A Practical Guide to Force Field Selection for Protein Simulations: From Foundations to Future-Proofing
Accurate molecular dynamics simulations are paramount for advancing research in drug design and understanding protein function, yet their predictive power is fundamentally limited by the choice of force field.
A Practical Guide to Force Field Selection for Protein Simulations: From Foundations to Future-Proofing
Abstract
Accurate molecular dynamics simulations are paramount for advancing research in drug design and understanding protein function, yet their predictive power is fundamentally limited by the choice of force field. This article provides a comprehensive framework for researchers and scientists to navigate force field selection for diverse protein types. We cover the foundational principles of major biomolecular force fields (AMBER, CHARMM, GROMOS, OPLS-AA), detail their specific applications and recommended combinations for different protein systems, address common pitfalls and optimization strategies, and present rigorous validation and comparative analysis techniques. By synthesizing the latest research, this guide aims to empower professionals to make informed, project-specific decisions that enhance the reliability and impact of their computational work.
Understanding the Force Field Landscape: AMBER, CHARMM, GROMOS, and OPLS-AA
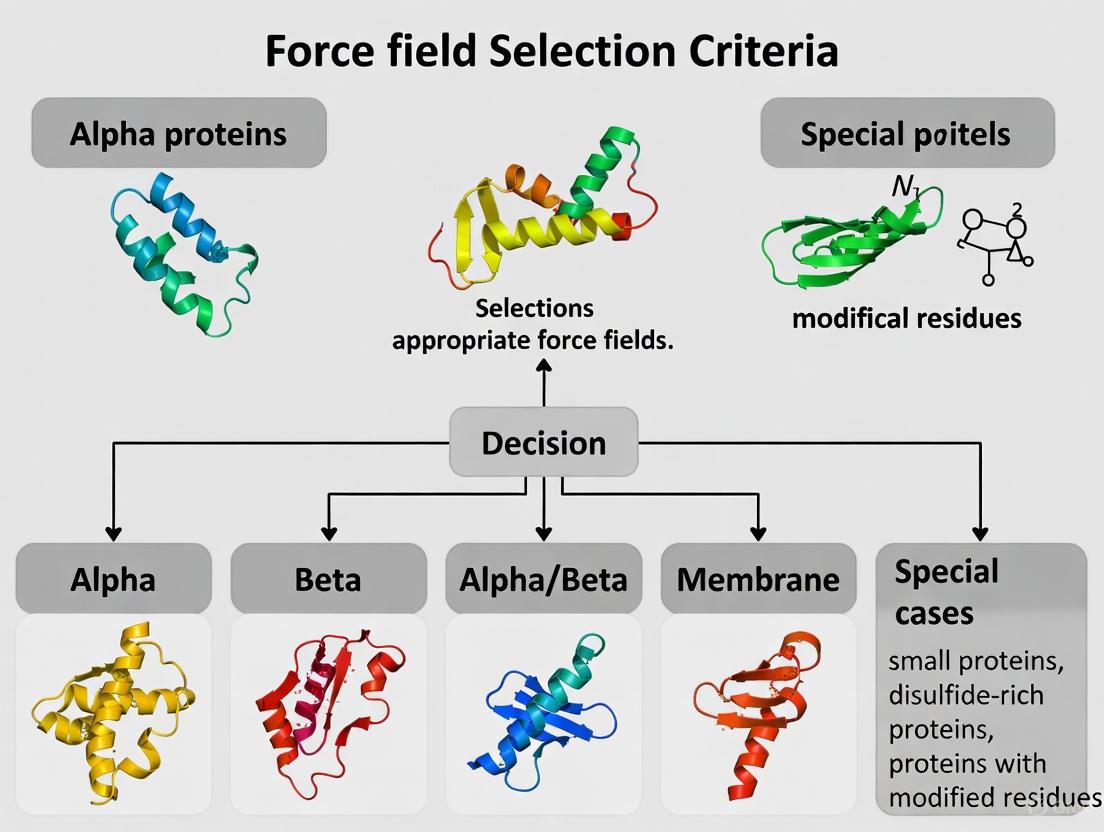
In molecular dynamics (MD) simulations, a force field refers to the computational model that describes the potential energy of a system of atoms and molecules. It is built from two core components: the potential functions, which are the mathematical equations used to calculate energies and forces, and the parameter sets, which are the specific numerical values fed into those equations [1]. The choice of an appropriate force field is foundational to the accuracy and reliability of simulations, particularly in biological research involving proteins, where different force fields are tuned for specific protein types, such as folded proteins versus intrinsically disordered proteins (IDPs) [2]. This guide details these components and provides troubleshooting advice for researchers.
FAQ: Understanding Force Field Components
Q1: What are the core components of a force field? A force field consists of two distinct parts:
- Potential Functions (Functional Form): This is the set of mathematical equations that define how the potential energy of a system depends on the positions of its atoms. This form is generally standardized for a given force field [3] [1].
- Parameter Sets: These are the specific numerical constants (e.g., atomic charges, bond force constants, equilibrium lengths) used within the potential functions. Parameters are derived from experimental data and quantum mechanical calculations and are specific to different types of atoms and molecules [3] [1].
Q2: What is the difference between an all-atom and a united-atom force field?
- All-atom force fields provide parameters for every atom in a system, including hydrogen atoms. Examples include OPLS-AA and AMBER99SB [3] [1].
- United-atom force fields treat the hydrogen atoms bound to carbon atoms as a single interaction center, thereby reducing the computational cost. An example is GROMOS-96 [3] [1].
Q3: My simulation of an Intrinsically Disordered Protein (IDP) is producing overly compact conformations. What might be wrong? Traditional force fields tuned for folded proteins often predict overly compact conformations for IDPs [2]. This is an active area of development. Troubleshooting steps include:
- Check your force field and water model: Recent improvements involve reweighting dihedral angle corrections (CMAPs) in protein force fields and using new water models with increased water-water dispersion interactions. The combination of the AMBER ff19SB protein force field and the OPC water model has been shown to predict both ordered and disordered polyampholyte sequences in good agreement with experimental Small Angle X-Ray Scattering (SAXS) data [2].
- Validate with experimental data: Use techniques like SAXS to compare the radius of gyration (Rg) and other ensemble properties of your simulation results against experimental data [2].
Q4: Is it safe to mix parameters from different force fields? No. Force fields are self-consistent sets of functional forms and parameters. The parameters from one force field are not transferable to another because they were derived together to work as a cohesive unit. Making ad hoc changes to a subset of parameters is dangerous and can lead to unpredictable and inaccurate results [3] [1].
Q5: What are the main energy terms in a typical molecular mechanics force field? The total potential energy ((E{total})) is generally composed of bonded and non-bonded interactions, and can be summarized as [3]: (E{total} = E{bonded} + E{nonbonded}) Where:
- (E{bonded} = E{bond} + E{angle} + E{dihedral})
- (E{nonbonded} = E{electrostatic} + E_{van der Waals})
Key Potential Functions in a Force Field
The table below summarizes the fundamental potential functions that make up a classical force field.
Table 1: Common Potential Energy Functions in Molecular Force Fields.
| Energy Term | Functional Form | Description |
|---|---|---|
| Bond Stretching | (E{bond} = \frac{k{ij}}{2}(l{ij} - l{0,ij})^2) | Harmonic potential describing the energy cost of stretching a bond between atoms (i) and (j) from its equilibrium length, (l{0,ij}). (k{ij}) is the force constant [3]. |
| Angle Bending | (E{angle} = \frac{k{\theta}}{2}(\theta{ijk} - \theta{0,ijk})^2) or cosine-based | Harmonic potential describing the energy cost of bending the angle between three bonded atoms (i-j-k) from its equilibrium value, (\theta_{0}) [3] [1]. |
| Torsional Dihedral | (E{dihedral} = \sum{n} \frac{V_n}{2} [1 + cos(n\phi - \gamma)]) | Periodic potential describing the energy barrier for rotation around the central bond of four sequentially bonded atoms. (V_n) is the barrier height, (n) is the periodicity, and (\gamma) is the phase [3]. |
| van der Waals | (E{vdW} = 4\epsilon{ij} \left[ \left(\frac{\sigma{ij}}{r{ij}}\right)^{12} - \left(\frac{\sigma{ij}}{r{ij}}\right)^{6} \right]) | Lennard-Jones potential describing attractive (dispersion) and repulsive (electron cloud overlap) interactions between non-bonded atoms. (\epsilon) is the well depth and (\sigma) is the collision diameter [3]. |
| Electrostatic | (E{Coulomb} = \frac{1}{4\pi\varepsilon0} \frac{qi qj}{r_{ij}}) | Coulomb's law describing the interaction between atomic partial charges (qi) and (qj) separated by a distance (r_{ij}) [3]. |
Troubleshooting Guide: Force Field Selection and Errors
Table 2: Common Force Field Issues and Recommended Solutions.
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Overly compact IDP ensembles | Force field biased towards folded proteins; water model with insufficient dispersion [2]. | Switch to a force field and water model validated for IDPs, such as AMBER ff19SB with the OPC water model [2]. |
| Unphysical bond lengths or angles | Incorrect assignment of atom types or parameters; a corrupted topology file. | Re-run the topology generation process. Manually check the parameters for the affected bonds/angles against the force field's documentation. |
| Unstable simulation (energy blow-up) | Overlapping atoms in the initial structure; incorrect treatment of cut-offs for long-range interactions. | Perform more thorough energy minimization before dynamics. Ensure the non-bonded interaction cut-off scheme (e.g., PME for electrostatics) is appropriate for your force field. |
| Incorrect density for a solvent | Parameter set not developed or tested for the specific molecule. | Use a component-specific parameter set developed for that solvent, or a transferable force field that includes validated parameters for it [3]. |
| Poor agreement with experimental data (e.g., SAXS) | The force field is not generalizable for your specific system or property of interest [2]. | Validate the force field against a known experimental property for a similar system before application. Consider using a detailed scattering model like SWAXS-AMDE for direct comparison [2]. |
Experimental Protocols: Validating a Force Field with SAXS
For researchers validating force fields, particularly for IDPs, comparison to Small Angle X-Ray Scattering (SAXS) data is a powerful method. The following workflow, used in a recent study of polyampholyte peptides, provides a detailed protocol [2].
Protocol: Isolating the Role of Force Field and Water Model using SAXS [2]
Sample Preparation:
- Synthesize sequence-precise peptides (e.g., EK polyampholytes like (EK)₁₆) using Fmoc-based solid-phase peptide synthesis (SPPS).
- Purify the samples using reversed-phase HPLC (RP-HPLC).
- Ion exchange, dialyze, and lyophilize the samples to ensure monodispersity and reduce counterion effects.
- Prepare SAXS sample solutions at a low concentration (e.g., 0.5 wt%) to minimize solute-solute interactions.
Data Collection:
- Conduct SAXS experiments at a synchrotron beamline (e.g., NSLS-II LiX).
- Use a flow cell setup and collect static scattering measurements at multiple exposures.
- Perform data reduction, averaging, and background subtraction using provided software (e.g., BioXTAS RAW).
Molecular Dynamics Simulation:
- Set up MD simulations of the peptide in explicit solvent.
- Run simulations using different combinations of protein force fields (e.g., AMBER ff19SB) and water models (e.g., OPC) to isolate their individual contributions.
Computational Scattering Analysis:
- Use an advanced scattering model like SWAXS-AMDE (Small and Wide Angle X-ray Scattering for All Molecular Dynamics Engines).
- Process the MD simulation trajectories with SWAXS-AMDE to compute the background-subtracted scattering profiles. This model accounts for solvent density changes in atomic detail and incorporates the effect of a thermally fluctuating solute, which is critical for IDPs.
Validation and Analysis:
- Directly compare the simulated scattering profiles to the experimental SAXS data.
- Analyze additional ensemble properties from the simulation, such as the radius of gyration (Rg) and dihedral angle distributions, to understand the conformational preferences predicted by the force field.
Table 3: Key Resources for Force Field-Based Research.
| Item | Function / Description | Example Tools & Databases |
|---|---|---|
| MD Simulation Software | Engine to perform the molecular dynamics calculations. | GROMACS [1], AMBER [2], NAMD [2], OpenMM [2]. |
| Force Field Parameter Databases | Repositories providing digitally available parameter sets. | MolMod (molecular and ionic force fields) [3], openKim (interatomic potentials) [3], TraPPE (transferable force fields for organic molecules) [3]. |
| Scattering Analysis Software | Translates MD trajectories into experimental observables for validation. | SWAXS-AMDE (open-source, works with multiple MD engines) [2], CRYSOL, WAXSiS [2]. |
| Coarse-Graining Tools | Assists in parameterizing coarse-grained models for larger systems. | VOTCA (Versatile Object-oriented Toolkit for Coarse-Graining Applications) [1]. |
| Specialized Force Fields | Parameter sets designed for specific molecules or simulation types. | MARTINI (coarse-grained for biomolecules) [1], AMOEBA (polarizable force field) [4], PLUM (solvent-free protein-membrane model) [1]. |
Additive all-atom force fields are the foundation of modern molecular dynamics (MD) simulations, providing the computational models that describe the interactions between all atoms in a biomolecular system. These force fields, characterized by fixed partial atomic charges and pairwise additive nonbonded interactions, enable researchers to study protein folding, ligand binding, and conformational changes at an atomic level. This technical support center addresses common challenges and questions researchers face when working with these essential tools, particularly within the CHARMM and AMBER force field ecosystems, to help you select and implement the appropriate parameters for your specific protein research.
Frequently Asked Questions (FAQs)
What is the fundamental difference between additive and polarizable force fields?
Additive all-atom force fields, such as CHARMM36 and AMBER FF14SB, use a fixed partial charge assigned to each atom. Electrostatic interactions are calculated using a pairwise additive approximation, meaning the total interaction is the sum of individual atom-atom interactions [5] [6]. This makes them computationally efficient and the most widely used for routine biomolecular simulations.
Polarizable force fields, like the CHARMM Drude model, introduce a more sophisticated treatment of electrostatics by allowing the charge distribution of atoms to change in response to their local chemical environment. This is often achieved by attaching virtual particles (Drude oscillators) to atoms via a harmonic spring [6]. While polarizable force fields are inherently more accurate for simulating systems with varying dielectric environments (e.g., membrane proteins, ion channels), they come at a significantly higher computational cost [5] [6].
How do I handle non-natural amino acids or chemical modifications in my protein?
Simulating proteins with non-natural residues or post-translational modifications (PTMs) is a common challenge. The standard force field files only include parameters for the 20 standard amino acids. To incorporate non-standard elements:
- Parameter Generation: You typically need to generate new topology and parameter entries for your non-standard residue. This can be done using tools like the
CGenFFprogram for CHARMM orAntechamberfor AMBER force fields [7] [5]. - Force Field Integration: After generating the parameters for the non-natural residue, you must add them to your simulation topology and include the new bonded and nonbonded parameters (bonds, angles, dihedrals, charges, etc.) in the force field parameter file (often
ffbonded.itpandffnonbonded.itpin GROMACS, or similar in other packages) [7]. - Caution: A frequent error, especially when porting methods between force fields, is missing parameters for specific interactions. If you encounter errors about missing "Bond types," "U-B types," or "Dih. types," you must ensure all necessary parameters for your new residue have been correctly added to the force field files [7].
My simulation fails with "Unknown CMAP torsion" errors. What does this mean and how can I fix it?
The CMAP (Correction Map) term is an extension to the protein backbone potential in force fields like CHARMM. It is a grid-based correction that significantly improves the accuracy of backbone conformational sampling, particularly for proteins [6] [8].
An "Unknown CMAP torsion" error occurs when the simulation software cannot find the CMAP parameters for a specific set of backbone atoms in your topology. This can happen if:
- The force field files are not correctly linked or included in your simulation setup.
- You are using a topology file generated for one force field (e.g., CHARMM) with the parameters of another (e.g., AMBER) that does not use the CMAP term.
- The topology for a non-standard residue is missing a CMAP assignment.
Solution: Ensure you are using a consistent and complete set of force field files. For CHARMM36 simulations, your topology must include the correct #include statements for the CMAP parameters. When working with non-standard residues, you may need to define a CMAP cross-term or ensure the residue is correctly patched into a polypeptide chain.
Which software packages are compatible with CHARMM and AMBER additive force fields?
Most modern MD packages support the major additive force fields. CHARMM-GUI, a widely used web-based platform for building simulation systems, generates inputs for many popular programs [9]:
| MD Software Package | CHARMM Force Field | AMBER Force Field |
|---|---|---|
| NAMD | Yes | Yes |
| GROMACS | Yes | Yes |
| AMBER | Yes | Yes |
| OpenMM | Yes | Yes |
| CHARMM/OpenMM | Yes | No |
| GENESIS | Yes | Yes |
| Desmond | Yes | Yes |
| LAMMPS | Yes | Yes |
How do I choose the right water model and ions for my force field?
Force fields are parametrized in conjunction with specific water models to reproduce experimental condensed-phase properties. Using the correct water model is critical for simulation accuracy.
- CHARMM36: Typically used with a modified TIP3P water model [8].
- AMBER: Often used with TIP3P, but TIP4P-EW and OPC are also supported options in tools like CHARMM-GUI [9].
Ion parameters are also force-field specific. CHARMM-GUI, for instance, allows for the use of standard ions or specialized "12-6-4" ion models for more accurate behavior [9]. Always use the ion parameters that are part of your chosen force field's distribution.
Troubleshooting Guides
Problem 1: Parameterization Errors for Novel Ligands or Cofactors
Symptoms: Simulation initialization fails with errors about missing atom types, bond types, or improper dihedrals for a non-standard molecule.
Solution Protocol:
- Generate Parameters: Use a dedicated parameter generation tool. For CHARMM, use the
CGenFFprogram. For AMBER, useAntechamberwith theGAFForGAFF2force field [7] [9]. - Validate Parameters: Always check the penalty scores provided by tools like
CGenFF. High penalties indicate that the analogy used to generate parameters is poor, and you may need to perform quantum mechanical (QM) calculations for more accurate parametrization. - Incorporate into Simulation:
- Append the generated topology for the ligand to your protein topology file.
- Add the new bonded and nonbonded parameters to your force field parameter file (e.g.,
ffbonded.itpandffnonbonded.itpin GROMACS).
- Test and Validate: Run a short energy minimization and equilibration of the ligand in water to ensure stability. Compare the QM-calculated and MM-calculated geometries of the ligand to validate the parameters.
The following diagram illustrates the workflow for this protocol:
Problem 2: System Instability During Equilibration
Symptoms: Simulation crashes during the first steps of equilibration with errors like "LINCS warning," "bond length violation," or atoms flying off to infinity. This often indicates bad contacts or incorrect topology.
Solution Protocol:
- Check for Overlaps: Visualize the initial structure (e.g., in VMD or PyMOL). It is normal for automatically assembled systems (e.g., from CHARMM-GUI) to have minor bad contacts, as these are typically removed during initial minimization [9].
- Verify Topology: Ensure all residues in your structure are correctly assigned and that there are no missing atoms or residues in your topology relative to your coordinate file.
- Gradual Equilibration: Do not jump directly to production dynamics. Follow a multi-step equilibration protocol:
- Energy Minimization: Heavily minimize the system to remove any steric clashes.
- Gradual Heating: Heat the system slowly to the target temperature (e.g., from 0 K to 300 K over 100-200 ps) while applying position restraints on solute (protein/ligand) heavy atoms.
- Pressure Coupling: After heating, slowly release the position restraints while coupling the system to a barostat to achieve the correct density.
The following diagram illustrates the step-by-step equilibration protocol:
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key computational "reagents" and tools essential for setting up and running simulations with additive all-atom force fields.
| Research Reagent / Tool | Function / Purpose in Simulation |
|---|---|
| CHARMM36 Force Field | An additive all-atom force field providing parameters for proteins, nucleic acids, lipids, and carbohydrates; optimized to reproduce experimental condensed-phase properties [6] [8]. |
| AMBER FF14SB Force Field | Another widely used additive force field, considered the gold standard for protein simulations within the AMBER ecosystem [9]. |
| TIP3P Water Model | A 3-site water model that is the standard companion for the CHARMM36 and many AMBER force fields, used to solvate biomolecules [8]. |
| CHARMM-GUI | A web-based suite that automates the construction of complex simulation systems (proteins, membranes, solutions) and generates input files for multiple MD engines [9]. |
| GAFF/GAFF2 (General Amber Force Field) | A force field designed to generate parameters for a wide variety of small organic molecules, making it compatible with AMBER protein force fields [9]. |
| CGenFF (CHARMM General Force Field) | The CHARMM-equivalent of GAFF, used to obtain parameters for drug-like molecules and connect them to the CHARMM force field [6]. |
| ION Parameters | Specific nonbonded parameters for ions (e.g., Na+, K+, Cl-) that are compatible with the chosen force field and water model, crucial for simulating physiological conditions [9]. |
Frequently Asked Questions (FAQs)
Q1: What are the main differences between the ff14SB and ff19SB force fields?
The ff19SB force field represents an evolution from ff14SB, with improvements in the backbone torsion potentials parameterized using a high-quality quantum mechanics dataset. While ff14SB remains highly reliable and widely used, ff19SB aims to provide a more accurate description of protein backbone conformations. Both can be supplemented with specialized parameters for non-standard amino acids, such as the recently developed phosaa14SB and phosaa19SB parameters for phosphorylated amino acids [10].
Q2: Which AMBER force field should I use for simulating intrinsically disordered proteins (IDPs)? For IDPs or proteins with intrinsically disordered regions (IDRs), the ff14IDPSFF force field is recommended. It is a specialized variant of ff14SB that incorporates backbone dihedral corrections for all 20 naturally occurring amino acids to better sample the conformational ensembles of disordered proteins [11]. Generic force fields like ff14SB or ff19SB, when combined with standard water models like TIP3P, can sometimes lead to an artificial structural collapse of disordered regions, which ff14IDPSFF helps to correct [12] [11].
Q3: How does the choice of water model impact my simulation results? The water model is critical. Studies have shown that the TIP3P water model, for example, can contribute to an artificial collapse of disordered protein regions [12]. The TIP4P-D water model has been demonstrated to significantly improve the reliability of simulations for proteins containing both structured and disordered regions when combined with biomolecular force fields like Amber99SB-ILDN, CHARMM22*, or CHARMM36m [12]. The influence of solvent models may be less pronounced when using an IDP-specific force field like ff14IDPSFF [11].
Q4: Can I use ff14IDPSFF for folded, structured proteins? Yes. Validation simulations indicate that ff14IDPSFF can still accurately model the structural and dynamical properties of folded proteins, with some tests showing slightly better agreement in loop regions compared to the base ff14SB force field [11].
Q5: Where can I find parameters for phosphorylated amino acids?
The standard ff14SB and ff19SB force fields do not contain parameters for phosphorylated amino acids. However, you can use the recently developed phosaa14SB and phosaa19SB parameter sets, which provide compatible parameters for these important post-translational modifications, allowing you to maintain accuracy comparable to simulations of standard amino acids [10].
Troubleshooting Guides
Issue 1: Unrealistic Structural Collapse in Disordered Regions
Problem: During a simulation of a protein with an intrinsically disordered region, the IDR collapses into an overly compact state that does not match experimental data (e.g., from NMR or SAXS).
Solutions:
- Check your Force Field and Water Model Combination: A common cause is using a generic force field with a standard water model.
- Recommended Action: Switch to a force field and water model combination validated for disordered systems.
- Option A (AMBER-focused): Use the IDP-specific force field ff14IDPSFF with TIP3P water [11].
- Option B (Water Model-focused): Use a force field like Amber99SB-ILDN paired with the TIP4P-D water model, which has been shown to improve results for hybrid proteins with both ordered and disordered domains [12].
Validation Protocol:
- Predict Experimental Observables: From your simulation trajectory, calculate parameters such as the radius of gyration (Rg) and NMR relaxation data [12].
- Compare with Experiments: Compare the calculated Rg with data from Small-Angle X-ray Scattering (SAXS) experiments. Compare predicted NMR chemical shifts and relaxation rates with experimental NMR data [12].
- Diagnose: A poor match, especially for NMR relaxation parameters which are highly sensitive to dynamics, indicates a problem with the force field/water model combination [12].
Issue 2: Inaccurate Secondary Structure Propensities
Problem: Your simulation fails to maintain a known transient helical motif or shows incorrect propensity for beta-sheet formation.
Solutions:
- Verify Force Field Balance: Some force fields may have biases towards or against certain secondary structure types.
- Recommended Action: Benchmark multiple force fields if possible. The choice of force field has been shown to be critical for retaining transient helical motifs observed in NMR experiments [12]. Ensure you are using a modern force field like ff14SB or ff19SB, which have better balances than older versions.
Validation Protocol:
- Analyze Secondary Structure Propensity: Use tools like
cpptrajto analyze the percentage of time your protein or peptide spends in helical or sheet conformations over the simulation trajectory. - Compare with Chemical Shifts: Predict chemical shifts from your trajectory and compare them with experimental NMR chemical shifts. Deviations can indicate inaccurate secondary structure populations [11].
Issue 3: Setting Up Simulations with Specialized Parameters
Problem: You need to incorporate parameters for non-standard residues (e.g., phosphorylated amino acids) into your AMBER topology.
Solutions:
- Use Supplemental Parameter Files: For phosphorylated amino acids, use the provided
phosaa14SBorphosaa19SBlibrary and parameter files [10]. - Modify Topology Files: Use the
LEaPmodule to load the standard force field (ff14SB or ff19SB) and then the supplemental parameter files. The AMBER community often provides Perl scripts to facilitate the revision of standard topology files to incorporate these specialized parameters [11].
Force Field Comparison and Selection Table
The table below summarizes key AMBER force fields and their target applications to guide your selection.
| Force Field | Basis | Target Application | Key Strengths | Special Notes |
|---|---|---|---|---|
| ff14SB | ff99SB | General purpose proteins [13] | Balanced accuracy for folded proteins; widely validated [11] [13] | Compatible with phosaa14SB and ff14IDPSFF [11] [10] |
| ff19SB | New QM data | General purpose proteins [10] | Improved backbone torsion potentials [10] | Compatible with phosaa19SB parameters [10] |
| ff14IDPSFF | ff14SB | Intrinsically Disordered Proteins (IDPs) [11] | Corrects backbone dihedrals for all 20 amino acids; improves agreement with NMR data [11] | Can still model folded proteins accurately [11] |
Experimental Validation Workflow
The following diagram outlines a robust protocol for validating your force field selection against experimental data, particularly for proteins containing disordered regions.
Force Field Validation Workflow
Detailed Methodology for Validation Protocol:
- System Setup: Prepare your protein structure in a solvated box with ions, using the
LEaPmodule in AMBER to generate topologies and coordinates with your chosen force field and water model [12] [11]. - MD Simulation: Run a production molecular dynamics simulation. For meaningful validation of conformational ensembles, simulations should be as long as computationally feasible (e.g., reaching microsecond timescales for smaller systems) [12] [14].
- Trajectory Analysis:
- Radius of Gyration (Rg): Use the
cpptrajmodule in AMBER to calculate the Rg over the trajectory. This measures the overall compactness of the protein [12]. - NMR Chemical Shifts: Employ tools like
SHIFTX2or similar to predict chemical shifts from the MD trajectory [12] [11]. - NMR Relaxation Parameters: Use analytical methods to predict NMR relaxation rates (R1, R2, and NOE) from the simulated dynamics [12].
- Radius of Gyration (Rg): Use the
- Comparison with Experiment:
- Compare the calculated Rg profile with experimental data from Small-Angle X-ray Scattering (SAXS). The molecular form factor from SAXS can be used for direct comparison [12].
- Perform a quantitative comparison of the predicted and experimental NMR chemical shifts and relaxation rates. NMR relaxation is particularly sensitive to the choice of force field and can reveal unrealistic dynamics even when the average Rg appears correct [12].
The Scientist's Toolkit: Essential Research Reagents and Materials
| Item | Function in Force Field Research |
|---|---|
| AMBER Software Suite | A primary software package for MD simulations, containing tools for system setup (LEaP), simulation (pmemd), and trajectory analysis (cpptraj) [13]. |
| ff14SB / ff19SB Force Field Parameters | The core parameter sets defining the bonded and non-bonded interactions for standard amino acids in proteins [10] [13]. |
| Specialized Parameter Sets (e.g., ff14IDPSFF, phosaa14/19SB) | Supplemental parameters that extend the core force fields to handle specific systems like intrinsically disordered proteins or post-translationally modified residues [11] [10]. |
| Explicit Solvent Models (TIP3P, TIP4P-D) | Water models that define the properties of the solvent environment. TIP4P-D is particularly noted for improving simulations of disordered proteins [12]. |
| NMR Spectroscopy Data | Experimental data used for critical validation of force fields. Key metrics include chemical shifts, residual dipolar couplings (RDCs), and relaxation rates (R1, R2, hetNOE) [12] [14]. |
| Small-Angle X-Ray Scattering (SAXS) Data | Experimental data providing a solution-state measurement of a protein's radius of gyration (Rg) and overall shape, used to validate structural compaction [12]. |
The CHARMM (Chemistry at HARvard Macromolecular Mechanics) force fields are a family of all-atom potential energy functions widely used for molecular dynamics simulations of biological macromolecules. The CHARMM36 (C36) update represents a significant refinement over its predecessors (C27 and C27r), addressing systematic flaws to better reproduce experimental data for diverse biomolecular systems [15]. A central innovation in modern CHARMM protein force fields is the CMAP (energy map) correction, a grid-based method that directly adjusts the potential energy surface for protein backbone dihedrals (φ and ψ angles) to achieve better agreement with quantum mechanical data and experimental observables [16] [17].
The Role of CMAP Backbone Corrections
The CMAP correction was introduced to rectify inaccuracies in the backbone conformational preferences of the earlier CHARMM22 (C22) force field. Its inclusion has led to substantial improvements in simulating both structured and intrinsically disordered protein regions [12].
- Enhanced Structural Accuracy: Simulations using C22/CMAP demonstrated significantly lower Cα atom positional root-mean-square differences from crystal structures (0.9 ± 0.1 Å) compared to C22 alone (1.8 ± 0.2 Å) in studies of hen lysozyme [16].
- Improved Dynamics: The CMAP correction yields more accurate backbone dynamics, bringing NMR-derived order parameters (S²) and root-mean-square fluctuations into closer agreement with experimental measurements [16].
- Increased Cooperativity: The revised CMAP potential results in enhanced cooperativity of α-helix and β-hairpin formation, better reproducing the experimental temperature dependence of helix formation and parameters for helix nucleation and elongation [17].
Figure 1: CMAP Correction Workflow. The CMAP correction takes CHARMM22 as input and produces an improved force field by directly adjusting backbone torsion potentials, secondary structure balance, and protein dynamics.
Frequently Asked Questions (FAQs) and Troubleshooting Guides
Force Field Selection and Compatibility
Q1: Which CHARMM force field should I use for my specific biomolecular system?
The optimal CHARMM force field depends on your system composition and research goals. The following table summarizes recommended force fields for different biomolecular systems:
Table 1: CHARMM Force Field Selection Guide for Different Biomolecular Systems
| System Type | Recommended Force Field | Key Improvements and Applications |
|---|---|---|
| Lipid Bilayers | CHARMM36 (C36) | Corrects surface area per lipid (within 2% of experiment); improved deuterium order parameters, area compressibility modulus, and density profiles [15]. |
| Proteins with Structured Domains | CHARMM22/CMAP or CHARMM36 | Improved backbone dihedral distributions; better agreement with NMR order parameters and crystallographic B-factors [16]. |
| Intrinsically Disordered Proteins (IDPs) | CHARMM36m | Modified TIP3P water model with strengthened protein-water interactions (H ε = 0.1 kcal/mol) prevents artificial collapse of disordered regions [12] [18]. |
| Membrane Proteins | CHARMM36 for both protein and lipid components | Consistent parameterization ensures proper protein-lipid interactions; compatible with the tensionless ensemble (NPT) simulations [15]. |
| Nucleic Acids | CHARMM36 | Includes updated nucleic acid model compounds and parameters; compatible with the latest CGenFF for small molecules [18]. |
Q2: I am using GROMACS with CHARMM36. What are the recommended MD simulation parameters?
When using CHARMM36 in GROMACS, employ these settings in your .mdp file [19]:
Note that dispersion correction should not be applied for lipid bilayer simulations, though it may be necessary for lipid monolayers [19]. The switching distance (0.8-1.2 nm) can be lipid-dependent, so consult literature for your specific lipids [19].
Q3: How do I handle terminal patches for N-terminal MET residues in CHARMM36 with GROMACS?
When using pdb2gmx for a polypeptide with an N-terminal MET residue, you must interactively choose the appropriate terminal patch. The program may erroneously select a carbohydrate-specific patch due to name matching. Use the -ter flag and manually select NH3+ for standard polypeptide termini [18].
Common Errors and Solutions
Q4: I encounter the error "atom C1 not found in building block 1MET while combining tdb and rtp" when using pdb2gmx with CHARMM36. How can I fix this?
This error occurs because the COO- terminus entry in aminoacids.c.tdb incorrectly uses atom "CB" as a control atom for adding OT1 and OT2 to C-terminal glycine residues. Until officially fixed, edit the relevant line in aminoacids.c.tdb [18]:
- Change from:
2 8 OT C CA CB - Change to:
2 8 OT C CA N
This modification replaces the reference to the non-existent glycine CB atom with the backbone N atom.
Q5: My CHARMM36 lipid bilayer simulation yields incorrect structural properties. What should I check?
First, verify that you are using the correct combination of force field files and simulation parameters. The CHARMM36 lipid force field has been specifically optimized to reproduce key experimental properties [15]:
Table 2: Benchmark Values for CHARMM36 Lipid Bilayer Validation
| Property | Target Experimental Agreement | Common Issues with Older Force Fields |
|---|---|---|
| Surface Area per Lipid | Within 2% of experimental values | C27 and C27r yielded smaller areas, gel-like structures above transition temperature [15]. |
| Deuterium Order Parameters (Scd) | Proper splitting for aliphatic carbon adjacent to carbonyl | C27r failed to reproduce fine structure at lipid-water interface [15]. |
| Area Compressibility Modulus (Kₐ) | Agreement with experimental measurements | C27 and C27r underestimated Kₐ [15]. |
| Simulation Ensemble | Tensionless ensemble (NPT) | Earlier versions required artificial surface tension to maintain area [15]. |
If issues persist, ensure you're using the latest version of the force field port, as periodic updates address identified issues (e.g., missing NBFIX terms for Ca²⁺ ions in earlier releases) [18].
Q6: When simulating hybrid proteins with ordered and disordered regions, which force field and water model combination is recommended?
For hybrid systems, CHARMM36m with the TIP4P-D water model has demonstrated superior performance. This combination prevents the artificial collapse of disordered regions while maintaining the structural integrity of ordered domains [12]. The TIP4P-D water model, combined with CHARMM36m, significantly improved reliability compared to TIP3P, which could cause unrealistic structural collapse and inaccurate NMR relaxation properties [12].
Experimental Protocols and Validation
Protocol: Validation of CHARMM36 Force Field for Lipid Bilayers
This protocol outlines the key steps for validating the CHARMM36 force field for phospholipid bilayer simulations, based on the methodology used in the original parameterization [15].
1. System Setup
- Select phospholipids of interest (e.g., DPPC, DMPC, POPC, DOPC, POPE).
- Build hydrated bilayers with at least 64 lipids per leaflet and sufficient water (∼50 waters/lipid for high hydration).
- Generate initial coordinates and topology using CHARMM-GUI Membrane Builder or similar tools.
2. Simulation Parameters
- Employ the NPT ensemble (constant particle number, pressure, and temperature) with semi-isotropic pressure coupling.
- Use a 2 fs time step with LINCS constraints for all bonds involving hydrogen.
- Apply the recommended cut-off schemes as specified in Section 2.1.
- Maintain temperature at 310 K (or relevant physiological temperature) and pressure at 1 atm.
3. Production Simulation and Analysis
- Run simulations for sufficient duration to ensure equilibration (typically ≥100 ns).
- Calculate the following properties for comparison with experimental data:
- Surface area per lipid: Compare with X-ray and neutron scattering data.
- Deuterium order parameters: Calculate SCD profiles and compare with NMR measurements.
- Electron density profiles: Compare with X-ray diffraction experiments.
- Area compressibility modulus: Derive from area fluctuations.
4. Validation Metrics
- Target agreement with experimental surface area within 2%.
- Verify proper splitting of SCD for the aliphatic carbon adjacent to the carbonyl.
- Ensure density profiles match neutron and X-ray diffraction data.
Protocol: Assessing CMAP Corrections for Protein Simulations
This protocol describes how to evaluate the impact of CMAP corrections on protein simulations [16] [17].
1. System Preparation
- Select a well-characterized model protein (e.g., hen lysozyme, ubiquitin).
- Generate starting structure from crystallographic coordinates (e.g., PDB: 6LYT).
- Solvate in a rectangular box with explicit solvent (TIP3P or TIP4P-D), ensuring minimum distance between protein and box edge.
2. Simulation Details
- Perform comparative simulations with and without CMAP correction (C22 vs. C22/CMAP).
- Use particle mesh Ewald electrostatics with periodic boundary conditions.
- Run simulations for sufficient time to converge dynamics (≥25 ns for lysozyme).
- Conduct multiple independent runs with different initial velocities for error analysis.
3. Analysis and Comparison with Experiment
- Calculate Cα root-mean-square deviations from crystal structures.
- Compute backbone NMR order parameters (S²) from N-H bond vector fluctuations.
- Determine root-mean-square fluctuations (RMSF) and compare with crystallographic B-factors.
- Analyze φ,ψ dihedral angle distributions and compare with database statistics.
4. Expected Outcomes with CMAP
- Reduced RMSD from crystal structures (e.g., from 1.8 Å to 0.9 Å for lysozyme).
- Improved agreement with experimental S² values (RMS deviation from experiment reduced from 0.179 to 0.067).
- Better correspondence with RMSF from crystallographic B-factors.
- Elimination of hyperflexibility in loop and turn regions.
Figure 2: CMAP Validation Workflow. Comparative simulation protocol for assessing the impact of CMAP corrections on protein structural and dynamic properties.
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Computational Tools for CHARMM Force Field Simulations
| Item | Function/Purpose | Availability |
|---|---|---|
| CHARMM36 Force Field Files | Provides parameters for lipids, proteins, nucleic acids, and carbohydrates | MacKerell Lab website (http://mackerell.umaryland.edu/charmm_ff.shtml) [18] |
| CHARMM-GUI | Web-based platform for building complex biomolecular systems and generating simulation inputs | https://charmm-gui.org/ [9] |
| GROMACS Simulation Package | High-performance molecular dynamics software supporting CHARMM force fields | http://www.gromacs.org/ [19] |
| AMBER/CHARMM Conversion Tools | Utilities for converting between different molecular dynamics formats (amb2gmx.pl, ACPYPE) | Distributed with AmberTools or separately [19] |
| CHARMM36m Force Field | Specialized version for intrinsically disordered proteins and structured/disordered hybrids | Included in current CHARMM36 distributions [12] [18] |
| Modified TIP3P Water Model | Enhanced water model for CHARMM36m with H ε = 0.1 kcal/mol | Apply define = -DUSE_MODIFIED_TIP3P_EPS in GROMACS .mdp file [18] |
| LJ-PME Parameters for Lipids | Alternative nonbonded treatment for lipids compatible with PME electrostatics | Separate force field port in current CHARMM36 distributions [18] |
Frequently Asked Questions (FAQs)
Q1: What are the primary use cases for the GROMOS-96 force field? GROMOS-96 is a united-atom force field, meaning it does not include explicit aliphatic (non-polar) hydrogens [19]. It is generally recommended for united-atom setups and has been historically used for the simulation of proteins, nucleotides, and sugars in aqueous solutions [20] [21]. However, it is not recommended for use with long alkanes and lipids [20].
Q2: My simulation with GROMOS-96 yields unexpected physical properties, like incorrect densities. What could be wrong? This is a known critical issue. The GROMOS force fields were parametrized using a physically incorrect multiple-time-stepping scheme for a twin-range cut-off [22] [23] [19]. When used with modern, correct single-range cut-off schemes or integrators, properties like density can deviate from intended values [22]. You should verify that your simulation parameters are compatible with the force field's original parametrization or consult current literature for correction strategies [19].
Q3: Can I use the GROMOS-96 43a1 parameter set for studying glycoproteins? Yes, research has indicated that GROMOS96 43a1, when enhanced with Löwdin HF/6-31G-derived atomic charges, can provide an accurate conformational description of glycoproteins in aqueous solution, showing agreement with experimental data [21].
Q4: Is it safe to mix different force field versions for my protein-ligand system? No, it is generally not advised. Using different force fields (e.g., GROMOS54a7 for a ligand and GROMOS96 43a1 for a protein) can result in inconsistent dynamics because the parameters were not developed to be compatible with each other [24]. You should use the same force field and parameter set for all components of your system.
Q5: What is the minimum Lennard-Jones cut-off (rvdw) I should use with GROMOS-96?
The GROMOS-96 force field was parameterized with a Lennard-Jones cut-off of 1.4 nm [20] [22]. You should ensure your rvdw parameter is set to at least this value. A larger cut-off is acceptable as the potential is negligible beyond 1.4 nm [20].
Troubleshooting Guides
Issue: Unphysical Bond Length Error During Topology Generation
Problem
When trying to generate a topology for a ligand using pdb2gmx or an online server like the Automated Topology Builder (ATB), you encounter an error such as: "Unsupported bond length encountered: 0.34nm (C23-H19). This often indicates there is an error in the submitted structure" [24].
Solution This error typically indicates a problem with the initial ligand geometry.
- Geometry Optimization: Optimize the 3D structure of your ligand before submitting it for topology generation. The initial coordinates from your
.pdbfile may contain unphysical bond lengths or angles. - Use Optimization Software:
Issue: "Residue not found in residue topology database" Error
Problem
The command gmx pdb2gmx -f ligand.pdb -o ligand_processed.gro -p ligand.top fails with the error: "Residue ‘UNK’ not found in residue topology database" [24].
Solution This error occurs because standard force field databases do not contain parameters for non-standard residues or novel ligands.
- Generate Ligand Parameters Separately: You cannot use
pdb2gmxfor unknown molecules. You need to derive parameters for your ligand separately and include them in your system's topology. - Utilize Specialized Servers/Tools: Use the Automated Topology Builder (ATB) or other tools like ACPYPE to generate topology files for your ligand that are compatible with GROMACS and your chosen force field [24].
- Manual Inclusion: Once you have the ligand's topology file (e.g.,
ligand.itp), you must manually include it in your main topology file (*.top) using the#include "ligand.itp"directive.
Issue: Choosing the Correct Parameter Set for Your System
Problem GROMOS-96 has multiple parameter sets (e.g., 43A1, 53A6, 54A7), and it can be confusing to select the right one for your specific research.
Solution Your choice should be guided by the biomolecule you are studying and the properties you wish to reproduce. The table below summarizes common GROMOS-96 parameter sets and their recommended applications.
Table: GROMOS-96 Parameter Sets and Applications
| Parameter Set | Recommended Application and Notes |
|---|---|
| 43A1 | Can be used for glycoprotein conformational studies when combined with specific atomic charges [21]. |
| 53A5 | Optimized to reproduce thermodynamic properties of pure liquids of small polar molecules and solvation free enthalpies in cyclohexane [25]. |
| 53A6 | A refinement of 53A5 where partial charges were adjusted to reproduce hydration free enthalpies in water. Recommended for simulations of biomolecules in explicit water [25]. |
| 54A7 | A later version based on 53A6, with adjustments to torsional angles for better helical propensities and improved parameters for charged amino acid side chains and ions [25]. It is generally advised to use this newer version over older ones for ligand topology if possible [24]. |
Quantitative Data and Methodologies
GROMOS-96 Force Field Parameter Sets
The following table provides a consolidated list of GROMOS-96 parameter sets included in GROMACS distributions.
Table: GROMOS-96 Parameter Sets in GROMACS
| Parameter Set | Status in GROMACS | Key Features / Intended Use |
|---|---|---|
| 43A1 | Included [19] | - |
| 43A2 | Included [20] [22] | Development version with improved alkane dihedrals [20]. |
| 45A3 | Included [20] [19] | Suitable for lipid aggregates, membranes, micelles, and apolar systems [25]. |
| 53A5 | Included [20] [19] | Optimized for condensed-phase properties of proteins and small molecules [20]. |
| 53A6 | Included [20] [19] | Optimized for condensed-phase properties of proteins and small molecules [20]. Recommended for biomolecules in explicit water [25]. |
| 54A7 | Included [19] | Includes improved torsional terms and new charges for charged amino acids and ions [25]. |
Force Field Validation Protocol
The methodology below, derived from a 2024 validation study, provides a framework for assessing the performance of a protein force field like GROMOS-96 [26].
Objective: To validate a protein force field against a curated set of experimental structures by comparing a range of structural metrics.
Curated Test Set:
- A set of 52 high-resolution protein structures (39 X-ray, 13 NMR) [26].
Simulation Setup:
- Software: GROMACS.
- Force Field: GROMOS-96 parameter set of choice.
- System: Protein solvated in explicit water.
- Replicates: Multiple independent simulation replicates are recommended to ensure statistical significance [26].
Validation Metrics to Calculate and Compare:
- Structural Metrics:
- Number of backbone hydrogen bonds.
- Number of native hydrogen bonds.
- Polar and nonpolar solvent-accessible surface area (SASA).
- Radius of gyration.
- Prevalence of secondary structure elements.
- Positional root-mean-square deviation (RMSD) from the experimental structure.
- Distribution of backbone φ and ψ dihedral angles.
- NMR-Derived Metrics (if applicable):
- J-coupling constants.
- Nuclear Overhauser effect (NOE) intensities.
Analysis: Statistically compare the average values of each metric from your simulations against the experimental data or between different force fields. Note that improvement in one metric may be offset by a loss of agreement in another, so a holistic view is necessary [26].
Diagram 1: Force field validation workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Components for GROMOS-96 Simulations
| Item / Resource | Function / Description |
|---|---|
| GROMACS Software | The molecular dynamics simulation package used to run the simulations [20] [19]. |
| GROMOS-96 Parameter Files | The set of parameters (e.g., *.itp, *.rtp) distributed with GROMACS that define the force field for your molecules [20]. |
| Automated Topology Builder (ATB) | An online server used to generate topologies and parameters for ligands and small molecules that are not in the standard force field database [24]. |
pdb2gmx Tool |
A GROMACS program used to process coordinate files and generate topologies for proteins and other molecules that are present in the force field's residue database [20]. |
| VOTCA Toolkit | A versatile toolkit for coarse-graining applications; useful for parameterization and analysis, with a well-tested interface to GROMACS [20] [22]. |
| Geometry Optimization Software (e.g., Avogadro, Gaussian) | Used to optimize the 3D structure of ligands prior to topology generation to avoid unphysical bond lengths and angles [24]. |
Diagram 2: Topology generation decision pathway.
Troubleshooting Guides
FAQ 1: How does OPLS-AA perform for intrinsically disordered proteins (IDPs) and unfolded states compared to other force fields?
Issue: Simulations of unfolded or disordered proteins result in overly compact structures that do not match experimental dimensions.
Explanation: Many traditional all-atom force fields, including earlier versions of AMBER and CHARMM, were primarily optimized for folded protein structures and exhibit a tendency to overly favor collapsed unfolded states. This is often due to insufficiently favorable protein-water interactions, causing the simulated chain to collapse in on itself [27].
Solutions:
- Consider OPLS-AA/M for RNA: The OPLS-AA/M force field, a reparameterization of OPLS-AA for RNA, has shown improved performance in reproducing accurate dihedral distributions and avoiding unphysical states in nucleotide simulations [28]. Its parameterization strategy, which includes fitting to high-level quantum mechanical scans, makes it a reliable choice for nucleic acid systems.
- Evaluate System Composition: For simulations involving organic liquids, lipids, or small drug-like molecules, OPLS-aa remains a well-validated choice as it was originally optimized for organic liquid and gas-phase interactions [29].
- Consult Comparative Studies: When simulating heterogeneous systems like asphalt mixtures, studies have found that both OPLS-aa and CHARMM can predict similar properties such as density and diffusion coefficient, though they may differ in specific predictions like molecular crystallization [29].
FAQ 2: What are the key differences in functional form between OPLS-AA and other major force fields?
Issue: A user needs to understand the fundamental differences in the energy functions of OPLS-AA versus CHARMM to make an informed selection.
Explanation: While both OPLS-AA and CHARMM are classified as Class I force fields and use harmonic functions for bonds and angles, they differ in their handling of dihedral and non-bonded interactions, as well as their parameterization philosophy [29].
Solutions:
- Review the potential energy functions as shown in the table below.
- For biomolecules in aqueous solution, note that CHARMM charges are derived from fitting solute-water interaction energies to ab initio data, thus optimizing them for aqueous environments [29].
- For organic molecules, OPLS-AA parameters are optimized to reproduce thermodynamic properties of pure organic liquids, such as density and heat of vaporization [29].
Table 1: Comparison of Force Field Energy Functions
| Force Field Component | OPLS-AA [29] | CHARMM [29] |
|---|---|---|
| Bonds & Angles | Harmonic potential | Harmonic potential |
| Dihedrals | Sum of Fourier series terms (( V1, V2, V_3 )) | ( K_\phi[1 + cos(n\phi - \delta)] ) |
| Non-bonded | Standard Lennard-Jones and Coulombic | Standard Lennard-Jones and Coulombic |
| Special Terms | Not present in standard form | Includes Urey-Bradley term |
| Parametrization Focus | Organic liquid thermodynamics & gas-phase structures | Protein chemistry & solute-water interactions |
FAQ 3: What software supports the OPLS-AA force field and what are the key setup considerations?
Issue: A user wants to set up a simulation with OPLS-AA in a common MD software package.
Explanation: OPLS-AA is widely implemented in major molecular dynamics software. The standard implementations are the BOSS and MCPRO programs from the Jorgensen group, but it is also available in other popular packages [19].
Solutions:
- GROMACS: GROMACS supports the OPLS-AA force field and its latest version, OPLS-AA/M [19].
- LAMMPS: The OPLS-aa force field has been used in LAMMPS for simulations of systems like model asphalt mixtures [29].
- NAMD: The CHARMM force field is also available in NAMD, which is relevant for comparisons or multi-force field studies [29].
- Verification: Always consult the specific documentation for your simulation software to ensure correct implementation and for any recommended settings (e.g., cut-off schemes).
Experimental Protocols
Protocol: Validation of Backbone Dihedral Parameters for RNA (Based on OPLS-AA/M Development)
This protocol outlines the key steps for deriving and validating new torsional parameters, as demonstrated in the development of the OPLS-AA/M force field for RNA [28].
1. System Preparation:
- Model Compounds: Select small model compounds that represent the chemical moieties of interest. For RNA backbone parameters, this involved dinucleotide fragments [28].
- Initial Conformations: Generate starting structures (e.g., in an initial A-form geometry for nucleotides) using molecular visualization or builder software [28].
- Solvation and Ions: Place the system in a cubic periodic water box with explicit water molecules (e.g., ~1200 water molecules for a dinucleotide). Add counterions (e.g., Na⁺, Cl⁻) to achieve charge neutrality. For systems with known metal ion binding sites, include these ions explicitly [28].
2. Quantum Mechanical (QM) Reference Calculations:
- Method: Perform QM potential energy surface scans. For OPLS-AA/M, this was done using Gaussian 09 at the ωB97X-D/6-311++G(d,p) level of theory in vacuum [28].
- Scanning: For complex backbones like RNA, scan two-dimensional dihedral surfaces (e.g., α/γ and ε/ζ) in 15° increments, while holding other backbone dihedrals and sugar puckering fixed at specific conformations (e.g., C3' or C2' endo) [28].
3. Parameterization and Force Field Optimization:
- Objective: Fit the molecular mechanics (MM) dihedral parameters to reproduce the QM potential energy surface.
- Technique: Use a Boltzmann-weighted error function (e.g., with a weighting temperature of 2000 K) to bias the fitting towards low-energy regions. Adjust the Fourier coefficients (V1, V2, V3) in the dihedral term to minimize the error between the MM and QM energies [28].
4. Validation via Molecular Dynamics (MD) Simulation:
- Systems: Run MD simulations on a range of systems, from dinucleotides to larger structured RNAs (e.g., tetranucleotides, tetraloops) [28].
- Conditions: Perform simulations at a constant temperature (e.g., 300 K or 275 K for compatibility with NMR data) and pressure (1 atm) using a Langevin thermostat and barostat. Use a 2 fs time-step with constraints on all bonds [28].
- Comparison to Experiment: Calculate observables from the simulation trajectories and compare them to experimental data. A key validation metric is the comparison of calculated ³J-couplings from dihedral angle distributions against values determined by NMR spectroscopy [28].
The workflow is summarized in the diagram below.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Force Fields for Molecular Simulations
| Item Name | Function / Purpose | Relevant Context |
|---|---|---|
| GROMACS | A high-performance molecular dynamics simulation package. | Supports OPLS-AA and OPLS-AA/M force fields natively [19]. |
| NAMD | A parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. | Often used with the CHARMM force field; can be used for comparative studies [29]. |
| LAMMPS | A classical molecular dynamics code with a focus on materials modeling. | Has been used with the OPLS-aa force field for complex mixtures like asphalt [29]. |
| CHARMM Force Field | A set of force fields originally developed for proteins, with later extensions to other molecules. | A primary alternative to OPLS-AA; optimized for biomolecules in aqueous solution [29]. |
| AMBER Force Field | A family of force fields for biomolecular simulations. | Includes modern versions (e.g., ff03ws) corrected for IDP properties; a key benchmark for protein studies [27]. |
| TIP3P / TIP4P Water Models | Explicit solvent models representing water molecules. | Critical for solvation; the choice of water model (TIP3P, TIP4P/2005) significantly impacts simulation outcome, especially for unfolded states [27]. |
| Gaussian 09 | A software package for electronic structure modeling (Quantum Chemistry). | Used for generating high-level QM potential energy surfaces for force field parameterization, as in OPLS-AA/M development [28]. |
| BOSS / MCPRO | Molecular simulation programs from the Jorgensen group. | The standard implementation platforms for the OPLS force field family [19]. |
FAQs: Core Concepts and Selection Criteria
Q1: What is the fundamental difference between a polarizable force field and a standard, non-polarizable (additive) force field?
Standard additive force fields use a fixed, unchanging partial charge assigned to each atom to represent electrostatic interactions. These charges are parameterized to represent an average polarization state in a specific environment (like water). In contrast, polarizable force fields allow the charge distribution of a molecule to respond to its immediate electrostatic environment. This means the electrostatics can change as a molecule moves from a hydrophobic protein core to a hydrophilic solvent or interacts with ions, providing a more realistic physical model [30] [31].
Q2: Why should I consider using a polarizable force field for my protein simulations?
Polarizable force fields can provide a more accurate description of interactions in heterogeneous environments. This is crucial for modeling processes where electrostatic response is important, such as:
- Membrane-protein interactions: The varying dielectric environment between lipid bilayers and aqueous solution.
- Ion and ligand binding: Polarization effects are significant when charged molecules bind to active sites.
- Intrinsically disordered proteins (IDPs): These proteins sample diverse environments, and their charge distribution must adapt accordingly [30] [5]. While some studies show non-polarizable force fields perform well for folding of small, structured proteins [32], polarizable models offer a systematic path to improved accuracy for a wider range of biophysical phenomena.
Q3: What are the main polarizable models available, and how do I choose between them?
The two most established polarizable models for biomolecular simulations are the Drude model and the AMOEBA model.
- The Drude Model: Also known as the "charge-on-spring" model, it represents polarization by attaching a negatively charged, massless "Drude particle" to a parent atom via a harmonic spring. The displacement of this particle in an electric field creates an induced dipole moment [30] [33] [34]. It is conceptually simpler and is implemented in packages like CHARMM and OpenMM.
- The AMOEBA Model: This model uses a more complex electrostatic representation. It employs permanent atomic multipoles (charge, dipole, quadrupole) to describe the permanent charge distribution and induced atomic dipoles to model polarization [30] [33]. It aims for high accuracy by better capturing anisotropic electronic effects.
Table: Comparison of Drude and AMOEBA Polarizable Force Fields
| Feature | Drude Model | AMOEBA Model |
|---|---|---|
| Core Polarization Method | Drude oscillators (charged particles on springs) [33] [34] | Inducible point dipoles [33] |
| Permanent Electrostatics | Atom-centered point charges [31] | Atomic multipoles (charges, dipoles, quadrupoles) [33] |
| Anisotropy | Can be modeled using anisotropic spring constants and off-center virtual sites (lone pairs) [34] | Inherently captured via atomic multipole moments [33] |
| Computational Cost | ~4x higher than additive force fields [34] | Typically higher than the Drude model due to more complex electrostatics [30] |
| Common Implementations | CHARMM Drude [34] [31], OpenMM [34] | AMOEBA (in Tinker, OpenMM) [34] [31] |
Q4: I've heard polarizable simulations are computationally expensive. Are they practical for routine research today?
Yes, they are becoming increasingly practical. Implementations on modern Graphics Processing Units (GPUs) have dramatically reduced simulation times. For example, the Drude force field in OpenMM allows for microsecond-long polarizable simulations of biomolecular systems to be completed in a month or less on consumer-grade desktop GPU hardware [34]. While still more expensive than additive models (approximately 4-fold for Drude [34]), this brings them within reach for many research applications where their improved physical accuracy is critical.
Troubleshooting Guides
Issue 1: Instability in Simulations at Short Time Steps
Problem: The simulation crashes or becomes unstable, often with errors related to the Drude particles or high-energy interactions.
Solutions:
- Use a Dual-Length Time Step Integrator: This is a standard practice for Drude simulations. A short time step (e.g., 0.5 fs) is used for the Drude particle vibrations and bonded interactions, while a longer time step (e.g., 1-2 fs) is used for other forces [34]. This maintains stability without a prohibitive cost.
- Employ a "Hard Wall" Constraint: The Drude implementation includes a "hard wall" potential that prevents the Drude particle from over-polarizing and collapsing onto a neighboring atom, which is a common source of instability [34]. Ensure this feature is enabled.
- Check the Thole Screening Parameters: Incorrect or missing Thole screening parameters for 1-2 and 1-3 interactions can lead to over-polarization. Verify that your parameter files (e.g.,
toppar_drude_master_protein_2013e.str) are loaded correctly and contain the necessaryNBTHOLEsections [34].
Issue 2: Inaccurate Treatment of Lone Pairs and Anisotropic Polarization
Problem: The model fails to correctly reproduce interaction energies or geometries for hydrogen-bonding atoms or halogens involved in halogen bonding.
Solutions:
- Utilize Lone Pair (Virtual) Sites: Both Drude and AMOEBA models can use off-atom charged sites to represent electron lone pairs on atoms like oxygen or nitrogen. In the Drude model, these are implemented as virtual sites [34]. For AMOEBA, the multipole expansion naturally captures directional effects [33]. Ensure your topology correctly includes these sites for relevant atoms.
- Enable Anisotropic Polarization (Drude): For atoms where polarization is not uniform in all directions (e.g., in carbonyl groups), the isotropic harmonic spring can be replaced with an anisotropic tensor. This is defined in the force field parameter files and should be activated for the appropriate atom types [34].
Issue 3: Force Field Parameterization for Novel Molecules or Residues
Problem: Standard Drude or AMOEBA protein force fields lack parameters for a non-standard residue, cofactor, or small molecule ligand in your system.
Solutions:
- Leverage Automated Parameterization Tools: For the Drude force field, tools like DGenFF (Drude General Force Field) are available to generate initial parameters for drug-like molecules [5] [31]. These should be carefully validated.
- Consult the Parametrization Workflow: Polarizable parameters are typically derived by fitting to quantum mechanical (QM) data. This includes:
- Electrostatic Potential (ESP): Deriving permanent electrostatic parameters (charges or multipoles) from QM calculations [30].
- Polarizability Fitting: Assigning atomic polarizabilities, often based on QM molecular polarizability tensors decomposed into atomic contributions [30] [35].
- Validation: Testing against QM interaction energies and conformational energies, as well as experimental condensed-phase properties [31].
The Scientist's Toolkit
Table: Essential Research Reagents and Computational Tools
| Item | Function in Research |
|---|---|
| CHARMM/OpenMM Software | A molecular dynamics package with a robust implementation of the Drude polarizable force field, optimized for GPU computing [34]. |
| Tinker-HP Software | A molecular modeling package specifically designed for high-performance computing with the AMOEBA polarizable force field [5]. |
| Drude Prepper Tool | A utility (often within CHARMM) to automatically add Drude particles and lone pairs to a molecular topology, preparing it for a polarizable simulation [34]. |
| Quantum Chemistry Software (e.g., Gaussian, ORCA) | Essential for performing ab initio calculations to derive target data (electrostatic potential, polarizabilities) for parameterizing new molecules [30] [31]. |
| Drude & AMOEBA Parameter Sets | The curated force field files (e.g., toppar_drude_master_protein_2013e.str for Drude) containing all necessary parameters for proteins, nucleic acids, lipids, and water models [34]. |
Experimental Protocol: Setting Up a Basic Protein Simulation with the Drude Force Field
This protocol outlines the key steps for setting up a simulation of a small protein, like the villin headpiece or CLN025 [32], using the Drude polarizable force field in a package like OpenMM [34].
Workflow Overview
Step-by-Step Methodology
Initial System Preparation:
- Obtain an initial protein structure from a PDB file.
- Use a tool like
pdb2gmx(GROMACS) or theCharmmGuito generate a topology and coordinates with a non-polarizable force field (e.g., CHARMM36). This ensures standard protonation states and disulfide bonds.
Drude-Specific Topology Generation:
- This is a critical step. Use the Drude Prepper utility (in CHARMM) or a similar tool to process the initial topology.
- The tool will:
- Add Drude particles to all non-hydrogen atoms.
- Assign atomic polarizabilities and Drude charge based on the force field parameters (
qD = sqrt(α * kD)) [34]. - Introduce lone pair virtual sites on electronegative atoms where defined.
- Apply Thole screening parameters between bonded atoms.
Solvation and Ionization:
- Place the protein in a simulation box (e.g., a rhombic dodecahedron) and solvate it with a polarizable water model, such as the SWM4-NDP water model for Drude [34].
- Add ions to neutralize the system and achieve the desired physiological concentration. Use polarizable ion parameters (e.g., Li, Na, K, Cl) that are compatible with the Drude force field.
Energy Minimization and Equilibration:
- Minimization: Perform steepest descent or conjugate gradient minimization to remove any bad contacts introduced during system building.
- Thermalization: Gradually heat the system to the target temperature (e.g., 300 K) over hundreds of picoseconds using a Langevin thermostat.
- Dual-Time Step Integration: Use an integrator like
LangevinMiddleIntegratorin OpenMM with a 2 fs time step for non-Drude forces and a 0.5 fs step for the Drude-related degrees of freedom [34]. - Apply Constraints: Use constraints (like SHAKE or LINCS) on bonds involving hydrogen to allow for the 2 fs time step.
Production Molecular Dynamics:
- Run a multi-nanosecond to microsecond simulation with constant temperature and pressure control (e.g., NPT ensemble).
- Ensure that the "hard wall" constraint is active to maintain Drude particle stability [34].
- Configure the non-bonded interactions with a cutoff (e.g., 12 Å) for Lennard-Jones and real-space electrostatic interactions, and use a particle-mesh Ewald (PME) method for long-range electrostatics.
Matching Force Fields to Protein Types: A Practical Application Guide
Frequently Asked Questions (FAQs)
FAQ 1: Why can't I just use the default water model (like TIP3P) for all my protein simulations? Using the default TIP3P model for all simulations is not recommended because different biological systems have specific hydration requirements that a single model cannot accurately capture. For instance, while TIP3P is computationally efficient, it can lead to an artificial structural collapse in intrinsically disordered proteins (IDPs) and produce unrealistic dynamics [12]. Furthermore, extensive benchmarking shows that no single model is best for all properties; recent three-site models like OPC show significant progress, but the best agreement with experimental data over a wide temperature range is often achieved with four-site models [36]. The choice of water model directly impacts critical outcomes like protein folding, ligand transport kinetics, and the stability of secondary structures.
FAQ 2: How does the choice of water model affect simulations of intrinsically disordered proteins (IDPs) or hybrid proteins with both ordered and disordered regions? IDPs and hybrid proteins are particularly sensitive to the water model. Studies comparing force fields for such proteins found that the TIP3P water model led to an artificial structural collapse and resulted in unrealistic NMR relaxation properties [12]. The TIP4P-D water model, when combined with modern protein force fields, significantly improved the reliability of these simulations. It was better able to reproduce experimental data such as radii of gyration and chemical shifts, and was capable of retaining transient helical motifs observed in NMR experiments [12].
FAQ 3: My research focuses on protein-glycosaminoglycan (GAG) complexes. Which water model should I use? Most studies on protein-GAG complexes have historically used the TIP3P model. However, systematic evaluations suggest that more advanced models can provide better accuracy. One study found that the OPC and TIP5P models showed the best agreement with experimental data for describing the structural features of heparin [37]. Another study focusing on chondroitin sulfate suggested TIP4P and TIP4PEw as the most appropriate [37]. Given the highly charged nature of GAGs and the importance of interfacial water molecules, moving beyond TIP3P is often warranted for these systems.
FAQ 4: I am simulating enzyme tunnels and ligand transport. Does the water model matter? Yes, the water model can significantly influence the observed characteristics of enzyme tunnels. Simulations of a haloalkane dehalogenase (LinB) and its variants revealed that while the overall tunnel topology was similar between TIP3P and OPC models, the geometrical characteristics of auxiliary tunnels and the stability of open tunnels differed [38]. TIP3P can be a valid choice for general topology assessment, especially with limited computational resources. However, OPC is preferable for calculations requiring an accurate description of transport kinetics, as it may provide a more reliable picture of tunnel dynamics and bottlenecks [38].
FAQ 5: For long-timescale protein folding simulations, how accurate are our current physical models? Long molecular dynamics simulations can now fold and unfold proteins multiple times, allowing for direct assessment of force field and water model accuracy. These simulations have shown that current physical models can accurently reproduce folding rates, free energies, and the structure/dynamics of the folded state [39]. However, they often still struggle with reproducing folding enthalpies and the characteristics of the unfolded state [39]. In some cases, force fields can exhibit a bias towards non-native states, such as favoring helical structures over the native beta-sheet in a WW domain, explaining folding failures [40].
Troubleshooting Guides
Issue: Unrealistic Collapse of Intrinsically Disordered Proteins (IDPs)
Problem: Your simulation of an IDP or a protein with a long disordered region shows an unnaturally compact structure, inconsistent with experimental data (e.g., from SAXS or NMR).
Diagnosis: This is a known issue with certain water model and force field combinations. The TIP3P model, in particular, has been identified as a key contributor to this problem [12].
Solution:
- Switch your water model: Replace TIP3P with a model parameterized for disordered proteins, such as TIP4P-D [12].
- Re-evaluate your protein force field: Ensure you are using a force field known to perform well for IDPs, such as CHARMM36m [12] [41].
- Validate with experimental data: Compare simulation outputs like the radius of gyration or NMR chemical shifts against known experimental data to confirm the improvement [12].
Issue: Inaccurate Description of Protein-GAG Complexes
Problem: Your molecular dynamics simulation of a protein-glycosaminoglycan complex fails to maintain the experimental binding pose or shows unstable binding.
Diagnosis: The highly charged and periodic nature of GAGs makes their interactions with proteins highly dependent on an accurate treatment of electrostatics and solvation. The default TIP3P model may not be sufficient [37].
Solution:
- Benchmark advanced water models: Test models like OPC, TIP4P, or TIP5P for your specific system [37].
- Analyze multiple descriptors: Don't rely on a single metric. Assess the binding pose stability, hydrogen bonding patterns, and free energy of binding across different water models.
- Consider computational cost: If resources are limited for extensive sampling with a 4- or 5-point model, OPC has been shown to be a good balance of accuracy and cost for carbohydrate systems [37].
Issue: Failure to Fold a Protein or Preference for Non-Native States
Problem: Long-term folding simulations do not reach the native state or get trapped in stable, non-native (e.g., helical) conformations.
Diagnosis: This could be a force field bias, where the potential energy function incorrectly stabilizes non-native structures over the native one. This has been observed in simulations of beta-sheet proteins like the Pin1 WW domain [40].
Solution:
- Identify the misfolded state: Use clustering and free energy calculations to characterize the non-native states that are being populated.
- Calculate free energy differences: Employ methods like deactivated morphing (DM) to quantify the free energy difference between the misfolded state and the native state. This can confirm a force field bias [40].
- Consult the literature: Investigate if others have reported similar issues with your protein of interest or fold type, and see if solutions like using a different force field variant or water model have been proposed.
Quantitative Comparison of Water Models
The table below summarizes key performance characteristics of common water models based on recent benchmarking studies. A "Yes" in the "Good for IDPs?" column indicates evidence of improved performance for intrinsically disordered proteins over TIP3P.
Table 1: Benchmarking Performance of Common Explicit Water Models
| Water Model | Number of Sites | Key Strengths and Weaknesses | Good for IDPs? | Recommended Use Cases |
|---|---|---|---|---|
| TIP3P [12] [38] | 3 | Fast, but can cause artificial collapse of IDPs; common default. | No | General purpose (when IDPs not involved); compatibility. |
| TIPS3P [12] | 3 | A modified TIP3P model; performance similar to TIP3P for proteins. | No | Similar to TIP3P. |
| TIP4P-D [12] | 4 | Significantly improves reliability for proteins with disordered regions. | Yes | Simulations of IDPs and hybrid ordered/disordered proteins. |
| TIP4PEw [37] | 4 | Often provides a good fit for water structure; recommended for some GAG systems. | Not Specified | Chondroitin sulfate complexes; general structural studies. |
| OPC [36] [37] [38] | 3 | High accuracy for bulk properties and structure; good for GAGs and enzyme tunnels. | Not Specified | Ligand transport kinetics; heparin/GAG complexes; general high-accuracy work. |
| TIP5P [37] | 5 | Excellent water structure; high computational cost; good for some GAGs. | Not Specified | Heparin structure analysis. |
Experimental Protocols & Workflows
Protocol: Benchmarking Water Models for a New Protein System
When studying a new protein system, especially one with unique characteristics like intrinsic disorder or bound carbohydrates, it is prudent to benchmark water models. The following workflow provides a methodology for a systematic comparison.
Diagram: Water Model Benchmarking Workflow
Title: Water Model Evaluation Protocol
Detailed Methodology:
- Select Candidate Water Models: Choose a set of 2-4 models that represent different classes and trade-offs. A typical set might include TIP3P (baseline), OPC (modern 3-site), and TIP4P-D (for disordered regions) or TIP4PEw (for structural accuracy) [36] [12].
- Set Up Molecular Dynamics Simulations:
- Use the same protein force field (e.g., CHARMM36m, AMBER ff19SB) and initial protein structure across all simulations.
- Solvate the protein in boxes of identical size and shape using each candidate water model.
- Neutralize the system with ions and add salt to the same physiological concentration (e.g., 100 mM NaCl) using ion parameters compatible with the chosen water model [12].
- Use identical simulation parameters for temperature and pressure control.
- Run Equilibration and Production Runs:
- Perform the same minimization and equilibration protocol for all systems.
- Run production simulations for a duration sufficient to observe the properties of interest (e.g., hundreds of nanoseconds to microseconds). Use the same simulation length for all models to allow fair comparison.
- Analyze Key Outputs: Compare trajectories using multiple quantitative descriptors:
- Structural Properties: Root-mean-square deviation (RMSD) of the protein backbone, radius of gyration (Rg), and secondary structure content [12] [39].
- Dynamic Properties: Root-mean-square fluctuation (RMSF) of residues, and if possible, NMR relaxation parameters, which are highly sensitive to water model choice [12].
- System-Specific Properties: For enzyme tunnels, analyze bottleneck radii and tunnel opening durations [38]. For protein-GAG complexes, monitor binding pose stability and hydrogen bonds [37].
- Compare with Experimental Data: The final and most critical step is to compare the simulation results with available experimental data. This can include:
The water model that best reproduces the experimental observables for your specific system should be selected for further production simulations.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Computational Tools and Models for Water Model Benchmarking
| Item | Function in Research | Example / Note |
|---|---|---|
| Explicit Water Models | Atomistically represent solvent molecules in MD simulations to capture solvation effects and hydrogen bonding. | TIP3P, SPC/E, OPC, TIP4P, TIP4P-D, TIP5P [36] [12] [37]. |
| Implicit Solvent Models | Approximate solvent as a continuous medium to speed up calculations; less accurate for specific solvation effects. | Generalized Born (GB) models in AMBER (IGB=1,2,5,7,8) [37]. |
| Biomolecular Force Fields | Define the potential energy function and parameters for proteins, nucleic acids, and lipids. | AMBER (e.g., ff14SB), CHARMM (e.g., C36m), OPLS-AA [12] [37] [41]. |
| MD Simulation Software | Software packages to perform energy minimization, dynamics, and analysis. | NAMD, GROMACS, AMBER [40]. |
| Trajectory Analysis Tools | Programs and scripts to analyze MD trajectories and compute properties of interest. | VMD, GROMACS tools, MDAnalysis, CPPTRAJ [40]. |
| Validation Databases | Public repositories of experimental data used to validate computational results. | PDB (structures), BMRB (NMR data), SASBDB (SAXS data) [12]. |
Recommended Force Field/Water Model Combinations for Binding Free Energy Calculations
Frequently Asked Questions (FAQs)
What is the most important consideration when choosing a force field for binding free energy calculations? The most critical consideration is achieving a balanced representation of all molecular interactions within your specific system. No single force field universally outperforms others. The choice depends on the nature of your protein (e.g., folded vs. intrinsically disordered), the properties of the ligand, and the compatibility between the protein force field, ligand parameters, and water model. A combination that excels for one system may perform poorly for another [42] [43].
Why does my simulated protein unfold or become unstable during binding free energy calculations?
This instability can arise from an imbalance in protein-protein versus protein-water interactions. Some modern force fields, particularly those developed to better model intrinsically disordered proteins (IDPs) like ff03ws, strengthen protein-water interactions. This can sometimes compromise the stability of folded domains [43]. If simulating a folded protein, consider using a force field like ff99SBws or ff19SB that has demonstrated better stability for folded structures [43].
My simulations of intrinsically disordered regions (IDRs) appear overly collapsed. How can I fix this? Overly collapsed IDRs are a known issue with older force fields paired with simple water models like TIP3P [12]. To resolve this, use a force field and water model combination specifically refined for IDPs. This includes:
- Using a modified water model (e.g., TIP4P-D, OPC) that enhances protein-water interactions [12] [43].
- Selecting a force field parameterized with these improved water models, such as
CHARMM36m,ff99SB-disp, orff99SBws[12] [43].
How significant is the water model's influence on binding thermodynamics? The water model is highly significant. Research has shown that the choice of water model (TIP3P, TIP4Pew, SPC/E, OPC) can dramatically impact the calculated binding free energies and enthalpies. In some cases, the water model had a greater influence on the accuracy of the results than the method used to assign partial charges to the ligand [42].
Troubleshooting Guides
Problem: Inaccurate Binding Enthalpies or Free Energies
Description Calculated binding free energies or enthalpies do not agree with experimental measurements, such as Isothermal Titration Calorimetry (ITC) data.
Possible Causes & Solutions
- Cause: Incompatible Force Field and Water Model. The combination may be incorrectly balanced for your specific host-guest or protein-ligand system [42]. Solution: Systematically test different validated combinations. For cyclodextrin-guest systems, studies have found that some force field/water model combinations can yield accurate free energies but poor enthalpies, and vice-versa [42].
- Cause: Poor Ligand Partial Charges. The method for assigning atomic charges to the small molecule is inaccurate. Solution: Use robust methods for partial charge assignment, such as RESP or AM1-BCC, which are commonly employed in force fields like AMBER [42].
- Cause: Underlying Force Field Limitations. The force field may have inherent inaccuracies in representing key interactions in your system.
Solution: Consider using a force field refined for a specific protein type. For example, for collagen simulations, the AMBER
ff99SBfamily has shown better performance for backbone dihedrals, while CHARMM36 required targeted scaling of CMAP terms for proline, hydroxyproline, and glycine to achieve accuracy [44].
Problem: Unrealistic Protein or Peptide Conformations
Description Simulated proteins show deviation from known experimental structures (e.g., from crystallography or NMR), or disordered peptides do not exhibit expected dimensions or secondary structure propensities.
Possible Causes & Solutions
- Cause: Incorrect Force Field for Protein Type. Using a force field designed for folded proteins to simulate IDPs, or vice-versa [12].
Solution: Select a force field validated for your system's characteristics.
CHARMM36mand refined AMBER force fields likeff99SBwshave been shown to provide a better balance for proteins containing both structured and disordered regions [12] [43]. - Cause: Primitive Water Model. The use of a simple three-point water model like TIP3P can lead to excessive protein-protein interactions and overly collapsed IDP ensembles [12] [43].
Solution: Switch to a more advanced, four-site water model. The TIP4P-D water model, in particular, has been successful in improving the conformational description of IDPs when paired with force fields like
ff99SB-ILDNandCHARMM36m[12] [43].
Force Field and Water Model Combinations
The table below summarizes recommended force field and water model combinations for different biological systems, based on recent research.
Table 1: Recommended Force Field/Water Model Combinations for Various Systems
| Protein/System Type | Recommended Force Field | Recommended Water Model | Key Evidence and Performance |
|---|---|---|---|
| Cyclodextrin Host-Guest | Varies by study [42] | TIP3P, TIP4Pew, SPC/E, OPC [42] | Performance is highly dependent on the specific combination; some yield accurate free energies but poor enthalpies. Requires systematic testing [42]. |
| Proteins with Structured and Intrinsically Disordered Regions (IDRs) | CHARMM36m [12], AMBER ff99SBws [43] | TIP4P-D [12], Modified TIP3P (for CHARMM36m) [12] | Improves reliability of IDR simulations and retains transient helical motifs while maintaining structured domain stability [12]. |
| Collagen Triple Helix | AMBER ff99SB [44], CHARMM36mGP (modified) [44] | Not specified in study [44] | AMBER ff99SB accurately reproduces collagen dihedrals, side-chain torsions, and SAXS data. CHARMM36 requires CMAP term scaling for accuracy [44]. |
| General Folded Proteins & Protein-Protein Complexes | AMBER ff99SBws [43], AMBER ff19SB [43] | TIP4P2005 [43], OPC [43] | Maintains stability of folded proteins (e.g., Ubiquitin, Villin HP35) and provides accurate protein-protein association profiles [43]. |
| Advanced Polarizable Simulations | Novel polarizable mean-field model [45] | Model-specific polarizable water [45] | Designed to be compatible with standard non-polarizable force fields (AMBER, CHARMM) while explicitly treating water polarizability in different environments [45]. |
Experimental Protocols for Validation
Protocol: Validating Force Fields Against Experimental Scattering Data
Methodology: Small-Angle X-Ray Scattering (SAXS) Purpose: To validate that a force field produces realistic structural ensembles, particularly for intrinsically disordered proteins or large complexes, by comparing simulated and experimental data [12] [44].
- Simulation: Run multiple independent molecular dynamics simulations of the protein in its native solution conditions to generate a conformational ensemble.
- Theoretical SAXS Calculation: From the simulation trajectory, calculate the theoretical SAXS scattering profile (form factor). This often involves averaging snapshots from the simulation and computing the scattering intensity using specialized software [44].
- Data Comparison: Compare the theoretical SAXS profile and the calculated radius of gyration (Rg) directly with the experimental SAXS data.
- Analysis: A force field is considered to perform well if the theoretical form factor and Rg from the simulation ensemble match the experimental data within error margins [44].
Protocol: Validating Force Fields Against NMR Observables
Methodology: Nuclear Magnetic Resonance (NMR) Spectroscopy Purpose: To assess the accuracy of a force field in capturing local backbone conformations, dynamics, and secondary structure propensities [12] [43].
- Simulation: Run molecular dynamics simulations to generate a structural ensemble of the protein.
- Prediction of NMR Parameters: From the simulation trajectory, predict key NMR observables, including:
- Chemical Shifts: Using empirical shift calculators (e.g., SHIFTX2).
- Residual Dipolar Couplings (RDCs): Calculating molecular alignment and RDCs from simulation structures.
- Spin Relaxation Rates (R1, R2) and heteronuclear NOE: To probe backbone dynamics on picosecond-to-nanosecond timescales [12].
- Validation: Compare the predicted NMR parameters with the experimentally measured values. A strong correlation indicates that the force field accurately captures the protein's conformational landscape and dynamics [12] [43].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Software and Computational Tools for Force Field Validation
| Tool/Reagent | Function in Research |
|---|---|
| GROMACS | A high-performance molecular dynamics package used to run simulations with force fields from AMBER, CHARMM, and GROMOS families [44]. |
| AMBER Tools | A suite of programs for molecular dynamics simulations and analysis, particularly with AMBER force fields [42]. |
| CHARMM | A versatile program for energy minimization, dynamics, and analysis of molecular systems using CHARMM force fields. |
| SAXS Analysis Software (e.g., from ATSAS suite) | Used to process experimental SAXS data and calculate parameters like the radius of gyration (Rg) for comparison with simulation results [12]. |
| NMR Chemical Shift Predictors (e.g., SHIFTX2) | Software that calculates expected NMR chemical shifts from a 3D structure, enabling direct comparison between simulation ensembles and experimental NMR data [12]. |
Workflow: Force Field Selection Logic
The diagram below outlines a logical workflow for selecting and validating a force field combination for binding free energy calculations.
Frequently Asked Questions (FAQs) on Force Field Selection
FAQ 1: Why does my simulation of chignolin populate a misfolded state, and how is this related to my force field choice?
It is not uncommon for simulations of the chignolin peptide to populate a stable, out-of-register misfolded state. Research has shown that this specific misfold is characterized by Gly-7 adopting a left-handed polyproline II (βPR) backbone conformation instead of the native αL conformation [46]. The propensity to observe this state is highly dependent on the force field. Studies using AMBER ff03*, ff03w, and ff99SB force fields found this misfolded state constituted 20–50% of the ordered structures, whereas it was not observed with the CHARMM22/CMAP force field [46]. This difference is largely attributed to variations in how different force fields parameterize glycine, particularly its propensity to form the βPR conformation [46].
FAQ 2: For proteins containing both structured domains and intrinsically disordered regions (IDRs), which force field should I use?
Simulating hybrid proteins with both structured and disordered regions presents a unique challenge. Standard force fields optimized for globular proteins often cause artificial collapse in IDRs. A 2020 benchmarking study recommends the CHARMM36m force field as a reliable choice for such systems [12]. The study also highlighted that the choice of water model is critical; the TIP4P-D water model, when combined with force fields like Amber99SB-ILDN, CHARMM22*, or CHARMM36m, significantly improved the reliability of simulations by preventing unrealistic collapse and better reproducing experimental NMR parameters [12].
FAQ 3: What are the key differences between all-atom, united-atom, and coarse-grained force fields, and when should I use them?
The choice depends on your research question and computational resources.
- All-Atom (AA): These force fields, such as AMBER and CHARMM, explicitly parameterize every atom, including hydrogen. They provide the most detailed representation and are essential for studying specific interactions like hydrogen bonding but are computationally expensive [41].
- United-Atom (UA): Force fields like CHARMM19 and AMBER-UA treat nonpolar carbons and their bonded hydrogens as a single interaction site. This reduces the number of particles and computational cost, offering a balance between speed and atomic detail [41].
- Coarse-Grained (CG): Models like MARTINI group several atoms into a single "bead." This allows for simulating large systems and long timescales (microseconds to milliseconds) but sacrifices atomic-level resolution. They are ideal for studying large-scale conformational changes or protein-protein interactions [41].
FAQ 4: How can I validate my force field choice for a fast-folding simulation?
Validation against experimental data is crucial. For fast-folding peptides like chignolin and CLN025, you can compare your simulation results with several NMR-derived observables [46]:
- Back-calculated NMR chemical shifts.
- Scalar coupling constants (³JHNHα).
- Proton-proton distances derived from Nuclear Overhauser Effect (NOE) measurements. Agreement between the ensemble-averaged back-calculated parameters from your simulation and the experimental data indicates a well-parameterized force field. For intrinsically disordered systems, additional validation against parameters like Radius of Gyration (from SAXS) and NMR relaxation data is highly recommended [12].
Troubleshooting Common Simulation Problems
Problem 1: Unrealistic Secondary Structure Collapse in Disordered Regions
- Symptoms: A disordered protein region or loop collapses into a compact, non-native globule during simulation. NMR back-calculations from the trajectory show poor agreement with experimental data.
- Possible Cause: The use of a force field and water model combination that is not optimized for disordered systems. The standard TIP3P water model, in particular, has been identified as a cause of artificial structural collapse [12].
- Solution:
- Switch to a force field designed for IDPs, such as CHARMM36m.
- Use a water model like TIP4P-D, which was specifically designed to improve the solvation of disordered proteins [12].
- Validate your simulation against experimental NMR relaxation or SAXS data to ensure the conformational ensemble is correct [12].
Problem 2: Persistent Misfolding of a Glycine-Rich Turn Motif
- Symptoms: In a peptide like chignolin, a stable, non-native state is highly populated, often involving a glycine residue in the turn region.
- Possible Cause: Inaccurate glycine parameters in your chosen force field, which incorrectly bias the backbone dihedral angles (e.g., towards βPR over αL) [46].
- Solution:
- Benchmark Force Fields: Test multiple force fields (e.g., AMBER ff99SB vs. CHARMM22/CMAP) and compare the populations of native and misfolded states.
- Validate with Experiment: Compare your results with experimental NMR data. For chignolin, including the misfolded state in the ensemble can sometimes slightly improve agreement with some NMR observables, making careful validation essential [46].
- Consider Mutagenesis: If the misfold is driven by a specific glycine (like Gly-7 in chignolin), simulating a mutant (e.g., G7K) where the misfold is sterically prohibited can serve as a control. If the mutant simulation agrees better with experiment, it confirms the force field's glycine parameters are the issue [46].
Problem 3: Stereochemical Errors Causing Structural Artifacts
- Symptoms: Unexplained kinks, bends, or complete loss of secondary structure (α-helices, β-sheets) during a simulation.
- Possible Cause: The initial structure contains flipped chiral centers or incorrect peptide bond isomers (cis instead of trans). Force fields will not correct these errors, and they can severely disrupt the simulation [47].
- Solution:
- Validate Input Structure: Before simulation, use structure validation tools like MolProbity or the SAVES server (which runs PROCHECK) to check for chirality and cis-trans peptide bond errors [47].
- Correct Errors: Use tools like the Chirality and Cispeptide plugins in VMD to identify and correct these errors, followed by local energy minimization [47].
Key Experimental Protocols
Protocol: Replica Exchange Molecular Dynamics (REMD) of a Fast-Folding Peptide
This protocol is adapted from the methodology used to study chignolin folding [46].
System Preparation:
- Obtain the initial structure from a database like the PDB (e.g., 1UAO for chignolin).
- Solvate the peptide in a truncated octahedron simulation box with a minimum face distance of 3.5 nm, containing approximately 1030 water molecules.
- Add ions (e.g., Na⁺ and Cl⁻) to neutralize the system's charge and simulate physiological ionic strength.
Equilibration:
- Perform energy minimization to remove steric clashes.
- Equilibrate the system first at constant pressure (1 atm) and temperature (300 K) to achieve the correct solvent density.
REMD Simulation:
- Use 32 replicas to span a temperature range (e.g., 278–595 K).
- Perform simulations at constant volume.
- Use the Particle-Mesh Ewald method for long-range electrostatics with a 9 Å real-space cutoff.
- Propagate the system using Langevin dynamics with a friction coefficient of 1 ps⁻¹.
- Attempt replica exchanges every 10 ps.
- Run each system for at least 200 ns per replica to ensure sufficient sampling.
Analysis of Results:
- Cluster Analysis: Use a tool like
g_clusterin GROMACS to identify dominant conformational states from the 300 K replica trajectory. A cutoff of 0.6 Å with the single linkage method is typical [46]. - NMR Observable Calculation:
- Cluster Analysis: Use a tool like
Data Presentation
Table 1: Force Field Performance for Different Protein Systems
| Protein / Peptide Type | Recommended Force Fields | Key Strengths | Reported Limitations |
|---|---|---|---|
| Fast-Folding β-Hairpins (e.g., Chignolin, CLN025) | CHARMM22/CMAP [46], AMBER ff99SB [46] | CHARMM22/CMAP avoided a specific Gly-7 misfold in chignolin; ff99SB reproduces native population well [46]. | AMBER ff03*, ff03w, ff99SB can populate a misfolded state (20-50%) due to glycine parameters [46]. |
| Proteins with Structured & Disordered Regions | CHARMM36m + TIP4P-D water [12] | Reliably reproduces Rg, chemical shifts, PRE, and NMR relaxation data without artificial collapse [12]. | TIP3P water model leads to unrealistic collapse and poor relaxation properties [12]. |
| Membranes | CHARMM36 Lipid [41], LIPID21 [41] | Parameters specifically tailored for lipid bilayers and their interactions. | Not optimized for soluble proteins or disordered regions. |
| Small Molecule Docking | AutoDock4 Force Field [41] | Grid-based approach enables fast, efficient scoring of ligand poses. | Not suitable for long-timescale dynamics; lacks full atomic detail. |
Table 2: Key Research Reagents and Computational Tools
| Reagent / Tool | Function in Research | Example Use Case |
|---|---|---|
| GROMACS | Molecular dynamics simulation package. | Running REMD simulations for fast-folding peptides [46]. |
| AMBER Force Fields (e.g., ff99SB, ff14SB) | Set of parameters for simulating biomolecules. | Simulating protein folding and dynamics [46] [41]. |
| CHARMM Force Fields (e.g., C22/CMAP, C36m) | Set of parameters for simulating biomolecules. | Studying structured/IDP hybrid proteins and avoiding specific misfolds [46] [12]. |
| VMD | Molecular visualization and analysis program. | Trajectory analysis, correcting stereochemical errors with plugins [47]. |
| SPARTA+ | Program for calculating chemical shifts from protein structures. | Validating simulation ensembles against experimental NMR data [46]. |
| MolProbity / SAVES | Structure validation server. | Checking for stereochemical errors prior to simulation setup [47]. |
Workflow and Relationship Visualizations
Force Fields for Intrinsically Disordered Proteins (IDPs) and Flexible Systems
Intrinsically Disordered Proteins (IDPs) are widely distributed across eukaryotic cells, lacking stable tertiary structures yet playing crucial roles in molecular recognition, signaling, and regulation. IDPs are associated with numerous diseases, including cancers, cardiovascular diseases, and neurodegenerative disorders such as Alzheimer's and ALS [48] [49]. Unlike folded proteins, IDPs exist as dynamic structural ensembles, sampling a diverse continuum of conformations [50].
This inherent flexibility makes characterizing IDPs experimentally challenging and complicates their simulation. Traditional molecular dynamics force fields were parameterized and optimized for folded proteins with stable structures [51] [49]. Consequently, they often fail to accurately capture the conformational landscapes of IDPs, typically over-stabilizing secondary structures like α-helices and β-sheets that should be transient in disordered states [48]. This limitation has driven the development of specialized force fields to address the unique requirements of IDP simulation.
Force Field Selection Guide
Selecting an appropriate force field is critical for obtaining reliable simulation data for IDPs and flexible systems. The table below summarizes key force fields and their characteristics.
Table 1: Force Fields for IDP and Flexible System Simulations
| Force Field Family | Specific Variants | Key Features/Strategies | Reported Performance |
|---|---|---|---|
| AMBER | ff99IDPs, ff14IDPs [51] | Residue-specific CMAP correction for disorder-promoting amino acids [51] [48]. | Improved agreement with NMR chemical shifts for IDPs; maintains performance on folded proteins [51]. |
| AMBER | ff99SB, ff03 [48] [50], ff99SB-disp [43] | Global adjustment of backbone torsional parameters (ff99SB*); rebalanced protein-water LJ interactions (ff99SB-disp) [48] [43]. | Better balance of secondary structure propensities; improved IDP chain dimensions vs. experiment [43]. |
| AMBER | ff03ws, ff99SBws [43] | Upscaled protein-water van der Waals interactions paired with 4-site water models [43]. | Accurately reproduces IDP dimensions; may destabilize some folded proteins (ff03ws) [43]. |
| CHARMM | CHARMM36m (C36m) [50] | Refined backbone CMAP potentials and added NBFIX terms to fix overpopulated left-handed helices [50]. | Balanced performance for both folded proteins and IDPs; good transferability [49] [50]. |
| CHARMM | CHARMM36m2021s3p [49] | Recent variant using a modified TIP3P water model. | Top-ranked in R2-FUS-LC benchmark; balanced sampling of compact/extended states [49]. |
| OPLS | OPLS-AA/M, OPLS3 [48] [50] | Reparameterization of backbone and side-chain dihedrals against QM torsional scans [50]. | Improved for proline/glycine peptides (AA/M); good for protein-ligand binding (OPLS3) [48]. |
Experimental Protocols and Validation
Workflow for Force Field Evaluation
The diagram below outlines a general protocol for evaluating and validating force fields for IDP systems.
Key Methodological Steps
System Preparation:
- Construct the initial coordinates for the IDP or peptide, which can be an extended structure or based on a known fragment if available.
- Use tools like the LEaP module in AMBER to solvate the system in a truncated octahedron water box with a minimum buffer distance (e.g., 10 Å). Add counter-ions to achieve system neutrality [51].
Simulation Setup:
- Employ periodic boundary conditions.
- Treat long-range electrostatics with the Particle Mesh Ewald (PME) method [51].
- Apply constraints to bonds involving hydrogen atoms using algorithms like SHAKE [51].
- Perform energy minimization, followed by gradual heating and equilibration in the NVT or NPT ensemble.
Production Simulation and Sampling:
- Run multiple relatively short, independent trajectories (e.g., 10 trajectories of 100-140 ns each) to sample diverse conformational states, as this can be more effective for some IDPs than a single long simulation [51].
- For enhanced sampling, consider replica-exchange molecular dynamics (REMD), which facilitates escape from local energy minima and improves convergence of the conformational ensemble [35].
Ensemble Analysis: Analyze the simulation trajectories by calculating a variety of experimental and structural observables for validation:
- Radius of Gyration (Rg): Measures the global chain compactness [49].
- Secondary Chemical Shifts: Calculate from simulation ensembles and compare directly to NMR experimental data [51].
- Secondary Structure Propensity: Analyze using definitions like DSSP to quantify transient structural elements [51] [49].
- Intra-molecular Contact Maps: Reveal preferential long-range or short-range interactions within the chain [49].
- Small-Angle X-Ray Scattering (SAXS): Compute theoretical scattering profiles from MD snapshots and compare to experimental data [43].
Validation Against Experiment:
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Item/Tool | Function/Description | Relevance to IDP Studies |
|---|---|---|
| AMBER | A suite of biomolecular simulation programs. | Widely used for MD simulations with the AMBER family of force fields [51]. |
| GROMACS | High-performance MD simulation package. | Used for simulations with force fields like OPLS-AA and CHARMM [51]. |
| CHARMM | Molecular simulation program with MM, MD, MC, etc. capabilities. | Native environment for CHARMM force fields [48]. |
| DSSP | Algorithm for assigning secondary structure of protein structures. | Used to define coil fragments from PDB for force field training and analysis [51]. |
| CMAP Method | Grid-based energy correction map for backbone dihedrals. | Key strategy in ff14IDPs and CHARMM to correct backbone dihedral distributions [51] [48]. |
| TIP3P, TIP4P/2005, OPC | Water models of varying complexity (3-site, 4-site). | Critical for balanced protein-water interactions; 4-site models often improve IDP sampling [43] [50]. |
Frequently Asked Questions (FAQs)
Q1: Why do general-purpose protein force fields often fail for IDPs? General force fields were primarily trained on and validated with databases of stable, folded globular proteins. They often contain an implicit bias towards forming stable secondary and tertiary structures. This leads to over-stabilization of native-like interactions (like hydrogen bonds and hydrophobic contacts) that can cause IDPs to appear overly structured or collapsed in simulation, inconsistent with experimental observations that show they are dynamic and extended [49] [50].
Q2: What are the main strategies used to improve force fields for IDPs? Two major strategies are employed:
- Refining Backbone Dihedral Potentials: Adjusting the parameters governing backbone torsion angles (φ and ψ) to better match the distributions seen in disordered regions, often using coil library data from the PDB or QM calculations. This includes methods like CMAP corrections (in ff14IDPs and CHARMM) or global scaling of torsional barriers (ff99SB*) [51] [48] [50].
- Rebalancing Protein-Water Interactions: Adjusting the Lennard-Jones parameters between protein atoms and water to slightly strengthen their interaction. This helps prevent the protein from collapsing by making the solvation of exposed hydrophobic residues more favorable, better mimicking the solution environment of IDPs [43] [50].
Q3: My folded protein becomes unstable during an IDP-optimized force field simulation. What could be wrong? This is a known trade-off. Some force fields optimized for IDPs (e.g., ff03ws) achieve more realistic IDP chain dimensions by strengthening protein-water interactions, which can sometimes destabilize native hydrophobic cores in folded proteins [43]. If your system contains both folded and disordered domains, consider using a more balanced force field like CHARMM36m or ff99SB-disp, which are designed to handle both types of proteins [43] [49] [50]. Always validate the stability of the folded domain in your system.
Q4: How do I know if my IDP simulation is accurate and has converged? Convergence and accuracy are separate challenges. For convergence, run multiple independent replicas and check if they sample similar distributions of key observables (like Rg). For accuracy, validate your simulation results directly against experimental data. Key comparisons include:
- NMR chemical shifts and scalar couplings [51] [50].
- Experimental SAXS or FRET profiles [43].
- For systems like R2-FUS-LC, compare sampled compact states to known reference structures from NMR or cryo-EM [49].
Q5: Are machine-learned or coarse-grained force fields a viable option for studying IDPs? Yes, this is a rapidly advancing area. Machine-learned coarse-grained (CG) models trained on all-atom simulation data can predict metastable states of folded, unfolded, and intermediate structures and capture IDP fluctuations at a fraction of the computational cost [35]. They show promise for studying large-scale conformational changes and aggregation. However, all-atom simulations with explicitly modeled water remain the standard for detailed mechanistic studies of local interactions and for direct comparison to atomically-resolved experimental data like NMR [35].
Core Concepts and Performance Comparison
Small organic molecules, or "ligands," are major players in chemical processes from material science to biology, and are crucial in drug discovery where most marketed drugs are small molecules [52]. These molecules present a unique challenge for molecular dynamics (MD) simulations because their diverse scaffolds and functional groups make it unfeasible to catalog them as done for standard amino acids in proteins [52]. To address this, several specialized force fields have been developed:
- GAFF/GAFF2 (Generalized Amber Force Field): Widely used AMBER-compatible force fields designed for pharmaceutical molecules [52] [53].
- CGenFF (CHARMM General Force Field): The CHARMM family's equivalent for small molecules [52].
- OpenFF (Open Force Fields): Modern, open-source force fields using the SMIRNOFF (SMIRKS Native Open Force Field) approach, which employs SMARTS patterns for direct chemical perception [54] [55].
These force fields provide the bonded parameters (bonds, angles, dihedrals) and non-bonded parameters (charges, van der Waals) needed to simulate ligand behavior within host systems like proteins.
Comparative Performance in Binding Affinity Prediction
Accuracy in predicting protein-ligand binding affinity is a critical benchmark. A 2024 evaluation of six open-source small-molecule force fields on 598 ligands and 22 protein targets revealed key performance differences [56].
Table 1: Force Field Accuracy in Relative Binding Free Energy Calculations
| Force Field | Overall Accuracy | Key Characteristics | Notable Strengths/Limitations |
|---|---|---|---|
| OPLS3e | Significantly more accurate | Proprietary force field | Highest accuracy in benchmark study [56] |
| OpenFF Sage | Comparable to GAFF & CGenFF | Open-source, SMIRNOFF format | Performance varies for specific chemistries; bespoke parametrization available [56] [55] |
| GAFF | Comparable to Sage & CGenFF | Well-established, AMBER-compatible | Mature community tools (e.g., antechamber) [53] [56] |
| CGenFF | Comparable to Sage & GAFF | CHARMM-compatible | Different charge derivation than AMBER/OpenFF [52] [56] |
| Consensus (Sage, GAFF, CGenFF) | Comparable to OPLS3e | Combined output of three force fields | Mitigates individual force field outliers [56] |
The study found that lower accuracy could be attributed not only to force field parameters but also to input preparation and insufficient sampling convergence in MD simulations [56]. This highlights that parametrization strategy is one part of a larger workflow requiring careful execution at every stage.
Practical Parametrization Methodologies
Traditional Approach: Parametrization with GAFF2 using AmberTools
This established protocol uses the antechamber and parmchk2 programs from AmberTools to generate parameters compatible with the AMBER family of protein force fields [53].
Table 2: Essential Research Reagents and Software for GAFF Parametrization
| Item/Software | Function | Key Notes |
|---|---|---|
| AmberTools | Software suite | Provides antechamber, parmchk2, and LEaP for topology generation [53]. |
| antechamber | Automates parameter generation | Assigns atom types, calculates charges, generates residue topology files [53]. |
| parmchk2 | Checks for missing parameters | Identifies undefined bonds, angles, or dihedrals and suggests values by analogy [53]. |
| GAFF2 Datatype | Force field parameter file | Contains the base atom types and parameters ($AMBERHOME/dat/leap/parm/gaff2.dat) [53]. |
| Input Structure File | Defines initial ligand geometry | Typically a PDB or MOL2 file with correct protonation states and 3D coordinates [53]. |
Step-by-Step Workflow:
Generate Point Charges and Assign Atom Types: Use
antechamberto process your ligand structure file (here, a PDB file). The-c bccflag uses the AM1-BCC2 method for fast charge calculation, and-at gaff2assigns GAFF2 atom types [53].This command produces a MOL2 file (
GWS.mol2) containing the 3D structure, GAFF2 atom types, and partial charges.Check for Missing Parameters: Use
parmchk2to identify any bonds, angles, or dihedrals in your ligand that lack pre-defined parameters in the GAFF2 library. This tool will attempt to fill gaps with reasonable values based on similar chemical moieties [53].The output file (
GWS.frcmod) must be carefully reviewed. Parameters flagged with "ATTN: needs revision" indicate missing values you must manually define [53].Incorporate Parameters into the System: The generated MOL2 and FRCMOD files are loaded into the
LEaPprogram (part of AmberTools) alongside your protein topology and the GAFF2 parameter file to create a complete system topology for simulation [53].
Modern Approach: Parametrization with OpenFF and Interchange
The Open Force Field (OpenFF) ecosystem offers a modern, Python-driven workflow with SMIRNOFF force fields, which can be more easily automated and integrated with other tools.
Step-by-Step Workflow:
Prepare Components: Load the protein, ligand, and force fields as separate components using the OpenFF Toolkit and
Interchange[57].Combine System Components: Use
Interchangeto combine the parameterized ligand with a separately parameterized protein system. This is powerful for building complex systems from modular parts [57].Solvation and Counter-Ions: Use helper functions to solvate the system and add ions to neutralize the total charge [57]. The
Interchangeobject can then export the fully parameterized system to various simulation engines like OpenMM, GROMACS, and AMBER via ParmEd.
Advanced Strategy: Bespoke Parametrization
For cases where standard force fields prove inadequate, bespoke parametrization optimizes parameters for a specific ligand. The OpenFF ecosystem provides BespokeFit, which automates the creation of tailored torsion parameters by fitting them to quantum mechanical (QM) calculations [55]. These bespoke parameters can be directly integrated into simulation workflows, such as relative binding free energy (RBFE) calculations performed with OpenFE [58].
Troubleshooting Common Issues
FAQ 1: Why does my simulation fail with atom naming or missing parameter errors?
- Cause: These are among the most common errors. Input structures may contain hydrogen atoms with names that don't match the force field's convention, or the ligand may contain chemical moieties with incomplete parameter coverage in the chosen force field [59] [53].
- Solution:
- For protein atoms, use the
-ignhflag withgmx pdb2gmxto ignore existing hydrogens and let the tool add them correctly [59]. - For ligands, always check the output of
parmchk2. Manually define any parameters flagged with "ATTN: needs revision" using literature values or custom QM calculations [53]. - Ensure ligand and protein chain IDs are distinct in your initial structure file if they are separate molecules [59].
- For protein atoms, use the
FAQ 2: How do I handle dihedral parameter inaccuracies for conjugated systems?
- Cause: Torsion angle parameterization is a known weakness, especially for ligands with π-electron conjugated systems. Standard force field dihedrals may not capture quantum effects that dictate the correct molecular conformation [52].
- Solution:
- Refit Torsion Parameters: Use a combination of QM calculations and atomistic free-energy calculations to achieve an improved parametrization, as demonstrated for the benzamidine/trypsin system [52].
- Use Bespoke Parametrization: Employ
OpenFF BespokeFitto automatically generate improved, ligand-specific torsion parameters, which has been shown to enhance the accuracy of binding free energy calculations [55] [58].
FAQ 3: My ligand topology has incorrect partial charges. What should I do?
- Cause: Different force fields and programs use different methods for assigning partial charges. Using incompatible charges (e.g., CGenFF charges with an AMBER/OpenFF protein force field) will lead to incorrect electrostatic interactions [52].
- Solution:
- Maintain Consistency: Use the charge derivation method intended for your force field. For GAFF/OpenFF, the RESP method fitted to HF/6-31G* electrostatic potentials is standard [52]. OpenFF also offers machine learning-based methods like
EspalomaChargefor ultra-fast charge assignment [55]. - Check Total Charge: Always verify that the total charge of your system is an integer value and add the appropriate number of counter-ions to neutralize the simulation box [57].
- Maintain Consistency: Use the charge derivation method intended for your force field. For GAFF/OpenFF, the RESP method fitted to HF/6-31G* electrostatic potentials is standard [52]. OpenFF also offers machine learning-based methods like
Troubleshooting Guides and FAQs
Frequently Asked Questions (FAQs)
Q1: I'm getting an error every time I try to save a file or run a job, leading to a pop-up that says "An error has occurred." What should I do? This issue is very likely that your Working Directory is set to a directory you do not have 'write access' to, such as the course-data folder. Change your working directory to a different folder (e.g., the Course folder on your Desktop) to resolve this [60].
Q3: The pH is noted as N.N. instead of the expected value (e.g., 7.4). Is this a problem? This is a known issue in older Maestro releases. Manually toggling or editing the pH in the Global Settings menu at the top of the Protein Preparation Workflow panel will correct the display [60].
Q4: I have a different number of LigPrep output entries in the Entry List than expected. Why? Additional LigPrep output structures can result from software improvements or changes to default settings in new releases. Double-check your calculation input and proceed with your output as usual [60].
Q5: When I try to open my Maestro project, I get a message saying, "The requested project is currently in use by another Maestro session." What does this mean? This message appears when a previous Maestro session was not closed properly before logging off the virtual cluster. Click "Yes" to proceed. For best practices, always close Maestro before logging off [60].
Advanced Troubleshooting Guide
Issue: Poor Sampling and Convergence
- Symptoms: High cycle error, significant hysteresis, poor overlap between lambda windows, and unreliable free energy predictions [61].
- Diagnosis and Resolution:
- Link Plots: Use link plots to visualize free energy differences (via TI and MBAR) and contributions from each lambda window. Discrepancies between TI and MBAR results or unusual patterns indicate insufficient sampling or slow conformational transitions [61].
- Cycle Error and Hysteresis Analysis: Perform cycle error analysis to identify discrepancies between direct and reverse transformation paths. Hysteresis indicates the system may not have reached equilibrium [61].
- Overlap Matrices: Plot overlap matrices for transformations. Visually identify areas of poor overlap to refine simulation parameters for specific links [61].
Issue: Inaccurate Ligand Pose or Unrealistic Geometries
- Symptoms: Incorrect binding modes or strained ligand conformations during simulation, leading to biased results [61].
- Diagnosis and Resolution:
- 3D Visualization: Use the 3D window to inspect the final frame of the trajectory for the beginning (λ=0) and end (λ=1) of both forward and backward transformations, ensuring no unrealistic geometries are adopted [61].
Issue: Inadequate Hydration of Binding Site
- Symptoms: Poor sampling of key solvation events affecting free energy accuracy [61].
- Diagnosis and Resolution:
- GCNCMC Water Sampling: Employ Grand Canonical Non-Equilibrium Candidate Monte Carlo (GCNCMC) to rehydrate the binding site by inserting and deleting water molecules within a user-defined sphere around the ligand, efficiently capturing solvent effects [61].
Table 1: Key Statistical Metrics for FEP+ Validation
| Metric | Description | Ideal Value/Outcome |
|---|---|---|
| Pearson R² | Measures the linear correlation between experimental and predicted binding affinities. | Close to 1.0 |
| Mean Unsigned Error (MUE) | Average of the absolute differences between experimental and predicted values. | As low as possible (e.g., < 1.0 kcal/mol) |
| Cycle Closure Error | The free energy difference between a direct transformation path and its reverse. | < 0.5 kcal/mol |
| Hysteresis | Indicates the system has not reached equilibrium, leading to inaccurate free energy estimates. | Minimal or non-existent |
| Overlap Matrix Score | Assesses the quality of sampling between neighboring lambda states. | High overlap (values close to 1) |
Table 2: Force Field Comparison for Protein Simulation (Based on a 52-Protein Test Set)
| Structural Property | GROMOS 43A1 | GROMOS 53A5 | GROMOS 53A6 | Key Finding |
|---|---|---|---|---|
| Backbone Hydrogen Bonds | Data not specified | Data not specified | Data not specified | Statistically significant but small differences between force fields were detected. |
| Native Hydrogen Bonds | Data not specified | Data not specified | Data not specified | Improvements in one metric were often offset by loss of agreement in another. |
| Radius of Gyration | Data not specified | Data not specified | Data not specified | No single force field performed best across all properties simultaneously. |
| Backbone φ/ψ Dihedral Distribution | Data not specified | Data not specified | Data not specified | Validation requires a diverse protein set and multiple structural criteria. |
Experimental Protocols
Detailed Methodology: Relative Binding FEP+ Calculation
Protocol 1: System Preparation for Relative Binding FEP+
- Protein Preparation:
- Load the protein structure into Maestro.
- Run the Protein Preparation Wizard to add missing hydrogen atoms, assign bond orders, and correct for missing side chains.
- Optimize the hydrogen-bonding network.
- Manually set the pH to the desired value (e.g., 7.4) in the Global Settings menu to avoid the "N.N." display issue [60].
- Conduct a restrained minimization to remove steric clashes.
- Ligand Preparation:
- Prepare ligand structures using LigPrep.
- Generate possible ionization states and tautomers at the target pH.
- Ensure proper chirality and generate low-energy ring conformations.
- Align all ligands to a common reference structure within the binding site [62].
Protocol 2: Running and Validating a Flare FEP Experiment
- FEP Graph Generation:
- Automatically generate the FEP graph using LOMAP to define alchemical transformations between ligands.
- The software intelligently inserts intermediate molecules for complex transformations to ensure computational feasibility and accuracy [61].
- Calculation Execution:
- Parameterize the system using a selected forcefield (e.g., Amber or OpenFF). OpenFF allows for the derivation of custom torsion parameters for ligands to enhance accuracy [61].
- Use the adaptive lambda window algorithm to run short MD calculations in the free phase, determining the optimal number of lambda windows for convergence and computational efficiency [61].
- Activate GCNCMC water sampling with a user-defined sphere radius around the ligand to capture critical solvation effects in the binding site [61].
- Results Analysis:
- Assess accuracy using the activity plot (experimental vs. predicted ΔG) and statistics (R², MUE) [61].
- Use sub-graph analysis to identify molecule subsets with lower internal error [61].
- Check for sampling issues using link plots, cycle error, hysteresis analysis, and overlap matrices [61].
- Visually inspect ligand conformations in the 3D viewer throughout the transformation to ensure realistic geometries [61].
Workflow and Signaling Pathway Diagrams
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for FEP Calculations
| Item | Function / Application |
|---|---|
| Maestro (Schrödinger) | A comprehensive graphical user interface for running molecular modeling and simulations, including FEP+ [60] [62]. |
| FEP+ (Schrödinger) | A specific tool for running relative binding free energy calculations within Maestro, used for lead optimization in drug design [60] [62]. |
| Flare FEP (Cresset) | A software solution for running accurate and efficient FEP calculations, featuring automated graph generation and advanced analysis tools [61]. |
| Protein Preparation Wizard | A workflow within Maestro for preparing and optimizing protein structures for subsequent simulations [60]. |
| LigPrep | A tool for preparing ligand structures, generating possible states, and ensuring proper geometry before an FEP run [60]. |
| Amber Forcefield | A widely used force field for parameterizing proteins and small molecules in molecular dynamics simulations [61]. |
| OpenFF | An open-source force field that allows for the derivation of custom parameters, particularly useful for novel ligand torsions [61]. |
| GROMOS Forcefield | A family of force fields parameterized for biomolecular simulations; selection depends on the specific protein system and target properties [26]. |
Technical Support Center: Troubleshooting Guides and FAQs
General Questions
| Question | Answer |
|---|---|
| Can I mix Martini 2 and Martini 3 models? | No. They are not compatible due to fundamental differences in force field parametrization and bead types, which can lead to inconsistencies in simulations [63]. |
| Can Martini simulate protein folding? | No. The secondary structure is an input parameter and remains fixed. Tertiary structural changes are allowed, but changes in secondary structure elements are beyond its scope [63]. |
| How should I interpret the time scale? | Martini dynamics are faster. A standard conversion factor of 4 is typically used, but the effective speed-up can vary between 3-8 depending on the system and process. Interpretation requires caution [63]. |
| How to reintroduce atomistic detail? | Several tools are available, including Backward, cg2at, and ezAlign, to convert a coarse-grained structure back to an all-atom representation [63]. |
Input Parameters and Simulation Setup
| Question | Answer |
|---|---|
| What time step should I use? | Martini is parameterized for time steps of 20-40 fs. A time step of 20-30 fs is recommended for most applications. Structural and thermodynamic properties are generally robust within this range [63]. |
| Can I do free energy calculations? | Yes, free energy calculations are possible with the Martini force field, and example tutorials are available [63]. |
Topologies and Molecule Types
| Question | Answer |
|---|---|
| How do I create a topology for a new molecule? | In-depth, step-by-step guides for parameterizing new molecules are provided in the official tutorial sections for both Martini 2 and Martini 3 [63]. |
| Are nucleotides available in Martini 3? | Not yet. A model for nucleotides is currently in development. The available Martini 2 model should be used for nucleotides [63]. |
| Is polarizable water available in Martini 3? | Not yet. It is currently in development. The polarizable water model for Martini 2 is available as an alternative for specific applications [63]. |
Frequent Simulation Problems
| Problem | Solution |
|---|---|
| My simulation keeps crashing. | - Reduce the time step to 20 fs.- Increase the neighbourlist update frequency/cutoff.- For proteins, try adding an elastic network (e.g., ELNEDYN).- Check topology for conflicting bonded potentials [63]. |
| My water is freezing. | Unwanted freezing is a known issue in Martini 2, as the model's freezing point is around 290K. A pragmatic solution is the use of "antifreeze" particles [63]. |
| My IDPs are too compact. | This is a known limitation in standard Martini 3. Use the specialized Martini3-IDP model, which features optimized bonded parameters for intrinsically disordered proteins to yield more expanded conformations [64]. |
| Transmembrane peptides insert incorrectly. | This is a recognized challenge in Martini 3. Current solutions may involve careful manual adjustment or the use of specialized parameters, as this is an active area of development [65]. |
Quantitative Data and Model Comparison
Effective Time Scale Conversion in Martini Simulations
The following table summarizes the speed-up factors for different processes, which should be used as a guideline for interpreting simulation time scales [63].
| Process / Molecule | Typical Speed-up Factor | Notes |
|---|---|---|
| Water Diffusion | 4 | Standard reference value. |
| Lipid Aggregation & Configurational Sampling | ~4 | Similar to water. |
| Protein Translational/Rotational Diffusion | ~4 | Based on simulations of rhodopsin. |
| Charged Molecules | >4 | Can be much larger. |
Experimental Protocols: Detailed Methodologies
Workflow for Setting Up a Soluble Protein Simulation
This protocol outlines the key steps for simulating a soluble globular protein (e.g., T4-lysozyme) using Martini 3 [66] [67].
Step-by-Step Guide
Obtain and Prepare the Atomistic Structure
Coarse-Graining and Topology Generation
- Use the
martinize2script to convert the all-atom structure to a coarse-grained model and generate the topology. - A typical command is:
- The
-dsspoption can be used to automatically assign secondary structure if a DSSP binary is available. Alternatively, provide a secondary structure string manually using the-ssflag [66] [67]. - This step generates a coarse-grained structure file (
.pdb), a master topology file (.top), and a protein topology file (.itp).
- Use the
System Assembly and Energy Minimization
- Use
gmx editconfto place the protein in a simulation box (e.g., dodecahedron) with a minimum 1.0 nm distance to the box edge. - Perform a brief energy minimization in vacuum (10-100 steps) to remove bad contacts [66].
- Use
Solvation and Ion Addition
- Solvate the system using
gmx solvatewith an equilibrated Martini water box (water.gro). Use-radius 0.21to avoid initial clashes. - Add ions to neutralize the system and to achieve a desired ionic strength (e.g., 0.15 M NaCl). Update the topology file (
.top) with the number of added water and ion beads [66].
- Solvate the system using
Equilibration and Production Run
- Perform energy minimization of the solvated system.
- Run a position-restrained (NPT) equilibration. The
-p backboneflag inmartinize2automatically generates the necessary position restraint file. - Start the production simulation without position restraints. Use
-maxwarn 1if needed to bypass a warning about the barostat and velocity generation [66].
Specialized Protocol: Working with Disordered Proteins
For intrinsically disordered proteins (IDPs), the standard Martini 3 model tends to produce overly compact conformations. The recommended solution is to use the Martini3-IDP model [64].
Key Improvement of Martini3-IDP
- Cause of Collapse: The standard model lacks specific dihedral potentials for coil regions, leading to unrestricted side-chain movements and unrealistic chain collapse [64].
- Solution: Martini3-IDP introduces optimized backbone and side-chain bonded parameters derived from fitting to a diverse set of atomistic (CHARMM36m) IDP simulations. This results in more accurate local configurations and expanded global dimensions that agree with experimental data like radii of gyration from SAXS [64].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key software tools and resources essential for setting up and running Martini simulations.
| Tool Name | Function | Application Notes |
|---|---|---|
| Martinize2 | Python program for converting atomistic protein structures into Martini coarse-grained models and topologies. | The primary tool for protein setup. Requires Python >3.9 [66] [67]. |
| DSSP | Program for assigning secondary structure from atomic coordinates. | Often called by martinize2 via the -dssp flag. Requires version 3.1.4 or older [68] [66]. |
| INSANE | Python script for building complex membrane systems with mixed lipids and proteins. | Used for generating lipid bilayers and embedding membrane proteins. Supports Martini 3 [69]. |
| Backward / cg2at | Tools for converting a coarse-grained structure back to an all-atom representation ("backmapping"). | Used to reintroduce atomistic detail after CG simulation for further analysis [63]. |
| cg_bonds.tcl | A Tcl script for VMD to visualize coarse-grained bonds and elastic network constraints. | Essential for visualizing the connectivity in Martini systems [69]. |
| GROMACS | The primary molecular dynamics software package for which Martini is most extensively implemented. | Features and implementation might differ in other MD packages like NAMD or CHARMM [63] [70]. |
Visualizing the MARTINI Ecosystem and Model Evolution
The diagram below illustrates the core MARTINI methodology and the specialized model variants available for different protein types.
Troubleshooting Force Field Bias and Optimization Strategies
Troubleshooting Guides and FAQs
FAQ: Understanding and Diagnosing Force Field Bias
Q1: What is force field bias, and how can I detect it in my protein folding simulations?
Force field bias occurs when the mathematical model (force field) used in molecular dynamics simulations inaccurately represents the physical interactions, leading to a preference for non-native protein structures. This systematic error can cause proteins to misfold or populate incorrect intermediate states.
To detect force field bias, researchers should:
- Compare with Experimental Data: Regularly validate simulation results against experimental structures from the Protein Data Bank and experimental observables such as NMR chemical shifts, scalar couplings, and SAXS data [43].
- Monitor Structural Metrics: Track key indicators like root-mean-square deviation, fraction of native contacts, and radius of gyration throughout simulations.
- Identify Recurring Non-native Motifs: Look for persistent, unrealistic structural elements. A documented example is the CHARMM22 force field's incorrect stabilization of helical structures in the Pin1 WW domain, which natively has a β-sheet architecture [40].
- Perform Free Energy Calculations: Use methods like deactivated morphing to calculate free energy differences between native and misfolded states. A significant free energy preference for a non-native state indicates a thermodynamic bias in the force field [40].
Q2: My simulation of a β-sheet protein is forming stable helices. Is this a known force field issue?
Yes, this is a documented issue in certain force fields. For example, a 10-μs simulation of the human Pin1 WW domain (a three-stranded β-sheet protein) using the CHARMM22 force field with CMAP corrections resulted in the formation of stable, non-native helical structures instead of the correct folded state. Free energy calculations revealed that these helical states were favored over the native state by 4.4–8.1 kcal/mol, confirming a substantial force field bias [40].
Q3: How does force field choice influence the observed protein folding mechanism, even if the native state is correct?
Even when a force field correctly identifies the native state as the global free energy minimum and reproduces experimental folding rates, the folding pathway can be highly force field-dependent.
A comparative study on the villin headpiece simulated with four different force fields (AMBER ff03, AMBER ff99SB-ILDN, CHARMM27, and CHARMM22) demonstrated this phenomenon [71]. While all force fields reproduced the correct native structure and folding rate, they showed significant differences in:
- Unfolded State Ensemble: The intrinsic stability of individual secondary structure elements in the unfolded state varied greatly.
- Order of Helix Formation: The preferential pathway and the order in which structural elements form differed across force fields.
The table below summarizes the key differences observed:
Table 1: Force Field Dependence of Folding Mechanism for the Villin Headpiece
| Force Field | Simulation Temp. (K) | Folding Time (μs) | Unfolded State Helicity (Helix 1/2/3) | Key Folding Mechanism Observation |
|---|---|---|---|---|
| AMBER ff03 | 390 | 0.8 ± 0.1 | 30%/52%/85% | Helix 1 unstable; forms last |
| AMBER ff99SB*-ILDN | 380 | 3.0 ± 0.4 | 22%/17%/59% | Helix 1 unstable; forms last |
| CHARMM27 | 430 | 0.9 ± 0.1 | 73%/33%/90% | Diffusion-collision mechanism |
| CHARMM22* | 360 | 2.6 ± 0.5 | 41%/9%/44% | Most heterogeneous folding pathway |
This indicates that matching a single native structure and folding rate does not guarantee a correct description of the folding free energy landscape [71].
Troubleshooting Guide: Correcting for Force Field Bias
Problem: Observed misfolding or excessive stabilization of non-native secondary structures.
Solution 1: Consider Switching to a Modern, Balanced Force Field
Older force fields often have known biases, such as an over-stabilization of helical content. Modern force fields are refined to provide a better balance for both folded and disordered proteins.
- Recommended Options:
- AMBER ff99SBws-STQ′ and ff03w-sc: These refined variants incorporate optimized protein-water interactions and torsional parameters. They accurately reproduce intrinsically disordered protein (IDP) dimensions while maintaining folded protein stability, addressing the collapse often seen in older force fields with simple water models [43].
- CHARMM22*: A helix-coil-balanced variant that showed good performance in villin headpiece simulations, producing a heterogeneous and realistic folding mechanism [71].
- AMBER ff99SB-disp: This force field is reparameterized with a modified water model to enhance protein-water interactions and backbone hydrogen bonding, showing state-of-the-art performance for both folded and disordered proteins [43].
Solution 2: Employ Advanced Sampling and Free Energy Methods
If changing the force field is not feasible, you can use advanced computational techniques to quantify and correct for the bias.
- Deactivated Morphing (DM): This method can calculate the free energy difference between native and misfolded states observed in your simulation. The workflow involves applying harmonic restraints, "deactivating" interactions, and morphing the system from one state to another to compute the free energy difference. This quantifies the thermodynamic bias and can be used to test corrections to the force field [40].
- Parallel Tempering/Replica-Exchange: These methods enhance conformational sampling, helping the simulation escape from deep, non-native energy minima where it might otherwise be trapped due to force field bias.
Solution 3: Utilize a Machine-Learned Coarse-Grained Model
As an alternative to all-atom force fields, consider newly developed transferable coarse-grained models.
- Machine-Learned Coarse-Grained Force Fields: These models are trained on diverse all-atom simulation data and can be several orders of magnitude faster. A recent model demonstrated the ability to predict metastable folded, unfolded, and intermediate states for various proteins, successfully handling sequences not used during training. This approach can bypass specific biases inherent in traditional all-atom force fields [35].
Experimental Protocols
Protocol 1: Detecting Thermodynamic Bias with Deactivated Morphing
Objective: Quantify the free energy difference between a native state and a misfolded state observed in simulations [40].
Methodology:
- Define Reference States: Obtain high-quality reference structures for the native state (e.g., from PDB) and the misfolded state (from your simulation trajectory via clustering).
- Calculate Free Energy Difference ((\Delta G)):
The deactivated morphing process follows a multi-step path to compute the free energy difference between two unrestrained conformational ensembles, E(A) and E(B).
- From the unrestrained ensemble of state A, E(A), apply harmonic restraints to all protein atoms to create a tightly restrained state, K1(A).
- "Deactivate" the non-bonded interactions in state A by turning off atomic charges and van der Waals parameters, creating a dummy state, D(A).
- Morph the coordinates of the dummy state from conformation A to conformation B along a least-squares path, resulting in state D(B).
- "Reactivate" the non-bonded interactions in state B, creating state K1(B).
- Remove the harmonic restraints to arrive at the unrestrained ensemble of state B, E(B).
- Interpretation: A negative (\Delta G) value favoring the misfolded state confirms a thermodynamic force field bias.
The following diagram illustrates the deactivated morphing free energy calculation workflow.
Protocol 2: Comparative Force Field Validation for a New Protein System
Objective: Evaluate multiple force fields to select the most appropriate one for simulating your protein of interest.
Methodology:
- System Setup:
- Prepare the native structure of your protein.
- Solvate it in a cubic water box with ~10,000 water molecules and neutralize the system with ions (e.g., 30 mM NaCl) [40].
- Use tools like VMD or CHARMM-GUI for setup.
- Simulation Parameters:
- Use a periodic boundary conditions.
- Employ a cutoff for short-range non-bonded interactions (e.g., 8.0–10.0 Å).
- Treat long-range electrostatics with Particle Mesh Ewald.
- Maintain temperature using a Langevin thermostat or Nosé-Hoover method.
- Maintain pressure using a Langevin piston or Parrinello-Rahman barostat.
- Multiple Production Runs:
- Run a set of microsecond-timescale simulations for the same protein system using different force fields (e.g., AMBER ff19SB, CHARMM36m, etc.).
- For each simulation, initiate from both the native state and unfolded states to test stability and foldability.
- Analysis and Comparison:
- Native Stability: Calculate the Cα-RMSD and fraction of native contacts from simulations started from the native state.
- Foldability: For simulations started from unfolded states, check if the protein folds to the native state and calculate the folding time.
- Mechanism: Analyze the formation of secondary structure elements and the order of folding events.
- Validation: Compare all results against available experimental data (NMR, SAXS, folding rates).
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Force Fields for Protein Folding Studies
| Item Name | Type | Primary Function | Example Use Case |
|---|---|---|---|
| CHARMM | Software Suite & Force Field | Performs molecular dynamics simulations; includes parameters for proteins, nucleic acids, lipids, etc. | Simulating protein folding with the CHARMM36m force field [72]. |
| AMBER | Software Suite & Force Field | A package for molecular dynamics simulations, particularly with AMBER-family force fields. | Testing protein stability with the ff19SB force field [43]. |
| GROMACS | Molecular Dynamics Engine | A high-performance MD engine capable of running simulations with various force fields. | Comparative force field studies due to its speed and efficiency. |
| NAMD | Molecular Dynamics Engine | A parallel MD code designed for high-performance simulation of large biomolecular systems. | Running long-timescale folding simulations [40]. |
| VMD | Molecular Visualization & Analysis | Visualizing, analyzing, and animating molecular trajectories; preparing simulation systems. | Solvating a protein in a water box and neutralizing the system [40]. |
| AMBER ff99SBws-STQ′ | Force Field | A refined force field with improved torsional parameters for glutamine; balanced for folded/IDPs. | Simulating systems with polyglutamine tracts or seeking a balanced force field [43]. |
| AMBER ff03w-sc | Force Field | A force field with selectively optimized protein-water interactions for stability and accuracy. | Studying folded protein stability and IDP chain dimensions simultaneously [43]. |
| CHARMM22* | Force Field | A helix-coil balanced variant of CHARMM22 for more realistic folding mechanisms. | Investigating protein folding pathways [71]. |
| Machine-Learned CG Model | Coarse-Grained Force Field | A transferable model for rapid exploration of protein conformational landscapes. | Quickly screening folding behavior of new protein sequences [35]. |
Frequently Asked Questions (FAQs)
FAQ 1: Why does my simulation of a beta-sheet protein get stuck in helical structures? This is a documented issue known as helical bias, where a force field incorrectly stabilizes non-native helical intermediates over the native beta-sheet structure. A seminal study on the human Pin1 WW domain (a three-stranded beta-sheet protein) found that under the CHARMM22 force field with CMAP corrections, helical misfolded states were more stable than the native state by 4.4–8.1 kcal/mol. This thermodynamic bias explains why long simulations failed to fold the protein correctly, instead trapping it in non-native helical states [40] [73].
FAQ 2: My simulation didn't fold. Is it a sampling problem or a force field problem? It can be either, or both. Long-timescale simulations are required to observe folding events, and insufficient sampling (kinetic trapping) is a common issue [73]. However, advanced free energy calculations, such as the deactivated morphing (DM) method, can help distinguish between the two. If the free energy of misfolded states is lower than that of the native state, the problem is likely a force field deficiency rather than a lack of sampling [40].
FAQ 3: Are there force fields less prone to helical bias? Force field development is a continuous effort. Newer approaches, including machine-learned coarse-grained (CG) models and polarizable force fields, aim to correct such inaccuracies [35] [5]. Some studies have also succeeded in folding proteins using distributed implicit-solvent simulations with different force fields, though inaccuracies may persist [73]. Always consult the latest literature for your specific protein type.
FAQ 4: What are the best practices for testing a force field's accuracy for my system? Whenever possible, compare your simulation results with experimental data. If available, use advanced sampling techniques to calculate the free energy landscape of folding. A reliable force field should have the experimentally known native state as the global free energy minimum. For new methods like machine-learned force fields, ensure they have been validated on proteins with low sequence similarity to your target to confirm transferability [35] [74].
Troubleshooting Guide
Issue 1: Force Field Bias and Misfolding
Problem: The simulation consistently populates incorrect, stable non-native states, indicating a potential thermodynamic bias in the force field.
Solution Protocol: Quantifying Bias with Free Energy Calculations The following protocol, based on the study of the Pin1 WW domain, uses deactivated morphing to calculate free energy differences between states [40].
Identify Reference Structures:
- Native State (Sheet): Obtain from an experimental structure (e.g., PDB).
- Misfolded States (e.g., helixU, helixL, helixV): Extract representative structures from your simulation trajectories using cluster analysis.
Calculate Free Energy Differences: The deactivated morphing method calculates the free energy difference between the unrestrained ensembles of two reference conformations (A and B) by following a defined path. The schematic pathway is: E(A) → K(A) → Q(A) → D(A) → D(B) → Q(B) → K(B) → E(B) where:
- E: Unrestrained ensemble of structures near a reference conformation.
- K: State with harmonic restraints applied to all protein atoms (κ = 1000 kcal/mol Ų).
- Q: "Deactivated" state with all protein atoms restrained to their reference coordinates.
- D: "Dummy" state with uniform van der Waals parameters and charges [40].
- Each transition is subdivided to ensure sufficient overlap between states, and free energy calculations are performed in the NPT ensemble.
Analysis and Interpretation:
- A negative free energy difference (ΔG) for a misfolded state relative to the native state confirms a force field bias.
- Perform error analysis using block averaging to ensure statistical significance [40].
Issue 2: Inadequate Sampling of Folding Events
Problem: The simulation fails to reach the native state within a practical timeframe.
Solution Protocol: Employing Enhanced Sampling and Specialized Hardware
- Leverage Structure-Based Models (SBMs): For large proteins, Gō-like models or other SBMs that bias interactions toward the native contacts can be highly effective. These models reduce computational cost by several orders of magnitude and are well-suited for predicting folding pathways and intermediates [75].
- Use Advanced Sampling Techniques: Implement methods like replica exchange molecular dynamics (REMD) to improve sampling over energetic barriers [75].
- Utilize High-Performance Computing: Exploit specialized supercomputers (e.g., ANTON) or distributed computing projects (e.g., Folding@home) to achieve the necessary microsecond-to-millisecond timescales for all-atom simulations [73] [75].
Experimental Protocols
Detailed Methodology: All-Atom Folding Simulation and Analysis
This protocol is adapted from methods used in studies of fast-folding proteins like the Pin1 WW domain and villin headpiece [40] [73].
1. System Preparation
- Protein Structure: Obtain the initial unfolded state. This can be a fully extended structure (e.g., all (ϕ, ψ) angles set to (-135,135)) or a thermally denatured state generated by a short simulation at high temperature (e.g., 490 K for 100 ns) [40].
- Solvation: Solvate the protein in a cubic box of explicit water molecules (e.g., TIP3P model).
- Neutralization: Add ions (e.g., NaCl) to neutralize the system and achieve a physiological concentration (e.g., 30 mM) [40].
2. Simulation Parameters (Example using NAMD/CHARMM)
- Force Field: CHARMM22 with CMAP corrections [40].
- Electrostatics: Particle-Mesh Ewald (PME) method for long-range interactions.
- Non-bonded interactions: Apply a cutoff (e.g., 8.0 Å) with a switching function.
- Constraints: Constrain bonds involving hydrogen atoms (e.g., using RATTLE/SETTLE algorithms).
- Thermostat: Use a Langevin thermostat with a damping constant (e.g., 0.1 ps⁻¹) to maintain temperature (e.g., 337 K for folding studies).
- Barostat: For NPT simulations, use a Nosé-Hoover Langevin piston barostat.
- Timestep: 2.0 fs [40].
3. Trajectory Analysis
- Cluster Analysis: Use tools like the GROMOS method in GROMACS to identify dominant conformational states from the trajectory [40].
- Root-Mean-Square Deviation (RMSD): Calculate Cα-RMSD to the native structure to measure folding progress.
- Fraction of Native Contacts (Q): Measure the fraction of native contacts formed; a value of ~1 indicates the native state [35].
- Free Energy Landscape: Construct a landscape using reaction coordinates like RMSD and Q to visualize metastable states and barriers [35].
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Key Computational Tools and Resources for Protein Folding Simulations.
| Item | Function | Example Use Case |
|---|---|---|
| All-Atom Force Fields (Additive) | Empirically derived potentials for atomic interactions; fixed partial charges. | Routine simulation of biomolecules; test for known biases (e.g., helical bias in CHARMM22/AMBER variants) [40] [5]. |
| Structure-Based Models (SBM/Gō) | Knowledge-based potentials that bias the simulation toward the native structure. | Efficiently explore folding pathways and intermediates of large proteins; understand topology-based folding [75]. |
| Machine-Learned Coarse-Grained (CG) Models | Bottom-up CG force fields trained on all-atom data; highly computationally efficient. | Extrapolative MD on new sequences; predict metastable states and folding free energies orders of magnitude faster [35]. |
| Polarizable Force Fields | Advanced models incorporating electronic polarization effects. | Improve accuracy in modeling electrostatic interactions, such as in ion channels or nucleic acids [5]. |
| Enhanced Sampling Algorithms | Methods to accelerate the exploration of configuration space. | Overcome free energy barriers to observe rare events like folding/unfolding (e.g., replica exchange, metadynamics) [75]. |
Visualizing the Workflow and Solutions
The following diagram illustrates the logical process for diagnosing and addressing common pitfalls in protein folding simulations.
The landscape of force fields is continuously evolving. The emergence of machine-learned, transferable coarse-grained models demonstrates the feasibility of universal models that can predict metastable states and relative folding free energies for unseen sequences, addressing key pitfalls like bias at a fraction of the computational cost of all-atom simulations [35] [5]. When selecting a force field, researchers must weigh the trade-offs between all-atom detail, computational efficiency, and the specific biases known for their protein class, leveraging the growing toolkit to navigate and correct these common challenges.
Frequently Asked Questions (FAQs)
FAQ 1: What is force field consensus scoring and why is it used in computational drug discovery?
Force field consensus scoring is a strategy where results from multiple molecular mechanics force fields are combined to improve the reliability and accuracy of binding affinity predictions, such as relative binding free energies (ΔΔGbind). It is used because the performance of individual force fields can be system-dependent; by averaging results from several force fields, researchers can mitigate the risk of poor predictions from any single model and obtain more robust outcomes. Studies have shown that while there may be no statistically noticeable performance difference among many different force field combinations, using a consensus approach provides valuable suggestions for force field selection and improves the predictability of alchemical free energy calculations [76].
FAQ 2: Which force field combinations are typically used in consensus scoring for protein-ligand binding?
Consensus scoring studies often test various combinations of force fields for the protein, the ligand, and the water model. A typical setup may include:
- Protein Force Fields: AMBER ff14SB [76], AMBER ff19SB [76].
- Ligand Force Fields: GAFF2.2 [76], OpenFF (e.g., versions 1.3.0, 2.0.0) [76] [77].
- Water Models: TIP3P [76], TIP4PEW [76], OPC [76]. One study found that a combination of ff14SB for the protein, GAFF2.2 for the ligand, and TIP3P for water performed best among 12 tested combinations, but consensus averaging across multiple force fields is recommended to enhance reliability [76].
FAQ 3: What are the key metrics for evaluating the success of a consensus scoring protocol?
The success of a consensus scoring protocol is typically evaluated by comparing the computationally predicted binding affinities against experimental values. Key statistical metrics include:
- Mean Unsigned Error (MUE): The average of the absolute differences between predicted and experimental values. An MUE of 0.87 kcal/mol was reported for a top-performing single force field combination [76].
- Root-Mean-Square Error (RMSE): Measures the magnitude of the average prediction error (e.g., 1.22 kcal/mol for a top performer) [76].
- Correlation Coefficients: Pearson’s (e.g., 0.64), Spearman’s (e.g., 0.73), and Kendall’s (e.g., 0.54) correlations assess the rank-ordering agreement between predictions and experiment [76]. A successful protocol should yield low MUE and RMSE values and high correlation coefficients.
Troubleshooting Guides
Issue 1: High Variance in Free Energy Estimates Between Different Force Fields
| Potential Cause | Solution |
|---|---|
| Insufficient sampling in the alchemical transformation. | Ensure adequate simulation time per lambda window. Research suggests that 5 ns per window with 12 lambda windows is sufficient for reliable results. Using 4 independent runs is also recommended [76]. |
| An outlier force field with poor performance for your specific system. | Implement a robust consensus method, such as averaging the results from multiple force fields. This can smooth out errors from any single, poorly performing force field [76] [78]. |
| Incorrect system setup (e.g., protonation states, missing residues). | Carefully prepare the molecular system using reliable tools (e.g., CHARMM-GUI Free Energy Calculator) and validate the initial structure [76]. |
Issue 2: Poor Correlation with Experimental Binding Data
| Potential Cause | Solution |
|---|---|
| The force field is not suitable for the specific protein-ligand system under study. | Do not rely on a single force field. Use a consensus of multiple force fields (e.g., combining different protein, ligand, and water models) to improve transferability and accuracy [76]. |
| The ligand partial charges are not assigned optimally. | For GAFF2.2, use the AM1-BCC method for charge assignment, as it has been shown to perform well without a clear need for more complex methods like RESP in this context [76]. For OpenFF, charges are often assigned via AM1-Mulliken [76]. |
| Inadequate conformational sampling of the ligand or protein. | Extend simulation time or employ enhanced sampling techniques. For standard practice, ensure you are using a 4 fs timestep with hydrogen mass repartitioning to improve efficiency [76]. |
Issue 3: Inconsistent Results When Repeating Simulations
| Potential Cause | Solution |
|---|---|
| Lack of statistical independence and convergence. | Perform multiple independent simulation runs (e.g., 4 runs as done in benchmark studies) and report the mean and standard error of the predictions [76]. |
| Uncontrolled initial conditions or velocities. | Use different random seeds for each independent run to ensure proper sampling of phase space. |
| Software or hardware instability. | Use standardized, well-tested simulation packages (e.g., AMBER20's pmemd.cuda) and verify consistent results across different computational resources where possible [76]. |
Experimental Protocols & Data
Key Protocol: AMBER-TI for Relative Binding Free Energy Calculation
This protocol is adapted from large-scale benchmark studies [76].
1. System Preparation:
- Select your protein-ligand complex from a reliable source (e.g., PDB codes: 4DJW for BACE1, 4GIH for TYK2).
- Use CHARMM-GUI Free Energy Calculator to generate inputs for the complex and ligand-only systems for alchemical transformation.
- Select the desired force field combinations (e.g., ff14SB/ff19SB for protein, GAFF2.2/OpenFF for ligand, TIP3P/TIP4PEW/OPC for water).
2. Simulation Parameters:
- Alchemical Transformation: Use the unified protocol with a 4 fs timestep and hydrogen mass repartitioning.
- Lambda Windows: Use 12 lambda windows. A suggested set is based on a linear scheme: 0.000, 0.091, 0.182, 0.273, 0.364, 0.455, 0.545, 0.636, 0.727, 0.818, 0.909, 1.000.
- Softcore Potential: Apply the second-order smoothstep softcore potential, SSC(2), with parameters α=0.2 and β=50 Ų.
- Nonbonded Interactions: Use Particle Mesh Ewald (PME) for long-range electrostatics. Set van der Waals cutoff to 10 Å.
- Ensemble: Perform simulations in the NPT ensemble at 300 K and 1 atm.
3. Execution and Analysis:
- Run simulations using the
pmemd.cudamodule of AMBER20. - Perform 4 independent runs for each transformation to ensure statistical robustness.
- For a 5 ns simulation, use the last 4 ns of data for analysis. For a 10 ns simulation, use the last 5 ns.
- Calculate the free energy using thermodynamic integration (TI) and the trapezoidal rule.
- Compute the relative binding free energy (ΔΔGbind) as: ΔΔGbind = ΔGcomplex - ΔGligand, where ΔGcomplex and ΔGligand are the alchemical transformation free energies in the complex and solution states, respectively.
Performance Data of Different Force Field Combinations
The table below summarizes the performance of selected force field combinations from a benchmark study on 80 alchemical transformations [76]. The values for the "Consensus" row are illustrative of the expected improvement from averaging.
| Force Field Combination (Protein/Ligand/Water) | Mean Unsigned Error (MUE) [kcal/mol] | Root-Mean-Square Error (RMSE) [kcal/mol] | Pearson's Correlation |
|---|---|---|---|
| ff14SB + GAFF2.2 + TIP3P | 0.87 | 1.22 | 0.64 |
| ff19SB + OpenFF1.3.0 + TIP4PEW | Data not explicitly stated in results; performance was not statistically noticeably different from the best performer [76]. | ||
| Consensus (Averaged) | Can yield lower error and higher correlation than most individual combinations [76]. |
Workflow Visualization
Consensus Scoring Workflow
Research Reagent Solutions
The table below lists essential tools and resources for implementing force field consensus scoring.
| Reagent / Resource | Function / Purpose | Reference / Source |
|---|---|---|
| CHARMM-GUI Free Energy Calculator | Web-based tool for preparing input files and scripts for alchemical free energy calculations (e.g., for AMBER-TI). | https://www.charmm-gui.org [76] |
| AMBER20 | Molecular dynamics software package. The pmemd.cuda module is used for running GPU-accelerated TI simulations. |
https://ambermd.org [76] |
| Force Fields: AMBER ff14SB, ff19SB | Provides parameters for the protein in the system. | Included in AMBER suite [76] |
| Force Fields: GAFF2.2 | Provides parameters for small molecule ligands. Charges are assigned with AM1-BCC. | https://ambermd.org/antechamber/gaff.html [76] |
| Force Fields: OpenFF 1.3.0, 2.0.0 | Alternative open-source force field for small molecules. Charges are assigned with AM1-Mulliken. | https://openforcefield.org [76] [77] |
| Water Models: TIP3P, TIP4PEW, OPC | Solvent models used to complete the solvated system. | Included in AMBER suite [76] |
| Benchmark Sets (e.g., JACS Set) | Standardized sets of protein-ligand systems with known experimental binding data for validation. | PDB Codes: 4DJW, 4GIH, 1H1Q, etc. [76] |
FAQ: Troubleshooting Common Simulation Issues
1. My simulation shows an unnatural structural collapse in an intrinsically disordered protein. What might be wrong?
This is a common issue when the force field and water model are not well-suited for disordered systems. Simulations using the TIP3P water model, for instance, are known to cause an artificial collapse of IDPs, leading to overly compact structures and unrealistic NMR relaxation properties [12]. To resolve this:
- Solution: Switch to a water model designed for disordered proteins, such as TIP4P-D [12]. Furthermore, ensure you are using a modern force field that has been validated for IDPs, such as CHARMM36m [12].
2. How do I determine the optimal distance cutoff for analyzing side-chain interactions in a Protein Structure Network?
The distance cutoff is critical to avoid a network that is either too fragmented or overly connected.
- Problem: Cutoffs below 5 Å result in too few residue hubs (fewer than four). Cutoffs above 5 Å cause the entire network to merge into a single, uninformative cluster [79].
- Solution: A distance cutoff of 5 Å between the centers of mass of residue side chains provides a robust description of network hubs and connected components. This value is generally robust across different protein folds, sizes, and MD force fields [79].
3. What is the maximum feasible simulation length, and why is there a limit?
The simulation length is limited by both practical and theoretical numerical constraints.
- Practical Limit: The need for femtosecond-scale time steps to accurately resolve atomic vibrations means that simulating biologically relevant timescales (microseconds to milliseconds) requires billions of integration steps, which is computationally demanding [80].
- Theoretical/Numerical Limit: Even with stable integration methods like Verlet, the accumulation of finite precision errors over billions of steps can cause the simulation to become inaccurate or unstable [80]. This is why coarse-grained models, which use larger time steps, can access much longer timescales [80] [35].
4. I am adding a new molecule to my simulation. The residue is not found in the force field database. What should I do?
The pdb2gmx tool (and similar tools in other software packages) relies on residue definitions in the force field's database.
- Solution: You cannot use automated topology generation if the residue is missing [81]. Your options are to:
- Manually parameterize the residue, which is complex and requires expert knowledge [81].
- Find a pre-existing topology file (
.itp) for the molecule from a reliable source and include it in your system's topology [81]. - Search the literature for published parameters compatible with your force field [81].
5. My simulation fails with an "Out of memory" error. How can I fix this?
This error occurs when the system demands more memory than is available.
- Solution: You can:
Optimizing Key Parameters: Protocols and Data
Cut-off Distances
The non-bonded cut-off distance is a critical parameter for balancing computational cost and simulation accuracy. The table below summarizes recommendations for different applications.
Table 1: Recommended Distance Cutoffs for Different Applications
| Application | Recommended Cut-off | Rationale and Notes |
|---|---|---|
| Protein Structure Network (PSN) Analysis | 5.0 Å (between side-chain centers of mass) | Provides a robust balance between identifying hub residues and avoiding a single, uninformative cluster. Validated across multiple proteins and force fields [79]. |
| General Non-bonded Interactions | Typically 1.0 - 1.2 nm (10 - 12 Å) | Standard for most biomolecular force fields (e.g., CHARMM, AMBER, GROMOS). Must be specified in the force field parameters. |
| LJ and Coulomb Interactions | Defined by the specific force field | The optimal value is force-field dependent. Always consult the force field's original publication for recommended settings. |
Experimental Protocol: Determining an Optimal PSN Cutoff To determine the optimal cutoff for your specific Protein Structure Network analysis, follow this protocol [79]:
- Generate Ensemble: Perform an MD simulation to obtain a conformational ensemble of your protein.
- Build Networks: Construct a series of PSNs from the trajectory using a range of distance cutoffs (e.g., from 4.0 Å to 7.0 Å in 0.5 Å increments).
- Calculate Properties: For each network, calculate fundamental properties like the number of hub residues (nodes with a degree ≥ 3) and the size distribution of connected components (clusters).
- Identify Optimal Value: The optimal cutoff is the value at which the number of hubs is meaningful (e.g., >4) and the network is not dominated by a single giant component. Studies show this is consistently near 5 Å [79].
λ Windows for Alchemical Free Energy Calculations
Alchemical free energy calculations require careful selection of λ values to ensure proper sampling and overlap between states.
Table 2: Guidelines for Setting Up λ Windows
| Parameter | Recommendation | Purpose and Consideration |
|---|---|---|
| Number of Windows | 12 - 24 windows | More complex transformations (e.g., charge changes, large conformational shifts) require more windows to ensure smooth transitions. |
| Spacing | Non-linear spacing is often beneficial. | Denser sampling is typically needed in regions where the potential energy changes rapidly as a function of λ (e.g., when Lennard-Jones interactions are turned on/off). |
| Soft-Core Potentials | Essential. | Prevents singularities and numerical instabilities when particles are created or annihilated at λ endpoints (0 and 1). |
| Validation | Monitor energy overlap. | Check that the potential energy distributions of adjacent λ windows have sufficient overlap. Poor overlap indicates the need for more windows or closer spacing. |
Experimental Protocol: Setting Up a Lambda Schedule A general protocol for setting up a λ schedule for a thermodynamic integration (TI) or free energy perturbation (FEP) calculation is:
- Define End States: Clearly define the initial (λ=0) and final (λ=1) states of your alchemical transformation.
- Choose Number of Windows: Start with a larger number of windows (e.g., 16-20) for a new system to ensure safety.
- Select Coupling Scheme: Decide which components (e.g., Coulombic, Lennard-Jones, bonded terms) are coupled to λ and in what order. Typically, Coulombic terms are transformed before Lennard-Jones terms.
- Run Test Simulations: Perform short simulations at each proposed λ value.
- Analyze Overlap: Calculate the overlap of the Hamiltonian energy distributions between successive windows. If the overlap is poor (<5%), add additional windows in that region.
- Production Run: Once a satisfactory schedule is confirmed, proceed with long production runs.
Simulation Length
Achieving sufficient sampling is one of the greatest challenges in MD simulations. The required length depends entirely on the process being studied.
Table 3: Simulation Length Guidelines for Different Biological Processes
| Process of Interest | Recommended Simulation Length | Technical Notes |
|---|---|---|
| Local Side-Chain Motions | Nanoseconds (ns) to hundreds of ns | Relatively fast and can often be sampled with standard GPU-accelerated simulations [82]. |
| Loop Dynamics | Hundreds of ns to microseconds (µs) | Depends on loop size and flexibility. |
| Domain Hingeing | Microseconds (µs) to milliseconds (ms) | Often beyond the reach of routine all-atom simulation; may require enhanced sampling or coarse-graining [35]. |
| Protein Folding/Unfolding | Microseconds (µs) to seconds | For all-atom MD, this typically requires specialized supercomputing or machine-learned coarse-grained models [35]. |
| Validation | Replicate simulations are crucial. | Run multiple independent simulations (with different initial velocities) to ensure observed events are reproducible and not artifacts of a single trajectory. |
Experimental Protocol: Assessing Simulation Convergence Before drawing conclusions, you must assess if your simulation is long enough to have sampled a representative ensemble.
- Run Multiple Replicas: Initiate at least 3-5 independent simulations from the same starting structure but with different random velocity seeds.
- Monitor Root-Mean-Square Deviation (RMSD): Plot the RMSD of the protein backbone over time. The RMSD should plateau and fluctuate around a stable average, indicating the simulation has equilibrated.
- Calculate Observables as a Function of Time: Block averaging is a powerful technique. Calculate a key observable (e.g., radius of gyration, SASA) by averaging over the first 10%, 20%, ... up to 100% of the trajectory. The value should become stable as the block length increases.
- Check for Drift: Monitor potential energy, temperature, and pressure to ensure the system remains stable and well-controlled throughout the production run.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Software and Force Fields for Biomolecular Simulation
| Tool / Resource | Type | Function and Application |
|---|---|---|
| CHARMM36m | All-Atom Force Field | A widely used and updated force field for proteins, shown to perform well for both globular proteins and intrinsically disordered regions (IDRs) [12]. |
| AMBER (e.g., ff19SB) | All-Atom Force Field | A leading family of force fields, continually improved with a focus on protein backbone and side-chain dihedral accuracy [83]. |
| GROMACS | MD Simulation Software | A high-performance, open-source software package for simulating Newtonian equations of motion. Known for its extreme speed and rich analysis tools [81]. |
| NAMD | MD Simulation Software | A parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems [83]. |
| CHARMM General FF (CGenFF) | Force Field Parameter Set | Provides parameters for a wide range of drug-like small molecules, enabling simulation of protein-ligand complexes [83]. |
| TIP4P-D | Water Model | An explicitly modeled water designed to improve the simulation of intrinsically disordered proteins and prevent unnatural collapse [12]. |
| Martini | Coarse-Grained Force Field | A popular coarse-grained force field that allows simulation of larger systems and longer timescales, excellent for membranes and protein-protein interactions [35]. |
| Machine-Learned Coarse-Grained Models | Coarse-Grained Force Field | Emerging models trained on all-atom simulation data that aim for transferability and high speed while maintaining predictive accuracy for folding and dynamics [35]. |
Diagnosing Issues in Thermodynamics and Kinetics of Protein Folding
Frequently Asked Questions (FAQs)
FAQ 1: My simulations show incorrect folding rates and free energies. Could the force field be the cause? Yes, the physical model (force field) is a primary factor. Advances in hardware now allow simulations to access folding timescales, enabling direct comparison of simulated rates and free energies with experimental data. Inaccuracies in these quantitative metrics often point to force field limitations. For instance, while some modern force fields can reproduce folding rates and free energies within a reasonable range, they may still struggle with other thermodynamic properties like folding enthalpies or heat capacity [84] [39].
FAQ 2: Why does my simulated protein structure collapse into an overly compact shape? This is a common issue, particularly when simulating Intrinsically Disordered Proteins (IDPs) or flexible regions. Many traditional force fields (e.g., standard AMBER or CHARMM versions) were parametrized and validated using structured, globular proteins. When applied to disordered systems, they tend to overestimate intramolecular attraction, leading to unrealistic compact structures. This problem is often exacerbated by the use of certain water models, like TIP3P [85] [12].
FAQ 3: How can I validate my force field choice for a protein with both structured and disordered regions? Simulating these "hybrid" proteins is particularly challenging. A robust validation should go beyond simple structural metrics (like RMSD) and compare a wide range of predicted parameters against experimental data. Key observables include:
- Radius of gyration (Rg): Sensitive to overall compactness.
- NMR parameters: Chemical shifts, Residual Dipolar Couplings (RDCs), Paramagnetic Relaxation Enhancement (PRE), and especially NMR relaxation data, which are highly sensitive to dynamics and force field choice [12].
- Small-Angle X-Ray Scattering (SAXS) data: Provides solution-state structural information [12].
FAQ 4: What are the best practices for force field selection and validation?
- Match the force field to your protein type: Use force fields specifically developed or optimized for your system of interest (e.g., IDPs vs. globular proteins).
- Prioritize water model selection: The solvent model is critical. Evidence suggests that models like TIP4P-D can significantly improve performance for disordered and hybrid systems [12].
- Benchmark against multiple experimental observables: Do not rely on a single metric. Use a curated test set of diverse structural properties to avoid overfitting and to ensure balanced performance [26].
- Ensure adequate sampling and statistics: Perform multiple replicates and ensure simulation length is sufficient for meaningful statistical comparison, as variations between replicates can be large [26].
Troubleshooting Guide
Problem 1: Inaccurate Thermodynamic Properties
Issue: Calculated folding free energies (ΔGf), enthalpies (ΔHf), or heat capacities (ΔCv) deviate significantly from experimental measurements.
| Possible Cause & Diagnosis | Solution & Recommended Action |
|---|---|
| Cause: Systematically biased force field energetics. This can be due to imperfect parametrization of bonded or non-bonded terms [39].Diagnosis: Compare multiple thermodynamic properties from long equilibrium simulations with many folding/unfolding events to experimental data. Check if the error is consistent across different properties [84]. | Action: Switch to a modern, refined force field. For villin headpiece simulations, the Amber ff99SB*-ILDN force field demonstrated a reasonably accurate calculation of folding free energies and melting temperatures [84]. Ensure you use the latest version of a force field that has been validated for folding studies. |
| Cause: Inadequate simulation sampling. Thermodynamic properties require the system to reach equilibrium and sample the folded and unfolded states extensively [26].Diagnosis: Run multiple long simulations and check for consistency in folded/unfolded populations. A single short trajectory may not yield reliable statistics. | Action: Increase simulation time and the number of replicates. For quantitative thermodynamics, you need to observe numerous spontaneous folding and unfolding events—ideally 30 or more per trajectory [84]. |
Problem 2: Unrealistic Structural Properties of Disordered States
Issue: Intrinsically Disordered Proteins (IDPs) or regions appear too compact in simulation, failing to match experimental SAXS or NMR data.
| Possible Cause & Diagnosis | Solution & Recommended Action |
|---|---|
| Cause: Excessive protein-protein interactions in the force field. Traditional force fields often lack sufficient protein-water dispersion, leading to overly "sticky" intramolecular interactions [85] [12].Diagnosis: Calculate the radius of gyration (Rg) and SAXS profile from your simulation. Compare directly with experimental values. | Action: Adopt an IDP-optimized force field. Consider DES-amber or ff99SBws, which have shown good performance for the disordered protein COR15A [85]. Other options include CHARMM36m or Amber ff99SB-disp. |
| Cause: Incompatible water model. Standard water models like TIP3P can contribute to the artificial collapse of IDPs [12].Diagnosis: If using TIP3P with an otherwise good force field and still observing collapse, the water model is likely a key factor. | Action: Re-run simulations using a water model designed to improve hydration, such as TIP4P-D [12]. The combination of a protein force field like CHARMM22* or Amber99SB-ILDN with TIP4P-D has been shown to improve reliability for proteins containing disordered regions [12]. |
Problem 3: Incorrect Kinetics and Pathways
Issue: The simulated folding rate is orders of magnitude off, or the protein follows a non-native folding pathway.
| Possible Cause & Diagnosis | Solution & Recommended Action |
|---|---|
| Cause: Inaccurate energy landscape topography. The force field may create incorrect free energy barriers between states, affecting rates and mechanisms [84] [39].Diagnosis: Calculate the folding rate (τf) and pre-exponential factor (k0) from multiple transitions. Compare with experimental kinetic measurements. Analyze Φ-values to see if mutation effects are reproduced [84]. | Action: Use a force field that has been quantitatively tested on fast-folding proteins. Long equilibrium simulations of villin headpiece variants with Amber ff99SB*-ILDN successfully reproduced the 5-fold faster folding of a norleucine double mutant, supporting the accuracy of its kinetic predictions [84]. |
| Cause: Lack of statistical certainty in kinetic analysis. Folding is a stochastic process, and inferring rates from too few events is unreliable [26].Diagnosis: The calculated folding time changes significantly between different simulation replicates. | Action: Dramatically increase the number of observed folding/unfolding events. One study ensured each trajectory contained between 30 and 150 such events to directly compute kinetic quantities without building approximate models [84]. |
Experimental Protocols for Validation
Protocol 1: Validating Thermodynamics from Long Equilibrium MD
This protocol is adapted from studies on the villin headpiece to directly compute thermodynamic properties [84].
System Setup:
- Protein Preparation: Obtain the initial coordinates for your protein (e.g., from the PDB).
- Solvation: Solvate the protein in an explicit water box, ensuring a minimum distance between the protein and box edges.
- Neutralization: Add ions (e.g., Na+/Cl-) to neutralize the system's charge and simulate physiological ionic strength.
Simulation Details:
- Force Field: Use a modern force field like Amber ff99SB*-ILDN.
- Water Model: Select an appropriate model (e.g., TIP4P-D for disordered systems).
- Software: Use a molecular dynamics package like GROMACS, AMBER, or NAMD.
- Conditions: Run multiple long simulations (e.g., >300 µs aggregate time) at different temperatures. Employ a thermostat (e.g., Nosé-Hoover) and a barostat (e.g., Parrinello-Rahman) to maintain constant temperature and pressure.
Data Analysis:
- State Assignment: Use a "transition-based assignment" on a reaction coordinate (e.g., Cα-RMSD to the native structure) to partition the trajectory into folded and unfolded states.
- Free Energy: Calculate the folding free energy (ΔGf) directly from the ratio of the populations: ΔGf = -kBT ln(Nfolded / Nunfolded).
- Enthalpy & Heat Capacity: Compute the folding enthalpy (ΔHf) from the temperature dependence of ΔGf (van't Hoff analysis) or as the difference in average potential energy between states. Estimate heat capacity (ΔCv) from energy fluctuations.
Protocol 2: Benchmarking Force Fields for Disordered Proteins
This protocol outlines a comparative approach for testing force fields against NMR and SAXS data [85] [12].
Initial Screening:
- System Preparation: Generate multiple (e.g., 10) independent starting structures for the IDP.
- Multi-Force Field Setup: Prepare identical simulation systems for each starting structure using a panel of different force field and water model combinations (see Table 1).
- Short Simulations: Run relatively short simulations (e.g., 200 ns) for all systems. Discard the initial equilibration period (e.g., first 120 ns).
Primary Validation against SAXS:
- From the production runs, calculate the radius of gyration (Rg) and the SAXS form factor.
- Compare these directly with experimental SAXS data. Select the few best-performing force fields for extended simulation.
Detailed Validation against NMR:
- Extended Simulations: Run longer simulations (e.g., >1 µs) for the selected top-performing force fields.
- Calculate NMR Observables: From these trajectories, predict:
- Chemical shifts
- Residual Dipolar Couplings (RDCs)
- NMR Relaxation Parameters (highly sensitive to dynamics)
- Quantitative Comparison: Systematically compare all predicted observables with experimental NMR data. The best force field will consistently match across all metrics and may also capture subtle differences, like helicity changes in point mutants [85].
The Scientist's Toolkit: Research Reagent Solutions
The table below lists key resources for conducting and validating protein folding simulations.
| Research Reagent | Function & Application |
|---|---|
| Amber ff99SB*-ILDN | A physics-based force field validated for simulating the folding kinetics and thermodynamics of fast-folding globular proteins like the villin headpiece [84]. |
| CHARMM36m | An all-atom force field incorporating adjustments to improve the description of disordered and structured regions, making it suitable for "hybrid" proteins [12]. |
| DES-amber | An example of an IDP-optimized force field. It was identified as the best model for reproducing the structure and dynamics of the disordered protein COR15A in aqueous solution [85]. |
| TIP4P-D Water Model | A modified water model designed to correct the over-collapse of IDPs by adjusting protein-water interactions. Can be paired with several protein force fields [12]. |
| Villin Headpiece (HP-35) | A well-studied, ultrafast-folding protein domain. Serves as a key benchmark system for testing a force field's ability to reproduce correct folding kinetics and pathways [84]. |
Force Field Selection and Validation Workflow
The following diagram outlines a logical pathway for diagnosing force field issues and selecting an appropriate model for your protein system.
Strategies for Handling Post-Translational Modifications and Non-Standard Residues
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary challenges when simulating post-translationally modified proteins or non-standard amino acids?
Simulating these proteins presents two main categories of challenges. Experimental and Detection Challenges include the low stoichiometry and dynamic regulation of many PTMs, making them difficult to detect without specific enrichment strategies [86]. Furthermore, the small size of certain modifications (e.g., methylation) complicates the generation of high-affinity antibodies for detection and isolation [86]. Computational and Force Field Challenges involve the need for accurate parameterization, as standard force fields lack parameters for many non-standard residues [87]. The chemical complexity and diversity of PTMs and NSAAs also require specialized parametrization protocols to correctly represent their unique energetic and electrostatic properties [87] [88].
FAQ 2: My simulation of a protein with a non-standard residue is unstable, leading to bond breaks or unrealistic conformations. What should I check?
This is a common parameterization issue. First, verify that all force field parameters for the non-standard residue are complete and accurate, especially the partial atomic charges, torsion angle parameters, and Lennard-Jones terms [87] [88]. These should be optimized against quantum mechanical (QM) data [87]. Second, ensure you are using an appropriate model compound for parameterization. For instance, for a modified amino acid, use a capped dipeptide (e.g., ACE-X-NME) rather than the isolated residue to properly represent the chemical environment [88]. Finally, check for covalent bonds between different residues; these may require defining the connected residues as a single new residue to prevent the force field from incorrectly breaking the bond during minimization [88].
FAQ 3: How can I efficiently incorporate multiple distinct non-standard amino acids into a single protein?
Simultaneous incorporation of multiple distinct NSAAs is a significant challenge. The most effective strategy involves using an Orthogonal Translation System (OTS) with optimized components [89]. This includes engineering the orthogonal aminoacyl-tRNA synthetase/tRNA pair for enhanced activity and selectivity, and engineering the elongation factor Tu (EF-Tu) for improved delivery of NSAA-charged tRNAs to the ribosome [89]. Cell-free protein synthesis (CFPS) is highly recommended for this purpose, as it avoids host cell toxicity issues, allows for direct control over the reaction environment, and enables the supplementation of multiple orthogonal components and NSAAs simultaneously [89].
FAQ 4: For global, unbiased analysis of post-translational modifications, what enrichment and mass spectrometry techniques are recommended?
A robust strategy involves a multi-step process. For enrichment, use modification-specific methods to isolate low-abundance PTM peptides from a complex background. Common techniques include antibody-based affinity purification, immobilized metal affinity chromatography (IMAC) for phosphopeptides, and lectin-based enrichment for glycopeptides [86] [90]. For mass spectrometry analysis, Data-Independent Acquisition (DIA) provides broad proteome coverage and excellent reproducibility for comparative studies, while targeted methods like Parallel Reaction Monitoring (PRM) offer high-sensitivity quantification for predefined modified peptides [90].
Troubleshooting Guides
Problem 1: Inadequate Force Field Parameters for Non-Standard Residues
Issue: Simulations crash, produce unrealistic geometries, or fail to maintain the integrity of non-standard residues due to missing or inaccurate parameters.
Solution: Implement a systematic parameterization protocol.
| Step | Action Description | Key Considerations |
|---|---|---|
| 1. Model Selection | Select a small molecule that accurately represents the chemical environment of the non-standard residue within a protein [87]. | For a side-chain modification, use a capped dipeptide (e.g., ACE-Residue-NME). This includes the backbone atoms adjacent to the modified site [88]. |
| 2. Quantum Mechanics (QM) Calculation | Perform geometry optimization and adiabatic potential energy surface (PES) scans on the model compound [87]. | Use high-level QM methods (e.g., MP2/6-31G(d)) to generate target data for charge derivation and torsion parameter optimization [87]. |
| 3. Charge Optimization | Derive partial atomic charges to reproduce QM-calculated electrostatic potentials (ESP) and dipole moments [87]. | Ensure the total charge of the model compound matches its protonation state at physiological pH (e.g., zwitterionic for a stand-alone amino acid) [87]. |
| 4. Parameter Fitting | Optimize bonded parameters (especially dihedral angles) against the QM PES scans [87]. | Prioritize parameters assigned high "penalty scores" by analogy tools, as these are the most uncertain and critical for accurate dynamics [87]. |
| 5. Validation | Run molecular dynamics simulations of a protein containing the non-standard residue and compare the conformation to experimental data (e.g., crystal structure) [87]. | Check for stability, realistic bond lengths/angles, and agreement with known structural data (e.g., RMSD to a reference structure) [87]. |
Problem 2: Low Detection Sensitivity for PTMs in Proteomics Experiments
Issue: Modified peptides are not detected by mass spectrometry due to low abundance and signal suppression from unmodified peptides.
Solution: Employ targeted enrichment strategies prior to MS analysis.
| Strategy | Principle | Applicable PTMs |
|---|---|---|
| Antibody-based Enrichment | Uses mono-/polyclonal antibodies to immunoaffinity purify PTM-containing peptides [86] [90]. | Phosphorylation, Acetylation, Ubiquitination, Methylation [86] [90]. |
| IMAC/MOAC | Relies on affinity between metal ions (e.g., Fe³⁺, TiO₂) and the phosphate group [90]. | Phosphorylation (Ser, Thr, Tyr) [90]. |
| Lectin-based Enrichment | Utilizes lectin proteins to bind specific carbohydrate structures [90]. | Glycosylation (N-linked, O-linked) [90]. |
Problem 3: Poor Efficiency when Incorporating Multiple Non-Standard Amino Acids
Issue: Low protein yield, high proportion of truncated products, or mis-incorporation of standard amino acids when attempting multi-site NSAA incorporation.
Solution: Coordinate optimization of the entire orthogonal translation system.
| System Component | Optimization Goal | Methodologies |
|---|---|---|
| Aminoacyl-tRNA Synthetase (aaRS) | Increase activity and selectivity for the target NSAA [89]. | Directed evolution (e.g., PACE) [89]. |
| tRNA | Enhance charging by aaRS and recognition by EF-Tu [89]. | Engineering anti-codon and acceptor stem domains [89]. |
| Elongation Factor Tu (EF-Tu) | Improve binding and delivery of NSAA-charged tRNA [89]. | Mutagenesis of the aminoacyl-binding domain [89]. |
| System Integration | Balance expression levels of OTS components [89]. | Use polycistronic vectors or optimize promoter strength [89]. |
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Tool | Function in PTM/NSAA Research |
|---|---|
| Pan-Specific PTM Antibodies | Immunoaffinity enrichment of modified peptides (e.g., phospho-tyrosine) for mass spectrometry [86] [90]. |
| Orthogonal aaRS/tRNA Pairs | Enable the site-specific incorporation of NSAAs during protein translation; commonly derived from M. jannaschii (Tyr) or M. barkeri (Pyl) [89]. |
| Cell-Free Protein Synthesis (CFPS) System | An open in vitro system for protein production that allows direct control over NSAA incorporation, bypassing cellular toxicity and delivery issues [89]. |
| Stable Isotope Labeling (SILAC) | A quantitative proteomics method that uses heavy isotope-labeled amino acids in cell culture to accurately measure dynamic changes in PTM levels [90]. |
| Isobaric Tags (TMT, iTRAQ) | Enable multiplexed quantitative comparison of PTM levels across multiple samples (e.g., 10-plex with TMT) in a single MS run [90]. |
| Parametrization Tools (e.g., ParamChem, R.E.D.) | Web servers and software that assist in generating initial guesses for force field parameters (charges, bonds, angles, dihedrals) for non-standard molecules [87]. |
Benchmarking and Validation: Ensuring Force Field Accuracy and Reliability
Frequently Asked Questions (FAQs)
FAQ 1: What are the most sensitive metrics for validating simulations of proteins with both structured and disordered regions?
For hybrid proteins containing structured domains and intrinsically disordered regions (IDRs), NMR relaxation parameters are particularly sensitive for validation. These parameters are more effective at revealing force field imperfections than commonly used metrics like root-mean-square deviation (RMSD). Other key observables include residual dipolar couplings (RDCs), paramagnetic relaxation enhancement (PRE), chemical shifts, and the radius of gyration (Rg) obtained from small-angle X-ray scattering (SAXS). The combination of these metrics provides a comprehensive picture of both local and global protein dynamics [12].
FAQ 2: My simulation shows a good average structure but unrealistic dynamics. What could be wrong?
This is a known challenge, often traced to the water model and non-bonded interaction parameters. For instance, the TIP3P water model has been shown to cause an artificial structural collapse in disordered protein regions, which in turn leads to unrealistic NMR relaxation properties. Switching to a model like TIP4P-D can significantly improve the reliability of the simulated dynamics. It is also crucial to ensure that the biomolecular force field parameters (e.g., CHARMM36m, Amber99SB-ILDN) are compatible with the chosen water model [12].
FAQ 3: How can I be sure my force field validation is statistically meaningful and not overfitted?
Relying on a single protein or a narrow set of observables for validation is risky. To ensure statistical meaning:
- Use a diverse protein test set: Validate against a curated set of multiple proteins with different topologies and sequences [26].
- Compare a wide range of metrics: Examine multiple structural and dynamic properties simultaneously. Improvement in one metric (e.g., RDCs) should not come at the cost of agreement in another (e.g., hydrogen bonding or radius of gyration) [26].
- Perform multiple replicates: Run several independent simulations to assess convergence and ensure results are not due to limited sampling [91].
FAQ 4: Which force field should I use for my specific protein system?
There is no universal "best" force field; the choice depends on your system and research question. The table below summarizes common recommendations based on the search results:
| Protein System | Recommended Force Field(s) | Key Considerations |
|---|---|---|
| Structured Globular Proteins | CHARMM36, AMBER ff99SB-ILDN, GROMOS (validated sets) | Good performance on structural criteria like hydrogen bonds, SASA, and radius of gyration [26] [91]. |
| Proteins with Intrinsically Disordered Regions (IDRs) | CHARMM36m | Specifically optimized for hybrid proteins with both ordered and disordered regions; improves reliability when paired with TIP4P-D water [12] [41]. |
| Membranes & Lipids | CHARMM36 Lipid, LIPID21 | Tailored to capture the unique dynamics of lipid bilayers [41]. |
| General Biomolecules (Proteins, Nucleic Acids) | AMBER, CHARMM | Extensively validated and reliable for biological simulations [41]. |
Troubleshooting Common Validation Issues
Problem: Simulated protein is too compact compared to experimental SAXS data.
- Potential Cause 1: The force field and water model combination may be over-stabilizing non-specific interactions, leading to collapse. This is a known issue with some force fields when simulating IDRs [12].
- Solution: Test a different force field proven for disordered systems (e.g., CHARMM36m) and use a modified water model like TIP4P-D [12].
- Potential Cause 2: Inadequate sampling. The simulation may be trapped in a low-energy, compact state.
- Solution: Run longer simulations or multiple replicates to ensure adequate sampling of the conformational landscape [91].
Problem: Poor agreement between back-calculated and experimental NMR parameters (e.g., RDCs, relaxation).
- Potential Cause: The underlying conformational ensemble generated by the force field is incorrect. For example, it may not properly retain transient secondary structures or may have incorrect chain dynamics [12] [26].
- Solution: Validate against multiple NMR observables simultaneously (relaxation, RDCs, PRE). NMR relaxation rates are highly sensitive to dynamics and can pinpoint issues that other metrics miss. Ensure the simulation protocol and force field can reproduce the experimental helical propensity in disordered regions if it exists [12].
Problem: Simulation becomes unstable, or the protein unfolds unrealistically.
- Potential Cause 1: Intrinsic limitations of the force field or its parameters, leading to unphysically large forces [92].
- Solution: Consult recent literature and community benchmarks for stable force fields for your system type. This is a particular challenge for some universal machine learning force fields, which can fail on complex systems despite good performance on benchmarks [92].
- Potential Cause 2: Incompatibility between the force field, water model, and simulation parameters (e.g., treatment of long-range electrostatics) [91].
- Solution: Use "best practice" parameters recommended by the force field and software developers. Particle Mesh Ewald (PME) is typically recommended for long-range electrostatics over straight cutoffs [91].
Key Metrics and Experimental Protocols
For a robust validation, compare your simulation data to a variety of experimental observables. The table below summarizes the key metrics and their structural interpretation.
| Metric Category | Specific Observables | What It Measures | Experimental Protocol Summary |
|---|---|---|---|
| NMR - Dynamics | Relaxation rates (R₁, R₂), heteronuclear NOE | Ps-ns timescale backbone dynamics and flexibility [12]. | Measured using standard pulsed field gradient NMR experiments on 15N-labeled protein. R₁ and R₂ rates are fitted from exponential decays at various relaxation delays [12]. |
| NMR - Structure | Residual Dipolar Couplings (RDCs) | Orientation of bond vectors relative to a global alignment tensor; sensitive to long-range structural order [12]. | Splitting measured in IPAP spectra of proteins in stretched polyacrylamide gel vs. isotropic medium [12]. |
| NMR - Distance | Paramagnetic Relaxation Enhancement (PRE) | Long-range distance restraints (up to ~20 Å) for low-populated states or transient conformations [12]. | Requires site-directed spin labeling of the protein (e.g., with a nitroxide radical). The enhancement of relaxation rates is measured [12]. |
| Scattering | Radius of Gyration (Rg) | Overall compactness and shape of the molecule in solution [12]. | Derived from SAXS data via Guinier analysis at low angles on a dedicated synchrotron or lab-based instrument [12]. |
| Structural Analysis | Hydrogen Bonds, SASA, Secondary Structure | Local and secondary structure stability, solvation. | Derived from a simulation trajectory or experimental structure. SASA calculated using a rolling probe sphere; hydrogen bonds defined by distance and angle criteria [26]. |
| Overall Structure | Root-mean-square deviation (RMSD) | Average distance of atoms between simulated and reference (e.g., crystal) structure after alignment [26]. | Calculated by comparing atomic positions in the simulation frame to a reference structure after optimal superposition. |
Experimental Workflow for Validation
The following diagram outlines a general workflow for validating molecular dynamics simulations against experimental data.
Research Reagent Solutions
The table below lists essential materials and computational tools used in the validation of molecular dynamics simulations.
| Item | Function in Validation |
|---|---|
| Isotopically Labeled Proteins (15N, 13C) | Enables advanced NMR spectroscopy for measuring chemical shifts, relaxation rates, RDCs, and PREs [12]. |
| Paramagnetic Spin Labels | Covalently attached to specific protein sites (e.g., cysteine residues) to measure long-range distance restraints via PRE [12]. |
| Stretched Polyacrylamide Gels | Used to weakly align proteins in solution for the measurement of Residual Dipolar Couplings (RDCs) by NMR [12]. |
| SAXS Instrumentation | Provides low-resolution structural data in solution, including the radius of gyration (Rg) and overall molecular shape [12]. |
| MD Software (GROMACS, NAMD, AMBER) | Packages used to run the molecular dynamics simulations with built-in analysis tools [91]. |
| NMR Prediction Software | Back-calculates NMR observables (e.g., shifts, relaxation, J-couplings) from simulation trajectories for direct comparison with experiment [12] [26]. |
Systematic Performance Evaluation of AMBER ff14SB vs. ff19SB with Different Water Models
Molecular dynamics (MD) simulations have become an indispensable tool in structural biology and drug development, providing atomistic insights into protein structure, dynamics, and interactions that complement experimental findings. The accuracy of these simulations critically depends on the empirical force fields used to describe atomic interactions. Among the most widely used protein force fields are the AMBER family, with ff14SB and ff19SB representing significant milestones in parameterization. However, force field performance is intrinsically linked to the water model employed in simulations, creating a complex parameter space that researchers must navigate. This technical evaluation provides a systematic comparison of AMBER ff14SB and ff19SB when paired with different water models, offering evidence-based guidance for researchers conducting simulations across diverse protein systems, from folded domains to intrinsically disordered proteins (IDPs) and protein complexes.
Quantitative Performance Comparison
Table 1: Overall Performance Characteristics of AMBER Force Fields with Different Water Models
| Force Field | Recommended Water Model | Best Application Areas | Strengths | Limitations |
|---|---|---|---|---|
| AMBER ff14SB | TIP3P, TIP4P2005 | Folded proteins, Collagen triple helices, Protein-GAG complexes | Good folded state stability, Established validation, Lower computational cost with TIP3P | Overly collapsed IDP ensembles with TIP3P, Excessive protein-protein association |
| AMBER ff19SB | OPC | Intrinsically disordered proteins, Balanced folded/disordered systems | Accurate IDP dimensions, Improved backbone torsions, Reduced protein-protein association | Potential instability in some folded proteins, Higher computational cost with OPC |
| AMBER ff03w-sc | Custom (scaled) | Systems requiring balanced protein-water interactions | Maintains folded stability while accurately modeling IDPs | Less established parameter set |
| AMBER ff99SBws-STQ′ | Custom (scaled) | Polyglutamine tracts and similar sequences | Corrects overestimated helicity in polyQ tracts | Specialized application |
Table 2: Quantitative Performance Metrics from Validation Studies
| Force Field Combination | Folded Protein Stability (RMSD) | IDP Chain Dimensions | Protein-Protein Association | Secondary Structure Accuracy |
|---|---|---|---|---|
| ff14SB-TIP3P | Low (<0.2-0.3 nm) for folded domains | Overly collapsed for many IDPs | Over-stabilized | Accurate for folded proteins |
| ff19SB-OPC | Variable (may show instability) | Experimentally accurate | Balanced, slightly weak | Excellent for disordered regions |
| ff99SB-disp | High stability | Slightly expanded | Underestimated | Balanced overall |
| CHARMM36m-TIP3P-modified | High stability | Slightly compact | Overestimated | Accurate for most systems |
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for Force Field Evaluation
| Item | Function/Description | Example Use Cases |
|---|---|---|
| AMBER ff14SB Force Field | Protein force field with improved side-chain torsions | General purpose protein simulations, Folded protein systems |
| AMBER ff19SB Force Field | Protein force field with updated backbone and side-chain parameters | IDP simulations, Systems requiring modern parameterization |
| TIP3P Water Model | Standard 3-site water model, computationally efficient | Large systems, Resource-limited simulations |
| OPC Water Model | Optimized 4-site water model with improved accuracy | Systems requiring high solvent accuracy, IDP simulations |
| TIP4P2005 Water Model | Accurate 4-site water model for biomolecular systems | Balanced protein-water interactions |
| GLYCAM06 Force Field | Carbohydrate-specific parameters for GAG simulations | Protein-glycosaminoglycan complexes |
| GROMACS Simulation Package | High-performance MD simulation software | Production simulations, Force field validation |
| AMBER Simulation Package | Comprehensive MD simulation suite | Integrated force field development and testing |
| SAXS Analysis Tools | Small-angle X-ray scattering data processing | Experimental validation of IDP dimensions |
| NMR Analysis Software | Nuclear magnetic resonance data analysis | Validation of local structure and dynamics |
Experimental Protocols for Force Field Validation
Protocol 1: Validation of Folded Protein Stability
Objective: Assess the ability of force field/water model combinations to maintain the native structure of folded proteins over microsecond timescales.
Methodology:
- System Preparation: Obtain crystal structures of benchmark proteins (e.g., Ubiquitin PDB: 1D3Z, Villin HP35 PDB: 2F4K) and prepare simulation systems using standard protonation states.
- Simulation Setup: Solvate proteins in appropriate water models (TIP3P for ff14SB, OPC for ff19SB) with neutralizing ions using a minimum 1.0 nm padding distance.
- Equilibration: Perform energy minimization followed by stepwise equilibration under NVT and NPT ensembles (100 ps each) with position restraints on protein heavy atoms.
- Production Simulation: Run multiple independent simulations (≥2.5 μs each) without restraints using a 2-fs time step at 300 K and 1 bar pressure.
- Analysis: Calculate backbone RMSD, radius of gyration, and per-residue RMSF. Monitor secondary structure content (DSSP) and hydrogen bonding patterns.
Expected Results: Stable force fields (e.g., ff14SB-TIP3P) should maintain RMSD <0.2-0.3 nm, while problematic combinations may show significant structural deviations or unfolding events [43].
Protocol 2: Validation of Intrinsically Disordered Protein Ensembles
Objective: Evaluate the performance of force fields in reproducing the extended conformational ensembles of intrinsically disordered proteins and polyampholytes.
Methodology:
- System Preparation: Construct initial extended structures of benchmark IDPs (e.g., FUS, RS peptide) or polyampholytes (EK sequences).
- Simulation Setup: Solvate in appropriate water models with neutralizing ions, using a minimum 1.5 nm padding to accommodate chain expansion.
- Enhanced Sampling: Employ replica-exchange MD or multiple long conventional MD simulations (≥10 μs aggregate sampling) to ensure adequate conformational sampling.
- Experimental Comparison: Calculate theoretical SAXS profiles from simulation ensembles and compare with experimental data using χ² statistics.
- Analysis: Compute radius of gyration (Rg), end-to-end distance, and scalar coupling constants (³J(HN-HCα)) for comparison with NMR data.
Expected Results: Accurate force fields (e.g., ff19SB-OPC) should reproduce experimental Rg values and SAXS profiles within error margins, while older combinations may produce overly compact chains [93].
Protocol 3: Assessment of Protein-Protein and Protein-GAG Interactions
Objective: Quantify the accuracy of force fields in modeling protein complex formation and protein-glycosaminoglycan interactions.
Methodology:
- System Preparation: Prepare benchmark systems including ubiquitin dimerization, Fused in Sarcoma (FUS) RRM domain, and protein-GAG complexes (e.g., heparin-FGF2).
- Simulation Setup: Solvate complexes in different water models (TIP3P, OPC, TIP4P, TIP5P) using appropriate force field combinations.
- Binding Analysis: For protein-protein systems, perform multiple simulations to assess dimerization propensity. For protein-GAG complexes, monitor binding pose stability and interaction fingerprints.
- Free Energy Calculations: Use MM/PBSA or umbrella sampling to quantify binding affinities where applicable.
- Validation: Compare results with experimental data on complex stability, known binding interfaces, and affinity measurements.
Expected Results: Performance varies significantly across systems, with ff19SB-OPC showing balanced protein-protein association, while ff14SB may over-stabilize certain complexes [43] [37].
Troubleshooting Guides
FAQ: Folded Protein Instability in Simulation
Q: My folded proteins show significant unfolding during microsecond-timescale simulations with ff19SB-OPC. What might be causing this and how can I address it?
A: This is a documented issue where enhanced protein-water interactions in some modern force fields can compromise folded state stability. A recent study observed significant instability in Ubiquitin and Villin HP35 with ff03ws (a related force field), while ff99SBws maintained structural integrity [43].
Solutions:
- Consider switching to ff14SB with TIP4P2005 for better balance between folded stability and disordered chain dimensions.
- Test the newly refined ff03w-sc or ff99SBws-STQ′ force fields that incorporate selective protein-water scaling to maintain folded stability [43].
- Increase simulation replicates to distinguish between rare unfolding events and systematic instability.
- Validate your simulation setup with a well-characterized protein like Ubiquitin before studying your target system.
FAQ: Disordered Proteins Appear Overly Compact
Q: My IDP simulations with ff14SB-TIP3P produce overly compact chains that don't match experimental SAXS data. How can I improve agreement with experiments?
A: This is a known limitation of older force fields with three-site water models, which tend to underestimate protein-water interactions, leading to collapsed IDP ensembles.
Solutions:
- Switch to ff19SB-OPC, which demonstrates improved accuracy for IDP chain dimensions and secondary structure propensities [93].
- Consider using ff99SB-disp or CHARMM36m with modified water models if ff19SB-OPC shows limitations for your specific system.
- Implement the SWAXS- scattering model for more accurate comparison with experimental SAXS data, as it accounts for hydration layer effects [93].
- Ensure adequate sampling through replica-exchange MD or multiple long simulations, as IDPs require extensive conformational sampling.
FAQ: Inconsistent Electroosmotic Flow in Nanopore Simulations
Q: I'm observing significant differences in electrohydrodynamic properties in biological nanopore simulations depending on my force field choice. Which combination is most reliable?
A: A comparative study of CytK and MspA nanopores found that while Amber-ff14SB, Amber-ff19SB, and CHARMM36 generally agree on selectivity patterns, they produce quantitatively different electroosmotic flow rates, particularly in narrow pores [94].
Solutions:
- For narrow pores like MspA, test multiple force fields as differences can be substantial enough to yield qualitatively different outcomes (e.g., detectable electroosmosis vs. none) [94].
- Consider using Amber-ff19SB with OPC water for most cases, as it represents the most modern parameterization.
- Validate against any available experimental data on ionic conductance or streaming potential for your specific nanopore system.
- Report force field dependencies as a sensitivity analysis in publications, as no single force field universally outperforms others in all nanopore systems.
FAQ: Force Field Selection for Collagen and Specialized Proteins
Q: Which force field provides the most accurate structure and dynamics for collagen triple helices and other repetitive protein motifs?
A: For collagenous domains like POG10 homotrimers, AMBER ff99SB-based force fields (including ff14SB) most accurately reproduce backbone dihedrals, side-chain torsions, and experimental SAXS data compared to CHARMM or GROMOS families [44].
Solutions:
- Use AMBER ff99SB or ff14SB with TIP3P or TIP4P2005 water for collagen simulations.
- If using CHARMM36, apply scaled CMAP terms for Pro, Hyp, and Gly residues (CHARMM36mGP) to improve accuracy [44].
- Validate against available crystal structure data, NMR relaxation measurements, and SAXS form factors for your specific collagen sequence.
- For collagen mutations associated with disease, ensure your force field reproduces wild-type behavior before introducing mutations.
Technical Diagrams and Workflows
Force Field Selection Workflow for Different Protein Systems
Force Field Validation Protocol with Multiple Benchmarks
Based on comprehensive evaluation across multiple protein systems and experimental validations, we provide these specific recommendations for force field selection:
For general purpose protein simulations involving both folded and disordered regions: AMBER ff19SB with OPC water provides the most balanced performance, though folded stability should be verified for your specific system.
For primarily folded proteins and collagen systems: AMBER ff14SB with TIP4P2005 offers reliable stability and accurate structural properties.
For intrinsically disordered proteins and polyampholytes: AMBER ff19SB-OPC consistently reproduces experimental chain dimensions and secondary structure propensities.
For protein-glycosaminoglycan complexes: FF14SB with OPC or TIP5P provides improved performance over standard TIP3P simulations.
When computational resources are limited: FF14SB with TIP3P remains serviceable for folded proteins, though with recognized limitations for disordered systems.
Researchers should implement the validation protocols outlined in this guide for their specific systems of interest, as force field performance can be system-dependent. The ongoing development of refined force fields like ff03w-sc and ff99SBws-STQ′ promises further improvements in balancing protein-protein, protein-water, and protein-solute interactions for more accurate molecular simulations across diverse biological contexts.
Comparative Analysis of Force Field Performance in Reproducing Folding Enthalpies and Unfolded States
Quantitative Force Field Performance Data
The table below summarizes the documented performance of various physical models in reproducing key protein folding properties, based on data from long molecular dynamics simulations [40] [39].
Table 1: Documented Performance of Physical Models in Reproducing Protein Folding Properties
| Protein/Folding Property | Force Field / Model | Reported Performance | Key Quantitative Findings |
|---|---|---|---|
| Human Pin1 WW Domain | CHARMM22 with CMAP [40] | Failed to fold to native state; favored misfolded helical states. | Misfolded states favored over native state by 4.4–8.1 kcal/mol [40]. |
| Folding Enthalpies | Current Physical Models (General) [39] | Reproduced poorly. | Long simulations can accurately reproduce rates and free energies of folding, but folding enthalpies are reproduced poorly [39]. |
| Characteristics of Unfolded State | Current Physical Models (General) [39] | Reproduced poorly. | The structure and dynamics of folded proteins can be accurately described, but characteristics of the unfolded state are reproduced poorly [39]. |
| Chignolin, TRPcage, Villin | Machine-Learned CG Model (CGSchNet) [35] | Successfully predicted folded states and metastable folding/unfolding transitions. | Predicted folded states with fraction of native contacts (Q) close to 1 and low Cα RMSD [35]. |
| BBA (β-β-α protein) | Machine-Learned CG Model (CGSchNet) [35] | Captured native state as a local minimum, but with performance issues. | Highlighted difficulty with proteins containing both helical and anti-parallel β-sheet motifs [35]. |
Troubleshooting Guides & FAQs
Frequently Asked Questions (FAQs)
FAQ 1: My simulation consistently fails to fold the protein to its known native structure. What is the most likely cause? The failure is likely due to a force field bias or deficiency. Evidence from studies on the human Pin1 WW domain shows that some force fields can incorrectly favor non-native states (e.g., helical structures for a native beta-sheet protein) by a significant free energy margin (4.4–8.1 kcal/mol) [40]. Kinetic trapping is another possibility, but thermodynamic bias must be ruled out first [40].
FAQ 2: Which aspects of protein folding do current physical models typically struggle with? According to assessments based on long MD simulations, current models often reproduce folding enthalpies poorly and provide an inaccurate description of the unfolded state ensemble [39]. However, they can more accurately predict folding rates, free energies, and the structure/dynamics of the folded state [39].
FAQ 3: Are there efficient alternatives to all-atom force fields for studying protein folding landscapes? Yes, machine-learned coarse-grained (CG) models are emerging as a powerful alternative. These models are trained on all-atom simulation data and can be several orders of magnitude faster, while still successfully predicting folded states, metastable intermediates, and folding/unfolding transitions for unseen protein sequences [35].
FAQ 4: My simulation folds the protein, but the native state is not the global free energy minimum. What does this indicate? This indicates that the force field has a thermodynamic inaccuracy. While the model can capture the native structure as a metastable state, it incorrectly assigns a lower free energy to a non-native or misfolded state. This was observed in a CG model for the BBA protein, where the native basin was a local, not global, minimum [35].
Troubleshooting Guide: Force Field Artifacts and Solutions
Problem: Observation of Non-Native Secondary Structure (e.g., Helices in a β-Sheet Protein)
- Description: During folding simulations, the protein population is trapped in stable, non-native secondary structures, such as the array of helical structures observed in simulations of the beta-sheet Pin1 WW domain [40].
- Underlying Cause: A known bias in the force field's parameters (e.g., torsional potentials) that over-stabilizes certain secondary structure elements [40].
- Solution Steps:
- Confirm Thermodynamic Bias: Use advanced free energy calculation methods, like Deactivated Morphing (DM), to quantify the free energy difference between the misfolded and native states [40].
- Switch Force Fields: Consider testing a different, more modern force field that has been benchmarked on proteins with similar topology.
- Consider Coarse-Grained Models: For initial landscape screening, use a transferable machine-learned CG model to see if it correctly predicts the native state as stable [35].
Problem: Inaccurate Characterization of the Unfolded State Ensemble
- Description: The simulated unfolded state does not match experimental expectations in terms of dimensions or dynamics [39].
- Underlying Cause: This is a recognized weakness of many current physical models, which may have inaccuracies in solvation or non-native interaction potentials [39].
- Solution Steps:
- Acknowledge the Limitation: Be transparent that this is a known challenge in the field.
- Utilize Enhanced Sampling: Combine long simulations with enhanced sampling techniques to achieve better convergence of the unfolded state ensemble.
- Validate with Experiment: Where possible, compare simulation results with experimental data from techniques like FRET or NMR to gauge the severity of the inaccuracy.
Experimental Protocols & Methodologies
Protocol: Calculating Free Energy Differences Between Misfolded and Native States
This protocol is based on the Deactivated Morphing (DM) method used to diagnose force field bias in the Pin1 WW domain [40].
Objective: To quantitatively determine if a force field thermodynamically favors a misfolded state over the native state.
Workflow Overview:
Detailed Steps:
- Define Reference Structures: Obtain the experimentally determined Native State (A) and a representative Misfolded State (B) from your simulation trajectories [40].
- Calculate Free Energy along DM Path: The free energy difference between the unrestrained ensembles E(A) and E(B) is calculated by breaking the process into a series of steps with overlapping states to ensure convergence [40]:
- Path from E(A) to D(A): Apply progressively stronger harmonic restraints (κ = 1000 kcal/mol Å) to all protein atoms, "deactivating" the force field by restraining atoms to their reference coordinates, and finally creating a 'dummy' state with uniform parameters [40].
- Morph from D(A) to D(B): The coordinates are morphed from reference A to reference B. The free energy change for this step is zero by definition [40].
- Path from D(B) to E(B): Reverse the process of applying restraints to arrive at the unrestrained ensemble around state B [40].
- Sum Free Energy Changes: The total free energy difference ΔG between the native and misfolded states is the sum of the free energy changes for all steps along the path [40].
- Error Analysis: Perform block averaging of the data (e.g., splitting into 10 blocks) to estimate the standard error of the calculated free energy difference [40].
Protocol: Assessing Force Field Accuracy with Long MD Simulations
This protocol outlines the methodology for benchmarking a force field's performance against known experimental or high-quality simulation data [39].
Objective: To systematically evaluate a force field's ability to reproduce key folding properties.
Workflow Overview:
Detailed Steps:
- System Setup:
- Production Simulations:
- Trajectory Analysis:
- Folded State: Calculate the Cα Root-Mean-Square Deviation (RMSD) and fraction of native contacts (Q) to assess structural accuracy. Compute Root-Mean-Square Fluctuations (RMSF) to evaluate dynamics [35].
- Unfolded State: Analyze the radius of gyration and secondary structure content in the unfolded ensemble [39].
- Free Energy Landscape: Construct landscapes using collective variables like RMSD and Q to identify metastable states and folding pathways [35].
- Thermodynamics and Kinetics: Extract folding free energies (ΔG), enthalpies (ΔH), and rates from the simulations [39].
- Validation: Compare all calculated properties against available experimental data or highly converged reference simulations to identify force field deficiencies [39].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Force Fields for Protein Folding Simulations
| Tool Name | Type | Primary Function | Key Consideration |
|---|---|---|---|
| NAMD [40] | Molecular Dynamics Software | Performs all-atom and coarse-grained MD simulations. | Scalable for large systems on parallel computers; supports various force fields. |
| GROMACS [40] | Molecular Dynamics Software | High-performance MD simulation package. | Known for its exceptional speed and efficiency for biomolecular systems. |
| CHARMM22/CMAP [40] | All-Atom Force Field | Empirical energy function for proteins. | Has known bias towards helical structures; requires benchmarking for beta-proteins [40]. |
| Machine-Learned CG Model (e.g., CGSchNet) [35] | Coarse-Grained Force Field | Machine-learned potential for efficient protein dynamics. | Transferable across sequences; orders of magnitude faster than all-atom MD [35]. |
| VMD [40] | Analysis & Visualization | System setup, trajectory analysis, and visualization. | Integral for solvating systems, analyzing RMSD, RMSF, and visualizing states. |
| Deactivated Morphing (DM) [40] | Free Energy Method | Calculates free energy differences between distinct conformations. | Critical for diagnosing thermodynamic bias in a force field [40]. |
| GROMOS Clustering [40] | Analysis Algorithm | Identifies dominant conformational states in a trajectory. | Used for cluster analysis to identify representative native and misfolded states. |
Using Markov State Models (MSMs) to Recover Thermodynamics and Kinetics
Frequently Asked Questions (FAQs) and Troubleshooting
FAQ 1: What are the most common sources of error in an MSM, and how can I diagnose them?
Diagnosing errors is crucial for building a valid model. The table below summarizes common issues, their symptoms, and diagnostic actions.
| Error Source | Symptoms | Diagnostic Checks |
|---|---|---|
| Poor State Discretization [95] [96] | • Implied timescales do not converge.• Low VAMP scores.• Unphysical, rapid cycling between states. | • Perform a Chapman-Kolmogorov test [95].• Check for convergence of implied timescales with increasing lag time [96]. |
| Insufficient Sampling [95] [97] | • Large uncertainty in transition probabilities (small counts).• Failure to discover known metastable states. | • Use Bayesian MSMs to estimate confidence intervals [96].• Check if the equilibrium distribution is stable with more data. |
| Non-Markovianity (Lag Time Too Short) [95] [96] | • Implied timescales increase with lag time.• Model fails the Chapman-Kolmogorov test. | • Plot implied timescales vs. lag time and choose a lag time where they plateau [96]. |
FAQ 2: How do I choose an appropriate lag time (τ) for my model?
Selecting the lag time is a critical step. A lag time that is too short violates the Markov assumption, while one that is too long obscures important kinetic details [95] [96]. The standard validation procedure is to compute the implied timescales as a function of the lag time [96]. The implied timescales are calculated from the eigenvalues (λᵢ) of the transition matrix as tᵢ = -τ / ln(λᵢ(τ)). You should choose a lag time where these timescales are relatively constant (a "plateau" region) [96]. This indicates that the model has captured the slow, kinetically relevant processes and that faster processes have been averaged out, satisfying the Markov assumption.
FAQ 3: My system has a rare, long-lived "trap" state that is poorly connected. Can I still include it in the MSM?
Yes. Standard MSM construction often involves trimming the network to the largest ergodic subset (where every state is reachable from every other state), which can discard these important states [98]. To include them, you can model them as absorbing "sink" states [98]. In this framework, the stable trap is treated as a state from which the system cannot escape on the simulation timescale. This allows you to compute key properties, such as the mean first-passage time into this trap or its infinite-time population, providing insight into misfolding or aggregation mechanisms [98].
FAQ 4: How can MSMs inform my choice of force field for a specific protein type?
MSMs provide a powerful framework for force field validation by enabling the comparison of simulated kinetics and thermodynamics with experimental data [99]. For example, you can:
- Compare Thermodynamics: Compute the stationary distribution (equilibrium populations) of your MSM and check if the folded state population matches experimental measurements [99].
- Compare Kinetics: Calculate the slowest implied timescales from your MSM and check if they match experimentally known folding timescales [99].
- Validate with NMR: Compute ensemble-averaged experimental observables (e.g., chemical shifts, J-couplings) from your MSM states and compare directly with solution-state NMR data [99].
A force field that produces an MSM yielding better agreement with a broader array of such experiments is generally more reliable for that class of protein [99].
FAQ 5: Are there alternatives to RMSD for clustering that might be better for my system?
Yes. While Root-Mean-Square Deviation (RMSD) is a common metric, it can sometimes group structures that are structurally similar but separated by a large energy barrier [98]. Alternative metrics can offer improved kinetic discrimination:
- Contact Maps: Define states based on the pattern of residue-residue contacts. This is highly relevant for processes like folding and domain swapping [98]. A coarse-grained contact map can make this more manageable [98].
- Backbone Dihedral Angles: Using phi/psi angles as features can effectively capture local conformational changes that are kinetically relevant [98].
The choice of metric should be guided by the specific biological process you are studying.
Diagram: MSM Construction and Validation Workflow
Experimental Protocols
Protocol 1: Building and Validating a Basic MSM
This protocol outlines the core steps for constructing an MSM from molecular dynamics trajectories [95] [100].
- Featurization: Start by transforming the raw Cartesian coordinates of your MD trajectories into features that capture relevant structural changes. Common choices include dihedral angles, inter-residue distances, or contact maps [100].
- Dimensionality Reduction (Optional but Recommended): Project the high-dimensional features into a lower-dimensional space using Time-lagged Independent Component Analysis (TICA). TICA finds the directions of maximum autocorrelation, which often correspond to slow collective variables [100].
- Clustering into Microstates: Cluster the projected data (or the original features) into a large number (e.g., thousands) of conformational microstates using an algorithm like k-means. This creates a fine-grained discretization of the conformational space [95] [100].
- Assigning Data and Counting Transitions: Assign every frame of your MD trajectories to one of the microstates. Then, choose a lag time (τ) and count the number of transitions observed between every pair of states (i and j) over that time interval. This produces a count matrix, Cᵢⱼ(τ) [95] [96].
- Estimating the Transition Matrix: The maximum likelihood estimate for the transition probability matrix, T(τ), is calculated by normalizing the count matrix: Tᵢⱼ = Cᵢⱼ / Σₖ Cᵢₜ [96]. Bayesian methods can also be used to estimate uncertainties [96].
- Validation: This is a critical step.
- Implied Timescales Test: Plot the model's implied timescales against different lag times. A valid MSM will show a plateau, indicating the Markov assumption holds [96].
- Chapman-Kolmogorov Test: Compare the model's predicted population decay for a state with the actual observed decay from the data. A good model will have close agreement [95].
Protocol 2: Using an MSM for Force Field Evaluation
This protocol describes how to use MSMs as part of a force field selection or validation pipeline [99].
- Simulation Set-up: Perform extensive MD sampling (e.g., using distributed computing platforms like Folding@home) for a benchmark protein (e.g., the mini-protein chignolin) using multiple different force fields (e.g., AMBER14SB, CHARMM36, etc.) [99].
- Build and Validate Individual MSMs: For the data generated by each force field, independently build and validate an MSM using the protocol above. Ensure all models are built to the same standard (e.g., pass the same validation checks) [99].
- Compute Experimental Observables:
- Thermodynamics: From each MSM's stationary distribution (π), compute the equilibrium population of the native folded state [99].
- Kinetics: From each MSM's eigenvalues, compute the slowest implied timescale, which corresponds to the folding/unfolding rate [99].
- Structural Observables: Calculate ensemble-averaged experimental observables like NMR chemical shifts or scalar couplings from the structures in each MSM state [99].
- Quantitative Comparison: Create a table comparing the MSM-derived properties with known experimental values. The force field whose MSM most accurately reproduces the experimental folding population, folding time, and other structural observables can be considered more reliable for that specific protein type [99].
Diagram: Relationship Between Discretization and Model Quality
The Scientist's Toolkit: Key Research Reagents and Software
The following table lists essential computational tools and methodological components for building and analyzing Markov State Models.
| Tool / Component | Function | Relevance to Research |
|---|---|---|
| MSMBuilder [96] | A software package for building MSMs from MD trajectories. It automates featurization, clustering, and transition matrix estimation. | Core software for constructing and performing maximum likelihood and Bayesian estimation of MSMs [96]. |
| PyEMMA [100] | (Emma: Easy Markov Model Analysis) A software suite for kinetic analysis of MD data. Provides TICA, clustering, MSM estimation, and validation. | Key alternative to MSMBuilder; includes comprehensive tutorials and methods for advanced analysis like Hidden Markov Models [100]. |
| Markov State Model (MSM) [95] | A coarse-grained kinetic model comprising discrete states and transition probabilities between them. | The core object of study; used to predict long-timescale kinetics and thermodynamics from short simulations [95]. |
| Bayesian Markov State Model [96] | An extension of the MSM that provides a distribution over transition matrices, allowing estimation of uncertainty. | Critical for diagnosing sampling errors and understanding the confidence in predictions like timescales and state populations [96]. |
| Time-lagged Independent Component Analysis (TICA) [100] | A dimensionality reduction technique that finds slow collective variables by maximizing autocorrelation. | Improves the kinetic meaningfulness of the subsequent clustering step, leading to more robust MSMs [100]. |
| Implied Timescales [96] | Timescales of relaxation processes computed from the eigenvalues of the MSM transition matrix. | The primary diagnostic for choosing a valid lag time and validating the Markov assumption [96]. |
Validation Against NMR Data, Native Contacts, and Secondary Structure Propensity
This technical support center provides troubleshooting guides and FAQs for researchers using molecular dynamics (MD) simulations, framed within the critical context of force field selection for different protein types.
➤ Frequently Asked Questions (FAQs)
1. What are the best practices for validating my simulation against NMR data? A robust method is ANSURR (Accuracy of NMR Structures using Random Coil Index and Rigidity). It compares local rigidity derived from NMR chemical shifts (using Random Coil Index, RCI) with rigidity calculated from your structure (using mathematical rigidity theory with the FIRST software). This provides a correlation score (assesses secondary structure placement) and an RMSD score (measures overall rigidity), offering a direct comparison to experimental data that is less manipulated than traditional restraint-based validation [101].
2. My simulation of an Intrinsically Disordered Protein (IDP) is overly collapsed. What force field should I use? This is a common issue with older force fields. Use modern force fields and water models specifically reparameterized for disordered regions. The CHARMM36m force field is a reliable choice, as it was designed to handle proteins containing both structured and intrinsically disordered regions. Furthermore, pairing protein force fields with more accurate water models like TIP4P-D has been shown to significantly improve the conformational ensemble of IDPs by rebalancing protein-water interactions [12] [43].
3. How can I use native contacts to analyze protein folding in simulations? Native contacts (NCs) define the similarity of a protein conformation to its native state and are vital for folding studies. You can compute the time evolution of NCs from your MD trajectory. Furthermore, deep learning models like Long Short-Term Memory (LSTM) networks can be trained on this MD data to predict the future evolution of native contacts, providing insight into the folding process [102] [103].
4. Where can I find updated amino acid propensity scales for secondary structure prediction? Revised propensity scales for six secondary structures (α-helix, β-strand, 310-helix, π-helix, turn, and coil) have been calculated using exhaustive, non-redundant PDB datasets. These updated scales, which can be considered a modern standard, were assigned using robust algorithms like DSSP and STRIDE [104].
➤ Troubleshooting Guides
Issue 1: Poor Correlation with NMR Chemical Shifts and Relaxation Data
This indicates a potential inaccuracy in your simulated protein's dynamics or local structure.
Diagnosis Questions:
- Have you assigned the correct protonation states for your residues?
- Is your system fully equilibrated before production run?
- Does your chosen force field accurately model backbone dihedral angles and side-chain rotamers?
Solutions:
- Run the ANSURR Analysis: Follow the workflow in the diagram below to quantitatively validate your structure against backbone chemical shifts [101].
- Validate with Multiple NMR Observables: Compare your simulation not only to chemical shifts but also to residual dipolar couplings (RDCs) and NMR relaxation parameters. These are particularly sensitive to the choice of force field and water model [12].
- Check Force Field and Water Model:
- For proteins with disordered regions, avoid using standard force fields like AMBER99SB-ILDN with the TIP3P water model, as this combination can cause artificial structural collapse and yield unrealistic NMR relaxation properties [12].
- Prefer modern, balanced force fields like CHARMM36m or amber ff99SBws-STQ' with water models such as TIP4P-D or TIPS3P [12] [43].
Issue 2: Incorrect Secondary Structure Propensities
Your simulation shows unrealistic helix, sheet, or coil content.
Diagnosis Questions:
- Are you using an outdated force field (e.g., pre-2010 versions)?
- Does the force field have a known bias (e.g., over-stabilizing α-helices)?
Solutions:
- Consult Updated Propensity Scales: Use the revised amino acid propensity scales derived from exhaustive PDB data as a benchmark for expected behavior [104].
- Refine Your Force Field: Consider using refined force fields like amber ff99SBws-STQ', which includes targeted torsional refinements to correct overestimated helicity in specific sequences like polyglutamine tracts [43].
- Analyze at the Individual Protein Level: Remember that propensities can vary across different protein structures and folds. A residue's preference is primarily dictated by its side chain's structural features, but the tertiary environment also plays a role [104].
Issue 3: Instability of Folded Proteins or Unrealistic IDP Dimensions
The protein unfolds spontaneously or the IDP chain dimensions do not match experimental data (e.g., from SAXS).
Diagnosis Questions:
- Is the imbalance between protein-protein and protein-water interactions causing instability or collapse?
- Have you run multiple independent replicates to confirm the observed behavior?
Solutions:
- Select a Balanced Force Field: The core challenge is finding a force field that stabilizes folded proteins without collapsing IDPs. Recent refinements like amber ff03w-sc (which incorporates selective upscaling of protein-water interactions) and amber ff99SBws-STQ' are designed to achieve this balance, maintaining folded stability while accurately reproducing IDP dimensions [43].
- Prioritize Validation with Multiple Data Types: A force field should be tested against both SAXS (for global dimensions) and NMR data (for local structure) to ensure overall accuracy [43].
➤ Quantitative Data for Force Field Selection
The table below summarizes key metrics and recommendations from recent studies to guide force field selection.
Table 1: Force Field Performance Guide for Different Protein Types
| Force Field | Recommended For | Key Strengths | Reported Limitations | Key Validation Metrics |
|---|---|---|---|---|
| CHARMM36m [12] [43] | Proteins with structured and disordered regions (Hybrid proteins). | Retains transient helical motifs in IDRs; reliable for folded domains. | Protein-protein association can be too strong in some cases [43]. | NMR relaxation, Rg (SAXS), residual dipolar couplings. |
| amber ff99SBws-STQ′ [43] | Balanced accuracy for folded proteins, IDPs, and protein complexes. | Maintains folded state stability; accurate IDP dimensions; corrects polyQ helicity bias. | A refined variant, indicating ongoing optimization in the field. | SAXS data, NMR chemical shifts, folded state RMSD. |
| amber ff03w-sc [43] | Systems requiring balanced protein-water interactions. | Selective water interaction scaling improves folded stability & IDP ensembles. | Performance should be verified for specific protein complexes. | Stability of folded proteins (RMSD), IDP chain dimensions. |
| AMBER ff99SB-ILDN [12] | Folded globular proteins (without significant disorder). | Well-established and extensively validated for structured systems. | Tends to cause artificial collapse of IDPs when used with TIP3P water [12]. | Backbone dihedral distributions, stability of folded proteins. |
➤ The Scientist's Toolkit: Essential Research Reagents & Software
Table 2: Key Resources for Experimental Validation and Analysis
| Resource Name | Type | Primary Function |
|---|---|---|
| ANSURR [101] | Software/Method | Validates protein structure accuracy by comparing NMR chemical shift data to structural rigidity. |
| DSSP & STRIDE [104] | Software Algorithm | Assigns secondary structure elements from 3D protein coordinates. |
| FIRST [101] | Software | Performs rigidity analysis on protein structures using mathematical graph theory. |
| Random Coil Index (RCI) [101] | Analytical Method | Predicts local protein backbone flexibility from NMR chemical shifts. |
| LSTM/GRU Networks [102] [103] | Deep Learning Model | Predicts the time-series evolution of structural parameters like native contacts from MD data. |
| TIP4P-D [12] [43] | Water Model | An advanced 4-site water model that improves IDP ensemble modeling by enhancing protein-water interactions. |
| Native Contacts (NCs) [102] | Analytical Metric | Quantifies the similarity of a simulated protein conformation to its native folded state. |
Research Reagent Solutions
Table 1: Essential computational reagents for Relative Binding Free Energy (RBFE) calculations.
| Reagent Category | Specific Examples | Function in RBFE Workflow |
|---|---|---|
| Molecular Dynamics Engines | NAMD, OpenMM, ChemShell QM/MM [83] | Performs the molecular dynamics simulations that generate conformational ensembles for free energy analysis. |
| Free Energy Calculation Tools | gmx_MMPBSA [105] | Automates the calculation of binding free energies from MD simulation trajectories. |
| Classical Fixed-Charge Force Fields | AMBER ff99SB*, ff99SB-ILDN, CHARMM C36 [83] [106] | Models molecular interactions for proteins and ligands; the default choice for most RBFE studies due to computational efficiency. |
| Polarizable Force Fields | Drude, AMOEBA [83] [106] | Provides a more accurate representation of electrostatics by including electronic polarization; used for limited benchmarking. |
| Semiempirical Quantum Methods | g-xTB, GFN2-xTB [107] | Offers a low-cost, quantum-mechanically informed method for calculating protein-ligand interaction energies. |
| Neural Network Potentials (NNPs) | UMA-m, UMA-s, AIMNet2 [107] | Machine-learned models that promise near-DFT accuracy at a fraction of the computational cost for energy calculations. |
| Ligand Charge Parameterization | AM1-BCC, CGenFF, RESP-HF, RESP-DFT [105] | Assigns partial atomic charges to ligands, which is critical for accurate electrostatic energy calculations. |
| Implicit Solvent Models | Generalized Born (GB), Poisson-Boltzmann (PB) [105] [108] | Approximates solvation effects without explicit water molecules, significantly speeding up calculations. |
Frequently Asked Questions & Troubleshooting Guides
FAQ: Force Field Selection
Q: Which force field should I use for RBFE calculations on multimeric ATPases? For large, complex systems like multimeric ATPases, fixed-charge force fields like AMBER are the most practical and widely validated choice [106]. While polarizable force fields (AMOEBA, Drude) offer a more sophisticated physical model, their prohibitive computational cost makes them unsuitable for the extensive sampling required for RBFE on these systems [106]. A large-scale benchmark on 55 interfacial sites across six ATPase classes demonstrated that AMBER could successfully predict binding preferences in 91% of sites for systems with low structural variability [106].
Q: When should I consider using a polarizable force field? Consider polarizable force fields like AMOEBA or the CHARMM Drude model when you are studying systems where electronic polarization is critical and computational resources are less constrained [83] [106]. This includes processes involving ion binding, interactions with highly charged groups, or systems where the electrostatic environment changes significantly. They have been shown to provide a more proper treatment of dielectric constants, which is essential for accurate modeling of hydrophobic solvation and other electrostatic phenomena [83].
Troubleshooting Common RBFE Problems
Table 2: Common issues, their diagnoses, and solutions in RBFE calculations.
| Problem Symptom | Affected System Types | Likely Cause | Recommended Solution |
|---|---|---|---|
| Poor prediction accuracy (high error) | Systems with high structural variability (e.g., Rho, FtsK) [106] | 1. Inadequate sampling of slow conformational relaxations.2. Structural instability or high flexibility. | 1. Extend sampling to >20 ns per alchemical window [106].2. Use structural validation tools (e.g., AlphaFold3) to model a more stable complex [106]. |
| Inaccurate ranking of ligands | Soluble & membrane proteins with charged ligands [105] | Incorrect assignment of ligand charges and poor handling of halogen bonds. | Benchmark charge methods (e.g., RESP-DFT-EP, AM1-BCC) and use extra points (EP) for halogens [105]. |
| Unstable protein-ligand complex | Systems with large conformational change upon binding [108] | The "one-average" (1A) approach ignores receptor/ligand reorganization. | Switch to the "two-average" (2A) approach, which includes sampling of the free ligand to account for reorganization energy [108]. |
| Slow convergence of free energy | Systems with highly charged, flexible ligands (e.g., nucleotides) [106] | Slow relaxation of long-range electrostatic interactions and ligand conformations. | Increase the number and duration of alchemical windows (λ states) to ensure proper equilibration at each step [106]. |
| Systematic over- or under-binding | General protein-ligand systems [107] | Underlying inaccuracies in the force field or interaction energy method. | For NNPs, apply a systematic correction (Δ-learning). For physical models, benchmark against high-level quantum data (e.g., PLA15) and adjust parameters [107]. |
FAQ: Methods & Benchmarking
Q: How does RBFE performance compare to other binding affinity methods like MM/GBSA? Large-scale benchmarks show that RBFE is generally more accurate and reliable than MM/PB(GB)SA and is competitive with the more rigorous Free Energy Perturbation (FEP) [105]. While MM/PB(GB)SA is faster, its performance is highly system-dependent and relies on crude approximations, such as frequently neglecting the entropic term or mishandling the reorganization of the protein and ligand upon binding [105] [108]. RBFE, by contrast, more rigorously accounts for the pathway between related ligands, leading to more robust predictions.
Q: What is the most critical factor for achieving accurate RBFE results with charged ligands? Extensive conformational sampling is the most critical factor. For highly charged and flexible ligands like nucleotides, the slow relaxation associated with long-range electrostatic interactions necessitates prolonged simulation times. Benchmarking studies have determined that >20 ns of sampling per alchemical window is often required for convergence in these challenging cases [106]. Using fewer or shorter windows risks inaccurate results due to poor sampling.
Experimental Protocols
Detailed Methodology: Alchemical RBFE for Multimeric ATPases
This protocol is adapted from a large-scale benchmarking study on 55 interfacial binding sites across six classes of oligomeric ATPases (F1-ATPase, MalK, MCM, Rho, FtsK, gp16) [106].
1. System Preparation
- Structure Source: Obtain starting coordinates from experimental (cryo-EM, X-ray) or high-quality predicted structures (e.g., from AlphaFold3). For unstable structures, AF3 can be used to generate a more stable model for simulation [106].
- System Building: Solvate the protein-ligand complex in a periodic box of explicit water molecules. Add ions to neutralize the system's charge and achieve a physiologically relevant salt concentration.
2. Force Field and Parameter Selection
- Primary Choice: Employ a fixed-charge force field like AMBER for all RBFE calculations due to its balance of accuracy and efficiency for large systems [106].
- Ligand Parameters: Assign charges and torsional parameters for the nucleotide ligands (ATP, ADP) using the force field's standard protocol. Ensure parameters are consistent with the chosen protein force field.
- Comparative Studies (Optional): For a limited set of systems, run unbiased MD simulations with a polarizable force field like AMOEBA to compare the stability of the binding site and ligand pose against the fixed-charge model [106].
3. Simulation Setup & Equilibration
- Energy Minimization: Heavily minimize the solvated system to remove bad contacts.
- Thermalization & Pressurization: Gradually heat the system to the target temperature (e.g., 300 K) and then equilibrate the pressure to 1 bar.
- Equilibration MD: Run extensive equilibrium molecular dynamics (multiple nanoseconds) until the system energy, temperature, pressure, and root-mean-square deviation (RMSD) of the protein backbone have stabilized.
4. Alchemical RBFE Simulation
- λ-Window Setup: Create a series of non-physical intermediate states (typically 12-24 windows) that alchemically transform one ligand into another (e.g., ATP to ADP).
- Enhanced Sampling: Use a soft-core potential to avoid singularities as atoms are decoupled.
- Extended Sampling: For each λ window, run a minimum of 20 ns of dynamics to ensure adequate sampling of the slow conformational relaxations associated with the charged phosphate groups of the nucleotides [106].
- Free Energy Estimator: Use the Multistate Bennett Acceptance Ratio (MBAR) or Thermodynamic Integration (TI) to compute the free energy difference from the data collected across all λ windows.
5. Analysis and Validation
- Error Analysis: Compute the standard error of the free energy estimate using bootstrapping or other statistical methods.
- Experimental Comparison: Validate the predicted relative binding free energies (ΔΔG) against experimentally observed nucleotide binding preferences where available.
- Diagnostic Checks: Monitor structural fidelity (RMSD), ligand pose stability, and the preservation of key binding interactions throughout the alchemical transformation to identify sources of error [106].
Workflow Visualization
RBFE Implementation and Troubleshooting Workflow: This diagram outlines the key steps for running a successful Relative Binding Free Energy (RBFE) calculation, incorporating critical decision points based on large-scale benchmarking studies. It highlights the recommendation to use fixed-charge force fields like AMBER for large systems and the necessity of extended sampling (>20 ns/λ window) for convergence, especially with charged ligands. The troubleshooting loop provides pathways to address poor accuracy, such as re-evaluating force field choice or improving the initial structural model.
Conclusion
The selection of an appropriate force field is not a one-size-fits-all decision but a critical, project-specific choice that directly determines the accuracy and reliability of molecular dynamics simulations. As this guide illustrates, a successful strategy involves understanding the foundational principles of major force fields, applying them correctly to specific protein types, being vigilant for potential biases, and rigorously validating results against experimental data. The future of force fields points toward increased incorporation of polarizability, automated parametrization through machine learning, and better handling of complex chemical modifications. For researchers in drug development and structural biology, adopting a systematic and informed approach to force field selection is essential for generating meaningful, predictive insights that can accelerate biomedical discovery and clinical translation.
References
- 1. Force field - GROMACS 2024.2 documentation [https://manual.gromacs.org/documentation/2024.2/reference-manual/functions/force-field.html]
- 2. The Protein Force Field Plays a Crucial Role in Obtaining ... [https://arxiv.org/html/2508.18570v1]
- 3. Force field (chemistry) [https://en.wikipedia.org/wiki/Force_field_(chemistry)]
- 4. Force Field Parameter Sets — Tinker User's Guide ... [https://dasher.wustl.edu/tinker/distribution/doc/sphinx/tinker/_build/html/text/parameters.html]
- 5. Review The continuous evolution of biomolecular force fields [https://www.sciencedirect.com/science/article/abs/pii/S0969212625001911]
- 6. CHARMM additive and polarizable force fields for biophysics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4334745/]
- 7. Cannot add non-natural residues in charmm36 forcefield [https://gromacs.bioexcel.eu/t/cannot-add-non-natural-residues-in-charmm36-forcefield/1372]
- 8. Optimization of the additive CHARMM all-atom protein force ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3549273/]
- 9. FAQ [https://charmm-gui.org/faq]
- 10. phosaa14SB and phosaa19SB: Updated Amber Force ... [https://pubmed.ncbi.nlm.nih.gov/39151116/]
- 11. IDP-Specific Force Field ff14IDPSFF Improves the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5731455/]
- 12. Choice of Force Field for Proteins Containing Structured and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7136338/]
- 13. Navigating AMBER Force Fields [https://diphyx.com/stories/amber-force-fields/]
- 14. Systematic Validation of Protein Force Fields against ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0032131]
- 15. Update of the CHARMM all-atom additive force field for lipids [https://pmc.ncbi.nlm.nih.gov/articles/PMC2922408/]
- 16. Importance of the CMAP Correction to the CHARMM22 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1367299/]
- 17. Inclusion of Many-Body Effects in the Additive CHARMM ... [https://www.sciencedirect.com/science/article/pii/S0006349512008557]
- 18. Newest CHARMM36 port for GROMACS [https://gromacs.bioexcel.eu/t/newest-charmm36-port-for-gromacs/868]
- 19. Force fields in GROMACS [https://manual.gromacs.org/current/user-guide/force-fields.html]
- 20. Force field - GROMACS 2021.7 [https://manual.gromacs.org/documentation/2021-current/reference-manual/functions/force-field.html]
- 21. GROMOS96 43a1 performance on the characterization of ... [https://www.sciencedirect.com/science/article/abs/pii/S0008621508006113]
- 22. Force field - GROMACS 2026-beta documentation [https://manual.gromacs.org/documentation/2026-beta/reference-manual/functions/force-field.html]
- 23. Force field - GROMACS 2025.2 documentation [https://manual.gromacs.org/documentation/2025.2/reference-manual/functions/force-field.html]
- 24. GROMOS96 43a1 force field for ligand topology [https://gromacs.bioexcel.eu/t/gromos96-43a1-force-field-for-ligand-topology/8606]
- 25. GROMOS [https://en.wikipedia.org/wiki/GROMOS]
- 26. On the Validation of Protein Force Fields Based on Structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11103706/]
- 27. Evolution of all-atom protein force fields to improve local ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7507668/]
- 28. Development and Testing of the OPLS-AA/M Force Field for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6585454/]
- 29. Comparison of CHARMM and OPLS-aa forcefield ... [https://www.sciencedirect.com/science/article/abs/pii/S0950061819330296]
- 30. Polarizable force fields for biomolecular simulations [https://pmc.ncbi.nlm.nih.gov/articles/PMC6520134/]
- 31. Pairwise-additive and polarizable atomistic force fields for ... [https://www.sciencedirect.com/science/article/abs/pii/S1877117319302157]
- 32. Protein folding, unfolding, and misfolding [https://pubmed.ncbi.nlm.nih.gov/33187403/]
- 33. Molecular Simulation/Polarizable force fields [https://en.wikibooks.org/wiki/Molecular_Simulation/Polarizable_force_fields]
- 34. Molecular Dynamics Simulations Using the Drude Polarizable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6031474/]
- 35. Navigating protein landscapes with a machine-learned ... [https://www.nature.com/articles/s41557-025-01874-0]
- 36. Comparison of water models for structure prediction [https://www.sciencedirect.com/science/article/pii/S0167732225011523]
- 37. Benchmarking Water Models in Molecular Dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10934818/]
- 38. Impact of water models on the structure and dynamics ... [https://www.sciencedirect.com/science/article/pii/S2001037024003714]
- 39. Assessing the accuracy of physical models used in protein ... [https://www.sciencedirect.com/science/article/pii/S0959440X13002157]
- 40. Force Field Bias in Protein Folding Simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2711430/]
- 41. The Force Field Frenzy: Choosing the Right One for Your ... [https://drugmarvel.wordpress.com/2023/12/20/the-force-field-frenzy-choosing-the-right-one-for-your-molecular-simulations/]
- 42. Evaluating Force Performance in Thermodynamic Field ... Calculations [https://pubmed.ncbi.nlm.nih.gov/28696692/]
- 43. Optimized protein-water interactions and torsional ... [https://www.nature.com/articles/s41467-025-65603-4]
- 44. Evaluation and refinement of all-atom force fields for ... [https://www.sciencedirect.com/science/article/pii/S000634952500373X]
- 45. Polarizable Mean-Field Model of Water for Biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4285689/]
- 46. Force-Field Dependence of Chignolin Folding and Misfolding [https://pmc.ncbi.nlm.nih.gov/articles/PMC3328722/]
- 47. Stereochemical errors and their implications for molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3124434/]
- 48. Recent Force Field Strategies for Intrinsically Disordered ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8256680/]
- 49. Benchmarking of force fields to characterize the intrinsically ... [https://www.nature.com/articles/s41598-023-40801-6]
- 50. Force Field Development and Simulations of Intrinsically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5826789/]
- 51. ff14IDPs Force Field Improving the Conformation Sampling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5480619/]
- 52. Improving Small-Molecule Force Field Parameters in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8711133/]
- 53. Parameterising your ligands [https://docs.bioexcel.eu/2020_06_09_online_ambertools4cp2k/04-parameters/index.html]
- 54. Examples using SMIRNOFF with the toolkit [https://docs.openforcefield.org/projects/toolkit/en/stable/examples.html]
- 55. Publications [https://openforcefield.org/science/publications/]
- 56. Current State of Open Source Force Fields in Protein-Ligand ... [https://pubmed.ncbi.nlm.nih.gov/38895959/]
- 57. Protein-ligand-water systems with Interchange [https://docs.openforcefield.org/en/latest/examples/openforcefield/openff-interchange/protein_ligand/protein_ligand.html]
- 58. Using bespoke force field parameters with OpenFE protocols [https://docs.openfree.energy/en/latest/cookbook/bespoke_parameters.html]
- 59. Fatal error while doing Protein-ligand simulation using ... [https://gromacs.bioexcel.eu/t/fatal-error-while-doing-protein-ligand-simulation-using-gromacs-2023/7515]
- 60. Free Energy Calculations for Drug Design with FEP+ Online ... [https://www.schrodinger.com/free-energy-calculations-for-drug-design-with-fep-online-course-faq/]
- 61. Make the molecules that matter with accurate, efficient, and ... [https://cresset-group.com/science/science-resources/accurate-efficient-and-easy-to-run-fep/]
- 62. Free energy calculations for drug design with FEP+ [https://www.schrodinger.com/life-science/learn/education/courses/smdd-fep/]
- 63. Frequently Asked Questions [https://cgmartini.nl/docs/faq/]
- 64. Martini3-IDP: improved Martini 3 force field for disordered ... [https://www.nature.com/articles/s41467-025-58199-2]
- 65. Assessing the Martini 3 protein model: A review of its path ... [https://www.sciencedirect.com/science/article/abs/pii/S1570963924000219]
- 66. Proteins - Part I: Basics and Martinize 2 [https://cgmartini.nl/docs/tutorials/Legacy/martini3/ProteinsI/]
- 67. Proteins - Part I: Basics and Martinize 2 [https://cgmartini.nl/docs/tutorials/Legacy/martini3/ProteinsI_Jan2025/]
- 68. Proteins [https://cgmartini.nl/docs/tutorials/Legacy/martini2/proteins.html]
- 69. Legacy Tools – Martini Force Field Initiative [https://cgmartini.nl/docs/downloads/tools/legacy.html]
- 70. MARTINI [https://en.wikipedia.org/wiki/MARTINI]
- 71. How Robust Are Protein Folding Simulations with Respect ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3149239/]
- 72. Development of force field methods - TEL - Thèses en ligne [https://theses.hal.science/tel-04194169v1]
- 73. Challenges in protein folding simulations: Timescale, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3032381/]
- 74. Best practices for machine-learned force fields [https://www.vasp.at/wiki/index.php/Best_practices_for_machine-learned_force_fields]
- 75. Successes and challenges in simulating the folding of large ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6952611/]
- 76. Practical Guidance for Consensus Scoring and Force Field ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9772090/]
- 77. Collaborative assessment of molecular geometries and ... [https://openforcefield.org/community/news/science-updates/benchmarking-openff-2022-04-15/]
- 78. Exponential consensus ranking improves the outcome in ... [https://www.nature.com/articles/s41598-019-41594-3]
- 79. An optimal distance cutoff for contact-based Protein ... [https://www.nature.com/articles/s41598-017-01498-6]
- 80. Time length molecular dynamics [https://physics.stackexchange.com/questions/160443/time-length-molecular-dynamics]
- 81. Common errors when using GROMACS [https://manual.gromacs.org/2024.4/user-guide/run-time-errors.html]
- 82. Molecular dynamics simulation for all - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6209097/]
- 83. Current Status of Protein Force Fields for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4554537/]
- 84. Protein folding kinetics and thermodynamics from atomistic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3497772/]
- 85. Molecular Dynamics of the Intrinsically Disordered Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12461933/]
- 86. Modification-specific proteomics: Strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2892724/]
- 87. Additive CHARMM36 Force Field for Nonstandard Amino Acids [https://pmc.ncbi.nlm.nih.gov/articles/PMC8207570/]
- 88. [AMBER] Help with parameterization of non-standard amino acid [http://archive.ambermd.org/202010/0387.html]
- 89. Incorporation of non-standard amino acids into proteins [https://pmc.ncbi.nlm.nih.gov/articles/PMC6449208/]
- 90. Common Research Strategies for Post-Translational ... [https://www.mtoz-biolabs.com/common-research-strategies-for-post-translational-modification.html]
- 91. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 92. Evaluating Universal Machine Learning Force Fields ... [https://arxiv.org/html/2508.05762v1]
- 93. Researchers Advance Protein Force Fields To Accurately ... [https://quantumzeitgeist.com/protein-advance-force-fields-accurately-model-disordered-conformations-solution/]
- 94. Comparison among Amber-ff14SB, Amber-ff19SB, and ... [https://pubmed.ncbi.nlm.nih.gov/41233298/]
- 95. Everything you wanted to know about Markov State Models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2933958/]
- 96. Markov state models (MSMs) [http://msmbuilder.org/3.6.0/msm.html]
- 97. Independent Markov decomposition: Toward modeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8346863/]
- 98. Markov state models of protein misfolding - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4760979/]
- 99. An Evaluation of Force Field Accuracy for the Mini-Protein ... [https://chemrxiv.org/engage/chemrxiv/article-details/65e3a9bee9ebbb4db9c5c2f0]
- 100. Learn PyEMMA — PyEMMA 2.5.7 documentation [http://www.emma-project.org/latest/tutorial.html]
- 101. A method for validating the accuracy of NMR protein ... [https://www.nature.com/articles/s41467-020-20177-1]
- 102. Predicting the evolution of number of native contacts of a ... [https://www.sciencedirect.com/science/article/pii/S1476927122000056]
- 103. Predicting the evolution of number of native contacts of a ... [https://pubmed.ncbi.nlm.nih.gov/35042082/]
- 104. Amino acid propensities for secondary structures and its ... [https://www.sciencedirect.com/science/article/pii/S1476927124000719]
- 105. MM/PB(GB)SA benchmarks on soluble proteins and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9751045/]
- 106. Benchmarking Alchemical Relative Binding Free Energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12591046/]
- 107. Benchmarking Protein–Ligand Interaction Energy | Rowan [https://www.rowansci.com/blog/benchmarking-protein-ligand-interaction-energy]
- 108. The MM/PBSA and MM/GBSA methods to estimate ligand- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4487606/]