Bridging Scales in Drug Discovery: Molecular Dynamics Simulations and Ising Models in Computational Biology
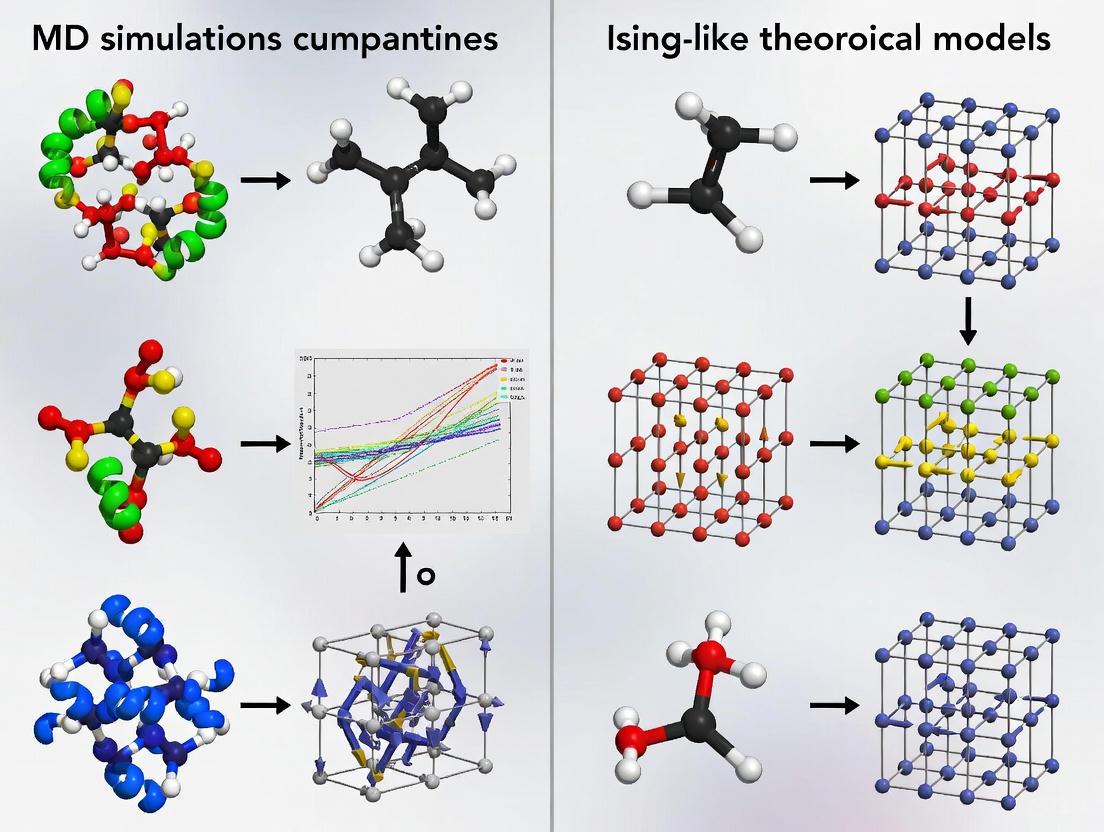
This article provides a comprehensive comparison between Molecular Dynamics (MD) simulations and Ising-like theoretical models, two powerful computational approaches in modern drug discovery and biomedical research.
Bridging Scales in Drug Discovery: Molecular Dynamics Simulations and Ising Models in Computational Biology
Abstract
This article provides a comprehensive comparison between Molecular Dynamics (MD) simulations and Ising-like theoretical models, two powerful computational approaches in modern drug discovery and biomedical research. Aimed at researchers, scientists, and drug development professionals, it explores the foundational principles of both methods, contrasting MD's atomistic, dynamical insights into protein-ligand interactions and drug delivery systems with the statistical mechanics framework of Ising models for analyzing collective behaviors and phase transitions. The scope extends to methodological applications in target validation, lead optimization, and the study of allosteric mechanisms, while also addressing shared computational challenges such as sampling efficiency and force field accuracy. The article further examines validation strategies and the emerging synergy of integrating both approaches with artificial intelligence to enhance predictive power and accelerate the development of targeted therapies.
First Principles: Contrasting Atomistic Dynamics with Statistical Mechanics Frameworks
In the computational study of biological and material systems, two powerful frameworks have emerged: Molecular Dynamics (MD) simulations and Ising-like theoretical models. At a fundamental level, these approaches offer complementary perspectives. MD simulations provide a high-resolution, physics-driven view of atomic motion, directly applying Newton's laws to predict the trajectory of every atom in a system over time [1]. In contrast, Ising-like models offer a coarse-grained, probabilistic framework, often derived from statistical mechanics, to capture the essential state transitions and collective behaviors of complex systems, from neuronal firing patterns to protein conformational changes [2] [3].
The choice between these tools is not merely technical but conceptual, influencing the nature of the questions a researcher can ask and the insights they can gain. This guide provides an objective comparison of their performance, supported by experimental data and detailed methodologies, to inform researchers and drug development professionals in selecting the appropriate tool for their specific challenges.
Core Principles and Methodologies
Molecular Dynamics: A Bottom-Up Physics-Based Approach
The core principle of MD is to simulate the physical movements of atoms and molecules over time. The system evolves by numerically solving Newton's equations of motion. The force on each atom, calculated as the negative gradient of the potential energy, determines its acceleration. The potential energy is described by a molecular mechanics force field, which includes terms for bond stretching, angle bending, torsional rotations, and non-bonded interactions (van der Waals and electrostatics) [1].
- Time Integration: The simulation progresses in discrete, femtosecond-scale time steps. At each step, forces are computed and used to update atomic positions and velocities. This makes simulations computationally demanding, often requiring millions of steps to model biologically relevant processes [1].
- Advanced MD: For processes involving chemical reactions (e.g., bond breaking/formation), Quantum Mechanics/Molecular Mechanics (QM/MM) simulations are used, where a small reactive region is treated quantum mechanically while the surroundings are handled classically [1].
Ising-like Models: A Top-Down Probabilistic Approach
Ising-like models describe a system as a network of simple units (e.g., spins, neurons) that can exist in a few discrete states (e.g., ±1, active/inactive). The system's configuration is governed by a Hamiltonian (energy function), and its dynamics are explored through the lens of statistical mechanics.
- The Hamiltonian: The typical form for a pairwise Ising model is: (\mathscr{H} = -\sum{i=1}^N hi si - \sum{i > j}^N J{ij} si sj) Here, (si) represents the state of unit (i), (hi) is an external field influencing unit (i), and (J{ij}) is the coupling strength between units (i) and (j) [3].
- The Inverse Ising Problem (Pairwise Maximum Entropy Method): Often, the model parameters ((hi), (J{ij})) are not known beforehand. They are inferred from experimental data—such as binarized neuronal firing patterns—by finding the parameters that reproduce the observed average states ((\langle si \rangle)) and pairwise correlations ((C{ij})) while maximizing the entropy of the system. This results in a Boltzmann probability distribution: (P({s}) \sim e^{-\mathscr{H}/T}) [2] [3].
Workflow Visualization
The diagram below illustrates the contrasting workflows for building and applying MD and Ising-like models.
Performance and Applicability Comparison
The table below summarizes the fundamental characteristics, strengths, and limitations of MD simulations and Ising-like models across key dimensions relevant to scientific research.
Table 1: Core Methodological Comparison between MD and Ising-like Models
| Feature | Molecular Dynamics (MD) | Ising-like Models |
|---|---|---|
| Fundamental Principle | Newton's laws of motion; classical physics [1] | Statistical mechanics; maximum entropy inference [2] [3] |
| System Representation | All atoms with explicit positions and velocities [1] | Coarse-grained, discrete states (e.g., spins ±1) [3] |
| Temporal Resolution | Femtoseconds (10⁻¹⁵ s) [1] | Discrete time steps (dependent on data sampling) |
| Primary Output | Atomic-level trajectory (a "3D movie") [1] | Energy landscape, state probabilities, and correlations [2] |
| Typical System Size | ~10⁴ - 10⁶ atoms (subject to computational limits) [1] | ~10 - 10³ units (limited by parameter inference) [2] |
| Key Strength | High-resolution, direct physical interpretation [1] | Captures collective behavior from limited data; computationally efficient post-inference [2] [3] |
| Key Limitation | Extremely computationally expensive; force field approximations [1] | Loss of molecular detail; often requires data for parameterization [2] |
Experimental Protocols and Benchmarking Data
Key Experimental Protocols
To objectively compare these tools, it is essential to understand the standard protocols for their application and validation.
Protocol 1: MD Simulation of a Biomolecular System [1] [4]
- System Preparation: Obtain an initial atomic structure from experimental data (e.g., PDB). Place the molecule (e.g., protein, DNA) in a simulation box with explicit water molecules and ions to mimic a physiological environment.
- Force Field Selection: Choose an appropriate molecular mechanics force field (e.g., OPLS4, used in high-throughput studies [5]).
- Energy Minimization: Remove steric clashes by minimizing the system's energy.
- Equilibration: Run simulations with position restraints on the solute, gradually releasing them to allow the solvent and system to equilibrate at the target temperature and pressure.
- Production Run: Perform an unrestrained simulation, saving atomic coordinates at regular intervals to generate a trajectory. Modern simulations leveraging GPUs or specialized hardware can now reach microsecond to millisecond timescales [1].
- Analysis: Analyze the trajectory to compute properties of interest, such as root-mean-square deviation (RMSD), radius of gyration, interaction energies, or free energy profiles.
Protocol 2: Constructing an Energy Landscape from Data via the Ising Model [2] [3]
- Data Acquisition & Binarization: Collect multivariate time series data (e.g., from fMRI, EEG, or spike recordings). Binarize each variable at each time point into one of two states (e.g., -1/+1 or 0/1).
- Compute Observables: Calculate the time-averaged activity, (\langle si \rangle^{obs}), for each unit and the pairwise covariance, (C{ij}^{obs}), from the binarized data.
- Model Inference (Inverse Problem): Find the parameters (hi) and (J{ij}) of the Ising model (Eq. 4) such that the model's averages and covariances match the observed ones. This is typically done using an iterative algorithm (e.g., gradient descent with Monte Carlo sampling) [3].
- Energy Landscape Analysis: With the inferred model, compute the energy for every possible system state. Construct a disconnectivity graph that maps out the stable states (local energy minima) and the energy barriers between them [2].
- Validation: Validate the model by checking its prediction of higher-order statistics (e.g., triple correlations) not used during the inference process [3].
Benchmarking with Experimental Data
Both methods are validated by their ability to reproduce and predict quantitative experimental measurements.
MD Performance Benchmarks:
- Structural Properties: MD simulations of DNA have been used to derive sequence-dependent flexibility models. A recent study showed that a model incorporating multimodality and nearest-neighbor coupling provided a ~0.03 kcal/mol per degree of freedom improvement over a simpler harmonic model when evaluated against MD data [4].
- Thermodynamic Properties: High-throughput MD of over 30,000 solvent mixtures demonstrated strong correlation with experimental density (R² ≥ 0.98) and heat of vaporization (R² ≥ 0.97) [5].
- Drug Design: MD simulations have proven valuable in deciphering protein functional mechanisms and optimizing small molecules and peptides, frequently appearing in experimental structural biology papers [1].
Ising Model Performance Benchmarks:
- Neuronal Networks: An Ising model inferred from in vitro neuronal spike data successfully reproduced the observed pairwise correlations. Crucially, it also predicted third-order correlations that were not constrained during the fitting process, demonstrating its predictive power for collective neural activity [3].
- Protein-DNA Binding: An advanced Ising-based model for DNA deformability provided better estimation of sequence-dependent deformation energies in protein-DNA complexes and more accurate correlation with experimental binding affinities compared to a unimodal harmonic model [4].
Table 2: Quantitative Benchmarking Against Experimental and Simulated Data
| Application Context | MD Performance Metric | Ising-like Model Performance Metric |
|---|---|---|
| Molecular Properties | R² > 0.97 for density & ΔHvap vs. expt. [5] | Not directly applicable (coarse-grained) |
| Nucleic Acid Flexibility | 0.03 kcal/mol/DOF improvement with advanced model [4] | Improved prediction of protein-DNA binding affinity [4] |
| Neuronal Activity | Not the primary tool for spike data | Accurately predicts unconstrained 3rd-order correlations [3] |
| Computational Cost | High (CPU/GPU hours, system size limited) [1] | Low post-inference; inference scales with system size & data [2] |
The following table details key computational "reagents" and resources essential for working with either MD or Ising-like models.
Table 3: Essential Resources for MD and Ising Model Research
| Resource Type | Specific Examples & Functions | Relevance |
|---|---|---|
| Force Fields | OPLS4, AMBER, CHARMM; define interatomic potentials for MD [5] | MD |
| Neural Network Potentials (NNPs) | eSEN, UMA models; ML-based force fields for accurate & faster MD [6] | MD / ML-MD |
| MD Software & Hardware | GPU-accelerated codes (e.g., OpenMM, GROMACS); specialized hardware (e.g., Anton2) [1] | MD |
| Experimental Datasets | RCSB PDB (biomolecular structures), neuronal spike recordings (e.g., multielectrode arrays) [2] [3] [6] | Both |
| Reference Datasets | OMol25 (quantum chemical calculations), SPICE, ANI-2x (for ML potential training) [6] | MD / ML-MD |
| Inference Algorithms | Boltzmann machine learning; Monte Carlo methods for solving the inverse Ising problem [3] | Ising |
| Analysis & Visualization | Software for constructing disconnectivity graphs and analyzing energy landscapes [2] | Ising |
Integrated Approaches and Future Outlook
The distinction between MD and Ising-like models is increasingly blurred by integrated approaches. For instance, all-atom MD simulations of DNA are used to parameterize coarse-grained Ising-type models that capture the sequence-dependent prevalence of BI/BII backbone substates, combining atomic-level detail with the computational efficiency of a statistical model [4].
Furthermore, machine learning (ML) is acting as a powerful unifying force. ML-driven spin-lattice dynamics simulations now achieve semi-to-full quantitative agreement with ab initio methods while reducing computational cost by about 80% [7]. Frameworks like TSPIN integrate symplectic integration with machine-learning potentials to accurately and efficiently handle coupled spin-lattice degrees of freedom [8]. The release of massive datasets like OMol25 and pre-trained universal models (UMA) is poised to dramatically accelerate the development and accuracy of ML-potentials for MD, making high-level quantum chemical accuracy more accessible [6].
For the researcher, this convergence means the toolkit is expanding. The choice is no longer just between a bottom-up physical simulation and a top-down statistical model, but increasingly involves hybrid strategies that leverage the strengths of each to tackle multi-scale challenges in molecular biology and materials science.
This guide provides an objective comparison between Molecular Dynamics (MD) simulations and Ising-like theoretical models, two cornerstone methods in computational science. It examines their performance in bridging the critical gap between atomistic resolution and macroscopic phenomena, supported by current experimental data and methodologies.
A central goal of computational materials science and drug discovery is to predict the physical properties and behaviors of complex systems using fundamental physical laws. However, a significant challenge lies in the vast disparity between the temporal and spatial scales of atomistic interactions and those of observable macroscopic phenomena. Molecular Dynamics (MD) simulations track the trajectories of every atom in a system over time, providing high-resolution insights but often at a prohibitive computational cost for large-scale systems. In contrast, Ising-like theoretical models are coarse-grained representations that sacrifice atomistic detail to access much larger spatial and temporal scales, making them powerful tools for studying phase transitions and collective behavior [9] [2]. This guide objectively compares the performance, applications, and recent advancements of these two approaches, with a particular focus on how emerging machine learning (ML) and accelerated computing methodologies are reshaping the field.
Core Principles and Methodologies
Molecular Dynamics Simulations
MD simulations model how atoms and molecules move over time by numerically solving Newton's equations of motion [10]. The "force fields" that describe interatomic interactions have historically been a limitation; traditional classical force fields struggle with describing chemical reactions, while highly accurate quantum mechanical methods are too computationally expensive for large systems [11].
- Recent Advancements: Machine learning has emerged as a transformative solution. Neural Network Potentials (NNPs), such as the Deep Potential (DP) scheme and the recently developed EMFF-2025 for energetic materials, are trained on high-quality quantum mechanical data. They can perform MD simulations at near-Density Functional Theory (DFT) accuracy but at a fraction of the computational cost, effectively breaking the traditional trade-off between accuracy and efficiency [11].
- Hardware: MD simulations heavily depend on high-performance computing. Graphics Processing Units (GPUs) are particularly advantageous due to their ability to handle many parallel calculations simultaneously, significantly speeding up the simulation process [12].
Ising-like Theoretical Models
The Ising model is a canonical mathematical model for representing systems as a network of discrete variables (spins) that interact with their neighbors. Its energy landscape analysis allows researchers to comprehend system dynamics as stochastic transitions between stable states, with transitions governed by energy barriers [2].
- The Parallelization Challenge: A key limitation of traditional Monte Carlo (MC) simulations for Ising-like models is their intrinsic sequential nature, which hinders large-scale parallelization [9].
- Recent Advancements: New algorithms like the Scalable Monte Carlo at eXtreme (SMC-X) method have been developed to overcome this. SMC-X is a generalized checkerboard algorithm designed for modern accelerator hardware like GPUs. Its implementation, SMC-GPU, harnesses massive parallelism to enable billion-atom simulations when combined with machine-learning surrogates of DFT, opening new avenues for exploring nanostructure evolution in complex materials like high-entropy alloys [9] [13].
Performance Comparison: Quantitative Data and Experimental Protocols
The following tables summarize the key performance characteristics and experimental protocols for MD simulations and Ising-like models, particularly in their modern, accelerated forms.
Table 1: Comparative Analysis of Temporal and Spatial Scaling
| Feature | Traditional MD Simulations | Modern ML-Accelerated MD | Traditional Ising/MC Models | Modern ML-Accelerated MC (SMC-GPU) |
|---|---|---|---|---|
| Typical System Size | Hundreds to thousands of atoms [9] | Millions of atoms [9] | Lattice-dependent | Over 1 billion atoms [9] |
| Temporal Scale | Picoseconds to nanoseconds [10] | Extended to microsecond and beyond [11] | Algorithm-limited sequential updates [9] | Massively parallel trial moves |
| Spatial Resolution | Full atomistic detail (Ångström scale) | Full atomistic detail (Ångström scale) | Coarse-grained (lattice site) | Coarse-grained or mesoscopic |
| Accuracy vs. DFT | Varies by force field; MLPs achieve near-DFT accuracy [11] | High (MAE: ~0.1 eV/atom for energy, ~2 eV/Å for force) [11] | Not directly comparable | High, when using ML surrogates [9] |
| Key Application | Protein folding, ligand binding, chemical reactions [10] | Complex reaction chemistry, explosive decomposition [11] | Phase transitions, magnetic ordering [2] | Nanostructure evolution in complex alloys [9] |
Table 2: Summary of Key Experimental Protocols and Validation Methods
| Method | Core Computational Protocol | Key Validation Metrics | Common Experimental Cross-Validation |
|---|---|---|---|
| ML-MD (e.g., EMFF-2025) | 1. Train NNP on DFT dataset.2. Run MD simulations using the potential.3. Analyze properties (mechanical, thermal).4. Map chemical space with PCA [11]. | Mean Absolute Error (MAE) of energy and forces against DFT; prediction of crystal structures and mechanical properties benchmarked against experimental data [11]. | Comparison with bulk RNA sequencing, multiplex immunofluorescence, and histological staining for biological systems [14]. |
| ML-MC (e.g., SMC-GPU) | 1. Define ML energy model (e.g., using local SRO parameters).2. Use SMC-X algorithm for parallel MC trials.3. Sample configurations to study phase transitions or nanostructures [9]. | Quantitative analysis of nanostructure size, composition, and morphology; direct comparison with atom-probe tomography (APT) and electron microscopy [9]. | Atom-probe tomography (APT), electron microscopy [9]. |
Workflow and Logical Relationships
The diagram below illustrates the typical workflows for MD and modern Ising/MC simulations, highlighting their parallel paths in leveraging machine learning and high-performance computing to bridge scales.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table details key computational tools and frameworks essential for research in this field.
Table 3: Key Research Reagent Solutions for Multi-Scale Simulations
| Tool/Solution Name | Type | Primary Function | Key Feature |
|---|---|---|---|
| DP-GEN (Deep Potential Generator) [11] | Software Framework | Automates the construction of accurate and transferable neural network potentials. | Integrates active learning to efficiently sample the configuration space. |
| EMFF-2025 [11] | Neural Network Potential | A general-purpose NNP for predicting mechanical and chemical properties of C, H, N, O-based high-energy materials. | Leverages transfer learning for high accuracy with minimal DFT data. |
| SMC-GPU [9] | Algorithm/Code | A GPU-accelerated Monte Carlo implementation for arbitrary short-range interactions. | Enables billion-atom simulations by overcoming sequential updating bottlenecks. |
| STORMM Libraries [12] | Molecular Simulation Engine | A next-generation engine optimized for performance on CPUs and GPUs. | Designed for high-throughput simulations, crucial for drug discovery applications. |
| Ising Model ELA (Energy Landscape Analysis) [2] | Analysis Method | Constructs energy landscapes and dynamics from multivariate time series data. | Captures system dynamics as state transitions on an energy landscape, useful for fMRI, microbiome data, etc. |
Both Molecular Dynamics simulations and Ising-like theoretical models are indispensable for connecting atomic-scale interactions to macroscopic behavior. The choice between them is not a matter of superiority but of appropriateness for the specific research question. MD simulations, especially when powered by ML potentials, are unparalleled for investigating processes where atomistic detail is non-negotiable, such as chemical reaction mechanisms and specific molecular recognition events in drug discovery [11] [10]. In contrast, modern, highly parallelized Ising/MC frameworks like SMC-GPU are the tool of choice for probing statistical phenomena and long-timescale evolutionary processes in complex materials, such as phase separation and nanostructure formation in high-entropy alloys [9].
The frontier of computational science lies in the continued integration of these approaches. Future research will likely focus on developing more sophisticated multiscale simulation methodologies that seamlessly couple different levels of description [15], further exploration of high-performance computing technologies like specialized AI chips, and a tighter integration of experimental and simulation data for validation and model construction [9] [14]. These efforts will further empower researchers and drug development professionals to solve increasingly complex problems across materials science, chemistry, and biomedicine.
The pursuit of new therapeutics and a deeper understanding of biological systems relies heavily on advanced computational methods that can predict molecular interactions and model complex collective behaviors. Among these, Molecular Dynamics (MD) simulations provide an atomic-resolution "movie" of physical movements over time, making them a powerful tool for directly investigating drug-target interactions [16] [17]. In parallel, Ising-like theoretical models, originally developed to describe magnetic spins in physics, have found novel applications in biology for analyzing collective behaviors and energy landscapes within proteins and neural systems [18] [2]. While both are valuable tools in computational biophysics, they operate at different scales and are designed to answer fundamentally different biological questions. This guide provides a objective comparison of these methodologies, detailing their key applications, experimental protocols, and performance benchmarks to inform researchers in selecting the appropriate tool for their specific research objectives.
Comparative Analysis: Molecular Dynamics vs. Ising Models
The table below summarizes the core characteristics, applications, and performance data for these two distinct computational approaches.
Table 1: Core Characteristics and Applications of MD Simulations and Ising Models
| Feature | Molecular Dynamics (MD) Simulations | Ising-like Theoretical Models |
|---|---|---|
| Fundamental Principle | Numerical simulation of physical atom movements based on Newtonian mechanics and empirical force fields [16]. | Statistical physics model of binary-state systems (+1/-1) interacting via pairwise couplings [19] [2]. |
| Primary Scale & Resolution | Atomic-to-molecular scale (Ångströms to nanometers); High spatial resolution [16]. | Coarse-grained scale (residues, brain regions); Binary or pattern-based states [18] [2]. |
| Typical System Size | ~10⁴ - 10⁶ atoms [16] | Up to ~180 residues/protein contacts or N~10-100 variables for multivariate analysis [18] [2]. |
| Key Applications in Biomedicine | - Binding free energy (affinity) calculation [16]- Binding pose prediction & validation [17]- Virtual screening & lead optimization [16] [17]- Investigation of (un)binding kinetics & pathways [20] | - Protein contact formation analysis & energy landscape mapping [18] [2]- Analysis of multivariate brain data (fMRI, EEG) [2]- Modeling collective opinion dynamics in public health [19]. |
| Representative Performance | - Binding free energy RMS errors: 1-2 kcal/mol (sufficient to guide synthesis) [16]- Can discriminate compounds for a 3-fold reduction in synthesis testing [16]. | - Reproduces contact frequencies from input MD trajectories [18]- Identifies meta-stable states and transition patterns in brain activity [2]. |
| Dominant Challenges | - Sampling: High energetic/entropic barriers limit conformational exploration [16].- Force Field Accuracy: Inaccuracies in energy functions affect predictions [16] [17].- System Representation: Correct protonation states, flexible loops [16].- Timescales: Microsecond simulations may miss slow, relevant processes [20]. | - Scalability: Computation becomes infeasible for large proteins (>180 residues) or many variables [18].- Data Requirements: Requires long, high-quality multivariate time series relative to variable count [2].- Oversimplification: Binary states may lack biochemical detail for specific mechanistic insights. |
Experimental Protocols and Workflows
A clear understanding of the methodological workflows is essential for their application and for interpreting results. The following diagrams and descriptions outline the standard protocols for each approach.
Molecular Dynamics for Drug-Target Binding
MD simulations for drug discovery typically follow a multi-stage process to predict how a small molecule (ligand) interacts with a biological target.
Diagram 1: Typical MD Simulation Workflow for Drug-Target Binding.
The core methodology involves [16] [17] [21]:
- System Preparation: A high-quality 3D structure of the target biomolecule (e.g., from X-ray crystallography or AlphaFold2) is prepared. The small molecule ligand is parameterized for the chosen force field (e.g., CHARMM, AMBER). The system is solvated in a water box and ions are added to neutralize charge and mimic physiological concentration.
- Energy Minimization and Equilibration: The system's energy is minimized to remove unfavorable atomic clashes. It is then gradually heated to the target temperature (e.g., 310 K) and the pressure is adjusted to the target value (e.g., 1 bar) in a series of short, constrained simulations.
- Production Simulation: This is the core data-generating phase, where a long, unconstrained simulation is performed using software like GROMACS, AMBER, or NAMD. Multiple replicates or ensemble simulations are often used to improve sampling.
- Trajectory Analysis: The resulting trajectory is analyzed to compute properties of interest. For binding, this includes calculating binding free energies via alchemical (e.g., FEP, TI) or physical pathway (e.g., Umbrella Sampling) methods [16]. Interaction Fingerprints (IFPs) can be derived to summarize key protein-ligand contacts over time [21].
Ising Model Analysis for Biomolecular Systems
The application of Ising models to biomolecular systems, such as for analyzing protein contact formation or neural activity patterns, follows a distinct workflow centered on fitting a statistical model to binary data.
Diagram 2: Ising Model Energy Landscape Analysis Workflow.
The core methodology involves [18] [2]:
- Data Input and Binarization: The input is a multivariate time series. For protein contact analysis, this could be an MD trajectory where distances between residues are binarized into "contact" or "no contact" [18]. For neural data, fMRI signals are binarized into "high" or "low" activity states [2]. The resulting activity pattern is an N-dimensional vector of +1/-1.
- Model Fitting (Inference): The Ising model parameters are inferred from the binarized data. The goal is to find the external fields ((hi)) and pairwise couplings ((J{ij})) of the model (the Pairwise Maximum Entropy Model, or PMEM) that best reproduce the observed single and pairwise frequencies in the data. This is typically done using maximum likelihood estimation.
- Energy Landscape Construction and Analysis: Using the fitted parameters, the energy (H(\vec{\sigma}) = - \sumi hi \sigmai - \sum{i
- Validation and Sampling: The model is validated by confirming that when sampled (e.g., using Monte Carlo methods), it reproduces the statistical properties of the original input data. The dynamics of the original time series are then interpreted as a "ball" moving between the basins of attraction on this calculated energy landscape.
Performance and Validation Data
Quantitative performance metrics are critical for assessing the reliability and utility of computational methods.
Performance of MD Simulations in Drug Discovery
MD and associated free energy calculations have matured to a point where they provide significant value in drug discovery projects, particularly for lead optimization.
Table 2: Performance Benchmarks for MD and Free Energy Calculations
| Application / Metric | Reported Performance | Context & Implications |
|---|---|---|
| Binding Affinity Prediction | RMS error of ~1-2 kcal/mol with current force fields [16]. | Accuracy is sufficient to prioritize compound synthesis, reducing experimental testing by ~3-fold [16]. |
| Alchemical Relative Free Energy | Successfully applied in prospective drug discovery campaigns [16] [17]. | Most robust when ligands are structurally similar; performance drops with significant chemical changes or high protein conformational barriers [16]. |
| Targeting Specific Protein Families | Performance varies; some systems like kinases show good results [16]. | "Domain of applicability" is not universal; results depend on force field, sampling, and system preparation [16]. |
| Interaction Analysis (IFPs) | IFPAggVis enables systematic aggregation and comparison of interactions from MD trajectories [21]. | Moves beyond frame-by-frame analysis, allowing identification of persistent and transient interaction patterns across simulations [21]. |
Performance of Ising Models in Biomedical Analysis
Ising models are validated by their ability to reproduce the statistics of the input data and to provide novel insights into the system's stable states and dynamics.
Table 3: Performance and Capabilities of Ising-like Models
| Application / Metric | Reported Performance / Capability | Context & Implications |
|---|---|---|
| Protein Contact Analysis | Accurately reproduces single and pairwise contact frequencies from input MD trajectories [18]. | Validates that the inferred Ising model captures essential energetics of contact formation from the simulation data. |
| Energy Landscape Analysis (ELA) | Successfully identifies meta-stable states and transition patterns in fMRI brain data [2]. | Provides a coarse-grained model of brain dynamics, linking spatial activity patterns to an underlying energy landscape. |
| Computational Scalability | Practical for proteins with ~180 residues or less due to quartic scaling of pairwise terms [18]. | A major limitation for large proteins or systems with many variables; analysis becomes computationally infeasible. |
| Temporal Data Requirements | Requires long multivariate time series relative to the number of variables (N) [2]. | For reliable parameter inference, the data must be "long enough" to adequately sample the (2^N) possible states. |
Essential Research Reagent Solutions
The following table details key software tools and resources essential for implementing the methodologies discussed in this guide.
Table 4: Key Research Reagents and Computational Tools
| Item Name | Type | Primary Function / Application | Relevant Citations |
|---|---|---|---|
| GROMACS / AMBER / NAMD | MD Simulation Software | High-performance software suites for running MD simulations, including energy minimization, equilibration, and production runs. | [16] [21] |
| GPU Computing Cluster | Hardware | Essential for achieving the microsecond-to-millisecond timescales needed for simulating biomolecular processes in a reasonable time. | [16] |
| Force Fields (e.g., CHARMM, AMBER) | Parameter Set | Empirical potential functions defining bond, angle, dihedral, and non-bonded interaction energies for atoms in the simulation. | [16] [17] |
| ProLIF (Protein-Ligand Interaction Fingerprints) | Software Library (Python) | Calculates interaction fingerprints (IFPs) from MD simulation data, docking poses, or experimental structures. | [21] |
| IFPAggVis | Software Library (Python) | A Python library for the systematic aggregation, visualization, and comparison of IFPs derived from MD simulations. | [21] |
| run_ising | Software Executable (C++) | A specialized tool for implementing an Ising-like model using Boltzmann machine learning to analyze protein contact formation from MD trajectories. | [18] |
| GNU Scientific Library (GSL) | Software Library (C/C++) | A numerical library for C and C++ programmers, providing a wide range of mathematical routines used by scientific software. | [18] |
| Armadillo C++ Library | Software Library (C++) | A high-quality linear algebra library for C++, enabling fast and expressive matrix mathematics used in model fitting. | [18] |
From Theory to Therapy: Methodological Approaches in Target Modeling and Lead Optimization
Molecular dynamics (MD) simulations have become an indispensable tool in computational chemistry and drug design, providing atomic-level insights into biomolecular processes. A significant challenge in this field is accurately capturing protein flexibility, predicting ligand binding poses, and calculating binding free energies—all critical for structure-based drug design. These dynamic processes in biological molecules can be conceptually framed within the context of phase transitions in complex physical systems, such as the Ising model. The Ising model, a cornerstone of statistical physics, describes how systems can undergo radical changes in state (e.g., from disordered to ordered) upon crossing a critical point. Similarly, proteins can undergo functional transitions, such as folding or binding, which are governed by similar principles of collective behavior and critical fluctuations [22].
This guide objectively compares the performance of various MD methodologies and computational protocols in addressing these challenges. We focus on providing supporting experimental data, detailing the requisite protocols, and identifying the essential tools for researchers, thereby bridging theoretical concepts from physics with practical applications in computational biology.
Comparative Analysis of Methodologies and Performance
The accurate prediction of binding affinities remains a primary goal of structure-based drug design. The table below summarizes the performance of alchemical free energy methods, a leading approach for this task, when applied to different protein-ligand systems.
Table 1: Performance of Alchemical Binding Free Energy Calculations
| Protein / System Type | Ligand Type | Computational Method | Key Performance Metric (Mean Absolute Error) | Notable Challenges |
|---|---|---|---|---|
| MDMX [23] | p53 Inhibitors | Absolute Binding Free Energy (ABFE) | 0.816 kcal/mol | System rigidity allows for high accuracy |
| MDM2 [23] | p53 Inhibitors | Absolute Binding Free Energy (ABFE) | 3.08 kcal/mol (1.95 kcal/mol with FEL integration) | High protein flexibility requires enhanced sampling |
| Multiple Kinases, GTPases, ATPases [24] | Nucleotides (ATP, ADP, etc.) | ABFE & Relative Binding Free Energy (RBFE) | ABFE: ±2 kcal/mol (87.5% of cases); RBFE: ±3 kcal/mol (88.9% of cases) | Handling charged ligands and divalent ions (e.g., Mg²⁺) |
Key Insights from Comparative Data
- Protein Flexibility is a Primary Challenge: The data clearly shows that high protein flexibility, as seen in MDM2, directly impacts the accuracy of binding free energy calculations, leading to significantly higher errors compared to more rigid systems like MDMX [23]. Accommodating this flexibility is therefore a central focus in method development [25].
- Alchemical Methods are Reliable for Specific Cases: For a diverse set of proteins binding nucleotides, alchemical free energy simulations can reliably reproduce experimental binding affinities, provided the proteins do not undergo large conformational changes upon binding [24].
- Limitations with Divalent Ions: A significant limitation of widely used fixed-charge force fields is their inability to accurately capture interactions involving divalent ions like Mg²⁺, which are often crucial for nucleotide binding. This can lead to substantial inaccuracies [24].
Experimental Protocols for Key Applications
Protocol for Absolute Binding Free Energy Calculations
This protocol is adapted from studies on flexible proteins like MDM2 and its inhibitors [23].
System Preparation:
- Obtain the initial protein-ligand complex structure from crystallography or docking.
- Parameterize the ligand using standard force fields (e.g., GAFF for AMBER).
- Solvate the system in a water box (e.g., TIP3P) and add neutralizing ions.
Equilibration:
- Perform energy minimization to remove steric clashes.
- Gradually heat the system to the target temperature (e.g., 300 K) under an NVT ensemble.
- Equilibrate the density of the system under an NPT ensemble (1 atm pressure) for at least 100 ps.
Production and Analysis:
- Run Standard MD: Conduct multiple, independent MD simulations (≥100 ns) to sample protein-ligand conformations.
- Calculate Absolute Binding Free Energy (ABFE): Use Free Energy Perturbation (FEP) or Thermodynamic Integration (TI) to alchemically annihilate the ligand in both the solvated complex and in bulk solvent.
- Integrate with Free Energy Landscape (FEL): For highly flexible proteins, construct the FEL from the MD trajectories to identify metastable states. The ABFE can then be calculated for each state and weighted by their probability, improving accuracy for systems like MDM2 [23].
Protocol for the Giant-Component-Based DNB (GDNB) Method
This protocol is used for detecting critical transition states in complex systems, such as protein folding, by leveraging concepts from percolation theory [22].
Data Input and Preprocessing:
- Input data should be an array of system observations (e.g., from MD trajectories), with dimensions:
mobservation points (e.g., time),nvariables (e.g., dihedral angles, distances), andsreplicates per point.
- Input data should be an array of system observations (e.g., from MD trajectories), with dimensions:
Candidate Variable Selection:
- At each observation point
i, calculate the relative fluctuation (RF) for each variablej:RF_ij = (1/mean(x_j)) * standard_deviation(x_ijk). - Use a one-sample t-test (e.g., p < 0.05) to select variables with significantly large relative fluctuations. This group is denoted as
F_i[22].
- At each observation point
Variable Clustering:
- For the selected variables in
F_i, perform hierarchical clustering based on the absolute value of the Pearson's Correlation Coefficient (PCC) between all variable pairs. - Determine the number of clusters using a predefined correlation threshold.
- For the selected variables in
Transition Core Identification:
- Select the largest cluster (the "giant component,"
G_i) at each observation point as the transition core. - Calculate a Composite Index (CI) to quantify the early-warning signal strength:
CI_i = GC_i * RF_i * |PCC_i|, whereGC_iis the size of the giant component,RF_iis the average relative fluctuation withinG_i, and|PCC_i|is the average absolute correlation withinG_i. A peak in the CI indicates a critical transition point [22].
- Select the largest cluster (the "giant component,"
Workflow and Conceptual Diagrams
GDNB Method for Detecting Phase Transitions
Diagram Title: GDNB Workflow for Critical Point Detection
Free Energy Calculation and Protein Flexibility
Diagram Title: Addressing Protein Flexibility in Free Energy Calculations
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of MD-based drug design projects requires a suite of specialized software and hardware. The table below details key components of a modern computational researcher's toolkit.
Table 2: Essential Research Reagent Solutions for MD Simulations
| Tool Category | Specific Examples | Function and Application |
|---|---|---|
| Specialized MD Software | AMBER, GROMACS, NAMD, OpenMM, Tinker-HP | Core simulation engines for running MD; each has strengths in different sampling algorithms, force fields, or hardware acceleration [26]. |
| Free Energy Calculation Tools | PLUMED, FEP+, various in-house suites | Enable advanced sampling and alchemical free energy calculations (ABFE, RBFE) crucial for predicting binding affinities [23] [24]. |
| Neural Network Potentials (NNPs) | FeNNix-Bio1(M), FeNNol, Deep-HP | Foundation models providing near-quantum mechanical accuracy at a fraction of the cost of ab initio methods, accelerating simulations [27]. |
| High-Performance Computing (HPC) | NVIDIA GPUs (RTX 4090, L40S, H200), BIZON Z Series Workstations | GPU acceleration is critical for throughput. Choices balance raw speed (ns/day) and cost-efficiency (cost/100 ns) [26] [28]. |
| Force Fields | CHARMM, AMBER, GAFF, AMOEBA (polarizable) | Parameter sets defining the energy function and atomic interactions. Selection depends on the system (proteins, nucleotides) and required accuracy [24]. |
Molecular dynamics simulations provide a powerful framework for capturing the intricate details of biomolecular behavior, directly addressing the challenges of protein flexibility, binding pose prediction, and free energy calculation. The comparative data and protocols presented here demonstrate that while significant progress has been made—particularly with alchemical methods and advanced sampling techniques—challenges remain in simulating highly flexible systems and interactions involving charged species and ions. Framing these biological processes through the lens of physical models like the Ising model enriches our understanding of their underlying dynamics, suggesting that critical fluctuations and collective behavior govern functional transitions in proteins. The continued development of neural network potentials, enhanced sampling protocols, and cost-effective high-performance computing hardware promises to further bridge the gap between theoretical models and predictive, industrially relevant drug design.
The rational design of advanced drug delivery systems, such as carbon nanotubes (CNTs) and lipid-based nanocarriers, relies on understanding complex molecular interactions that are difficult to observe experimentally. Molecular dynamics (MD) simulations have emerged as a powerful computational tool that provides atomic-level resolution of nanocarrier behavior, complementing traditional theoretical approaches like Ising-like models. While Ising models offer valuable insights into general phase behavior and cooperative effects through simplified lattice-based representations, MD simulations capture the specific atomistic details, molecular conformations, and dynamic processes essential for predicting drug loading, stability, and release profiles.
This comparison guide examines how MD simulations outperform Ising-like models in capturing the complex, non-equilibrium behavior of nanocarrier systems, providing researchers with quantitative data to guide the selection of computational approaches for specific drug delivery challenges. We present experimental data, detailed methodologies, and comparative analyses to illustrate the distinct advantages and limitations of each modeling paradigm in the context of carbon nanotube and lipid bilayer-based drug delivery systems.
Comparative Performance: MD Simulations vs. Ising-like Theoretical Models
Table 1: Performance comparison between MD simulations and Ising-like models for nanocarrier design.
| Feature | MD Simulations | Ising-like Models |
|---|---|---|
| Spatial Resolution | Atomic-level (Å scale) [29] [30] | Lattice/coarse-grained (nm-μm scale) |
| Temporal Range | Nanoseconds to microseconds [31] | Effectively infinite (statistical mechanics) |
| Physical Realism | High (atomistic force fields) [32] | Low (simplified spin interactions) |
| Application to CNTs | Predicts thermal conductivity enhancement (4.05x increase with CNTs) [29] | Limited to electronic properties or adsorption isotherms |
| Application to Lipid Bilayers | Quantifies area per lipid, bending rigidity, order parameters [33] [32] | Models phase transitions (fluid-gel) |
| Drug Interaction Modeling | Specific binding energies, diffusion pathways [34] | Non-specific partitioning coefficients |
| Computational Cost | High (requires HPC) [31] [34] | Low (analytical or minimal computing) |
| Handling Dynamics | Explicit time evolution of all atoms [32] | Equilibrium properties, master equations |
Table 2: Quantitative performance data from MD simulations of nanocarrier systems.
| Nanocarrier System | Simulation Type | Key Performance Metric | Result | Reference |
|---|---|---|---|---|
| CNT/Paraffin CPCM | All-atom MD | Thermal conductivity | 0.81 W·m⁻¹·K⁻¹ (4.05x pure paraffin) [29] | |
| Ti-doped CNT H₂ Storage | Multiscale MD | Hydrogen storage density | 8.04 wt.% at 77K [30] | |
| DOPC Lipid Bilayer | All-atom MD | Area per lipid (APL) under EHorz | 0.650 ± 0.006 nm² (2.6% decrease) [33] | |
| Lipid Bilayer Mechanics | All-atom/CG-MD | Bending rigidity (kc) | 10-30 kBT for fluid phases [32] | |
| Functionalized CNTs | All-atom MD | Drug-loading capacity & stability | High for anticancer drugs [34] |
Methodologies: Experimental Protocols in MD Simulations
MD Simulation of Carbon Nanotube-Based Composites
System Setup: The protocol begins with constructing a CNT model defined by its chiral vector (n,m). The diameter is calculated as d = a√(n² + m² + nm)/π, where the graphene lattice constant a = 2.46 Å [30]. For composite studies, such as CNT/paraffin systems, N-docosane is often used as a paraffin substitute. CNTs are incorporated into the matrix at specific mass fractions (e.g., 4 wt%), with parameters like length and dispersion carefully controlled [29].
Force Field Selection: Interactions are typically governed by a combination of potential functions. A critical step is parameterizing the dopant-specific interactions; for example, Ti-H interactions in Ti-doped CNTs for hydrogen storage use a pressure-modified Lennard-Jones (LJ) potential, with parameters refined against Density Functional Theory (DFT) calculations to ensure quantum mechanical accuracy [30].
Simulation Execution: Simulations are performed under the NPT (constant Number of particles, Pressure, and Temperature) ensemble. Temperature and pressure are regulated using algorithms like the Langevin thermostat and the virial stress tensor, respectively. The pressure is often set to a specific value relevant to the application (e.g., 3 MPa for hydrogen storage) [30]. Production runs typically extend for hundreds of nanoseconds to ensure proper equilibration, with data collected from the stable trajectory phase [33] [30].
Analysis: Key properties analyzed include:
- Thermal conductivity: Calculated using non-equilibrium MD or the Green-Kubo method, often showing a positive correlation with CNT length [29].
- Adsorption capacity: For hydrogen storage, the gravimetric density (wt.%) is tracked [30].
- Interfacial properties: The interfacial thermal resistance between CNT and matrix is a critical output [29].
MD Simulation of Lipid Bilayers and LNPs
System Setup: A lipid bilayer is built with specific lipid compositions, such as pure DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) or mixtures with cholesterol (e.g., 0-30 mol%). The system is solvated in water models (e.g., TIP3P) and neutralized with ions (e.g., 150 mM KCl) [33]. For Lipid Nanoparticles (LNPs), the model includes ionizable lipids, helper lipids, cholesterol, and PEG-lipids, often requiring constant pH molecular dynamics (CpHMD) to accurately model the environment-dependent protonation states of ionizable lipids [31].
Force Field and Equilibration: Standard biomolecular force fields like CHARMM36 are used [33]. The system is energy-minimized and equilibrated with position restraints on lipids before the production run. The area per lipid (APL) is monitored to confirm equilibration, which typically occurs after ~100 ns [33].
Application of External Stimuli: To study mechanical or electrostatic responses, external electric fields can be applied. For instance, a horizontal field (EHorz) of 0.05 V/nm can be used to study in-plane membrane contraction [33].
Analysis: Key properties analyzed include:
- Structural properties: Area per lipid (APL), bilayer thickness, and mass density profiles [33] [32].
- Dynamic properties: Lipid diffusion coefficients, order parameters (SCD) for acyl chains [33].
- Mechanical properties: Bending rigidity (kc) calculated from thermal fluctuations or stress-strain analysis, and area compressibility modulus [32].
Conceptual Workflow and Signaling Pathways
The following diagram illustrates the integrated multiscale computational workflow for simulating nanocarriers, highlighting the complementary roles of MD and Ising-like models.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key research reagents and computational tools for MD simulations of nanocarriers.
| Item Name | Function/Description | Example Use Case |
|---|---|---|
| CHARMM36 Force Field | A set of empirical interaction parameters for lipids, proteins, and nucleic acids. | Simulating structural properties of DOPC lipid bilayers [33]. |
| GROMACS | A versatile software package for performing MD simulations. | Simulating lipid bilayer responses to electric fields [33]. |
| Martini Coarse-Grained Model | A reduced-resolution force field that groups 2-4 heavy atoms into a single bead. | Studying self-assembly of Lipid Nanoparticles (LNPs) on longer timescales [31]. |
| Constant pH MD (CpHMD) | A specialized MD method that allows protonation states to change dynamically. | Modeling ionizable lipids in LNPs for accurate pKa prediction [31]. |
| DOPC Lipid | A common phospholipid with two unsaturated chains, creating a fluid lipid bilayer. | A standard model system for studying basic membrane properties [33]. |
| TIP3P Water Model | A widely used 3-site model for representing water molecules in simulations. | Solvating lipid bilayer and CNT systems [33]. |
| Langevin Thermostat | An algorithm to control temperature in MD simulations by adding friction and noise. | Maintaining constant temperature during NPT simulations of Ti-CNTs [30]. |
Molecular dynamics simulations provide an indispensable toolkit for the rational design of next-generation nanocarriers, offering atomic-level insights that are simply inaccessible to Ising-like models. While Ising models retain value for rapid screening and understanding universal phase behavior, MD simulations deliver the specific, quantitative, and dynamic data needed to optimize complex performance metrics such as drug-loading capacity, thermal conductivity, and structural responses to biological stimuli. The future of nanocarrier design lies in multiscale frameworks that intelligently integrate the strengths of both approaches—using Ising-like models for initial system exploration and MD simulations for detailed mechanistic investigation—to accelerate the development of more effective and targeted drug delivery systems.
The Ising model, a seminal concept in statistical mechanics, has evolved from its origins in modeling ferromagnetism into a powerful, interdisciplinary framework for understanding cooperative effects in complex systems. At its core, the model captures how simple, local interactions between discrete components can give rise to sophisticated macroscopic behaviors and phase transitions [35]. This comparative guide examines how Ising-type theoretical models serve as complementary and sometimes alternative approaches to Molecular Dynamics (MD) simulations for researching cooperative phenomena in molecular systems and biological networks.
The model's fundamental ingredients are remarkably simple: constituent units that occupy only two discrete states (typically represented as +1 or -1), with interactions occurring primarily between nearest neighbors [36]. Despite this simplicity, these two elements prove sufficient for explaining long-range ordering and emergent collective behavior across diverse domains. Originally conceived by Wilhelm Lenz in 1920 and analyzed by his student Ernst Ising in 1924, the model demonstrated no phase transition in one dimension but was later shown by Rudolf Peierls in 1936 to exhibit spontaneous ordering in two and three dimensions [36]. This breakthrough established the Ising model as a fundamental paradigm for cooperative phenomena.
In contemporary research, Ising models provide a conceptual and mathematical foundation for interpreting data from sophisticated MD simulations and experimental techniques. Their ability to distill complex system behaviors into essential interaction parameters makes them particularly valuable for studying allosteric regulation, molecular self-assembly, and cellular decision-making processes where cooperative effects play a decisive functional role [37] [38].
Methodological Comparison: Ising Models Versus Molecular Dynamics Simulations
Fundamental Approaches and Underlying Principles
Molecular Dynamics (MD) Simulations operate at high spatial and temporal resolution, numerically solving Newton's equations of motion for all atoms in a molecular system. This approach captures detailed conformational landscapes and transient intermediate states through explicit simulation of atomic interactions over time [38]. Advanced implementations now integrate AI-driven methods to enhance sampling and accelerate convergence, with recent applications ranging from SARS-CoV-2 spike protein dynamics to membrane transporter mechanisms [38].
In contrast, Ising-like Theoretical Models employ a coarse-grained representation that abstracts molecular components or systems into discrete states with defined interaction rules. This approach focuses on identifying emergent statistical regularities and phase behavior rather than atomic-level detail [35]. The power of Ising models lies in their ability to reduce complex cooperative phenomena to essential interaction parameters, making them particularly effective for studying how local interactions produce system-wide transitions.
Table 1: Fundamental Methodological Differences Between MD Simulations and Ising-Type Models
| Feature | Molecular Dynamics (MD) Simulations | Ising-Type Theoretical Models |
|---|---|---|
| Representation | Atomistic/All-atom detail | Coarse-grained discrete states |
| Time Evolution | Numerical integration of equations of motion | Markov processes/Monte Carlo sampling |
| Key Parameters | Force fields, partial charges, solvent models | Coupling constants (Jij), external fields (hj) |
| Observables | Atomic coordinates, energies, forces | Order parameters, correlation functions, critical temperatures |
| Cooperative Effects | Emerge from explicit atomic interactions | Built into Hamiltonian through spin-spin couplings |
| Computational Scaling | O(N2) to O(N) with approximations | Varies with lattice size and algorithm |
Experimental and Computational Protocols
Molecular Dynamics Protocol for Studying Biomolecular Cooperation
Advanced MD protocols for investigating cooperative effects in molecular systems typically involve multiple stages of increasingly refined simulation:
System Preparation: Build molecular assembly using structural data from cryo-EM, X-ray crystallography, or AlphaFold predictions [38]. Add explicit solvent ions, and parametrize force fields.
Equilibration Phase: Perform energy minimization followed by gradual heating to target temperature (typically 300K) with position restraints on heavy atoms, then release restraints under constant temperature and pressure conditions.
Production Simulation: Run extended simulations (nanoseconds to microseconds, sometimes longer with enhanced sampling) using high-performance computing resources. For studying allostery or cooperativity, multiple replicates with different initial conditions are essential.
Analysis Phase: Identify conformational states via dimensionality reduction (PCA, t-SNE); quantify populations and transition rates between states; calculate free energy landscapes; identify correlated motions through mutual information analysis or dynamical network analysis.
Recent implementations integrate machine learning for automated state identification and analysis, with methods like VAE (Variational Autoencoder) being used to convert trajectory data into one-dimensional representations suitable for quantifying cooperative transitions [39].
Ising Model Protocol for Molecular and Network Systems
Implementing Ising-type models for studying cooperative effects follows a distinct computational pathway:
System Mapping: Identify the binary or discrete states relevant to the molecular system (e.g., folded/unfolded, bound/unbound, active/inactive) [39]. Define the interaction network (lattice topology or graph structure).
Parameter Estimation: Determine coupling constants (Jij) from experimental data or MD simulations. For molecular systems, these may correspond to effective energies of cooperative interactions.
Monte Carlo Simulation: Implement sampling algorithm (typically Metropolis or Glauber dynamics) [40]. The basic steps include:
- Initialize system configuration
- Randomly select a site to flip
- Compute energy change ΔE for the flip
- Accept or reject the flip based on Metropolis criterion: accept if ΔE < 0 or with probability exp(-ΔE/kBT) otherwise
- Repeat for sufficient Monte Carlo steps to ensure equilibration
Analysis: Calculate order parameters (e.g., magnetization), correlation functions, specific heat, and susceptibility. Identify phase transitions through finite-size scaling analysis.
The following diagram illustrates the logical workflow for applying Ising models to molecular cooperation studies:
Comparative Applications in Molecular and Biological Systems
Protein Folding and Allostery
Both MD simulations and Ising-type models provide complementary insights into protein folding and allosteric regulation, though through fundamentally different approaches.
MD simulations approach these phenomena through explicit representation of atomic interactions, as demonstrated in studies of the WT-HP35 protein's folding and unfolding transitions. Researchers employed distance matrices between residues and used Variational Autoencoders (VAE) to reduce dimensionality before clustering conformational states [39]. This approach revealed not just folded and unfolded states, but also various intermediate forms, with the relative populations shifting under different conditions.
Ising models conceptualize protein folding and allostery through coarse-grained representations, where amino acids or structural domains are treated as interacting binary units. This abstraction successfully captures the cooperative nature of folding transitions and allosteric propagation. The model's ability to describe how local perturbations (e.g., ligand binding, mutations) can induce global conformational changes aligns with the observation that "through allosteric control of the dynamics of single molecules, different interactors can thus be selected, generating supramolecular units with different functions" [38].
Molecular Self-Assembly and Surface Networks
The formation of self-assembled molecular networks (SAMNs) at liquid/solid interfaces represents an area where both approaches have yielded complementary insights, with the Ising model providing a thermodynamic framework for interpreting experimental observations.
Scanning Tunneling Microscopy (STM) studies of alkoxylated dimethylbenzene derivatives at the heptanoic acid/HOPG interface have revealed concentration-dependent formation of SAMNs [37]. These experiments demonstrated that even molecular analogues lacking hydrogen bonding capacity (where carboxylic acid groups are replaced by methyl groups) still exhibit cooperative network formation, though with altered structural parameters and periodicity.
The Ising model has been successfully adapted to describe this cooperative assembly through a 2D Ising-type formulation that treats molecular adsorption as a function of concentration and interaction energies [37]. This modeling approach connects "classical and statistical thermodynamics" to explain experimental results "in terms of molecular behaviour using the nearest-neighbour model approach, thus revealing the free energy contributions to the total free energy of the SAMN formation arising from the molecule–molecule and molecule–surface interactions" [37].
Table 2: Comparison of Approach to Molecular Self-Assembly Studies
| Aspect | Experimental STM Approach | Ising Model Interpretation |
|---|---|---|
| Primary Data | High-resolution molecular images | Coverage vs. concentration curves |
| Interaction Quantification | Structural parameters from images | Coupling constants J from fits |
| Cooperativity Assessment | Sharpness of coverage transition | Cooperativity parameter σ |
| Free Energy Components | Indirect through temperature dependence | Explicit decomposition possible |
| Limitations | Surface defects, limited sampling | Simplified interaction topology |
Biological Networks and Cellular Decision Making
At the cellular level, Ising models provide a conceptual framework for understanding how collective behaviors emerge in protein interaction networks and signaling systems. MD simulations face significant challenges in simulating systems at this scale due to computational constraints, though advances in coarse-grained MD and AI-assisted methods are beginning to bridge this gap [38].
The Ising model's application to protein-protein interaction networks illustrates how local interaction rules can generate switch-like behaviors in cellular signaling. This approach conceptualizes proteins as existing in "dynamic ensembles with conformational distributions that may change in response to varying cellular conditions" [38], with the Ising model describing how these ensembles collectively transition between functional states.
This conceptual framework has been particularly valuable in understanding allosteric mechanisms where "the redistribution of conformational populations in response to a perturbation causes functional changes at distant sites within the protein" [38]. The model successfully captures how small inputs can lead to complex cellular outputs through modification of "the internal dynamics of the complexes" [38].
Performance Comparison: Quantitative Benchmarks
The relative performance and application ranges of MD simulations and Ising-type models can be quantitatively compared across several dimensions:
Table 3: Quantitative Comparison of MD Simulations and Ising-Type Models
| Performance Metric | MD Simulations | Ising-Type Models |
|---|---|---|
| System Size Capability | ~100,000 atoms for conventional MD; millions with coarse-graining [39] | Virtually unlimited with Monte Carlo methods |
| Timescale Access | Nanoseconds to milliseconds for all-atom; longer with enhanced sampling [39] | Direct access to equilibrium properties and steady states |
| Phase Transition Resolution | Requires extensive sampling near critical points | Naturally suited for critical behavior analysis |
| Cooperative Interaction Mapping | From explicit atomic contacts | From coupling parameters Jij |
| Computational Resource Demand | High (exascale computing for complex systems) [38] | Moderate (often feasible on workstations) |
| Parameterization Requirements | Extensive (force fields, solvent models) | Minimal (coupling constants, fields) |
| Experimental Connection | Direct structural comparison | Macroscopic observables (binding curves, coverage) |
Integration and Synergistic Applications
The most powerful applications emerge when MD simulations and Ising-type models are used synergistically rather than as competing alternatives. Two successful integration patterns have emerged:
First, MD simulations can parameterize Ising models by providing quantitative estimates of effective coupling constants between molecular components. For example, all-atom simulations of protein domains can quantify the energetic coupling between different sites, which can then be used to parameterize a coarse-grained Ising model that captures the system's larger-scale collective behavior [39].
Second, Ising models can provide conceptual frameworks for interpreting MD results. When MD simulations reveal complex conformational ensembles, Ising models can help identify whether these represent distinct phases or fluctuate around a single minimum. As noted in protein studies, "the variation in the populations of the conformational ensembles caused by some of the above-described perturbations determines the type of structures that are presented to partners for interaction at specific points in time" [38] – a perspective naturally formalized through Ising models.
The following diagram illustrates this synergistic relationship:
Research Reagent Solutions for Experimental Studies
For experimental investigation of cooperative effects in molecular systems:
- Scanning Tunneling Microscopy (STM): Enables nanoscale visualization of self-assembled molecular networks at liquid/solid interfaces [37].
- Alkoxylated Molecular Analogues: Structural variants (e.g., DMBOC18 vs. ISAOC18) allow systematic study of specific interactions like hydrogen bonding on cooperativity [37].
- Functionalized Graphite Surfaces (HOPG): Provide standardized substrates for studying 2D molecular self-assembly with minimal defects [37].
- Ising Model Reference Systems: Simple physical systems (e.g., iron magnets) for validating computational implementations and methodologies [36].
For computational studies of cooperative effects:
- Monte Carlo Simulation Codes: Custom implementations (e.g., Metropolis algorithm) for sampling Ising model configurations [40].
- Molecular Dynamics Packages: GROMACS, AMBER, or NAMD for all-atom simulations of molecular systems [38].
- Variational Autoencoders (VAE): Machine learning tools for dimensionality reduction of conformational data from MD trajectories [39].
- Clustering Quality Metrics: Physically interpretable scoring metrics (V-Measure, Silhouette Index) for evaluating state identification in complex molecular data [39].
Molecular Dynamics simulations and Ising-type theoretical models offer complementary approaches to studying cooperative effects in molecular systems and networks. MD provides high-resolution dynamical information but faces challenges in accessing longer timescales and capturing emergent collective behavior directly. Ising models excel at identifying universal principles of cooperation and phase transitions but sacrifice molecular detail for conceptual clarity.
The most promising future direction lies in further integration of these approaches, particularly through AI-driven methods that can bridge scales and extract essential cooperative parameters from detailed simulations. As one research team noted regarding biomolecular simulations, "integrating AI and Machine Learning with multiscale MD methods will enhance fundamental understanding for systems of ever-increasing complexity" [38]. Similarly, Ising models continue to evolve through connections to graph theory and network science, expanding their applicability to increasingly complex biological systems.
For researchers and drug development professionals, the strategic combination of both approaches – using MD to parameterize simplified models and Ising frameworks to interpret simulation results – offers the most powerful path toward understanding and harnessing cooperative effects in molecular systems. This synergistic methodology promises to unlock new opportunities in drug discovery, materials design, and fundamental biology by revealing how local interactions give rise to sophisticated collective behaviors across scales.
This guide provides a comparative analysis of two powerful computational approaches used in cancer research: Molecular Dynamics (MD) simulations for studying drug-carrier interactions and Ising-like theoretical models for analyzing tumor growth dynamics. It is structured to offer researchers, scientists, and drug development professionals an objective comparison of their performance, supported by experimental data and detailed methodologies.
Molecular Dynamics Simulation of Doxorubicin with a Graphene Oxide Nanocarrier
Experimental Protocol & Workflow
This case study is based on research that used Molecular Dynamics (MD) simulations to investigate the loading and dynamics of Doxorubicin (DOX) on Graphene Oxide (GO) and poly(ethylene glycol) decorated GO (PEGGO) nanocarriers [41]. The simulations were performed to reveal atomic-level details of drug adsorption as a function of PEG chain length.
Key Experimental Steps [41]:
- System Preparation: A GO sheet (40×40 Ų) was built and decorated with hydroxyl, epoxide, and carboxylic acid functional groups, with protonation states set to physiological pH (7.4). PEG chains of two lengths (15 and 30 repeat units) were covalently attached to create short-chain (Sh-PEGGO) and long-chain (L-PEGGO) systems.
- Force Field & Parameterization: The systems were geometry-optimized using the Dreiding force field. Parameters for GO and PEGGO were taken from the CHARMM36 force field, while parameters for DOX were derived from the Merck Molecular Force Field (MMFF) via the SwissParam interface.
- Simulation Setup: The equilibrated nanocarriers were placed in a simulation cell with 10 DOX molecules initially positioned 20 Å from the carrier surface. The system was solvated with TIP3P water molecules and neutralized with sodium ions.
- Production Run: NPT (constant Number of atoms, Pressure, and Temperature) simulations were run for 20 ns at 310 K (human body temperature) and 1 atm pressure using the NAMD (v2.12) software. A time step of 2 fs was used, with long-range interactions calculated with a 12 Å cut-off.
- Data Analysis: Key analyzed properties included DOX-nanocarrier interaction energy, PEG-DOX solvent-accessible contact area, water density around the nanocarrier, and DOX aggregation behavior.
The workflow for this protocol is summarized in the following diagram:
Key Quantitative Findings from MD Simulations
The MD simulations generated quantitative data on the interactions and loading behavior of DOX. The table below summarizes the primary findings for the three systems studied [41].
Table 1: Comparison of DOX Loading and Dynamics on GO and PEGylated GO Nanocarriers
| System Description | Total DOX-Nanocarrier Interaction Energy | PEG-DOX Solvent-Accessible Contact Area (Ų) | Key Observation on Drug Distribution |
|---|---|---|---|
| DOX/GO (Control) | Same within margin of error | Not Applicable (No PEG) | Drug molecules are less aggregated [41] |
| DOX/Sh-PEGGO (Short PEG Chains) | Same within margin of error | ~50% of L-PEGGO value | DOX migration to solvated,\nPEG-free GO surface is more pronounced [41] |
| DOX/L-PEGGO (Long PEG Chains) | Same within margin of error | ~100% (Baseline) | Increased water density acts as a barrier;\nbenefits DOX loading on nanocarrier [41] |
Ising Model Analysis of Tumor Growth and Immunoediting
Experimental Protocol & Workflow
This case study examines the use of an Ising-model Hamiltonian, implemented within an Agent-Based Model (ABM), to simulate and characterize the process of Cancer Immunoediting (CI) in the Tumor Micro-Environment (TME). This approach formalizes the complex interactions between tumor and immune cells as an energy-based system to study phase transitions [42].
Key Experimental Steps [42]:
- Model Formalization: The TME is represented as a grid where each cell is an agent. The state of the system is described using an Ising-model analogue, where the overall "energy" or Hamiltonian reflects the collective interaction between agents (e.g., cancer cells and immune cells).
- Agent Definition: Key agents include Cancer Cells (CCs) and various Immune System cells (NK cells, macrophages (M𝜑s), neutrophils (Ns), dendritic cells (DCs), CD4+ T, CD8+ T, and Treg cells).
- Rule Definition: Probabilistic rules govern agent interactions and state transitions (e.g., a cancer cell may be eliminated by an NK cell). Tumor cell recruitment and growth are often modeled using logistic functions or Gompertzian growth dynamics.
- Simulation Execution: The model is run for a sufficient number of time steps to observe the emergence of the three CI phases. Simulations are often performed in platforms like NetLogo.
- System Characterization: The Ising-model Hamiltonian is calculated throughout the simulation. The temporal evolution of this "energy" is used as a signature to identify and characterize the different phases of immunoediting.
The following diagram illustrates the logical relationships and transitions between the three phases of Cancer Immunoediting:
Key Quantitative Findings from Ising Model Simulations
The ABM linked to the Ising-model Hamiltonian successfully reproduces the three phases of CI, with the Hamiltonian providing a clear, quantifiable signature for each phase [42].
Table 2: Ising-Model Characterization of Cancer Immunoediting Phases
| Immunoediting Phase | System Description (IS vs. Tumor Balance) | Ising-Model Hamiltonian ("Energy") Signature | Key Simulated Outcome |
|---|---|---|---|
| Elimination | Immune System (IS) prevails over tumor cells. | Energy favors the "IS state"; patterns indicate dominant immune activity. | Most abnormal cells are successfully eliminated [42]. |
| Equilibrium | Balanced state between IS activities and tumor cell proliferation. | Energy is in a dynamic balance; fluctuations correspond to ongoing immune pressure and tumor cell adaptation. | Tumor cell variants with increased immune evasion capabilities are selected for [42]. |
| Escape | Tumor cells triumph over the IS. | Energy shifts to favor the "tumor state"; patterns indicate breaking of immune control. | Tumor growth becomes clinically apparent without hindrance [42]. |
Comparative Performance Analysis: MD Simulations vs. Ising-like Models
The following table provides a direct, objective comparison of the two methodologies based on their performance, output, and application.
Table 3: Performance and Application Comparison of MD Simulations and Ising-like Models
| Aspect | Molecular Dynamics (MD) Simulations | Ising-like Theoretical Models |
|---|---|---|
| Primary Research Focus | Drug-carrier interactions, loading dynamics, and atomic-level energetics [41] [43]. | Tumor growth dynamics, immunoediting phases, and population-level interactions [44] [42]. |
| System Scale | Atomic and molecular scale (Nanoscale). | Cellular and tissue scale (Micro to Macroscale). |
| Key Output Data | Interaction energies, solvent-accessible surface areas, molecular conformations, and diffusion coefficients [41]. | System "energy" (Hamiltonian), phase transitions, population counts, and spatial organization of cells [42]. |
| Temporal Resolution | Picoseconds to nanoseconds. | Days to years (model-dependent). |
| Strengths | Provides high-resolution, atomistic detail on molecular interactions; can predict properties difficult to measure experimentally [41]. | Captures emergent behavior from simple local rules; highly flexible for incorporating different cell types and therapies; powerful for conceptual understanding [44] [42]. |
| Limitations | Computationally expensive; limited in spatial and temporal scale compared to biological processes; accuracy depends on force field parameters [41]. | Highly abstracted; lacks molecular-level mechanistic detail; requires calibration with biological data for quantitative predictions [44]. |
The Scientist's Toolkit: Essential Research Reagents & Solutions
This section lists key computational tools and resources essential for conducting research in MD simulations and Ising-model analysis of cancer systems.
Table 4: Essential Research Reagents and Computational Tools
| Tool/Resource | Function/Application | Relevance to Case Studies |
|---|---|---|
| NAMD | A parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. | Used for running the MD simulations of DOX with GO/PEGGO nanocarriers [41]. |
| GROMACS | A versatile package for performing MD simulations, with high performance and extensive analysis tools. | Employed in other MD studies of anticancer drug delivery systems [45]. |
| CHARMM Force Field | A widely used set of parameters for MD simulations of proteins, nucleic acids, lipids, and small molecules. | Provided parameters for the GO and PEGGO structures in the DOX loading study [41]. |
| NetLogo | A programmable modeling environment for simulating natural and social phenomena, ideal for ABMs. | A common platform for implementing agent-based models, including those for tumor growth [42]. |
| Ising-Model Hamiltonian | A mathematical function from statistical mechanics that calculates the energy of a system based on interactions between its components. | Used to characterize the global state and phase transitions in the Cancer Immunoediting ABM [42]. |
Overcoming Computational Hurdles: Sampling, Force Fields, and Scalability
Molecular dynamics (MD) and Monte Carlo (MC) simulations are indispensable tools for investigating the structural and dynamic properties of molecular systems, from proteins and nucleic acids to complex materials. However, a fundamental challenge plagues these simulations: the sampling limitations that arise due to the high dimensionality of molecular systems and the multiple minima on their energy landscapes. These limitations are particularly acute when simulating rare events or systems with rough energy landscapes, where conventional simulations tend to become trapped in local minima, failing to explore the full configuration space within feasible computational time. Enhanced sampling techniques have emerged as powerful approaches to overcome these limitations, enabling more thorough exploration of energy landscapes and accelerating the convergence of simulations.
This review provides a comprehensive comparison of enhanced sampling methodologies across MD and MC simulations, with a specific focus on their applications to Ising-like theoretical models and biomolecular systems. We examine the theoretical foundations, performance metrics, and practical implementations of these techniques, providing researchers with the necessary framework to select appropriate methods for their specific sampling challenges.
Theoretical Foundations of Enhanced Sampling
The Sampling Problem in Molecular Simulations
In conventional MD simulations, the system evolves according to Newton's equations of motion, sampling configurations from the Boltzmann distribution. However, as noted in studies of protein folding and dynamics, "conventional simulations of biomolecules suffer from the multiple-minima problem: The canonical fixed-temperature simulations at low temperatures tend to get trapped in a huge number of local-minimum-energy states" [46]. Similarly, in MC simulations, local updates lead to critical slowing down, especially near phase transitions, severely limiting sampling efficiency.
The core issue is that biological and physical systems often exhibit energy landscapes characterized by high barriers separating metastable states. Crossing these barriers requires thermal activation, which occurs infrequently on the timescales accessible to standard simulations. This problem manifests as poor convergence of equilibrium properties and inability to observe functionally relevant transitions.
Generalized-Ensemble Algorithms
Generalized-ensemble algorithms address the sampling problem by modifying the sampling distribution to facilitate barrier crossing. In these approaches, "each state is weighted by an artificial, non-Boltzmann probability weight factor so that a random walk in potential energy space and/or other physical quantities may be realized" [46]. This random walk enables the simulation to escape from energy local minima and sample much wider conformational space than conventional methods.
Key generalized-ensemble methods include:
- Multicanonical Algorithm (MUCA): Samples configurations according to a non-Boltzmann weight designed to yield a flat energy distribution [46]
- Replica-Exchange Method (REM): Parallel simulations at different temperatures exchange configurations, allowing high-temperature replicas to overcome barriers [46]
- Metadynamics: Uses a history-dependent bias potential to discourage revisiting previously sampled configurations [46]
These methods have been successfully applied to protein folding, biomolecular binding, and phase transitions in complex systems.
Enhanced Sampling in Molecular Dynamics
Advanced MD Sampling Techniques
Recent advances in MD sampling have introduced sophisticated algorithms that leverage machine learning and advanced statistical methods. The Timewarp method represents a particularly innovative approach that "uses a normalising flow as a proposal distribution in a Markov chain Monte Carlo method targeting the Boltzmann distribution" [47]. This method learns to make large steps in time (simulating 10⁵-10⁶ fs) and demonstrates transferability between molecular systems, providing wall-clock acceleration of sampling compared to standard MD [47].
Other notable MD enhanced sampling approaches include:
- Parallel Tempering: Similar to replica-exchange but with coordinates exchanged between temperatures
- Accelerated MD: Modifies the potential energy surface to lower energy barriers
- Variational Enhanced Sampling: Uses machine learning to identify collective variables and enhance sampling along them
A critical challenge in MD simulations is force field accuracy, as "approximations built into the mathematical forms of MD force fields and their associated parameterizations give rise to the accuracy problem" [48]. Even with perfect sampling, inaccurate force fields will yield incorrect results, highlighting the need for continued force field refinement alongside sampling improvements.
Validation of MD Sampling Methods
Validating enhanced sampling methods requires careful comparison with experimental data. Studies have shown that "although four molecular dynamics simulation packages reproduced a variety of experimental observables for two different proteins equally well overall at room temperature, there were subtle differences in the underlying conformational distributions and the extent of conformational sampling obtained" [48]. This ambiguity underscores the challenge of method validation when experimental data cannot provide detailed information about underlying conformational ensembles.
Table 1: Comparison of MD Sampling Performance Across Simulation Packages
| Simulation Package | Force Field | Sampling Efficiency | Agreement with Experiment | Unfolding Behavior at High T |
|---|---|---|---|---|
| AMBER | ff99SB-ILDN | Moderate | Good | Partial unfolding |
| GROMACS | ff99SB-ILDN | Moderate | Good | Partial unfolding |
| NAMD | CHARMM36 | Moderate | Good | Varies |
| ilmm | Levitt et al. | Moderate | Good | Restricted unfolding |
The table above summarizes findings from comparative studies, revealing that while overall agreement with experimental data may be similar across packages, significant differences emerge in their ability to sample large-amplitude motions and unfolding processes [48].
Enhanced Sampling in Monte Carlo for Ising Models
Cluster Algorithms for Critical Slowing Down
In the context of Ising models, conventional local-update MC algorithms suffer from critical slowing down near phase transitions, where correlation times diverge with system size. Cluster algorithms were developed specifically to address this limitation. The Swendsen-Wang algorithm and its variant, the Wolff algorithm (a single-cluster method), dramatically reduce autocorrelation times by updating clusters of spins simultaneously [49].
Comparative studies of these algorithms in the 2D and 3D Ising models at criticality found that "the new algorithm decorrelates faster in all cases and gains about an order of magnitude on a 64³ lattice" [49]. Remarkably, the Wolff algorithm exhibits practically negligible critical slowing down, possibly completely absent in three dimensions [49], representing a significant advancement for studying critical phenomena.
Ising Models in Biological Applications
Ising models have found applications beyond traditional statistical physics, particularly in modeling biological systems. For DNA flexibility, researchers have developed models that "combine a harmonic coupled oscillator description with an Ising model to include all possible substates of a DNA duplex" [4]. This approach gives improved prediction of the equilibrium distribution of DNA helical variables and better agreement with experimental protein-DNA binding affinities compared to unimodal harmonic models [4].
Table 2: Performance Comparison of DNA Flexibility Models
| Model Type | Nearest-Neighbor Coupling | Multimodality | Accuracy (kcal/mol/degree) | Experimental Agreement |
|---|---|---|---|---|
| Unimodal Harmonic Model | Yes | No | Baseline | Moderate |
| Multimodal Monte Carlo | Partial | Yes | +0.02 | Good |
| Multivariate Ising Model | Yes | Yes | +0.03 | Excellent |
The table above compares different approaches to modeling DNA flexibility, demonstrating that the multivariate Ising model, which incorporates both nearest-neighbor coupling and multimodality, provides the most accurate description of DNA deformability [4].
Comparative Analysis: Performance Metrics and Applications
Quantitative Comparison of Sampling Efficiency
When comparing enhanced sampling methods, several metrics are essential for evaluating performance:
- Autocorrelation Time: Measures how quickly configurations become decorrelated; lower values indicate better sampling [49]
- Wall-clock Acceleration: Practical speedup in real time achieved by enhanced methods [47]
- Convergence Rate: Speed at which equilibrium properties approach their true values
- Transferability: Ability of methods trained on one system to perform well on unseen systems [47]
For the Ising model, cluster algorithms reduce autocorrelation times by approximately an order of magnitude compared to local update methods [49]. In MD simulations, methods like Timewarp can accelerate sampling by effectively simulating 10⁵-10⁶ fs steps while maintaining correct equilibrium distributions [47].
Application-Specific Considerations
The choice of enhanced sampling method depends strongly on the specific application:
- Protein Folding: Generalized-ensemble algorithms like MUCA and REM have proven effective for studying folding landscapes [46]
- Protein-DNA Interactions: Ising-based models successfully capture sequence-dependent DNA deformability relevant for protein binding [4]
- RNA Dynamics: Integration of experimental data with MD simulations improves ensemble generation for flexible RNAs [50]
- Critical Phenomena: Cluster MC algorithms are indispensable for studying phase transitions in Ising models [49]
A significant advantage of modern machine learning approaches is their transferability; for instance, Timewarp "generalises to unseen small peptides at all-atom resolution, exploring their metastable states" [47], suggesting potential for broader application across molecular systems.
Experimental Protocols and Methodologies
Implementation of Multicanonical Algorithm
The multicanonical algorithm follows these key steps [46]:
- Initialization: Define the energy range of interest and initial weights for flat energy distribution
- Iterative Weight Optimization: Perform short simulations to refine weights until energy distribution becomes approximately flat
- Production Run: Conduct extended simulation with optimized weights
- Reweighting: Use weighted histogram analysis to recover canonical distributions at temperatures of interest
The algorithm requires careful tuning but provides comprehensive sampling of energy landscapes, making it particularly valuable for biomolecular systems with complex rugged energy landscapes.
Implementation of Cluster Monte Carlo for Ising Models
The Wolff single-cluster algorithm implementation involves [49]:
- Seed Selection: Randomly select a initial spin as cluster seed
- Cluster Growth: Examine neighboring spins; add to cluster with probability P = 1 - exp(-2βJ) if aligned with seed spin
- Cluster Flip: Flip all spins in the completed cluster
- Repetition: Repeat process for updated configuration
This algorithm dramatically reduces critical slowing down compared to local update methods, with studies showing "critical slowing down is practically negligible and possibly completely absent in three dimensions" [49].
Protocol for Validating MD Simulations
Comprehensive validation of MD simulations against experimental data involves [48]:
- System Preparation: Obtain initial coordinates from experimental structures (e.g., PDB)
- Simulation Setup: Employ multiple force fields and simulation packages with consistent settings
- Triplicate Sampling: Perform simulations in triplicate to assess variability
- Comparison Metrics: Calculate experimental observables (NMR, SAXS) from simulations
- Statistical Analysis: Quantify agreement between simulation and experiment
This protocol highlights that "correspondence between simulation and experiment does not necessarily constitute a validation of the conformational ensemble" [48], as different ensembles may yield similar averages.
Integration of Simulations with Experimental Data
Strategies for Experimental Integration
Combining molecular simulations with experimental data enhances both the validation and refinement of computational models. Several integration strategies have emerged [50]:
- Quantitative Validation: Using experimental data to assess and select force fields
- Qualitative Restraints: Employing experimental data to generate initial models or restrain simulations
- Maximum Entropy Methods: Reweighting simulation ensembles to match experimental data
- Force Field Improvement: Using experimental data to refine force field parameters
These approaches are particularly valuable for RNA systems, where "MD simulations can be fruitfully applied also when current force fields are not predictive, by suitable integration of experimental data" [50].
Workflow for Experimental Data Integration
The following diagram illustrates the iterative process of integrating experimental data with molecular simulations:
This workflow demonstrates how experimental data can be used to validate, refine, and improve molecular simulations, creating a cycle of iterative improvement that enhances both the accuracy and interpretability of computational models [50].
Research Reagent Solutions: Essential Tools and Methods
Table 3: Essential Research Reagents and Computational Tools
| Tool/Method | Type | Primary Function | Key Applications |
|---|---|---|---|
| AMBER | MD Software Package | Biomolecular MD simulations | Protein folding, drug binding |
| GROMACS | MD Software Package | High-performance MD simulations | Large biomolecular systems |
| CHARMM36 | Force Field | Empirical energy function | Protein and lipid simulations |
| ff99SB-ILDN | Force Field | Optimized for proteins | Protein folding and dynamics |
| Multicanonical Algorithm | Enhanced Sampling | Flat energy distribution | Protein folding landscapes |
| Replica-Exchange MD | Enhanced Sampling | Temperature-based sampling | Biomolecular stability |
| Swendsen-Wang Algorithm | Cluster MC | Collective spin updates | Ising model critical behavior |
| Wolff Algorithm | Cluster MC | Single-cluster updates | Efficient critical sampling |
| Timewarp | ML-Accelerated MD | Normalizing flow proposals | Transferable acceleration |
| WHAM | Analysis Tool | Reweighting ensembles | Free energy calculations |
Enhanced sampling methods have dramatically improved our ability to explore complex energy landscapes in both molecular dynamics and Monte Carlo simulations. For Ising models, cluster algorithms like the Wolff method have essentially solved the critical slowing down problem, enabling efficient study of phase transitions. In biomolecular simulations, generalized-ensemble algorithms and machine learning approaches have significantly expanded the accessible timescales and system sizes, though challenges remain in force field accuracy and validation.
The integration of experimental data with simulations has emerged as a powerful paradigm for validating and refining computational models, particularly for complex systems like RNA where force fields remain imperfect. As methods like Timewarp demonstrate transferability between systems, we move closer to general, transferable algorithms for accelerating molecular simulations across diverse scientific domains.
The continued development and comparison of enhanced sampling methods will be crucial for addressing increasingly complex questions in molecular biophysics, materials science, and drug discovery, where thorough sampling of configuration space remains a fundamental challenge.
Molecular dynamics (MD) simulation has become an indispensable computational tool for investigating the structural dynamics of biomolecules, including proteins and RNA, at an atomistic scale. The accuracy of these simulations is fundamentally governed by the molecular mechanics force fields (FFs)—empirical potential energy functions and parameters used to calculate the potential energy of a system as a function of its atomic coordinates. Force fields approximate complex quantum mechanical interactions with simpler mathematical functions, making simulations of large biological systems computationally tractable. A typical biomolecular force field consists of both bonded terms (for bond stretching, angle bending, and torsion rotations) and non-bonded terms (for van der Waals and electrostatic interactions) [51]. The physical realism of simulations predicting protein folding, drug binding, or RNA dynamics hinges on the precision and balance of these force field parameters.
Despite their widespread use, a significant challenge persists: most classical force fields were originally developed and parameterized for stable, globular proteins, and their performance can vary significantly when applied to other biomolecular systems such as intrinsically disordered proteins (IDPs) or nucleic acids [52] [53]. This article provides a comparative guide to the performance of modern force fields, objectively assessing their accuracy against experimental data. Furthermore, we frame this evaluation within the context of theoretical models for protein folding, particularly Ising-like models, which offer a simplified but powerful lens through which to understand the relationship between force field parameterization and the emergent behavior of biomolecules.
Force Field Fundamentals and Parameterization Philosophy
Mathematical Underpinnings of Force Fields
The potential energy function of a force field is a sum of terms describing various atomic interactions. For Class 1 force fields—which include widely used families like AMBER, CHARMM, and OPLS—the energy is calculated as follows [51]:
( U(\vec{r}) = \sum{U{bonded}}(\vec{r}) + \sum{U{non-bonded}}(\vec{r}) )
The bonded interactions are further decomposed into:
- Bond stretching: ( V{Bond} = kb(r{ij}-r0)^2 )
- Angle bending: ( V{Angle} = k\theta(\theta{ijk}-\theta0)^2 )
- Torsion rotation: ( V{Dihed} = k\phi(1+cos(n\phi-\delta)) )
The non-bonded interactions comprise:
- van der Waals forces: Typically described by the Lennard-Jones potential: ( V_{LJ}(r)=4\epsilon\left[\left(\frac{\sigma}{r}\right)^{12}-\left(\frac{\sigma}{r}\right)^{6}\right] )
- Electrostatics: Governed by Coulomb's law: ( V{Elec}=\frac{q{i}q{j}}{4\pi\epsilon{0}\epsilon{r}r{ij}} )
The accurate parametrization of these terms, particularly the partial atomic charges ((q)), torsion potentials ((k_\phi, n, \delta)), and van der Waals parameters ((\epsilon, \sigma)), is crucial for generating physically realistic simulations [54] [51].
Parameterization and Validation Philosophies
Force field development involves a delicate balancing act. Parameters are often initially fit to reproduce quantum mechanical calculations on small model compounds and are then adjusted to agree with experimental data, such as crystallographic structures and thermodynamic properties [55]. A critical concept is that a well-parameterized force field must be balanced, meaning it can accurately reproduce molecular behavior at the atomic scale while also capturing macroscopic thermodynamic quantities [54].
Validation is a crucial final step. The utility of a parameter set is evaluated by its ability to reproduce experimental measures outside its training set. For protein force fields, this often involves testing their ability to fold proteins into native conformations or to reproduce experimental NMR observables like chemical shifts and scalar couplings [54] [55]. This process is complicated by the fact that errors in different parameters can cancel each other out, allowing an imperfect force field to still fit target data—a phenomenon known as parameter correlation [54].
Comparative Performance of Modern Force Fields
Performance Benchmarking for Globular Proteins
Systematic evaluations of force fields against extensive NMR data have revealed significant differences in their performance. One comprehensive study benchmarked 55 combinations of 11 force fields and 5 water models against 524 NMR measurements (chemical shifts and J-couplings) across dipeptides, tripeptides, tetra-alanine, and the protein ubiquitin [55].
Table 1: Force Field Performance for Globular Proteins (Ubiquitin) and Peptides
| Force Field | Overall Accuracy (χ²) | Strengths | Key Limitations |
|---|---|---|---|
| ff99sb-ildn-nmr | Highest | Excellent agreement with NMR data on both small peptides and full proteins [55]. | Accuracy approaches limit of NMR prediction uncertainty [55]. |
| ff99sb-ildn-phi | Highest | Excellent performance across system sizes; modified backbone torsion [55]. | Moderate errors in 3J(HαC′) and 3J(HNHα) couplings [55]. |
| CHARMM27 | Intermediate | Reasonable agreement on short timescales [56]. | Conformational drift in long simulations reduces accuracy [56]. |
| ff03/ff03* | Intermediate | -- | Substantial conformational drift in long simulations [56]. |
| OPLS-AA | Lower | -- | Significant conformational drift in multi-microsecond simulations [56]. |
The study concluded that force fields combining recent side-chain and backbone torsion modifications—specifically ff99sb-ildn-nmr and ff99sb-ildn-phi—achieved the highest accuracy, with calculation errors comparable to the uncertainty in the experimental comparison itself [55]. This suggests that extracting further force field improvements from NMR data may require more accurate J-coupling and chemical shift prediction models.
Benchmarking for Intrinsically Disordered Proteins
IDPs lack a stable folded structure and sample a diverse ensemble of conformations, presenting a distinct challenge for force fields parameterized for globular proteins. A 2023 study benchmarked 13 force fields by simulating the R2 region of the FUS-LC domain, an IDP implicated in ALS, and scoring them based on the compactness (radius of gyration, Rg), secondary structure propensity, and intra-peptide contact maps of the resulting ensembles [52].
Table 2: Force Field Performance for Intrinsically Disordered Proteins (R2-FUS-LC)
| Force Field | Final Score (Group) | Rg Preference | Contact Map Accuracy |
|---|---|---|---|
| c36m2021s3p | Top (*) | Balanced sampling of compact and extended states [52]. | High |
| a99sb4pew | Top (*) | Bias towards more compact conformations [52]. | Medium |
| a19sbopc | Top (*) | Balanced sampling of compact and extended states [52]. | Medium |
| c36ms3p | Top (*) | Bias towards flexible, extended conformations [52]. | Medium |
| a14sb3p | Bottom (#) | Strong bias, poor fit to reference Rg's [52]. | Relatively good |
| c27s3p | Bottom (#) | Poor performance across all Rg measures [52]. | Low |
The results demonstrated a clear trend: CHARMM FFs generally produced more extended conformations, while AMBER FFs tended to generate more compact states [52]. The top-performing force field, CHARMM36m2021 with the mTIP3P water model, was identified as the most balanced for studying the R2-FUS-LC system. Furthermore, the choice of water model proved critical; the standard TIP3P model was found to cause an artificial structural collapse in disordered regions, whereas the TIP4P-D water model significantly improved reliability when combined with modern protein force fields [53].
Performance in RNA Simulations
RNA molecules possess unique structural motifs and a high density of negative charges, making them particularly difficult to simulate accurately. Comparisons between simulation and experimental data such as NMR and SAXS have been essential for identifying force field deficiencies and guiding improvements [57] [50]. For instance, simulations of RNA tetraloops and junctions have been used to test corrections to non-bonded terms, where RNA-RNA and RNA-solvent interactions are tuned separately to better match experimental data [50]. These studies highlight that no single force field is universally superior for all RNA systems, and validation against system-specific experimental data is often necessary.
Integration with Experiments and Ising-like Theoretical Models
Experimental Integration Strategies
Experimental data is not only used for final validation but is increasingly integrated directly into the simulation process to improve accuracy. The strategies for integrating MD simulations with experimental data can be categorized as follows [57] [50]:
- Validation and Force Field Selection: Experimental data is used as a quantitative benchmark to select the most trustworthy force field for a given system.
- Ensemble Refinement: Experimental data is used to improve simulated ensembles, either through qualitative restraints or quantitative methods like the maximum entropy principle, which reweights simulation trajectories to match experimental observables.
- Force Field Improvement: Experimental data is used to directly refine force field parameters, creating improved and transferable models for future simulations.
Table 3: Experimental Techniques for Validating and Refining MD Simulations
| Experimental Technique | Spatio-Temporal Resolution | Key Validated Observables |
|---|---|---|
| Nuclear Magnetic Resonance (NMR) | Atomic, µs-ms | Chemical shifts, J-couplings, Residual Dipolar Couplings (RDCs), Relaxation parameters [57] [55]. |
| Small-Angle X-Ray Scattering (SAXS) | Molecular, ns-ms | Radius of gyration (Rg), overall shape and compactness [52] [57]. |
| Cryo-Electron Microscopy (Cryo-EM) | Near-atomic, ms+ | Low-resolution 3D density maps for large complexes [57]. |
| Single-Molecule FRET | Nanometer, ms | Inter-dye distances and dynamics [57]. |
Connection to Ising-like Theoretical Models
A powerful, simplified approach to understanding protein folding is the use of Ising-like theoretical models. These models are "native-centric," meaning they consider only interactions present in the protein's final, folded structure, effectively assuming a "perfectly funneled" energy landscape [58]. In one such model, each amino acid residue can exist in one of two states—native (n) or coil (c)—and folding proceeds by the growth of a limited number of segments of contiguous native residues [58].
All-atom MD simulations have been used to test the key assumptions of these models. For the villin headpiece subdomain, analysis of MD transition paths (the segments where folding/unfolding actually occurs) revealed that structure indeed grows in only a few regions of the amino acid sequence, strongly supporting a major simplifying assumption of the Ising-like model [58]. This synergy between detailed MD and simplified models is mutually beneficial: MD simulations can validate the assumptions of theoretical models, and the success of the theoretical models helps identify the essential physical principles governing folding, which can in turn inform future force field development.
The following diagram illustrates the workflow for validating force fields and their connection to theoretical models.
Diagram 1: Workflow for force field development, validation, and integration with theoretical models.
Table 4: Key Software, Force Fields, and Resources for Biomolecular MD
| Tool/Resource | Type | Primary Function | Relevance to Challenge |
|---|---|---|---|
| AMBER | Software Suite | Simulation & analysis of biomolecules [54]. | Provides force fields (ff99SB-ILDN, ff19SB) and tools for running/test simulations. |
| CHARMM | Software Suite | Simulation & analysis of biomolecules [54]. | Provides force fields (CHARMM36, CHARMM36m) and simulation tools. |
| GROMACS | Software Suite | High-performance MD simulation engine [55]. | Widely used for benchmarking due to its speed and efficiency. |
| LAMMPS | Software Suite | General-purpose MD simulator [59]. | Used for complex systems, e.g., with ReaxFF for material interfaces. |
| ff99sb-ildn-nmr | Force Field | Optimized for proteins vs. NMR data [55]. | Top performer for folded proteins; benchmark against this for accuracy. |
| CHARMM36m | Force Field | Optimized for folded & disordered proteins [52]. | Top performer for IDPs; good balance of accuracy for structured/disordered regions. |
| TIP4P-D | Water Model | Modified water model for disordered proteins [53]. | Critical for preventing collapse of IDPs; often paired with CHARMM36m. |
| ReaxFF | Force Field | Reactive force field for chemical reactions [59]. | Essential for simulating processes like adsorption or bond formation. |
The quest for highly accurate and universally applicable biomolecular force fields is ongoing. Systematic benchmarking reveals that while modern force fields like ff99sb-ildn-nmr and CHARMM36m achieve remarkable accuracy for their target systems (folded proteins and IDPs, respectively), no single force field is optimal for all biomolecules and scenarios. The choice of force field must therefore be guided by the specific system under investigation, with careful validation against available experimental data.
Future improvements will likely come from several fronts. First, the development of polarizable force fields (Class 3), which allow the molecular charge distribution to respond to the local environment, promises a more physical description of electrostatics [51]. Second, the integration of machine learning with MD simulations is emerging as a powerful tool to either accelerate simulations or directly learn accurate potential energy surfaces from quantum mechanical data [59]. Finally, the continued close dialogue between all-atom simulations, simplified theoretical models, and a growing body of high-resolution experimental data will ensure that force field development remains grounded in the essential physics of biomolecular folding and function.
Molecular dynamics (MD) simulations are indispensable in computational chemistry, biophysics, and materials science, enabling the study of physical atomic and molecular movements over time [60]. Similarly, the study of Ising-like theoretical models provides fundamental insights into phase transitions and critical phenomena in statistical physics. Both fields face escalating computational demands. The current landscape is defined by a "compute crisis" characterized by unsustainable energy consumption, prohibitive training costs, and the approaching limits of conventional CMOS scaling [61]. This guide objectively compares the performance of specialized hardware—GPUs, ASICs, and emerging architectures—for accelerating these computational workloads, providing researchers with experimentally-validated data to inform hardware selection and methodological approaches.
Hardware Landscape: A Comparative Analysis
GPU Dominance in General-Purpose Acceleration
Graphics Processing Units (GPUs) have become the cornerstone of modern high-performance computing due to their massively parallel architecture. For MD simulations, GPUs dramatically accelerate calculations, allowing for the simulation of larger systems or longer timescales [62].
Performance Characteristics:
- Architecture: Contain thousands of computing cores (e.g., RTX 4090: 16,384 CUDA cores) for massive parallel processing [63] [64].
- Memory: High-bandwidth memory (e.g., H100: 3.35 TB/s bandwidth with 80GB HBM3) crucial for large model training and simulation [64].
- Precision: Modern GPUs support mixed-precision operations, balancing speed and accuracy [62].
For Ising model research, which involves extensive Monte Carlo simulations and statistical sampling, GPUs offer tremendous advantages through parallel computation of spin updates and energy calculations.
ASICs: Specialized Efficiency
Application-Specific Integrated Circuits (ASICs) represent the extreme of specialized computing, designed for optimal performance on specific tasks.
Performance Characteristics:
- Efficiency: Extreme performance and power efficiency for dedicated tasks [63]. Google's TPU v5 reduces per-unit computing costs by 70% compared to general GPUs [63].
- Throughput: In Bitcoin mining, ASIC hashrate per watt is over 2 million times that of GPUs [63].
- Limitations: Fixed functionality with high initial development costs (approximately $50 million for 7nm process) [63].
Table 1: ASIC vs GPU Performance Metrics Comparison
| Metric | ASIC | GPU | Advantage |
|---|---|---|---|
| Single-task Performance | 100% | 10-20% | ASIC |
| Power Efficiency | 90% | 30% | ASIC |
| Development Cost | $50M+ | $0 | GPU |
| Flexibility | Extremely Low | Extremely High | GPU |
| Lifespan | 2-3 years | 4-6 years | GPU |
| Application Range | Single | Broad | GPU |
Physics-Based ASICs: An Emerging Paradigm
A transformative approach comes from physics-based ASICs that directly harness intrinsic physical dynamics for computation rather than enforcing idealized digital abstractions [61]. By relaxing conventional constraints like enforced statelessness, unidirectionality, determinism, and synchronization, these devices aim to operate as exact realizations of physical processes [61]. This approach is particularly promising for Ising model simulations, where the hardware physics can directly mirror the mathematical formalism of the physical system being studied.
Experimental Performance Data
Molecular Dynamics Benchmarking
Comprehensive benchmarking of AMBER 24 across NVIDIA GPU architectures provides critical performance data for hardware selection.
Table 2: AMBER 24 Single GPU Performance (ns/day) Across Various Molecular Systems
| GPU Model | STMV (1M atoms) | Cellulose (408K atoms) | FactorIX (90K atoms) | DHFR (23K atoms) | Myoglobin GB (2.5K atoms) |
|---|---|---|---|---|---|
| RTX 5090 | 109.75 | 169.45 | 529.22 | 1655.19 | 1151.95 |
| RTX 6000 Ada | 70.97 | 123.98 | 489.93 | 1697.34 | 1016.00 |
| H100 PCIe | 74.50 | 125.82 | 410.77 | 1532.08 | 1094.57 |
| B200 SXM | 114.16 | 182.32 | 473.74 | 1513.28 | 1020.24 |
| GH200 Superchip | 101.31 | 167.20 | 191.85 | 1323.31 | 1159.35 |
Key observations from AMBER benchmarking:
- In larger systems (>100,000 atoms), newer architectures like Blackwell show significant performance gains [65].
- For cost-effective performance with smaller systems, the RTX PRO 4500 Blackwell matches higher-end GPUs at a fraction of the cost [65].
- Data center GPUs like B200 SXM and H100 PCIe offer top performance but at a premium price that may not be justified for MD alone [65].
AI Accelerator Performance
For machine learning workloads increasingly integrated with both MD and Ising model research, specialized AI accelerators show remarkable performance.
Table 3: AI Training GPU Comparison for Large Language Models
| GPU Model | Architecture | VRAM | Memory Bandwidth | FP32 Performance | Ideal Model Size |
|---|---|---|---|---|---|
| RTX 4090 | Ada Lovelace | 24GB GDDR6X | ~1 TB/s | 82.6 TFLOPS | <36B parameters |
| A100 | Ampere | 80GB HBM2e | 2 TB/s | ~19.5 TFLOPS | <70B parameters |
| H100 | Hopper | 80GB HBM3 | 3.35 TB/s | ~60 TFLOPS | >70B parameters |
| H200 | Hopper | 141GB HBM3e | 4.8 TB/s | Similar to H100 | >100B parameters |
The H100 delivers 2-3x faster training than A100 with 67% more memory bandwidth, though both deliver strong performance for most AI workloads [64].
Machine Learning Integration in Molecular Dynamics
ML-IAP-Kokkos Interface Methodology
The ML-IAP-Kokkos interface represents a significant advancement in integrating PyTorch-based machine learning interatomic potentials (MLIPs) with the LAMMPS MD package [66]. This interface, developed through collaboration between NVIDIA, Los Alamos National Lab, and Sandia National Lab, enables scalable MD simulations by bridging Python and C++/Kokkos LAMMPS, ensuring end-to-end GPU acceleration [66].
Experimental Protocol:
- Environment Setup: LAMMPS must be built with Kokkos, MPI, and ML-IAP support, alongside a Python environment with PyTorch and trained MLIP model [66].
- Interface Development: Researchers implement the
MLIAPUnifiedabstract class from LAMMPS, defining thecompute_forcesfunction to infer pairwise forces and energies using data passed from LAMMPS [66]. - Model Serialization: The model object is saved using PyTorch's save function, creating a "pickled" Python object that LAMMPS can load [66].
- Simulation Execution: LAMMPS runs with Kokkos on GPUs, calling the Python interpreter during simulation to execute the native Python model [66].
This approach supports message-passing MLIP models, utilizing LAMMPS's built-in communication capabilities for efficient data transfer between GPUs—crucial for large-scale simulations [66].
Precision Considerations in Computational Workflows
A critical methodological consideration is precision selection, which significantly impacts both performance and accuracy across MD and Ising model simulations.
Quick Diagnostic Checks for Precision Requirements:
- Your code defaults to double precision and warns or fails with mixed precision [62].
- Published benchmarks or documentation specify "double precision only" or "accuracy requires FP64" [62].
- Results drift, blow up, or fail validation when moving from double to single/mixed precision [62].
Methodological Guidance:
- Molecular Dynamics: GROMACS, AMBER, NAMD, and LAMMPS have mature GPU paths using mixed precision effectively [62].
- Double Precision-Dominated Codes: Quantum ESPRESSO, VASP, and similar codes often mandate true double precision (FP64) and benefit from high FP64 throughput hardware [62].
- AI/ML Workloads: NVIDIA Tensor Cores support FP64, TF32, FP32, FP16, INT8, and FP8, reducing memory usage while maintaining accuracy for LLMs [64].
Diagram 1: Hardware Selection Workflow for Computational Research. This diagram outlines the decision process for selecting appropriate computing hardware based on algorithmic needs and precision requirements.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Computational Research Tools and Platforms
| Tool Category | Specific Solutions | Function & Application |
|---|---|---|
| MD Software Suites | AMBER, GROMACS, NAMD, LAMMPS | Specialized molecular dynamics simulation packages with GPU acceleration support [62] [60] [65]. |
| ML-MD Integration | ML-IAP-Kokkos Interface | Bridges PyTorch-based machine learning interatomic potentials with LAMMPS for scalable MD simulations [66]. |
| Consumer GPUs | NVIDIA RTX 4090/5090 Series | Cost-effective acceleration for mixed-precision MD simulations and models up to 36B parameters [64] [65]. |
| Data Center GPUs | NVIDIA H100/H200, A100 | High-performance acceleration for large-scale MD and AI training workloads requiring FP64 precision [64] [65]. |
| Workstation GPUs | RTX 6000/5000 Ada, PRO Blackwell Series | Balanced performance for professional research environments with larger VRAM capacities [65]. |
| Specialized Accelerators | Physics-Based ASICs, FPGAs | Emerging architectures offering potential orders-of-magnitude efficiency gains for specific physical simulations [61] [67]. |
| Cloud Platforms | Hyperbolic, AWS, Azure | Provides access to high-end computational resources without capital investment [64]. |
The computational landscape for molecular dynamics and Ising model research is rapidly evolving. Key trends include the widespread adoption of GPU acceleration across diverse fields, continued innovation in precision capabilities, and the emergence of novel architectures like physics-based ASICs [62] [61]. These advances promise to enable larger simulations, faster training times, and more accurate results while managing costs.
For MD simulations, benchmarking clearly indicates that consumer GPUs like the RTX 4090 and RTX 5090 offer exceptional value for most workloads, while data center GPUs provide necessary capabilities for the largest simulations [65]. For Ising model research, the emerging paradigm of physics-based ASICs presents particularly exciting opportunities by directly mapping computational physics to hardware physics [61].
Diagram 2: Computational Research Hardware Ecosystem. This diagram illustrates the relationship between research methodologies, hardware platforms, and the integrating role of machine learning in advancing scientific discovery.
The integration of machine learning with both MD simulations and statistical physics models represents perhaps the most significant methodological shift, enabling new research approaches that leverage the complementary strengths of physical simulation and data-driven modeling. As hardware continues to evolve, researchers who strategically leverage these advances while maintaining rigorous methodological standards will be best positioned to push the boundaries of computational science.
Long-range interactions, where components of a system influence one another over substantial distances without direct contact, represent a fundamental challenge across scientific disciplines. In biomolecular systems, this phenomenon is exemplified by allostery, a quintessential mechanism of biological regulation where a perturbation at one site, such as ligand binding, affects the functional dynamics of a distant active site [68]. Similarly, in magnetic systems, long-range spin correlations dictate collective phenomena such as magnetic ordering and phase transitions [69]. Despite the stark differences in their physical manifestations—information transmission in proteins versus spin alignment in materials—these systems share a common mathematical language for describing their cooperative behaviors.
The Ising model, a cornerstone of statistical physics, provides a powerful unifying framework for investigating these seemingly disparate phenomena. Originally developed to describe ferromagnetism, its application has expanded to become an indispensable tool for conceptualizing and quantifying long-range interactions in biology. This guide objectively compares the performance of detailed, all-atom Molecular Dynamics (MD) simulations with simpler, Ising-like theoretical models in navigating the complex landscape of long-range interactions. We focus on their respective capacities to predict system behavior, capture thermodynamic properties, and scale computationally, providing researchers with a clear basis for selecting the appropriate tool for their specific investigation.
Theoretical Frameworks: A Tale of Two Approaches
Molecular Dynamics Simulations: An Atomistic Bottom-Up View
Molecular Dynamics simulations operate on a first-principles approach, calculating the trajectories of every atom in a system by numerically solving Newton's equations of motion. The fundamental Hamiltonian is atomistically detailed, and the methodology aims to capture the full complexity of a system's dynamics without prior assumptions about the nature of its collective states [70] [15].
- Key Strengths: The primary strength of MD is its high resolution. It provides an unbiased, atomic-level view of dynamics, allowing for the discovery of unexpected pathways and states. It can explicitly model the effects of solvent, ions, and specific chemical modifications, making it a powerful tool for detailed mechanistic studies when the atomistic details of a interaction are critical [15].
- Inherent Challenges: This high resolution comes at a tremendous computational cost, which severely limits the accessible timescales and system sizes. Furthermore, the "curse of dimensionality" means that as system size increases, the number of globally distinct metastable states can grow exponentially, making it infeasible to sample each configuration and the transitions between them [70]. This is a fundamental bottleneck for studying large biomolecular complexes where long-range interactions are paramount.
Ising-Like Theoretical Models: A Coarse-Grained Top-Down View
Ising-like models adopt a coarse-grained, top-down perspective. They abstract a complex system into a network of discrete, interacting components, or "spins." In the Allosteric Ising Model (AIM) formulation for proteins, these components can represent coarse-grained structural units like helices, sheets, or entire domains, each capable of occupying a limited number of discrete states (e.g., "on" or "off," "active" or "inactive") [68] [71].
The Hamiltonian for such a system is given by: [ H = -\sum{i=1}^{n} \sum{j=1}^{n} \frac{J{ij} \sigmai \sigmaj}{2} - \sum{i=1}^{n} hi \sigmai ] where ( \sigmai ) represents the state of the ( i )-th component, ( J{ij} ) is the coupling constant between components ( i ) and ( j ) that encodes the strength and nature (ferromagnetic/positive or antiferromagnetic/negative) of their interaction, and ( h_i ) represents the influence of an external field on the ( i )-th component [68].
- Key Strengths: The greatest advantage of Ising models is their computational efficiency and conceptual clarity. By drastically reducing the system's degrees of freedom, they allow for the exploration of long-timescale behavior and the systematic investigation of how interaction networks (( J_{ij} )) give rise to emergent phenomena like allostery. They directly connect local interactions to global thermodynamics [68] [70].
- Inherent Challenges: This approach requires a priori knowledge or assumptions about the system's relevant states and their connectivity. The model's accuracy is contingent on the quality of the coarse-graining scheme and the parameterization of the ( J_{ij} ) couplings, which can be derived from theory, experiment, or underlying MD simulations [71].
Comparative Performance Analysis
Table 1: Quantitative Comparison of MD Simulations and Ising-like Models for Studying Long-Range Interactions.
| Performance Metric | Molecular Dynamics (MD) Simulations | Ising-like Theoretical Models |
|---|---|---|
| Spatial Resolution | Atomic-scale (Ångström) [15] | Coarse-grained (domains, spins) [68] |
| Typical Timescale | Nanoseconds to microseconds [39] | Effectively infinite (equilibrium sampling) |
| Computational Cost | Extremely high (CPU/GPU-intensive) [70] | Very low (often analytical or quick numerical solutions) |
| Treatment of Long-Range Interactions | Explicitly modeled via force fields (electrostatics, etc.) [15] | Encoded in the coupling matrix (J_{ij}) [68] |
| Handling of System Size | Poor scaling; exponential state growth [70] | Excellent scaling with number of components [70] |
| Primary Output | Atomistic trajectories, time-dependent properties | Equilibrium probabilities, phase diagrams, order parameters [69] |
| Key Advantage | Discovery of unanticipated pathways and states | Identifies fundamental principles and thermodynamic limits |
| Best Suited For | Detailed mechanism studies; specific ligand binding | Exploring network design principles; mapping phase behavior |
Table 2: Application-Based Comparison for Specific Biological and Magnetic Problems.
| Research Application | MD Simulation Performance & Findings | Ising Model Performance & Findings |
|---|---|---|
| Protein Allostery | Can reveal specific atomic rearrangements but may struggle to sample all functionally relevant states for large proteins [70]. | AIMs successfully quantify allosteric efficacy (\alpha) and map signaling pathways in GPCRs like the dopamine D2 receptor [68]. |
| Protein Folding | Microsecond all-atom simulations of villin subdomain folding are possible [71]. | Ising-like model for villin subdomain produced a similar distribution of folding mechanisms to MD, with far less computational effort [71]. |
| Spin Lattice Dynamics | Not typically applied to pure magnetic systems. | Accurately predicts phenomena like dynamic phase transitions, compensation temperatures, and magnetocaloric effects in 2D lattices [69]. |
| Large Complexes (e.g., Viruses) | All-atom simulation is possible but parameterizing a global kinetic model from the data is currently infeasible [70]. | Markov Field Models (MFMs), an Ising generalization, are proposed as the scalable solution for modeling domain-level dynamics [70]. |
The data reveals a clear trade-off. MD simulations excel in providing high-resolution, mechanistic insights for systems of manageable size, where atomistic detail is non-negotiable. In contrast, Ising-like models shine in their ability to elucidate fundamental principles, explore large-scale thermodynamic behavior, and handle system sizes that are currently prohibitive for all-atom approaches. The study on protein folding is particularly telling: a simple Ising-like model was able to recapitulate the distribution of folding mechanisms observed in extensive MD simulations, demonstrating that the essential physics of that process was captured by the coarse-grained model [71].
Experimental Protocols and Methodologies
Protocol for Ising Model Analysis of Allostery
This protocol outlines the steps for building and analyzing an Allosteric Ising Model (AIM) to study a biomolecular system, based on the methodology described by [68].
- System Decomposition: Decompose the protein or complex of interest into
ndiscrete, coherent structural components (e.g., domains, secondary structure elements). The choice of components depends on the system and the allosteric question being asked. - State Definition: Assign each component
ia discrete state variableσ_i(e.g.,+1for "active,"-1for "inactive"). - Parameterization: Define the parameters of the Ising Hamiltonian:
- Local Fields (
h_i): Represent the intrinsic preference of a component for one state over another, independent of its neighbors. This can be informed by knowledge of a component's stability. - Coupling Constants (
J_ij): Represent the interaction energy between components. Strong positiveJ_ijfavors alignment (both +1 or both -1), while negativeJ_ijfavors anti-alignment. These can be derived from statistical analysis of structural databases, MD simulations, or experimental data.
- Local Fields (
- Compute the Partition Function: Calculate
Z = Σ exp(-βH), summing over all possible configurations of the system. This is the core statistical mechanical task from which all equilibrium properties are derived. - Calculate Observable Quantities:
- Allosteric Efficacy (
α): Compute the change in the equilibrium constant for a global functional shift (e.g., active vs. inactive) upon a perturbation (e.g., ligand binding at a specific site). This is directly related to the free energy difference via-RT log(α) = ΔG[68]. - Correlation Functions: Calculate
<σ_i σ_j>to quantify the degree of cooperativity between distant componentsiandj.
- Allosteric Efficacy (
- Validation: Compare model predictions, such as the magnitude of the allosteric effect or the identity of key pathway components, with experimental data (e.g., binding assays, FRET measurements, mutational studies).
Protocol for MD Simulations with Markov State Modeling (MSM)
This protocol describes a modern MD approach to study long-timescale dynamics, such as allostery or folding, by combining many short simulations [70].
- System Preparation: Construct the all-atom model of the protein in its relevant environment (explicit solvent, ions, ligands). Equilibrate the system using standard MD protocols.
- Enhanced Sampling: Run a large ensemble of hundreds to thousands of relatively short, independent MD simulations, often initiated from different conformations. Biasing potentials (e.g., metadynamics) can be used to encourage exploration of specific reaction coordinates.
- Dimensionality Reduction: Project the high-dimensional trajectory data (atomic coordinates) into a lower-dimensional space of relevant collective variables (CVs) using algorithms like t-Distributed Stochastic Neighbor Embedding (t-SNE) or Principal Component Analysis (PCA) [39].
- Clustering: Group the molecular conformations from the projected space into a finite number of discrete states or "microstates" based on structural similarity (e.g., using k-means clustering).
- MSM Construction: Build a Markov State Model (MSM)—a matrix of transition probabilities between the identified microstates over a defined lag time
τ. This model must satisfy the Markov assumption, meaning transitions depend only on the current state. - Model Validation: Validate the MSM by checking its self-consistency (e.g., implied timescales are independent of the lag time
τ) and, if possible, by comparing its predictions to experimental observables. - Analysis: Use the validated MSM to compute kinetic rates between functional states, identify metastable states, and extract mechanistic pathways of allosteric communication or folding.
The following workflow diagram illustrates the starkly different logical approaches of these two methodologies.
Table 3: Key Computational and Theoretical "Reagents" for Studying Long-Range Interactions.
| Tool / Resource | Function / Description | Primary Use Case |
|---|---|---|
| Allosteric Ising Model (AIM) [68] | A coarse-grained model that maps a biomolecule onto a network of interacting two-state components to quantify allostery. | Theoretical studies of allosteric signaling pathways and efficacy. |
| Markov Field Models (MFMs) [70] | A generalization of Ising models; an umbrella term for models that describe global dynamics via coupled local domain dynamics. | Scaling kinetic modeling to very large biomolecular complexes and machines. |
| Variational Autoencoder (VAE) [39] | A deep learning technique for non-linear dimensionality reduction of high-dimensional simulation data. | Preprocessing MD trajectories for clustering before MSM construction. |
| Markov State Models (MSMs) [70] | A kinetic model built from short MD simulations that describes dynamics as transitions between discrete states. | Extracting long-timescale kinetics and pathways from massive sets of short MD runs. |
| Independent Markov Decomposition (IMD) [70] | A specific MFM that approximates global dynamics as a Kronecker product of independent local domain dynamics. | Initial, simplified modeling of large systems where domain couplings are assumed weak. |
| Principal Component Analysis (PCA) [11] | A linear statistical method for identifying the most important directions of motion in a dataset. | Analyzing the essential dynamics and collective motions from MD trajectories. |
| Effective Field Theory (EFT) [69] | A theoretical framework used to approximate the behavior of complex many-body systems, like spin lattices. | Studying dynamic magnetic behavior and magnetocaloric effects in 2D lattices. |
The navigation of long-range interactions remains a central challenge in both biomolecular and magnetic sciences. This comparison demonstrates that the choice between detailed Molecular Dynamics simulations and simpler Ising-like models is not a matter of identifying a superior tool, but of selecting the right tool for the specific question at hand. MD simulations provide an unmatched, high-resolution lens for mechanistic discovery in systems of tractable size. In contrast, Ising-like models offer a powerful, scalable framework for understanding the fundamental thermodynamic and design principles of cooperative systems.
The future of the field lies not in the exclusive use of one approach over the other, but in their strategic integration. Promising directions include the use of MD simulations to parameterize the coupling constants (J_ij) for Ising models of specific proteins, thereby grounding the coarse-grained model in atomistic reality [68] [71]. Furthermore, the development of multi-scale frameworks like Markov Field Models (MFMs) represents a concerted effort to overcome the scalability limits of pure MD while retaining more physical detail than a simple Ising model [70]. As these integrative methodologies mature, they will profoundly accelerate our ability to not only understand but also to rationally design molecular systems and materials with tailored long-range interactions.
Benchmarking and Synergy: Validating Predictions and Integrating Multi-Scale Models
Molecular dynamics (MD) simulations have become an indispensable tool in computational chemistry and materials science, providing atomic-level insights into complex dynamic processes. However, the predictive power and quantitative accuracy of these simulations are entirely dependent on the rigorous validation of their results against experimental data. This guide objectively compares the performance of different MD simulation and theoretical modeling approaches by examining their validation against wet-lab and observational data, with a specific focus on the context of Ising-like theoretical models research. The convergence of computational and experimental data is the cornerstone of reliable scientific discovery, enabling researchers to cross-verify results from virtual simulations with tangible laboratory findings.
Comparative Analysis of MD Validation Performance
Table 1: Performance Comparison of MD Simulation Packages in Reproducing Experimental Observables
| Simulation Package | Force Field | Test System | Agreement with Experiment | Key Strengths | Identified Limitations |
|---|---|---|---|---|---|
| AMBER | ff99SB-ILDN | EnHD, RNase H [48] | High for room-temperature native state dynamics [48] | Accurate reproduction of NMR chemical shifts and J-couplings [48] | Performance varies with target protein and simulation temperature [48] |
| GROMACS | ff99SB-ILDN | EnHD, RNase H [48] | High for room-temperature native state dynamics [48] | Reproduces conformational distributions well [48] | Subtle differences in underlying conformational ensembles vs. other packages [48] |
| NAMD | CHARMM36 | EnHD, RNase H [48] | High for room-temperature native state dynamics [48] | Good overall reproduction of experimental observables [48] | Larger divergence in simulating thermal unfolding [48] |
| ilmm | Levitt et al. | EnHD, RNase H [48] | High for room-temperature native state dynamics [48] | Effective for native state sampling [48] | May fail to allow protein unfolding at high temperature [48] |
| LAMMPS | ReaxFF | Ethanol on Al slab [59] | Validated against QM calculations and experimental data [59] | Capable of simulating chemical reactions and adsorption processes [59] | Computationally expensive; requires massive data processing [59] |
Table 2: Validation Outcomes Across Application Domains
| Application Domain | Validation Experimental Data | MD Simulation Findings | Correlation with Experiment | Key Validation Metrics |
|---|---|---|---|---|
| Grain Growth in Polycrystalline Nickel [72] | Experimental initial microstructure [72] | Absence of correlation between grain boundary velocity and curvature [72] | Broad match with experimental grain growth characteristics [72] | Grain size distribution, growth kinetics, boundary curvature [72] |
| Drug Solubility Prediction [73] | Experimental aqueous solubility (logS) [73] | ML models using MD properties (SASA, DGSolv) predict solubility with R²=0.87 [73] | High predictive accuracy for diverse drug classes [73] | Solvent Accessible Surface Area (SASA), Coulombic interactions, Lennard-Jones potential [73] |
| Polymer Micelle Drug Delivery [74] | In vitro drug release, cell viability [74] | PFuCL hydrophobic block showed highest polymer-drug interactions [74] | Enhanced drug loading and controlled release confirmed [74] | Drug loading capacity, release kinetics, cellular uptake [74] |
| Ethanol Adsorption on Aluminum [59] | Binding energy, reaction kinetics [59] | Bayesian GPR model predicted adsorption rates within seconds [59] | Accurate prediction validated against MD and experimental data [59] | Number of adsorbed molecules, binding energies, temperature effects [59] |
Experimental Protocols for Cross-Verification
Protocol 1: Validation of Protein Dynamics Simulations
Objective: To validate MD simulations of protein dynamics against experimental spectroscopic data [48].
Methodology:
- System Preparation: Initialize simulations with high-resolution X-ray crystal structures from Protein Data Bank (e.g., PDB ID: 1ENH for EnHD, 2RN2 for RNase H). Remove crystallographic solvent and add explicit hydrogen atoms [48].
- Simulation Parameters: Perform triplicate 200-nanosecond simulations for each protein using multiple MD packages (AMBER, GROMACS, NAMD, ilmm). Employ periodic boundary conditions, explicit water models (TIP4P-EW), and 'best practice parameters' as defined by software developers. Maintain temperature at 298 K and pH consistent with experimental conditions [48].
- Experimental Comparison: Compare simulation results with nuclear magnetic resonance (NMR) derived observables including scalar J-couplings, chemical shifts, and residual dipolar couplings. Compute ensemble averages from simulation trajectories for direct comparison with experimental data [48].
- Analysis: Evaluate reproduction of native state dynamics and conformational distributions. Assess divergence in thermal unfolding simulations at 498 K [48].
Protocol 2: Computational-Experimental Pipeline for Drug Delivery Systems
Objective: To computationally design and experimentally validate polymer micelles for drug delivery [74].
Methodology:
- In Silico Design:
- Conduct all-atom MD simulations of amphiphilic diblock copolymer micelles with varied hydrophobic blocks.
- Calculate solvent-accessible surface area, water shell, hydrogen bonding, radius of gyration, and polymer-drug interaction energies using linear interaction energy analysis [74].
- Identify promising candidate (PEG-b-PFuCL) based on strongest polymer-drug interactions [74].
- Wet-Lab Synthesis and Validation:
- Synthesize selected polymer via ring-opening polymerization of FuCL monomer [74].
- Self-assemble polymers in aqueous media to form micelles [74].
- Load doxorubicin (DOX) and determine drug-loading capacity (4.25 wt% achieved) [74].
- Perform in vitro drug release studies at pH 5.0 and 7.4 to simulate physiological and acidic environments [74].
- Conduct cell viability assays using MDA-MB-231 cells to confirm cytotoxicity and cellular uptake of drug-loaded micelles [74].
Protocol 3: Machine Learning Accelerated Molecular Adsorption
Objective: To train machine learning models on MD simulation data for predicting ethanol adsorption on aluminum surfaces [59].
Methodology:
- MD Simulation Setup: Perform reactive MD simulations using LAMMPS with ReaxFF force field. Model ethanol adsorption on Al slab for various temperatures, velocities, and concentrations. Use triclinic simulation cells with periodic boundary conditions [59].
- Data Collection: Extract number of adsorbed molecules over time from MD trajectories. Calculate bond orders using distance-dependent bond-order function with empirical parameters validated against quantum mechanics calculations [59].
- Machine Learning Training: Develop 28 different regression models including Gaussian Process Regression, support vector machines, decision trees, and neural networks. Train models to predict adsorption rates from simulation conditions [59].
- Validation: Compare ML-predicted adsorption rates with full MD simulation results and experimental data. Evaluate accuracy and computational efficiency gains [59].
Workflow Visualization
Diagram 1: MD-Experimental Cross-Validation Workflow. This diagram illustrates the parallel paths of simulation and experimental approaches that converge at the cross-verification stage.
Diagram 2: ML-Enhanced MD Validation Framework. This workflow shows how machine learning models are trained on MD simulation data and validated against experimental results.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Tools for MD-Experimental Validation
| Tool/Reagent | Function in Validation | Example Applications |
|---|---|---|
| GROMACS [48] [73] | Open-source MD simulation package for biomolecular systems | Protein dynamics validation [48], solubility prediction [73] |
| LAMMPS [59] | Large-scale atomic/molecular massively parallel simulator | Reactive force field simulations, adsorption studies [59] |
| AMBER [48] | Suite of biomolecular simulation programs | Protein folding, nucleic acids simulations [48] |
| ReaxFF Force Field [59] | Reactive force field for chemical reactions | Ethanol adsorption on aluminum [59] |
| GROMOS 54a7 [73] | Biomolecular force field for MD simulations | Drug solubility predictions [73] |
| Bayesian Gaussian Process Regression [59] | Machine learning for predicting molecular behavior | Inferring adsorption rates from MD data [59] |
| PEG-b-PFuCL Polymer [74] | Amphiphilic diblock copolymer for drug delivery | Micelle formation, controlled drug release systems [74] |
| DoXorubicin (DOX) [74] | Model chemotherapeutic drug for delivery studies | Testing drug loading and release kinetics [74] |
The integration of molecular dynamics simulations with experimental validation represents a powerful paradigm for accelerating scientific discovery across materials science, drug development, and molecular engineering. As demonstrated by the comparative data, while different MD packages show generally good agreement with experimental observables for native state dynamics, their performance can diverge significantly for larger conformational changes or extreme conditions. The emergence of machine learning approaches trained on MD data offers promising pathways to overcome traditional limitations in time and length scales, enabling rapid predictions of molecular behavior while maintaining physical accuracy. The consistent theme across all successful applications is the rigorous cross-verification cycle: initial MD simulations inform experimental design, experimental results validate and refine computational models, and the iterative process leads to increasingly accurate predictive capabilities. This validation framework ensures that computational insights translate into tangible advances in drug delivery systems, material design, and molecular-level understanding of complex phenomena.
In computational science, selecting the appropriate model is paramount to obtaining meaningful, reliable results. Molecular Dynamics (MD) and Ising-like models represent two fundamentally different approaches, each with distinct strengths, limitations, and ideal application domains. MD simulations are a cornerstone of computational chemistry and biophysics, providing atomistically detailed trajectories of molecular systems by numerically solving Newton's equations of motion. In contrast, the Ising model is a highly simplified mathematical abstraction from statistical mechanics, originally conceived to explain ferromagnetism by representing systems as discrete variables (spins) on a lattice. While both are used to study complex systems, their philosophies differ: MD aims for a detailed, physical representation, whereas the Ising model seeks to capture the universal behavior of cooperative phenomena through minimalism. This guide provides a structured comparison to help researchers, particularly in drug development and materials science, make an informed choice between these powerful techniques.
Core Principles and Methodologies
The Molecular Dynamics (MD) Approach
MD simulations model the time-dependent behavior of a system by calculating the forces between atoms and integrating them to update atomic positions and velocities. The potential energy of the system is described by a force field, which includes terms for bonded interactions (bonds, angles, dihedrals) and non-bonded interactions (van der Waals and electrostatic forces) [75]. A key strength of MD is its ability to model specific molecular interactions, such as the role of partial atomic charges in hydrocarbons, which can be critical for accurately simulating solvation in aqueous environments [75]. The workflow involves energy minimization, equilibration, and finally a production run from which thermodynamic and dynamic properties are extracted.
The Ising-like Model Approach
The Ising model abstracts a system into a collection of discrete variables (spins, ( \sigmai ), that can be in one of two states, typically +1 or -1). These spins are arranged on a graph or lattice and interact with their neighbors. The Hamiltonian (energy function) for the classic Ising model is: [ H(\sigma) = -\sum{\langle ij \rangle} J{ij} \sigmai \sigmaj - \mu \sumj hj \sigmaj ] where ( J{ij} ) is the interaction strength between neighboring spins ( i ) and ( j ), ( hj ) is an external field, and ( \mu ) is the magnetic moment [76]. The model's power lies in its simplicity; it ignores most microscopic details to focus on how local alignment rules can give rise to macroscopic phase transitions and collective behavior. Configurations are typically sampled using the Metropolis-Hastings algorithm, a Monte Carlo method that accepts or rejects random spin flips based on the resulting change in energy to achieve a Boltzmann distribution [77].
Comparative Analysis: Strengths, Weaknesses, and Ideal Use Cases
The following table summarizes the core characteristics of each modeling approach to facilitate comparison.
Table 1: Fundamental Characteristics of MD and Ising-like Models
| Feature | Molecular Dynamics (MD) | Ising-like Models |
|---|---|---|
| System Representation | Atomistic/All-atom. Atoms and molecules with explicit positions in 3D space. [75] | Coarse-grained lattice. Discrete spins on a graph or lattice. [76] |
| Governed by | Newton's equations of motion; empirical force fields. [75] | Statistical mechanics of a defined Hamiltonian. [76] |
| Key Outputs | Thermodynamic properties, time-evolution trajectories, conformational changes, diffusion constants. [75] | Phase diagrams, critical exponents, correlation functions, magnetization. [76] [78] |
| Computational Cost | High (scales with number of atoms and simulation time). | Low to moderate (scales with number of spins and Monte Carlo steps). |
| Time Dimension | Explicit, femtoseconds to milliseconds. | Abstract, non-physical "Monte Carlo steps". |
The strategic choice between MD and Ising-like models depends heavily on the research question. Their respective strengths and optimal applications are detailed below.
Table 2: Comparative Strengths, Weaknesses, and Primary Applications
| Aspect | Molecular Dynamics (MD) | Ising-like Models |
|---|---|---|
| Primary Strengths | - High physical fidelity for specific molecules.- Provides dynamical information.- Can model complex, heterogeneous environments (e.g., protein in solvent). [75] | - Captures universal phenomena and phase transitions. [76]- Computationally efficient for large-scale collective behavior.- Highly interpretable and analytically tractable in some cases. |
| Inherent Limitations | - Computationally expensive, limiting system size and timescale.- Accuracy depends on the force field parameterization. [75] | - Lacks molecular specificity and explicit dynamics.- Often too simplistic for quantitative predictions in complex molecular systems. |
| Ideal Use Cases | - Drug-target binding affinity and kinetics.- Protein folding and conformational dynamics.- Predicting properties of specific materials or solvents. [79] [75] | - Studying critical phenomena and universality classes. [76]- Modeling social dynamics, opinion formation, and neural networks. [80] [78]- Analyzing lattice-based adsorption or alloy ordering. |
Decision Framework and Experimental Protocols
A Workflow for Model Selection
The following diagram outlines a logical decision process for choosing between MD and Ising-like models based on the nature of the research question.
Key Experimental and Simulation Protocols
To ensure reproducibility, it is essential to document the core methodologies for both approaches.
Protocol 1: Typical MD Simulation Workflow [79] [75]
- System Preparation: Obtain or build the initial 3D coordinate file (e.g., .pdb) for the molecule(s) of interest. Place the molecule in a simulation box and solvate it with explicit water molecules (e.g., TIP3P model) or other solvents.
- Force Field Selection and Assignment: Choose an appropriate force field (e.g., CHARMM, AMBER, OPLS). Assign atom types, partial charges, and bonded parameters to all atoms in the system. For example, in hydrocarbon modeling, the presence or absence of partial charges must be defined [75].
- Energy Minimization: Perform an energy minimization to remove any steric clashes or unrealistic geometry in the initial structure, using algorithms like steepest descent or conjugate gradient.
- Equilibration: Run short MD simulations in canonical (NVT) and isothermal-isobaric (NPT) ensembles to bring the system to the desired temperature and pressure. Positional restraints are often applied to the solute during initial equilibration.
- Production Run: Conduct an unrestrained MD simulation for a sufficiently long timescale (nanoseconds to microseconds) to sample the properties of interest. The trajectory (atomic positions and velocities over time) is saved for analysis.
- Analysis: Analyze the saved trajectory to compute properties such as density, diffusion coefficients, radial distribution functions, root-mean-square deviation (RMSD), or binding free energies.
Protocol 2: Monte Carlo Simulation of an Ising Model [78] [77]
- Lattice Initialization: Define a lattice (e.g., 2D square grid of size L x L). Initialize each spin ( \sigma_i ) to a random state (+1 or -1).
- Parameter Definition: Set the interaction strength ( J ) (ferromagnetic: ( J > 0 ), antiferromagnetic: ( J < 0 )) and the reduced inverse temperature ( \beta = 1/k_B T ). The external field ( h ) is often set to zero.
- Metropolis-Hastings Monte Carlo Step: a. Propose: Randomly select a spin ( i ) for a flip trial (( \sigmai \rightarrow -\sigmai )). b. Compute Energy Change: Calculate the change in energy ( \Delta E ) associated with the flip. For a 2D square lattice with nearest-neighbor interactions, ( \Delta E = 2 J \sigmai \sum{\text{neighbors}} \sigma_j ). c. Accept/Reject: If ( \Delta E \leq 0 ), accept the flip. If ( \Delta E > 0 ), accept the flip with probability ( \exp(-\beta \Delta E ) ).
- Sampling: Repeat step 3 for a large number of Monte Carlo steps (MCS), often on the order of ( 10^5 - 10^6 ) MCS, to ensure the system reaches equilibrium and is sampled adequately. One MCS is typically defined as N flip attempts (where N is the total number of spins).
- Measurement: Calculate average properties, such as the magnetization per spin ( M = \langle \frac{1}{N} \sumi \sigmai \rangle ) or the energy per spin ( E = \langle H \rangle / N ), once the system has equilibrated.
Essential Research Reagents and Computational Tools
The "research reagents" for computational studies are the software packages, force fields, and datasets that form the foundation of the work.
Table 3: Key Research Reagent Solutions for MD and Ising Modeling
| Tool / Reagent | Type | Primary Function & Application |
|---|---|---|
| GROMACS [75] | MD Software | A high-performance molecular dynamics package for simulating proteins, lipids, and nucleic acids. Used for calculating properties like hydration free energy and diffusion constants. [75] |
| AMBER/CHARMM | Force Field | Empirical force fields providing parameters (masses, charges, bond stiffness) for MD simulations of biomolecules. |
| Adaptive Force Matching (AFM) [75] | Fitting Method | A method for developing highly accurate force fields based on electronic structure calculations (e.g., MP2), crucial for modeling challenging systems like hydrocarbons. [75] |
| R Statistical Software [78] | Programming Language | Used for simulating Ising model dynamics, statistical analysis of results, and network visualization in psychological or social applications of the Ising model. [78] |
| Ising Model Simulator (Custom Code) [77] | Simulation Code | Custom C++ or Python code, often implementing the Metropolis algorithm, for sampling Ising configurations on lattices or complex networks. [77] |
| ZINC/CHEMBL [81] | Chemical Database | Large-scale databases of commercially available and bioactive compounds, used for training AI models and validating computational predictions. [81] |
MD and Ising-like models are complementary, not competing, tools in the computational scientist's arsenal. The choice hinges on the level of abstraction required to answer the scientific question effectively. MD simulations are the tool of choice when the research demands atomistic resolution, specific molecular interactions, and dynamical information, as in rational drug design or predicting the properties of a novel material. Ising-like models are superior for investigating fundamental statistical mechanics, uncovering universal principles of phase transitions, and modeling large-scale collective behavior in systems ranging from magnetic materials to social networks. A promising future direction lies in multi-scale modeling, where insights from large-scale, coarse-grained Ising-like studies can inform the setup and focus of more detailed, resource-intensive MD simulations, creating a powerful synergistic loop for scientific discovery.
In computational science, a significant challenge lies in connecting phenomena that occur across vastly different spatial and temporal scales. Multi-scale modeling emerges as a critical framework for addressing this challenge, strategically linking models at separate scales to provide a comprehensive understanding of complex systems. This guide focuses on the specific interaction between two powerful modeling approaches: Ising-type theoretical models and Molecular Dynamics (MD) simulations. Ising models provide a coarse-grained, statistical representation of systems, often capturing critical behavior and phase transitions in a computationally efficient manner. In contrast, MD simulations offer an atomistic, high-resolution view of molecular interactions and dynamics, albeit at a substantially higher computational cost. The integration of these approaches enables researchers to leverage the strengths of each method, creating a more powerful and computationally tractable framework for investigating scientific problems that span multiple scales.
The core principle of this hybrid framework is the bidirectional flow of information. Ising model outputs, such as identified critical regions or dominant interaction modes, can inform MD simulations by highlighting specific thermodynamic conditions or molecular configurations worthy of atomistic investigation. Conversely, MD simulations can provide precise, first-principles data on interaction energies and molecular conformations to parameterize and validate coarse-grained Ising models. This guide objectively compares the performance, applications, and implementation protocols of this integrated approach against traditional, single-scale methods, providing researchers with the data and methodologies needed to apply these techniques effectively in their work, particularly in materials science and drug development.
Comparative Analysis of Modeling Approaches
The table below provides a systematic comparison of the core characteristics of MD simulations, Ising-like models, and their powerful hybrid integration.
Table 1: Comparison of Molecular Dynamics, Ising-like Models, and the Hybrid Approach
| Feature | Molecular Dynamics (MD) Simulations | Ising-like Theoretical Models | Hybrid MD/Ising-informed Framework |
|---|---|---|---|
| Spatial Resolution | Atomistic/All-Atom [57] [82] | Coarse-grained (Lattice-based) | Multi-scale, bridging atomistic to coarse-grained |
| Temporal Scale | Nanoseconds to Microseconds [59] | Effectively infinite (Equilibrium states) | Extends effective timescale via coarse-graining |
| Computational Cost | Very High [83] | Low | Moderate to High (dependent on iteration level) |
| Primary Output | Atomistic trajectories, energies, forces [57] | Free energy, phase diagrams, critical points | Validated multi-scale properties and mechanisms |
| Key Strengths | High-resolution dynamical insight [82], Detailed interaction mechanisms [57] | Captures critical behavior, Phase transitions, Computational efficiency | Enhanced transferability, Reproducible and accurate estimation [84], Bridges scales |
| Common Applications | RNA folding [57] [50], Protein-ligand binding, Polymer dynamics [15] | Magnetic systems, Generic phase transitions, Lattice gases | Boiling point estimation [84], Carbon film growth [83], RNA dynamics [57] |
Workflow for Hybrid and Multi-Scale Modeling
The integration of Ising-like models and MD simulations can be implemented through several distinct workflows. The following diagram visualizes the two primary pathways for information exchange between these modeling scales.
Diagram 1: Bidirectional Workflow between Ising and MD Models
Pathway 1: From Coarse-Grained to Atomistic
In this pathway, the Ising model acts as a computationally efficient scout. It identifies key regions in a phase diagram or critical points of interest. These large-scale insights then strategically inform where to deploy more computationally expensive MD simulations.
- Ising Model Outputs: The primary outputs from the Ising model that guide MD simulations include identification of phase boundaries and critical points, which suggest specific temperatures and pressures for atomistic simulation to study transition mechanisms [84]. Ising models can also predict dominant interaction modes (e.g., ferromagnetic vs. antiferromagnetic ordering), helping to select initial configurations for MD systems.
- Informing MD Simulations: The results from the Ising model are used to set up MD simulations. This involves defining the thermodynamic ensemble (e.g., NPT, NVT) based on the phase of interest identified by the Ising model. The simulation box size and composition can be constructed to match the correlation lengths observed near critical regions in the Ising model. Furthermore, running multiple, shorter MD simulations can be strategically deployed across the parameter space identified as most relevant by the Ising scan.
Pathway 2: From Atomistic to Coarse-Grained
This pathway uses detailed MD simulations to build accurate and physically grounded Ising-type models.
- MD Simulation Outputs: Key quantitative data extracted from MD trajectories include interaction energies between molecular components or spins, which can be directly used to parameterize the coupling constants (J) in the Ising Hamiltonian. MD also reveals the free energy landscapes and relative stability of different states, which the Ising model's equilibrium statistics must reproduce. The dynamics and relaxation times observed in MD can inform the choice of update rules (e.g., Metropolis, Glauber) in dynamic Ising models.
- Parameterizing the Ising Model: The data from MD is integrated into the Ising model. The coupling constants (J) in the Ising Hamiltonian are fitted to reproduce the potential of mean force or interaction energies calculated from MD simulations [59]. In more complex systems, MD can be used to derive and validate the coarse-grained mapping, defining what an "effective spin" represents in the molecular system.
Experimental Protocols and Performance Data
Case Study 1: Boiling Point Estimation via Hybrid MD-ML
This case demonstrates a hybrid MD-Machine Learning (ML) framework, where ML acts as a sophisticated intermediary, learning from MD data to create a rapid prediction tool—a function analogous to a parameterized Ising model.
Experimental Protocol:
- MD Simulation Setup: Equilibrium MD simulations are performed for target aromatic fluids (e.g., biphenyl) using force fields like OPLS-AA. The system is simulated across a range of temperatures [84].
- Property Calculation: The liquid density is calculated from the MD trajectories at each temperature. The boiling point is estimated by identifying a density threshold or by applying a thermodynamically rigorous inflection-point method on the density-temperature curve [84].
- Machine Learning Training: Key features from the MD simulations are used to train regression models. This includes the temperature, estimated boiling point, and corresponding density. Three common models are trained and evaluated: Nearest Neighbours Regression (NNR), Neural Network (NN), and Support Vector Regression (SVR) [84].
- Validation: The best-performing ML model is validated against the original MD data and available experimental data to assess its predictive accuracy [84].
Performance Data: Table 2: Performance of MD-ML Framework for Boiling Point Prediction [84]
| Model | Predicted Boiling Point (K) | Predicted Density (g/cm³) | Key Outcome |
|---|---|---|---|
| Nearest Neighbours Regression (NNR) | 524.97 K | 0.064 | Best match with MD data |
| Neural Network (NN) | 525.3 K | Overestimated | Accurate temperature, poor density |
| Support Vector Regression (SVR) | Underestimated | Underestimated | Lowest accuracy |
| Experimental Reference | ~508.18 K (onset) | N/A | ~3.3% deviation between onset and completion |
Case Study 2: Carbon Film Deposition via Active Learning
This protocol showcases a tight integration between MD and machine learning to create a highly accurate and efficient neuroevolution potential (NEP), which can be viewed as a complex, high-dimensional successor to traditional Ising-type force fields.
Experimental Protocol:
- Initial Data Generation: An initial dataset of diverse carbon structures is compiled, often from existing sources like the GAP-20 dataset [83].
- Active Learning Loop: An iterative process begins:
- Step A: An NEP is trained on the current dataset.
- Step B: The NEP is coupled with LAMMPS to perform hybrid MD/Time-stamped force-biased Monte Carlo (tfMC) simulations of carbon atom deposition. The tfMC method is critical for accelerating the sampling of rare events like surface diffusion and nucleation [83].
- Step C: Structures are sampled from the trajectories and their energies/forces are recalculated with high-fidelity DFT.
- Step D: Structures with high prediction errors are added to the training set. The loop repeats until model convergence [83].
- Transferability Testing: The final, validated NEP is applied to simulate growth on different substrates (e.g., Cu(111), Al2O3(0001)) to test its transferability [83].
Performance Data: The active learning workflow produced an NEP with high predictive accuracy. The model converged after approximately 200,000 generations, achieving a root-mean-square error (RMSE) for the test dataset of 57.752 meV/atom for energy and 552.347 meV/Å for forces [83]. This high accuracy enabled the discovery of a new growth mechanism: adhesion-driven growth at low energies and peening-induced densification at high energies of carbon atoms on Si(111) substrates [83].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The successful implementation of hybrid multi-scale modeling relies on a suite of software tools and computational methods.
Table 3: Essential Research Tools for Hybrid Modeling
| Tool / Solution | Function | Example Use Case |
|---|---|---|
| LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) | A highly versatile and widely used MD simulator [83] [59]. | Performing the atomistic deposition simulations in the carbon film growth study [83]. |
| Neuroevolution Potential (NEP) | A machine-learning potential that offers high computational efficiency while maintaining near-DFT accuracy [83]. | Describing interatomic interactions during carbon deposition on various substrates [83]. |
| ReaxFF (Reactive Force Field) | A bond-order based force field that allows for chemical reactions during MD simulations [59]. | Simulating the adsorption and reaction of ethanol molecules on an aluminium surface [59]. |
| Time-stamped Force-Biased Monte Carlo (tfMC) | An enhanced sampling method to accelerate structural relaxation and sample rare events [83]. | Improving the sampling efficiency of carbon surface diffusion and ring formation in deposition simulations [83]. |
| Gaussian Process Regression (GPR) | A Bayesian non-parametric regression method well-suited for small datasets and providing uncertainty estimates [59]. | Creating a fast surrogate model to predict ethanol adsorption rates on an Al slab from MD data [59]. |
| Maximum Entropy Reweighting | A quantitative method to bias simulated ensembles to match experimental data [57] [50]. | Refining an MD-simulated ensemble of an RNA molecule to match NMR data [50]. |
The integration of Ising-like models and Molecular Dynamics simulations represents a powerful paradigm for multi-scale modeling. As the case studies demonstrate, this hybrid approach is not merely a theoretical concept but a practical strategy that delivers reproducible and accurate estimation of complex properties like boiling points and reveals atomistic growth mechanisms. The bidirectional flow of information between coarse-grained and atomistic models creates a framework that is greater than the sum of its parts, enhancing transferability and providing a more complete picture of system behavior from the molecular to the macroscopic scale. For researchers in drug development and materials science, adopting these protocols and tools can significantly accelerate insight and innovation.
The Role of AI and Machine Learning in Unifying and Enhancing Both Frameworks
The study of complex molecular systems relies on two powerful, yet historically distinct, computational frameworks: detailed, all-atom Molecular Dynamics (MD) simulations and simplified, conceptual Ising-like theoretical models. MD simulations provide high-resolution, physicochemical accuracy by numerically solving Newton's equations of motion for all atoms in a system, capturing structural and dynamic details at the atomic level [85]. In contrast, Ising-like models offer a coarse-grained representation, where system components (e.g., amino acid residues, atoms, or spins) occupy discrete states, and their interactions are governed by a simplified Hamiltonian. This makes them exceptionally valuable for studying collective behaviors, phase transitions, and long-timescale processes that remain challenging for standard MD [85].
Despite their complementary strengths, a significant gap has persisted between these frameworks. MD simulations are often hamstrung by immense computational costs and limited sampling of rare events, especially for large systems and intrinsically disordered proteins (IDPs) that exist as dynamic ensembles [85]. Ising models, while computationally efficient, traditionally lacked the atomic-level physical realism provided by MD. Artificial Intelligence (AI) and Machine Learning (ML) are now playing a transformative role in unifying these two approaches. By serving as a "translational layer," AI methods can extract the essential physics from detailed MD data to parameterize and inform accurate Ising-like models, and conversely, use the statistical insights from Ising models to guide and accelerate MD sampling. This review explores how this AI-mediated synergy is creating a powerful, unified computational framework for biomolecular research and drug development.
AI-Enhanced Molecular Dynamics: Overcoming Traditional Limitations
The Sampling and Timescale Challenge of Conventional MD
Traditional MD simulations provide high-resolution data but struggle with the vast conformational space of biomolecular systems, particularly IDPs. As noted in a 2025 review, "MD simulations, though accurate and widely used, are computationally expensive and struggle to sample rare, transient states" [85]. Capturing the full ensemble of IDP conformations requires simulations spanning microseconds to milliseconds, demanding immense computational resources that are often impractical [85]. This limitation means that MD alone may miss functionally crucial, but transient, molecular states.
Deep Learning for Conformational Sampling and Analysis
Deep learning (DL) offers a data-driven solution to the sampling problem. DL models can learn the complex, non-linear relationships between a protein's sequence and its possible structures from large-scale datasets, enabling the efficient generation of diverse conformational ensembles without the constraints of traditional physics-based simulations [85].
Key Application: A notable example is the Distributional Graphormer (DiG), a deep learning framework designed to predict the equilibrium distribution of molecular systems. DiG uses deep neural networks to transform simple input distributions into the complex equilibrium distributions of molecular systems, demonstrating high performance in tasks like protein conformation sampling and ligand structure sampling [86]. This approach directly addresses the sampling challenge of IDPs, with studies showing that "such DL approaches have been shown to outperform MD in generating diverse ensembles with comparable accuracy" [85].
AI-Augmented Analysis: Furthermore, ML techniques have become indispensable for analyzing the enormous datasets generated by MD simulations. A 2025 tutorial illustrates how supervised ML models—including logistic regression, random forest, and multilayer perceptron (MLP)—can be applied to MD trajectory data to identify key residues that impact the stability of protein complexes, such as the SARS-CoV-2 spike protein receptor binding domain (RBD) with the ACE2 receptor [87]. This moves analysis beyond simple observation to a quantitative, feature-based identification of critical molecular determinants.
Neural Network Potentials and Accelerated ab Initio MD
A revolutionary advancement is the development of neural network potentials (NNPs), which replace classical force fields. These AI-driven potentials are trained on data from high-level quantum mechanical calculations, enabling them to achieve near-quantum accuracy at a fraction of the computational cost. This approach is often termed Machine Learning-accelerated MD (MLMD) or AI-accelerated ab initio MD (AI2MD) [88].
Case Study - The ElectroFace Dataset: The ElectroFace dataset is a prime example of AI2MD in action. It is a collection of over 60 distinct ab initio MD and MLMD trajectories for electrochemical interfaces. The workflow involves:
- Running a short, reference ab initio MD simulation with CP2K/QUICKSTEP code [88].
- Using an active learning loop (e.g., with the DP-GEN package) to iteratively generate a robust machine learning potential (MLP) with the DeePMD-kit [88].
- Once trained, the MLP is deployed in the LAMMPS MD code to run long-timescale simulations (nanoseconds) with ab initio accuracy, which would be computationally prohibitive for pure ab initio methods [88].
This methodology bridges the gap between the high accuracy of quantum methods and the need for extensive sampling, directly tackling the timescale limitation of conventional ab initio MD.
The Ising Model Framework and Its Connection to MD
The Ising model is a cornerstone of statistical physics, originally developed to describe magnetic moments (spins) on a lattice. Its utility has expanded to model a vast array of biological phenomena, including protein folding, allostery, and peptide aggregation. In a biological context, the "spins" can represent discrete states of amino acid residues (e.g., hydrophobic/hydrophilic, folded/unfolded, or specific dihedral angle states), and the interactions between them capture cooperative effects.
The power of the Ising-like framework lies in its simplicity and its computational tractability for simulating long-timescale behaviors and phase transitions of large systems. However, a significant challenge has been the parameterization of its Hamiltonian—the rules defining the energetic coupling between components. AI and ML are now enabling this parameterization to be directly informed and validated by data from detailed MD simulations, creating a more physically-grounded and predictive coarse-grained model.
AI as a Unifying Bridge: From MD Data to Informed Coarse-Grained Models
The unification process involves using AI to extract the essential, collective variables and effective interaction energies from atomistic MD data, which can then be used to parameterize a highly accurate Ising-like model. The following workflow diagram illustrates this synergistic cycle.
Workflow for AI-Mediated Unification
The diagram above shows a cyclic workflow for integrating MD and Ising models via AI:
- Detailed MD Simulation: An all-atom MD simulation of the system is performed, generating high-resolution trajectory data on atomic movements and interactions [85].
- AI/ML Analysis: AI models, such as those based on manifold learning or deep neural networks, are applied to the MD trajectories. These models identify the system's collective variables—the low-dimensional representation of the system's essential dynamics—and quantify the effective energy landscape and residue-residue interactions in this reduced space [87].
- Parameterized Ising-like Model: The insights from the AI analysis are used to define the states and parameterize the interaction strengths (the J-couplings and fields) of an Ising-like Hamiltonian.
- Prediction of Collective Behavior: The now-physically-informed Ising model is used to run rapid, large-scale simulations, predicting collective behaviors, stability, and long-timescale dynamics that are difficult to access with MD alone.
- Experimental Validation and Refinement: Predictions from the unified model are validated against experimental data from techniques like NMR spectroscopy or Small-Angle X-Ray Scattering (SAXS) [85]. Discrepancies can be used to refine the MD sampling or the Ising model parameters, creating a closed loop for continuous improvement.
Comparative Performance Analysis
Quantitative Comparison of Sampling Methods
The table below summarizes a comparison of the key characteristics of traditional MD, AI-enhanced MD, and Ising-like models, demonstrating the performance gains achieved through their AI-mediated unification.
Table 1: Performance Comparison of Traditional MD, AI-Enhanced MD, and Ising-like Models
| Feature | Traditional MD | AI-Enhanced MD (MLMD/NNP) | Ising-like Models (AI-Parameterized) |
|---|---|---|---|
| Spatial Resolution | All-atom (Ångström scale) | All-atom / Near-quantum accuracy [88] | Coarse-grained (residue/bead level) |
| Timescale Access | Nanoseconds to microseconds [85] | Nanoseconds to milliseconds (effectively) [88] | Effectively infinite (equilibrium properties) |
| Sampling Efficiency | Low; struggles with rare events [85] | High; generative DL and enhanced sampling overcome barriers [85] [86] | Very High; by design, focuses on essential states |
| Physical Basis | Classical force fields | AI-trained potentials on ab initio data [88] | Simplified Hamiltonian, parameterized from MD/experiments |
| Computational Cost | Very High | High (training), Lower (inference) [86] | Low |
| Best For | Atomic-level mechanism studies, detailed interaction maps | Accurate, long-timescale dynamics; ab initio property prediction | Studying collective behavior, phase transitions, and large-scale system properties |
Case Study: SARS-CoV-2 Spike Protein Dynamics
Research by Pavlova et al., as detailed in a 2025 tutorial, provides a concrete example of this unified approach in action. The goal was to determine which residues in the SARS-CoV-2 spike protein's RBD were most critical for its increased binding affinity to the ACE2 receptor compared to SARS-CoV [87].
- MD Data Generation: MD simulations were performed for both the SARS-CoV and SARS-CoV-2 RBD-ACE2 complexes.
- AI-Driven Feature Extraction: Three ML models (logistic regression, random forest, and a multilayer perceptron) were trained to classify from which virus an MD snapshot originated, based on the distances between residue pairs in the complex.
- Identification of Key Determinants: The feature weights (coefficients in logistic regression, feature importance in random forest) from the trained models directly identified the specific residue-residue interactions that most significantly distinguished the two viruses, pinpointing the molecular determinants of higher affinity binding [87].
This workflow exemplifies how AI can extract the most "important" features—the effective "interactions" in a conceptual Ising-like model—from complex MD data, providing sharp, biophysical insights.
The Scientist's Toolkit: Essential Research Reagents and Software
To implement the methodologies discussed, researchers can leverage the following suite of key software tools and datasets.
Table 2: Essential Tools for AI-Unified Simulations
| Tool Name | Type | Primary Function | Relevance to Framework Unification |
|---|---|---|---|
| DeePMD-kit [88] | Software Package | Training and running neural network potentials (NNPs). | Enables ML-accelerated MD (MLMD) by providing near-ab initio accuracy at lower cost. |
| DP-GEN [88] | Software Package | An active learning platform for generating robust NNPs. | Automates the creation of training datasets for NNPs, ensuring reliability across conformational space. |
| LAMMPS [88] | MD Simulator | A highly versatile molecular dynamics simulator. | The primary engine for running production MD simulations using NNPs from DeePMD-kit. |
| CP2K [88] | Ab Initio MD Code | Performs ab initio molecular dynamics simulations. | Generates the high-quality reference data used to train NNPs. |
| ElectroFace Dataset [88] | Research Dataset | A curated collection of AI2MD trajectories for electrochemical interfaces. | Serves as a benchmark and training resource for developing and validating MLMD methods for interfaces. |
| TensorFlow/PyTorch [89] | ML Framework | Libraries for building and training deep learning models. | The foundation for creating custom DL models for trajectory analysis, state classification, and generative sampling. |
The artificial dichotomy between high-resolution MD simulations and coarse-grained Ising-like models is being dismantled by Artificial Intelligence and Machine Learning. AI does not merely improve each framework independently; it actively unifies them. It serves as a powerful bridge, distilling the physical insights from detailed MD trajectories to create statistically rigorous and predictive Ising-like models, and using the conceptual guidance of coarse-grained models to drive more efficient MD sampling. This synergistic integration, as evidenced by platforms like DeePMD-kit and methodologies for analyzing complex protein interactions, provides a more holistic and powerful computational lens. For researchers and drug development professionals, this AI-unified framework offers a robust path to tackle problems that were previously intractable, from decoding the conformational landscapes of disordered proteins to accelerating the rational design of novel therapeutics.
Conclusion
Molecular Dynamics simulations and Ising-like theoretical models, though operating on different physical principles and scales, offer powerful and complementary lenses for tackling complex problems in drug discovery. MD provides unparalleled, high-resolution insights into the dynamic interactions between drugs and their targets, directly informing lead optimization and the design of advanced delivery systems. Ising models, conversely, excel at revealing emergent collective behaviors and thermodynamic properties in complex biological networks. The future of computational biomedical research lies in strategically integrating these approaches into multi-scale frameworks, a convergence that will be profoundly accelerated by continued advances in high-performance computing and artificial intelligence. This synergy promises to unlock deeper understanding of disease mechanisms and accelerate the development of precise, effective therapeutics, ultimately bridging the gap from atomic-level interactions to clinical outcomes.
References
- 1. Molecular dynamics simulation for all - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6209097/]
- 2. Energy landscape analysis based on the Ising model: Tutorial ... [https://journals.plos.org/complexsystems/article?id=10.1371/journal.pcsy.0000039]
- 3. Ising-like model replicating time-averaged spiking ... [https://www.nature.com/articles/s41598-024-55922-9]
- 4. Accurate modeling of DNA conformational flexibility by a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8053917/]
- 5. Leveraging high-throughput molecular simulations and ... [https://www.nature.com/articles/s41524-025-01552-2]
- 6. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 7. A machine-learning framework for accelerating spin-lattice ... [https://www.nature.com/articles/s41524-025-01547-z]
- 8. Symplectic Spin-Lattice Dynamics with Machine-Learning ... [https://arxiv.org/html/2506.12877v1]
- 9. Revealing nanostructures in high-entropy alloys via ... [https://www.nature.com/articles/s41524-025-01762-8]
- 10. for All Molecular Dynamics Simulation [https://pubmed.ncbi.nlm.nih.gov/30236283/]
- 11. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 12. Advancements in Molecular for Simulations Discovery Drug [https://scisimple.com/en/articles/2025-11-04-advancements-in-molecular-simulations-for-drug-discovery--ak2zgwn]
- 13. Revealing Nanostructures in High-Entropy Alloys via ... [https://arxiv.org/html/2503.12591v3]
- 14. Comparison of imaging based single-cell resolution spatial ... [https://www.nature.com/articles/s41467-025-63414-1]
- 15. Frontier advances in molecular dynamics simulations for the ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra00059a?page=search]
- 16. Predicting binding free energies: Frontiers and benchmarks [https://pmc.ncbi.nlm.nih.gov/articles/PMC5544526/]
- 17. Molecular dynamics-driven drug discovery [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp00380f]
- 18. GitHub - bestlab/ Ising - Model : Code for analyzing contact formation in... [https://github.com/bestlab/Ising-Model]
- 19. Sociophysics models inspired by the Ising model [https://arxiv.org/html/2506.23837v1]
- 20. Predicting and understanding drug-target binding kinetics ... [https://www.cecam.org/workshop-details/predicting-and-understanding-drug-target-binding-kinetics-via-molecular-simulations-1292]
- 21. Systematic analysis, aggregation and visualisation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10935884/]
- 22. Data-driven detection of critical points of phase transitions ... [https://www.nature.com/articles/s42005-023-01429-0]
- 23. Absolute Binding Free Energy Calculations for Highly Flexible Protein MDM2 and Its Inhibitors - PubMed [https://pubmed.ncbi.nlm.nih.gov/32635537/]
- 24. Absolute and Relative Binding Free Energy Calculations of Nucleotides to Multiple Protein Classes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11859759/]
- 25. Accommodating protein flexibility for structure-based drug design - PubMed [https://pubmed.ncbi.nlm.nih.gov/20939792/]
- 26. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOoo0rZU1vZp5MPtROy2B9Sunw37P_MULLsbrPMFgXEYAZ4yvKz7t]
- 27. Accelerating Molecular Dynamics Simulations with ... [https://arxiv.org/html/2510.06562v1]
- 28. Benchmarking GPUs for MD Simulations [https://www.linkedin.com/pulse/benchmarking-gpus-md-simulations-speed-cost-insights-barbosa-farias-ynoke]
- 29. An experimental and molecular dynamics simulation study ... [https://www.sciencedirect.com/science/article/abs/pii/S0735193325014496]
- 30. Multiscale molecular dynamics investigation of high ... [https://www.nature.com/articles/s41598-025-26005-0]
- 31. Challenges and opportunities in computational studies for ... [https://www.nature.com/articles/s44386-025-00024-3]
- 32. Exploration of lipid bilayer mechanical properties using ... [https://www.sciencedirect.com/science/article/abs/pii/S000398612400273X]
- 33. Distinct Structural Responses of Lipid Bilayers to Horizontal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12536441/]
- 34. Molecular Dynamics Simulations and Their Novel ... [https://pubmed.ncbi.nlm.nih.gov/41023346/]
- 35. The Ising model celebrates a century of interdisciplinary ... [https://www.nature.com/articles/s44260-024-00012-0]
- 36. Origins of the Ising model [https://arxiv.org/html/2509.00632v1]
- 37. On the origin of cooperativity effects in the formation of self ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11041291/]
- 38. Advancing Molecular Simulations: Merging Physical Models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010417/]
- 39. Improving Clustering Quality in Biomolecular Simulations [https://scisimple.com/en/articles/2025-06-04-improving-clustering-quality-in-biomolecular-simulations--ak4614q]
- 40. GitHub - JonathanFrassineti/2D- Ising - Model - simulation : Repository... [https://github.com/JonathanFrassineti/2D-Ising-Model-simulation]
- 41. Molecular Insights into the Loading and Dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7731932/]
- 42. Modeling cancer immunoediting in tumor microenvironment ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04731-w]
- 43. Computational approaches to delivery of anticancer drugs ... [https://www.sciencedirect.com/science/article/pii/S2001037023002842]
- 44. Toward an Ising Model of Cancer and Beyond - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3151151/]
- 45. Combinatorial MD/QM studies to develop novel ionic liquid ... [https://www.nature.com/articles/s41598-024-74250-6]
- 46. Protein structure predictions by enhanced conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6976031/]
- 47. Timewarp: Transferable Acceleration of Molecular ... [https://proceedings.neurips.cc/paper_files/paper/2023/hash/a598c367280f9054434fdcc227ce4d38-Abstract-Conference.html]
- 48. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 49. Comparison between cluster Monte Carlo algorithms in the ... [https://www.sciencedirect.com/science/article/abs/pii/0370269389915633]
- 50. Integrating experimental data with molecular simulations to ... [https://arxiv.org/html/2207.08622v4]
- 51. Force Fields and Interactions - GitHub Pages [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/01-Force_Fields_and_Interactions/index.html]
- 52. Benchmarking of force fields to characterize the intrinsically ... [https://www.nature.com/articles/s41598-023-40801-6]
- 53. Choice of Force Field for Proteins Containing Structured ... [https://www.sciencedirect.com/science/article/pii/S0006349520301685]
- 54. Force fields for simulating the interaction of surfaces with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4686237/]
- 55. Are Protein Force Fields Getting Better? A Systematic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3383641/]
- 56. Comparing Molecular Dynamics Force Fields in the Essential ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0121114]
- 57. Integrating experimental data with molecular simulations to ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X22001828]
- 58. Comparing a simple theoretical model for protein folding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3816406/]
- 59. Assessment of machine learning models trained by ... [https://www.nature.com/articles/s41598-024-71007-z]
- 60. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOooajfC1ckyXAaPO_RQGTDYDWjLuwG3bYvlolxB5pgWBLNvfzMPK]
- 61. Solving the compute crisis with physics-based ASICs [https://arxiv.org/html/2507.10463v1]
- 62. Scientific modeling on cloud GPUs: fit guide for 2025 [https://compute.hivenet.com/post/scientific-modeling-cloud-gpus-fit-guide]
- 63. What are ASIC Chips? A Detailed Comparison with GPUs and ... [https://ai-stack.ai/en/asic-vs-gpu]
- 64. GPU Comparison Guide for AI Workloads in 2025 [https://www.hyperbolic.ai/blog/gpu-comparison]
- 65. AMBER 24 NVIDIA GPU Benchmarks | B200, RTX PRO ... [https://www.exxactcorp.com/blog/molecular-dynamics/amber-molecular-dynamics-nvidia-gpu-benchmarks]
- 66. Enabling Scalable AI-Driven Molecular Dynamics ... [https://developer.nvidia.com/blog/enabling-scalable-ai-driven-molecular-dynamics-simulations/]
- 67. FASDA: An FPGA-Aided, Scalable and Distributed ... [https://www.pnnl.gov/publications/fasda-fpga-aided-scalable-and-distributed-accelerator-range-limited-molecular-dynamics]
- 68. AIM for Allostery: Using the Ising Model to Understand ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4652859/]
- 69. Magnetocaloric effects and dynamic magnetic behavior in ... [https://www.sciencedirect.com/science/article/abs/pii/S0304885325009138]
- 70. Markov field models: Scaling molecular kinetics ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X22001373]
- 71. Comparing a simple theoretical model for protein folding with ... [https://pubmed.ncbi.nlm.nih.gov/24128764/]
- 72. Comparing molecular dynamics simulations of grain ... [https://www.sciencedirect.com/science/article/abs/pii/S1359646224004640]
- 73. Machine learning analysis of molecular dynamics ... [https://www.nature.com/articles/s41598-025-11392-1]
- 74. Computational design to experimental validation [https://pubs.rsc.org/en/content/articlelanding/2025/tb/d4tb02789b]
- 75. Exploring the promise and limitations of point-charge-free ... [https://www.nature.com/articles/s41598-025-06558-w]
- 76. Ising model - Wikipedia [https://en.wikipedia.org/wiki/Ising_model]
- 77. Simulating an Ising Model — Computing in Physics (498CMP) [https://courses.physics.illinois.edu/phys498cmp/sp2022/Ising/IsingModel.html]
- 78. The Theoretical and Statistical Ising Model: A Practical Guide in R [https://www.mdpi.com/2624-8611/3/4/39]
- 79. Advancements in molecular simulation for understanding ... [https://www.sciencedirect.com/science/article/pii/S0167732224015708]
- 80. Ising Model: Recent Developments and Exotic Applications II [https://www.mdpi.com/journal/entropy/special_issues/Ising_Model_II]
- 81. Foundation models for materials discovery – current state ... [https://www.nature.com/articles/s41524-025-01538-0]
- 82. Molecular Simulations Integrated with Experiments for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11242915/]
- 83. Generalized modeling of carbon film deposition growth via ... [https://www.nature.com/articles/s41524-025-01781-5]
- 84. A hybrid molecular dynamics–machine learning framework ... [https://www.sciencedirect.com/science/article/pii/S2214157X2500944X]
- 85. Use of AI-methods over MD simulations in the sampling ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1542267/full]
- 86. How AI Supercharge Molecular Simulation and ... [https://capestart.com/resources/blog/how-ai-has-supercharged-molecular-simulation-and-molecular-dynamics/]
- 87. Using Machine Learning to Analyze Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12147208/]
- 88. An artificial intelligence accelerated ab initio molecular ... [https://www.nature.com/articles/s41597-025-05338-5]
- 89. A beginner's approach to deep learning applied to VS and MD ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-00985-7]