/science/article/pii/S0264410X24005991]
- 44. State of the Art of Protein Structure Prediction as of 2025 ... [https://medium.com/advances-in-biological-science/state-of-the-art-of-protein-structure-prediction-as-of-2025-as-evaluated-by-casp16-0c423636bc97]
- 45. Computational Modeling for Targeted Protein Degraders – 2024/ 2025 ... [https://www.linkedin.com/pulse/computational-modeling-targeted-protein-degraders-20242025-vakal-t6bdf]
- 46. Determining accurate conformational ensembles of ... [https://www.nature.com/articles/s41467-025-64098-3]
- 47. Modeling protein conformational ensembles by guiding ... [https://pubmed.ncbi.nlm.nih.gov/40753333/]
- 48. A hybrid -kMC algorithm for MD in explicit solvent folding proteins [https://pubmed.ncbi.nlm.nih.gov/24499973/]
- 49. Molecular Dynamics Studies on the Conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3712241/]
- 50. Conformational dynamics of adenylate kinase in crystals - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10883716/]
- 51. Mapping the Dynamics Landscape of Conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4572606/]
- 52. Dynamic energy conversion in protein catalysis [https://www.sciencedirect.com/science/article/pii/S2001037025003149]
- 53. A Benchmark of Protein Folding Models [https://sciety-labs.elifesciences.org/articles/by?article_doi=10.1101/2025.11.04.686486]
- 54. Heterogeneous path ensembles for conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3108504/]
- 55. Exploring Conformational Change of Adenylate Kinase by ... [https://www.sciencedirect.com/science/article/pii/S0006349520300023]
- 56. in Force - fields 2025.3 documentation GROMACS GROMACS [https://manual.gromacs.org/2025.3/user-guide/force-fields.html]
- 57. Force field comparison between CHARMM and AMBER99SB ... [https://gromacs.bioexcel.eu/t/force-field-comparison-between-charmm-and-amber99sb-ildn/184]
- 58. Lessons learned from comparing molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5581938/]
- 59. Evaluation of nine condensed-phase force of the GROMOS... fields [https://pubs.rsc.org/en/content/articlelanding/2021/cp/d1cp00215e]
- 60. Assessing the accuracy of physical models used in protein ... [https://www.sciencedirect.com/science/article/pii/S0959440X13002157]
- 61. Comprehensive analysis of experimental and AlphaFold 2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12149446/]
- 62. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 63. Successes and challenges in simulating the folding of large ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6952611/]
- 64. Convergence and equilibrium in molecular dynamics ... [https://www.nature.com/articles/s42004-024-01114-5]
- 65. Ab initio characterization of protein molecular dynamics ... [https://www.nature.com/articles/s41586-024-08127-z]
- 66. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 67. Investigating whether deep learning models for co-folding ... [https://www.nature.com/articles/s41467-025-63947-5]
- 68. Why co-folding models are here to stay [https://www.apheris.com/resources/blog/why-co-folding-models-are-here-to-stay]
- 69. Why AI Models for Drug Design Struggle with Physics [https://bioengineer.org/why-ai-models-for-drug-design-struggle-with-physics/]
- 70. Benchmarking of AlphaFold2 accuracy self-estimates as ... [https://pubmed.ncbi.nlm.nih.gov/39115813/]
- 71. Advances in AI for Protein Structure Prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC10968151/]
- 72. Beyond evolution: De novo designed protein toolkit ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12624534/]
- 73. Accurate prediction of protein folding mechanisms by ... [https://www.nature.com/articles/s41467-023-41664-1]
- 74. Best Practices for Foundations in Molecular Simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6884151/]
- 75. Comparison of implicit solvent models and force fields in ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261411011353]
- 76. Comparison of various implicit solvent models in molecular ... [https://pubmed.ncbi.nlm.nih.gov/12632469/]
- 77. A compact guide to performing Molecular Dynamics ... [https://www.sciencedirect.com/science/article/pii/S0167273825001869]
- 78. Best practices for running molecular dynamics simulations on ... [https://aws.amazon.com/blogs/hpc/best-practices-for-running-molecular-dynamics-simulations-on-aws-graviton3e/]
- 79. Protein Folding Transition Path Times from Single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5826754/]
- 80. Hydrogen-deuterium exchange mass spectrometry ... [https://www.nature.com/articles/s41467-020-20032-3]
- 81. Hydrogen-Deuterium Exchange Mass Spectrometry [https://www.mdpi.com/2075-1729/10/11/286]
- 82. Interpretation of HDX Data by Maximum-Entropy Reweighting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7136279/]
- 83. Tutorials and Applications of the HDXer Ensemble ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9835200/]
- 84. How Robust Are Protein Folding Simulations with Respect ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3149239/]
- 85. What can we learn from transition path time distributions for ... [https://pubs.rsc.org/en/content/articlelanding/2021/cp/d1cp03296h]
- 86. Hydrogen Deuterium Exchange ( HDX )... | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/proteomics-mass-spectrometry/protein-structure-analysis-mass-spectrometry/hydrogen-deuterium-exchange-hdx-protein-structure-ms.html]
- 87. Quantitative Evaluation of Native Protein Folds and Assemblies by... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6318237/]
- 88. Artificial intelligence-based HDX (AI-HDX) prediction ... [https://www.sciencedirect.com/science/article/pii/S2589004223003590]
- 89. Protein structure prediction with mass spectrometry data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9672978/]
- 90. Systematic Validation of Protein Force Fields against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3285199/]
- 91. On the Origins of the Weak Folding Cooperativity of a ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000998]
- 92. WW domain [https://en.wikipedia.org/wiki/WW_domain]
- 93. Structure-function-folding relationship in a WW domain [https://pubmed.ncbi.nlm.nih.gov/16807295/]
- 94. Structural insights into the functional versatility of WW domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4374002/]
- 95. Benchmarking AlphaFold2 on Peptide Structure Prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC9883802/]
- 96. pLDDT: Understanding local confidence | AlphaFold [https://www.ebi.ac.uk/training/online/courses/alphafold/inputs-and-outputs/evaluating-alphafolds-predicted-structures-using-confidence-scores/plddt-understanding-local-confidence/]
- 97. how to plot the prediction quality metrics with AlphaFold2 [https://www.mindwalkai.com/blog/explained-how-to-plot-the-prediction-quality-metrics-with-alphafold2]
- 98. AlphaFold2 models indicate that protein sequence determines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9226352/]