A Comprehensive Guide to RESP Charge Derivation: Protocols for Accurate Biomolecular Force Fields
This article provides a complete guide to the Restrained Electrostatic Potential (RESP) charge derivation protocol, a cornerstone of modern molecular mechanics force fields.
A Comprehensive Guide to RESP Charge Derivation: Protocols for Accurate Biomolecular Force Fields
Abstract
This article provides a complete guide to the Restrained Electrostatic Potential (RESP) charge derivation protocol, a cornerstone of modern molecular mechanics force fields. Aimed at researchers and drug development professionals, we cover the foundational theory behind ESP-fitting, a detailed step-by-step methodology using tools like R.E.D. and Gaussian, and advanced troubleshooting for common pitfalls. The guide also explores next-generation approaches like RESP2 and provides rigorous validation techniques to ensure derived charges yield accurate results in biomolecular simulations, particularly for drug-like molecules and novel chemical entities.
Understanding RESP Charges: The Foundation of Modern Molecular Simulation
What Are RESP Charges and Why Are They Crucial for Force Fields?
Restrained Electrostatic Potential (RESP) charges represent a cornerstone of modern molecular mechanics force fields, providing the atomic partial charges that are essential for accurately modeling electrostatic interactions in biomolecular simulations. This application note details the fundamental principles, evolving methodologies, and standardized protocols for RESP charge derivation, with particular emphasis on the latest RESP2 advancement that incorporates both gas- and aqueous-phase electronic structure calculations. Designed for researchers engaged in force field parameterization for drug development, this guide provides comprehensive computational workflows, quantitative comparisons of charge derivation strategies, and essential toolkits for implementing these methods in research practice. By establishing rigorous standards for charge generation, RESP methodologies enable more reliable simulations of protein-ligand interactions, solvation phenomena, and condensed-phase behavior critical to pharmaceutical applications.
The Role of Partial Charges in Molecular Mechanics
Atomic partial charges are fundamental parameters in empirical force fields used for molecular dynamics (MD) simulations, serving as the primary representation of electronic distribution for calculating electrostatic energies and forces [1]. Unlike quantum mechanical methods that explicitly treat electrons, classical molecular mechanics employs fixed point charges centered on atomic nuclei to model Coulombic interactions. The accuracy of these partial charges critically influences a simulation's ability to reproduce experimentally observed properties, including molecular geometries, interaction energies, and thermodynamic behavior [1] [2]. For polar molecules and biological macromolecules, electrostatic forces largely determine the strengths of hydrogen bonds and molecular recognition events, making charge parameterization particularly crucial for simulating drug-receptor interactions [1].
Limitations of Early Charge Derivation Methods
Various methods exist for deriving atomic partial charges, including empirical fitting to experimental properties, quantum mechanical wavefunction partitioning (e.g., Mulliken, Bader, and distributed multipole analyses), and electrostatic potential (ESP) fitting [1] [3]. Early ESP-derived charges, while offering good reproduction of intermolecular properties, exhibited several pathological behaviors including excessive polarity, large charge values for buried atoms, and significant dependence on molecular orientation and conformation [3]. These limitations stemmed from the statistical nature of the fitting process and the poor representation of atoms with limited surface exposure [3].
The RESP Methodology: Fundamental Principles
Theoretical Foundation
The Restrained Electrostatic Potential (RESP) approach, introduced by Bayly et al., addresses the limitations of unrestrained ESP fitting by incorporating harmonic penalty functions to attenuate charge magnitudes while maintaining accuracy in reproducing the quantum mechanical electrostatic potential [1] [4]. The fundamental objective function minimized in RESP fitting combines two components:
[ \chi^2 \left( \mathbf{x}{resp} \right) = \chi{esp}^2 \left( \mathbf{x}{resp} \right) + \chi{restr}^2 \left( \mathbf{x}_{resp} \right) ]
where (\chi{esp}^2) represents the squared deviations between the classical and quantum mechanical electrostatic potentials, and (\chi{restr}^2) represents the hyperbolic restraint function that penalizes large charge magnitudes [4]. The ESP term is defined as:
[ \chi{esp}^2 \left( \mathbf{x}{resp} \right) = \left\| \mathbf{A}\mathbf{x}{resp} - \mathbf{b} \right\|2^2 ]
where (\mathbf{A}) is the design matrix encoding inverse distances from atoms to grid points, (\mathbf{x}_{resp}) is the vector of unique charges, and (\mathbf{b}) is the reference quantum mechanical ESP values [4]. The restraint term takes the form:
[ \chi{restr}^2 \left( \mathbf{x}{resp} \right) = a \sum^{N{heavy}}{j} \left( x_j^2 + b^2 \right)^{\frac{1}{2}} - b ]
where (a) and (b) control the strength and tightness of the hyperbolic restraint, respectively, and (N_{heavy}) is the number of heavy atoms [4].
The Two-Stage RESP Protocol
The standard RESP implementation employs a two-stage fitting process [4]:
Stage 1: All heavy atoms except methyl/methylene groups are weakly restrained with strength (a = 0.0005), while methyl hydrogens are constrained to be equivalent and methylene hydrogens are constrained to be equivalent.
Stage 2: Only non-methyl/methylene heavy atoms are restrained with strength (a = 0.001), with methyl and methylene hydrogens constrained to be equivalent within their groups.
This protocol, combined with the use of the HF/6-31G* quantum mechanical method, which fortuitously overestimates gas-phase polarity by approximately the right amount to approximate hydration effects, has become the standard for the AMBER force field and related biomolecular parameter sets [1] [2] [3].
Advanced RESP Methodologies
RESP2: Integrating Environment Polarization Effects
The RESP2 method represents a next-generation approach that explicitly addresses electronic polarization in different environments by combining gas- and aqueous-phase electrostatic potentials [2] [5]. Rather than relying on the fortuitous overpolarization of HF/6-31G* calculations, RESP2 computes ESPs using higher-level quantum mechanical methods (e.g., PW6B95/aug-cc-pV(D+d)Z) in both gas phase and aqueous continuum solvent, then generates final charges as a linear combination:
[ q{RESP2} = \delta \cdot q{aqueous} + (1 - \delta) \cdot q_{gas} ]
where the mixing parameter (\delta) typically ranges from 0.5 to 0.6 (60% aqueous, 40% gas-phase) [2]. This approach decouples charge derivation from the arbitrary overpolarization pattern of HF/6-31G* and provides a physically motivated framework for modeling environmental polarization effects in fixed-charge force fields.
Table 1: Comparison of RESP Methodologies
| Method | QM Level | Polarization Treatment | Optimal Applications | Key Parameters |
|---|---|---|---|---|
| RESP | HF/6-31G* | Implicit via basis set overpolarization | General biomolecules in aqueous solution | Restraint strength (a=0.0005-0.001) |
| RESP2 | PW6B95/aug-cc-pV(D+d)Z | Explicit via gas/aqueous mixing | Condensed-phase properties, dielectric constants | Mixing parameter (δ=0.5-0.6) |
| IPolQ | MP2/cc-pV(T+d)Z | Explicit via QM/MM averaging | Polarizable force field development | 50:50 gas:aqueous weighting |
Technical Implementation Advances
Recent computational advances have addressed RESP's significant computational costs, particularly for large systems. The DF-RESP method combines density fitting techniques with RESP derivation to dramatically accelerate calculations while maintaining accuracy [6]. This approach achieves excellent accuracy with mean absolute errors in charges below 0.003 e for benchmark systems and achieves up to 14-fold speedups for protein-sized systems, making RESP practical for drug discovery applications involving protein-ligand complexes [6].
Automation tools have also streamlined the complex, error-prone process of RESP charge derivation. The R.E.D. Tools (RESP and ESP charge Derive) enable automatic charge derivation for molecules containing elements up to bromine, interfacing with multiple quantum mechanical programs and ensuring charge reproducibility across computational platforms with accuracy of 0.0001 e [3]. The accompanying RESP ESP charge DDataBase (R.E.DD.B.) provides curated charge parameters and force field libraries for over 80 whole molecules and 94 molecular fragments, facilitating force field development [7].
Computational Protocols for RESP Charge Derivation
Standardized Workflow for Molecular Parameterization
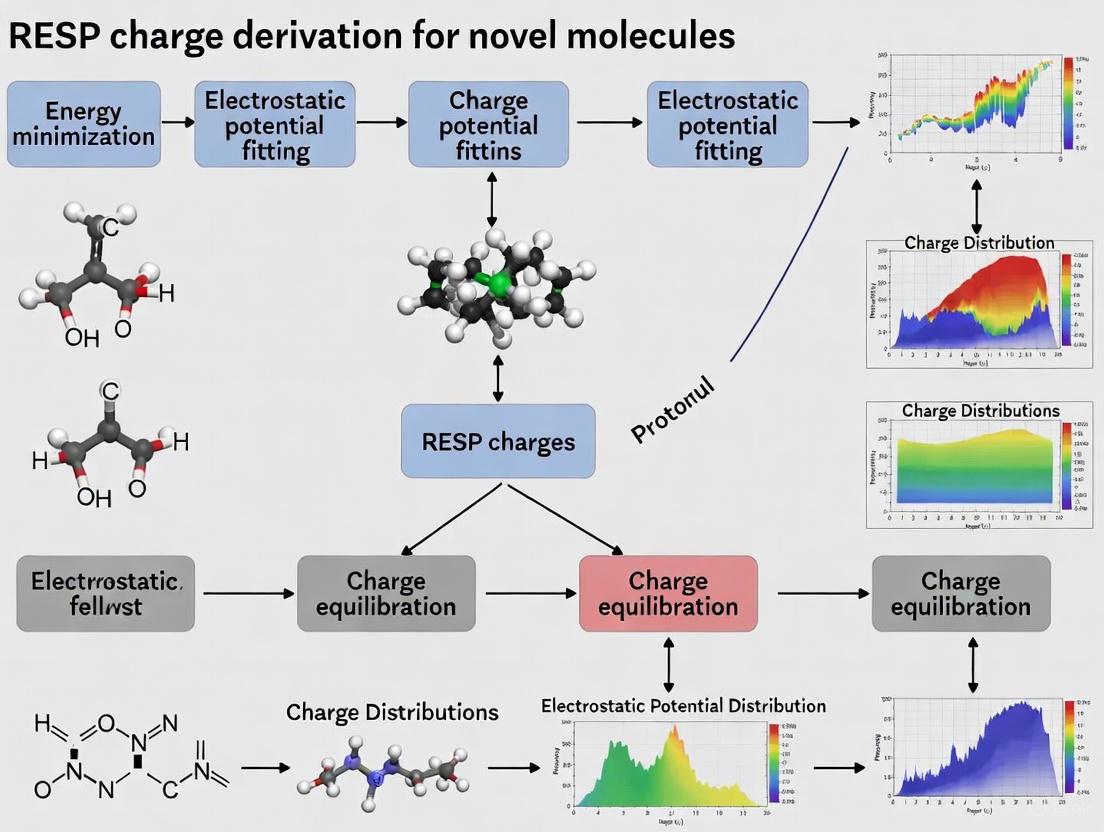
The following diagram illustrates the comprehensive workflow for deriving RESP charges for novel molecules, incorporating multiple conformation sampling and force field library generation:
Quantum Mechanical Calculation Specifications
Conformation Sampling and Geometry Optimization:
- Select representative conformations spanning the molecule's accessible conformational space [3]
- Perform geometry optimization at the HF/6-31G* level for standard RESP or PW6B95/aug-cc-pV(D+d)Z for RESP2 [2] [3]
- Define tight optimization criteria (energy gradient < 0.0001 hartree/bohr) to ensure precise molecular geometries [3]
Electrostatic Potential Calculation:
- Compute molecular electrostatic potential using the CHELPG algorithm or Connolly surface points [1] [3]
- Generate a sufficient number of points (typically >10,000) outside the van der Waals surface to ensure adequate sampling [1] [3]
- For RESP2, perform separate calculations in gas phase and aqueous continuum solvent (e.g., IEF-PCM) [2]
RESP Fitting Procedure with Constraints
Charge Fitting Implementation:
- Implement the restrained fitting according to the Lagrangian formulation described in Section 2.1
- Apply constraint matrices to maintain molecular total charge and enforce chemical equivalence [4]
- For multiple conformations, combine ESP data from all conformers with appropriate weighting [3]
- Execute the two-stage fitting protocol with the specified restraint strengths [4]
Convergence Criteria:
- Iterate until charge sets converge to within 0.1×10⁻⁵ between iterations [4]
- Verify that fitted charges reproduce the QM electrostatic potential with RMSD < 2-5 kcal/mol
- Ensure physical reasonable charge values (typically |q| < 0.8 e for non-metal atoms)
Table 2: RESP Restraint Parameters and Fitting Options
| Parameter | Stage 1 Value | Stage 2 Value | Purpose | Alternative Options |
|---|---|---|---|---|
| Hyperbolic restraint strength (a) | 0.0005 | 0.001 | Limits charge magnitudes | 0.001-0.01 for carbohydrates [1] |
| Hyperbolic tightness (b) | 0.1 | 0.1 | Controls restraint shape | Fixed parameter |
| Equivalent charge constraints | Methyl & methylene H | Methyl & methylene H | Enforce chemical symmetry | User-defined atom lists [8] |
| Total charge constraint | Molecular charge | Molecular charge | Maintain electroneutrality | Fixed via constraint matrix [4] |
Computational Software and Databases
Table 3: Essential Research Reagent Solutions for RESP Charge Derivation
| Resource | Type | Function | Access |
|---|---|---|---|
| R.E.D. Tools | Software | Automated RESP/ESP charge derivation and force field library building | http://q4md-forcefieldtools.org/RED/ [3] |
| R.E.DD.B. | Database | Curated RESP/ESP charges and force field libraries for 80+ molecules | https://upjv.q4md-forcefieldtools.org/REDDB/ [7] |
| CP2K | Software | Periodic and nonperiodic RESP fitting for condensed phase systems | https://manual.cp2k.org/ [8] |
| DF-RESP | Method | Accelerated RESP with density fitting for large systems | Implementation-specific [6] |
| ForceBalance | Software | Systematic optimization of RESP2 and LJ parameters | Implementation-specific [2] |
Validation and Application Protocols
Crystal Structure Validation:
- Implement molecular dynamics simulations of crystal structures using derived charges [1]
- Monitor unit cell dimensions and hydrogen bonding geometry during simulation
- Compare with experimental neutron diffraction data to assess charge set accuracy [1]
Liquid Property Validation:
- Calculate liquid-state properties (density, heat of vaporization, dielectric constant) from MD simulations [2]
- Optimize Lennard-Jones parameters in conjunction with RESP charges to avoid error cancellation [2]
- Validate against experimental measurement to ensure transferability to condensed phases
RESP charge methodology has evolved from a specialized technique for biomolecular force fields to a sophisticated toolkit for electrostatic parameterization in drug discovery and materials science. The ongoing development of RESP2, automated tools like R.E.D., and accelerated algorithms such as DF-RESP demonstrates the continued vitality of this approach. For researchers parameterizing novel molecules, adherence to the protocols outlined herein—including proper conformational sampling, quantum mechanical level selection, and rigorous validation against experimental data—ensures the derivation of physically meaningful charges that yield accurate predictions in molecular simulations. As force field development continues advancing toward explicitly polarizable models, the principles underlying RESP will continue informing next-generation electrostatic parameterization for computational chemistry and drug development.
Atomic partial charges are a cornerstone of empirical force fields used in molecular mechanics and dynamics simulations. While not quantum mechanical observables, these charges are crucial for computing electrostatic interactions, which dominate inter-molecular phenomena such as hydrogen bonding, solute-solvent interactions, and molecular recognition events in drug design [3]. The challenge in charge derivation stems from the need to represent the continuous electron density distribution of a molecule with a discrete set of point charges on atomic centers. Among various approaches, charges derived from the Molecular Electrostatic Potential (MEP) have gained widespread acceptance because the MEP is a quantum mechanical property directly related to how a molecule is "seen" by its external environment, such as a protein binding pocket or solvent molecules [3] [1].
The Restrained Electrostatic Potential (RESP) method represents a refinement of this approach, designed to mitigate the tendency of standard ESP-derived charges to overestimate bond polarities and produce excessively large charges for buried atoms, which can lead to artifacts in molecular dynamics simulations [1]. This application note details the theoretical foundation, computational protocols, and practical tools for deriving RESP charges, providing a standardized workflow for researchers engaged in force field development for novel molecules, particularly in the context of drug discovery.
Theoretical Foundation: MEP and the RESP Formalism
The Molecular Electrostatic Potential (MEP)
The Molecular Electrostatic Potential (MEP), ( V(\vec{r}) ), at a point ( \vec{r} ) in space is defined by the following equation, which incorporates contributions from the nuclear charges and the electron density:
Here, ( ZA ) represents the charge of nucleus ( A ) located at ( \vec{R}A ), and ( \rho(\vec{r}\,') ) is the molecule's electron density [3] [1]. The MEP is a physically significant property that can be computed accurately from ab initio or Density Functional Theory (DFT) calculations. In practice, ( V(\vec{r}) ) is evaluated at a large number of points (typically thousands) located on a molecular surface surrounding the van der Waals envelope of the molecule.
Fitting Atomic Charges to the MEP
The core objective is to determine a set of atomic partial charges ( {qj} ) that best reproduce the quantum mechanically computed MEP at the grid points ( {i} ). This is achieved by minimizing the least-squares fit error, ( \chi{esp}^2 ):
where ( Vi ) is the QM-derived MEP at point ( i ), and ( \hat{V}i ) is the classical electrostatic potential generated by the atomic point charges: ( \hat{V}i = \sumj \frac{qj}{r{ij}} ) [1].
The RESP Restraint
A well-known issue with this straightforward fitting procedure is that it can lead to anomalously high charge magnitudes for atoms (like carbons in hydrocarbon groups) that are buried within the molecular structure and thus poorly sampled by the MEP grid [3] [1]. The RESP model addresses this by adding a hyperbolic restraint term, ( \chi_{rstr}^2 ), to the minimization function, thereby penalizing large charge values without fixing them to zero. The total function minimized in the RESP fit is:
The parameter ( b ) defines the tightness of the hyperbolic restraint, and ( k_{rstr} ) is the restraint weight, which controls the strength of the penalty [1]. This restraint effectively attenuates charge magnitudes, leading to a more balanced and transferable charge set that performs better in condensed-phase simulations.
Computational Protocols and Methodologies
Standard RESP Charge Derivation Workflow
The following diagram illustrates the multi-stage workflow for deriving RESP charges, incorporating best practices for ensuring robustness and accuracy.
Detailed Experimental Protocols
Protocol 1: Standard Single-Conformation RESP Derivation
This protocol is suitable for relatively rigid molecules.
Geometry Optimization
- Software: Use quantum chemical software like Gaussian, GAMESS-US, NWChem, or CP2K [3].
- Method and Basis Set: A common starting point is the Hartree-Fock (HF) method with the 6-31G* basis set [3] [1]. This level of theory is known to produce dipole moments about 10% larger than experimental gas-phase values, which is often considered desirable for implicitly accounting for polarization in aqueous-phase simulations within additive force fields [3].
- Objective: Fully optimize the molecular geometry to a local energy minimum, confirming the absence of imaginary frequencies if a frequency calculation is performed.
Molecular Electrostatic Potential (MEP) Calculation
- Single-Point Calculation: Using the optimized geometry, perform a single-point energy calculation at the same theory level (e.g., HF/6-31G*) to obtain the wavefunction [3].
- Grid Generation: Compute the MEP at a large number of points (e.g., several thousand) on a molecular surface. The Connolly surface or algorithms like CHELPG are typically used to define the grid points outside the van der Waals radius [3] [1].
RESP Charge Fitting
- Software: Use the
RESPorFITCHARGEprogram, often accessed through tools likeR.E.D. ToolsorAntechamber[3]. - Restraint Parameters: A two-stage fitting process is often employed. In the first stage, a restraint weight (( k{rstr} )) of 0.0005 is applied to all atoms. In the second stage, a stronger restraint (e.g., ( k{rstr} = 0.001 )) is applied only to non-hydrogen atoms, while the charges of hydrogen atoms are equivalenced (constrained to be equal) based on their chemical type [1].
- Software: Use the
Protocol 2: Multiple-Conformation RESP for Flexible Molecules
For flexible molecules, deriving charges from a single conformation is insufficient. This protocol enhances transferability across conformational space [3].
- Conformational Sampling: Generate an ensemble of low-energy conformations representative of the molecule's accessible space. Methods include systematic torsional scanning, molecular dynamics simulations, or stochastic searching.
- QM Calculation per Conformation: For each unique, optimized conformation, perform a geometry optimization and MEP calculation as described in Protocol 1.
- Simultaneous Fitting: The RESP fit is performed simultaneously against the MEP grids from all conformations in the ensemble. This produces a single set of atomic charges that represent a Boltzmann average over the conformations, crucial for obtaining accurate energetics in molecular dynamics simulations [3].
Protocol 3: Validation via Crystal Molecular Dynamics
A powerful validation method for carbohydrate and other polar molecules involves simulating crystal structures [1].
- Crystal Structure Preparation: Obtain a high-resolution (preferably neutron diffraction) crystal structure of the molecule.
- Building the Crystal Model: Construct a simulation box containing multiple unit cells (e.g., 2x2x4) to avoid imaging artifacts from non-bonded cutoffs [1].
- Molecular Dynamics Simulation: Perform an MD simulation in the NPT ensemble (e.g., 50 ps total, analyzing the last 30 ps) using the newly derived RESP charges and the target force field.
- Analysis: Monitor the unit cell dimensions (a, b, c) and energies. A successful charge set will maintain the experimental crystal geometry and intermolecular hydrogen-bonding network, whereas a poor charge set will lead to significant distortion or collapse of the crystal lattice [1].
Advanced Method: DF-RESP for Large-Scale Systems
A recent advancement, DF-RESP, combines Density Fitting (DF) techniques with the RESP formalism to dramatically accelerate calculations for large biomolecular systems [6].
- Principle: DF-RESP uses the density fitting MEP (DF-MEP) method to compute the electrostatic potential, reducing the computational cost associated with the conventional MEP calculation that requires dense grid sampling [6].
- Performance: For the 1493-atom protein 1h59, DF-RESP achieved a 14-fold speedup while maintaining accuracy comparable to conventional RESP [6].
- Accuracy: On the S22 benchmark set, DF-RESP produced atomic charges with a mean absolute error (MAE) below 0.003 e compared to conventional RESP. For electrostatic interaction energies in androgen receptor-ligand complexes, deviations were under 0.1 kcal/mol [6].
Table 1: Comparison of RESP Charge Derivation Methods
| Method | Key Feature | Best For | Computational Cost | Key Consideration |
|---|---|---|---|---|
| Standard RESP | Single-conformation fitting | Rigid molecules, initial testing | Low | Charge values may not be transferable across conformations. |
| Multi-Conf. RESP | Fitting to an ensemble of conformers | Flexible molecules, MD simulations | High | Ensures robust performance across conformational landscape. |
| DF-RESP | Density fitting for MEP calculation | Large biomolecules (proteins, complexes) | Significantly Lower | Maintains high accuracy (~0.003 e MAE) with large speedups (14x) [6]. |
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Software Tools for RESP Charge Derivation and Validation
| Tool Name | Type | Primary Function | Notes |
|---|---|---|---|
| R.E.D. Tools [3] | Software Suite | Automates RESP/ESP charge derivation and force field library generation. | Handles multiple orientations, conformations, and molecules; ensures high reproducibility. |
| Gaussian [3] | QM Program | Performs geometry optimization and MEP computation. | A proprietary, widely used standard in the field. |
| GAMESS-US / NWChem [3] | QM Program | Open-source alternatives for geometry optimization and MEP computation. | May produce slightly different results than Gaussian. |
| Antechamber [3] | Software Tool | Derives charges for organic molecules automatically. | Part of the AmberTools package; less customizable than R.E.D. |
| CP2K [9] | QM Program | Performs DFT calculations to obtain electrostatic potential. | Can be used with batoms (Python) for MEP visualization [9]. |
| Amber / GROMACS | MD Engine | Validates charges via MD simulation of crystals or solution. | Critical for assessing charge performance in a simulated condensed phase [1]. |
Results and Data Presentation
The choice of restraint weight (( k_{rstr} )) is critical. As demonstrated in MD simulations of α-D-glucopyranose crystal structures, different restraint weights lead to significantly different outcomes [1].
Table 3: Impact of RESP Restraint Weight on Crystal Simulation Quality
| Charge Type / Restraint Weight (( k_{rstr} )) | Simulation Outcome Category | Description | Suitability for Condensed Phase |
|---|---|---|---|
| Unrestrained ESP charges | Category C | Poor maintenance of crystal geometry and interactions. | Poor - Overestimates bond polarities. |
| RESP, ( k_{rstr} = 0.01 ) | Category A | Good agreement with neutron diffraction structure. | Good - Optimal for carbohydrates in GLYCAM [1]. |
| RESP, ( k_{rstr} = 0.001 ) | Category B | Maintained hydrogen bonds but distorted unit cell. | Moderate - May require further tuning. |
| Mulliken/Distributed Multipole | Category C | Failed to maintain geometry and interactions. | Poor - Not recommended for this application. |
Workflow Visualization: From Molecule to Force Field
The complete pathway from a novel molecule to a fully parameterized force field library, integrating the concepts and protocols discussed, is summarized below.
In molecular simulations utilizing empirical force fields (FFs), the accuracy of computed properties depends critically on the parameters defining non-bonded interactions, particularly the partial atomic charges assigned to each atom. Among the most respected methods for deriving these charges is the Restrained Electrostatic Potential (RESP) approach [3]. A key innovation in the RESP model is the introduction of hyperbolic restraints to mitigate the problem of overpolarization and generate charges that are both chemically reasonable and effective for condensed-phase simulations [3]. This application note details the role of these restraints within the broader protocol for RESP charge derivation, providing researchers with the methodologies and tools necessary for its application to novel molecules.
Theoretical Foundation: The Challenge of Overpolarization
The ESP Charge Derivation Paradigm
Electrostatic Potential (ESP) derived charges are obtained by fitting atomic-centered charges to reproduce the Molecular Electrostatic Potential (MEP) computed from quantum mechanical (QM) calculations [3]. The MEP is calculated at a large number of points on a three-dimensional surface surrounding the molecule. While these charges optimally handle inter-molecular properties essential for solute-solvent and solvent-solvent interactions, they suffer from several artifacts [3]:
- Poorly Defined Buried Atoms: Atoms located within the molecular core, such as carbons in hydrocarbons, are represented by few MEP points, making their charges poorly defined during the fitting process.
- Conformational and Orientational Dependence: Resulting charges can exhibit significant variability depending on the molecule's initial conformation and orientation in the QM calculation.
- Overpolarization: The fitting process can produce excessively large charge values on buried atoms in an attempt to minimize the error in the MEP reproduction. This is the core problem addressed by RESP restraints [3].
The RESP Solution with Hyperbolic Restraints
The RESP model enhances the basic ESP fitting by adding a penalty function to the objective function being minimized [3]. The total function becomes:
Q = χ² + R(a)
Where:
- χ² is the sum of squares of the differences between the QM-derived MEP and the MEP generated by the fitted charges.
- R(a) is the restraint penalty function that depends on the charges a.
Instead of a simple harmonic restraint, the RESP method employs a hyperbolic restraint of the form R(a) = w * sqrt(a² + b²), where w is a weight and b is a constant [3]. This form effectively restrains charges from becoming too large without imposing an unduly harsh penalty on smaller, chemically reasonable charges, thus directly countering overpolarization.
Evolution of RESP Charge Models
The following table summarizes the key developments in RESP-based charge models, highlighting the progression in addressing overpolarization and improving accuracy for biomolecular simulations.
Table 1: Evolution of RESP Charge Models
| Model | QM Theory Level | Key Innovation | Primary Application |
|---|---|---|---|
| RESP (or RESP1) [2] [3] | HF/6-31G* [3] | Hyperbolic restraints on charges to prevent overpolarization [3]. | Condensed-phase simulations with AMBER force fields [3]. |
| RESP2 [2] | PW6B95/aug-cc-pV(D+d)Z (Recommended) [2] | Charges derived as a linear combination of gas- and aqueous-phase ESPs (tuned by parameter δ, ~0.6), moving beyond fortuitous HF/6-31G* overpolarization [2]. | Improved accuracy for liquid properties and hydration free energies [2]. |
| IPolQ/IPolQ-Mod [2] | MP2/cc-pV(T+d)Z [2] | Charges from a 50:50 average of QM charges in gas phase and explicit/implicit solvent reaction field [2]. | Polarizable charge models for condensed phases [2]. |
| DF-RESP [6] | Various | Uses density fitting to accelerate MEP calculation, drastically reducing computational cost for large systems [6]. | Large-scale biomolecular systems (e.g., protein-ligand complexes) [6]. |
Core Protocol for RESP Charge Derivation with Hyperbolic Restraints
The derivation of RESP charges for a novel molecule is a multi-step process. The workflow below outlines the key stages from initial geometry preparation to the final force field library.
Protocol Steps
Geometry Optimization and Conformational Sampling
- Objective: Generate a representative set of low-energy molecular geometries.
- Methodology: Use a QM program (e.g., Gaussian, GAMESS-US, PSI4) for geometry optimization. For a robust charge set, perform optimization and subsequent charge fitting on multiple conformations to avoid bias toward a single structure [3]. Tight optimization criteria are recommended.
Molecular Electrostatic Potential (MEP) Calculation
- Objective: Compute the reference MEP for the optimized geometry.
- Methodology: Using the same QM program, compute the MEP on a dense grid of points, typically defined on a Connolly or similar van der Waals surface [3]. The historical and widely used theory level for non-polarizable force fields like AMBER is Hartree-Fock (HF) with the 6-31G* basis set [3]. The fortuitous overpolarization of this method implicitly accounts for some polarization effects in aqueous solution [2] [3]. The advanced RESP2 method recommends more accurate levels like PW6B95/aug-cc-pV(D+d)Z for the underlying gas- and aqueous-phase calculations [2].
RESP Charge Fitting with Hyperbolic Restraints
- Objective: Fit atomic charges to the MEP while restraining atom charge magnitudes.
- Methodology: Use the RESP program to fit the charges by minimizing the objective function Q = χ² + R(a).
- χ² Fitting: Standard least-squares fit to the MEP.
- Hyperbolic Restraint Application: Apply the restraint R(a) = w * sqrt(a² + b²) to all non-hydrogen atoms. The weight w and constant b are parameters; common values are w=0.0005 (a weak restraint) and b=0.1 for the original RESP model [3]. This specifically counteracts overpolarization by making very large charges energetically unfavorable without strongly impacting smaller charges.
Force Field Library Generation
- Objective: Incorporate the derived charges into a usable force field file.
- Methodology: Use tools like the R.E.D. Tools (RESP and ESP charge Derive) or Antechamber to format the charges and other force field parameters (bonds, angles, dihedrals) into a library file (e.g., in Tripos mol2 format) compatible with molecular dynamics packages like AMBER, CHARMM, or GROMACS [3].
Validation via Molecular Dynamics Simulation
- Objective: Assess the performance of the new parameters.
- Methodology: Run simulations to compute target experimental properties such as density, heat of vaporization for pure liquids, hydration free energies, and liquid dielectric constants [2]. Co-optimization of Lennard-Jones (LJ) parameters alongside charges, as done in the RESP2 development, can lead to significant improvements in accuracy [2].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Tools and Resources for RESP Charge Derivation
| Tool/Resource | Type | Function & Application |
|---|---|---|
| R.E.D. Tools [3] | Software Suite | Automates the multi-step process of RESP charge derivation, handles multiple conformations and molecules, and generates force field libraries. Essential for complex systems [3]. |
| Antechamber [3] | Software Program | Part of AMBER tools. Rapidly generates parameters and RESP charges for organic molecules, though less suited for molecular fragments than R.E.D. [3]. |
| ForceBalance [2] | Software Tool | Optimizes force field parameters (like the RESP2 δ parameter and LJ terms) against experimental condensed-phase data [2]. |
| RESP ESP charge DDataBase (R.E.DD.B.) [3] | Database | Repository of pre-calculated atomic charges, optimized coordinates, and force field libraries for over fifty molecular systems [3]. |
| HF/6-31G* [3] | QM Method | Historical standard for RESP1; provides a baseline level of theory with consistent overpolarization [3]. |
| PW6B95/aug-cc-pV(D+d)Z [2] | QM Method | Recommended level for RESP2 for more accurate ESPs, balancing speed and accuracy [2]. |
| DF-RESP [6] | Computational Method | Density-Fitting RESP accelerates MEP calculation for large biomolecules (e.g., proteins) with minimal accuracy loss [6]. |
Advanced Application: The RESP2 Protocol for Mitigating Overpolarization
The recent RESP2 model represents a significant shift from relying on the fortuitous errors of a single QM method. Its protocol explicitly addresses electronic polarization:
- Dual-Phase ESP Calculation: Perform two separate MEP calculations for the molecule of interest:
- One in the gas phase.
- One in the aqueous phase (using an implicit solvation model like IEF-PCM).
- Charge Fitting and Mixing: Independently fit two sets of ESP charges (e.g., using the advanced PW6B95/aug-cc-pV(D+d)Z level).
- Linearly Combine Charges: Generate the final charge set as a linear combination of the gas- and aqueous-phase charges: q_final = δ * q_aq + (1-δ) * q_gas.
- System-Specific Tuning: The parameter δ (found to be ~0.6 for optimal performance in many cases) can be co-optimized with LJ parameters against liquid properties using the ForceBalance software, leading to a more physically grounded and accurate non-polarizable force field [2].
Atomic partial charges are a fundamental component of empirical force fields used in molecular dynamics (MD) simulations and computational drug discovery. These charges represent the distribution of electron density in a molecule and are critical for accurately modeling electrostatic interactions, which govern key biological processes such as hydrogen bonding, molecular recognition, and protein-ligand binding. Unlike covalent parameters, atomic charges are highly context-dependent and must be derived for each new molecular system. The reliability of MD simulations is profoundly dependent on the underlying force field and the quality of the atomic charge parameters used [10].
Several computational methods have been developed to derive these charges, each with distinct theoretical foundations and practical implications. The three predominant approaches are the Electrostatic Potential (ESP) fitting method, the Restrained Electrostatic Potential (RESP) model, and the semi-empirical AM1 with Bond Charge Correction (AM1-BCC) method. ESP charges are derived by directly fitting atomic point charges to reproduce the quantum mechanically calculated molecular electrostatic potential. The RESP approach extends this by introducing mathematical restraints to mitigate overfitting and produce more chemically reasonable charges. In contrast, AM1-BCC utilizes fast semi-empirical calculations followed by empirical corrections to approximate high-level quantum mechanical results [11] [3] [1]. This article provides a comprehensive comparative overview of these charge derivation methods, focusing on their theoretical basis, practical implementation, and applicability in modern computational chemistry and drug development workflows.
Theoretical Foundations and Methodologies
ESP (Electrostatic Potential) Charges
The ESP charge derivation method operates on the principle that atomic point charges should reproduce the molecular electrostatic potential (MEP) computed from a quantum mechanical wavefunction. The MEP is calculated at a large number of points on a three-dimensional grid surrounding the molecule of interest. A least-squares fitting procedure is then employed to determine the set of atomic charges that minimizes the difference between the classical Coulomb potential from the point charges and the quantum mechanical MEP [3] [1]. The objective function for this fit is:
[ \chi{esp}^2 = \sumi (Vi - \hat{V}i)^2 ]
where (Vi) is the quantum mechanical MEP at point (i), and (\hat{V}i = \sumj \frac{qj}{r{ij}}) is the classical electrostatic potential at point (i) resulting from atomic point charges (qj) [1].
A significant limitation of the basic ESP approach is that atoms buried within the molecular framework (such as carbon atoms in hydrocarbon chains) have their electrostatic potential poorly sampled by grid points outside the van der Waals surface. This can lead to overestimation of charge magnitudes, instability in the fitting procedure, and poor transferability of charges between conformations. Furthermore, ESP charges are known to be sensitive to molecular orientation and conformation, sometimes resulting in non-reproducible values even for the same molecule [12] [3].
RESP (Restrained Electrostatic Potential) Charges
The RESP model was developed to address the limitations of the basic ESP approach. Introduced by Bayly et al., RESP incorporates a hyperbolic restraint penalty term that discouragively large atomic charges without significantly compromising the quality of the electrostatic potential fit [1] [4]. This restraint helps produce more chemically reasonable charges and improves transferability between molecular environments.
The RESP objective function incorporates an additional restraint term:
[ \chi{resp}^2 = \chi{esp}^2 + \chi_{restr}^2 ]
where the restraint term (\chi_{restr}^2) is defined as:
[ \chi{restr}^2 = a \sum{j}^{N{heavy}} \left( \sqrt{qj^2 + b^2} - b \right) ]
Here, (a) controls the strength of the restraint, (b) defines the tightness of the hyperbola, and the summation typically runs over all heavy atoms [1] [4]. The hyperbolic restraint gently penalizes large charge magnitudes while having minimal effect on smaller charges, effectively reducing overfitting without resorting to hard constraints that could degrade the electrostatic potential fit.
The RESP procedure is typically conducted in two stages to enhance charge transferability. In the first stage, methyl and methylene hydrogens are not equivalenced (i.e., their charges can vary independently), and a stronger restraint is applied. In the second stage, these hydrogen atoms are equivalenced to have identical charges within their symmetry groups, and a weaker restraint is used [4]. This two-stage approach, combined with the multi-conformational and multi-orientational fitting capabilities of tools like R.E.D., results in highly reproducible charges that are suitable for condensed-phase simulations [12] [3].
AM1-BCC (Austin Model 1 with Bond Charge Correction) Charges
The AM1-BCC method provides a computationally efficient alternative to RESP charges while aiming to reproduce target HF/6-31G* electrostatic potentials. This approach first calculates preliminary charges using the semi-empirical AM1 method, which captures underlying electronic structure features including formal charge and electron delocalization. Bond charge corrections (BCCs) are then applied to these AM1 atomic charges to produce the final AM1-BCC charges [11].
The BCC parameters were globally parameterized by fitting to the HF/6-31G* ESP of a large training set of over 2700 molecules, encompassing most organic functional groups and a wide variety of cyclic and fused bicyclic heteroaryl systems [11]. This parameterization allows the AM1-BCC method to handle virtually all types of organic compounds found in major chemical databases while achieving accuracy comparable to RESP charges at a fraction of the computational cost. Validation studies have demonstrated that AM1-BCC charges reproduce hydrogen-bonded dimer energies to within 0.95 kcal/mol RMS deviation from ab initio values and relative free energies of solvation to within 0.69 kcal/mol of experimental values [11].
Table 1: Comparison of Key Characteristics of RESP, ESP, and AM1-BCC Charge Models
| Characteristic | RESP | ESP | AM1-BCC |
|---|---|---|---|
| Theoretical Basis | QM ESP fitting with restraints | QM ESP fitting without restraints | Semi-empirical with bond charge corrections |
| Computational Cost | High | High | Low |
| Charge Reproducibility | High (with multi-orientation) | Variable | High |
| Handling of Buried Atoms | Good (due to restraints) | Poor | Good (via parameterization) |
| Basis Set Dependence | HF/6-31G* (typical) | HF/6-31G* (typical) | Parameterized to HF/6-31G* ESP |
| Multi-Conformation Support | Yes | Possible but sensitive | Yes (implicit in parameterization) |
| Primary Application Domains | Protein, nucleic acids, general biomolecules | General molecules | Organic small molecules, drug-like compounds |
Experimental Protocols and Implementation
RESP Charge Derivation Workflow
The derivation of RESP charges for a novel molecule involves a multi-step process that integrates quantum mechanical calculations and charge fitting. The following protocol outlines the key steps, which can be automated using tools such as the R.E.D. (RESP ESP charge Derive) tools [12] [3].
- Molecular Structure Preparation: Begin with a 3D structure of the target molecule in PDB format. The structure should include explicit hydrogens, which can be added using tools like
AddHin UCSF Chimera [13]. For molecules with ionizable groups, ensure proper protonation states representative of the physiological pH condition of interest. - Geometry Optimization: Perform a quantum mechanical geometry optimization to determine a stable energy minimum. This step is typically conducted at the HF/6-31G* theory level, though DFT methods like B3LYP with moderate basis sets are also used [10] [3]. The R.E.D. tools can interface with various quantum chemistry packages including Gaussian, GAMESS-US, and Firefly for this step [12].
- Molecular Electrostatic Potential (MEP) Calculation: Using the optimized geometry, compute the MEP on a three-dimensional grid surrounding the molecule. The Connolly surface algorithm (used in AMBER) or the CHELPG algorithm (used in GLYCAM) can be employed to define the grid points [3]. The same quantum chemistry program and theory level (typically HF/6-31G*) used for optimization are generally recommended for MEP calculation.
- Charge Fitting with Restraints: The computed MEP grid is used as input to the RESP program, which performs the least-squares fitting with hyperbolic restraints. The R.E.D. tools automate this process, applying the two-stage fitting procedure with appropriate charge constraints for chemically equivalent atoms [12] [4]. For robust results, especially for flexible molecules, it is advisable to perform a multi-conformation RESP fit using several low-energy conformers.
- Validation and Force Field Integration: The derived RESP charges are output in Tripos mol2 file format, which serves as a precursor for AMBER OFF, CHARMM RTF, or other force field libraries [12] [3]. Validation should include checking the molecular dipole moment against QM calculations and ensuring the total charge sums to the correct integer value.
Diagram 1: RESP Charge Derivation Workflow. This diagram outlines the key steps for deriving RESP charges, from initial structure preparation to final validation.
ESP Charge Derivation Protocol
The protocol for deriving ESP charges follows a similar path to RESP but omits the restraint step:
- Structure Preparation and Optimization: As with RESP, begin with a properly protonated 3D structure and perform quantum mechanical geometry optimization at an appropriate theory level (e.g., HF/6-31G*).
- MEP Calculation: Calculate the MEP on a 3D grid using a quantum chemistry program.
- Direct ESP Fitting: Perform an unconstrained least-squares fit of the atomic charges to the MEP. This can be done using the
FITCHARGEprogram or similar utilities [3]. - Analysis: Examine the resulting charges for chemical reasonableness, as ESP charges are prone to exaggerated values for buried atoms.
AM1-BCC Charge Derivation Protocol
The AM1-BCC method offers a significantly streamlined workflow:
- Structure Preparation: Provide a 3D molecular structure with explicit hydrogens.
- Semi-Empirical Calculation: Perform a single-point AM1 calculation using programs like MOPAC or the Antechamber module of AMBER tools [11] [13].
- Apply Bond Charge Corrections: Automatically apply pre-parameterized BCCs to the AM1 population charges to generate the final AM1-BCC charges. This entire process is implemented in tools like Antechamber and can be accessed through UCSF Chimera's
Add Chargetool [13].
Table 2: Software Tools for Charge Derivation and Their Capabilities
| Tool Name | Supported Methods | QM Program Interfaces | Key Features | Typical Use Cases |
|---|---|---|---|---|
| R.E.D. Tools [12] [3] | RESP, ESP | Gaussian, GAMESS, Firefly | Multi-orientation, multi-conformation, multi-molecule fitting | High-quality charge derivation for force field development |
| Antechamber (in AMBER) [13] | AM1-BCC, RESP | MOPAC, Gaussian | Automated workflow, fast | High-throughput charge assignment for small molecules |
| OpenFF Recharge [4] | RESP, AM1BCC-type models | Various via QC data | Modern, programmable framework | Developing new charge models and force fields |
| ESPsim [14] | ESP similarity scoring | Psi4 (for RESP charges) | Molecular similarity based on ESP | Virtual screening, molecular alignment |
| UCSF Chimera [13] | AM1-BCC, Gasteiger, RESP (via Antechainer) | Integrated | User-friendly graphical interface | Visualization, rapid charge assignment for modeling |
Comparative Analysis and Applications
Performance in Molecular Dynamics Simulations
The performance of different charge models has been systematically evaluated in comparative studies. In one such investigation focusing on amino acids, RESP charges derived using both HF and B3LYP theories showed excellent agreement with literature values from the AMBER FF14SB force field. The study found that all analyzed charge derivation methods (RESP and AM1-BCC) reproduced benchmark values with sufficient accuracy for parameterizing novel species, with subtle differences emerging in their ability to restrain backbone charges to predefined values during fragmentation approaches for capped amino acids [10].
For carbohydrate simulations, RESP charges have demonstrated superior performance in maintaining crystal geometries during molecular dynamics simulations compared to unrestrained ESP charges. One study found that a restraint weight (krstr) of 0.01 provided the best agreement with the neutron diffraction structure of α-d-glucopyranose, while unrestrained ESP charges performed poorly, as did charges from Mulliken and distributed multipole analyses [1]. This highlights the importance of restraints for simulating condensed phases, where over-polarized charges can distort intermolecular interactions.
Application in Drug Discovery and Virtual Screening
Charge models play a critical role in computer-aided drug design, particularly in virtual screening and molecular similarity assessments. The ESPsim software package leverages electrostatic potential similarities to identify potential drug candidates [14]. This tool can calculate shape and ESP similarities between molecules using various charge methods, including Gasteiger, MMFF94, machine-learned charges, and quantum-mechanically derived RESP charges. The ability to rapidly compare electrostatic properties enables more effective virtual screening, as electrostatic complementarity is a key determinant of binding affinity [14].
Recent advances in artificial intelligence have further integrated these charge models into generative drug discovery pipelines. For instance, DiffSMol—a generative AI model for creating 3D structures of potential drug molecules—can generate novel molecules conditioned on the shapes of known ligands [15]. While not directly using traditional charge derivation methods, such approaches still rely on accurate electrostatic properties to predict binding characteristics and optimize drug-like properties.
Table 3: Key Research Reagent Solutions for Charge Derivation
| Resource | Type | Function | Availability |
|---|---|---|---|
| R.E.D. Tools [12] | Software Suite | Automates RESP/ESP charge derivation with multi-orientation/conformation fitting | http://q4md-forcefieldtools.org/RED/ |
| Antechamber [13] | Software Tool | Automates AM1-BCC and RESP charge derivation for small molecules | Part of AMBER Tools |
| GAFF (General Amber Force Field) [11] | Force Field | Provides parameters for organic molecules; often used with AM1-BCC charges | Part of AMBER |
| ESPsim [14] | Software Package | Calculates shape and ESP similarity for molecular comparison | https://github.com/hesther/espsim |
| HF/6-31G* | Basis Set | Standard QM level for computing MEP for RESP charges in condensed phase | In Gaussian, GAMESS, etc. |
| q4md-forcefieldtools Mailing List [12] | Community Support | Forum for discussion and troubleshooting of R.E.D. tools and charge derivation | q4md-fft@q4md-forcefieldtools.org |
The comparative analysis of RESP, ESP, and AM1-BCC charge models reveals a landscape of complementary approaches, each with distinct advantages for specific applications in computational chemistry and drug discovery. RESP charges, with their restraint-based fitting and support for multiple conformations, provide high-quality, reproducible charges suitable for biomolecular simulations in the condensed phase. ESP charges offer a direct physical interpretation but require careful handling due to their sensitivity to molecular orientation and tendency to produce exaggerated charges for buried atoms. AM1-BCC strikes an exceptional balance between computational efficiency and accuracy, making it ideal for high-throughput applications involving drug-like small molecules.
Future developments in charge derivation will likely focus on several emerging areas. Machine learning approaches are already being employed to predict accurate partial charges rapidly, as seen in the ML charge model implemented in ESPsim [14]. The integration of generative AI with electrostatic property optimization, exemplified by models like DiffSMol, points toward a future where charge models are seamlessly incorporated into de novo molecular design pipelines [15]. Furthermore, the development of next-generation polarizable force fields will demand more sophisticated charge models that can respond to changing electronic environments, moving beyond the static point-charge paradigm that dominates current biomolecular simulation practices [16]. As these computational approaches continue to evolve, the fundamental principles underlying RESP, ESP, and AM1-BCC will remain essential for understanding and modeling molecular interactions with high fidelity.
Biomolecular simulation serves as a "computational microscope," providing atomistic-level insights into the structure, dynamics, and interactions of biological macromolecules that are often difficult to capture through experimental methods alone [17]. This computational approach has expanded significantly in scope, demonstrating practical value in investigating biological systems and contributing to interdisciplinary research at the interfaces between physics, chemistry, and biology [17]. The field has progressed from theoretical promise to practical application, with simulations increasingly making uniquely detailed contributions to understanding biological macromolecules and their functions [17].
The growing impact of biomolecular simulation is evidenced by its diverse applications across biological research and drug development. These computational methods allow researchers to investigate processes that occur across vastly different timescales, from rapid molecular vibrations to slow conformational changes that might be inaccessible to direct experimental observation. As the field continues to mature, integration with experimental data has become increasingly important for validating predictions and providing a more complete understanding of complex biological systems [17].
Table 1: Key Biomolecular Types and Their Simulation Applications
| Biomolecule Class | Key Simulation Applications | Representative System Features |
|---|---|---|
| Proteins | Protein folding & conformational changes [17], enzyme catalysis [17], ion channel dynamics [17] | Complex internal motions, secondary & tertiary structure, active sites |
| Nucleic Acids | Macromolecular interactions [17], structure-based drug design [17] | Double helix, base pairing, groove geometry, backbone flexibility |
| Carbohydrates | Molecular interactions in food systems [18] | Glycosidic linkages, ring conformations, hydrogen bonding networks |
| Drug-like Molecules | Ligand binding & free energy calculations [17], association with proteins [17] | Molecular weight <500, balanced polarity, drug-receptor complementarity |
Fundamental Principles and Force Fields
The Critical Role of Partial Atomic Charges
At the heart of accurate biomolecular simulations lies the precise description of electrostatic interactions, which are predominantly governed by the assignment of partial atomic charges. These charges represent the distribution of electron density across a molecule and fundamentally influence its reactive behavior, molecular recognition, and intermolecular interaction energies [2]. Until recently, the accurate determination of atomic partial charges presented a significant challenge, as this concept lacks a precise quantum-mechanical definition and has been difficult to quantify experimentally [19].
The restrained electrostatic potential (RESP) approach has emerged as a highly regarded method for assigning partial charges in molecular simulations [2]. This method generates partial charges designed to reproduce the electrostatic potentials (ESPs) of molecules, typically computed at the quantum-mechanical level [2]. The RESP approach specifically addresses the challenge of deriving chemically reasonable charges that balance computational accuracy with physical transferability, often employing hyperbolic restraints to avoid excessively large charges on equivalent atoms [6].
Advances in Charge Derivation Methods
Recent years have witnessed significant methodological advances in partial charge determination. The RESP2 method represents a next-generation approach that addresses limitations in traditional RESP by tuning charge polarity through a parameter (δ) that scales contributions from gas- and aqueous-phase calculations [2]. This approach decouples the calculation of ESP charges from reliance on the arbitrary and inconsistent pattern of overpolarization afforded by Hartree-Fock calculations with the 6-31G* basis set, which was historically considered the de facto standard for RESP [2].
A groundbreaking development in the field came with the introduction of ionic scattering factors (iSFAC) modeling, which enables experimental determination of partial charges using electron diffraction [19]. This method provides absolute values for the partial charge of each atom in a crystallographic structure and has demonstrated strong correlation with quantum chemical computations (Pearson correlation of 0.8 or higher for organic compounds) [19]. The iSFAC approach integrates seamlessly into standard electron crystallography workflows and has been successfully applied to diverse compounds including antibiotics, amino acids, and zeolites [19].
Further innovation arrived with DF-RESP, which combines density fitting of molecular electrostatic potentials with RESP charge derivation to dramatically accelerate calculations while maintaining accuracy [6]. This approach achieves excellent accuracy with a mean absolute error in charges below 0.003 e for benchmark systems and achieves a 14-fold speedup for large biomolecules while maintaining comparable accuracy [6].
Application Notes by Biomolecular Class
Protein Simulations
Protein simulations have demonstrated tremendous value in elucidating the relationship between protein dynamics and biological function. Molecular dynamics simulations have been instrumental in revealing how proteins flex and undergo complex internal motions that in some cases are directly related to their functional mechanisms [17]. These simulations provide critical insights that complement experimental structural biology approaches.
Key Application Areas:
- Protein Folding and Unfolding: Simulations have proven valuable in interpreting experimental data and complementing experiments focused on protein folding pathways and stability [17].
- Enzyme Catalysis: Biomolecular dynamics play crucial roles in enzyme catalysis, with simulations helping to elucidate the relationship between protein motions and catalytic efficiency [17].
- Membrane Proteins: Simulations of ion channels and other membrane proteins have provided insights into transport mechanisms across biological membranes [17].
- Conformational Changes: Studies of protein conformational changes have revealed how structural transitions enable functional mechanisms in biological systems [17].
Nucleic Acid Simulations
Nucleic acid simulations focus on understanding the structural dynamics, recognition mechanisms, and interactions of DNA and RNA molecules with proteins, small molecules, and other biological partners. While specific applications to nucleic acids were less prominently featured in the search results, their importance in macromolecular interactions and structure-based drug design is well-established [17]. These simulations typically employ similar force field principles as protein simulations, with careful attention to the representation of phosphate groups, nucleobases, and sugar puckering conformations.
Carbohydrate Simulations
Carbohydrate simulations present unique challenges due to the structural complexity, branching patterns, and diverse chemical modifications found in these biomolecules. Molecular simulation technology has been applied to analyze interactions between food molecules, assessing structural changes of biomolecules and mechanisms of physical and chemical property alterations [18]. These applications provide deeper understanding of molecular interaction mechanisms in complex carbohydrate systems, helping to overcome limitations of purely experimental approaches.
Drug-like Molecules and Pharmaceutical Applications
Simulations of drug-like molecules have made significant contributions to pharmaceutical research, particularly in understanding drug-receptor interactions and optimizing therapeutic compounds. Applications include studies of protein association with small molecules, computation of binding free energies for ligands, and structure-based drug design [17]. The accurate simulation of these interactions relies heavily on proper parameterization of drug-like molecules, including the assignment of high-quality partial atomic charges.
Recent Breakthrough Applications:
- Google collaborated with Boehringer Ingelheim to demonstrate quantum simulation of Cytochrome P450, a key human enzyme involved in drug metabolism, with greater efficiency and precision than traditional methods [20].
- Quantum computing approaches have been applied to molecular systems such as butyronitrile, a molecule with emerging importance in battery and solar cell research [21].
- Pharmaceutical companies are increasingly exploring hybrid quantum-machine learning approaches for biologics research and drug discovery [22].
Table 2: Performance Metrics for Advanced Charge Derivation Methods
| Method | Computational Efficiency | Charge Accuracy (MAE) | Key Application Strength |
|---|---|---|---|
| RESP1 [2] | Baseline reference (HF/6-31G*) | Varies by molecule | Established benchmark; extensive validation |
| RESP2 (δ=0.6) [2] | ~20x slower than RESP1 (implicit solvent) | Optimized against liquid properties | Superior liquid property prediction |
| DF-RESP [6] | 14x faster than RESP (1493-atom system) | <0.003 e (S22 benchmark) | Large-scale biomolecular systems |
| iSFAC Modelling [19] | Experimental approach | ~0.8 Pearson correlation with QM | Absolute charge values; experimental validation |
Experimental Protocols
RESP Charge Derivation with PyPE_RESP
Protocol Objective: Derivation of partial atomic charges using the Restrained Electrostatic Potential (RESP) approach via the PyPE_RESP tool, which facilitates and standardizes the charge derivation process for novel molecules.
Materials and Reagents:
- PyPE_RESP software (distributed with AmberTools) [23]
- Quantum chemistry package (e.g., psi4 for electrostatic potential calculation) [2]
- Molecular structure files in formats compatible with RDKit [23]
- Computational resources appropriate for the system size
Procedure:
- Input Preparation: Prepare molecular structure files containing the novel molecule of interest. For multiconformer RESP fitting, generate multiple reasonable conformers representing the molecule's conformational space [23].
Constraint Group Definition: Comprehensively define charge constraint groups through multiple methods available in PyPE_RESP. This step ensures chemically equivalent atoms receive identical charges [23].
Electrostatic Potential Calculation: Compute the molecular electrostatic potential using the specified quantum chemical method. While HF/6-31G* has been historical standard, consider more accurate methods such as PW6B95/aug-cc-pV(D+d)Z for improved accuracy [2].
Charge Fitting: Perform the RESP fitting procedure with appropriate hyperbolic restraints to avoid excessively large charges on buried atoms. PyPE_RESP allows for both additive and polarizable force field charge derivation [23].
Validation and Output: Review the output constraint group and non-constraint group charges to assess the fit result. Integrate the derived charges with other force field parameters for subsequent molecular simulations [23].
Experimental Partial Charge Determination via iSFAC Modeling
Protocol Objective: Experimental determination of partial atomic charges using ionic scattering factors (iSFAC) modeling based on crystal structure analysis with three-dimensional electron diffraction.
Materials and Reagents:
- Crystalline sample of the target compound suitable for electron diffraction
- Transmission electron microscope with electron diffraction capabilities
- Crystallographic software capable of iSFAC modeling implementation
- Reference compounds for method validation if needed
Procedure:
- Crystal Preparation: Grow high-quality crystals of the target compound. For organic compounds, consider radiation-resistant materials or cryogenic conditions to minimize beam damage [19].
Data Collection: Collect three-dimensional electron diffraction data using standard electron crystallography workflows. Ensure adequate data completeness and resolution for reliable charge determination [19].
iSFAC Modeling Implementation: For each atom in the crystallographic structure, refine one additional parameter that describes the fraction of the ionic scattering factor, equivalent to its charge, along with the conventional nine parameters (three coordinates and six atomic displacement parameters) [19].
Scattering Factor Balance: Balance the contribution of the ionic form with the contribution of the neutral form for each atom using scattering factors based on the Mott-Bethe formula, which places the resulting partial charges on an absolute scale [19].
Validation and Analysis: Validate the results by comparing with quantum chemical computations where possible. Analyze the charge distribution for chemical reasonableness, noting characteristic patterns such as negative charges on carbon atoms in carboxylate groups due to electron delocalization [19].
Diagram 1: RESP charge derivation workflow. The protocol encompasses molecular structure input, conformer generation, quantum mechanical electrostatic potential calculation, RESP fitting with constraints, and validation through molecular dynamics simulation.
Diagram 2: Experimental iSFAC charge determination. The workflow involves crystal growth, three-dimensional electron diffraction data collection, iSFAC modeling implementation, and validation through comparison with quantum mechanical calculations.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Tools for Biomolecular Simulation and Charge Derivation
| Tool/Resource | Function | Application Context |
|---|---|---|
| PyPE_RESP [23] | Facilitates and standardizes RESP charge derivation | Command-line tool for batch processing of novel molecules |
| DF-RESP [6] | Accelerates RESP calculations via density fitting | Large-scale biomolecular systems (>1000 atoms) |
| iSFAC Modeling [19] | Experimental partial charge determination | Crystalline compounds requiring experimental validation |
| Quantum Chemistry Packages [2] | Computes reference electrostatic potentials | RESP and RESP2 charge derivation protocols |
| AmberTools [23] | Provides simulation preparation and analysis tools | Force field parameterization and molecular dynamics |
| Quantum Computing Platforms (e.g., IQM Resonance, InQuanto) [21] [24] | Enables complex quantum chemistry calculations | Molecular systems challenging for classical computation |
Emerging Technologies and Future Directions
The field of biomolecular simulation is rapidly evolving with the integration of emerging computational technologies. Quantum computing has demonstrated particular promise for advancing molecular simulations, with recent breakthroughs indicating its potential to address problems beyond the reach of classical computers [20] [21]. Companies including Quantinuum, IBM, and IonQ have made significant progress in developing quantum computing approaches for chemical simulation, with some already demonstrating quantum advantage for specific chemistry applications [22].
Quantum error correction represents another critical area of advancement, addressing what has historically been a fundamental barrier to practical quantum computing [20]. In 2025, progress in this area has been dramatic, with companies including Google, IBM, and Microsoft achieving exponential error reduction as qubit counts increase [20]. These developments have pushed error rates to record lows and reduced quantum error correction overhead by up to 100 times, substantially moving forward timelines for practical quantum computing applications in chemical simulation [20].
The emerging integration of artificial intelligence with quantum computing approaches further accelerates potential applications in biomolecular simulation. Companies are developing transformer-based Generative Quantum AI approaches that use AI models to efficiently synthesize circuits for preparing molecular ground states on quantum computers [24]. These hybrid approaches have demonstrated orders-of-magnitude speedups in generating training data for complex molecules, pointing toward a future where AI, quantum computing, and classical simulation work synergistically to advance biomolecular research [24].
A Step-by-Step RESP Derivation Protocol: From Molecule to Force Field Library
Within the overarching protocol for deriving RESP charges for novel molecules, the initial stage of Molecular Geometry Optimization and Conformational Sampling is a critical determinant of success. The accuracy of partial atomic charges, such as those calculated by the Restrained Electrostatic Potential (RESP) method, is intrinsically dependent on the quality and representativeness of the molecular geometries used in the quantum mechanical (QM) calculations. Fixed-charge force fields, which are essential for molecular dynamics (MD) simulations in drug design, rely on these charges to model electrostatic interactions implicitly. Consequently, a robust and thorough conformational sampling ensures that the derived RESP charges are not artifacts of a single, potentially non-representative geometry but accurately reflect the molecule's electrostatic behavior across its accessible conformational space. This step lays the foundational molecular framework upon which all subsequent charge derivation and validation procedures are built.
The necessity of this stage is further emphasized by the fact that solvent environment can induce significant conformational changes. A molecule might adopt a folded conformation in the gas phase but an open form in an aqueous solution [25]. Forcing the RESP fitting procedure to use an electrostatic potential (ESP) derived from a gas-phase geometry for a molecule that will be simulated in water can introduce a systematic bias. Therefore, the generation of a conformationally diverse set of structures, ideally representative of the target solvent environment, is a crucial prerequisite for obtaining transferable and accurate force field parameters.
Computational Methodologies for Sampling
Two primary computational approaches, each with distinct advantages, are commonly employed for exploring the conformational landscape of molecules: Molecular Dynamics (MD) and Monte Carlo (MC) based methods. The choice between them depends on factors such as system size, desired sampling speed, and the specific properties of interest.
Molecular Dynamics (MD) Simulations
MD simulations model the time evolution of a molecular system by numerically integrating Newton's equations of motion. This provides a trajectory of atomic coordinates that samples the potential energy surface.
Detailed MD Protocol [26]:
- System Preparation: Start with an initial 3D structure of the molecule (e.g., from a crystal structure or model building).
- Energy Minimization: Perform an energy minimization of the structure to remove any steric clashes or unphysical geometries using a steepest descent or conjugate gradient algorithm.
- Equilibration:
- NVT Ensemble: Equilibrate the system in the canonical ensemble (constant Number of particles, Volume, and Temperature) for approximately 100-200 picoseconds to stabilize the temperature, typically using a thermostat like Nosé-Hoover or Berendsen.
- NPT Ensemble: Further equilibrate in the isothermal-isobaric ensemble (constant Number of particles, Pressure, and Temperature) for 100-200 picoseconds to stabilize the density, using a barostat such as Parrinello-Rahman.
- Production Run: Execute an MD simulation in the NPT or NVT ensemble for a duration sufficient to observe the conformational transitions of interest. For enhanced sampling, a temperature of 350 K can be used to accelerate barrier crossing [26].
- Restraints (Optional): To focus sampling on a specific region (e.g., a ligand binding pocket), apply positional restraints to atoms outside a defined radius (e.g., 12 Å) from the region of interest [26].
Monte Carlo (MC) Backrub Sampling
The Monte Carlo backrub algorithm, as implemented in software like Rosetta, provides an alternative, efficient method for conformational sampling. It uses a Metropolis Monte Carlo criterion to accept or reject random moves, which typically involve localized rotations around pivots (like Cα atoms) and side-chain adjustments [26].
Detailed Backrub Monte Carlo Protocol [26]:
- Input Structure: Provide a starting PDB file for the complex or molecule.
- Parameter Setup:
- Define the number of Monte Carlo trials (e.g., 50,000 per complex).
- Set the
kTvalue for the Metropolis criterion (e.g., 0.35 and 1.2). - Specify pivot atoms (e.g., Cα atoms for all protein and peptide residues).
- Define the backrub segment size (minimum of 3 atoms, maximum of 64 atoms).
- Set probabilities for sampling side-chain and backbone torsions to default values.
- Simulation Execution: Run the simulation, which generates an ensemble of conformations. The output is a set of structures that can be clustered to select representative geometries for subsequent QM calculations.
Table 1: Key Parameters for Conformational Sampling Methods [26].
| Parameter | Molecular Dynamics (MD) | Monte Carlo (MC) Backrub |
|---|---|---|
| Software | GROMACS, AMBER, NAMD | Rosetta |
| Key Control Parameters | Simulation time (e.g., 20 ns), Temperature (e.g., 350 K), Restraints | Number of MC trials (e.g., 50,000), kT value, Pivot atoms |
| Sampling Driver | Natural dynamics via atomic forces | Stochastic moves with Metropolis acceptance |
| Typical Output | Trajectory (time-series of coordinates) | Ensemble of decoy structures |
| Strengths | Physically realistic dynamics, explicit solvent | Computationally efficient for local conformational changes |
Workflow Visualization
The following diagram illustrates the integrated workflow for geometry optimization and conformational sampling, highlighting how different methods feed into the final selection of conformers for RESP charge derivation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Computational Tools for Geometry Optimization and Sampling.
| Tool / Reagent | Function in Protocol | Specific Application Note |
|---|---|---|
| GROMACS [26] | A high-performance MD package for simulating molecular dynamics. | Used for running explicit-solvent MD simulations with parameters like 20 ns duration and 350 K temperature for accelerated sampling [26]. |
| Rosetta Commons [26] | A software suite for macromolecular modeling, including Monte Carlo methods. | Provides the backrub algorithm for efficient side-chain and local backbone conformational sampling [26]. |
| PyPE_RESP [23] | A tool for standardizing the derivation of RESP charges. | Can be integrated into batch processes to derive charges for multiple conformers, ensuring a comprehensive electrostatic model [23]. |
| ForceBalance [2] | A software for systematic force field optimization. | Used to co-optimize LJ parameters and RESP2 charge mixing parameter (δ) against experimental liquid data for a balanced non-bonded force field [2]. |
| FlexiSol Benchmark [25] | A benchmark set of solvation data for flexible molecules. | Provides experimental solvation energies and partition ratios for validating conformational ensembles and derived charges in a drug-like chemical space [25]. |
Advanced Sampling and Force Field Considerations
For complex biomolecules or systems with high energy barriers, advanced sampling techniques are often necessary. Generalized Ensemble methods, such as Replica Exchange MD (REMD) and its variants, enhance sampling efficiency. Techniques like REST2 (Replica Exchange with Solute Tempering) belong to a class of methods that dynamically modulate atomic parameters like charges in selected regions to accelerate conformational changes [27]. The feasibility of combining such parameter-variable methods with efficient electrostatic calculators like the Zero-Multipole Summation Method (ZMM) has been demonstrated, enabling more extensive sampling without introducing significant bias [27].
The choice of partial charge model is also paramount. The next-generation RESP2 approach addresses a key limitation of traditional RESP (RESP1) by deriving charges as a linear combination (tuned by a parameter δ, typically ~0.6) of gas-phase and aqueous-phase QM calculations [2]. This produces charges with more accurate polarity, as it moves away from relying on the fortuitous overpolarization of the HF/6-31G* method. Critically, the LJ parameters of the force field must be co-optimized with the new charge model to achieve a balanced and accurate non-bonded force field [2].
The Molecular Electrostatic Potential (MEP) is a fundamental property in computational chemistry that provides a detailed mapping of the electrostatic landscape around a molecule. It is defined as the force acting on a positive test charge (a proton) at a specific point in space due to the molecule's unperturbed charge distribution from its electrons and nuclei [28]. In the context of deriving Restrained Electrostatic Potential (RESP) charges for novel molecules, the accurate calculation of the MEP is the critical second stage that forms the foundation for all subsequent charge fitting procedures. This step determines how the molecule will interact with its environment, making it paramount for reliable simulations in drug design and materials science [29] [30] [31].
For years, the Hartree-Fock (HF) method with the 6-31G basis set has been the de facto standard for MEP calculations used in RESP charge derivation. Its widespread adoption is not due to its superior quantum mechanical accuracy, but rather to its fortuitous overpolarization of electron density. This error systematically mimics the polarization a molecule experiences in a condensed phase (like water), making the resulting charges suitable for use in fixed-charge, non-polarizable force fields without any explicit treatment of polarization [2] [1]. However, the field is evolving. This protocol details the established HF/6-31G method while also exploring advanced strategies that leverage more modern quantum chemical methods to achieve greater accuracy and physical rigor.
Theoretical Background
The MEP, V(r), at a point r in space is given by:
where ZA is the charge of nucleus A located at RA, and ρ(r') is the molecule's electron density [31]. In practice, this potential is visualized by mapping its values onto a surface representing the molecular boundary, such as a constant electron density surface (e.g., 0.001 a.u.) or a van der Waals surface [28].
The MEP is a powerful tool for predicting molecular reactivity. Regions of negative MEP (often colored red in visualizations) indicate areas where a molecule is susceptible to electrophilic attack, as they are stabilized by interaction with a positive charge. Conversely, regions of positive MEP (blue) indicate an affinity for nucleophiles [28] [31]. This makes MEP analysis invaluable for understanding drug-receptor interactions, where the electrostatic complementarity between two species is a key determinant of binding affinity [29] [30].
Computational Methods for MEP Calculation
The Standard Method: HF/6-31G*
The HF/6-31G* model chemistry remains the benchmark for RESP charge derivation. The " * " signifies that this is a polarized basis set, meaning it includes d-type polarization functions on all non-hydrogen atoms (e.g., Li to Kr). This polarization is essential for accurately modeling the distortion of electron density in bonds and lone pairs [32] [33].
Rationale for Use:
- Systematic Overpolarization: The HF method lacks electron correlation, and the 6-31G* basis set is relatively small and lacks diffuse functions. Together, this combination exaggerates molecular dipole moments in a way that, empirically, yields partial charges well-suited for simulating molecules in aqueous solution [2] [1].
- Transferability and Consistency: Its long history of use has created a vast dataset of parameters derived from this method, ensuring consistency across molecular libraries in force fields like AMBER and GLYCAM [1].
Limitations:
- The overpolarization is arbitrary and can be inconsistent across different chemical classes [2].
- It provides only an implicit, approximate treatment of polarization and is not a path toward more physically realistic, polarizable force fields.
Advanced Strategies Beyond HF/6-31G*
For researchers requiring higher accuracy or a more physically grounded starting point, several advanced strategies are available.
Table 1: Advanced Quantum Mechanical Methods for MEP Calculation
| Method | Description | Advantages | Disadvantages |
|---|---|---|---|
| HF/6-31G | Adds p-type polarization functions to hydrogen atoms [33]. | Improved description of X-H bonds (e.g., O-H, N-H). Modest increase in cost. | Does not address the core limitations of HF/6-31G*. |
| HF/6-31+G* | Adds diffuse functions to heavy atoms [33]. | Critical for anions or molecules with lone pairs in extended systems (e.g., carboxylates). | Higher computational cost. Can be unstable for neutral molecules. |
| Density Functional Theory (DFT) | Uses a functional (e.g., PW6B95) to account for electron correlation [2]. | More accurate electron densities and dipole moments than HF. A better physical model. | Results depend on the chosen functional. Removes the "convenient error" of HF/6-31G*. |
| Implicit Solvent (e.g., IPolQ, RESP2) | MEP is computed in gas phase and aqueous continuum solvent; charges are derived from a weighted mix [2]. | Explicitly accounts for solvent polarization. RESP2 (δ=0.6) has shown superior accuracy in liquid simulations. | Significantly more computationally expensive (up to 20x slower than HF/6-31G*) [2]. |
The move toward methods like RESP2 is particularly noteworthy. This next-generation approach uses a higher-level QM method (e.g., PW6B95/aug-cc-pV(D+d)Z) and calculates the MEP in both gas and aqueous phases. The final partial charges are a linear combination of the two, with a mixing parameter δ (typically 0.6, or 60% aqueous, 40% gas phase). This decouples the charge derivation from the arbitrary errors of HF/6-31G* and allows for a more systematic, physically motivated treatment of environmental polarization [2].
Experimental Protocol: Calculating the MEP
This section provides a detailed, step-by-step protocol for calculating the MEP, adaptable to various computational chemistries.
Step 1: Geometry Optimization
Objective: Obtain a stable, energetically minimized molecular structure. Procedure:
- Start with a reasonable initial 3D geometry.
- Perform a geometry optimization using the chosen level of theory (e.g.,
#P Becke3LYP/6-31G* opt scf=tight). - Verify that the optimization has converged and that the resulting structure has no imaginary frequencies (if a frequency calculation is performed), confirming it is a true minimum.
Step 2: Single-Point Energy Calculation for MEP
Objective: Calculate the electron density and electrostatic potential using the pre-optimized geometry. Procedure:
- Using the optimized geometry, perform a single-point energy calculation to generate a formatted checkpoint file (
.fchk). This file contains the wavefunction information necessary for property analysis.- Example Gaussian Input (Traditional RESP):
- The
FormCheckkeyword in Gaussian directs the creation of the formatted checkpoint file.
Step 3: Generate the Electron Density Grid and MEP Cube Files
Objective: Use the cubegen utility to compute the MEP and electron density on a 3D grid around the molecule.
Procedure:
- Generate the electron density cube file:
- Generate the MEP cube file:
- Arguments Explained:
0: Automatic memory allocation.Density=SCForPotential=SCF: Property to calculate.imidazole.fchk: Input formatted checkpoint file.imidazole_den.cub/imidazole_esp.cub: Output cube files.0: Requests a medium-sized grid (~80 points per dimension). Use-4for a finer grid or-2for a coarser one.h: Include a header in the cube file [28].
- Arguments Explained:
Step 4: Visualize and Analyze the MEP
Objective: Map the MEP onto an molecular surface to identify key reactive sites. Procedure (using GaussView):
- Load the electron density cube file (
imidazole_den.cub). - From the
Resultsmenu, selectSurfaces.... Choose an appropriate isodensity value (e.g., 0.001 a.u.) to generate the molecular surface. - Load the MEP cube file (
imidazole_esp.cub) using theLoad...button in the Surfaces menu. - With the density surface selected, choose
Map values from a 2nd surface. - Adjust the color scale (e.g., red for negative V, blue for positive V) to interpret the electrostatic landscape [28].
The following workflow diagram summarizes this four-step protocol for both the standard and advanced methods:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for MEP Calculation and RESP Derivation
| Tool / "Reagent" | Function / Purpose | Notes |
|---|---|---|
| Gaussian | A general-purpose quantum chemistry software package. | Industry standard for running HF and DFT calculations to generate wavefunctions and MEP data. |
| GaussView | A graphical user interface for Gaussian. | Used for building molecules, setting up calculations, and (as per the protocol) visualizing the MEP mapped onto molecular surfaces. |
| cubegen | A utility program included with Gaussian. | Generates the 3D grid-based data (cube files) for the electron density and electrostatic potential from a checkpoint file. Essential for the MEP calculation workflow [28]. |
| 6-31G* Basis Set | A polarized double-zeta basis set. | The traditional "reagent" for RESP charges. Provides a balance of speed and the necessary systematic error for condensed phase simulations [32] [1]. |
| aug-cc-pV(D+d)Z | A correlation-consistent basis set with diffuse functions. | A high-quality "reagent" for advanced MEP calculations, as used in the RESP2 method. Designed for more accurate property prediction [2]. |
| RESP / RESP2 Fitting Code | Program to perform the restrained fit of charges to the MEP. | Fits the atomic partial charges to reproduce the quantum mechanical MEP while applying restraints to avoid overfitting. RESP2 incorporates a weighting between gas and aqueous phase MEPs [2] [1]. |
Case Study: MEP Analysis of Imidazole
The MEP analysis of imidazole, a common heterocycle in biochemistry, perfectly illustrates the predictive power of this technique. Imidazole contains two nitrogen atoms: one is a "pyrrole-like" nitrogen (N1) that is part of an NH group, and the other is a "pyridine-like" nitrogen (N3) with a lone pair.
When the MEP is calculated (at the HF/6-31G* level) and mapped onto its electron density isosurface, a clear and unambiguous picture emerges. The region around the pyridine-like nitrogen (N3) shows a strong negative MEP (red), identifying it as the prime site for electrophilic attack. In contrast, the area around the pyrrole-like NH nitrogen shows a positive or neutral MEP [28].
This MEP prediction is confirmed by experimental chemistry: imidazole is indeed protonated at the pyridine-like nitrogen (N3). This case demonstrates how MEP calculation serves as a powerful guide for assessing molecular reactivity and predicting protonation sites, a critical step in understanding the behavior of novel molecules in a biological context.
The Restrained Electrostatic Potential (RESP) method is a highly regarded approach for deriving partial atomic charges for molecular simulations. These charges are fundamental parameters in empirical force fields, enabling the accurate calculation of electrostatic interactions in molecular dynamics studies, a critical aspect in computational drug development. The core objective of the RESP procedure is to assign atomic charges that best reproduce the quantum mechanically (QM) derived molecular electrostatic potential (MEP) outside the van der Waals surface of the molecule. This reproduction is vital for modeling intermolecular interactions, such as those between a drug candidate and its protein target. The "restrained" component of the method involves applying a harmonic restraint during the fitting process, which discourages the generation of excessively large charges on buried atoms, thereby enhancing the transferability and physical realism of the resulting charge set. Historically, RESP charges derived from Hartree-Fock (HF) calculations with the 6-31G* basis set have been widely successful; this method fortuitously overpolarizes molecules, approximately accounting for the polarization effects experienced in a condensed phase, such as in water, despite being a gas-phase calculation [2].
Theoretical Background and Evolution
The foundational principle of the RESP method is the fitting of atomic partial charges to a precomputed molecular electrostatic potential (MEP). The standard RESP (sometimes called RESP1) fit minimizes a penalty function that contains two key terms [2]:
- The primary term is the sum of squares of the differences between the QM-calculated MEP and the MEP generated by the point charges at a large number of points around the molecule.
- A restraint term that applies a hyperbolic function to penalize charges that deviate significantly from zero, preventing overfitting and improving the model's transferability.
A significant advancement in this field is the development of RESP2, a next-generation approach that addresses the inconsistency of HF/6-31G* overpolarization across different molecular systems. The RESP2 model generates partial charges as a linear combination of charges derived from gas-phase and aqueous-phase QM calculations, scaling their contributions by a parameter, δ [2]:
RESP2_charge = δ * Charge_aqueous + (1 - δ) * Charge_gas
Extensive parametrization against experimental condensed-phase data has demonstrated that a value of δ ≈ 0.6 (representing a 60% weighting of the aqueous-phase charge and 40% of the gas-phase charge) yields optimal accuracy for a wide range of organic molecules in liquid simulations [2]. This hybrid approach more systematically accounts for the polarization environment, moving beyond the fortuitous cancellation of errors in the original method.
Further innovation focuses on computational efficiency. The conventional RESP method can be prohibitively expensive for large systems due to the cost of dense MEP sampling. The DF-RESP (Density Fitting RESP) method tackles this bottleneck by combining the density fitting MEP (DF-MEP) method with RESP charge derivation [6]. This integration dramatically accelerates the calculation—achieving up to a 14-fold speedup for a ~1500-atom protein system—while maintaining high accuracy, with a mean absolute error (MAE) in charges below 0.003 e for standard benchmarks [6].
Workflow for RESP Charge Derivation
The following diagram illustrates the logical workflow and decision points involved in deriving partial charges using the RESP method and its variants.
Computational Protocols
Standard RESP Protocol with HF/6-31G*
This protocol outlines the traditional and widely supported method for deriving RESP charges [2].
- Step 1: Molecular Structure Preparation. Obtain a well-minimized 3D molecular geometry. It is critical that the structure represents a realistic, low-energy conformation.
- Step 2: Quantum Mechanical Calculation. Perform a single-point energy calculation at the HF/6-31G* level of theory. The primary output required is the molecular electrostatic potential (MEP). A popular software package for this step is Gaussian. A sample input command for Gaussian is provided below.
#P HF/6-31G(d) Pop=MK IOp(6/33=2,6/41=10,6/42=17) - Step 3: RESP Fitting. Use the
antechamberprogram (from the AMBER tools package) to perform the restrained fit. The command reads the checkpoint file from Gaussian and outputs the RESP charges.antechamber -i molecule.log -fi gout -o molecule_resp.mol2 -fo mol2 -c resp - Step 4: Validation. Visually inspect the fitted charges for physical reasonableness (e.g., more negative charges on electronegative atoms) and verify that the MEP generated by the point charges closely matches the QM-derived MEP.
Advanced Protocol: RESP2 with PW6B95/aug-cc-pV(D+d)Z
This protocol utilizes a higher-level QM method and the modern RESP2 approach for improved accuracy and consistency [2].
- Step 1: Molecular Structure Preparation. Identical to the standard protocol.
- Step 2: Gas-Phase QM Calculation. Perform a single-point energy calculation to generate the MEP using the PW6B95/aug-cc-pV(D+d)Z level of theory (or a similar accurate method) in the gas phase.
- Step 3: Aqueous-Phase QM Calculation. Perform a second single-point energy calculation at the same level of theory, but using an implicit solvation model (e.g., the SMD model) with water as the solvent to generate an aqueous-phase MEP.
- Step 4: RESP2 Fitting. Perform two separate RESP fits, one for the gas-phase MEP and one for the aqueous-phase MEP. The final RESP2 charge set is calculated as a linear combination:
Q_final = 0.6 * Q_aqueous + 0.4 * Q_gas. This process can be automated with scripting or specialized tools.
Performance Comparison of RESP Methods
The table below summarizes key quantitative data for the RESP methods discussed, providing a clear comparison of their performance and characteristics [6] [2].
Table 1: Performance and Characteristics of RESP Charge Methods
| Method | QM Level | Recommended δ | Charge Error (MAE) | Speedup Factor | Key Application |
|---|---|---|---|---|---|
| Standard RESP | HF/6-31G* | N/A | Baseline | 1.0x | General purpose, small molecules |
| RESP2 | PW6B95/aug-cc-pV(D+d)Z | 0.6 | Improved consistency vs exp. | ~0.14x (slower) | High-accuracy liquid simulations |
| DF-RESP | Dependent on base method | N/A | < 0.003 e (vs RESP) | 14.0x (for 1h59 protein) | Large-scale biomolecular systems |
Research Reagent Solutions
The following table details the essential software tools and resources required for performing RESP charge derivation.
Table 2: Essential Research Reagents and Software for RESP Charge Derivation
| Item Name | Function/Brief Explanation | Resource Type |
|---|---|---|
| Quantum Chemistry Software | Software packages like Gaussian, PSI4 [2], or ORCA are required to perform the initial electronic structure calculation and generate the Molecular Electrostatic Potential (MEP). | Software |
| Force Field Parametrization Tools | Suites such as AMBER Tools (containing antechamber and resp) or Open Force Field Toolkit are used to execute the RESP fitting procedure itself. |
Software Suite |
| RESP2 & DF-RESP Scripts | Custom scripts or modified versions of standard codes are needed to implement the advanced RESP2 weighting or the density-fitting acceleration, as they are not always default features. | Algorithm/Code |
| LJ Parameter Set | Non-bonded interactions require Lennard-Jones (LJ) parameters alongside charges. Using a self-consistent set (e.g., from GAFF, AMBER) is critical for simulation accuracy [2]. | Parameter Library |
Validation and Best Practices
Conformational Dependence and Charge Fitting
A single, static geometry is typically used for RESP fitting. However, it is crucial to understand that partial charges can be conformationally dependent. For molecules with significant flexibility, it is a recommended best practice to derive charges for multiple low-energy conformers and examine the variation. The RESP2 method has been shown to precisely capture conformational energy variations, for instance, in tripeptide dynamics simulations [6]. For a robust force field, consider deriving a single set of charges from a representative conformation or averaging charges across multiple conformers, though the latter requires careful consideration of the relative conformational energies and populations.
Interaction Energy Validation
A critical validation step is to compare the intermolecular electrostatic interaction energies calculated using the RESP-fitted charges against those computed directly with high-level QM methods for molecular dimers or complexes. An effective validation protocol involves [6]:
- Selecting benchmark complexes relevant to the system of interest (e.g., the S22 dataset of non-covalent interactions).
- Calculating the QM-level interaction energies for these complexes.
- Calculating the interaction energies using the force field with the newly derived RESP charges.
- Quantifying the agreement using metrics like Mean Absolute Error (MAE). The DF-RESP method, for example, demonstrates excellent performance here, with electrostatic interaction energy deviations under 0.1 kcal/mol for standard benchmarks [6].
Condensed-Phase Property Validation
The ultimate test for charges intended for molecular simulation is their performance in reproducing experimental condensed-phase properties. This is particularly important when using advanced methods like RESP2, where the parameter δ was optimized against such data [2]. Key properties to simulate and compare include:
- Densities and heats of vaporization of pure organic liquids.
- Hydration Free Energies (HFE), which are highly sensitive to electrostatic interactions.
- Dielectric constants of pure liquids. Agreement with these experimental observables indicates that the charge model, in concert with the LJ parameters, correctly captures the balance of intermolecular interactions in the condensed phase.
Deriving atomic charges and building a force field library for a new molecule are key steps in developing force fields required for conducting structural and energy-based analysis using molecular mechanics. Among various approaches, the Restrained Electrostatic Potential (RESP) charge model has emerged as a particularly influential method, widely recognized for its effectiveness in condensed phase molecular dynamics simulations [3]. The derivation of popular RESP charges for a set of residues is inherently a complex and error-prone procedure, as it depends on numerous input parameters and requires seamless integration of multiple computational steps [3]. Traditionally, this process has been tedious, time-consuming, and susceptible to human-induced variability—a challenge that parallels the broader reproducibility concerns prevalent in life science research [34].
The fundamental procedure for RESP and ESP (Electrostatic Potential) charge derivation involves three critical steps: First, the molecule of interest undergoes geometry optimization to determine a stable minimum using quantum chemical software. Next, this minimized structure is used to calculate a Molecular Electrostatic Potential (MEP) on a three-dimensional grid. Finally, this grid is processed by the RESP program to fit atom-centered charges to the MEP [12]. This multi-step process demands significant expertise and meticulous attention to detail, as even minor deviations can lead to substantial inconsistencies in the resulting charge distributions.
Automation in computational biochemistry addresses several critical challenges. It minimizes human-induced variability, enhances reproducibility, and increases the efficiency of research workflows [34]. The R.E.D. Tools (RESP and ESP charge Derive) were developed specifically to overcome these barriers by performing charge derivation in an automatic and straightforward way [3]. By implementing rigorous control over molecular orientation and conformer selection, R.E.D. Tools ensure that charge values can be reproduced across different computational platforms with an exceptional accuracy of 0.0001 e, establishing a new standard of reliability in force field parameterization [3].
Development History and Version Capabilities
The R.E.D. Tools have undergone significant evolution since their initial release, with each version introducing enhanced capabilities for charge derivation and force field development:
R.E.D. version I (Released December 2003) introduced the foundational automation framework, enabling highly reproducible RESP and ESP charge values (± 0.0001 e) through control of molecular orientation during optimization [35]. This version established the multi-orientation RESP or ESP fit procedure to limit charge uncertainty associated with single-orientation approaches.
R.E.D. version II expanded the toolkit's capabilities by implementing multiple conformations of a single molecule in the fitting procedure [12]. This advancement enabled the derivation of more general charge values suitable for molecular dynamics simulations where extensive conformational space must be sampled. The combination of multi-conformation and multi-orientation approaches significantly enhanced the transferability of derived parameters.
R.E.D. version III.x (Initial release April 2007, with updates through III.5 in February 2012) introduced sophisticated control of charge constraints for atoms and groups of atoms within and between molecules [36]. This critical development enabled the derivation of RESP and ESP atomic charges for molecular fragments and sets of molecules, facilitating force field development for complex biomolecular systems [12].
The latest iteration, R.E.D. version IV, continues this trajectory of innovation, further refining the tools' capabilities for handling increasingly complex molecular systems [12]. This evolutionary progression has transformed R.E.D. from a specialized utility into a comprehensive platform for force field development, serving diverse research communities in computational chemistry and biology.
Key Technological Features
R.E.D. Tools incorporate several innovative features that address longstanding challenges in charge parameterization. The implementation of multiple molecular orientations during the fitting process effectively eliminates the dependence of derived charges on initial coordinate selection, ensuring consistent results regardless of molecular positioning in the computational space [3]. The multi-conformation fitting capability allows researchers to derive charges that represent an average over relevant conformational states, producing more transferable parameters for molecular dynamics simulations [12].
For complex biomolecular systems, the constraint handling system enables charge derivation for molecular fragments with strict conservation of charge groups, which is essential for maintaining appropriate charge distribution in partitioned molecules [36]. Furthermore, R.E.D. supports eight different charge derivation procedures utilizing various MEP computation algorithms (Connolly surface and CHELPG) and fitting approaches (with or without hyperbolic restraints), providing exceptional flexibility for method development and comparison [12].
Table 1: Evolution of R.E.D. Tools Capabilities
| Version | Release Timeline | Key Innovations | Compatible Force Fields |
|---|---|---|---|
| R.E.D. I | December 2003 [35] | Multi-orientation fitting, reproducibility ±0.0001 e | AMBER, CHARMM, GLYCAM, OPLS |
| R.E.D. II | Not specified | Multi-conformation fitting, elements up to Z=35 | AMBER, CHARMM, GLYCAM, OPLS |
| R.E.D. III.x | April 2007 - February 2012 [36] | Charge constraints for molecular fragments, multiple molecule fitting | AMBER, CHARMM, GLYCAM, OPLS |
| R.E.D. IV | Under development [12] | Enhanced fragment handling, expanded database | AMBER, CHARMM, GLYCAM, OPLS |
Theoretical Framework and Methodological Advances
Fundamental Principles of RESP and ESP Charges
Electrostatic potential-derived atomic charges are founded on the principle of reproducing the molecular electrostatic potential (MEP) outside the van der Waals surface through a set of atom-centered point charges [3]. The MEP represents a directly computable quantum mechanical property derived from self-consistent field (SCF) calculations, providing a physically meaningful target for charge parameterization [3]. ESP charges are determined by fitting to the MEP calculated at a large number of points defined on three-dimensional surfaces around the molecule, with different force fields employing variations in surface generation algorithms—Amber force fields typically use the Connolly surface, while GLYCAM employs the CHELPG algorithm [3].
The RESP (Restrained Electrostatic Potential) approach introduces hyperbolic restraints during the fitting process to mitigate issues associated with poorly defined buried atoms that lack sufficient MEP sampling [3]. These restraints effectively control large charge fluctuations without significantly impacting the quality of the electrostatic potential reproduction, addressing a key limitation of conventional ESP fitting [3]. The combination of multiple conformations in charge derivation with the RESP model has produced charge values widely used in Amber force field development, particularly valued for condensed phase molecular dynamics simulations [3].
Basis Set Selection and Theory Level Considerations
The selection of appropriate quantum mechanical theory levels represents a critical consideration in RESP charge derivation. Research has demonstrated that ESP charge values exhibit significant fluctuations with lower-level basis sets but tend to converge after reaching the 6-31G* basis set [3]. The Hartree-Fock method with the 6-31G* basis set has become a standard for condensed phase simulations in force fields like Cornell et al., as it yields dipole moments approximately ten percent larger than experimental gas-phase values—an effect that implicitly accounts for solute polarization in aqueous solution within additive force field models [3].
For gas-phase calculations or polarizable force fields, alternative approaches have been employed, including B3LYP exchange-correlation functionals with correlation-consistent basis sets (cc-pVTZ, cc-pVDZ, aug-cc-pVDZ) [3]. The R.E.D. Tools support these various theory levels, providing researchers with flexibility in selecting appropriate computational methods for their specific applications, whether employing the widely used Gaussian software or academic alternatives such as GAMESS-US, PC-GAMESS/Firefly, and NWChem [3].
R.E.D. Tools Workflow: Application Notes and Protocols
System Preparation with Ante_R.E.D.
The charge derivation process begins with careful system preparation using Ante_R.E.D., a specialized preprocessing tool that generates the necessary input files for R.E.D. execution [12]. The critical preparation steps include:
Molecular Structure Input: Researchers begin with an initial molecular structure, typically in PDB format. Ante_R.E.D. facilitates the creation of "P2N" files—an enhanced PDB format that incorporates a second column of atom names, which is essential for rigorous definition of atom identities and connectivity [12].
Topology and Chemical Equivalencing: The P2N file format rigorously defines key elements including atom names, molecular topology, and chemical equivalencing relationships necessary for building consistent force field libraries [12]. This step is particularly crucial for molecular fragments where charge groups must be properly defined and conserved.
Conformational Sampling: For multi-conformation fitting, researchers must prepare an ensemble of representative conformations that sample relevant low-energy states and functional geometries. This ensemble approach ensures derived charges provide adequate coverage of the conformational space explored during molecular dynamics simulations [3].
Proper system preparation establishes the foundation for successful charge derivation, ensuring that molecular identity is preserved throughout the automated workflow and that appropriate constraints are applied to maintain chemical rationality in the resulting parameters.
Quantum Chemical Calculations
The R.E.D. Tools automate the execution of quantum chemical software for geometry optimization and molecular electrostatic potential calculation [12]. The recommended protocol involves:
Geometry Optimization: Each molecular structure (or conformation) undergoes rigorous geometry optimization at an appropriate theory level, typically HF/6-31G* for compatibility with established biomolecular force fields [3]. The optimization process seeks stable minima on the potential energy surface that represent physically meaningful molecular structures.
Molecular Orientation Control: A key innovation of R.E.D. Tools is the implementation of multiple molecular orientations for each optimized structure [3]. By automatically generating and processing multiple orientations, R.E.D. eliminates the coordinate dependence that has historically plagued electrostatic potential fitting procedures.
MEP Computation: Following optimization, the molecular electrostatic potential is computed on a three-dimensional grid surrounding the molecule using either the Connolly surface or CHELPG algorithm [12]. The MEP computation employs the same theory level as the geometry optimization to ensure consistency between electronic structure and property calculation.
This stage seamlessly interfaces with popular quantum chemical packages including Gaussian, GAMESS-US, and Firefly, providing researchers with flexibility in software selection while maintaining consistent output formatting for subsequent charge fitting [12].
Charge Fitting and Force Field Library Generation
The final stage involves charge fitting and incorporation of parameters into force field libraries:
RESP Fitting: The RESP program performs least-squares fitting of atom-centered charges to reproduce the computed molecular electrostatic potential [3]. For multiple orientations and conformations, R.E.D. performs simultaneous fitting to all data points, yielding averaged charges that provide robust performance across conformational space.
Charge Constraint Application: During fitting, intra- and inter-molecular charge constraints can be applied to preserve charge groups, ensure proper fragment neutrality, and maintain chemical transferability between related molecular systems [36]. This capability is particularly valuable for developing consistent parameters across homologous series of molecules.
Library Generation: Following charge derivation, R.E.D. incorporates the parameters into force field libraries in Tripos mol2 format, which serve as precursors to AMBER OFF and CHARMM RFT or PSF libraries [3]. For complex molecular families, R.E.D. can generate entire force field topology databases (FFTopDB) ensuring internal consistency across related molecular entities.
Table 2: Supported Software and Computational Methods in R.E.D. Tools
| Component | Supported Options | Key Specifications |
|---|---|---|
| Quantum Chemistry Software | Gaussian, GAMESS-US, Firefly, NWChem [12] | Version compatibility: Gaussian 1994-2009, GAMESS-US, WinGAMESS [12] |
| Theory Levels | HF, DFT (B3LYP) [3] | Common basis sets: 6-31G*, cc-pVTZ, cc-pVDZ, aug-cc-pVDZ [3] |
| MEP Algorithms | Connolly Surface, CHELPG [12] | Surface generation: Connolly (Amber), CHELPG (GLYCAM) [3] |
| Fitting Approaches | Standard ESP, Restrained ESP (RESP) [12] | Hyperbolic restraints for buried atoms [3] |
| Chemical Elements | Up to bromine (Z=35) [12] | Periodic table coverage for biological molecules [12] |
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of R.E.D. Tools requires careful selection and configuration of computational resources and software components. The following table details essential "research reagent solutions" for automated RESP charge derivation:
Table 3: Essential Research Reagents for R.E.D.-Based Charge Derivation
| Reagent Solution | Function | Implementation Notes |
|---|---|---|
| R.E.D. Tools Software Suite | Core automation platform for charge derivation | Perl-based main program with Tcl/Tk GUI (X R.E.D.) [12] |
| Ante_R.E.D. Module | Input preparation and P2N file generation | Critical for defining atom names, topology, and chemical equivalencing [12] |
| Quantum Chemistry Software (Gaussian, GAMESS-US) | Geometry optimization and MEP computation | Gaussian requires license; GAMESS-US is academic alternative [12] |
| RESP Program | Charge fitting to molecular electrostatic potential | Implements restrained fitting with hyperbolic constraints [3] |
| Tripos mol2 File Format | Force field library precursor | Convertible to AMBER OFF and CHARMM RFT/PSF formats [3] |
| Molecular Structure Database (R.E.DD.B.) | Reference charges and validation data | Contains >50 model systems with derived charges [3] |
| Perl and Tcl/Tk Interpreters | Execution environment for R.E.D. Tools | Cross-platform compatibility (UNIX, Mac OS X, Windows) [12] |
Advanced Applications and Case Studies
Biomolecular Force Field Development
The R.E.D. Tools have been extensively applied in developing parameters for biomolecular simulations, demonstrating their versatility across diverse molecular systems:
Nucleic Acid Parameters: Projects F-45 and F-51 have generated RESP atomic charges based on the Connolly surface algorithm and HF/6-31G* theory level for 16 components of force field topology databases for modeling regular DNA and RNA, complete with conversion scripts for AMBER OFF files [36]. These projects highlight the capability of R.E.D. for handling complex biomolecular building blocks with multiple interchangeable fragments.
Amino Acid Fragments: For the dimethylalanine amino acid system, Project F-3 and related initiatives have derived RESP charges for central, terminal, and modified fragments using the Connolly surface algorithm at the HF/6-31G* theory level [36]. This case exemplifies the application of charge constraints in defining appropriate molecular partitions while maintaining chemical consistency across fragments.
Solvent Parameterization: Projects W-46 through W-49 have established RESP and ESP atomic charges for 10 solvent molecules using both Connolly surface and CHELPG algorithms at the HF/6-31G* theory level [36]. These implementations demonstrate the flexibility of R.E.D. in accommodating different charge derivation philosophies and algorithm preferences across research groups.
Integration with Drug Discovery Workflows
The automation provided by R.E.D. Tools aligns with broader trends in pharmaceutical research, where automated molecular design approaches are increasingly employed to accelerate drug development [37]. While traditional lead optimization relies heavily on manual modification of molecular structures by medicinal chemists, computational approaches can generate novel chemical entities with optimized properties more efficiently [37]. The machine-learned molecular graph-based model described by MIT researchers exemplifies this trend, generating chemically valid molecules with improved potency—a complementary approach to the force field parameterization enabled by R.E.D. Tools [37].
The high reproducibility of R.E.D.-derived charges (±0.0001 e) directly addresses reproducibility concerns that have plagued pharmaceutical and biomedical research [34]. By minimizing human-induced variability in charge parameterization, R.E.D. supports more robust and transferable force fields, facilitating reliable molecular dynamics simulations of drug-target interactions [3] [34].
Visual Guide to the R.E.D. Tools Workflow
The following diagram illustrates the comprehensive workflow for RESP charge derivation using the R.E.D. Tools, highlighting the integration of preparation, quantum calculation, and charge fitting stages:
R.E.D. Tools Automated Workflow for RESP Charge Derivation
This workflow visualization captures the staged automation implemented in R.E.D. Tools, demonstrating how molecular structures progress through preparation, quantum chemical computation, and parameterization stages to produce validated force field libraries. The color-coding differentiates between input preparation (green), quantum calculation (red), and charge fitting/force field generation (blue) phases of the protocol.
The R.E.D. Tools represent a significant advancement in automating the critically important but traditionally labor-intensive process of RESP charge derivation and force field library construction. By addressing key challenges in reproducibility, efficiency, and transferability, these tools have established themselves as essential resources for researchers engaged in molecular dynamics simulations and force field development [3]. The implementation of multi-orientation, multi-conformation, and multi-molecule fitting approaches has systematically addressed longstanding limitations in electrostatic potential-derived charges, producing parameters with enhanced reliability and broader applicability [3] [12].
Looking forward, the growing emphasis on automation in life science research suggests an expanding role for tools like R.E.D. in computational biochemistry and drug discovery [34] [37]. As molecular simulations address increasingly complex biological systems and processes, the demand for robust, automated parameterization methods will continue to grow. The R.E.D. Tools development trajectory—with its emphasis on reproducibility, flexibility, and comprehensive force field library generation—positions this platform as a continuing contributor to advances in computational molecular science [3] [12].
The restrained electrostatic potential (RESP) approach is a highly regarded method for deriving atomic partial charges, which are fundamental components of modern empirical force fields used in molecular dynamics (MD) simulations. These force fields, including AMBER, CHARMM, and OPLS-AA, enable researchers to study biological processes at the atomistic level, supporting applications in drug design, protein folding, and macromolecular interactions [38] [2]. Atomic partial charges primarily govern electrostatic interactions within these force fields, critically influencing the accuracy of simulated structural features, dynamic motions, and thermodynamic properties [1].
The RESP method originated as an improvement over unrestrained electrostatic potential (ESP) charge fitting. While standard ESP charges derived from quantum mechanical calculations tend to overestimate bond polarities in gas-phase computations, RESP incorporates a hyperbolic restraint function that attenuates charge magnitudes without significantly affecting molecular dipole moments [1]. This approach addresses the known over-polarization issue while maintaining an integral net charge on ions, making it particularly valuable for simulating biological molecules in condensed phases [1] [2]. The derivation of RESP charges begins with quantum mechanical calculation of the molecular electrostatic potential, followed by fitting atomic partial charges to this potential while applying restraints that control charge magnitudes [1].
Table 1: Key Force Fields and Their Charge Derivation Approaches
| Force Field | Primary Charge Derivation Method | Targeted Applications |
|---|---|---|
| AMBER | RESP charges fitted to HF/6-31G* ESP | Proteins, nucleic acids, carbohydrates |
| CHARMM | Bond-charge increment (BCI) rules | Diverse biomolecules and drug-like compounds |
| OPLS-AA | ESP charges with empirical adjustment | Liquid properties and thermodynamic observables |
| GLYCAM | RESP with specific restraint weights | Carbohydrates and glycoproteins |
Theoretical Foundations and Evolution of RESP Methodology
Fundamental RESP Equation
The RESP approach modifies the standard ESP charge fitting by incorporating a restraint term into the objective function. The fundamental equation is expressed as:
[ \chi{\text{RESP}}^2 = \chi{\text{ESP}}^2 + \chi_{\text{restraint}}^2 ]
where (\chi{\text{ESP}}^2) represents the standard sum of squared differences between the quantum mechanical electrostatic potential and the classical model, and (\chi{\text{restraint}}^2) is the hyperbolic restraint function defined as:
[ \chi{\text{restraint}}^2 = k{\text{restraint}} \sumj \left( \sqrt{qj^2 + b^2} - b \right) ]
In this equation, (b) determines the tightness of the hyperbola around its minimum, while (k_{\text{restraint}}) represents the restraint weight that controls the strength of the restraint function [1]. The restraint weight typically ranges from 0.001 to 0.01 for polar molecules, with studies on carbohydrates suggesting that a value of 0.01 provides optimal agreement with experimental neutron diffraction structures [1].
Advanced RESP2 Development
Recent advancements have led to RESP2, a next-generation approach that addresses the limitations of traditional RESP (now often called RESP1). While RESP1 relies on Hartree-Fock calculations with the 6-31G* basis set that fortuitously overpolarizes molecules approximately correctly for hydrated environments, this overpolarization is inconsistent across different molecular systems [2]. The RESP2 method introduces a parameter δ that scales the contributions from gas- and aqueous-phase calculations:
[ \text{RESP2 charge} = \delta \times \text{aqueous charge} + (1 - \delta) \times \text{gas-phase charge} ]
Extensive validation suggests that RESP2 with δ ≈ 0.6 (60% aqueous, 40% gas-phase) provides an accurate and robust method for generating partial charges when co-optimized with Lennard-Jones parameters [2]. This approach decouples charge derivation from the arbitrary overpolarization pattern of HF/6-31G* calculations, instead utilizing more accurate quantum mechanical methods such as PW6B95/aug-cc-pV(D+d)Z while explicitly accounting for solvation effects [2].
Figure 1: RESP2 Charge Derivation Workflow. This diagram illustrates the integrated process for deriving RESP2 charges, combining both gas-phase and aqueous-phase quantum mechanical calculations with an optimal mixing parameter of δ = 0.6.
Research Reagent Solutions for Force Field Development
Table 2: Essential Tools and Resources for RESP Charge Derivation and Force Field Implementation
| Tool/Resource | Function | Availability |
|---|---|---|
| R.E.DD.B. (RESP ESP charge DataBase) | Database of RESP/ESP charges and force field libraries | http://q4md-forcefieldtools.org/REDDB [39] |
| R.E.D. Tools | Software tools for RESP/ESP charge derivation and force field library building | Academic distribution [40] |
| Gaussian 94/09 | Quantum mechanical software for electrostatic potential calculation | Commercial license [1] |
| ForceBalance | Systematic parameter optimization against experimental data | Open source [2] |
| Antechamber | Automated parameterization tool for AMBER force field | Part of AMBER tools [41] |
| MATCH | Automated parameterization for CHARMM force field | Academic distribution [41] |
Protocol for RESP Charge Derivation for Novel Molecules
Quantum Mechanical Calculations
The initial step in RESP charge derivation involves quantum mechanical computation of molecular electrostatic potentials. The traditional approach utilizes Hartree-Fock calculations with the 6-31G* basis set, though RESP2 recommends more advanced methods:
- Method Selection: Employ PW6B95 functional with aug-cc-pV(D+d)Z basis set for improved accuracy in dipole moments and electrostatic potentials [2].
- Solvation Treatment: Perform calculations in both gas phase and aqueous phase using implicit solvation models such as IEF-PCM or SMD [2].
- Grid Generation: Use the CHELPG algorithm to generate points around the molecular van der Waals surface for electrostatic potential evaluation [1]. For α-D-glucopyranose, this typically involves 13,670 grid points [1].
RESP Fitting Procedure
The actual charge fitting involves optimizing atomic partial charges to reproduce the quantum mechanical electrostatic potential while applying magnitude restraints:
- Single-Stage vs. Two-Stage Fitting: For molecules with molecular or rotational symmetry, use two-stage fitting to adjust hydrogen charges. For asymmetric molecules like carbohydrates, single-stage fitting is appropriate [1].
- Restraint Parameters: Set the hyperbolic restraint parameter (b = 0.1) atomic units and restraint weight (k_{\text{restraint}} = 0.01) for carbohydrates, or 0.001 for more flexible molecules [1].
- RESP2 Implementation: For RESP2, combine gas-phase and aqueous-phase charges with δ = 0.6 using the linear combination formula [2].
Validation Methods
Validation of derived charges is essential before incorporation into force field libraries:
- Crystal Simulations: Perform MD simulations of crystal structures monitoring unit cell dimensions and hydrogen bonding patterns. For α-D-glucopyranose, simulations of 64 molecules in a 2×2×4 unit cell arrangement for 50 ps (with 30 ps analysis) have proven effective [1].
- Liquid Properties: Compute densities, heats of vaporization, and dielectric constants of pure organic liquids comparing to experimental values [2].
- Binding Affinities: For drug design applications, validate with relative binding free energy calculations using thermodynamic integration or free energy perturbation methods [41].
Force Field-Specific Integration Strategies
AMBER Force Field Integration
The AMBER force field employs RESP charges as its primary electrostatic model, with minimal empirical adjustments:
- Philosophy: Belief that accurate RESP charges reduce the need for extensive torsional parameter adjustments [38].
- Implementation: Use HF/6-31G* electrostatic potentials with two-stage RESP fitting and restraint weights of 0.0005 for non-hydrogen atoms and 0.001 for hydrogens in initial fitting [38].
- Validation Focus: Reproduction of conformational energies and hydrogen-bonded complex geometries [38].
CHARMM Force Field Integration
CHARMM employs a different strategy for partial charge assignment but can incorporate RESP-derived charges:
- Bond-Charge Increments: CHARMM typically uses bond-charge increment (BCI) rules where charges are transferred from model compounds to novel molecules based on chemical environment matching [41].
- RESP Integration Strategy: When BCIs are unavailable, derive RESP charges for molecular fragments using model compounds, then assemble complete molecules using transferable principles [41].
- Validation: Focus on reproduction of hydration free energies and liquid properties, with binding affinity calculations for drug-like compounds [41].
OPLS-AA Force Field Integration
OPLS-AA employs a philosophy balancing accurate electrostatic representation with experimental thermodynamic data:
- Charge Derivation: Originally based on ESP charges with empirical adjustments to reproduce liquid properties [38].
- RESP Compatibility: Can incorporate RESP charges with subsequent scaling or adjustment to maintain accuracy for thermodynamic observables [41].
- Validation Emphasis: Heats of vaporization, liquid densities, and free energies of solvation [38].
Table 3: Performance Comparison of Charge Methods Across Force Fields
| Force Field | Charge Method | Hydration Free Energy MUE (kcal/mol) | Binding Affinity MUE (kcal/mol) | Key Applications |
|---|---|---|---|---|
| AMBER/GAFF | RESP (HF/6-31G*) | ~1.0 | ~1.0 | Protein-ligand binding [41] |
| AMBER/GLYCAM | RESP (k=0.01) | N/A | N/A | Carbohydrate crystals [1] |
| CHARMM/CGenFF | BCI Rules | ~1.0 | 1.3 (TIBO compounds) | Drug design [41] |
| OPLS-AA | CM1A (scaled) | ~1.0 | 0.9-1.2 | TIBO/HIV-RT inhibitors [41] |
Application Notes and Practical Considerations
System-Specific Recommendations
Different chemical systems require tailored approaches to RESP charge derivation:
- Carbohydrates: Use single-stage RESP fitting with restraint weight of 0.01, validated against crystal structures [1].
- Protein Ligands: Implement RESP2 with δ = 0.6 for improved accuracy in binding free energy calculations [2].
- Small Organic Molecules: For rapid parameterization, consider AM1-BCC method which approximates RESP charges at lower computational cost [2].
Workflow Integration
Efficient integration of RESP charge derivation into force field development pipelines:
- Automation Tools: Utilize R.E.D. tools for automated RESP charge derivation and force field library building for multiple molecular dynamics packages [40].
- Database Utilization: Consult R.E.DD.B. database for pre-parameterized molecules and molecular fragments to ensure reproducibility [39] [42] [43].
- Parameter Optimization: Employ ForceBalance or similar tools for systematic optimization of partial charges and Lennard-Jones parameters against experimental data [2].
Figure 2: Force Field Integration Pathways. This diagram illustrates the distinct integration strategies for RESP charges across major force fields, with their respective validation priorities.
The successful integration of RESP charges into biomolecular force fields requires careful attention to both the theoretical foundations and practical implementation details specific to each force field's philosophy. The development of RESP2 represents a significant advancement by systematically balancing gas-phase and aqueous-phase electrostatics, addressing the arbitrary nature of traditional RESP's reliance on HF/6-31G* overpolarization. As force field development continues, several emerging trends warrant attention:
Future developments will likely focus on incorporating electronic polarization explicitly through methods like the AMOEBA force field, which includes atomic multipoles and polarization effects [38]. Additionally, geometry-dependent charge flux models that account for how atomic charges change with molecular geometry show promise for more accurate electrostatic representation [38]. The expansion of automated parameterization tools and comprehensive databases like R.E.DD.B. will further streamline the process of force field library building for novel molecules [39] [42] [43].
For researchers implementing these protocols, the key recommendations include: utilizing the RESP2 method with δ = 0.6 for new charge derivation projects, validating charges against multiple experimental observables relevant to the intended application, and leveraging existing database resources to ensure reproducibility and compatibility across force fields. Following these structured protocols will enhance the accuracy and reliability of molecular simulations across diverse chemical and biological systems.
Optimizing and Troubleshooting RESP Charges for Robust Performance
The derivation of RESP (Restrained ElectroStatic Potential) atomic charges is a foundational step in developing molecular mechanics force fields for computational chemistry and drug design. These charges are critical for accurately modeling intermolecular interactions, such as those between a drug candidate and its protein target. The selection of the quantum mechanical (QM) method—specifically the electronic structure theory and basis set—profoundly influences the quality and transferability of the resulting charges. This application note provides detailed protocols for selecting basis sets and density functionals for RESP charge derivation, framed within a research project on novel molecules. We focus on three prominent choices: HF/6-31G*, *cc-pVTZ, and the functional PW6B95, elucidating their specific roles and rationales within different research contexts.
Theoretical Background and Key Concepts
RESP Charge Derivation
Atomic charges are not quantum mechanical observables but are fitted parameters designed to reproduce the molecular electrostatic potential (MEP) outside the van der Waals surface. The RESP model fits atomic charges to the QM-derived MEP while applying hyperbolic restraints to mitigate overfitting and reduce the occurrence of large, unrealistic charge values on buried atoms. This approach is particularly well-suited for condensed-phase molecular dynamics simulations, as implemented in major force fields like AMBER. The procedure involves three key steps: (1) geometry optimization of the molecule of interest, (2) computation of the MEP around the optimized geometry, and (3) fitting of atomic charges to the computed MEP under restraint [3].
The Role of Basis Sets and Functionals
The accuracy of the MEP, and consequently the derived RESP charges, is highly dependent on the QM method used.
- Basis Sets: A basis set is a set of mathematical functions that describes the molecular orbitals. Its size and quality determine how accurately the electron density, and hence the MEP, can be represented.
- Density Functionals: In Density Functional Theory (DFT), the functional approximates the exchange-correlation energy. Different functionals offer varying balances of accuracy, cost, and applicability for different chemical systems.
Recommended Methods: Rationale and Applications
The choice of method is not one-size-fits-all; it depends on the target force field and the chemical nature of the system under study. The following table summarizes the primary recommended approaches.
Table 1: Summary of Recommended QM Methods for RESP Charge Derivation
| QM Method | Primary Use Case | Key Rationale | Performance & Notes |
|---|---|---|---|
| HF/6-31G* | Standard RESP for additive force fields (e.g., AMBER) | Produces an ~10% larger dipole moment, implicitly accounting for aqueous polarization in additive models [3] [44]. | Considered a best compromise of speed and accuracy [33]. The default for tools like Antechamber and R.E.D. |
| B3LYP/cc-pVTZ | RESP charges for use with specific force fields (e.g., ff03) | Uses an implicit solvation model (SCRF) during MEP computation to mimic a protein environment [44]. | Higher level of theory; requires consistency with the intended force field's parametrization strategy. |
| PW6B95-D3 | Systems involving transition metals or challenging bond activations | Benchmark studies identify it as a top-performing hybrid functional for reaction energies and barriers, including for Pd and Ni catalysts [45]. | Especially useful for organometallic drug candidates. A robust choice when DFT is required. |
Hartree-Fock with the 6-31G* Basis Set
The HF/6-31G* level of theory is the historical and most common standard for deriving RESP charges in force fields like AMBER. Its primary justification is the overestimation of the gas-phase dipole moment by approximately 10%. This systematic error serendipitously mimics the polarization of a solute molecule in an aqueous environment, providing an implicit solvation effect within the additive force field paradigm [3] [44]. This method is strongly recommended for researchers deriving charges for compatibility with standard AMBER, CHARMM, or GLYCAM force fields.
Correlation-Consistent Basis Sets: cc-pVTZ
The cc-pVTZ (correlation-consistent polarized Valence Triple-Zeta) basis set is part of a systematically improvable series designed for high-accuracy quantum chemistry. It offers a more complete and balanced description of the electron density compared to Pople-style basis sets like 6-31G* [33]. While it may be overkill for routine HF-based RESP calculations, it is the preferred basis for post-Hartree-Fock methods like coupled-cluster theory (CCSD(T)) [46]. Its use is indicated in scenarios demanding high-accuracy gas-phase charges or when following specific force field parametrization protocols that employ higher-level theories, such as the B3LYP/cc-pVTZ approach used in the development of the ff03 force field [44].
Hybrid Meta-GGA Functional: PW6B95
PW6B95 is a hybrid meta-GGA functional that incorporates a portion of exact Hartree-Fock exchange. When combined with an empirical dispersion correction (e.g., -D3), it has demonstrated top-tier performance in benchmark studies for calculating activation energies and reaction energies, particularly for reactions involving transition metals like Pd and Ni [45]. While not traditionally used for standard RESP derivation, PW6B95-D3 is an excellent functional choice for the initial geometry optimization of challenging systems, such as organometallic complexes or drug candidates containing transition metals, where its accuracy for structures and energetics is superior. It is also a suitable functional for the MEP computation step when deriving charges for a specialized, in-house force field targeting such systems.
Experimental Protocols
This section provides detailed, step-by-step workflows for deriving RESP charges using the R.E.D. Tools, which automate the process and ensure high reproducibility [3].
Standard Protocol for Organic Molecules (HF/6-31G*)
This protocol is designed for deriving RESP charges for standard organic molecules, such as drug-like ligands, for use with mainstream additive force fields.
Diagram 1: RESP derivation workflow for organic molecules
Step-by-Step Procedure:
- Molecule Preparation: Obtain the initial 3D structure of the molecule. Ensure correct protonation states at biological pH and remove any crystallographic artifacts. The structure should be in a recognized format like PDB or Mol2.
- Geometry Optimization: Perform a geometry optimization using the HF/6-31G* level of theory. This can be done with quantum chemistry packages like Gaussian, GAMESS, or NWChem. The R.E.D. Tools can interface with these programs to automate this step [3].
- Frequency Calculation: Run a frequency calculation on the optimized geometry to confirm that a true minimum has been found (no imaginary frequencies).
- MEP Calculation: Using the optimized geometry, compute the molecular electrostatic potential at the HF/6-31G* level. It is critical to use the same level of theory for both optimization and MEP calculation in this protocol. The R.E.D. Tools employ a multiple orientation strategy for this step to guarantee charge reproducibility across different computational platforms [3].
- RESP Fitting: Feed the computed MEP into the RESP program. For molecules with flexible dihedrals, it is good practice to perform a multi-conformational fit. The R.E.D. Tools automate this process, applying the standard hyperbolic restraints.
- Validation and Library Generation: Analyze the fitted charges for chemical reasonableness. Finally, use R.E.D. Tools to incorporate the charges into a force field library file (Tripos mol2 format) ready for use in MD simulation packages.
Advanced Protocol for Transition Metal Complexes
For systems containing transition metals (e.g., Cu(II), Pd, Ni), the electronic structure is more complex, often requiring higher-level methods for geometry optimization.
Diagram 2: RESP derivation workflow for transition metal complexes
Step-by-Step Procedure:
- Molecule Preparation: Construct the initial model, paying careful attention to the coordination geometry around the transition metal ion based on crystallographic data or chemical knowledge.
- Geometry Optimization: Optimize the geometry using a robust functional like PW6B95-D3 and a suitable basis set such as def2-TZVP. The def2 series are modern basis sets available for the entire periodic table and are a good choice for systems containing transition metals [46]. This step ensures an accurate geometric and electronic structure.
- Frequency Calculation: Confirm the optimized structure is a minimum on the potential energy surface via frequency analysis.
- MEP Calculation: The choice of method for the MEP calculation depends on the intended force field.
- For compatibility with standard force fields, use the HF/6-31G* level on the DFT-optimized geometry.
- For a more accurate gas-phase charge model or a specialized force field, the MEP can be computed at a higher level, such as B3LYP/cc-pVTZ.
- RESP Fitting: Perform the RESP fit. Due to the distinct electronic environment of the metal center, consider defining charge constraints for the metal and its directly coordinated atoms to ensure charge transfer is chemically reasonable. The multiple-molecule fitting capability of R.E.D. Tools is useful for fragments or homologous series [3].
- Validation and Library Generation: Critically assess the derived charges, particularly the charge on the metal center and the overall charge distribution on the organic ligand. Incorporate the final charges into a force field library.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Computational Tools for RESP Charge Derivation
| Tool Name | Type | Primary Function | URL / Reference |
|---|---|---|---|
| R.E.D. Tools | Software Server | Automates the multi-step process of RESP/ESP charge derivation and force field library generation. | http://q4md-forcefieldtools.org/RED/ [3] |
| RESP ESP charge DDataBase (R.E.DD.B.) | Database | Repository of pre-computed atomic charges and force field libraries for over 50 molecular systems. | [3] |
| Gaussian | Quantum Chemistry Software | Proprietary software widely used for geometry optimization and MEP computation. | [3] |
| GAMESS-US / NWChem | Quantum Chemistry Software | Free, academic quantum chemistry programs that can be interfaced with R.E.D. Tools. | [3] |
| Antechamber | Software Tool | Part of AMBERTools; assists in parametrizing organic molecules, including RESP charge derivation. | [3] |
The selection of a QM method for RESP charge derivation is a critical decision that directly impacts the accuracy of subsequent molecular simulations. For researchers working within established biomolecular force fields like AMBER, the HF/6-31G* protocol remains the benchmark, providing an implicit account of solvation effects. The cc-pVTZ basis set is the cornerstone for high-accuracy, gas-phase quantum chemistry, especially when paired with post-HF methods or specific DFT functionals like B3LYP. For challenging systems containing transition metals, the hybrid functional PW6B95-D3 offers superior performance for geometry optimization, ensuring a structurally sound foundation for subsequent charge derivation. By following the detailed protocols outlined in this document, scientists and drug development professionals can make informed, rational choices when deriving RESP charges for novel molecules, thereby enhancing the reliability of their computational research.
Managing Molecular Orientation and Conformation Dependence
In the realm of computational drug discovery, accurately predicting molecular behavior is paramount for successful outcomes in virtual screening and lead optimization. Molecular conformation and electrostatic characterization represent two fundamental pillars of this predictive capability [47]. The three-dimensional arrangement of atoms (conformation) and the distribution of partial atomic charges collectively determine how a molecule interacts with its biological target [48]. Managing these properties is particularly crucial for challenging drug discovery scenarios involving highly flexible molecules and novel chemical entities where experimental data may be limited [49]. This Application Note details integrated protocols for conformer generation using enhanced sampling methods and charge derivation using the advanced RESP2 approach, providing researchers with a robust framework for handling molecular orientation and conformation dependence within computational workflows.
Theoretical Background
The Critical Role of Conformation and Electrostatics in Drug Discovery
Molecular conformation directly influences binding affinity, specificity, and ultimately, therapeutic efficacy [50]. Small molecules exist as ensembles of interconverting conformers in solution, and the specific conformation that binds to a biological target may not correspond to the global energy minimum [49]. Similarly, partial atomic charges derived from electrostatic potential (ESP) calculations are essential for modeling key intermolecular interactions, including hydrogen bonding, ionic interactions, and polarization effects [2]. The restrained electrostatic potential (RESP) method has emerged as a gold standard for charge derivation in molecular simulations, though its computational demands have historically limited application to large-scale systems [6].
Table 1: Key Challenges in Molecular Representation for Drug Discovery
| Challenge | Impact on Drug Discovery | Traditional Limitations |
|---|---|---|
| Conformational Flexibility | Inaccurate binding pose prediction, poor activity prediction | Incomplete sampling of conformational space, especially for macrocycles and flexible linkers [49] |
| Electrostatic Representation | Errors in binding affinity prediction, unreliable interaction modeling | Fortuitous error cancellation in RESP charges, inadequate polarization treatment [2] |
| Computational Efficiency | Limited scope of virtual screening, restricted chemical space exploration | High cost of RESP calculations for large systems [6] |
| Integration Challenges | Disconnect between different computational methods | Lack of unified workflows combining conformation sampling and charge parameterization |
RESP Charge Evolution: From RESP to RESP2
The original RESP methodology (hereafter RESP1) calculates partial atomic charges by fitting to the quantum-mechanically derived electrostatic potential around a molecule using Hartree-Fock/6-31G* calculations, which fortuitously overpolarize to approximate hydration effects [2]. While widely successful, this approach suffers from inconsistent polarization treatment across diverse molecular structures. The RESP2 method addresses this limitation by computing charges as a weighted combination of gas-phase and aqueous-phase ESPs, scaled by a parameter δ (typically 0.6), thereby providing a more physically realistic account of solvation effects [2]. Recent advances like DF-RESP further accelerate these calculations through density fitting techniques, enabling application to larger systems including protein-ligand complexes [6].
Experimental Protocols
Protocol 1: Enhanced Conformer Generation with Moltiverse
Principle: Explore conformational space efficiently using enhanced sampling molecular dynamics to capture diverse bound-state conformations, particularly crucial for flexible drug-like molecules and macrocycles [49].
Materials and Software:
- Moltiverse package (novel protocol for conformer generation)
- Reference datasets for validation (Platinum Diverse Data set for drug-like molecules, Prime data set for macrocycles)
- Comparative software for benchmarking (RDKit, CONFORGE, Balloon, iCon, Conformator)
Procedure:
- System Preparation: Initialize molecular structure from standardized format (SMILES, SDF) with proper atom typing and bond orders.
- Collective Variable Selection: Define the radius of gyration (RDGYR) as the primary collective variable to drive conformational sampling.
- Enhanced Sampling Setup: Configure the extended adaptive biasing force (eABF) algorithm combined with metadynamics.
- Sampling Execution: Run molecular dynamics simulations with enhanced sampling to efficiently explore conformational landscape.
- Cluster Analysis: Process trajectory to identify unique conformational clusters using RMSD-based clustering algorithms.
- Representative Selection: Extract lowest-energy representative structures from each major cluster.
- Validation: Benchmark against reference crystal structures and known bioactive conformations.
Technical Notes:
- For macrocycles and highly flexible systems, extend sampling time to ensure adequate coverage of conformational space.
- The RDGYR collective variable efficiently captures global conformational changes while remaining computationally inexpensive.
- Moltiverse demonstrates particular effectiveness for challenging systems with high conformational flexibility, achieving highest accuracy among tested algorithms for macrocycles [49].
Protocol 2: RESP2 Charge Derivation with Density Fitting Acceleration
Principle: Generate accurate partial atomic charges that balance gas-phase and condensed-phase polarization effects while maintaining computational efficiency through density fitting techniques [2] [6].
Materials and Software:
- Quantum chemistry software (Psi4 v.1.3.2 or later recommended)
- DF-RESP implementation for accelerated charge calculation
- Force field parameterization tools (ForceBalance software for LJ parameter optimization)
Procedure:
- Initial Structure Preparation: Use optimized molecular geometry from conformational sampling protocol.
- QM Method Selection: Employ PW6B95 functional with aug-cc-pV(D+d)Z basis set for accurate ESP calculations.
- Dual-Phase ESP Calculation:
- Perform gas-phase ESP calculation
- Perform aqueous-phase ESP calculation using implicit solvation model
- RESP2 Charge Calculation: Combine gas-phase and aqueous-phase ESPs using weighting parameter δ = 0.6 (60% aqueous, 40% gas-phase) [2].
- Charge Fitting: Apply RESP restraints during fitting to maintain chemical transferability and prevent overfitting.
- Validation: Compare calculated dipole moments with higher-level theoretical references and experimental data where available.
Technical Notes:
- The RESP2δ=0.6 parameterization has been optimized against experimental condensed-phase properties including liquid densities, heats of vaporization, and hydration free energies [2].
- DF-RESP achieves approximately 14-fold speedup for medium-sized proteins (∼1500 atoms) while maintaining charge accuracy with mean absolute error below 0.003 e [6].
- For specific application contexts (e.g., membrane protein simulations), the δ parameter may be adjusted toward more gas-phase-like (lower δ) or aqueous-like (higher δ) character.
Protocol 3: Integrated Workflow for Conformation-Dependent Charge Parameterization
Principle: Account for conformational flexibility in charge derivation by generating context-specific partial charges for key conformational states, particularly important for molecules with significant charge transfer or polarization effects between conformers.
Procedure:
- Conformer Ensemble Generation: Apply Protocol 1 to produce representative conformational ensemble.
- Conformer Clustering: Group similar conformations using RMSD-based clustering with 1.0-1.5 Å cutoff.
- Charge Calculation for Representatives: Calculate RESP2 charges for lowest-energy representative of each major conformational cluster.
- Charge Comparison: Analyze charge variations across conformational states to identify significant differences.
- Dynamic Charge Assignment (Optional): Implement multi-conformer charge sets for advanced simulations where conformational dependence significantly impacts electrostatic properties.
Data Presentation and Analysis
Quantitative Performance Metrics
Table 2: Performance Comparison of Conformer Generation Algorithms
| Software/Method | Accuracy (RMSD Å) | Computational Speed | Macrocycle Performance | Key Application Context |
|---|---|---|---|---|
| Moltiverse | Highest accuracy in benchmarks | Moderate | Superior for flexible macrocycles | Challenging flexible systems, macrocyclic drugs [49] |
| RDKit | Moderate to high | Fast | Limited | High-throughput screening, initial conformer generation |
| CONFORGE | High | Moderate | Moderate | General drug discovery applications |
| Balloon | Variable | Fast | Limited | Rapid ensemble generation |
| iCon | High | Moderate | Good | Balanced accuracy and efficiency |
| Conformator | Moderate | Fast | Limited | Large-scale virtual screening |
Table 3: Accuracy and Efficiency of RESP Charge Methods
| Method | Charge Accuracy (MAE, e) | Speed Relative to RESP1 | Electrostatic Energy Error (kcal/mol) | Recommended Use Cases |
|---|---|---|---|---|
| RESP1 | Baseline | 1.0x | Baseline | General use, legacy compatibility |
| RESP2 (δ=0.6) | Improved accuracy | ~0.7x (no DF) | < 0.1 kcal/mol | Condensed-phase simulations, drug binding [2] |
| DF-RESP | Comparable to RESP1 (MAE <0.003 e) | 14x faster for 1500-atom systems | < 0.1 kcal/mol | Large systems, high-throughput workflows [6] |
| RESP2 with DF | Improved accuracy with efficiency | ~10x faster than RESP1 | < 0.1 kcal/mol | Production simulations requiring accuracy and efficiency |
The Scientist's Toolkit
Table 4: Essential Research Reagents and Computational Tools
| Tool/Resource | Function | Application Context |
|---|---|---|
| Moltiverse | Enhanced sampling conformer generation | Accurate conformational ensemble prediction for flexible molecules and macrocycles [49] |
| DF-RESP | Accelerated RESP charge calculation | Large-scale charge parameterization for big molecules and high-throughput screening [6] |
| RESP2 | Advanced partial charge derivation | Physically accurate charges for condensed-phase simulations [2] |
| Platinum Diverse Data set | Benchmarking conformer generation | Validation of conformational sampling algorithms [49] |
| ForceBalance | Force field parameter optimization | Systematic optimization of LJ parameters alongside charge sets [2] |
| PW6B95/aug-cc-pV(D+d)Z | QM method for ESP calculation | Accurate reference ESPs for charge fitting [2] |
Workflow Visualization
Integrated Workflow for Molecular Parameterization
Managing molecular orientation and conformation dependence through integrated protocols for conformer generation and charge parameterization represents a critical capability in modern computational drug discovery. The combination of Moltiverse for enhanced conformational sampling and RESP2 with density fitting acceleration provides researchers with a robust, physically grounded approach to molecular representation. These protocols enable more accurate prediction of binding poses, protein-ligand interactions, and physicochemical properties, particularly for challenging drug targets involving flexible molecules and novel chemotypes. By implementing these methodologies, researchers can significantly enhance the reliability of computational predictions throughout the drug discovery pipeline, from initial virtual screening to lead optimization.
The derivation of partial atomic charges is a cornerstone of molecular simulation, directly influencing the accuracy of calculated electrostatic properties and intermolecular interactions. The Restrained Electrostatic Potential (RESP) method has emerged as a highly regarded approach for assigning these charges, particularly within biomolecular force fields. The core challenge in RESP charge derivation lies in balancing two competing objectives: achieving a high-fidelity fit to the quantum mechanically calculated molecular electrostatic potential (MEP) while maintaining chemical reasonableness in the resulting charges. This balance is governed by the restraint weight parameter, krstr. This application note provides a detailed protocol for tuning krstr to optimize this critical trade-off, ensuring the production of robust and reliable partial charges for novel molecules, with a specific focus on applications in drug development.
Theoretical Foundation of RESP and the Role of krstr
The RESP method operates by fitting partial atomic charges to reproduce the MEP surrounding the molecule, typically derived from quantum mechanical calculations such as HF/6-31G* or more advanced levels like PW6B95/aug-cc-pV(D+d)Z [2]. The fitting process minimizes a target function, A(q), which contains two key terms:
A(q) = χ²(q) + R(q)
Here, χ²(q) represents the squared deviation between the computed MEP and the MEP generated by the partial charges, q. R(q) is the hyperbolic restraint term, R(q) = krstr * ∑ [sqrt(qi² + a²) - a], which penalizes chemically unreasonable charges. The parameter a provides a smooth transition near zero charge, while krstr acts as a weighting factor determining the penalty strength applied to large atomic charges.
A higher krstr value enforces greater "chemical reasonableness" by heavily penalizing large partial charges, often at the expense of MEP-fitting accuracy. Conversely, a lower krstr value prioritizes an accurate reproduction of the MEP, which can sometimes lead to exaggerated or non-intuitive charge values for buried atoms. Tuning krstr is therefore essential for achieving a charge set that is both physically meaningful and computationally useful.
Quantitative Guidelines for Restraint Weight Selection
Selecting an appropriate krstr value is not a one-size-fits-all process. The optimal value can depend on the molecule's chemical nature and the intended application. The following table summarizes recommended restraint weights and their typical applications based on current practices and literature:
Table 1: Guidelines for Restraint Weight (krstr) Selection
| Restraint Weight (krstr) | Fitting Philosophy | Best Use Cases | Performance Considerations |
|---|---|---|---|
| 0.001 a.u. | Weak restraint; prioritizes MEP reproduction. | Standard organic molecules; initial charge fitting where charge transfer is expected. | May produce large atomic charges for buried atoms; requires careful validation. |
| 0.0005 a.u. | Very weak restraint; maximum MEP accuracy. | Systems where electrostatic interactions are paramount (e.g., enzyme active sites). | High risk of chemically unreasonable charges; use with caution. |
| 0.01 - 0.1 a.u. | Strong restraint; prioritizes chemical reasonableness. | Polypeptides and proteins; avoiding over-polarization in condensed-phase simulations [2]. | Ensures charge stability but may reduce electrostatic accuracy. |
The choice of krstr is part of a larger parametric strategy. For instance, the next-generation RESP2 approach decouples charge derivation from the fortuitous overpolarization of HF/6-31G* by calculating ESPs as a linear combination of gas- and aqueous-phase charges, scaled by a parameter δ (typically 0.6) [2]. When adopting such advanced methods, krstr must be re-optimized in the context of the new charge derivation model.
Experimental Protocol for krstr Optimization
This protocol provides a step-by-step guide for systematically determining the optimal krstr value for a novel molecule.
The following diagram illustrates the iterative workflow for tuning the restraint weight:
Step-by-Step Procedure
Step 1: System Preparation and Quantum Mechanical Calculations
- Geometry Optimization: Perform a full geometry optimization of the novel molecule at an appropriate level of theory (e.g., B3LYP/6-31G*). Ensure the structure is at a true energy minimum by confirming the absence of imaginary frequencies.
- Electrostatic Potential (ESP) Calculation: Using the optimized geometry, compute the molecular electrostatic potential on a dense grid of points surrounding the molecule. A standard method is HF/6-31G*, but for higher accuracy, use a method like PW6B95/aug-cc-pV(D+d)Z [2]. The grid should be sufficiently dense (e.g., 1 point per Ų) to ensure a robust fit.
Step 2: Initial RESP Fitting
- Conduct an initial RESP fit using a standard restraint weight of
krstr = 0.001atomic units. - Use a two-stage fitting procedure:
- Fit all atoms with a weak restraint.
- Fit heavy atoms with a stronger restraint while relaxing hydrogens (a common practice to maintain chemical sense for hydrocarbon groups).
Step 3: Evaluation of the Initial Charge Set
- Quantitative Metrics:
- Calculate the root-mean-square error (RMSE) between the QM-calculated MEP and the MEP reproduced by the fitted charges.
- Compute the relative RMSE to assess the quality of the fit.
- Qualitative/Chemical Assessment:
- Inspect the magnitudes of the fitted charges. Charges on equivalent atoms (e.g., methyl group hydrogens) should be similar.
- Check for atoms with unusually large positive or negative charges that lack a clear chemical justification (e.g., buried carbon atoms with large charges).
- Compare the molecular dipole moment from the RESP charges to the QM-derived dipole moment.
Step 4: Iterative Adjustment and Validation
- If the MEP fit is poor (high RMSE) and charges appear chemically reasonable, decrease
krstr(e.g., to 0.0005) to allow for a closer fit. - If the MEP fit is good but charges are chemically unreasonable, increase
krstr(e.g., to 0.01 or 0.1) to enforce greater restraint. - Repeat the fitting and evaluation process until a satisfactory balance is achieved. This is often an iterative, subjective process guided by the researcher's chemical intuition.
Step 5: Condensed-Phase Validation (Critical Step)
- The ultimate test for a charge set is its performance in molecular dynamics (MD) simulations.
- Simulate the neat liquid of the compound or its solvation in water using the derived charges and an appropriate force field (e.g., GAFF, OPLS).
- Compare the simulated bulk properties—such as density (Δρ) and heat of vaporization (ΔHvap)—against experimental data. The table below shows target accuracy for a well-fitted model [2].
Table 2: Target Accuracy for Condensed-Phase Validation
| Property | Experimental Value (Example) | Target MUE for Validated Model |
|---|---|---|
| Density (g/cm³) | Compound-specific | < 0.02 g/cm³ |
| Heat of Vaporization (kcal/mol) | Compound-specific | < 0.8 kcal/mol |
| Hydration Free Energy (kcal/mol) | Compound-specific | < 1.0 kcal/mol |
The Scientist's Toolkit: Research Reagent Solutions
The following table details essential software and computational tools required for implementing the RESP charge derivation protocol.
Table 3: Essential Research Reagents and Software for RESP Charge Derivation
| Tool Name | Type | Primary Function in Protocol | Key Notes |
|---|---|---|---|
| GAUSSIAN | Software | QM geometry optimization and ESP calculation (HF/6-31G*, DFT) | Industry standard; provides grid data for RESP fit. |
| PSI4 | Software | Alternative QM package for ESP calculation (supports advanced methods like PW6B95) | Open-source; efficient for high-accuracy ESPs [2]. |
| Antechamber | Software Module | Automates RESP fitting and force field parameter assignment (e.g., for GAFF). | Part of AmberTools; uses krstr as an input parameter. |
| ForceBalance | Software | Systematic optimization of force field parameters (e.g., krstr, LJ parameters). |
Co-optimizes charges and LJ parameters against experimental data [2]. |
| ACPYPE | Script | Interface for Antechamber; simplifies topology generation for GROMACS. | Handles the RESP fitting process and file format conversion. |
Advanced Applications and Recent Developments
The principles of krstr tuning extend to advanced RESP methodologies and specialized applications:
- RESP2: This approach uses a parameter
δ(typically 0.6) to blend gas- and aqueous-phase charges, better representing polarization in condensed phases. When using RESP2,krstrmust be re-optimized in conjunction with theδparameter and Lennard-Jones parameters to avoid error cancellation [2]. - Large-Scale Biomolecular Systems: The new DF-RESP method addresses the high computational cost of traditional RESP by combining it with density fitting for MEP calculations. This allows application to large systems like the 1493-atom protein 1h59, achieving a 14-fold speedup while maintaining accuracy. Tuning
krstrremains essential in this context to ensure conformational energy variations are correctly captured during dynamics [51] [6]. - Non-Canonical Amino Acids (ncAAs): For drug design involving peptides with ncAAs, an initial charge derivation using AM1-BCC is often performed, followed by final refinement using RESP fitting. This two-step process is critical for generating generalizable features for machine learning models that predict peptide structures and properties [52].
The derivation of accurate Restrained Electrostatic Potential (RESP) charges has long been a cornerstone of reliable molecular simulations in drug discovery. Traditional approaches, however, have predominantly focused on single, static conformations, potentially overlooking the dynamic nature of biomolecular systems. The paradigm is now shifting from static structures to dynamic conformational ensembles, recognizing that protein function is not solely determined by static three-dimensional structures but is fundamentally governed by dynamic transitions between multiple conformational states [53]. This shift is particularly crucial for understanding the mechanistic basis of protein function and regulation, as many pathological conditions, including Alzheimer's disease and Parkinson's disease, stem from protein misfolding or abnormal dynamic conformations [53].
The emergence of deep learning, particularly AlphaFold, has revolutionized static protein structure prediction, marking a transformative milestone in structural biology [53]. However, the inherent flexibility of proteins and their ability to adopt multiple conformational states in response to environmental factors, ligand binding, or mutations necessitates advanced strategies that go beyond single-conformation approaches [53]. Multi-conformation and multi-molecule fitting addresses these limitations by incorporating structural heterogeneity directly into the charge derivation process, potentially leading to more physiologically relevant and transferable atomic partial charges for molecular simulations.
Methodological Foundations: From Single to Multiple Conformations
Theoretical Basis for Multi-Conformation RESP Fitting
The standard RESP approach fits atomic partial charges to reproduce the quantum mechanical (QM) electrostatic potential (ESP) at points around a molecular structure by minimizing the residual ( R_{\text{esp}} ):
[ R{\text{esp}}=\frac{1}{N}\sumk^N{(V{\text{QM}}(\mathbf{r}k)-V{\text{RESP}}(\mathbf{r}k))^2} ]
where ( V{\text{QM}} ) is the quantum mechanical potential and ( V{\text{RESP}} ) the potential generated by the RESP charges at points ( \mathbf{r}_k ) [8].
In multi-conformation RESP fitting, this formalism is extended to simultaneously fit charges to multiple conformational states or molecular complexes. The objective function becomes:
[ R{\text{esp}}^{\text{multi}} = \sum{i=1}^{M} \frac{wi}{Ni} \sumk^{Ni} (V{\text{QM}}^i(\mathbf{r}k) - V{\text{RESP}}^i(\mathbf{r}k))^2 ]
where ( M ) represents the number of conformations or complexes, ( wi ) are weighting factors for each structure, and ( Ni ) the number of grid points for each conformation ( i ). This approach ensures that the derived charges provide a balanced representation across the entire conformational landscape rather than being optimized for a single state.
Accelerating RESP with Density Fitting
Despite its accuracy, the conventional RESP method suffers from high computational cost due to dense molecular electrostatic potential sampling, particularly for large-scale systems [6]. The recently introduced DF-RESP method combines density fitting MEP (DF-MEP) with RESP charge derivation to dramatically accelerate calculations while maintaining accuracy [6].
Compared to conventional RESP calculations, DF-RESP achieves excellent accuracy with a mean absolute error (MAE) in charges below 0.003 e for the S22 benchmark and electrostatic interaction energy deviations under 0.1 kcal/mol [6]. For androgen receptor-ligand complexes, DF-RESP achieves an MAE of less than 0.06 kcal/mol while achieving a 14-fold speedup for the 1493-atom protein 1h59 [6]. This acceleration is particularly valuable for multi-conformation approaches that require numerous QM calculations.
Table 1: Performance Metrics of DF-RESP Acceleration Method
| System/Benchmark | Mean Absolute Error (MAE) | Speed Improvement | Key Applications |
|---|---|---|---|
| S22 Benchmark | <0.003 e | Not specified | Small molecule complexes |
| Androgen Receptor-Ligand Complexes | <0.06 kcal/mol | Not specified | Protein-ligand binding |
| Protein 1h59 (1493 atoms) | Comparable to standard RESP | 14-fold | Large biomolecules |
Advanced Sampling of Conformational Landscapes
AI-Driven Conformational Sampling with AlphaFold Variants
Recent advancements in artificial intelligence have revolutionized our ability to sample protein conformational landscapes. Several AlphaFold-based methods now enable the prediction of multiple conformations rather than single structures:
AFsample2 employs random multiple sequence alignment (MSA) column masking to reduce co-evolutionary signals, thereby enhancing structural diversity in AlphaFold2-generated models [54]. The method randomly masks MSA columns with "X" (denoting unknown residue) with a predefined probability (typically 15%), breaking covariance constraints that normally drive AF2 toward a single conformation [54]. This approach has demonstrated remarkable success, improving alternate state models in 9 out of 23 test cases without affecting preferred state generation, with TM-score improvements sometimes exceeding 50% (e.g., from 0.58 to 0.98) [54].
Targeted MSA Masking represents a more focused approach where specific columns in the MSA corresponding to structurally variable regions are masked. Research on engineered green fluorescent proteins has demonstrated that this approach can not only predict alternative conformations but also provide qualitative estimation of their population ratios [55]. In this strategy, the uniref90_hits.sto alignments are edited by replacing with gaps any amino acid in columns corresponding to alternative structural elements [55].
MSA Clustering and Subsampling techniques, such as those implemented in AF-cluster, generate diverse conformational states by clustering the MSA based on sequence similarity and using different clusters for separate predictions [56]. This approach can sample alternative states of metamorphic proteins and predict their response to point mutations [56].
AI-Driven Conformational Sampling Workflow
Molecular Dynamics for Enhanced Sampling
While AI methods excel at identifying stable states, molecular dynamics (MD) simulations remain invaluable for sampling continuous conformational transitions and local flexibility. Molecular dynamics uses Newton's equations of motion to simulate the time evolution of interacting atoms, with the net force and acceleration experienced by each atom determining its trajectory [57]:
[ Fi = mi ai = mi \frac{d^2ri}{dt^2} = -\nablai V ]
where ( Fi ) is the force on atom i with mass ( mi ) at position ( r_i ), and ( V ) is the potential energy [57].
Several specialized MD databases provide pre-computed trajectories for various protein families:
Table 2: Molecular Dynamics Databases for Conformational Sampling
| Database | System Types | Number of Trajectories | Time Scale | Applications |
|---|---|---|---|---|
| ATLAS | General proteins | 5841 | ns | Protein dynamics analysis |
| GPCRmd | G Protein-Coupled Receptors | 2115 | ns | GPCR functionality and drug discovery |
| SARS-CoV-2 | Coronavirus proteins | 300 | ns/μs | SARS-CoV-2 drug discovery |
| MemProtMD | Membrane proteins | 8459 | μs | Membrane protein folding and stability |
Advanced MD techniques such as replica exchange, metadynamics, and accelerated MD can enhance sampling of rare events and conformational transitions, providing valuable input structures for multi-conformation RESP fitting.
Multi-Molecule Complex Prediction and Modeling
Predicting Biomolecular Complexes with AlphaFold 3
The release of AlphaFold 3 represents a significant advancement in multi-molecule modeling, enabling the prediction of entire biomolecular complexes rather than single proteins [58]. AlphaFold 3 can jointly model proteins with DNA, RNA, small molecules (ligands), ions, and even post-translational modifications, predicting how these components fit together in 3D [58]. This expanded capability addresses a critical need in structural biology, as most biological processes involve multiple interacting molecules.
Benchmarking studies have demonstrated that AlphaFold 3 delivers a ≥50% accuracy improvement on protein-ligand and protein-nucleic acid interactions compared to prior methods [58]. This capability makes it particularly valuable for generating structural models of protein-ligand complexes for RESP charge derivation in drug discovery applications.
Integrated Structure and Affinity Prediction with Boltz-2
A landmark development in mid-2025 is Boltz-2, an open-source "biomolecular foundation model" that simultaneously predicts a protein's structure and how strongly a ligand will bind to it [58]. Boltz-2 can co-fold a protein-ligand pair and output both the 3D complex and a binding affinity estimate in about 20 seconds on a single GPU [58].
This unified approach tackles a longstanding bottleneck in drug discovery by providing both structural and functional information. Impressively, Boltz-2 achieved accuracy on par with gold-standard free-energy perturbation calculations while dramatically reducing computation time [58]. The model's training incorporated molecular dynamics simulations and "physical steering" to ensure predictions remain realistic and to avoid unphysical conformations, making it particularly suitable for generating structures for charge derivation [58].
Integrated Protocol for Multi-Conformation RESP Charge Derivation
Comprehensive Workflow for Charge Derivation
This protocol describes an integrated approach for deriving RESP charges that account for conformational and compositional diversity, suitable for both small molecules and biomolecular systems.
Multi-Conformation RESP Charge Derivation Workflow
Step-by-Step Experimental Procedure
Step 1: Conformational Ensemble Generation
- For small molecules: Use conformational search algorithms (systematic, stochastic, or knowledge-based) to identify low-energy conformers. Apply molecular dynamics with enhanced sampling techniques to explore conformational space.
- For proteins and complexes: Employ AI-based sampling methods:
Step 2: Representative Structure Selection
- Cluster all generated structures using RMSD-based clustering algorithms.
- For each major cluster, select representative structures based on:
- Cluster population (for MD trajectories)
- Prediction confidence (pLDDT for AlphaFold models)
- Structural diversity to cover the conformational landscape
- Typically, 5-20 representative structures provide a balance between comprehensiveness and computational feasibility.
Step 3: Quantum Mechanical Calculations
- For each representative structure, perform QM calculations to obtain reference electrostatic potentials:
- Geometry optimization at the DFT level with medium-sized basis sets (e.g., B3LYP/6-31G)
- Single-point energy calculation with larger basis sets (e.g., HF/6-31G) for ESP generation
- For large systems, consider fragment-based or QM/MM approaches
Step 4: Multi-Conformation RESP Fitting
- Implement the multi-conformation RESP fitting using the extended objective function:
- Apply appropriate constraints:
- Total molecular charge constraint
- Equivalence constraints for chemically equivalent atoms
- Weak harmonic restraints (β = 0.0001 a.u.) to prevent extreme charge values
- Utilize DF-RESP acceleration for large systems [6]
Step 5: Validation and Quality Control
- Back-validate fitted charges by calculating ESP for each conformation using derived charges
- Compare with QM reference: target RMSE < 10% of ESP range
- Check charge consistency across conformations and with chemical intuition
- For drug discovery applications, validate against experimental binding data if available
Research Reagent Solutions
Table 3: Essential Tools for Multi-Conformation RESP Charge Derivation
| Tool/Resource | Type | Function | Access |
|---|---|---|---|
| AlphaFold2/3 | AI Structure Prediction | Predict protein structures and complexes | Open source / AlphaFold Server |
| AFsample2 | Conformational Sampling | Generate alternative protein conformations | Open source [54] |
| Boltz-2 | Structure & Affinity Prediction | Co-fold protein-ligand pairs with affinity | Open source (MIT license) [58] |
| CP2K | Quantum Chemistry | QM calculations and RESP fitting | Open source [8] |
| DF-RESP | Charge Derivation | Accelerated RESP charge calculation | Method description [6] |
| GROMACS/AMBER | Molecular Dynamics | Conformational sampling via MD | Open source / Licensed |
| GPCRmd | Specialized Database | Pre-sampled conformations for GPCRs | Database [53] |
| ATLAS | MD Database | General protein MD trajectories | Database [53] |
Applications in Drug Discovery and Challenges
Practical Applications
The multi-conformation RESP approach finds particular utility in several drug discovery applications:
Protein-Ligand Binding Studies: By deriving charges that represent both bound and unbound states, researchers obtain more accurate predictions of binding affinities and mechanisms. Boltz-2 has demonstrated particular promise in this area, nearly doubling the performance of previous methods for binding affinity prediction [58].
Membrane Protein Modeling: Proteins such as GPCRs and transporters frequently adopt multiple conformational states during their functional cycles. The availability of specialized MD databases like GPCRmd provides valuable starting points for multi-conformation charge derivation for these important drug targets [53].
Allosteric Modulator Design: Allosteric regulation often involves conformational transitions between states. Multi-conformation charges can better capture the electrostatic changes associated with these transitions, facilitating the design of allosteric modulators.
Current Limitations and Future Directions
Despite these advances, several challenges remain in multi-conformation and multi-molecule RESP fitting:
Data Limitations: High-quality structural data for alternative conformations, particularly for multi-component complexes, remains limited [53]. While computational methods can generate models, experimental validation is still essential.
Sampling Completeness: Current methods may not fully sample rare but functionally important conformational states, particularly for large-scale conformational changes.
Computational Cost: Even with accelerations like DF-RESP, multi-conformation QM calculations remain computationally demanding for large systems.
Methodological Integration: Better integration between AI-based structure prediction, physical simulation methods, and experimental data is needed to improve the accuracy and reliability of generated conformational ensembles [53].
Future developments will likely focus on improved sampling algorithms, more efficient QM methods, and tighter integration of experimental data from techniques such as cryo-EM and NMR to guide and validate conformational sampling. As these methods mature, multi-conformation RESP charge derivation will become an increasingly standard approach for achieving higher accuracy in molecular simulations across pharmaceutical and biomedical research.
Application Note: RESP Charge Derivation for Carbohydrates and Macrocyclic Hosts
Accurate molecular dynamics (MD) simulations of carbohydrates and macrocyclic hosts are pivotal for advancements in drug delivery, supramolecular chemistry, and material science. The fidelity of these simulations is critically dependent on the quality of the atomic partial charges used in the force field. This note details the application of the Restrained Electrostatic Potential (RESP) method to derive optimized partial charges for such challenging, polar systems, enabling more reliable modeling of their behavior in condensed phases.
The Underlying Challenge: Electrostatic Interactions in Polar Systems
In biomolecular force fields, electrostatic forces are primarily computed using atomic partial charges. These forces are especially important for stabilizing structures like hydrogen bonds, which are ubiquitous in carbohydrates and macrocyclic hosts like cyclodextrins [59] [1]. The inherent flexibility of carbohydrates and the delicate balance between solute-solute and solute-solvent (typically water) interactions make them particularly sensitive to the choice of partial charges [59] [1].
A common practice is to derive charges by fitting a classical Coulomb model to the quantum mechanical molecular electrostatic potential (ESP-charges). However, ESP-charges computed with a standard 6-31G* basis set are known to overestimate bond polarities in the gas phase. This over-polarization can artificially skew the balance of intermolecular forces in condensed-phase simulations, leading to unrealistic dynamics and structural artifacts [59] [1].
The RESP Solution for Carbohydrates
The RESP methodology addresses over-polarization by incorporating a hyperbolic restraint function during the charge-fitting process. This attenuates the magnitudes of the charges without significantly affecting molecular dipole moments, offering a more physically realistic model for simulations [59] [1].
The key equation for the RESP fit is: [ \chi{\text{RESP}}^2 = \chi{\text{ESP}}^2 + \chi{\text{rstr}}^2 ] where (\chi{\text{ESP}}^2) is the standard ESP fit, and (\chi{\text{rstr}}^2) is the restraint function given by: [ \chi{\text{rstr}}^2 = k{\text{rstr}} \sumj \left( \sqrt{qj^2 + b^2} - b \right) ] Here, (k{\text{rstr}}) is the restraint weight and (b) defines the tightness of the hyperbola [1].
Table 1: Performance of Different Charge Sets in MD Simulations of an α-d-Glucopyranose Crystal [1]
| Charge Derivation Method | Restraint Weight (krstr) | Agreement with Neutron Diffraction Structure | Category of Result |
|---|---|---|---|
| RESP | 0.01 | Best Agreement | A: Geometry & interactions maintained |
| Unrestrained ESP | N/A | Poor | B/C: Geometry distorted and/or interactions lost |
| Mulliken Analysis | N/A | Poor | B/C: Geometry distorted and/or interactions lost |
| Distributed Multipole Analysis | N/A | Poor | B/C: Geometry distorted and/or interactions lost |
For carbohydrates, a restraint weight of 0.01 has been validated as optimal for use with the GLYCAM parameter set. This was determined by running MD simulations of a carbohydrate crystal and monitoring the stability of the experimental unit cell geometry, which is highly sensitive to non-bonded forces. Simulations using the RESP charge set with (k_{\text{rstr}}=0.01) successfully maintained the crystallographic structure, while other methods led to significant distortions [59] [1].
Experimental Protocol: RESP Charge Derivation for a Novel Carbohydrate
This protocol provides a step-by-step guide for deriving RESP charges for a novel carbohydrate molecule, using α-d-glucopyranose as a model system.
Workflow Diagram
The following diagram outlines the complete charge derivation and validation process.
Step-by-Step Procedure
Step 1: Initial Geometry Acquisition
- Objective: Obtain a reliable starting conformation for the carbohydrate.
- Procedure: For common monosaccharides like α-d-glucopyranose, use the high-precision neutron diffraction crystal structure as the initial coordinate set [1]. For novel molecules, begin with a geometry optimized at an appropriate quantum mechanical level (e.g., HF/6-31G* or B3LYP-D3/6-31G(d,p)).
Step 2: Quantum Mechanical Electrostatic Potential Calculation
- Objective: Generate the target electrostatic potential for charge fitting.
- Software: Gaussian 16 [60].
- Method:
- Perform a single-point energy calculation on the optimized geometry at the HF/6-31G* level of theory.
- Use the CHELPG algorithm to compute the molecular electrostatic potential at a large number of points (e.g., >13,000) around the van der Waals surface of the molecule [1].
Step 3: RESP Charge Fitting
- Objective: Derive the final set of restrained atomic partial charges.
- Software: AMBER suite
respprogram or equivalent [1]. - Parameters:
- Employ a single-stage fitting process (as rotational symmetry is often absent in carbohydrates).
- Set the restraint weight (
k_rstr) to 0.01. - Use the default value for the hyperbola tightness parameter
b[1].
- Output: A file containing the final restrained electrostatic potential (RESP) charges for each atom.
Step 4: Validation via Crystal MD Simulation
- Objective: Assess the performance of the new charge set in a condensed-phase environment.
- System Setup:
- Simulation Details:
- Software: AMBER or a compatible MD package.
- Conditions: Run under periodic boundary conditions (NPT ensemble) at 298 K and 1 atm for a minimum of 50 ps. Discard the first 20 ps for equilibration [1].
- Analysis: Monitor the average unit cell dimensions (A, B, C) over the final 30 ps of the simulation and compare them to the experimental neutron diffraction data. Successful validation is achieved when the simulated cell parameters closely match the experimental values (Category A result in Table 1) [1].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Computational Tools and Resources for RESP Charge Derivation and Validation
| Item Name | Function / Application | Specific Example / Note |
|---|---|---|
| GLYCAM Force Field | A specialized parameter set for simulating carbohydrates and glycoconjugates. | Used in conjunction with derived RESP charges for MD simulations [59] [1]. |
| AMBER Software Suite | A comprehensive package for biomolecular simulation. | Contains tools for RESP fitting (resp) and running MD simulations (sander, pmemd) [1]. |
| Gaussian 16 | Software for quantum chemical calculations. | Used for computing the molecular electrostatic potential at the HF/6-31G* level [60]. |
| Cambridge Structural Database (CSD) | A repository of experimentally determined small-molecule crystal structures. | Source for initial coordinates of molecules like β-cyclodextrin for conformational analysis and charge derivation [60]. |
| DFT (B3LYP-D3/6-31G(d,p)) | A higher-level quantum method for geometry optimization and conformational analysis. | Recommended for optimizing complex macrocyclic hosts like β-cyclodextrin and calculating their thermodynamic properties [60]. |
| PCM (Polarizable Continuum Model) | An implicit solvation model for quantum chemical calculations. | Used to approximate aqueous solvation effects during geometry optimization (ε = 78.54 for water) [60]. |
Validating and Comparing Charge Sets for Predictive Simulations
The accurate prediction of molecular crystal properties is a cornerstone of modern drug development. The existence of multiple crystal forms, or polymorphs, of a single Active Pharmaceutical Ingredient (API) can have profound implications on a drug's solubility, stability, and bioavailability. Computational models, particularly those employing Crystal Structure Prediction (CSP), are indispensable for navigating this complexity. However, the predictive power of these models hinges entirely on their rigorous benchmarking against experimental data. This application note details protocols for validating computational assessments of crystal lattice stability and liquid properties, with a specific focus on their critical role in deriving accurate Restrained Electrostatic Potential (RESP) charges for novel molecules.
Benchmarking Lattice Energy and Polymorph Stability
Accurately ranking the stability of polymorphs is a central challenge in solid-form drug development. The following protocol outlines a method for benchmarking computational lattice energy predictions against experimental stability data.
Experimental Benchmarking Protocol
Step 1: Curate a Benchmarking Dataset Assemble a set of flexible compounds with known polymorphism. The dataset should be divided into a validation subset (e.g., 6 compounds forming 7 polymorphic pairs) for method assessment and a test subset (e.g., 9 compounds forming 10 pairs) for final evaluation. Prefer experimental structures from neutron diffraction or low-temperature single-crystal X-ray diffraction for highest reliability [61] [62].
Step 2: Compute Lattice Energy Partitions For each crystal structure, calculate the total lattice energy (
E_latt-global) and partition it into its components using the equation [61]:E_latt-global = E_inter + E_intra-global = E_inter + (E_adjustment + ΔE_change-global)Here,E_interis the intermolecular interaction energy, andE_intra-globalis the intramolecular energy penalty, comprising the conformational adjustment energy (E_adjustment) and the global change energy (ΔE_change-global).Step 3: Select and Validate the Energy Model Systematically evaluate different computational methods. Studies have identified that combining an intermolecular treatment with PBE-MBD (for many-body dispersion corrections) and an intramolecular model using the B2PLYPD double hybrid functional achieves exceptional agreement with experiment, yielding a mean absolute deviation (MAD) as low as 2.3 kJ·mol⁻¹ across a set of polymorphic pairs [61]. This model can be designated as the Best Lattice Energy Model (BLEM).
Step 4: Analyze the Intra- to Intermolecular Energy Ratio Calculate the ratio of the intramolecular energy penalty (
E_intra-global) to the intermolecular stabilization energy (E_inter). An empirical trend, the "40% limit," has been observed where the probability of observing a high-energy conformation in the solid-state becomes negligible when this ratio exceeds 40% [61]. This metric is crucial for assessing the crystallizability of predicted structures.
Table 1: Benchmarking Performance of Selected Computational Models for Lattice Energies
| Intermolecular Method | Intramolecular Method | Mean Absolute Deviation (MAD) | Key Strengths |
|---|---|---|---|
| PBE-MBD | B2PLYPD | 2.3 kJ·mol⁻¹ | Best overall performance for flexible molecules [61] |
| PBE-MBD | ωB97XD | 2.4 kJ·mol⁻¹ | Excellent alternative to double-hybrid functionals [61] |
| PBE-TS | B2PLYPD | >2.3 kJ·mol⁻¹ | Highlights importance of many-body dispersion [61] |
Large-Scale CSP Validation
For comprehensive validation of a CSP workflow, a large-scale study on a diverse set of 66 molecules with 137 known polymorphic forms can be conducted. The protocol involves a hierarchical ranking method combining machine learning force fields (MLFF) and periodic DFT, which successfully reproduced all known polymorphs and ranked them within the top candidates [62]. This approach is vital for derisking drug development by identifying potentially missing, low-energy polymorphs that could appear late in the development process.
Benchmarking Liquid and Nanoconfined Water Properties
The behavior of water in confined spaces, such as within protein active sites or nanofluidic devices, differs dramatically from its bulk properties. Benchmarking computational models against experimental data for these systems is essential for simulations of biomolecular recognition and permeability.
Protocol for Characterizing Premelting State of Water
Step 1: Sample Preparation Synthesize a host system with well-defined, hydrophilic nanopores. For instance, grow hexagonal rod-like crystals with quasi-one-dimensional channels approximately 1.6 nm in diameter [63]. Fill these nanopores with heavy water (D₂O) to facilitate nuclear magnetic resonance (NMR) analysis.
Step 2: Solid-State Deuterium NMR Spectroscopy Use static solid-state deuterium NMR to probe the confined water molecules. The key is to measure the spin-lattice relaxation time, which quantifies the rotational mobility of water molecules [63].
Step 3: Temperature-Dependent Measurement Acquire NMR spectra while gradually heating the crystal from a low temperature. Monitor the spectra for distinct changes that indicate phase transitions [63].
Step 4: Data Analysis Analyze the NMR spectra and relaxation times. The emergence of a premelting state is characterized by a unique signature: water molecules exhibit solid-like positions (as seen in the spectral peaks) but liquid-like rotational motion (as seen in the correlation time close to that of bulk liquid water) [63]. This state represents a novel phase where frozen and mobile water layers coexist.
Diagram 1: Workflow for characterizing premelting state of water in nanopores.
Integrating Experimental Charge Determination for RESP Derivation
The derivation of RESP charges for novel molecules traditionally relies on quantum mechanical calculations on isolated molecules. However, the molecular charge distribution can be modified by its chemical environment, a factor that can now be experimentally quantified to inform and benchmark charge derivation protocols.
Protocol for Experimental Determination of Partial Charges
Step 1: Crystallization and Data Collection Grow a high-quality single crystal of the target compound. Collect a 3D electron diffraction dataset using standard crystallographic workflows [19].
Step 2: Ionic Scattering Factors (iSFAC) Modelling During the crystal structure refinement, introduce one additional parameter per atom. This parameter refines the fraction of the ionic scattering factor, which is equivalent to the atom's partial charge, alongside the usual atomic coordinates and displacement parameters [19]. This method is known as iSFAC modelling.
Step 3: Charge Assignment and Validation The iSFAC modelling outputs absolute partial charge values for every atom in the structure. These experimental charges should be compared with quantum chemical computations (showing Pearson correlations of >0.8 for organic compounds) to validate computational methods [19].
Table 2: Key Research Reagent Solutions for Featured Experiments
| Reagent / Material | Function in Protocol |
|---|---|
| Heavy Water (D₂O) | Probe for deuterium NMR spectroscopy; enables direct observation of water molecule dynamics in confinement [63]. |
| Molecular Crystals with Hydrophilic Nanopores | Host system for confining water; provides a well-defined nanoscale environment to study premelting phenomena [63]. |
| High-Quality Single Crystals | Essential for electron diffraction studies; enables accurate determination of electron density and partial charges via iSFAC modelling [19]. |
| Neural Network Potentials (NNPs) | Machine-learning force fields used in MD simulations to accurately and efficiently model thermal decomposition and lattice energies [64] [62]. |
Application to RESP Derivation
The experimentally determined partial charges serve as a critical benchmark for in silico charge derivation methods. For instance, the DF-RESP method, which accelerates charge calculation using density fitting, must be validated for its ability to reproduce charge distributions observed in real crystalline environments [6]. Furthermore, understanding environment-induced charge transfer, such as the negative charge found on carbon atoms in carboxylate groups in zwitterionic amino acids [19], provides essential physical insights for refining RESP parameterization and ensuring its transferability to condensed phases.
Diagram 2: Experimental charge determination workflow for benchmarking RESP.
Robust benchmarking against experimental data is non-negotiable for developing reliable computational protocols in drug development. The methodologies detailed herein—for validating lattice energy predictions, characterizing exotic states of nanoconfined water, and experimentally deriving atomic partial charges—provide a rigorous framework for enhancing the predictive power of molecular simulations. Integrating these benchmarked protocols, particularly into the RESP charge derivation pipeline for novel molecules, ensures that computational models are grounded in experimental reality, thereby de-risking the drug development process and accelerating the creation of effective therapeutics.
The accurate reproduction of the Quantum-Mechanically (QM) computed Electrostatic Potential (ESP) surrounding a material is a cornerstone in the development of reliable force fields for molecular simulations. In the context of force field parameterization, particularly for drug discovery applications, the Restrained Electrostatic Potential (RESP) method and its successors are widely used to derive partial atomic charges. These charges are critical for modeling non-bonded interactions, which largely determine the energetic landscape of molecular recognition, binding, and folding processes. The central challenge lies in developing electrostatic models that are not only accurate for a single, optimized geometry but also transferable across multiple geometric conformations that a molecule may adopt in dynamic simulations. Assessing the fidelity of these models requires robust, quantitative metrics. Among these, the Relative Root-Mean-Squared Error (RRMSE) has emerged as a key statistical measure for evaluating how well a computed ESP matches the reference QM-derived ESP across diverse molecular geometries.
This protocol details the application of RRMSE within a comprehensive framework for assessing charge models, specifically tailored for the derivation of RESP charges for novel molecules. It provides a standardized approach for researchers to benchmark the performance of charge assignment methods, ensuring the development of force fields that are both accurate and conformationally robust.
Theoretical Foundation: Key Concepts and Metrics
The Electrostatic Potential (ESP) in Force Fields
In fixed-charge force fields, the electrostatic interactions are modeled using a simple Coulombic potential between atom-centered point charges. The quality of this approximation hinges on how well these charges reproduce the true, QM-computed ESP around the molecule. The ESP is a physical observable that directly influences intermolecular interactions, making it a primary target for charge fitting procedures like RESP. A charge model that poorly reproduces the ESP will lead to inaccurate simulations of properties like solvation free energies, liquid densities, and binding affinities [2].
Root-Mean-Squared Error (RMSE) and Relative RMSE (RRMSE)
The Root-Mean-Squared Error (RMSE) is a standard metric for quantifying the difference between values predicted by a model and the values observed. In the context of ESP reproduction, it measures the average magnitude of the error in the electrostatic potential across a set of points surrounding the molecule [65].
Calculation of RMSE: For a given molecular conformation, the RMSE is calculated over a grid of points around the molecule. The formula is: ( RMSE = \sqrt{\frac{1}{N} \sum{i=1}^{N} (Vi^{\text{model}} - Vi^{\text{QM}})^2 } ) where ( N ) is the number of points, ( Vi^{\text{QM}} ) is the QM-computed ESP at point ( i ), and ( V_i^{\text{model}} ) is the ESP generated by the classical point-charge model.
Introduction of RRMSE: The Relative Root-Mean-Squared Error (RRMSE) normalizes the RMSE, providing a scale-independent metric that facilitates comparison across different molecules and systems with varying absolute ESP magnitudes [65]. It is often defined as the RMSE divided by the range or a norm of the reference QM ESP values. This normalization is crucial when comparing performance across a diverse set of materials, from organic molecules to transition metal complexes and nanoporous crystals.
Table 1: Key Quantitative Metrics for ESP Assessment
| Metric | Formula | Application in Charge Assessment |
|---|---|---|
| Root-Mean-Squared Error (RMSE) | ( \sqrt{\frac{1}{N} \sum{i=1}^{N} (Vi^{\text{model}} - V_i^{\text{QM}})^2 } ) | Quantifies the absolute average error in the reproduced ESP for a single conformation. |
| Relative RMSE (RRMSE) | ( \frac{RMSE}{\text{Norm}(V^{\text{QM}})} ) | Provides a normalized, scale-independent error metric for cross-system comparison. |
| Conformational Sensitivity | Standard deviation of RMSE/RRMSE across multiple conformations | Evaluates the transferability and robustness of a charge model against molecular geometry changes. |
Experimental Protocol for RRMSE Assessment
This protocol outlines the steps for evaluating charge derivation methods, using RRMSE as a primary metric, within the broader context of developing RESP charges for novel molecules.
Computational Setup and Workflow
A standardized computational workflow is essential for obtaining comparable and meaningful RRMSE values. The following diagram illustrates the key stages from molecule preparation to final evaluation.
Step-by-Step Methodological Details
Step 1: System Preparation and Conformational Sampling
For a novel molecule, begin with a geometry optimization at an appropriate level of theory (e.g., DFT with a medium-sized basis set). Subsequently, generate an ensemble of conformations to assess charge model transferability.
- Training Dataset: As demonstrated in recent studies, a robust assessment uses a training dataset containing 21 geometric conformations per material [65]. These should include the optimized ground-state geometry and non-equilibrium structures sampled via ab initio molecular dynamics (AIMD) or other conformation sampling methods.
- Validation Dataset: A separate validation dataset with 20 new geometric conformations per material should be used to test the model's predictive power on unseen geometries [65]. This rigorous splitting helps prevent overfitting and evaluates true conformational robustness.
Step 2: High-Quality Quantum Mechanical Reference Calculations
Perform single-point energy calculations on each conformation in the training and validation sets to generate the reference ESP.
- Recommended Level of Theory: For RESP2, the PW6B95 functional with the aug-cc-pV(D+d)Z basis set has been identified as providing an excellent balance of accuracy and computational cost for ESP calculations [2]. This represents a significant advancement over the traditional HF/6-31G* method, which relies on fortuitous error cancellation.
- ESP Grid Generation: Compute the QM ESP on a well-defined grid of points around the van der Waals surface of the molecule. The density and extent of this grid are critical for a representative RMSE calculation.
Step 3: Charge Assignment and Model ESP Calculation
Assign partial atomic charges using the method under investigation (e.g., RESP, RESP2, DDEC6, CM5).
- RESP2 Protocol: The next-generation RESP2 approach involves computing ESP charges in both the gas phase and an implicit aqueous solvent (e.g., using a Polarizable Continuum Model). The final charges are a linear combination:
Q_final = δ * Q_aqueous + (1-δ) * Q_gas. The parameter δ ≈ 0.6 (60% aqueous, 40% gas-phase) has been found to yield optimal accuracy for condensed-phase simulations when LJ parameters are co-optimized [2]. - Model ESP: Using the assigned partial charges, calculate the classical electrostatic potential at the same grid points used for the QM reference calculation.
Step 4: RRMSE Calculation and Analysis
For each conformation, calculate the RMSE between the model ESP and the QM ESP. Normalize the RMSE to obtain the RRMSE.
- Data Visualization: Use raincloud plots to visualize the resulting distribution of RRMSE (and RMSE) values across all conformations in the dataset [65]. These plots combine a density distribution, box plot, and raw data points, providing a comprehensive view of the model's performance and sensitivity.
- Performance Benchmarking: Compare the RRMSE distributions of different charge methods (e.g., RESP2, DDEC6, CM5, multiframe ESP methods) to identify which provides the best trade-off between accuracy and conformational transferability.
Comparative Data and Benchmarking of Charge Methods
Extensive benchmarking is vital for selecting an appropriate charge assignment method. Recent large-scale studies have evaluated multiple methods across diverse material types, including organic molecules, inorganic molecules, and transition metal complexes [65].
Table 2: Benchmarking Charge Assignment Methods via ESP RRMSE
| Charge Assignment Method | Typical RRMSE Performance | Conformational Sensitivity | Key Advantages and Limitations |
|---|---|---|---|
| Multiframe ESP Methods | Low RRMSE | Low | Best overall accuracy across conformations. Requires a training set with many geometries [65]. |
| QDR-DDEC6 / CM5 | Low to Moderate RRMSE | Low | Good transferability and accuracy even when trained on a single geometry [65]. |
| RESP2 (δ=0.6) | Moderate RRMSE | Moderate | Optimized for condensed-phase properties; improved over RESP1; depends on co-optimized LJ parameters [2]. |
| DDEC6_ad (with dipoles) | Very Low RRMSE | Low | Greatly improved ESP accuracy; more computationally intensive for force field simulations [65]. |
| QTAIM | Higher RRMSE | High | Not recommended for point-charge-only models; requires higher-order multipoles for accuracy [65]. |
The data reveals a key trade-off: while multiframe ESP methods achieve the highest accuracy, they require substantial computational resources for training. Methods like QDR-DDEC6 and CM5 offer a compelling balance, providing good accuracy and transferability with less conformational training data. Furthermore, including atomic dipoles (e.g., in the QDR-DDEC6_ad model) dramatically improves ESP reproduction beyond what is possible with atom-centered point charges alone [65].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Software and Computational Tools for ESP Assessment
| Tool / Resource | Type | Function in Protocol |
|---|---|---|
| psi4 | Quantum Chemistry Software | Performs QM geometry optimizations and reference ESP calculations at levels like PW6B95/aug-cc-pV(D+d)Z [2]. |
| ForceBalance | Optimization Software | Co-optimizes force field parameters (e.g., RESP2 δ and LJ parameters) against experimental condensed-phase data [2]. |
| RESP2 | Charge Assignment Protocol | Generates partial charges tuned for condensed-phase simulations by mixing gas- and aqueous-phase ESPs [2]. |
| DDEC6 | Electron Density Partitioning Method | Assigns atomic charges by partitioning the total electron density; known for good transferability [65]. |
| Latent Ewald Summation (LES) | Machine Learning Library | Infers atomic charges and long-range electrostatics from energy/force data; compatible with ML interatomic potentials [66] [67]. |
Advanced Considerations and Future Directions
Machine Learning and Latent Charge Models
The field is advancing with methods that infer electrostatics without explicit charge labels. The Latent Ewald Summation (LES) framework, for example, is a machine learning approach that infers hidden variables (interpreted as atomic charges) solely from total energy and atomic forces [67]. These "latent charges" are then used to compute long-range electrostatic interactions via Ewald summation. This approach demonstrates that it is possible to learn physically meaningful charges that reproduce dipole moments and Born effective charges without being explicitly trained on them, offering a promising path for future force field development [66] [67].
Beyond Point Charges: The Role of Multipoles
A fundamental limitation of atom-centered point-charge models is their inability to correctly reproduce the molecular multipole moments beyond the dipole in systems like homodiatomic molecules (H₂, N₂, O₂), which have a non-zero quadrupole moment [65]. This implies that for the highest accuracy, especially for modeling polarization and complex charge distributions, electrostatic models must incorporate atomic dipoles or quadrupoles, or use off-site charges. While this increases computational cost, methods like QDR-DDEC6_ad show it is a necessary step for pushing the accuracy frontier [65].
Integrated Workflow for Robust Charge Derivation
The following diagram synthesizes the core decision-making process for deriving and validating charges for a novel molecule, incorporating the key concepts of RRMSE assessment and method selection.
In the realm of computer-aided drug design and molecular dynamics (MD) simulations, the accurate representation of molecular electrostatic interactions is paramount. These interactions are predominantly governed by the assignment of atomic partial charges within the force fields used to simulate biological processes. The selection of an appropriate charge model is a critical step in the protocol for deriving charges for novel molecules, as it directly influences the accuracy of predicting properties such as hydration free energy, protein-ligand binding affinities, and bulk liquid properties. This application note provides a detailed comparison of four widely used charge models—RESP, RESP2, AM1-BCC, and CM5—framed within the context of deriving charges for novel chemical entities. We summarize quantitative performance data, outline detailed methodologies for charge derivation, and provide visual workflows to guide researchers in selecting and applying these models effectively.
Fundamental Principles of Each Model
RESP (Restrained Electrostatic Potential): RESP derives atomic partial charges by fitting a classical Coulomb model to the quantum mechanical (QM) molecular electrostatic potential (ESP) computed at the HF/6-31G* level of theory. A key feature is the use of a hyperbolic restraint function to attenuate the magnitudes of the charges, preventing over-polarization and improving agreement with experimental condensed-phase data [1]. It is considered a high-accuracy, though computationally expensive, method.
RESP2: A next-generation approach that addresses the inherent overpolarization of the original RESP method. RESP2 computes partial charges as a linear combination of charges derived from gas-phase and aqueous-phase QM calculations. The polarity is tuned by a parameter, δ (typically around 0.6, indicating 60% aqueous and 40% gas-phase), which aims to more accurately account for self-polarization in the condensed phase [2]. It uses a higher level of QM theory (e.g., PW6B95/aug-cc-pV(D+d)Z) for improved ESP accuracy [2].
AM1-BCC (Austin Model 1 - Bond Charge Corrections): This is a highly efficient semi-empirical method. It first calculates Mulliken charges using the AM1 Hamiltonian and then applies a set of pre-determined bond charge corrections (BCCs) to emulate the HF/6-31G* ESP [68]. Its primary advantage is speed, as it avoids expensive ab initio QM calculations, making it suitable for high-throughput applications.
CM5 (Charge Model 5): CM5 computes atomic charges through Hirshfeld population analysis derived from density functional theory (DFT) calculations [69] [70]. To make these charges suitable for condensed-phase simulations, an empirical scaling factor (e.g., 1.27) is often applied to account for polarization effects not captured in the gas-phase calculation [69].
Quantitative Performance Comparison
The following tables summarize the performance of these charge models in predicting key physicochemical properties as reported in the literature.
Table 1: Accuracy in Predicting Hydration Free Energies (HFEs)
| Charge Model | Force Field | Mean Unsigned Error (MUE) [kcal/mol] | Key Study Findings |
|---|---|---|---|
| AM1-BCC (Original) | GAFF2 | 1.03 | Baseline performance for neutral organic solutes [68]. |
| AM1-BCC (ABCG2) | GAFF2 | 0.37 | New BCC parameters developed for GAFF2 significantly improve HFE accuracy [68]. |
| RESP2 (δ=0.6) | SMIRNOFF99Frosst | ~0.8 - 1.0 | Shows improved accuracy for HFEs compared to RESP1 [2]. |
Table 2: Accuracy in Predicting Bulk Liquid Properties
| Charge Model | Force Field | Density Error (%) | Viscosity Error (%) | Heat of Vaporization (HOV) MUE (kcal/mol) | |
|---|---|---|---|---|---|
| RESP2 | GAFF | 2.1 - 3.07 | N/R | Performance is optimized when LJ parameters are co-optimized with charges [2]. | |
| AM1-BCC | GAFF | 2.1 - 3.07 | N/R | Performance similar to RESP2 for density with GAFF [69]. | |
| CM5 (scaled) | OPLS-AA | 0.1 - 0.5 | N/R | Excellent agreement with experimental density for acetic anhydride [69]. | |
| RESP1 | SMIRNOFF99Frosst | ~2.5 | N/R | ~0.8 | Baseline performance without LJ re-optimization [2]. |
| RESP2 (δ=0.6) | SMIRNOFF99Frosst (opt. LJ) | ~1.5 | N/R | ~0.5 | Significant improvement in density and HOV with optimized LJ parameters [2]. |
Table 3: Computational Cost and Typical Application Context
| Charge Model | Computational Cost | Typical Application Context | Key Software Tools |
|---|---|---|---|
| RESP | High | High-accuracy studies, small molecule sets, protein force fields (ff14SB) [71] | Gaussian, GAMESS, Psi4, resp in AmberTools [72] |
| RESP2 | High (~7x slower than RESP) | High-accuracy studies requiring condensed-phase accuracy, parameterization [2] | Psi4 [2] |
| AM1-BCC | Low | High-throughput screening, long MD simulations, database generation [68] [71] | ANTECHAMBER (AmberTools), UCSF Chimera [68] [71] |
| CM5 | Medium (DFT required) | Studies with OPLS-AA force field, where it shows excellent performance [69] | GAMESS-US, various DFT codes [69] |
Detailed Experimental Protocols
Protocol 1: RESP Charge Derivation
This protocol is used for deriving high-quality, conformationally robust partial charges.
- Conformational Sampling: For flexible molecules, generate multiple low-energy conformers. This ensures the derived charges are representative of the molecule's electrostatic character across its accessible conformational space.
- Quantum Mechanical Calculation: For each conformer, perform a geometry optimization and subsequent single-point energy calculation to compute the electrostatic potential (ESP). The traditional standard is the HF/6-31G* level of theory [1] [71], using programs like Gaussian or GAMESS.
- ESP Fitting with Restraints: The RESP fit is performed using the
resporpy_resp.pyprogram in AmberTools [72] [71]. This is typically a two-stage process:- Stage 1: Fit all atoms with a weak hyperbolic restraint (e.g., restraint weight of 0.0005 or 0.001) to determine heavy atom charges.
- Stage 2: Equate charges for chemically equivalent atoms (e.g., hydrogens in a methyl group) to maintain molecular symmetry, often using a stronger restraint [1].
Protocol 2: RESP2 Charge Derivation
RESP2 builds upon the RESP protocol to better account for environmental polarization.
- Dual-Phase QM Calculation: For the target molecule, perform two separate QM calculations to obtain the ESP:
- A gas-phase calculation.
- An aqueous-phase calculation using an implicit solvation model (e.g., IEF-PCM or SMD). Recommended Level of Theory: PW6B95 functional with the aug-cc-pV(D+d)Z basis set for a better accuracy-to-cost ratio [2].
- ESP Charge Fitting: Independently fit two sets of RESP charges (as in Protocol 1) from the gas-phase and aqueous-phase ESPs.
- Linear Combination: Generate the final RESP2 charges using the formula:
q_RESP2 = δ * q_aqueous + (1 - δ) * q_gaswhere the optimal mixing parameterδis approximately 0.6 for a wide range of organic liquids [2].
Protocol 3: AM1-BCC Charge Derivation
This protocol is designed for rapid charge generation without the need for ab initio QM.
- Molecular Input: Provide a 3D structure of the molecule in a format readable by ANTECHAMBER (e.g., MOL2).
- Semi-Empirical Calculation: The
antechamberprogram (part of AmberTools) automatically performs a semi-empirical calculation using the AM1 Hamiltonian to obtain Mulliken population analysis charges. - Apply Bond Charge Corrections (BCCs): The program then applies a library of pre-parameterized BCC terms to the AM1 Mulliken charges. For use with the GAFF2 force field, specify the new
abcg2parameter set to achieve higher accuracy for hydration free energies [68]. The command is typically:antechamber -i molecule.mol2 -fi mol2 -o molecule_bcc.mol2 -fo mol2 -c bcc -s 2.
Workflow Visualization
The following diagram illustrates the logical decision process for selecting and applying the appropriate charge model, integrating the protocols described above.
Decision Workflow for Charge Model Selection
The Scientist's Toolkit: Essential Research Reagents and Software
Table 4: Key Software Tools for Charge Derivation
| Tool Name | Function | Access/Reference |
|---|---|---|
| ANTECHAMBER | Automated molecular parameterization tool; implements AM1-BCC and interfaces with RESP. | Distributed with AmberTools [68]. |
| Psi4 | Open-source quantum chemistry package; includes integrated RESP and RESP2 plugins. | https://psicode.org/ [2] [72] |
| Gaussian | Quantum chemistry program for high-level ab initio and DFT calculations (e.g., for RESP ESP). | Commercial software [71]. |
| GAMESS-US | Quantum chemistry program for ab initio calculations. | Open-source [69]. |
| RESP Plugin | A dedicated plugin for Psi4 to perform RESP and RESP2 fittings. | Installed via Conda: conda install resp -c psi4 [72]. |
Table 5: Key Force Fields and Datasets
| Item | Description | Use Case |
|---|---|---|
| GAFF/GAFF2 | General Amber Force Field parameterized for organic molecules. | Primary force field for drug-like molecules with AM1-BCC or RESP charges [68]. |
| ff14SB | AMBER force field for proteins. | Uses RESP charges; serves as a benchmark for biomolecular simulations [71]. |
| OPLS-AA | Optimized Potentials for Liquid Simulations - All Atom. | Often paired with CM5 charges for organic liquids [69]. |
| ABCG2 Dataset | A set of 442 neutral organic solutes for validating HFEs. | Training and validation set for charge model development [68]. |
The choice between RESP, RESP2, AM1-BCC, and CM5 charge models involves a critical trade-off between computational cost, ease of use, and accuracy for specific properties. For high-throughput applications or long simulations, AM1-BCC offers an excellent balance of speed and reliability, especially with the newer ABCG2 parameters. For maximum accuracy in modeling condensed-phase behavior and hydration, RESP2 (with δ ≈ 0.6) represents the current state-of-the-art, though it requires significant computational resources and co-optimization of Lennard-Jones parameters for best results. The traditional RESP method remains a robust and accurate standard for systems where its computational cost is acceptable. Finally, the CM5 model, particularly when scaled, demonstrates exceptional performance with the OPLS-AA force field. By following the protocols and decision framework outlined in this application note, researchers can make informed, justified choices in their charge derivation pipeline for novel molecules.
The accuracy of binding affinity predictions is a central challenge in computational chemistry and rational drug design. Accurately predicting the binding free energies of small organic molecules to biological macromolecules can greatly accelerate drug discovery by reducing the number of compounds that must be synthesized [73]. A key source of error in these calculations is the force field, particularly the assignment of partial atomic charges to novel molecules [74]. The Restrained Electrostatic Potential (RESP) method is a widely adopted approach for deriving these charges, ensuring that the electrostatic potential generated by the point charges closely matches the quantum mechanical electrostatic potential [1].
Host-guest systems have emerged as powerful and tractable models for assessing force field inaccuracies, including those arising from charge derivation [74] [73]. These systems, which involve supramolecular hosts like cyclodextrins and cucurbiturils binding to small drug-like guests, reduce many complexities inherent to protein-ligand binding, such as slow conformational dynamics and ambiguous protonation states [74] [73]. This makes them ideal testbeds for benchmarking and validating computational protocols, such as RESP charge derivation, by providing well-defined systems with high-quality experimental binding data for comparison [74]. This case study examines the performance of RESP-derived charges in the context of host-guest binding thermodynamics, providing detailed protocols and quantitative benchmarks for researchers.
Performance Benchmarking of Force Fields in Host-Guest Systems
Quantitative benchmarking against experimental data is crucial for validating computational methods. Host-guest systems provide an excellent platform for this, as their binding thermodynamics can be measured with high accuracy using techniques like isothermal titration calorimetry (ITC) [74].
Binding Affinity Prediction Accuracy
The performance of force fields using RESP-derived charges (typically with the AM1-BCC method) has been evaluated in blinded community challenges. The Open Force Field Consortium, for instance, benchmarked its SMIRNOFF99Frosst force field against established force fields like GAFF on a set of 43 α- and β-cyclodextrin host-guest complexes [74].
Table 1: Performance of Force Fields on Cyclodextrin Host-Guest Binding Free Energies
| Force Field | Root Mean Square Error (RMSE) vs. Experiment | 95% Confidence Interval |
|---|---|---|
| SMIRNOFF99Frosst | 0.9 kcal/mol | [0.7, 1.1] kcal/mol |
| GAFF 1.7 | 0.9 kcal/mol | [0.7, 1.1] kcal/mol |
| GAFF 2.1 | 1.7 kcal/mol | [1.5, 1.9] kcal/mol |
Source: Adapted from [74]
The results demonstrate that the SMIRNOFF99Frosst and GAFF 1.7 force fields, both employing RESP/AM1-BCC charge models, perform competitively, achieving RMSEs of approximately 0.9 kcal/mol. This level of accuracy is significant for practical applications in drug discovery [74].
Thermodynamic Signature Analysis
Beyond binding free energies (ΔG°), host-guest systems allow for the dissection of the full thermodynamic signature of binding—enthalpy (ΔH) and entropy (ΔS°). The Attach-Pull-Release (APR) method, for example, can be used to compute all these quantities from molecular dynamics simulations [74]. Studies have shown that consistent, negative changes in heat capacity (ΔCp,b) are a unifying feature of host-guest complex formation in water, largely attributed to solvation effects [75]. Accurately capturing these nuanced thermodynamic components is a stringent test for any charge derivation method.
Protocols for RESP Charge Derivation and Application
The derivation of RESP charges is a critical step in preparing molecules for simulation. The following section outlines the core protocol and its application in host-guest studies.
Core Protocol for RESP Charge Derivation
The standard protocol for deriving RESP charges involves a series of quantum mechanical calculations and subsequent fitting procedures [76].
Diagram 1: Workflow for deriving RESP atomic partial charges.
Step 1: Geometry Optimization
- Begin with a 3D molecular structure, ensuring correct protonation states.
- Perform a quantum mechanical geometry optimization. A common method is B3LYP/6-31G* [76].
- For specific force fields, other levels may be required. For the polarizable ff02 force field, an optimization at HF/6-31G or B3LYP/DZpdf/6-31G (for transition metals) is recommended [76].
Step 2: Electrostatic Potential (ESP) Calculation
- Using the optimized geometry, perform a single-point energy calculation to compute the molecular electrostatic potential.
- The Pop=MK keyword in Gaussian is used for this purpose, along with specific IOp commands to control the density of sampling points (e.g.,
IOp(6/33=2,6/41=10,6/42=17)) [76]. - For the Amber ff94, ff99, ff14SB, ff19SB, and GAFF force fields, the ESP is typically calculated at the HF/6-31G* level. For ff02 and ff03, B3LYP/cc-pVTZ is used, with the latter also requiring a polarizable continuum model (IEFPCM) with a dielectric constant of 4 [76].
Step 3: RESP Charge Fitting
- The RESP fitting procedure restrains the magnitudes of the charges to avoid over-polarization, a known issue with unrestrained ESP charges [1]. The fit minimizes the function:
χ²_resp = χ²_esp + χ²_restraint, where the restraint term uses a hyperbolic function [1]. - This step is often automated using tools like
antechamberfrom the AmberTools suite [76]. A typical command is:antechamber -fi gout -fo prepi -c resp -i file.out -o res.in -rn MOL -at gaff -pf y - For advanced users, fitting for periodic systems or using density-fitting (DF-RESP) to accelerate calculations for large molecules is possible with software like CP2K [8] [6].
Protocol for Host-Guest Binding Thermodynamics
Once the host and guest molecules are parameterized with RESP charges, their binding thermodynamics can be computed using molecular dynamics simulations.
Table 2: Research Reagent Solutions for Host-Guest Studies
| Reagent / Tool | Function in Protocol | Application Note |
|---|---|---|
| Cyclodextrins (α, β) | Model host molecules | Neutral, well-characterized structures; ideal for benchmarking [74]. |
| Cucurbit[n]urils (CB7, CB8) | Model host molecules | Symmetric, rigid hosts; challenge methods with dewetting transitions [73]. |
| Octa-Acid (OA) & TEMOA | Deep-cavity host molecules | Low-symmetry hosts with hydrophobic binding sites; used in SAMPL challenges [73]. |
| AmberTools/antechamber | Automated RESP fitting | Streamlines charge derivation and force field parameter assignment [76]. |
| pAPRika Toolkit | Implements APR method | Calculates absolute binding free energies, enthalpies, and entropies [74]. |
| Open Force Field Toolkit | Assigns SMIRNOFF force fields | Uses SMIRKS-based atom typing, an alternative to traditional atom types [74]. |
Methodology: The Attach-Pull-Release (APR) Method The APR method is a highly reliable approach for predicting host-guest binding thermodynamics [74].
- System Setup: The host-guest complex is solvated in a water box (e.g., TIP3P) with counterions. The parameters for the host and guest are assigned using a force field with RESP charges (e.g., GAFF, SMIRNOFF).
- Umbrella Sampling: The guest is physically pulled from the binding site into the bulk solvent along a reaction coordinate. Multiple independent simulations (windows) are run, each with a harmonic restraint keeping the guest at a specific point along the pathway.
- Free Energy Calculation: The potential of mean force (PMF) along the pulling coordinate is constructed by combining data from all windows using the weighted histogram analysis method (WHAM). The standard binding free energy, ΔG°, is obtained from the PMF after applying an analytic correction for the standard state [74].
- Enthalpy and Entropy Calculation: The binding enthalpy (ΔH) is computed as the difference in the average potential energy of the solvated bound complex and the solvated dissociated complex. The binding entropy (TΔS°) is then derived from the relationship ΔG° = ΔH - TΔS° [74].
Discussion and Visual Synthesis
Host-guest systems are invaluable because they isolate force field error from other complexities. The reliable performance of force fields using RESP charges, as shown in Table 1, underscores the method's robustness for drug discovery applications.
Diagram 2: The logical role of host-guest systems in force field and charge validation.
The logic of using host-guest systems is summarized in Diagram 2. Their tractable nature allows researchers to isolate and identify deficiencies in force fields, such as inaccuracies in electrostatic models [73]. Subsequent community challenges, like those in the SAMPL series, then drive improvements in force fields and charge derivation protocols [73]. The development of new methods like DF-RESP, which dramatically accelerates charge calculation for large systems while maintaining accuracy, is a direct outcome of the need for efficient and reliable parameterization in drug discovery workflows [6].
The Restrained Electrostatic Potential 2 (RESP2) method represents a significant evolution in partial atomic charge derivation for fixed-charge force fields. Unlike its predecessor, RESP1, which relies on gas-phase quantum-mechanical (QM) calculations with fortuitous overpolarization, RESP2 integrates both gas-phase and aqueous-phase QM calculations to better mimic solvation effects. This protocol details the application of RESP2 for parametrizing novel molecules, emphasizing its superiority in reproducing condensed-phase properties (e.g., liquid densities, dielectric constants, and hydration free energies) when combined with optimized Lennard-Jones (LJ) parameters [2].
Quantitative Comparison: RESP1 vs. RESP2
RESP2 introduces a mixing parameter δ, which scales the contributions of gas-phase (δ) and aqueous-phase (1 – δ) charges. Optimized δ values (e.g., δ = 0.6) yield charges that balance polarization for accuracy in condensed-phase simulations [2]. Key performance metrics are summarized below:
Table 1: Accuracy of RESP2 vs. RESP1 for Condensed-Phase Properties
| Property | RESP1 (Baseline LJ) | RESP2 (δ = 0.6) + Optimized LJ | Experimental Reference |
|---|---|---|---|
| Density (g/cm³) MUE | 0.02–0.03 | 0.01–0.02 | Pure organic liquids [2] |
| Enthalpy of Vaporization MUE | 0.4–0.6 kcal/mol | 0.2–0.4 kcal/mol | Pure organic liquids [2] |
| Dielectric Constant MUE | 15–20% | 5–10% | Pure organic liquids [2] |
| Hydration Free Energy MUE | 1.0–1.5 kcal/mol | 0.5–1.0 kcal/mol | Hydration benchmarks [2] |
MUE = Mean Unsigned Error; LJ = Lennard-Jones parameters.
Table 2: QM Methods for Electrostatic Potential (ESP) Calculations
| QM Method | Dipole Moment Accuracy | ESP Accuracy | Speed vs. HF/6-31G* |
|---|---|---|---|
| HF/6-31G* (RESP1 baseline) | Low | Low | 1× (reference) |
| PW6B95/aug-cc-pV(D+d)Z | High | High | ~7× slower (gas) |
| Implicit Solvent (e.g., PCM) | N/A | N/A | ~20× slower (aqueous) |
PW6B95/aug-cc-pV(D+d)Z is recommended for RESP2 for optimal accuracy/speed balance [2].
Protocol for RESP2 Charge Derivation
The RESP2 charge derivation process integrates multiconformer ESP fitting and LJ parameter optimization. Below is a graphical representation of the workflow:
Step-by-Step Methodology
Multiconformer Generation
- Input: 3D molecular structure (e.g., SDF file).
- Tool: RDKit or OpenBabel for conformer sampling.
- Output: Ensemble of low-energy conformers (≥10 recommended).
QM ESP Calculations
- Gas Phase: Compute ESPs using PW6B95/aug-cc-pV(D+d)Z [2].
- Aqueous Phase: Repeat with an implicit solvent model (e.g., PCM).
- Software: Psi4 (v1.3.2 or higher) for QM calculations.
Charge Fitting with RESP2
- Fit charges to ESPs using RESP constraints (e.g., hyperbolic restraint).
- Calculate final charges as: RESP2δ = δ × Qgas + (1 – δ) × Qaq, where δ = 0.6 is optimal [2].
LJ Parameter Optimization
- Tool: ForceBalance software [2].
- Target Data: Liquid densities, enthalpies of vaporization, and dielectric constants.
- Output: Optimized LJ parameters (e.g., σ and ε).
Validation
- Test against experimental hydration free energies and liquid properties.
- Compare errors to baseline RESP1 (see Table 1).
The Scientist’s Toolkit: Research Reagent Solutions
Table 3: Essential Tools for RESP2 Implementation
| Tool/Software | Function | Application in RESP2 |
|---|---|---|
| Psi4 | Quantum-mechanical calculation of ESPs. | Computes gas-phase and aqueous-phase ESPs for charge fitting [2]. |
| PyPE_RESP | Automated RESP charge derivation. | Fits multiconformer/multimolecule charges; supports constraint groups [23]. |
| ForceBalance | Optimization of force field parameters. | Co-optimizes LJ parameters against experimental liquid data [2]. |
| RDKit | Cheminformatics and conformer generation. | Generates input conformers for QM calculations [23]. |
| AMBER Tools | Molecular simulation and parameterization. | Validates charges in MD simulations (e.g., hydration free energies) [2]. |
Visualizing the RESP2 Solvation Effect
RESP2 addresses electronic polarization by blending gas- and aqueous-phase charges. The diagram below illustrates this conceptual advancement:
RESP2 provides a systematic protocol for deriving partial charges that outperform RESP1 in condensed-phase simulations. By integrating QM calculations with implicit solvation and optimizing LJ parameters, researchers can achieve higher accuracy in drug design and molecular dynamics studies. Adoption of RESP2 (δ = 0.6) is recommended for novel molecule parametrization.
Conclusion
The RESP charge derivation protocol, especially when enhanced by modern tools and validation strategies, remains a powerful and essential method for creating reliable force fields. By understanding its theoretical foundation, meticulously applying its methodology, and rigorously validating outcomes against experimental data, researchers can generate high-quality parameters for novel molecules. Future directions point towards the increased use of implicitly polarized models like RESP2, which better account for solvation effects, and the ongoing development of automated, reproducible workflows. These advances will further improve the accuracy of molecular simulations, directly impacting rational drug design and the understanding of complex biomolecular interactions in biomedical research.
References
- 1. Restrained electrostatic potential atomic partial charges for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4191892/]
- 2. Non-bonded force field model with advanced restrained ... [https://www.nature.com/articles/s42004-020-0291-4]
- 3. The R.E.D. Tools: Advances in RESP and ESP charge ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2918240/]
- 4. RESP charges — OpenFF Recharge documentation [https://docs.openforcefield.org/projects/recharge/en/stable/users/resp.html]
- 5. Force Field Partial Charges with Restrained Electrostatic ... [https://chemrxiv.org/engage/chemrxiv/article-details/60c745a09abda22807f8c5ea]
- 6. Accelerating RESP Charge Calculation With Density Fitting [https://pubmed.ncbi.nlm.nih.gov/40965340/]
- 7. RESP ESP charge DDataBase: R.E.DD.B. [https://upjv.q4md-forcefieldtools.org/REDDB/]
- 8. RESP Charges [https://manual.cp2k.org/trunk/methods/properties/resp_charges.html]
- 9. Electrostatic potential maps - Beautiful Atoms - Read the Docs [https://beautiful-atoms.readthedocs.io/en/latest/tutorial/electrostatic_potential_map.html]
- 10. Comparison of Charge Derivation Methods Applied to Amino ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6641686/]
- 11. Fast, efficient generation of high-quality atomic charges. ... [https://pubmed.ncbi.nlm.nih.gov/12395429/]
- 12. RESP ESP charge Derive: Overview [https://upjv.q4md-forcefieldtools.org/RED/overview.php/resp/]
- 13. Add Charge [https://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/addcharge.html]
- 14. hesther/espsim: Scoring of shape and ESP similarity with ... [https://github.com/hesther/espsim]
- 15. AI Drug Discovery Enhanced by Novel 3D Molecule Model [https://www.technologynetworks.com/drug-discovery/news/ai-model-generates-realistic-3d-structures-of-potential-drug-molecules-399562]
- 16. Enhancing molecular design efficiency: Uniting language ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11026973/]
- 17. Biomolecular simulation and modelling: status, progress and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2706107/]
- 18. A review on application of molecular simulation technology ... [https://www.sciencedirect.com/science/article/pii/S2665927122001824]
- 19. Experimental determination of partial charges with electron ... [https://www.nature.com/articles/s41586-025-09405-0]
- 20. Quantum Computing Industry Trends 2025: A Year of ... [https://www.spinquanta.com/news-detail/quantum-computing-industry-trends-2025-breakthrough-milestones-commercial-transition]
- 21. Quantum Chemistry in the Cloud: Kvantify's QDK on IQM ... [https://meetiqm.com/blog/quantum-chemistry-in-the-cloud-kvantifys-qdk-on-iqm-resonance/]
- 22. Top quantum breakthroughs of 2025 [https://www.networkworld.com/article/4088709/top-quantum-breakthroughs-of-2025.html]
- 23. A Tool to Facilitate and Standardize Derivation of RESP ... [https://pubmed.ncbi.nlm.nih.gov/40285710/]
- 24. Unlocking Scalable Chemistry Simulations for Quantum- ... [https://www.quantinuum.com/blog/unlocking-scalable-chemistry-simulations-for-quantum-supercomputing]
- 25. A diverse and chemically relevant solvation model benchmark ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc06406f?page=search]
- 26. Conformational Sampling Optimization [https://bio-protocol.org/exchange/minidetail?id=9993209&type=30]
- 27. Feasibility of Combining Biomolecular Conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12308781/]
- 28. Molecular Electrostatic Potential (MEP) [https://www.cup.uni-muenchen.de/ch/compchem/pop/mep1.html]
- 29. Unveiling Drug Discovery Insights through Molecular ... [https://chemrxiv.org/engage/chemrxiv/article-details/6725dc987be152b1d0bce046]
- 30. The molecular electrostatic potential and drug design [https://www.sciencedirect.com/science/article/abs/pii/016612809287166W]
- 31. Molecular electrostatic potentials: an effective tool for the ... [https://pubmed.ncbi.nlm.nih.gov/2866089/]
- 32. Basis set (chemistry) [https://en.wikipedia.org/wiki/Basis_set_(chemistry)]
- 33. Basis Set FAQ [https://downloads.wavefun.com/FAQ/BasisSetFAQ.html]
- 34. Automation in the Life Science Research Laboratory - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7691657/]
- 35. AMBER: Resp Esp charge Derive (RED v-1.0) release [http://archive.ambermd.org/200312/0034.html]
- 36. RESP ESP charge Derive: R.E.D. III Tools release [https://upjv.q4md-forcefieldtools.org/RED/release3.php]
- 37. Automating molecule design to speed up drug development [https://news.mit.edu/2018/automating-molecule-design-speed-drug-development-0706]
- 38. Supplemental: Overview of the Common Force Fields [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/S01-Force_Fields_Overview/index.html]
- 39. RESP ESP charge DDataBase: R.E.DD.B. [http://q4md-forcefieldtools.org/REDDB/index.php]
- 40. advances in RESP and ESP charge derivation and force ... [https://pubs.rsc.org/en/content/articlelanding/2010/cp/c0cp00111b]
- 41. Validating CHARMM parameters and exploring charge ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2719862/]
- 42. a database for RESP and ESP atomic charges, and force field ... [https://pubmed.ncbi.nlm.nih.gov/17962302/]
- 43. a database for RESP and ESP atomic charges, and force field ... [https://www.hsls.pitt.edu/obrc/index.php?page=URL1209755497]
- 44. Re: [AMBER] RESP charge of the ligand [http://archive.ambermd.org/201210/0338.html]
- 45. Benchmark Study of the Performance of Density Functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3703816/]
- 46. Could Someone Give me Advice on Optimizing Basis Set ... [https://forum.psicode.org/t/could-someone-give-me-advice-on-optimizing-basis-set-selection-for-calculating-reaction-energies/3108]
- 47. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 48. Assessing molecular docking tools: understanding drug ... [https://fjps.springeropen.com/articles/10.1186/s43094-025-00862-y]
- 49. Moltiverse: Molecular Conformer Generation Using ... [https://pubmed.ncbi.nlm.nih.gov/40421608/]
- 50. BMCS 2nd Conformational Design in Drug Discovery 2025 [https://www.rscbmcs.org/events/conformationaldesign25/]
- 51. Accelerating RESP Charge Calculation With Density Fitting [https://ui.adsabs.harvard.edu/abs/2025JCoCh..46E0237Z/abstract]
- 52. Molecular Dynamics (MD)-Derived Features for Canonical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11863381/]
- 53. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 54. AFsample2 predicts multiple conformations and ensembles ... [https://www.nature.com/articles/s42003-025-07791-9]
- 55. AlphaFold2 Predicts Alternative Conformation Populations in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11423400/]
- 56. Structure prediction of alternative protein conformations [https://www.nature.com/articles/s41467-024-51507-2]
- 57. An Overview of Molecular Modeling for Drug Discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6539951/]
- 58. 2025 Guide to AI-Driven Protein Design & Machine Learning ... [https://nanohelix.ai/blog/ai-protein-design-guide-2025]
- 59. Restrained electrostatic potential atomic partial charges for ... [https://www.sciencedirect.com/science/article/abs/pii/S0166128000004875]
- 60. Conformational landscape of β-cyclodextrin [https://link.springer.com/article/10.1007/s10822-025-00694-1]
- 61. Lattice Energy Partitions in Crystals of Flexible Molecules ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576777/]
- 62. A robust crystal structure prediction method to support ... [https://www.nature.com/articles/s41467-025-57479-1]
- 63. Scientists finally capture water's hidden state that's both ... [https://www.sciencedaily.com/releases/2025/09/250922074936.htm]
- 64. Thermal Stability Ranking of Energetic Crystals via a ... [https://www.sciencedirect.com/science/article/pii/S2667134425000525]
- 65. How well do various QM-derived net atomic charges ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d4ra07900k?page=search]
- 66. A universal augmentation framework for long-range ... [https://arxiv.org/html/2507.14302v1]
- 67. Machine learning of charges and long-range interactions ... [https://www.nature.com/articles/s41467-025-63852-x]
- 68. A fast and high-quality charge model for the next generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7728379/]
- 69. A theoretical investigation of different point charges ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261420306229]
- 70. Quantum-based machine learning and AI models to ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1002535/full]
- 71. [AMBER] Partial charge discrepancy between AM1-BCC small ... [http://archive.ambermd.org/202207/0162.html]
- 72. A. Alenaizan's restricted electrostatic potential (RESP) ... [https://github.com/cdsgroup/resp]
- 73. Overview of the SAMPL6 host-guest binding affinity prediction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6301044/]
- 74. Binding thermodynamics of host-guest systems with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7328435/]
- 75. The temperature-dependence of host–guest binding ... [https://pubs.rsc.org/en/content/articlelanding/2023/sc/d3sc01975f]
- 76. How to run a RESP calculation [https://signe.teokem.lu.se/ulf/Methods/resp.html]