The Verlet Algorithm in Molecular Dynamics: A Complete Guide for Biomedical Researchers
This article provides a comprehensive exploration of the Verlet algorithm, a cornerstone of Molecular Dynamics (MD) simulations.
The Verlet Algorithm in Molecular Dynamics: A Complete Guide for Biomedical Researchers
Abstract
This article provides a comprehensive exploration of the Verlet algorithm, a cornerstone of Molecular Dynamics (MD) simulations. Tailored for researchers, scientists, and drug development professionals, we cover its foundational principles rooted in Newtonian mechanics, detailed implementation of its key variants (Velocity Verlet, Leap-Frog), and practical guidance for troubleshooting and optimizing simulations. The guide also includes a critical comparison with other numerical integrators and validates the algorithm's properties, highlighting its indispensable role in achieving accurate, stable, and energy-conserving simulations for biomolecular systems and drug discovery.
Understanding the Verlet Algorithm: From Newton's Laws to Numerical Integration
The Role of Numerical Integration in Molecular Dynamics Simulations
Molecular Dynamics (MD) simulation is a powerful computational technique that predicts how a system of atoms or molecules evolves over time by numerically solving Newton's equations of motion. The core of any MD simulation is the integration algorithm, a numerical procedure that calculates atomic trajectories by stepping forward in discrete time increments. The choice of integrator is critical, as it directly impacts the simulation's accuracy, stability, and ability to conserve energy—properties essential for producing physically meaningful results. Among the various algorithms available, the family of Verlet integrators has become the cornerstone of modern molecular dynamics due to its favorable numerical properties and computational efficiency. This article explores the fundamental role of numerical integration in MD, with a specific focus on the Verlet algorithm and its variants, providing a technical guide for researchers and scientists in computational drug development and related fields.
Fundamental Principles of Molecular Dynamics
In Molecular Dynamics, the motion of a system of N atoms is described by Newton's second law:
[ Fi = mi ai = mi \frac{\partial^2 r_i}{\partial t^2} ]
where ( Fi ) is the force acting on atom ( i ), ( mi ) is its mass, ( ai ) is its acceleration, and ( ri ) is its position. The force is also equal to the negative gradient of the potential energy ( V ) that governs the interatomic interactions:
[ Fi = -\frac{\partial V}{\partial ri} ]
The analytical solution to these equations is not feasible for systems with more than two atoms; therefore, MD relies on numerical integration to approximate the trajectories. The general MD procedure involves:
- Initialization: Defining initial atomic positions and velocities.
- Force Calculation: Computing forces on all atoms based on the current configuration (the most computationally expensive step).
- Integration: Using an integration algorithm to update atomic positions and velocities over a small time step ( \Delta t ) (typically on the order of femtoseconds, ( 10^{-15} ) s).
- Iteration: Repeating steps 2 and 3 for thousands to millions of time steps to generate a trajectory.
The integration algorithm must balance several requirements: accuracy in describing atomic motion, stability in conserving system energy, simplicity of implementation, and computational speed.
The Verlet Algorithm: A Family of Integrators
The original Verlet algorithm, first used in 1791 and rediscovered by Loup Verlet in the 1960s, is one of the most widely used integration methods in MD. Its popularity stems from its numerical stability, time-reversibility, preservation of the symplectic form on phase space, and relatively low computational cost compared to simpler methods like Euler integration.
Mathematical Foundation
Verlet integration is derived from Taylor expansions of the position ( r(t) ) around time ( t ):
[ r(t + \Delta t) = r(t) + \Delta t v(t) + \frac{1}{2} \Delta t^2 a(t) + \frac{1}{6} \Delta t^3 b(t) + \mathcal{O}(\Delta t^4) ]
[ r(t - \Delta t) = r(t) - \Delta t v(t) + \frac{1}{2} \Delta t^2 a(t) - \frac{1}{6} \Delta t^3 b(t) + \mathcal{O}(\Delta t^4) ]
where ( v(t) ) is velocity, ( a(t) ) is acceleration, and ( b(t) ) is the third derivative (jerk). Adding these two equations eliminates the velocity and odd-order terms, yielding the basic Verlet algorithm:
[ r(t + \Delta t) = 2r(t) - r(t - \Delta t) + a(t) \Delta t^2 + \mathcal{O}(\Delta t^4) ]
The remarkable property of this formulation is that the position update has a local error of ( \mathcal{O}(\Delta t^4) ), despite not explicitly involving velocity.
Limitations and Variants
The basic Verlet algorithm has two main drawbacks:
- Velocities are not explicitly calculated, yet are needed for computing kinetic energy.
- It is not self-starting, as it requires positions from the previous time step ( r(t - \Delta t) ).
To address these limitations, several variants have been developed. The following workflow illustrates the relationships between the core Verlet algorithms and their key characteristics:
Key Verlet Integration Methods
Standard Verlet Algorithm
The standard Verlet method, also known as the Størmer-Verlet method, uses the position-update formula derived above. Although velocities are not needed for the trajectory propagation, they can be approximated to ( \mathcal{O}(\Delta t^2) ) accuracy using the central difference formula:
[ v(t) = \frac{r(t + \Delta t) - r(t - \Delta t)}{2\Delta t} ]
This approach is simple and memory-efficient but suffers from the velocities being out of sync with the positions.
Velocity Verlet Algorithm
The Velocity Verlet algorithm is the most commonly used variant in modern MD codes because it explicitly calculates positions and velocities at the same time instant. The algorithm proceeds in three steps:
- Update positions using current velocities and accelerations: [ r(t + \Delta t) = r(t) + v(t) \Delta t + \frac{1}{2} a(t) \Delta t^2 ]
- Calculate velocities at the half-step and compute new accelerations from forces at the new positions: [ v(t + \frac{\Delta t}{2}) = v(t) + \frac{1}{2} a(t) \Delta t ] [ a(t + \Delta t) = \frac{1}{m} F(r(t + \Delta t)) ]
- Complete the velocity update using the new acceleration: [ v(t + \Delta t) = v(t + \frac{\Delta t}{2}) + \frac{1}{2} a(t + \Delta t) \Delta t ]
In practice, this is often implemented as a two-step process for the velocity, combining steps 2 and 3 into: [ v(t + \Delta t) = v(t) + \frac{1}{2} [a(t) + a(t + \Delta t)] \Delta t ]
Velocity Verlet is time-reversible, symplectic (which leads to good long-term energy conservation), and requires only one force evaluation per time step.
Leapfrog Algorithm
The Leapfrog algorithm is another popular variant where velocities and positions are updated at slightly different times. The algorithm is defined by:
[ v(t + \frac{1}{2}\Delta t) = v(t - \frac{1}{2}\Delta t) + a(t) \Delta t ] [ r(t + \Delta t) = r(t) + v(t + \frac{1}{2}\Delta t) \Delta t ]
If full-step velocities are needed, they can be approximated as: [ v(t) = \frac{1}{2} \left[ v(t - \frac{1}{2}\Delta t) + v(t + \frac{1}{2}\Delta t) \right] ]
Leapfrog is computationally efficient and numerically stable, but the non-synchronous updates of positions and velocities can be inconvenient.
Comparative Analysis of Verlet Integrators
The following table provides a quantitative comparison of the key Verlet integrators and their properties:
Table 1: Comparison of Verlet Integration Algorithms
| Method | Position Error | Velocity Calculation | Velocity Error | Key Advantages | Key Disadvantages |
|---|---|---|---|---|---|
| Standard Verlet | (\mathcal{O}(\Delta t^4)) | Not explicit | N/A | High position accuracy; Simple | Velocities not directly available |
| Störmer-Verlet | (\mathcal{O}(\Delta t^4)) | Derived from positions | (\mathcal{O}(\Delta t^2)) | Good for energy calculations | Lower velocity accuracy |
| Leapfrog Verlet | (\mathcal{O}(\Delta t^4)) | At half-steps | (\mathcal{O}(\Delta t^2)) | Economical memory use | Positions and velocities out of sync |
| Velocity Verlet | (\mathcal{O}(\Delta t^2)) | Explicit at full-step | (\mathcal{O}(\Delta t^2)) | Complete phase information; Self-starting | Slightly higher position error |
The performance of these integrators can also be compared against other common numerical methods:
Table 2: Performance and Stability Comparison of Integration Algorithms
| Method | Evaluations of Acceleration Function | Stability: Underdamped Systems | Stability: Overdamped Systems | Overall Accuracy |
|---|---|---|---|---|
| Euler | 1 | Poor | Excellent | Low |
| Velocity Verlet | 1 | Excellent | Good | High |
| Improved Euler | 2 | Good | Fair | Medium |
| Runge-Kutta 4 (RK4) | 4 | Good | Poor | Very High |
As shown in Table 2, Velocity Verlet provides an excellent balance between computational cost (only one force evaluation per step) and stability, particularly for the underdamped systems commonly encountered in molecular dynamics.
Advanced Applications and Optimizations
Molecular Dynamics with Constraints
In simulations of molecules with strong chemical bonds, the standard Verlet algorithm is often combined with constraint algorithms like SHAKE and RATTLE. These methods rigidly enforce bond lengths and angles, preventing numerically stiff forces from limiting the time step. The original Verlet algorithm works naturally with SHAKE, while Velocity Verlet requires RATTLE, which applies constraints to both positions and velocities. Recent research has focused on optimizing these constraint algorithms to reduce the computational overhead associated with the double calculation of Lagrange multipliers in standard implementations.
Optimized Verlet-like Algorithms
Research continues to develop improved versions of Verlet algorithms. For instance, one study introduced explicit velocity- and position-Verlet-like algorithms containing a free parameter that is optimized to minimize the influence of truncated terms. These optimized algorithms demonstrate improved accuracy in simulations of Lennard-Jones fluids while maintaining the same computational cost and preserving the symplectic and time-reversible properties of the original Verlet methods.
Experimental Protocol: Implementing Velocity Verlet
For researchers implementing MD simulations, the following detailed protocol outlines the Velocity Verlet integration process:
Research Reagent Solutions
Table 3: Essential Components for MD Simulations
| Component | Function |
|---|---|
| Initial Atomic Coordinates | Provides starting configuration of the molecular system (e.g., from protein data bank structures) |
| Force Field Parameters | Defines potential energy function (bonds, angles, dihedrals, non-bonded interactions) |
| Integration Time Step (( \Delta t )) | Discrete time increment (typically 0.5-2 fs) for numerical integration |
| Thermostat/Barrowstat | Maintains constant temperature/pressure (e.g., Nosé-Hoover, Berendsen) |
| Periodic Boundary Conditions | Simulates bulk environment while minimizing finite-size effects |
| Neighbor Lists | Optimizes non-bonded force calculations (e.g., Verlet lists) |
Step-by-Step Methodology
System Initialization:
- Read initial atomic positions ( r(0) ) and velocities ( v(0) ).
- Assign masses ( m_i ) to all atoms.
- Compute initial forces ( F(0) ) and accelerations ( a(0) = F(0)/m_i ).
Main Integration Loop (repeat for desired number of steps):
- Position Update: Calculate new positions using: [ r(t + \Delta t) = r(t) + v(t) \Delta t + \frac{1}{2} a(t) \Delta t^2 ]
- Apply Constraints: If using constraint algorithms like RATTLE, enforce distance constraints on new positions.
- Force Calculation: Compute forces ( F(t + \Delta t) ) and accelerations ( a(t + \Delta t) ) based on new positions (most computationally intensive step).
- Velocity Update: Complete the velocity step using: [ v(t + \Delta t) = v(t) + \frac{1}{2} [a(t) + a(t + \Delta t)] \Delta t ]
- Apply Thermostat/Barostat: If simulating canonical (NVT) or isothermal-isobaric (NPT) ensembles, adjust velocities and positions accordingly.
- Sample Data: Record positions, velocities, energies, and other properties at specified intervals.
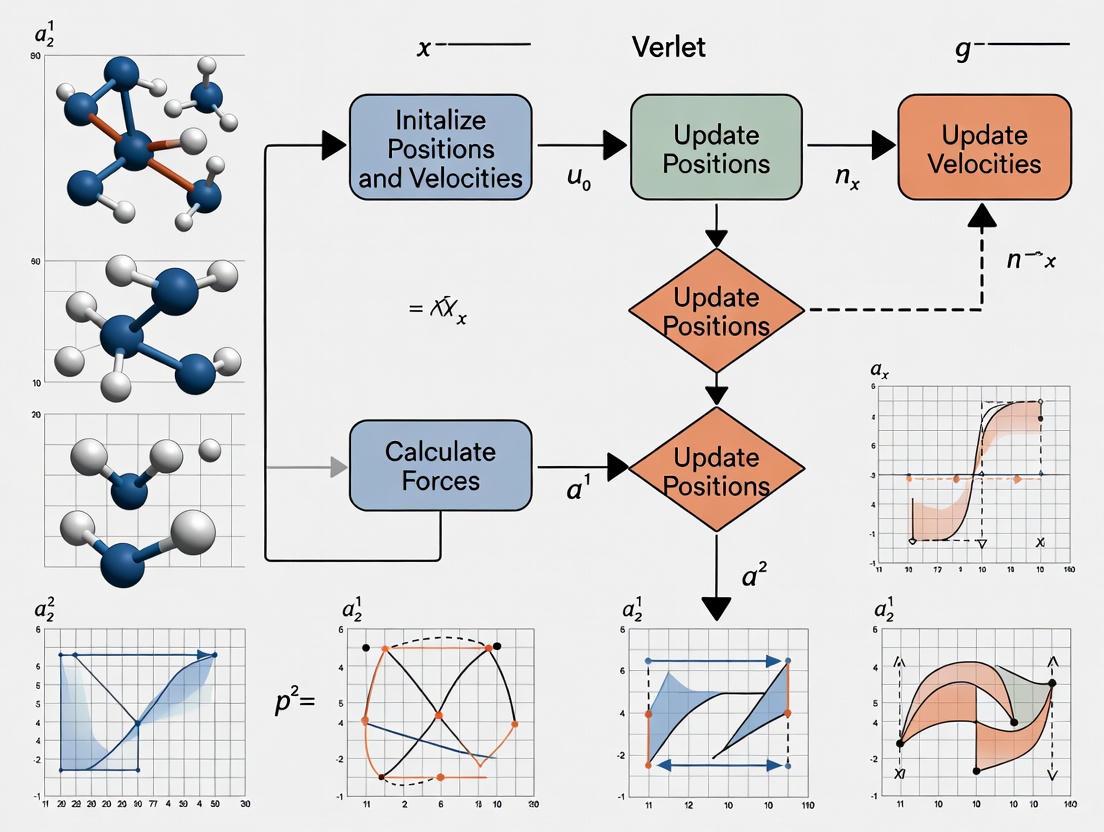
The following diagram illustrates the complete workflow of a molecular dynamics simulation using the Velocity Verlet integrator:
Numerical integration, particularly through the Verlet family of algorithms, plays an indispensable role in molecular dynamics simulations. The Velocity Verlet algorithm has emerged as the preferred method in most modern MD applications due to its combination of numerical stability, time-reversibility, symplectic properties, and explicit calculation of velocities. While the basic Verlet method provides excellent position accuracy, the need for velocity-dependent calculations in many applications makes Velocity Verlet the more versatile choice. Ongoing research continues to optimize these algorithms, particularly for handling constraints and improving accuracy without increasing computational cost. For researchers in drug development and molecular science, understanding these integration techniques is crucial for designing reliable simulations that can accurately predict molecular behavior and interactions.
Molecular dynamics (MD) is a computational technique that derives statements about the structural, dynamical, and thermodynamical properties of molecular systems by simulating their motion over time [1]. At the heart of every MD simulation lies the numerical integration of Newton's equations of motion, which describe how atomic positions evolve under the influence of interatomic forces [2]. The model system typically consists of a biomolecule such as a protein or enzyme immersed in an aqueous solvent, represented as a collection of interacting particles described as atoms [1].
In atomistic "all-atom" MD, the simulation box contains tens of thousands of atoms, and successive states at regular time intervals are stored in a trajectory for analysis [1]. The choice of integration algorithm is crucial, as it determines the numerical stability, accuracy, and conservation properties of the simulation [2]. This technical guide explores the mathematical foundation, implementation details, and practical applications of the Verlet family of algorithms, which have become the standard for MD simulations in drug discovery and pharmaceutical development.
Newton's Equations of Motion in Molecular Dynamics
Mathematical Foundation
Newton's equation of motion for conservative physical systems is expressed as:
[ \boldsymbol{M}\ddot{\mathbf{x}}(t) = \mathbf{F}(\mathbf{x}(t)) = -\nabla V(\mathbf{x}(t)) ]
For a system of N particles, this becomes:
[ mk\ddot{\mathbf{x}}k(t) = Fk(\mathbf{x}(t)) = -\nabla{\mathbf{x}_k}V(\mathbf{x}(t)) ]
Where:
- (t) represents time
- (\mathbf{x}(t) = (\mathbf{x}1(t), \ldots, \mathbf{x}N(t))) represents the positions of N particles
- (V) is the potential energy function
- (F) represents the forces acting on the particles
- (\boldsymbol{M}) is the diagonal mass matrix with elements (m_k) [2]
After transformation to bring mass to the right side, the equation simplifies to:
[ \ddot{\mathbf{x}}(t) = \mathbf{A}(\mathbf{x}(t)) ]
Where (\mathbf{A}(\mathbf{x})) represents the acceleration, with initial conditions (\mathbf{x}(0) = \mathbf{x}0) and (\mathbf{v}(0) = \dot{\mathbf{x}}(0) = \mathbf{v}0) [2].
Discretization for Numerical Solution
To discretize and numerically solve this initial value problem, a time step (\Delta t > 0) is chosen, and the time grid (tn = n\Delta t) is defined. The numerical approximation (\mathbf{x}n \approx \mathbf{x}(t_n)) is computed sequentially [2]. While Euler's method uses the forward difference approximation to the first derivative, Verlet integration employs the central difference approximation to the second derivative:
[ \frac{\Delta^2\mathbf{x}n}{\Delta t^2} = \frac{\mathbf{x}{n+1} - 2\mathbf{x}n + \mathbf{x}{n-1}}{\Delta t^2} = \mathbf{a}n = \mathbf{A}(\mathbf{x}n) ]
This formulation provides the mathematical basis for the Verlet algorithm [2].
The Verlet Integration Algorithm Family
Basic Størmer-Verlet Algorithm
The original Verlet algorithm uses positions at two successive time steps without explicitly storing velocities [2]. The procedure is as follows:
- Set (\mathbf{x}1 = \mathbf{x}0 + \mathbf{v}0\Delta t + \frac{1}{2}\mathbf{A}(\mathbf{x}0)\Delta t^2)
- For n = 1, 2, ... iterate: (\mathbf{x}{n+1} = 2\mathbf{x}n - \mathbf{x}{n-1} + \mathbf{A}(\mathbf{x}n)\Delta t^2) [2]
While velocities are not needed to compute trajectories, they can be estimated for calculating observables like kinetic energy using:
[ \mathbf{v}(t+\Delta t) = \frac{\mathbf{r}(t+\Delta t) - \mathbf{r}(t-\Delta t)}{2\Delta t} ]
The Verlet algorithm is time-reversible and energy-conserving, making it suitable for MD simulations [3].
Velocity Verlet Algorithm
The Velocity Verlet algorithm explicitly incorporates velocity, solving the problem of the first time step in the basic Verlet algorithm [3]. This algorithm maintains synchronous position and velocity vectors through these steps:
- (\mathbf{v}i(t + \frac{1}{2}\Delta t) = \mathbf{v}i(t) + \frac{\mathbf{F}i(t)}{2mi}\Delta t)
- (\mathbf{r}i(t + \Delta t) = \mathbf{r}i(t) + \mathbf{v}_i(t + \frac{1}{2}\Delta t)\Delta t)
- Compute forces (\mathbf{F}_i(t+\Delta t)) from the new positions
- (\mathbf{v}i(t + \Delta t) = \mathbf{v}i(t+\frac{1}{2}\Delta t) + \frac{\mathbf{F}i(t+\Delta t)}{2mi}\Delta t) [4]
Table 1: Comparison of Verlet Integration Algorithms
| Algorithm | Velocity Handling | Storage Requirements | Implementation Complexity | Conservation Properties |
|---|---|---|---|---|
| Original Verlet | Implicit, calculated retrospectively | Lower (positions only) | Moderate | Time-reversible, energy-conserving |
| Velocity Verlet | Explicit, synchronous with positions | Higher (positions + velocities) | Straightforward | Time-reversible, energy-conserving |
| Leap-Frog | Explicit, staggered half-step | Moderate (positions + half-step velocities) | Moderate | Time-reversible, energy-conserving |
Leap-Frog Variant
The leap-frog algorithm is a modified version where velocities are not calculated at the same time as positions [3]. Instead, positions and velocities are computed at interleaved time points:
- Derive (\mathbf{a}(t)) from the interaction potential using positions (\mathbf{r}(t))
- Compute (\mathbf{v}(t+\frac{\Delta t}{2}) = \mathbf{v}(t-\frac{\Delta t}{2}) + \mathbf{a}(t)\Delta t)
- Compute (\mathbf{r}(t+\Delta t) = \mathbf{r}(t) + \mathbf{v}(t+\frac{\Delta t}{2})\Delta t) [3]
Though the restart files differ between Leap-Frog and Velocity Verlet, they produce equivalent trajectories [3].
Discretization Error and Numerical Stability
Error Analysis
The time symmetry inherent in the Verlet method reduces local discretization errors by removing all odd-degree terms [2]. Using Taylor expansions:
[ \mathbf{x}(t+\Delta t) = \mathbf{x}(t) + \mathbf{v}(t)\Delta t + \frac{\mathbf{a}(t)\Delta t^2}{2} + \frac{\mathbf{b}(t)\Delta t^3}{6} + \mathcal{O}(\Delta t^4) ]
[ \mathbf{x}(t-\Delta t) = \mathbf{x}(t) - \mathbf{v}(t)\Delta t + \frac{\mathbf{a}(t)\Delta t^2}{2} - \frac{\mathbf{b}(t)\Delta t^3}{6} + \mathcal{O}(\Delta t^4) ]
Adding these expansions gives:
[ \mathbf{x}(t+\Delta t) = 2\mathbf{x}(t) - \mathbf{x}(t-\Delta t) + \mathbf{a}(t)\Delta t^2 + \mathcal{O}(\Delta t^4) ]
This formulation demonstrates that the Verlet algorithm has fourth-order accuracy in position despite using only second-order derivatives [2].
Stability Considerations
Verlet integrators are stable for time steps satisfying (\Delta t \leq \frac{1}{\pi f}), where (f) is the oscillation frequency of the fastest motions in the system [3]. In molecular systems, the fastest motions are typically:
- Bond vibrations involving hydrogens: period of ~10 fs
- Bond vibrations of heavy atoms and angles involving hydrogens: period of ~20 fs
Since the period of oscillation of a C-H bond is about 10 fs, Verlet integration is theoretically stable for time steps < 3.2 fs, though in practice, a time step of 1 fs is recommended to reliably describe this motion [3].
Practical Implementation in MD Simulations
Time Step Selection and Constraints
Selecting an appropriate time step involves balancing computational efficiency with numerical accuracy. Larger time steps allow faster simulation but decrease accuracy [3]. Several strategies can increase the usable time step:
- Constraining bonds with hydrogens: Allows doubling the time step to 2 fs
- Constraining all bonds and angles involving hydrogen atoms: Enables further increase
- Hydrogen mass repartitioning: Allows increasing time step to 4 fs by redistributing mass from heavy atoms to bonded hydrogens [3]
Table 2: Time Step Selection Guidelines for MD Simulations
| Constraint Method | Typical Time Step | Accuracy Considerations | Computational Overhead |
|---|---|---|---|
| No constraints | 1 fs | Accurately captures all bond vibrations | Lowest |
| H-bond constraints | 2 fs | Requires LINCS or SHAKE algorithms | Moderate |
| All-bond constraints | 2-3 fs | May affect certain conformational changes | Moderate to high |
| Hydrogen mass repartitioning | 4 fs | Alters dynamics but preserves thermodynamics | Low |
Constraint Algorithms
To constrain bond lengths without affecting system dynamics and energetics, specialized algorithms are employed:
- SHAKE: Iterative algorithm that resets all bonds to constrained values sequentially until desired tolerance is achieved. Simple and stable, applicable to large molecules, but slower than alternatives and difficult to parallelize [3].
- LINCS: Linear constraint solver, 3-4 times faster than SHAKE and easier to parallelize. P-LINCS allows constraining all bonds in large molecules but is not suitable for constraining both bonds and angles [3].
- SETTLE: Very fast analytical solution for small molecules, widely used to constrain bonds in water molecules [3].
Implementation in MD Software Packages
Different MD software packages implement Verlet integration with varying specifications:
GROMACS offers multiple integration algorithms specified in the run parameter mdp file [3]:
integrator = md(leap-frog algorithm)integrator = md-vv(velocity Verlet algorithm)integrator = md-vv-avek(velocity Verlet with averaged kinetic energy)integrator = sd(accurate leap-frog stochastic dynamics)integrator = bd(Euler integrator for Brownian dynamics)
NAMD uses Verlet integration as its only available method, with parameters specified in the configuration file [3]:
timestep = 2.0(time step in femtoseconds)nonbondedFreq = 2(frequency of nonbonded evaluations)fullElectFrequency = 4(frequency of full electrostatic evaluations)
Applications in Drug Discovery and Pharmaceutical Development
Role in Modern Drug Development
MD simulations have become increasingly useful in the modern drug development process [1]. Key applications include:
- Target validation: MD studies provide important insights into the dynamics and function of identified drug targets such as sirtuins, RAS proteins, and intrinsically disordered proteins [1].
- Antibody design: MD facilitates evaluation of binding energetics and kinetics of ligand-receptor interactions [1].
- Membrane protein studies: MD enables the study of G-protein coupled receptors and ion channels in their biological lipid bilayer environment [1].
- Formulation development: MD assists in studying crystalline and amorphous solids, stability of amorphous drug formulations, and drug solubility [1].
Experimental Protocols
A typical MD simulation protocol for drug discovery applications involves these key steps [5] [1]:
System Preparation
- Obtain protein structure from Protein Data Bank or through homology modeling
- Place the biomolecule in an appropriate simulation box
- Add solvent molecules and ions to mimic physiological conditions
- Energy minimization to remove steric clashes
Equilibration Phase
- Gradually heat the system to target temperature (e.g., 274K for Argon simulations [5])
- Apply position restraints on solute atoms during initial equilibration
- Achieve stable temperature and pressure through controlled dynamics
Production Simulation
- Run extended simulation with Verlet integration (typically Velocity Verlet or Leap-Frog)
- Use time steps of 1-4 fs depending on constraints
- Simulate for nanoseconds to microseconds depending on system size and research question
- Save trajectories at regular intervals for analysis
Analysis Phase
- Calculate structural properties (RMSD, RMSF)
- Analyze binding energies and interactions
- Study conformational changes and dynamics
- Compare with experimental data for validation
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Computational Tools for MD Simulations
| Item | Function/Purpose | Examples/Specifications |
|---|---|---|
| Force Fields | Define potential energy functions governing atomic interactions | CHARMM, AMBER, GROMOS; include bonded and non-bonded interaction terms [1] |
| MD Software Packages | Provide simulation engines with integration algorithms | GROMACS, AMBER, NAMD, CHARMM; implement Verlet algorithms [1] |
| Solvation Models | Represent aqueous environment surrounding biomolecules | TIP3P, SPC, TIP4P water models; periodic boundary conditions [1] |
| Long-Range Electrostatic Methods | Handle electrostatic interactions beyond cut-off distance | Particle Mesh Ewald (PME), Particle-Particle Particle-Mesh; essential for accuracy [1] |
| Temperature/Pressure Couplers | Maintain constant temperature and pressure | Nosé-Hoover, Berendsen, Parrinello-Rahman algorithms; mimic laboratory conditions [1] |
| Visualization Tools | Analyze and visualize simulation trajectories | VMD, PyMOL, Chimera; enable structural analysis and figure generation |
| (E)-2-Butenyl-4-methyl-threonine | (E)-2-Butenyl-4-methyl-threonine, CAS:81135-57-1, MF:C9H17NO3, MW:187.24 g/mol | Chemical Reagent |
| 9-Hydroxyspiro[5.5]undecan-3-one | 9-Hydroxyspiro[5.5]undecan-3-one, CAS:154464-88-7, MF:C11H18O2, MW:182.26 g/mol | Chemical Reagent |
Future Perspectives
With continuing advances in computing power, the role of MD simulations in facilitating the drug development process is likely to grow substantially [1]. Key developments include:
- Enhanced sampling methods: Improved algorithms for exploring configuration space more efficiently
- Force field improvements: More accurate parameterization for diverse molecular systems
- Hybrid quantum/classical methods: Combining accuracy of quantum mechanics with efficiency of classical MD
- Machine learning integration: Using neural networks to accelerate simulations and improve accuracy
- Large-scale simulations: Modeling increasingly complex systems such as entire viral envelopes or cellular components [1]
As these technical advancements mature, Verlet integration and its variants will continue to provide the fundamental numerical framework for propagating Newton's equations of motion in molecular systems, maintaining their position as the cornerstone of molecular dynamics simulations in pharmaceutical research and development.
The Verlet algorithm represents a cornerstone of molecular dynamics (MD) simulations, enabling researchers to numerically integrate Newton's equations of motion with exceptional stability and precision. Originally developed by Jean Baptiste Delambre in 1791, subsequently rediscovered multiple times, and formally established by Loup Verlet in the 1960s, this algorithm has revolutionized computational studies of biomolecular systems. Its mathematical formalism provides time-reversible integration with minimal computational overhead, making it particularly valuable for drug development professionals studying protein-ligand interactions, conformational changes, and thermodynamic properties. This technical guide examines the historical trajectory, mathematical foundations, and practical implementations of Verlet algorithms, with specific attention to their applications in modern molecular research and pharmaceutical development.
Molecular Dynamics (MD) has emerged as an indispensable methodology for simulating complex molecular systems, providing insights into structural biology, drug design, and materials science that are often inaccessible through experimental techniques alone [6]. At its core, MD solves Newton's equations of motion for atoms by iterating through small time steps, using numerical integration to predict new atomic positions and velocities based on calculated forces [7]. The revolutionary advances in computer technology since the first MD simulations in the late 1950s have been paralleled by algorithmic improvements, with the Verlet algorithm family standing as the most significant development in ensuring numerical stability and physical fidelity [6].
The Verlet integration scheme satisfies fundamental requirements for MD integration algorithms: accuracy in describing atomic motion, stability in conserving system energy and temperature, simplicity for programming implementation, speed in calculation, and economy in computing resources [7]. These characteristics have made Verlet algorithms the predominant choice for molecular dynamics programmers, particularly in pharmaceutical research where simulations of protein folding, ligand binding, and allosteric regulation provide critical insights for drug development [6].
Historical Development and Rediscovery
The mathematical foundations of what would become known as Verlet integration have experienced multiple independent discoveries throughout scientific history, reflecting its fundamental nature for solving second-order differential equations. This recurrent rediscovery pattern underscores the algorithm's intrinsic mathematical elegance and practical utility across diverse scientific domains.
Chronological Development Pathway
Table: Historical Timeline of Verlet Algorithm Development
| Year | Scientist | Contribution | Application Domain |
|---|---|---|---|
| 1791 | Jean Baptiste Delambre | First known use of the algorithm | Astronomical calculations |
| 1907 | Carl Størmer | Independent development | Trajectories of electrical particles in magnetic fields |
| 1909 | P. H. Cowell and A. C. C. Crommelin | Independent rediscovery | Computation of Halley's Comet orbit |
| 1967 | Loup Verlet | Formal derivation and popularization | Molecular dynamics simulations of classical fluids |
The algorithm's earliest known manifestation occurred in 1791 when French astronomer Jean Baptiste Delambre employed it for celestial calculations [2]. In the early 20th century, the method reappeared independently in the work of Carl Størmer (1907), who studied the trajectories of electrical particles in magnetic fields—an application that led to the alternative nomenclature "Størmer's method" [2]. Shortly thereafter, P. H. Cowell and A. C. C. Crommelin utilized the same mathematical approach in 1909 to compute the orbit of Halley's Comet with remarkable accuracy given the computational limitations of the era [2].
The algorithm entered its modern era when Loup Verlet derived and formalized it for molecular simulations in 1967, publishing his seminal work "Computer experiments on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules" in Physical Review [8]. Verlet's derivation focused specifically on molecular dynamics applications, establishing the mathematical framework that would become foundational to computational chemistry and biomolecular simulation. His formulation used third-order Taylor expansions for atomic positions, both forward and backward in time, to achieve a discretization error of order Δtⴠwhile avoiding explicit velocity dependence in the position update [8].
The Rediscovery Pattern
The repeated independent discovery of this integration method across centuries and scientific disciplines highlights its fundamental mathematical nature. Each rediscovery occurred when researchers confronted the challenge of numerically integrating second-order differential equations without introducing significant energy drift or numerical instability [2]. The algorithm's symmetry properties, which confer time-reversibility and symplectic structure preservation, make it naturally suited for physical systems obeying conservation laws [2].
The modern prevalence of Verlet algorithms in molecular dynamics began with Verlet's 1967 publication, which coincided with the increasing availability of computational resources for scientific simulation [8]. Unlike previous discoverers, Verlet explicitly derived the method for molecular systems and analyzed its numerical stability, establishing it as the standard approach for MD simulations just as the field began rapid expansion into biological applications [8].
Mathematical Foundations
The mathematical derivation of the Verlet algorithm begins with Newton's equations of motion for conservative physical systems. For a system of N particles, the equations take the form Máº(t) = F(x(t)) = -∇V(x(t)), where M represents the mass matrix, x(t) denotes atomic positions, F is the force vector, and V is the potential energy function [2]. The Verlet algorithm provides a numerical solution to these equations by employing a central difference approximation to the second derivative.
Core Mathematical Derivation
The fundamental Verlet algorithm derivation starts with two third-order Taylor expansions for the positions r(t) of particles, one forward and one backward in time [8]:
Forward expansion: r(t+Δt) = r(t) + v(t)Δt + (1/2)a(t)Δt² + (1/6)b(t)Δt³ + O(Δtâ´)
Backward expansion: r(t-Δt) = r(t) - v(t)Δt + (1/2)a(t)Δt² - (1/6)b(t)Δt³ + O(Δtâ´)
Adding these two expressions eliminates the velocity and third-derivative terms, yielding the basic Verlet position update equation [8]: r(t+Δt) = 2r(t) - r(t-Δt) + a(t)Δt² + O(Δtâ´)
where a(t) = F(t)/m represents the acceleration computed from the force at time t. The remarkable feature of this formulation is that although third derivatives explicitly appear in the Taylor expansions, they cancel out in the summation, resulting in a local truncation error of order Δtⴠ[8].
Discretization Error and Numerical Stability
The discretization error of the Verlet algorithm merits particular attention. The local truncation error for positions is O(Δtâ´), while the global error is proportional to Δt² [2]. This favorable error characteristic arises from the time symmetry inherent in the method, which eliminates all odd-degree terms in the discretization [2]. The algorithm is symplectic, meaning it preserves the phase-space volume, a property crucial for long-term stability in molecular dynamics simulations [2].
Velocities can be approximated from the positions using the central difference formula: v(t) = [r(t+Δt) - r(t-Δt)] / (2Δt) + O(Δt²)
However, this velocity approximation has a larger error (O(Δt²)) compared to the position update, and it introduces numerical precision issues because it computes velocities as the difference between two similar-valued position vectors [8]. These limitations motivated the development of improved Verlet variants that handle velocities more accurately and consistently.
Verlet Algorithm Variants
The basic Verlet algorithm, while mathematically elegant, presents practical implementation challenges including moderate accuracy, non-self-starting behavior, and imprecise velocity handling. These limitations spurred the development of several variants that retain the core mathematical structure while improving numerical properties and implementation convenience.
Comparative Analysis of Verlet Algorithm Family
Table: Comparison of Verlet Algorithm Variants
| Algorithm | Key Equations | Accuracy | Storage Requirements | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Basic Verlet | r(t+Δt) = 2r(t) - r(t-Δt) + a(t)Δt² | O(Δtâ´) positions, O(Δt²) velocities | 6N (two position sets) | Simple, modest storage | Not self-starting, imprecise velocities |
| Velocity Verlet | r(t+Δt) = r(t) + v(t)Δt + (1/2)a(t)Δt²v(t+Δt) = v(t) + (1/2)[a(t) + a(t+Δt)]Δt | O(Δtâ´) positions, O(Δt²) velocities | 9N (positions, velocities, accelerations) | Synchronous positions and velocities, self-starting | Additional force calculation |
| Leap-Frog | v(t+Δt/2) = v(t-Δt/2) + a(t)Δtr(t+Δt) = r(t) + v(t+Δt/2)Δt | O(Δtâ´) positions, O(Δt²) velocities | 6N (positions, half-step velocities) | Economical storage | Asynchronous positions and velocities |
Velocity Verlet Algorithm
The velocity Verlet algorithm addresses the primary limitations of the basic Verlet formulation by explicitly incorporating velocities and making them synchronous with positions [8]. This variant is currently the most widely used in production molecular dynamics codes, including popular packages like NAMD [9]. The algorithm decomposes into discrete steps:
- Position update: r(t+Δt) = r(t) + v(t)Δt + (1/2)a(t)Δt²
- Half-step velocity: v(t+Δt/2) = v(t) + (1/2)a(t)Δt
- Force computation: a(t+Δt) = -(1/m)∇V(r(t+Δt))
- Velocity completion: v(t+Δt) = v(t+Δt/2) + (1/2)a(t+Δt)Δt
This formulation requires 9N storage locations for positions, velocities, and accelerations but never needs simultaneous storage of values at two different times for any quantity [8]. The velocity Verlet algorithm provides the same trajectory as the basic Verlet method but with improved numerical properties and more convenient handling of velocities [7].
Leap-Frog Algorithm
The leap-frog algorithm represents an economical version that stores only one set of positions and one set of velocities while maintaining time-reversibility [7]. Its implementation follows:
- Velocity advance: v(t+Δt/2) = v(t-Δt/2) + a(t)Δt
- Position update: r(t+Δt) = r(t) + v(t+Δt/2)Δt
In this scheme, velocities "leap-frog" over positions, being defined at half-integer time steps while positions are defined at integer time steps [7]. If full-step velocities are required, they can be approximated as v(t) = [v(t-Δt/2) + v(t+Δt/2)]/2, though this introduces additional approximation error [7].
Practical Implementation and Research Applications
The transition from mathematical formulation to practical implementation requires careful consideration of numerical precision, force calculation efficiency, and integration with broader molecular dynamics workflows. The Velocity Verlet algorithm has become the de facto standard for production molecular dynamics codes across pharmaceutical and materials science research.
Research Reagent Solutions
Table: Essential Components for Molecular Dynamics Implementation
| Component | Function | Implementation Example |
|---|---|---|
| Integration Algorithm | Advances system through time steps | Velocity Verlet algorithm |
| Force Field | Computes potential energy and forces | CHARMM, AMBER, OPLS parameters |
| Thermostat | Regulates system temperature | Nose-Hoover thermostat, Langevin dynamics |
| Barostat | Maintains constant pressure | Andersen barostat, Parrinello-Rahman |
| Boundary Conditions | Mimics bulk environment | Periodic boundary conditions |
| Constraint Algorithms | Handles rigid bonds | SHAKE, LINCS, RATTLE |
| Parallelization Framework | Enables large-scale simulation | NAMD, GROMACS, AMBER |
Experimental Protocol: Velocity Verlet Implementation
Implementing the Velocity Verlet algorithm for molecular dynamics simulations follows a structured protocol that ensures numerical stability and physical accuracy:
Initialization:
- Set initial positions r(0) from experimental structures or previous simulations
- Initialize velocities v(0) from Maxwell-Boltzmann distribution at target temperature
- Compute initial forces F(0) = -∇V(r(0)) and accelerations a(0) = F(0)/m
Main Integration Loop:
- Update positions: r(t+Δt) = r(t) + v(t)Δt + (1/2)a(t)Δt²
- Calculate half-step velocities: v(t+Δt/2) = v(t) + (1/2)a(t)Δt
- Compute new forces: F(t+Δt) = -∇V(r(t+Δt))
- Calculate new accelerations: a(t+Δt) = F(t+Δt)/m
- Complete velocity update: v(t+Δt) = v(t+Δt/2) + (1/2)a(t+Δt)Δt
Property Calculation:
A critical implementation consideration involves the conservation of total energy E = K + V, where K represents kinetic energy and V potential energy. Monitoring energy conservation provides the most important verification that the MD simulation is proceeding correctly [8]. For biological systems, typical time steps range from 1-2 femtoseconds, constrained by the highest frequency vibrations (typically C-H bond stretches) [7].
Advanced Implementation: External Field Integration
Recent extensions to the Velocity Verlet algorithm have incorporated capacity for external fields, significantly expanding its application scope. For instance, researchers have implemented magnetic field forces within the NAMD package, modifying the algorithm to include Lorentz force contributions [9]. The implementation follows the Velocity Verlet structure but adds the magnetic force component Fₘ = q(v × B) to the total force calculation, where q represents charge, v is velocity, and B is the magnetic field vector [9].
This extension required careful treatment of the velocity dependence in the magnetic force, which necessitates implicit or iterative solution methods at each time step. The successful implementation enabled investigation of magnetic field effects on biomolecular systems, particularly water structure and ion behavior near proteins [9]. Such algorithmic extensions demonstrate the flexibility of the Verlet framework for incorporating increasingly complex physical interactions relevant to pharmaceutical research, including electric fields, shear flows, and other non-equilibrium conditions.
The historical journey of the Verlet algorithm—from Delambre's astronomical calculations to Verlet's molecular dynamics formalism—exemplifies how fundamental mathematical methods transcend their original domains to enable scientific progress across disciplines. The algorithm's symplectic nature, time-reversibility, and numerical stability have established it as the integration method of choice for molecular simulations, particularly in pharmaceutical research where accurate sampling of conformational space is essential for drug discovery.
The continued evolution of Verlet algorithms, including the velocity Verlet and leap-frog variants, addresses the demanding requirements of modern biomolecular simulation: energy conservation over nanosecond-to-microsecond timescales, efficient utilization of parallel computing architectures, and consistent sampling of thermodynamic ensembles. As molecular dynamics continues expanding into new research domains—including machine learning force fields, enhanced sampling techniques, and multi-scale modeling—the mathematical foundation established by Delambre, Størmer, Cowell, Crommelin, and Verlet remains central to extracting physically meaningful insights from computational experiments.
The central difference approximation is a foundational numerical method for estimating derivatives of functions, serving as the mathematical cornerstone of the Verlet integration algorithm. In molecular dynamics (MD) simulations, this principle enables the stable and efficient computation of particle trajectories by solving Newton's equations of motion. The Verlet algorithm, derived from this approximation, is ubiquitously employed in MD simulations within pharmaceutical research and material science, facilitating the study of complex molecular systems and their time-evolving properties. Its numerical stability and time-reversibility make it particularly suited for long-timescale simulations, such as those analyzing protein folding or drug-receptor interactions [2]. By providing a deterministic framework for modeling molecular interactions, the Verlet algorithm allows researchers to bridge atomic-level interactions with macroscopic observables like free energy and diffusion rates, which are critical for rational drug design [10].
Mathematical Foundation
Derivation of the Central Difference Approximation
The central difference approximation for the second derivative is derived from the Taylor series expansions of a function ( x(t) ) around time ( t ). The forward and backward Taylor expansions are given by:
[ x(t + \Delta t) = x(t) + v(t)\Delta t + \frac{a(t)\Delta t^2}{2} + \frac{b(t)\Delta t^3}{6} + \mathcal{O}(\Delta t^4) ]
[ x(t - \Delta t) = x(t) - v(t)\Delta t + \frac{a(t)\Delta t^2}{2} - \frac{b(t)\Delta t^3}{6} + \mathcal{O}(\Delta t^4) ]
where:
- ( x(t) ) represents the position at time ( t )
- ( v(t) = \dot{x}(t) ) is the velocity
- ( a(t) = \ddot{x}(t) ) is the acceleration
- ( b(t) = \dot{a}(t) ) is the third derivative
- ( \Delta t ) is the finite time step [2]
Adding these two expansions cancels the odd-order terms, including the velocity term, yielding:
[ x(t + \Delta t) + x(t - \Delta t) = 2x(t) + a(t)\Delta t^2 + \mathcal{O}(\Delta t^4) ]
Rearranging this expression isolates the acceleration term:
[ \frac{x(t + \Delta t) - 2x(t) + x(t - \Delta t)}{\Delta t^2} = a(t) + \mathcal{O}(\Delta t^2) ]
This establishes the central difference approximation for the second derivative, with a truncation error of order ( \Delta t^2 ). The cancellation of odd-order terms confers valuable numerical stability and time-reversibility properties to algorithms based on this formulation [2].
Connection to Newton's Equations of Motion
In molecular dynamics, particle motion is governed by Newton's second law:
[ mi \frac{d^2\mathbf{r}i}{dt^2} = \mathbf{F}_i ]
where:
- ( m_i ) is the mass of particle ( i )
- ( \mathbf{r}_i ) is its position vector
- ( \mathbf{F}_i ) is the force acting on it [11]
The force is derived from the potential energy function ( V ) through the relationship:
[ \mathbf{F}i = -\frac{\partial V}{\partial \mathbf{r}i} ]
Replacing the continuous second derivative with its central difference approximation produces the discretized equation of motion:
[ mi \frac{\mathbf{r}i(t + \Delta t) - 2\mathbf{r}i(t) + \mathbf{r}i(t - \Delta t)}{\Delta t^2} = \mathbf{F}_i(t) ]
This formulation provides the mathematical basis for the Verlet algorithm, enabling the numerical integration of particle trajectories in discrete time steps [11] [2].
The Verlet Integration Algorithm
Original Verlet Algorithm
The original Verlet algorithm follows directly from the central difference approximation. Solving the discretized equation of motion for the future position yields the update equation:
[ \mathbf{r}(t + \Delta t) = 2\mathbf{r}(t) - \mathbf{r}(t - \Delta t) + \frac{\mathbf{F}(t)}{m} \Delta t^2 ]
This formulation possesses several notable properties:
- It is time-reversible, mirroring the underlying physical laws
- It preserves the symplectic form on phase space, ensuring excellent energy conservation
- Its global error in position is of order ( \Delta t^3 ) [2] [12]
Despite these advantages, the original algorithm has practical limitations. It is not self-starting, requiring positions from two previous time steps. Velocity calculation is also problematic, as it must be estimated indirectly using:
[ \mathbf{v}(t) = \frac{\mathbf{r}(t + \Delta t) - \mathbf{r}(t - \Delta t)}{2\Delta t} ]
This approach suffers from numerical precision loss because it computes velocity as the difference between two similar-valued positions [12].
Velocity Verlet Variant
The velocity Verlet algorithm addresses these limitations while maintaining mathematical equivalence. It provides a self-starting formulation that explicitly tracks velocities:
[ \mathbf{r}(t + \Delta t) = \mathbf{r}(t) + \mathbf{v}(t)\Delta t + \frac{1}{2}\frac{\mathbf{F}(t)}{m} \Delta t^2 ]
[ \mathbf{v}(t + \Delta t) = \mathbf{v}(t) + \frac{\mathbf{F}(t) + \mathbf{F}(t + \Delta t)}{2m} \Delta t ]
This variant minimizes roundoff errors and is now widely implemented in production MD software like GROMACS [11] [12]. The algorithm proceeds in two phases per time step: position update followed by force computation and velocity update.
Diagram Title: Velocity Verlet Integration Workflow
Discretization Error Analysis
The Verlet algorithm's numerical accuracy stems from the cancellation of error terms in the central difference approximation. The following table summarizes its error characteristics:
| Component | Error Order | Description |
|---|---|---|
| Position (Local) | ( \mathcal{O}(\Delta t^4) ) | Error per time step in position |
| Position (Global) | ( \mathcal{O}(\Delta t^3) ) | Accumulated error over simulation |
| Velocity (Global) | ( \mathcal{O}(\Delta t^2) ) | Error in velocity estimation |
| Energy Conservation | Excellent | Due to symplectic property [2] |
The algorithm's stability is constrained by the time step ( \Delta t ), which must be sufficiently small to resolve the highest frequency motions in the system, particularly bond vibrations involving hydrogen atoms. In practice, this limits time steps to approximately 2 femtoseconds (fs) unless constraint algorithms are employed [11].
Implementation in Molecular Dynamics
Complete MD Integration Workflow
A complete molecular dynamics simulation integrates the Verlet algorithm with force computation and auxiliary procedures. The following workflow illustrates a single MD cycle:
Diagram Title: Complete MD Integration Cycle
Force Computation and Neighbor Searching
The force calculation phase dominates computational expense in MD simulations. Forces arise from various contributions:
- Non-bonded interactions: Lennard-Jones and Coulomb potentials between atom pairs
- Bonded interactions: Bond stretching, angle bending, and torsional potentials
- Restraints and external forces: Position restraints, pulling forces [11]
To optimize performance, MD codes like GROMACS employ neighbor searching algorithms with a buffered Verlet list. This approach creates a list of atom pairs within a cut-off radius ( r\ell = rc + rb ), where ( rc ) is the interaction cut-off and ( r_b ) is a safety buffer. The list is updated periodically rather than at every step, significantly reducing computational overhead [11].
The pair list generation employs a cluster-based approach where groups of 4-8 particles are processed together, improving computational efficiency on modern hardware with SIMD (Single Instruction Multiple Data) capabilities. Dynamic pair list pruning further optimizes performance by removing pairs that move beyond the interaction range between full list updates [11].
Energy Drift and Buffer Optimization
A critical consideration in Verlet-based MD is energy drift caused by particles diffusing into the interaction zone between list updates. The probability of this occurrence depends on the buffer size ( r_b ), time step ( \Delta t ), and particle displacement statistics.
For a canonical (NVT) ensemble, the atomic displacement distribution along one dimension follows a Gaussian with variance:
[ \sigma^2 = t^2 kB T \left( \frac{1}{m1} + \frac{1}{m_2} \right) ]
where ( k_B ) is Boltzmann's constant and ( T ) is temperature [11]. Modern MD packages can automatically determine the optimal buffer size given a tolerance for energy drift, typically targeting ~0.005 kJ/mol/ps per particle [11].
Applications in Drug Development
Solubility Prediction Framework
Molecular dynamics simulations employing Verlet integration have enabled significant advances in predicting key pharmaceutical properties. A recent study demonstrated the application of MD-derived properties for predicting aqueous solubility—a critical factor in drug bioavailability [10].
The research compiled a dataset of 211 drugs from diverse classes, conducted MD simulations using GROMACS, and extracted ten MD-derived properties as features for machine learning models. The following table summarizes the key molecular dynamics properties influencing drug solubility:
| Property | Symbol | Role in Solubility Prediction |
|---|---|---|
| Octanol-Water Partition Coefficient | logP | Measures lipophilicity/hydrophobicity |
| Solvent Accessible Surface Area | SASA | Quantifies surface exposed to solvent |
| Coulombic Interaction Energy | Coulombic_t | Electrostatic solute-solvent interactions |
| Lennard-Jones Interaction Energy | LJ | Van der Waals solute-solvent interactions |
| Estimated Solvation Free Energy | DGSolv | Thermodynamic driving force for dissolution |
| Root Mean Square Deviation | RMSD | Measures conformational flexibility |
| Average Solvation Shell Occupancy | AvgShell | Quantifies local solvent organization [10] |
Experimental Protocol for Solubility Prediction
Methodology:
- System Preparation: Obtain initial coordinates and generate topology files using the GROMOS 54a7 force field
- Simulation Setup: Conduct MD simulations in the isothermal-isobaric (NPT) ensemble using GROMACS
- Property Extraction: Calculate ten MD-derived properties from production trajectories
- Feature Selection: Apply statistical analysis to identify most influential properties
- Model Training: Employ ensemble machine learning algorithms (Random Forest, Extra Trees, XGBoost, Gradient Boosting) to predict logarithmic solubility (logS) [10]
Results: The Gradient Boosting algorithm achieved superior performance with a predictive R² of 0.87 and RMSE of 0.537 on the test set, demonstrating that MD-derived properties have comparable predictive power to traditional structural descriptors [10].
Research Reagent Solutions
The following table details essential computational tools and their functions in molecular dynamics studies for drug development:
| Research Reagent | Function in MD Simulation |
|---|---|
| GROMACS Software Package | Molecular dynamics simulation engine with Verlet integration [10] |
| GROMOS 54a7 Force Field | Mathematical representation of interatomic potentials [10] |
| Solvent Water Model | Explicit representation of aqueous environment |
| Thermodynamic Ensemble (NPT) | Maintains constant Number of particles, Pressure, and Temperature [10] |
| Verlet Neighbor Search | Efficient algorithm for non-bonded pair list generation [11] |
| Machine Learning Algorithms (GBR, XGBoost) | Predictive modeling of solubility from MD features [10] |
Advancements and Future Directions
The integration of Verlet-based molecular dynamics with data science approaches represents the cutting edge of computational drug discovery. Current research focuses on addressing several key challenges:
- Trajectory Databases: Initiatives are underway to create standardized repositories of MD simulations for various biomolecular systems, enabling meta-analyses and machine learning applications [13]
- FAIR Data Principles: Ensuring simulations are Findable, Accessible, Interoperable, and Reusable represents a paradigm shift from traditional isolated simulation approaches [13]
- Hybrid Methodologies: Combining Verlet integration with machine learning potential energy surfaces promises to enhance both accuracy and efficiency [10] [13]
These developments highlight the evolving role of the central difference approximation and Verlet algorithm in next-generation molecular simulation frameworks. As MD simulations generate increasingly large volumes of trajectory data, robust numerical integrators remain essential for producing reliable dynamics that can inform pharmaceutical development through advanced data science techniques [13].
The Verlet algorithm is a foundational numerical integration method central to Molecular Dynamics (MD) simulations, which are critical computational tools in fields ranging from drug development to materials science. It is used to solve Newton's equations of motion for a system of interacting particles, enabling the study of thermodynamic properties, structural changes, and dynamic processes at the atomic level [14] [2]. The algorithm's significance stems from its numerical stability and its ability to preserve important physical properties of continuous dynamic systems over long simulation times, even when discretized with a finite time step [2]. For molecular dynamics research, these characteristics—time-reversibility, symplectic nature, and energy conservation—are not merely mathematical curiosities; they are essential for producing physically meaningful and reliable simulation data that can guide scientific discovery and product development, including the design of novel pharmaceutical compounds.
Mathematical Formulation of the Verlet Algorithm
Core Integration Scheme
The standard Verlet integrator is derived from a central difference approximation to the second derivative. For a second-order differential equation of the type (\ddot{\mathbf{x}}(t) = \mathbf{A}(\mathbf{x}(t))), where (\mathbf{A}) is the acceleration (typically force divided by mass), the position at the next time step (\mathbf{x}_{n+1}) is calculated from the current and previous positions [2]:
[ \mathbf{x}{n+1} = 2\mathbf{x}n - \mathbf{x}{n-1} + \mathbf{a}n \Delta t^2 ] where (\mathbf{a}n = \mathbf{A}(\mathbf{x}n)), and (\Delta t) is the time step [2].
A more commonly used variant, the Velocity Verlet algorithm, explicitly calculates velocities and is self-starting. Its integration steps are as follows [15]:
- Update positions: (\mathbf{x}(t + \Delta t) = \mathbf{x}(t) + \mathbf{v}(t) \Delta t + \frac{1}{2} \mathbf{a}(t) \Delta t^2)
- Calculate forces and accelerations (\mathbf{a}(t + \Delta t)) at the new positions.
- Update velocities: (\mathbf{v}(t + \Delta t) = \mathbf{v}(t) + \frac{\mathbf{a}(t) + \mathbf{a}(t + \Delta t)}{2} \Delta t)
Discretization Error and Accuracy
The local truncation error of the Verlet integrator is of the order (O(\Delta t^4)) for positions and (O(\Delta t^2)) for velocities [2]. This favorable error profile arises because the central difference approximation cancels out the odd-order terms in the Taylor expansion [2]. The following table summarizes the error characteristics and computational cost.
Table 1: Discretization error and computational profile of the Verlet algorithm.
| Aspect | Description | Mathematical Expression |
|---|---|---|
| Position Update Error | Local truncation error is fourth-order [2]. | (O(\Delta t^4)) |
| Velocity Update Error | Local truncation error is second-order [2]. | (O(\Delta t^2)) |
| Global Error | Error over a fixed time span is one order lower [2]. | Positions: (O(\Delta t^2)) |
| Computational Cost | Requires one force calculation per time step, comparable to simple Euler method [2]. | (O(N)) per step (for N particles) |
Key Properties: Theoretical Foundations and Practical Implications
Time-Reversibility
Time-reversibility is a fundamental property of Newton's equations of motion. The Verlet algorithm, by virtue of its symmetric formulation, preserves this property in its discrete integration process [2]. If the signs of all velocities and the time direction are reversed, the system will retrace its trajectory back through the sequence of positions it came from.
This property is intrinsically linked to the algorithm's stability. Small errors do not tend to accumulate in a one-directional, destabilizing manner, which is crucial for long-term integration. The Velocity Verlet algorithm's sequence of operations—updating positions, then forces, then velocities—is specifically designed to maintain this symmetry in time [15].
Symplectic Nature
The Verlet integrator is symplectic, meaning it preserves the symplectic form on phase space [2]. In practical terms, this guarantees that the computed trajectory conserves the total energy and other invariants of the Hamiltonian system with remarkable accuracy over long simulation periods, even if there is a small phase error in the oscillations [16] [2].
This is a significant advantage over non-symplectic integrators (e.g., Runge-Kutta), which may exhibit a steady drift in the total system energy over time, leading to unphysical behavior [14]. The symplectic property ensures that the energy fluctuates around a stable mean value rather than diverging [2].
Energy Conservation
In the NVE (microcanonical) ensemble, where particle number, volume, and total energy are conserved, the Velocity Verlet algorithm demonstrates excellent long-term stability for the total energy [14]. The energy is not perfectly constant but oscillates within a small, bounded margin around the true value. The amplitude of these oscillations is highly dependent on the chosen time step [15].
Table 2: Summary of the three key properties of the Verlet algorithm.
| Property | Theoretical Foundation | Practical Implication in MD |
|---|---|---|
| Time-Reversibility | Symmetric form from central difference approximation [2]. | Enhanced numerical stability for long simulations [2]. |
| Symplectic Nature | Preserves the symplectic form on phase space [16] [2]. | Prevents long-term energy drift; essential for correct thermodynamic sampling [16] [14]. |
| Energy Conservation | Bounded energy oscillations due to symplectic property [14] [15]. | Enables reliable NVE ensemble simulations; accuracy depends on time step choice [14]. |
Experimental Protocols and Validation
Methodology for Validating Energy Conservation
A standard protocol for validating the energy-conserving properties of the Verlet algorithm involves simulating a simple, well-understood system and monitoring its total energy over time.
System Setup:
- Model System: A 1D harmonic oscillator or a simple pair of particles interacting via a spring potential, as this has a known analytical solution [15].
- Initial Conditions: Define initial particle positions and velocities. For a spring system, one can start with particles displaced from their equilibrium distance [15].
- Potential and Force: Use a Hookean spring potential, ( V(r) = \frac{1}{2}k(r - r0)^2 ), where ( k ) is the spring constant and ( r0 ) is the equilibrium distance. The force is ( F = -\nabla V ) [15].
Simulation Procedure:
- Integration: Apply the Velocity Verlet algorithm to update particle positions and velocities over a large number of steps (e.g., 1000) [15].
- Energy Calculation: At each time step, compute the total energy ( E{total} = E{kinetic} + E_{potential} ) [15].
- Analysis: Plot the total energy as a function of simulation time. A high-quality, symplectic integrator will show stable oscillations without a secular drift [15].
Workflow for a Molecular Dynamics Simulation
The following diagram illustrates the standard sequence of operations in a complete MD simulation step using the Velocity Verlet integrator.
Figure 1: A single iteration of an MD simulation using the Velocity Verlet algorithm.
The Scientist's Toolkit: Essential Components for MD Simulations
Table 3: Key software and computational reagents used in modern Molecular Dynamics simulations.
| Tool/Component | Function | Example Implementations |
|---|---|---|
| MD Engine | Core software that performs the simulation, handling integration, force calculations, and neighbor lists. | LAMMPS [17], ASE (Atomic Simulation Environment) [14], GROMACS, AMBER. |
| Force Field | A set of empirical potentials and parameters that describe the interatomic forces. | Lennard-Jones potential [16], Spring potential [15], AMBER, CHARMM. |
| Integrator | The numerical algorithm that solves the equations of motion. | Velocity Verlet [14] [15], Langevin (for NVT ensemble) [14]. |
| Neighbor List | An algorithmic optimization to efficiently identify particles within interaction range, reducing computation from (O(N^2)) [15]. | Hierarchical grid algorithm [17], Linked cell method [17]. |
| Analysis & Visualization | Tools for processing trajectory data to compute properties and generate visual representations. | MDAnalysis, MDTraj [15], VMD. |
| 1,1-Difluoro-1,3-diphenylpropane | 1,1-Difluoro-1,3-diphenylpropane | |
| Methyl 2-(2-methoxyethoxy)acetate | Methyl 2-(2-methoxyethoxy)acetate|148.16 g/mol | Methyl 2-(2-methoxyethoxy)acetate (CAS 17640-28-7) is a versatile ester and glycol ether solvent for organic synthesis and energy storage research. For Research Use Only. Not for human or veterinary use. |
Advanced Optimizations and Recent Developments
Optimized Verlet-like Algorithms
Research continues to refine the core Verlet algorithm. One approach involves an extended decomposition scheme that introduces a free parameter, which is optimized to minimize the influence of truncated terms [16]. These optimized algorithms are more efficient than the original Verlet versions while remaining symplectic and time-reversible, leading to improved accuracy at the same computational cost, as demonstrated in simulations of Lennard-Jones fluids [16].
Addressing Limitations in Polydisperse Systems
Recent work has identified a subtle flaw in the standard velocity-Verlet implementation within Discrete Element Method (DEM) codes when simulating systems with large particle-size ratios (e.g., in granular flows). The half-step difference between position and velocity updates can cause unphysical pendular motion of small particles trapped between larger ones [17].
An improved velocity-Verlet integration has been proposed to correct this. The key modification is to use the average velocity over the interval ([t, t+\Delta t]) for the position update, which more accurately represents the mean velocity during that step and eliminates the spurious oscillations [17]. This improvement has been integrated into the developer version of LAMMPS, enhancing simulation accuracy for systems with large size ratios [17].
Selecting the Time Step and Other Parameters
The choice of time step ((\Delta t)) is a critical trade-off between computational efficiency and physical accuracy [14] [15].
Table 4: Guidelines for selecting the MD simulation time step.
| System Type | Recommended Time Step | Rationale |
|---|---|---|
| Metallic Systems | ~5.0 fs [14] | A good balance for systems with moderate atomic masses and bond strengths. |
| Systems with Light Atoms (H) or Strong Bonds (C) | 1-2 fs [14] | Higher frequency vibrations require a smaller time step to maintain stability. |
| General Guideline | Must be small relative to the highest frequency vibration [15]. | A too-large time step causes a dramatic energy increase and system instability [14]. |
Furthermore, for simulations involving large size ratios, careful selection of contact stiffness and damping models is also crucial to avoid unphysical behavior and ensure accurate results [17].
The Verlet algorithm, particularly its Velocity Verlet variant, remains a cornerstone of molecular dynamics simulations due to its robust numerical properties: time-reversibility, symplectic nature, and excellent energy conservation. These characteristics are indispensable for generating reliable, physically meaningful data in scientific research and industrial applications, such as drug discovery. While the core algorithm is well-established, ongoing research continues to develop optimized and problem-specific variants, ensuring its relevance and accuracy for increasingly complex simulation challenges. As computational power grows and scientists tackle new domains, a thorough understanding of these foundational integration schemes is paramount to avoid subtle inaccuracies and push the boundaries of what molecular dynamics can reveal.
Molecular dynamics (MD) simulation represents a cornerstone technique in computational chemistry, physics, and drug development, enabling researchers to probe the temporal evolution of molecular systems at atomic resolution. The Verlet algorithm, first introduced by Loup Verlet in 1967 for computer experiments on classical fluids, has emerged as one of the most fundamental and widely-used numerical integration methods in this field [18] [12]. Its significance stems from an exceptional combination of numerical stability, computational efficiency, and desirable physical properties including time-reversibility and symplecticity (preservation of the geometric structure of Hamiltonian systems) [2]. Within the broader thesis of understanding molecular dynamics algorithms, analyzing the global error properties of integration schemes—specifically the third-order accuracy for position and second-order accuracy for velocity in the Verlet method—provides critical insights for researchers seeking to balance computational expense with numerical precision in simulating biomolecular systems, materials science, and drug-target interactions.
Mathematical Foundation of Verlet Integration
Fundamental Algorithm Derivation
The Verlet algorithm integrates Newton's equations of motion for a system of N particles governed by conservative forces derived from a potential function V(x). For a second-order differential equation of the type:
[ \ddot{\mathbf{x}}(t) = \mathbf{A}(\mathbf{x}(t)) ]
where (\mathbf{A}(\mathbf{x}(t)) = \mathbf{M}^{-1}\mathbf{F}(\mathbf{x}(t))) and (\mathbf{F}(\mathbf{x}) = -\nabla V(\mathbf{x})), the Verlet method employs a central difference approximation to the second derivative [2]:
[ \frac{\Delta^2\mathbf{x}n}{\Delta t^2} = \frac{\mathbf{x}{n+1} - 2\mathbf{x}n + \mathbf{x}{n-1}}{\Delta t^2} = \mathbf{a}n = \mathbf{A}(\mathbf{x}n) ]
This leads to the position update equation in the Størmer-Verlet form:
[ \mathbf{x}{n+1} = 2\mathbf{x}n - \mathbf{x}{n-1} + \mathbf{a}n \Delta t^2 ]
where (\mathbf{x}n \approx \mathbf{x}(tn)) represents the position at time (tn = t0 + n\Delta t), and (\Delta t) is the integration time step [2].
Velocity Formulation
Although the basic Verlet algorithm does not explicitly include velocities, they can be derived from the position trajectory:
[ \mathbf{v}n = \frac{\mathbf{x}{n+1} - \mathbf{x}_{n-1}}{2\Delta t} ]
However, this velocity calculation suffers from numerical precision issues due to the difference between two quantities of similar magnitude [12]. The velocity Verlet algorithm, a mathematically equivalent formulation, addresses this limitation by explicitly incorporating velocity updates:
[ \mathbf{x}(t + \Delta t) = \mathbf{x}(t) + \mathbf{v}(t)\Delta t + \frac{1}{2}\mathbf{a}(t)\Delta t^2 ]
[ \mathbf{v}(t + \Delta t) = \mathbf{v}(t) + \frac{1}{2}[\mathbf{a}(t) + \mathbf{a}(t + \Delta t)]\Delta t ]
This self-starting algorithm minimizes roundoff errors while maintaining the same error properties as the original Verlet method [12] [19].
Global Error Analysis Methodology
Taylor Series Expansion Framework
The global error analysis of the Verlet algorithm derives from Taylor series expansions of position around time t. The forward and reverse expansions are given by:
[ \mathbf{x}(t + \Delta t) = \mathbf{x}(t) + \mathbf{v}(t)\Delta t + \frac{\mathbf{a}(t)\Delta t^2}{2} + \frac{\mathbf{b}(t)\Delta t^3}{6} + \mathcal{O}(\Delta t^4) ]
[ \mathbf{x}(t - \Delta t) = \mathbf{x}(t) - \mathbf{v}(t)\Delta t + \frac{\mathbf{a}(t)\Delta t^2}{2} - \frac{\mathbf{b}(t)\Delta t^3}{6} + \mathcal{O}(\Delta t^4) ]
where (\mathbf{v} = \dot{\mathbf{x}}), (\mathbf{a} = \ddot{\mathbf{x}}), and (\mathbf{b} = \dddot{\mathbf{x}}) represent velocity, acceleration, and jerk, respectively [2].
Error Order Determination
Adding the forward and reverse Taylor expansions produces the Verlet position update:
[ \mathbf{x}(t + \Delta t) = 2\mathbf{x}(t) - \mathbf{x}(t - \Delta t) + \mathbf{a}(t)\Delta t^2 + \mathcal{O}(\Delta t^4) ]
The cancellation of the third-order terms ((\Delta t^3)) results in a local truncation error of (\mathcal{O}(\Delta t^4)) for the position. However, as this equation represents a difference scheme with cumulative error over multiple steps, the global error for position becomes third-order ((\mathcal{O}(\Delta t^3))) [12] [2].
For velocity, subtracting the reverse expansion from the forward expansion yields:
[ \mathbf{v}(t) = \frac{\mathbf{x}(t + \Delta t) - \mathbf{x}(t - \Delta t)}{2\Delta t} - \frac{\mathbf{b}(t)\Delta t^2}{6} + \mathcal{O}(\Delta t^3) ]
The leading error term of order (\Delta t^2) translates to a global error for velocity of second-order ((\mathcal{O}(\Delta t^2))) [12].
Table 1: Global Error Properties of Verlet Integration
| Variable | Local Truncation Error | Global Error | Key Mathematical Property |
|---|---|---|---|
| Position | (\mathcal{O}(\Delta t^4)) | (\mathcal{O}(\Delta t^3)) | Third-order accuracy |
| Velocity | (\mathcal{O}(\Delta t^2)) | (\mathcal{O}(\Delta t^2)) | Second-order accuracy |
| Algorithm | Time-symmetric | Time-reversible | Symplectic |
Comparative Analysis of Verlet Integration Variants
Algorithmic Implementations and Their Error Profiles
Various implementations of the Verlet algorithm have been developed to address specific numerical challenges while maintaining the core error properties:
The original Verlet algorithm (1967) provides excellent numerical stability and time-reversibility but suffers from numerical precision issues in velocity calculation and is not self-starting [18] [12]. The velocity Verlet algorithm (Swope, 1982) eliminates roundoff errors in velocity computation by explicitly including velocities in the update steps, making it self-starting while maintaining the same global error order [12] [19]. RATTLE (1983) extends Verlet integration to handle holonomic constraints through Lagrange multipliers, preserving the error order while enabling bond length and angle constraints [18]. Optimized Verlet-like algorithms (Omelyan, 2002) introduce free parameters to minimize the influence of truncated terms, achieving improved accuracy at the same computational cost while maintaining the fundamental error bounds [20].
Table 2: Comparison of Verlet Integration Variants
| Algorithm | Position Error | Velocity Error | Key Features | Computational Cost |
|---|---|---|---|---|
| Original Verlet | (\mathcal{O}(\Delta t^3)) | (\mathcal{O}(\Delta t^2)) | Excellent stability, not self-starting | Low |
| Velocity Verlet | (\mathcal{O}(\Delta t^3)) | (\mathcal{O}(\Delta t^2)) | Self-starting, minimal roundoff error | Low (2 force evaluations/step) |
| RATTLE | (\mathcal{O}(\Delta t^3)) | (\mathcal{O}(\Delta t^2)) | Handles constraints, time-reversible | Moderate |
| Beeman | (\mathcal{O}(\Delta t^3)) | (\mathcal{O}(\Delta t^2)) | Better energy conservation | Moderate |
| Optimized Verlet-like | (\mathcal{O}(\Delta t^3)) | (\mathcal{O}(\Delta t^2)) | Enhanced accuracy, parameter-controlled | Low |
Multiple Time Stepping and Resonance Instabilities
Advanced implementations of Verlet integration, particularly Verlet-I/r-RESPA (Reversible Reference System Propagator Algorithms), employ multiple time stepping to enhance computational efficiency for systems with forces acting at different time scales. However, these methods can suffer from linear and nonlinear resonance instabilities when the longest time step approaches half the period of the fastest motion in the system [21].
Mollified Verlet-I/r-RESPA (MOLLY) methods overcome these instabilities by applying mollification (smoothing) functions to the slow forces, allowing for time steps up to 50% longer than standard Verlet-I/r-RESPA for a given energy drift [21]. This extension maintains the fundamental error properties while improving numerical stability for complex biomolecular systems with steep potential gradients.
Experimental Protocols for Error Validation
Harmonic Oscillator Validation Methodology
The implementation and error properties of Verlet integration can be rigorously validated using the harmonic oscillator model, which provides an exact analytical solution for comparison [19]. The protocol involves:
System Setup: A one-dimensional harmonic oscillator with mass μ and force constant k, governed by the potential energy function (V(x) = \frac{1}{2}k(x - x_{eq})^2). Newton's equation of motion for this system is (\ddot{x}(t) = -\omega^2 x(t)), where (\omega = \sqrt{k/\mu}) is the oscillator frequency [19].
Initial Conditions: Initial displacement (x(0) = A\sin(\phi) + x{eq}) and initial velocity (v(0) = A\omega\cos(\phi)), where A is the oscillation amplitude, φ is the initial phase, and (x{eq}) is the equilibrium position.
Analytical Solution: The exact position as a function of time is given by (x(t) = A\sin(\omega t + \phi) + x_{eq}).
Numerical Integration: Application of the Velocity Verlet algorithm with position update: [ x(t + \Delta t) = x(t) + v(t)\Delta t + \frac{1}{2}a(t)\Delta t^2 ] and velocity update: [ v(t + \Delta t) = v(t) + \frac{1}{2}[a(t) + a(t + \Delta t)]\Delta t ] where (a(t) = -\omega^2(x(t) - x_{eq})) [19].
Error Quantification: Calculation of the root-mean-square deviation (RMSD) between numerical and analytical trajectories: [ \text{RMSD} = \sqrt{\frac{1}{N}\sum{i=1}^N (x{\text{numeric}}(ti) - x{\text{analytic}}(t_i))^2} ] across N time steps to verify the theoretical third-order error scaling for position.
Molecular Dynamics Validation with Lennard-Jones Fluids
For more complex systems, validation involves molecular dynamics simulations of Lennard-Jones fluids, which model noble gas atoms interacting through the pair potential: [ V(r) = 4\epsilon\left[\left(\frac{\sigma}{r}\right)^{12} - \left(\frac{\sigma}{r}\right)^6\right] ] where ε defines the well depth and σ the atomic diameter [20].
The experimental protocol includes:
- System Initialization: N particles placed in a cubic simulation box with periodic boundary conditions
- Equilibration Phase: System equilibrated to target temperature using Langevin dynamics or Nosé-Hoover thermostats
- Production Phase: Trajectory propagation using Verlet integration with time steps typically between 1-10 femtoseconds
- Conservation Monitoring: Tracking of total energy drift, which should be bounded for symplectic integrators like Verlet
- Structural Validation: Comparison of radial distribution functions with theoretical predictions or known results
- Temporal Error Analysis: Measurement of position and velocity errors through comparison with reference simulations using smaller time steps
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Verlet Integration Studies
| Tool/Resource | Function | Application Context |
|---|---|---|
| LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) | Molecular dynamics simulator with multiple integrators | High-performance MD simulations of materials and biomolecules [18] |
| CHARMM (Chemistry at HARvard Macromolecular Mechanics) | Biomolecular simulation program with Verlet implementations | Pharmaceutical research and biomolecular dynamics [21] |
| NAMD (Nanoscale Molecular Dynamics) | Parallel MD code designed for biomolecular systems | Large-scale simulation of protein folding and membrane systems [21] |
| ProtoMol | Object-oriented MD framework with multiple Verlet variants | Algorithm development and testing [21] |
| Particle Mesh Ewald (PME) | Method for efficient calculation of long-range electrostatic forces | Full electrostatics in molecular simulations [21] |
| Langevin Thermostat | Stochastic temperature control method | Maintaining constant temperature in MD simulations [21] |
| 2-(2-Aminopyridin-3-yl)acetonitrile | 2-(2-Aminopyridin-3-yl)acetonitrile|RUO|Supplier | Get 2-(2-Aminopyridin-3-yl)acetonitrile for your research. This aminopyridine building block is for research use only (RUO). Not for human or veterinary diagnostic or therapeutic use. |
| Barium(2+);2-ethylhexan-1-olate | Barium(2+);2-ethylhexan-1-olate, CAS:29170-99-8, MF:C16H34BaO2, MW:395.8 g/mol | Chemical Reagent |
Implications for Molecular Dynamics Research and Drug Development
The third-order position accuracy and second-order velocity accuracy of Verlet integration have profound implications for molecular dynamics research, particularly in drug development. The favorable error scaling enables researchers to use larger time steps (typically 2 femtoseconds vs. 0.5 femtoseconds for Euler method) while maintaining numerical stability, directly reducing computational costs for simulating biological processes that occur on microsecond to millisecond timescales, such as protein folding and ligand-binding events [18] [2].
In drug discovery, Verlet integration facilitates more accurate prediction of binding affinities and residence times through enhanced sampling of conformational space. The algorithm's symplectic property ensures excellent energy conservation, preventing artificial heating or cooling that could distort free energy calculations crucial for rational drug design [2]. Furthermore, the stability of Verlet integration enables longer simulation timescales, permitting observation of rare events and more reliable statistics for ensemble properties.
The development of optimized Verlet-like algorithms continues to push the boundaries of molecular simulation, allowing researchers to extract more biological insights from each computational cycle while maintaining the rigorous error bounds established through global error analysis [20]. As molecular dynamics expands into new domains such as machine learning potential and multiscale modeling, the fundamental understanding of Verlet integration error properties remains essential for validating novel methodologies and ensuring physical fidelity in computational predictions.
Implementing Verlet: Variants, Code, and Practical Applications in Biomedicine
Molecular Dynamics (MD) simulation serves as a computational microscope, revealing the physical motions of atoms and molecules over time. This technique, first introduced by Alder and Wainwright in the 1950s, has become indispensable across numerous scientific disciplines for understanding system behavior at the atomic level [22]. The foundation of any MD simulation lies in its integration algorithm—the mathematical procedure that advances the system by calculating new atomic positions and velocities based on the forces acting upon them. Among these algorithms, the Verlet integrator, developed by Loup Verlet in the 1960s, represents one of the most significant and enduring contributions to the field [12] [2]. Although the algorithm was first used by Delambre in 1791 and rediscovered multiple times, it was Verlet who popularized it for molecular dynamics simulations [2]. This technical guide examines the original Verlet algorithm in detail, exploring its mathematical formulation, key characteristics, and practical limitations within the context of modern molecular dynamics research, particularly for applications in drug development and biomolecular simulation.
Mathematical Foundation
Derivation from Taylor Series Expansions
The original Verlet algorithm derives from classical mechanics and Taylor series expansions of particle position. The algorithm calculates the trajectory of particles by solving Newton's second law of motion, F = ma, where the acceleration a is obtained from the force field representing the physical system [2] [22].
The derivation begins with two Taylor series expansions for position—one forward and one backward in time:
x(t + Δt) = x(t) + v(t)Δt + (a(t)Δt²)/2 + (b(t)Δt³)/6 + O(Δtâ´) [2]
x(t - Δt) = x(t) - v(t)Δt + (a(t)Δt²)/2 - (b(t)Δt³)/6 + O(Δtâ´) [2]
Where:
- x(t) represents the position at time t
- v(t) represents the velocity at time t
- a(t) represents the acceleration at time t
- b(t) represents the jerk (third derivative) at time t
- Δt represents the time step
- O(Δtâ´) represents the error term, indicating that the truncation error is proportional to Δtâ´
Adding these two equations cancels out the odd-order terms, including the velocity term, yielding the fundamental Verlet position update formula:
x(t + Δt) = 2x(t) - x(t - Δt) + a(t)Δt² + O(Δtâ´) [2]
This formulation provides a direct method to compute the new position without explicit velocity dependence. The elimination of odd-order terms through this symmetric formulation reduces local discretization errors and enhances numerical stability [2].
Velocity Calculation
Although the position update equation does not require velocity, it is often necessary to compute velocities for analyzing system properties such as kinetic energy. The velocity can be approximated from the position history using the central difference method:
v(t) = [x(t + Δt) - x(t - Δt)] / (2Δt) + O(Δt²) [12] [23]
This approximation provides second-order accuracy but introduces numerical challenges. The calculation involves subtracting two quantities of similar magnitude (x(t + Δt) and x(t - Δt)), which when performed with finite numerical precision, can result in significant roundoff errors and loss of numerical precision [12].
Key Properties and Characteristics
Numerical Stability and Time Reversibility
The Verlet algorithm exhibits exceptional numerical stability properties that account for its enduring popularity in molecular dynamics simulations. The method is time-reversible, meaning that if the signs of all velocities are reversed, the system will retrace its trajectory backward in time [2]. This property aligns with the time-reversible nature of Newton's equations of motion in classical mechanics.
Additionally, the Verlet algorithm is symplectic, meaning it preserves the geometric structure of phase space in Hamiltonian systems [2]. This characteristic is crucial for long-term energy conservation in molecular dynamics simulations, as it prevents artificial energy drift that can occur with non-symplectic integrators. The algorithm's stability allows for larger time steps compared to simpler integration methods like Euler integration, though the specific maximum stable time step depends on the highest frequencies present in the system being simulated [7].
Discretization Error and Precision
The Verlet algorithm achieves a favorable balance between computational efficiency and numerical accuracy. The position update maintains fourth-order local accuracy (O(Δtâ´)) through the cancellation of odd-order terms in the Taylor series expansion, while the global error for position is third-order (O(Δt³)) and for velocity is second-order (O(Δt²)) [12] [2].
However, the velocity calculation presents precision challenges. As noted by various sources, computing velocity as v(t) = [x(t + Δt) - x(t - Δt)] / (2Δt) involves subtracting two similar-sized numbers, which can lead to substantial roundoff errors in finite-precision arithmetic [12]. This limitation motivated the development of variant algorithms that handle velocity computation more robustly.
Table 1: Error Analysis of the Original Verlet Algorithm
| Component | Local Error | Global Error | Primary Sources of Error |
|---|---|---|---|
| Position Update | O(Δtâ´) | O(Δt³) | Truncation of higher-order terms |
| Velocity Calculation | O(Δt²) | O(Δt²) | Finite difference approximation and roundoff |
| Energy Conservation | N/A | O(Δt²) | Accumulated error from position and velocity |
Advantages and Limitations
Algorithmic Strengths
The original Verlet algorithm offers several compelling advantages that explain its widespread adoption and continued use in molecular dynamics simulations:
Simplicity and Computational Efficiency: The algorithm requires only one force evaluation per time step and involves relatively simple arithmetic operations, making it straightforward to implement and computationally efficient [7]. The position update formula x(t + Δt) = 2x(t) - x(t - Δt) + a(t)Δt² can be evaluated with minimal computational overhead.
Minimal Memory Requirements: The basic Verlet algorithm needs to store only two sets of positions—the current and previous time steps—for each particle in the system [7]. This modest memory footprint enables simulations of large systems with limited computational resources.
Long-term Stability: The symplectic nature of the algorithm ensures excellent energy conservation properties over long simulation timescales, which is essential for obtaining physically meaningful results in molecular dynamics simulations [2].
Time Reversibility: As previously mentioned, the algorithm is time-reversible, maintaining fundamental symmetry properties of the underlying physical laws being simulated [2].
Practical Limitations
Despite its advantages, the original Verlet algorithm suffers from several significant limitations that researchers must consider:
Not Self-Starting: The algorithm requires knowledge of positions at two previous time steps (x(t) and x(t - Δt)) to compute the new position (x(t + Δt)). At the beginning of a simulation (t=0), only initial positions and velocities are typically known, necessitating a different method to generate the first few time steps [12] [7].
Implicit Velocity Handling: The velocity does not participate directly in the integration of equations of motion and must be calculated separately from positions [12]. This complicates the implementation of thermostats and barostats that require velocity modifications.
Precision Issues with Velocity: The velocity calculation v(t) = [x(t + Δt) - x(t - Δt)] / (2Δt) is particularly susceptible to roundoff errors in finite-precision arithmetic because it involves subtracting two numbers of similar magnitude [12].
Inconvenient Output Timing: The algorithm produces positions and velocities at different time instances—positions are available at the current time step, while velocities are effectively one time step behind [7].
Table 2: Comparison of Verlet Algorithm Variants
| Algorithm | Velocity Handling | Self-Starting | Memory Requirements | Key Advantages |
|---|---|---|---|---|
| Original Verlet | Calculated separately from positions | No | Stores two position sets | Simplicity, minimal computation |
| Velocity Verlet | Explicitly integrated alongside positions | Yes | Stores positions, velocities, and forces | Convenient output, better velocity accuracy |
| Leapfrog Verlet | Defined at half-time steps | Yes | Stores positions and velocities | Good energy conservation, simple implementation |
Implementation in Molecular Dynamics
Basic Molecular Dynamics Workflow
The integration algorithm represents one component in a comprehensive molecular dynamics simulation workflow. A typical MD simulation follows these key steps:
Diagram 1: Molecular Dynamics Simulation Workflow
Research Reagent Solutions
Table 3: Essential Components for Molecular Dynamics Simulations
| Component | Function | Examples |
|---|---|---|
| Force Fields | Mathematical functions describing potential energy | AMBER, CHARMM, GROMOS [22] |
| Solvent Models | Represent water and other solvent molecules | SPC, SPC/E, TIP3P [22] |
| Constraint Algorithms | Maintain rigid bonds and angles | SHAKE, RATTLE [24] |
| Thermostats/Barostats | Control temperature and pressure | Nosé-Hoover, Berendsen [22] |
| Software Packages | Implement integration algorithms and analysis | LAMMPS, GROMACS, AMBER [15] |
Experimental Protocol for Implementing the Original Verlet Algorithm
For researchers implementing the original Verlet algorithm in molecular dynamics simulations, the following detailed protocol ensures proper initialization and execution:
System Initialization:
- Define initial particle positions x(0) typically derived from experimental structures or molecular modeling
- Assign initial velocities v(0) from Maxwell-Boltzmann distribution appropriate for the target temperature
- Calculate initial forces F(0) from the potential energy function: F = -∇V
Starting Procedure:
- Use the forward Euler method to generate the first time step: x(Δt) = x(0) + v(0)Δt + (a(0)Δt²)/2
- Store both x(0) and x(Δt) for the Verlet algorithm proper
Main Simulation Loop:
- For each time step n = 1, 2, 3, ...: a. Calculate forces F(n) based on positions x(n) b. Compute new positions: x(n+1) = 2x(n) - x(n-1) + a(n)Δt² c. If velocities are needed, compute: v(n) = [x(n+1) - x(n-1)] / (2Δt) d. Apply boundary conditions (periodic, reflective, etc.) e. Implement thermostats/barostats if required (may require velocity modifications) f. Record trajectory and thermodynamic data for analysis
Constraint Handling (for rigid bonds):
- Apply constraint algorithms such as SHAKE after position updates
- Iteratively adjust positions to satisfy constraint equations: σ = (rj - ri)² - d_ij² = 0 [24]
This protocol highlights the algorithm's non-self-starting nature, requiring special initialization, and demonstrates the additional steps needed for velocity computation and constraint satisfaction.
Evolution and Variants
Development of Verlet-Based Algorithms
The limitations of the original Verlet algorithm prompted the development of several mathematically equivalent variants that address its practical shortcomings while preserving its desirable numerical properties:
Diagram 2: Evolution of Verlet Integration Algorithms
The Velocity Verlet algorithm has become particularly popular in modern molecular dynamics software due to its self-starting nature and synchronized position-velocity output [7]. Its formulation:
x(t + Δt) = x(t) + v(t)Δt + (a(t)Δt²)/2 [15]
v(t + Δt) = v(t) + [a(t) + a(t + Δt)]Δt/2 [15]
provides mathematically equivalent trajectory to the original Verlet algorithm while offering improved numerical properties for velocity computation.
Recent research continues to refine Verlet-style algorithms. In 2002, optimized Verlet-like algorithms were introduced using an extended decomposition scheme with a free parameter that, when tuned appropriately, can reduce the influence of truncated terms and improve accuracy without additional computational cost [16].
The original Verlet algorithm represents a foundational methodology in molecular dynamics simulation that continues to influence computational science decades after its introduction. Its elegant mathematical formulation, combining forward and backward Taylor series expansions, yields exceptional numerical stability and conservation properties that make it well-suited for long-timescale molecular simulations. While the algorithm's limitations—particularly its non-self-starting nature and imprecise velocity handling—have led to the development of more convenient variants like the Velocity Verlet algorithm, the original formulation remains historically significant and practically relevant.
For researchers in drug development and biomolecular simulation, understanding the properties and limitations of the Verlet algorithm provides crucial insights into the inner workings of molecular dynamics software and the theoretical foundations of numerical integration in computational chemistry. As molecular dynamics continues to evolve, addressing challenges of longer timescales and more complex systems, the principles embodied in the Verlet algorithm continue to inform the development of next-generation simulation methodologies.
The Velocity Verlet algorithm stands as a cornerstone of modern molecular dynamics (MD) simulations, providing a robust, self-starting, and numerically stable framework for integrating Newton's equations of motion. Its significance extends across diverse fields, from fundamental physics to applied drug development, where accurate trajectory calculation is paramount for predicting molecular behavior. This technical guide delineates the algorithm's mathematical foundations, detailed implementation protocols, and quantitative analysis of its performance characteristics, contextualized within the broader scope of Verlet-type integration methods in molecular dynamics research. The algorithm's capacity for long-term energy conservation and time-reversibility makes it particularly indispensable for generating reliable NVE (microcanonical) ensembles in pharmaceutical research, where understanding molecular interactions forms the basis of rational drug design [14] [19].
Within molecular dynamics research, the family of Verlet integration algorithms represents a class of numerical methods for solving Newton's equations of motion. While the basic Verlet algorithm was first used by Jean Baptiste Delambre in 1791 and subsequently rediscovered multiple times [2], its original formulation suffered from numerical limitations. The Velocity Verlet algorithm emerged as a mathematically equivalent but computationally superior variant that explicitly calculates velocities alongside positions, making it self-starting and minimizing roundoff errors [12] [2].
This evolution addressed critical shortcomings in the original Verlet method, which calculated velocities imprecisely as the difference between two large numbers and was not self-starting [12]. The Velocity Verlet algorithm's stability stems from its symplectic nature, meaning it preserves the phase-space volume in Hamiltonian systems, and its time-reversibility, both crucial properties for accurate long-term molecular dynamics simulations [2] [14]. These characteristics have established it as a gold standard in computational chemistry and molecular biology, particularly in drug development where simulating protein-ligand interactions requires both numerical stability and conservation of thermodynamic quantities.
Mathematical Formulation
The Velocity Verlet algorithm derives from Taylor series expansions of position and velocity, offering a discrete solution to Newton's second law of motion. For a system of N particles, Newton's equation is expressed as:
[ mk \ddot{\mathbf{x}}k(t) = Fk(\mathbf{x}(t)) = -\nabla{\mathbf{x}_k} V(\mathbf{x}(t)) ]
where (mk) is the mass of particle k, (\mathbf{x}k) its position, (F_k) the force acting on it, and (V) the potential energy function [2]. The algorithm achieves its numerical stability by employing central difference approximations to the second derivative, effectively canceling out first-order error terms [2].
The core mathematical operations can be summarized through the following fundamental equations:
Table 1: Core Mathematical Equations of the Velocity Verlet Algorithm
| Equation | Description | Purpose |
|---|---|---|
| (\mathbf{v}i(t + \frac{1}{2}\Delta t) = \mathbf{v}i(t) + \frac{\mathbf{F}i(t)}{2mi}\Delta t) | Half-step velocity update | Estimates velocity at mid-interval |
| (\mathbf{r}i(t + \Delta t) = \mathbf{r}i(t) + \mathbf{v}_i(t + \frac{1}{2}\Delta t)\Delta t) | Full-step position update | Advances position using half-step velocity |
| (\mathbf{F}i(t+\Delta t) = -\nabla V(\mathbf{r}i(t+\Delta t))) | Force computation | Calculates new forces at updated positions |
| (\mathbf{v}i(t + \Delta t) = \mathbf{v}i(t + \frac{1}{2}\Delta t) + \frac{\mathbf{F}i(t+\Delta t)}{2mi}\Delta t) | Complete velocity update | Finalizes velocity using average acceleration |
The discretization error of the Velocity Verlet algorithm is O(Δtâ´) for positions and O(Δt²) for velocities, making it superior to basic Euler integration methods [2] [23]. This high-order accuracy, combined with its symplectic properties, ensures excellent energy conservation over long simulation timescales - a critical requirement for meaningful molecular dynamics trajectories in drug development research.
Algorithm Implementation and Workflow
The Velocity Verlet algorithm implements a well-defined sequence of operations that advance a molecular system through discrete time steps. The complete workflow integrates force calculations with position and velocity updates in a carefully orchestrated sequence that maintains numerical stability. The following diagram illustrates this cyclic process:
Detailed Computational Protocol
The implementation protocol for the Velocity Verlet algorithm requires precise execution of each computational step:
Initialization:
- Input initial conditions: positions (\mathbf{r}(t0)) and velocities (\mathbf{v}(t0)) of all atoms [25].
- If velocities are not available, generate them from a Maxwell-Boltzmann distribution at the desired temperature [25]: [ p(vi) = \sqrt{\frac{mi}{2 \pi kT}}\exp\left(-\frac{mi vi^2}{2kT}\right) ]
- Compute initial forces (\mathbf{F}(t0) = -\nabla V(\mathbf{r}(t0))) and accelerations (\mathbf{a}(t0) = \mathbf{F}(t0)/m_i).
Main Integration Loop (repeat for required number of steps):
Half-step velocity update: [ \mathbf{v}i(t + \frac{1}{2}\Delta t) = \mathbf{v}i(t) + \frac{\mathbf{F}i(t)}{2mi}\Delta t ] This estimates velocities at the intermediate time point using current accelerations [4].
Full-step position update: [ \mathbf{r}i(t + \Delta t) = \mathbf{r}i(t) + \mathbf{v}_i(t + \frac{1}{2}\Delta t)\Delta t ] Positions are advanced using the half-step velocities [19] [4].
Force computation at new positions: [ \mathbf{F}i(t+\Delta t) = -\nabla V(\mathbf{r}i(t+\Delta t)) ] This is typically the most computationally intensive step, involving calculation of both bonded and non-bonded interactions [25].
Complete velocity update: [ \mathbf{v}i(t + \Delta t) = \mathbf{v}i(t + \frac{1}{2}\Delta t) + \frac{\mathbf{F}i(t+\Delta t)}{2mi}\Delta t ] Velocities are finalized using the average of the initial and new accelerations [19] [4].
Output and Analysis:
This implementation requires two force evaluations per time step - once at the beginning and once after updating positions [19]. The explicit velocity calculations make the algorithm self-starting (unlike the original Verlet method) and minimize roundoff errors by avoiding the subtraction of nearly equal numbers [12].
Stability and Error Analysis
The Velocity Verlet algorithm exhibits exceptional numerical stability properties that account for its widespread adoption in molecular dynamics simulations. A comprehensive analysis of its error characteristics and stability thresholds reveals why it performs exceptionally well for molecular systems.
Discretization Error and Energy Conservation
The local truncation error of the Velocity Verlet algorithm is O(Δtâ´) for positions and O(Δt²) for velocities [2] [23]. The global error in positions accumulates as O(Δt²) over the entire simulation. This favorable error propagation stems from the algorithm's time-symmetry, which cancels out odd-order terms in the Taylor expansion [2].
For a harmonic oscillator with angular frequency ω, the algorithm produces stable solutions when ωΔt < 2, though for practical accuracy, much smaller values (typically ωΔt < 0.01) are recommended [2]. The following table quantifies key stability and error metrics:
Table 2: Numerical Stability and Error Characteristics
| Property | Value/Description | Implications for MD Simulations |
|---|---|---|
| Position Update Error | O(Δtâ´) local, O(Δt²) global | High positional accuracy for moderate timesteps |
| Velocity Update Error | O(Δt²) local | Sufficient for thermodynamic properties |
| Energy Conservation | Excellent long-term stability | Minimal energy drift in NVE simulations |
| Time Reversibility | Preserved to O(Δt²) | Physically realistic trajectory propagation |
| Symplectic Property | Yes | Preserves phase-space volume |
| Stability Condition | ωΔt < 2 (theoretical) | Dictates maximum usable timestep |
Time Step Selection Guidelines
Choosing an appropriate time step (Δt) is critical for balancing computational efficiency and numerical accuracy. The following experimental protocols provide guidance for time step selection based on system characteristics:
- Standard metallic systems: 5 femtoseconds provides a good balance between stability and efficiency [14].
- Systems with light atoms (e.g., hydrogen): 1-2 femtoseconds due to higher vibrational frequencies [14].
- Systems with strong bonds (e.g., carbon): 1-2 femtoseconds to capture rapid bond oscillations [14].
- Validation methodology: Monitor total energy conservation in NVE simulations; significant energy increase indicates unstable integration [14].
In practice, the maximum usable time step is determined by the highest frequency vibration in the system. For biomolecular simulations containing hydrogen atoms, this typically limits time steps to 1-2 femtoseconds. Some implementations employ constraint algorithms for bond vibrations to enable larger time steps [25].
Comparative Analysis with Related Integrators
The Velocity Verlet algorithm exists within a family of related integration schemes, each with distinct characteristics and applications. Understanding its relative advantages informs appropriate algorithm selection for specific molecular dynamics scenarios.
Table 3: Comparison of Verlet-Type Integration Algorithms
| Algorithm | Position Accuracy | Velocity Accuracy | Self-Starting | Velocity Calculation | Key Advantages |
|---|---|---|---|---|---|
| Original Verlet | O(Δt²) | O(Δt²) via difference | No | Not explicit | Simple, memory efficient |
| Velocity Verlet | O(Δt²) | O(Δt²) | Yes | Explicit, synchronous | Self-starting, better velocity handling |
| Leap-Frog | O(Δt²) | O(Δt²) | No | Explicit, asynchronous | Computationally efficient |
| Beeman | O(Δt²) | O(Δt²) | No | Complex form | Better energy conservation |
The Velocity Verlet algorithm provides several distinct advantages over related methods:
- Superior to Original Verlet: Eliminates the numerical precision issues associated with calculating velocities as ( \mathbf{v}i(t) = \frac{\mathbf{r}i(t+\Delta t) - \mathbf{r}_i(t-\Delta t)}{2\Delta t} ), which involves subtracting nearby numbers [12] [2].
- Advantage over Leap-Frog: Maintains synchronous position and velocity values, unlike the leap-frog method where positions and velocities are offset by half a time step [4].
- Preferable to Beeman Algorithm: While Beeman may offer slightly better energy conservation in some cases, its more complex formulation and non-self-starting nature make Velocity Verlet more practical for general use [12].
Practical Applications in Molecular Dynamics
The Velocity Verlet algorithm serves as the integration engine for numerous molecular dynamics applications across computational chemistry, materials science, and drug development. Its implementation in major MD software packages underscores its fundamental importance to the field.
Essential Research Reagents and Computational Tools
Successful implementation of Velocity Verlet-based molecular dynamics requires a suite of computational "research reagents" - the essential software components and parameters that enable accurate simulations:
Table 4: Essential Research Reagents for Velocity Verlet MD Simulations
| Reagent/Solution | Function/Purpose | Implementation Example |
|---|---|---|
| Force Fields | Defines potential energy function V(ð«) | CHARMM, AMBER, OPLS parameters |
| Neighbor Lists | Efficient non-bonded force calculation | Verlet lists with buffer distance [25] |
| Periodic Boundary Conditions | Mimics bulk environment | Particle-mesh Ewald for electrostatics |
| Thermostats/Couplers | Temperature control (NVT ensemble) | Nosé-Hoover, Langevin, Bussi dynamics [14] |
| Constraint Algorithms | Handles rigid bonds (e.g., SHAKE, LINCS) | Enables larger time steps [25] |
| Parallelization Schemes | Accelerates force calculations | Domain decomposition, GPU implementations |
Applications in Drug Development
For pharmaceutical researchers, the Velocity Verlet algorithm enables critical investigations into:
- Protein-ligand binding dynamics: Simulating molecular recognition processes with stable energy conservation.
- Conformational sampling: Generating thermodynamically relevant structural ensembles of drug targets.
- Solvation effects: Modeling explicit solvent interactions over nanosecond to microsecond timescales.
- Drug-membrane interactions: Studying partitioning and diffusion of compounds in lipid bilayers.
The algorithm's numerical stability ensures that observed physical phenomena derive from the force field physics rather than integration artifacts, making it indispensable for reliable molecular predictions in drug design pipelines.
Recent Advances and Future Directions
While the core Velocity Verlet algorithm remains unchanged, ongoing research focuses on enhancing its efficiency and extending its applicability. Recent developments include:
- Improved neighbor list algorithms: Automatic buffering and dynamic pair list pruning to reduce computational overhead [25].
- Extended phase space methods: Enabling efficient sampling of rare events while maintaining numerical stability.
- Multi-time stepping schemes: Combining Velocity Verlet with RESPA (Reversible Reference System Propagator Algorithms) for handling forces with different time scales.
- Specialized variants for granular flows: Modified Velocity Verlet algorithms for systems with large particle size ratios, addressing unphysical results in discrete element method simulations [26].
- Integration with machine learning potentials: Coupling with neural network force fields for ab initio accuracy in large-scale systems.
These advances continue to expand the applicability of the Velocity Verlet framework while maintaining its fundamental numerical advantages, ensuring its continued relevance for future molecular simulation challenges in basic research and applied drug development.
Molecular Dynamics (MD) simulation serves as a crucial computational technique for understanding the physical movements of atoms and molecules over time, with applications spanning from drug development to materials science. At the heart of any MD simulation lies the integration algorithm—the mathematical scheme that numerically solves Newton's equations of motion to update particle positions and velocities. The Verlet algorithm, in its various forms, represents one of the most fundamental and widely adopted integration families in this field. First introduced in the late 1960s and rediscovered multiple times, the Verlet algorithm provides the foundation upon which modern MD simulations are built. Its popularity stems from favorable numerical properties, including time-reversibility, symplectic nature (phase-space volume preservation), and computational efficiency compared to other integration methods.
Within the family of Verlet integrators, the leap-frog algorithm emerges as a particularly significant variant that addresses specific computational challenges while introducing its own unique characteristics. This technical guide explores the leap-frog variant within the broader context of Verlet algorithms, focusing specifically on its defining features: computational economy achieved through strategic implementation and the implications of its staggered velocity formulation. As MD simulations continue to push the boundaries of system size and temporal scale, understanding these algorithmic nuances becomes increasingly critical for researchers aiming to optimize their computational workflows while maintaining physical accuracy. The following sections provide a comprehensive examination of the mathematical foundations, implementation details, and practical considerations of the leap-frog algorithm, with particular attention to its role in contemporary biomolecular simulation and drug development applications.
Mathematical Foundation: From Verlet to Leap-Frog
The Verlet Base Algorithm
The original Verlet algorithm operates directly on particle positions, eliminating explicit velocity dependence in the position update. The method derives from Taylor expansions of position around time t:
Adding these equations cancels the velocity terms and yields the Verlet position integrator:
where a(t) = F(t)/m represents acceleration computed from the force F(t) acting on a particle of mass m [2] [4]. This formulation provides excellent numerical stability and energy conservation properties but suffers from two practical limitations: it is not self-starting (requiring knowledge of positions at t-Δt), and velocities do not appear explicitly, making temperature calculation and velocity-dependent operations inconvenient.
Derivation of the Leap-Frog Variant
The leap-frog algorithm addresses the velocity limitation by introducing a staggered time grid where velocities are defined at half-integer time steps. This formulation emerges naturally from the Velocity Verlet algorithm, which is mathematically equivalent to the original Verlet method but updates positions and velocities synchronously:
The leap-frog variant reorganizes these calculations by evaluating velocities at midpoints between position updates. The algorithm can be derived by considering velocity updates at half-step intervals:
This formulation creates a leap-frogging pattern where positions and velocities alternate in their update sequence, giving the algorithm its name [4]. The mathematical equivalence to the original Verlet algorithm can be demonstrated by substituting the position update into the velocity update and rearranging terms, ultimately recovering the original Verlet position equation.
Table 1: Comparison of Verlet Algorithm Variants
| Algorithm | Position Update | Velocity Update | Velocity Availability | Self-Starting |
|---|---|---|---|---|
| Original Verlet | r(t+Δt) = 2r(t) - r(t-Δt) + a(t)Δt² |
Not explicit | Calculated as [r(t+Δt) - r(t-Δt)]/(2Δt) |
No |
| Velocity Verlet | r(t+Δt) = r(t) + v(t)Δt + (1/2)a(t)Δt² |
v(t+Δt) = v(t) + [a(t) + a(t+Δt)]Δt/2 |
Synchronous with positions | Yes |
| Leap-Frog | r(t+Δt) = r(t) + v(t+Δt/2)Δt |
v(t+Δt/2) = v(t-Δt/2) + a(t)Δt |
Staggered at half-steps | Yes |
The Leap-Frog Algorithm: Core Mechanics and Staggered Velocities
Algorithmic Workflow
The leap-frog algorithm implements a specific sequence of operations that alternate between updating velocities and positions at staggered time points. A complete integration step follows this precise workflow:
- Force Computation: Calculate forces
F_i(t)for all particles based on the current positionsr_i(t)using the appropriate force field potential energy functions [11]. - Velocity Update: Advance velocities by a full time step using the computed accelerations:
v_i(t+Δt/2) = v_i(t-Δt/2) + [F_i(t)/m_i]Δt[4]. - Position Update: Advance positions by a full time step using the newly updated velocities:
r_i(t+Δt) = r_i(t) + v_i(t+Δt/2)Δt[4]. - Time Advancement: Update the simulation time from
ttot+Δt, which effectively shifts the time reference for the velocities (what wasv(t+Δt/2)now becomesv(t-Δt/2)for the next iteration).
This sequence creates an interleaved update pattern where velocities always "leap" ahead of positions, then positions "leap" ahead of velocities, creating the characteristic staggered integration scheme.
The Staggered Velocity Scheme
The defining characteristic of the leap-frog algorithm is its treatment of velocities at half-time steps offset from positions. This staggered scheme means that at any integer time step t, the simulation has access to:
- Positions
r(t)at the current time - Velocities
v(t-Δt/2)from the previous half-step - Forces
F(t)computed from the current positions
The velocities at integer time steps are not directly available but can be approximated as:
This averaging approach provides a statistically reasonable estimate of the instantaneous velocity, though it introduces a slight phase shift in velocity-dependent properties [27]. Research has shown that for calculating thermodynamic properties like temperature and pressure, a more accurate approach is to average the kinetic energies at the previous and following half-steps rather than using the square of the average velocity, as the latter introduces a systematic bias in these measurements [27].
Computational Economy: Implementation Advantages
Performance and Memory Considerations
The leap-frog algorithm offers significant computational advantages that contribute to its widespread adoption in production MD codes, particularly for large-scale biomolecular simulations. These efficiency gains manifest in several key areas:
Reduced Memory Operations: The algorithm requires storing only one set of velocities (at half-steps) and updates them in place, minimizing memory transfer operations. This becomes particularly advantageous for massive parallel simulations where memory bandwidth often limits performance [11].
Optimized Force Computation: By updating all positions before recomputing forces, the algorithm enables efficient neighbor list construction and potential energy calculations. Modern MD implementations like GROMACS leverage this structure to optimize non-bonded force calculations using cluster-based approaches that process multiple particle interactions simultaneously, mapping efficiently to SIMD and SIMT architectures in CPUs and GPUs [11].
Minimal Floating Point Operations: Compared to other second-order integrators, leap-frog requires fewer floating point operations per time step. Each position update involves a single multiplication and addition per degree of freedom, while each velocity update similarly requires minimal arithmetic operations.
Integration with Modern MD Infrastructure
The computational economy of leap-frog integration extends beyond the core algorithm to its synergy with broader MD simulation infrastructure:
Neighbor List Optimization: The algorithm's structure accommodates efficient neighbor list updates, which typically occur less frequently than integration steps (every
nstliststeps). GROMACS implements a buffered Verlet list scheme with automatic pair-list pruning that leverages the leap-frog sequence to minimize unnecessary force calculations [11].Parallelization Efficiency: The clear separation of position and velocity updates creates natural synchronization points in parallel implementations. This regularity enables efficient domain decomposition and load balancing across processor cores, which is critical for scaling to thousands of processors in modern high-performance computing environments.
Temperature and Pressure Coupling: The staggered velocity scheme integrates effectively with standard thermostat and barostat algorithms. For example, the Berendsen thermostat applies velocity scaling as
v_{new} = v_{old} × λwhereλ = [1 + (Δt/τ)(T₀/T - 1)]^{1/2}, withTbeing the instantaneous temperature computed from the velocities [28]. This scaling can be applied cleanly during the velocity update step without disrupting the position integration.
Table 2: Computational Requirements Comparison for N Particles
| Operation | Leap-Frog | Velocity Verlet | Original Verlet |
|---|---|---|---|
| Position Updates per Step | N | N | N |
| Velocity Updates per Step | N | N | Not explicit |
| Force Computations per Step | 1 | 1 | 1 |
| Memory Requirements (Beyond Forces) | 2N (positions, velocities) | 2N (positions, velocities) | 2N (current, previous positions) |
| Velocity-Dependent Operations | Requires interpolation | Direct | Requires calculation from positions |
Practical Implementation in Biomolecular Research
Research Reagent Solutions: Computational Tools
Successful implementation of leap-frog integration in drug development research requires specialized computational tools and "reagent" solutions that form the infrastructure for MD simulations:
Table 3: Essential Research Reagent Solutions for MD Simulations
| Tool Category | Specific Examples | Function in Leap-Frog MD | Implementation Considerations |
|---|---|---|---|
| Force Fields | AMBER, CHARMM, GROMOS [22] | Define potential energy functions for force computation | Parameterization determines integration stability; heavier atoms permit larger time steps |
| Integration Packages | GROMACS, LAMMPS, NAMD [11] | Provide optimized leap-frog implementation | GROMACS uses cluster-based neighbor lists for CPU/GPU efficiency [11] |
| Thermostats | Berendsen, Nosé-Hoover, Langevin [14] [28] | Regulate temperature during simulation | Berendsen: v_{new} = v_{old}×[1 + (Δt/τ)(T₀/T - 1)]^{1/2} [28] |
| Barostats | Parrinello-Rahman, Berendsen [14] | Maintain constant pressure | Coupled to position updates through box scaling |
| Solvation Models | TIP3P, SPC, SPC/E [22] | Provide aqueous environment for biomolecules | Water model affects optimal time step (typically 1-2 fs for explicit water) |
| Analysis Suites | MDAnalysis, MDTraj, VMD [15] | Process trajectory data | Velocity analysis requires reconstruction from staggered values |
Experimental Protocol: System Setup and Execution
Implementing a biomolecular MD simulation using leap-frog integration follows a standardized protocol:
System Preparation:
- Obtain initial coordinates from experimental structure (X-ray, NMR) or homology modeling [22]
- Solvate the biomolecule in an appropriate water model (e.g., TIP3P) using a predefined box size with periodic boundary conditions [22]
- Add ions to neutralize system charge and achieve physiological concentration (e.g., 150mM NaCl)
Initialization and Minimization:
- Generate initial velocities from Maxwell-Boltzmann distribution at target temperature:
p(v_i) = √(m_i/(2πkT)) × exp(-m_iv_i²/(2kT))[11] - Perform energy minimization using steepest descent or conjugate gradient method to remove bad contacts
- Apply positional restraints on heavy atoms during initial equilibration
- Generate initial velocities from Maxwell-Boltzmann distribution at target temperature:
Equilibration Protocol:
- Run short simulation (50-100ps) with velocity rescaling thermostat (Berendsen or velocity scaling) to stabilize temperature [28]
- Gradually release positional restraints on non-solvent atoms
- For NPT ensemble, enable pressure coupling with barostat after temperature stabilization
Production Simulation:
- Switch to conservative thermostat (Nosé-Hoover or Langevin) for production dynamics [14]
- Set time step based on system characteristics: 1fs for all-atom systems with hydrogen, 2fs with constrained bonds, 4-5fs for coarse-grained systems [14]
- Configure trajectory output frequency (every 1-10ps) based on storage capacity and analysis requirements
- Run simulation for duration sufficient to observe phenomena of interest (nanoseconds to microseconds)
Analysis and Validation:
- Monitor stability of thermodynamic quantities (temperature, pressure, energy)
- Calculate root-mean-square deviation (RMSD) to assess structural stability
- Compute relevant biological properties (binding energies, distance measurements, fluctuation analyses)
Comparative Analysis and Research Applications
Algorithmic Performance in Biomolecular Systems
The leap-frog algorithm demonstrates specific performance characteristics across different biomolecular simulation scenarios:
Temperature Control Accuracy: While the staggered velocities introduce a small phase error in instantaneous temperature calculations, research has shown that proper implementation using kinetic energy averaging produces accurate thermodynamic sampling. The systematic bias mentioned in the literature emerges primarily when using the square of averaged velocities rather than averaging kinetic energies from half-steps [27].
Energy Conservation Properties: In microcanonical (NVE) ensemble simulations, the leap-frog algorithm exhibits excellent long-term energy conservation, with deviations typically smaller than those observed in non-symplectic integrators. This property makes it particularly valuable for studying energy flow and thermodynamic stability in biomolecular systems.
Discretization Error Accumulation: Like all Verlet-family algorithms, leap-frog exhibits fourth-order local error and second-order global error in positions. The error accumulation follows
ε ∠Δt², meaning that halving the time step reduces error by approximately a factor of four [15]. This relationship continues until numerical roundoff error dominates, which occurs at significantly smaller step sizes.
Table 4: Performance Metrics for Different Biomolecular Systems
| System Type | Recommended Time Step | Typical Simulation Duration | Stability Considerations |
|---|---|---|---|
| All-Atom Protein (explicit solvent) | 1-2 fs [14] | 100 ns - 1 μs | Constraints on bonds involving hydrogen essential for stability |
| Coarse-Grained Protein | 10-20 fs | 1-10 μs | Larger time steps possible with reduced degrees of freedom |
| Membrane Protein | 2 fs | 200 ns - 2 μs | Lipid dynamics may require longer equilibration |
| Protein-Ligand Complex | 1-2 fs | 50-500 ns | Ligand parameterization critical for accurate binding |
| Nucleic Acids | 1-2 fs | 100 ns - 1 μs | Counterion placement important for structure stability |
Applications in Drug Development Research
The computational economy of the leap-frog algorithm enables several critical applications in pharmaceutical research:
Binding Affinity Calculations: Multiple short simulations of drug-receptor complexes can be efficiently performed using leap-frog integration, enabling ensemble-based approaches to binding free energy estimation through methods such as MM/PBSA or thermodynamic integration.
Conformational Sampling: The algorithm's numerical stability facilitates extended simulations that capture rare biological events, such as protein folding transitions or allosteric conformational changes, which are critical for understanding drug mechanism of action.
Membrane Permeability Studies: Efficient simulation of drug compounds in lipid bilayers provides insights into bioavailability and membrane transport properties, with the leap-frog algorithm enabling the necessary multi-nanosecond timescales for adequate sampling.
Solvation and Hydration Analysis: The explicit solvent simulations enabled by leap-frog's computational efficiency help characterize drug solvation shells and hydration patterns, which influence solubility and formulation properties.
In each of these applications, the staggered velocity scheme provides a favorable balance between computational cost and physical accuracy, making it particularly suitable for the ensemble-based approaches increasingly employed in modern drug discovery pipelines. As MD simulations continue to expand into high-throughput virtual screening and multi-scale modeling, the economic advantages of robust, efficient integration algorithms like leap-frog become increasingly significant for pharmaceutical research.
Step-by-Step Implementation Guide for a Simple Molecular System
Molecular Dynamics (MD) simulation is a computational technique that uses numerical methods to simulate the physical movements of atoms and molecules over time. By applying classical mechanics, MD provides insights into the dynamic evolution of molecular systems, enabling researchers to study structural changes, thermodynamic properties, and kinetic behavior at an atomic level. The foundation of MD lies in numerically solving Newton's equations of motion for a system of interacting particles, a process that requires robust integration algorithms to ensure accuracy and stability over long simulation times [29] [22].
Among the various integration schemes available, the Verlet algorithm has emerged as a cornerstone of molecular dynamics simulations due to its favorable numerical properties. First introduced by Loup Verlet in the 1960s, this algorithm provides a time-reversible and symplectic method for integrating equations of motion, effectively conserving system energy and preserving the symplectic form on phase space [2]. The Verlet algorithm's combination of computational efficiency, numerical stability, and conservation properties has made it the integration method of choice in major MD software packages including GROMACS, NAMD, and ASE [29] [14] [3]. Its implementation is particularly crucial for simulating biomolecular systems where simulation accuracy and energy conservation are paramount for obtaining physically meaningful results.
Theoretical Foundation of the Verlet Algorithm
Mathematical Derivation
The Verlet algorithm derives from Taylor expansions of particle positions around time t. By writing third-order Taylor expansions for positions both forward and backward in time, we obtain:
$$\begin{equation} \mathbf{r}(t+\Delta t) = \mathbf{r}(t) + \mathbf{v}(t)\Delta t + \frac{1}{2}\mathbf{a}(t)\Delta t^2 + \frac{1}{6}\mathbf{b}(t)\Delta t^3 + O(\Delta t^4) \end{equation}$$
$$\begin{equation} \mathbf{r}(t-\Delta t) = \mathbf{r}(t) - \mathbf{v}(t)\Delta t + \frac{1}{2}\mathbf{a}(t)\Delta t^2 - \frac{1}{6}\mathbf{b}(t)\Delta t^3 + O(\Delta t^4) \end{equation}$$
Adding these two expressions eliminates the velocity and third-order terms, yielding the fundamental Verlet equation:
$$\begin{equation} \mathbf{r}(t+\Delta t) = 2\mathbf{r}(t) - \mathbf{r}(t-\Delta t) + \mathbf{a}(t)\Delta t^2 + O(\Delta t^4) \end{equation}$$
where $\mathbf{r}$ represents atomic positions, $\mathbf{v}$ velocities, $\mathbf{a}$ accelerations, and $\mathbf{b}$ the third derivatives of position with respect to time [8] [2]. The acceleration $\mathbf{a}(t)$ is computed from Newton's second law as $\mathbf{a}(t) = \mathbf{F}(t)/m$, where the force $\mathbf{F}(t)$ is determined from the interaction potential $V$ through $\mathbf{F}i = -\partial V/\partial \mathbf{r}i$ [29].
The remarkable property of this algorithm is that although it requires only forces and positions from previous time steps, it achieves fourth-order accuracy in position while using only second-order calculations. This efficiency, combined with its time-reversibility and symplectic nature, makes it particularly suitable for long-time MD simulations [2].
Velocity Verlet Variant
The original Verlet algorithm does not explicitly calculate velocities, which are necessary for computing kinetic energy and monitoring conservation of total energy. The Velocity Verlet algorithm addresses this limitation by explicitly incorporating velocity calculations while maintaining the accuracy and stability of the original method:
$$\begin{equation} \mathbf{r}(t+\Delta t) = \mathbf{r}(t) + \mathbf{v}(t)\Delta t + \frac{1}{2}\mathbf{a}(t)\Delta t^2 \end{equation}$$
$$\begin{equation} \mathbf{v}(t + \Delta t/2) = \mathbf{v}(t) + \frac{1}{2}\mathbf{a}(t)\Delta t \end{equation}$$
$$\begin{equation} \mathbf{a}(t + \Delta t) = -\frac{1}{m}\nabla V(\mathbf{r}(t+\Delta t)) \end{equation}$$
$$\begin{equation} \mathbf{v}(t + \Delta t) = \mathbf{v}(t + \Delta t/2) + \frac{1}{2}\mathbf{a}(t + \Delta t)\Delta t \end{equation}$$
This formulation provides numerically stable velocities at the same time points as positions and requires only one force evaluation per time step, making it highly efficient for MD simulations [8] [3]. The Velocity Verlet algorithm has become the most widely used integration scheme in modern MD simulations due to its simplicity, stability, and explicit velocity handling [3].
Discretization Error and Numerical Stability
The local truncation error of the Verlet algorithm is of order O(Δtâ´) for positions, though the global error accumulates as O(Δt²) over the simulation. The algorithm's time symmetry eliminates odd-order terms in the error expansion, contributing to its excellent long-term energy conservation properties [2]. Numerical stability requires that the time step Δt satisfies the condition Δt ≤ 1/(Ï€f), where f is the highest frequency vibration in the system. For bonds involving hydrogen atoms, which have vibrational periods of approximately 10 femtoseconds, this limits practical time steps to 1-2 fs unless constraints are applied [3].
Table 1: Comparison of Verlet Algorithm Variants
| Algorithm | Position Update | Velocity Update | Force Evaluations per Step | Memory Requirements |
|---|---|---|---|---|
| Original Verlet | r(t+Δt) = 2r(t) - r(t-Δt) + a(t)Δt² |
v(t) = [r(t+Δt) - r(t-Δt)]/(2Δt) |
1 | 6N (positions at t and t-Δt) |
| Velocity Verlet | r(t+Δt) = r(t) + v(t)Δt + ½a(t)Δt² |
v(t+Δt) = v(t) + ½[a(t) + a(t+Δt)]Δt |
1 | 9N (positions, velocities, accelerations) |
| Leap-Frog | r(t+Δt) = r(t) + v(t+Δt/2)Δt |
v(t+Δt/2) = v(t-Δt/2) + a(t)Δt |
1 | 6N (positions, half-step velocities) |
System Setup and Force Field Selection
Initial System Configuration
Implementing an MD simulation begins with defining the initial molecular system configuration. For a simple monatomic system such as argon gas, this involves placing atoms in a simulation box with defined dimensions. A common approach is initializing the system by placing atoms on a cubic lattice to avoid unrealistic atomic overlaps [5]. The system size is determined by the number of particles (N) and the target density. For argon gas under standard conditions (274 K, 1 atm), a density of approximately 1.774 kg/m³ corresponds to a numerical density of 2.66×10²ⵠatoms/m³ [5].
The initial velocities are typically assigned from a Maxwell-Boltzmann distribution at the desired temperature:
$$\begin{equation} p(vi) = \sqrt{\frac{mi}{2\pi kB T}}\exp\left(-\frac{mi vi^2}{2kB T}\right) \end{equation}$$
where $kB$ is Boltzmann's constant, $T$ is temperature, and $mi$ is the atomic mass [29]. After initial assignment, velocities are adjusted to ensure the center-of-mass velocity is zero, preventing overall drift of the system during simulation [29].
Force Field Parameters
The interatomic forces governing particle motions are derived from a force field, which mathematically describes the potential energy surface of the system. For simple monatomic systems like argon, the Lennard-Jones potential provides an effective description of van der Waals interactions:
$$\begin{equation} V{LJ}(r{ij}) = 4\epsilon\left[\left(\frac{\sigma}{r{ij}}\right)^{12} - \left(\frac{\sigma}{r{ij}}\right)^6\right] \end{equation}$$
where $r_{ij}$ is the distance between atoms i and j, ε is the potential well depth, and σ is the finite distance at which the interatomic potential is zero [5]. The corresponding force between two atoms is derived as the negative gradient of this potential.
Table 2: Lennard-Jones Parameters for Common Atoms
| Atom Type | Mass (amu) | ε (kJ/mol) | σ (nm) | Cut-off Radius (nm) |
|---|---|---|---|---|
| Argon | 39.95 | 0.996 | 0.340 | 1.0-1.2 |
| Carbon | 12.01 | 0.393 | 0.350 | 1.0-1.2 |
| Oxygen | 16.00 | 0.650 | 0.304 | 1.0-1.2 |
| Nitrogen | 14.01 | 0.712 | 0.325 | 1.0-1.2 |
For more complex biomolecular systems, additional force field terms are required to properly describe bonded interactions:
$$\begin{equation}
E{\text{total}} = E{\text{bonded}} + E{\text{non-bonded}} = \sum{\text{bonds}} Kr(r - r0)^2 + \sum{\text{angles}} K\theta(\theta - \theta0)^2 + \sum{\text{dihedrals}} \frac{Vn}{2}[1 + \cos(n\phi - \gamma)] + \sum{i
where the terms represent bond stretching, angle bending, torsional rotations, and non-bonded interactions (van der Waals and electrostatic), respectively [22].
Implementation Protocol
Molecular Dynamics Workflow
The following diagram illustrates the complete molecular dynamics simulation workflow, from system initialization to trajectory analysis:
Molecular Dynamics Simulation Workflow
Step-by-Step Implementation Code
The following C++ code snippet demonstrates the core Velocity Verlet integration algorithm for a simple molecular system:
This implementation follows the Velocity Verlet algorithm precisely, with separate stages for position updates, force calculations, and velocity updates. The force calculation uses a truncated Lennard-Jones potential with a defined cut-off radius to improve computational efficiency [5].
Neighbor List Optimization
For systems with large numbers of particles, calculating all pairwise interactions becomes computationally prohibitive (O(N²)). To address this, MD implementations typically use neighbor lists that track only particles within a certain cut-off distance. The Verlet buffer algorithm maintains a list with a cut-off radius slightly larger than the interaction cut-off, updating it periodically to account for particle diffusion:
$$\begin{equation} r\ell = rc + r_b \end{equation}$$
where $r\ell$ is the list cut-off, $rc$ is the interaction cut-off, and $r_b$ is the buffer size [29]. This approach reduces the complexity of force calculations to approximately O(N) while maintaining accuracy through careful buffer selection.
Simulation Parameters and Equilibration
Time Step Selection
Choosing an appropriate time step is critical for simulation stability and accuracy. The maximum time step is limited by the highest frequency vibration in the system, typically bond stretching involving hydrogen atoms:
$$\begin{equation} \Delta t \leq \frac{1}{\pi f_{\text{max}}} \end{equation}$$
where $f_{\text{max}}$ is the highest vibrational frequency [3]. For systems with C-H bonds (vibrational period ~10 fs), this limits time steps to approximately 3.2 fs, though in practice 1-2 fs is recommended for accurate integration [3].
Table 3: Recommended Time Steps for Different Systems
| System Type | Recommended Δt (fs) | Constraint Method | Rationale |
|---|---|---|---|
| All-atom, no constraints | 0.5-1.0 | None | Captures all bond vibrations including C-H |
| Bonds with H constrained | 2.0 | SHAKE/LINCS | Removes fastest vibrations (C-H bonds) |
| All bonds constrained | 3.0-4.0 | SHAKE/LINCS | Removes all bond vibrations |
| Hydrogen mass repartitioning | 4.0 | Modified masses | Increases hydrogen mass, decreasing vibration frequency |
Equilibration Protocol
Before production simulation, the system must undergo equilibration to reach the target temperature and eliminate artificial configurations from initial setup. A typical equilibration protocol includes:
- Energy Minimization: Steepest descent or conjugate gradient minimization to remove bad contacts and high-energy configurations
- Initial Thermalization: NVT simulation with velocity rescaling to reach target temperature
- Density Equilibration: NPT simulation to achieve correct system density
- Production Equilibration: Extended NVT or NPT simulation with proper thermostat/barostat
For the argon system described in the search results, equilibration at 274 K for at least 1000 steps with 1000 particles produces good agreement between simulated pressure (10953.4) and theoretical pressure (11008.9) [5].
Analysis and Validation
Energy Conservation
In microcanonical (NVE) ensemble simulations, the total energy should remain constant throughout the simulation. The Velocity Verlet algorithm exhibits excellent energy conservation properties when implemented with an appropriate time step. Energy conservation is typically monitored by calculating:
$$\begin{equation} \delta E(t) = \frac{|E(t) - E(0)|}{|E(0)|} \end{equation}$$
where $E(t)$ is the total energy at time t and $E(0)$ is the initial total energy [14]. For a well-equilibrated system, the energy should fluctuate around a stable mean value without significant drift.
Thermodynamic Properties
After equilibration, production simulations are used to compute thermodynamic averages and structural properties. Key validation metrics include:
- Temperature: Calculated from kinetic energy via the equipartition theorem:
$$\begin{equation} T(t) = \frac{2\langle K(t) \rangle}{(3N - Nc)kB} \end{equation}$$
where $N_c$ is the number of constraints [29]
- Pressure: For a Lennard-Jones system, pressure can be computed via the virial theorem:
$$\begin{equation}
P = \frac{NkB T}{V} + \frac{1}{3V}\left\langle \sum{i
- Diffusion Coefficient: Calculated from the mean-squared displacement:
$$\begin{equation} D = \lim{t\to\infty} \frac{1}{6t} \langle |\mathbf{r}i(t) - \mathbf{r}_i(0)|^2 \rangle \end{equation}$$
The following diagram illustrates the analysis workflow for validating simulation results:
Molecular Dynamics Analysis Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Tools and Algorithms for MD Simulations
| Tool Category | Specific Implementation | Function/Purpose | Key Applications |
|---|---|---|---|
| Integration Algorithms | Velocity Verlet [8] [3] | Time-reversible, symplectic integration | NVE ensemble simulations |
| Leap-Frog [3] | Efficient velocity update scheme | Large-scale production runs | |
| Constraint Algorithms | SHAKE [3] | Bond length constraints | Enabling larger time steps |
| LINCS [3] | Faster bond constraints | Parallel simulations | |
| SETTLE [3] | Water molecule constraints | Rigid water models | |
| Force Fields | Lennard-Jones [5] | Van der Waals interactions | Simple fluids, noble gases |
| AMBER/CHARMM [22] | Biomolecular force fields | Proteins, nucleic acids | |
| Thermostats | Langevin [14] | Stochastic temperature control | NVT ensemble |
| Nosé-Hoover [14] | Deterministic thermostat | Accurate NVT sampling | |
| Software Packages | GROMACS [29] [3] | High-performance MD | Biomolecular systems |
| ASE [14] | Python-based simulation | Prototyping, education | |
| 2-Morpholin-4-yl-8-nitroquinoline | 2-Morpholin-4-yl-8-nitroquinoline|CAS 292053-56-6 | High-purity (≥98%) 2-Morpholin-4-yl-8-nitroquinoline for cancer research and kinase studies. For Research Use Only. Not for human use. | Bench Chemicals |
| DOTA-GA(tBu)4 | DOTA-GA(tBu)4, MF:C35H64N4O10, MW:700.9 g/mol | Chemical Reagent | Bench Chemicals |
Advanced Applications and Optimizations
Enhanced Sampling Techniques
For complex biomolecular systems, standard MD simulations may be insufficient to overcome energy barriers and sample relevant conformational space. Enhanced sampling methods include:
- Temperature Replica Exchange: Parallel simulations at different temperatures with occasional exchanges
- Metadynamics: Adding bias potential to discourage revisiting sampled configurations
- Umbrella Sampling: Applying biasing potentials to specific reaction coordinates
These techniques enable the study of rare events and free energy landscapes that would be inaccessible through conventional MD.
Optimized Verlet-like Algorithms
Recent advances in integration schemes have led to optimized Verlet-like algorithms that maintain the symplectic property while improving accuracy. These methods introduce free parameters in the decomposition scheme that can be tuned to minimize truncation error, resulting in improved accuracy at equivalent computational cost [16]. Such optimizations are particularly valuable for simulations requiring high precision in energy conservation or long-time stability.
The Verlet algorithm represents a fundamental component of molecular dynamics simulations, providing an optimal balance between computational efficiency, numerical stability, and physical accuracy. This step-by-step implementation guide has detailed the theoretical foundation, practical implementation, and validation protocols for deploying Verlet integration in molecular systems. From simple monatomic fluids to complex biomolecular assemblies, the principles outlined here form the basis for accurate and reliable MD simulations.
The continued development of optimized integration schemes and advanced sampling techniques ensures that molecular dynamics will remain an essential tool for researchers investigating atomic-scale phenomena in chemical, biological, and materials systems. As computational power increases and algorithms become more sophisticated, MD simulations will continue to bridge the gap between theoretical predictions and experimental observations across diverse scientific disciplines.
Molecular Dynamics (MD) simulations have become an indispensable tool in computational chemistry, biophysics, and drug discovery, providing atomic-level insights into the behavior of biomolecular systems over time. The Verlet algorithm and its variants form the fundamental numerical framework that enables these simulations by solving Newton's equations of motion for systems ranging from small ligands to massive protein complexes. First introduced by Loup Verlet in 1967 for classical fluid simulations, this algorithm has evolved into the cornerstone of modern biomolecular simulation packages [12] [4]. Its stability, simplicity, and conservation properties make it particularly suited for studying biological processes such as protein folding, ligand binding, and solvation dynamics—processes essential for understanding disease mechanisms and developing novel therapeutics.
The significance of the Verlet algorithm extends beyond mere computational convenience; its time-reversible and symplectic nature ensures excellent energy conservation in microcanonical (NVE) ensembles, making it ideal for long-timescale simulations where numerical stability is paramount [4]. For researchers and drug development professionals, understanding the principles and applications of this algorithm is crucial for designing robust simulation protocols and interpreting results accurately. This technical guide explores the theoretical foundations, practical implementations, and specific applications of Verlet-based integration schemes in studying proteins, ligands, and solvation environments, providing both conceptual frameworks and detailed methodologies for implementing these techniques in cutting-edge biomolecular research.
Theoretical Foundations of the Verlet Algorithm
Mathematical Formulation and Variants
The Verlet algorithm derives from Taylor expansions of particle positions around time ( t ). The standard Verlet formulation calculates new positions using current and previous positions along with current acceleration [12] [4]:
[ \mathbf{r}{i}(t+\Delta t) = 2\mathbf{r}{i}(t)-\mathbf{r}{i}(t-\Delta t)+\frac{\mathbf{F}{i}}{2m}(t)\Delta t^{2}+\mathcal{O}(\Delta t ^{3}) ]
Where ( \mathbf{r}{i} ) represents the position of particle ( i ), ( \Delta t ) is the integration time step, ( \mathbf{F}{i} ) is the force acting on particle ( i ), and ( m ) is its mass. Although computationally efficient and memory-conserving (requiring only one set of force evaluations per step), the basic Verlet algorithm has limitations: it is not self-starting, and velocities are not directly available [12].
The velocity Verlet algorithm addresses these limitations by providing explicit velocity updates and being self-starting. It decomposes into these sequential steps [4] [30]:
- ( \mathbf{v}{i}(t + \frac{1}{2}\Delta t)=\mathbf{v}{i}(t)+\frac{\mathbf{F}{i}(t)}{2m{i}}\Delta t )
- ( \mathbf{r}{i}(t + \Delta t) = \mathbf{r}{i}(t) + \mathbf{v}_{i}(t + \frac{1}{2}\Delta t)\Delta t )
- Compute forces ( \mathbf{F}_{i}(t+\Delta t) ) at new positions
- ( \mathbf{v}{i}(t + \Delta t)=\mathbf{v}{i}(t+\frac{1}{2}\Delta t)+\frac{\mathbf{F}{i}(t+\Delta t)}{2m{i}}\Delta t )
This formulation maintains the symplectic property while providing synchronous position and velocity vectors [4] [30]. The leap-frog variant, another popular implementation, uses a slightly different approach where velocities are calculated at half-time steps [4]:
- Compute forces ( \mathbf{F}_{i}(t) )
- ( \mathbf{v}{i}(t + \frac{1}{2}\Delta t)=\mathbf{v}{i}(t- \frac{1}{2}\Delta t)+\frac{\mathbf{F}{i}(t)}{m{i}}\Delta t )
- ( \mathbf{r}{i}(t + \Delta t) = \mathbf{r}{i}(t) + \mathbf{v}_{i}(t + \frac{1}{2}\Delta t)\Delta t )
In this scheme, position and velocity vectors are asynchronous, but the algorithm remains computationally efficient and numerically stable [4].
Numerical Properties and Conservation Laws
The Verlet algorithm's superiority in biomolecular simulations stems from its symplectic nature, meaning it preserves the phase-space volume in Hamiltonian systems. This property leads to excellent long-term energy conservation, crucial for simulating thermodynamic properties accurately [4]. The global error in positions is third-order (( \mathcal{O}(\Delta t ^{3}) )), while velocity estimates are second-order accurate (( \mathcal{O}(\Delta t ^{2}) )) [12].
For biomolecular systems where simulations extend to microseconds or milliseconds, this numerical stability prevents energy drift and ensures physically meaningful trajectories. The algorithm's time-reversibility also mirrors the fundamental symmetry of Newton's equations, making it particularly suited for studying reversible processes in molecular systems [12] [4].
Implementation in Biomolecular Systems
Integration with Force Fields and Simulation Packages
The Verlet algorithm serves as the computational engine in major MD software packages used for biomolecular simulations. GROMACS, renowned for its high-performance MD capabilities, utilizes highly optimized Verlet integration schemes to achieve exceptional simulation speeds for proteins, nucleic acids, and lipid membranes [31] [32]. Similarly, NAMD and AMBER implement Verlet-based approaches with specialized enhancements for scalable biomolecular simulations on parallel architectures [33] [32].
These implementations leverage modern force fields—mathematical functions describing potential energy surfaces—to compute forces during simulations. Common force fields like CHARMM, AMBER, and GROMOS include terms for bonded interactions (bond stretching, angle bending, torsional rotations) and non-bonded interactions (van der Waals and electrostatic forces) [34]. The Verlet algorithm integrates these forces to propagate the system through time, typically using time steps of 1-2 femtoseconds for accurate sampling of bond vibrations and other fast motions [34].
Table 1: Key MD Software Packages Utilizing Verlet Integration
| Software | Specialization | Verlet Implementation | License | Notable Features |
|---|---|---|---|---|
| GROMACS | High-speed biomolecular simulations | Velocity Verlet, Leap-frog | Free open source (GPL) | Exceptional performance, extensive analysis tools [31] [32] |
| NAMD | Scalable biomolecular simulations | Velocity Verlet | Free academic use | Excellent parallel scaling, CUDA acceleration [33] [32] |
| AMBER | Biomolecular systems | Velocity Verlet | Proprietary, free open source | Comprehensive analysis tools [33] [32] |
| LAMMPS | Materials modeling | Velocity Verlet | Free open source (GPLv2) | Versatile for various system types [35] [32] |
| OpenMM | High performance, flexibility | Velocity Verlet | Free open source (MIT) | Highly flexible, Python scriptable [32] |
Parallelization and Performance Optimization
Modern biomolecular systems of interest often contain hundreds of thousands to millions of atoms, requiring efficient parallelization strategies. The Verlet algorithm's structure naturally accommodates spatial decomposition approaches, where the simulation domain is divided into regions assigned to different processors [36]. Recent research has demonstrated optimized Verlet implementations using OpenMP for shared-memory systems and hybrid approaches combining MPI with OpenMP or CUDA for GPU acceleration [36].
These parallelization strategies are particularly effective for biomolecular systems because they minimize communication overhead while maintaining load balance. Studies show that optimized Verlet-based parallelization can achieve significant computing time reductions for force and energy evaluations compared to serial implementations, enabling longer timescales and larger systems in protein-ligand and solvation studies [36].
Application to Protein Systems
Simulation Methodologies for Protein Dynamics
Proteins exhibit dynamic processes across multiple timescales, from femtosecond bond vibrations to millisecond conformational changes and folding events [37]. Verlet-based MD simulations can capture these motions through careful implementation of several key methodologies:
System Preparation Protocol:
- Initial Structure Acquisition: Obtain protein coordinates from experimental sources (PDB files from X-ray crystallography or NMR) or computational modeling [34].
- Solvation: Embed the protein in an explicit water box (e.g., TIP3P, SPC/E water models) with a minimum 1.0 nm distance between protein and box edges [34].
- Neutralization: Add counterions (Na+, Cl-) to neutralize system charge using Monte Carlo placement methods.
- Energy Minimization: Perform 5,000-10,000 steps of steepest descent or conjugate gradient minimization to remove atomic clashes and bad contacts [34].
- Equilibration:
- 100 ps NVT equilibration with position restraints on protein heavy atoms (force constant 1000 kJ/mol/nm²) at target temperature (commonly 300K) using velocity rescaling or Nosé-Hoover thermostats [34].
- 100 ps NPT equilibration with similar restraints using Parrinello-Rahman or Berendsen barostats to achieve target density (1 atm pressure) [34].
- Production MD: Run unrestrained simulation using Verlet integration with 2-fs time steps, LINCS constraints on bonds involving hydrogen, and particle mesh Ewald (PME) for long-range electrostatics [34].
Enhanced Sampling Techniques: For processes with high energy barriers (e.g., protein folding, conformational transitions), standard MD may be insufficient. Enhanced sampling methods combined with Verlet integration include:
- Umbrella Sampling: Apply biasing potentials along reaction coordinates to overcome energy barriers [34].
- Metadynamics: Add history-dependent bias potentials to explore free energy surfaces [34].
- Replica Exchange MD (REMD): Run parallel simulations at different temperatures, exchanging configurations periodically to enhance conformational sampling [34].
Case Study: Protein Folding and Dynamics Analysis
Verlet-based MD has provided crucial insights into protein folding mechanisms and native state dynamics. Simulations can track folding pathways, identify intermediate states, and characterize the free energy landscape of proteins [37]. Analysis of trajectories typically includes:
- Root Mean Square Deviation (RMSD): Measures structural stability and convergence.
- Radius of Gyration: Assesses compactness during folding.
- Secondary Structure Analysis (DSSP): Tracks formation of α-helices and β-sheets.
- Principal Component Analysis (PCA): Identifies essential collective motions.
- Free Energy Calculations: Determines stability of different conformational states.
These analyses help researchers understand the relationship between protein dynamics and function, with applications in enzyme mechanism studies, allosteric regulation, and protein design [37].
Application to Ligand Binding and Drug Discovery
Simulation Protocols for Protein-Ligand Systems
The Verlet algorithm enables detailed studies of ligand binding mechanisms, residence times, and binding free energies—critical parameters in drug discovery. Specialized protocols for protein-ligand systems include:
System Setup:
- Ligand Parameterization: Generate force field parameters for small molecules using programs like GAUSSIAN [33] for quantum mechanical calculations followed by RESP charge fitting, or utilize pre-parameterized libraries.
- Docking and Placement: Position ligands in binding sites using molecular docking software or based on experimental complex structures.
- Solvation and Neutralization: Similar to protein-only systems but with careful attention to binding site solvation.
Equilibration Strategy:
- Initial Minimization: 5,000 steps with heavy restraints on protein and ligand (1000 kJ/mol/nm²).
- Solvent Relaxation: 100 ps with restraints on protein and ligand heavy atoms.
- Side-chain Optimization: 200 ps with restraints on protein backbone and ligand.
- Full System Equilibration: 500 ps without restraints.
Binding Free Energy Calculations:
- Thermodynamic Integration (TI): Alchemically transform ligand to nothing or to another ligand.
- Free Energy Perturbation (FEP): Uses Zwanzig equation to compute free energy differences.
- MM/PBSA or MM/GBSA: Molecular Mechanics/Poisson-Boltzmann or Generalized Born Surface Area methods approximate binding free energies from single trajectories.
Analysis of Binding Kinetics and Mechanisms
Verlet-based MD simulations can elucidate atomic-level details of ligand binding pathways, residence mechanisms, and allosteric effects. Key analyses include:
- Contact Analysis: Identify specific protein-ligand interactions (hydrogen bonds, hydrophobic contacts, π-stacking).
- Binding Pathway Characterization: Use supervised MD or adaptive sampling to enhance rare event sampling.
- Residence Time Estimation: Perform multiple unbinding simulations to compute mean residence times.
- Allosteric Communication Analysis: Apply correlation analysis (DCCM, LINC) or community analysis (Graphs) to identify allosteric networks.
These simulations provide insights complementary to experimental techniques like X-ray crystallography and cryo-EM, offering dynamic information about binding processes that can guide lead optimization in drug discovery [37].
Application to Solvation and Hydration Effects
Modeling Explicit and Implicit Solvent Environments
Solvation critically influences biomolecular structure, dynamics, and function. Verlet-based MD simulations employ both explicit and implicit solvent models:
Explicit Solvent Methodologies:
- Water Models: TIP3P, TIP4P, SPC/E with modified Verlet cutoffs for improved performance.
- Ion Parameters: Customized ion parameters matching experimental solution activities.
- Long-Range Electrostatics: Particle Mesh Ewald (PME) with 1.0-1.2 nm real-space cutoff and 0.12-0.15 nm Fourier spacing.
- Protocol: Solvate biomolecules in water boxes with periodic boundary conditions; equilibrate water density before production runs.
Implicit Solvent Approaches:
- Generalized Born (GB) Models: Approximate solvent as dielectric continuum; faster but less accurate for specific solvent interactions.
- Poisson-Boltzmann Methods: Solve PB equation numerically for electrostatic contributions.
- Applications: Rapid sampling, constant pH MD, and enhanced sampling simulations.
Hydration Dynamics and Ion Binding Studies
Verlet integration enables detailed investigation of water structure and dynamics around biomolecules:
- Hydration Site Analysis: Identify specific water positions with long residence times.
- Water Networks: Characterize extended hydrogen-bonded water clusters in binding sites or protein interfaces.
- Ion Binding and Transport: Study ion coordination, selectivity, and permeation through channels.
- Dielectric Properties: Calculate position-dependent dielectric constants from water dipole fluctuations.
These studies reveal how water mediates biomolecular recognition, influences conformational changes, and affects catalytic activity—information crucial for understanding biological function and designing inhibitors that target hydration sites.
Advanced Integration: Enhanced Sampling and Free Energy Methods
Overcoming Timescale Limitations
While the Verlet algorithm provides numerical stability, many biomolecular processes occur on timescales beyond reach of standard MD. Enhanced sampling methods combined with Verlet integration address this limitation:
Umbrella Sampling Protocol:
- Reaction Coordinate Selection: Choose appropriate collective variables (CVs) such as distance, angle, or RMSD.
- Window Setup: Run independent simulations with harmonic biases along the CV (20-40 windows typically).
- Force Constant Optimization: Use sufficient force constants (typically 100-1000 kJ/mol/nm²) to ensure overlap between windows.
- Data Analysis: Combine results using Weighted Histogram Analysis Method (WHAM) or Multistate Bennet Acceptance Ratio (MBAR) to construct potential of mean force (PMF).
Metadynamics Implementation:
- Collective Variable Definition: Select 1-3 relevant CVs describing the process.
- Gaussian Deposition: Add Gaussians of height 0.5-2.0 kJ/mol and width 5-15% of CV range every 500-1000 steps.
- Convergence Monitoring: Track reconstructed free energy until fluctuations around mean are small.
- Bias Removal: Subtract deposited bias to recover unbiased free energy surface.
Accelerated Molecular Dynamics and Replica Exchange
Accelerated MD (aMD):
- Applies boost potential to smooth energy landscape when system potential drops below threshold.
- Allows more rapid transitions between conformational states.
- Requires careful reweighting for quantitative free energy calculations.
Replica Exchange MD (REMD):
- Simultaneously runs multiple replicas at different temperatures or Hamiltonians.
- Exchanges configurations between replicas periodically based on Metropolis criterion.
- Temperature REMD enhances sampling of folding/unfolding transitions.
- Hamiltonian REMD with solute tempering reduces computational cost.
These advanced methods, built upon Verlet integration foundations, significantly expand the scope of addressable biological questions in drug discovery and structural biology.
Quantitative Comparison of Integration Algorithms
Table 2: Performance Characteristics of Verlet Algorithm Variants in Biomolecular Simulations
| Algorithm | Global Error | Stability | Memory Requirements | Computational Cost | Best Applications |
|---|---|---|---|---|---|
| Verlet | ( \mathcal{O}(\Delta t ^{3}) ) (position) | Excellent | Low (stores r(t), r(t-Δt)) | Low | Standard biomolecular MD [12] |
| Velocity Verlet | ( \mathcal{O}(\Delta t ^{3}) ) (position) ( \mathcal{O}(\Delta t ^{2}) ) (velocity) | Excellent | Moderate (stores r(t), v(t), a(t)) | Moderate | Most applications, requires velocities [4] [30] |
| Leap-Frog | ( \mathcal{O}(\Delta t ^{3}) ) (position) ( \mathcal{O}(\Delta t ^{2}) ) (velocity) | Excellent | Low (stores r(t), v(t-Δt/2)) | Low | Large systems, constrained dynamics [4] |
| Beeman | ( \mathcal{O}(\Delta t ^{3}) ) | Good | High (stores r(t), v(t), a(t), a(t-Δt)) | Moderate | Better energy conservation [12] |
Table 3: Recommended Time Steps for Different Biomolecular Systems with Verlet Integration
| System Type | Recommended Δt (fs) | Constraints | Temperature Control | Notes |
|---|---|---|---|---|
| All-atom, explicit solvent | 2 | All bonds with LINCS/SHAKE | Nosé-Hoover or velocity rescaling | Standard for protein folding [34] |
| All-atom, implicit solvent | 2-4 | All bonds with LINCS/SHAKE | Langevin dynamics | Faster but less accurate solvation |
| Coarse-grained | 20-40 | None or distance restraints | Langevin dynamics | Extended spatial and temporal scales |
| Membrane systems | 2 | All bonds with LINCS/SHAKE | Nosé-Hoover semiisotropic pressure coupling | Special barostat for membrane pressure |
| Protein-ligand | 1-2 | All bonds with LINCS/SHAKE | Nosé-Hoover | Smaller Δt for flexible ligands |
The Scientist's Toolkit: Essential Research Reagents and Software
Table 4: Key Research Reagent Solutions for Biomolecular Simulations
| Item | Function/Description | Examples/Specifications |
|---|---|---|
| MD Simulation Software | Engine for running molecular dynamics simulations | GROMACS [31], NAMD [33], AMBER [33], LAMMPS [35], OpenMM [32] |
| Force Fields | Mathematical functions defining potential energy and forces | CHARMM [34] [32], AMBER [33] [32], GROMOS [32], OPLS-AA [32] |
| Visualization Tools | Analysis and visualization of trajectories | VMD [32], PyMOL, Chimera [32], OVITO [35] |
| Quantum Chemistry Software | Parameterization of novel molecules, QM/MM simulations | Gaussian [33], ORCA [33], Q-Chem [33] |
| Enhanced Sampling Packages | Accelerate rare events, calculate free energies | PLUMED, Colvars |
| Water Models | Represent solvent water molecules | TIP3P [34], TIP4P, SPC/E |
| Analysis Tools | Extract properties from trajectory data | MDAnalysis, GROMACS analysis tools [31] |
| 2-(2-Oxocyclohexyl)acetyl chloride | 2-(2-Oxocyclohexyl)acetyl chloride|High-Purity Building Block | 2-(2-Oxocyclohexyl)acetyl chloride is a versatile acyl chloride reagent for organic synthesis and pharmaceutical research. This product is For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
| 1,3,5-Tris(4-cyanophenyl)benzene | 1,3,5-Tris(4-cyanophenyl)benzene (TCPB)|CAS 382137-78-2 |
Workflow Visualization
Verlet-Based MD Workflow for Biomolecular Systems
Velocity Verlet Integration Sequence
Integration with Neighbor List Algorithms (Verlet Table, Linked-Cell) for Efficiency
In molecular dynamics (MD) simulations, integrating Newton's equations of motion to determine particle trajectories is a foundational task, most commonly accomplished using the Verlet algorithm and its variants [2] [4]. However, the computational cost of this integration is dominated by the calculation of short-range non-bonded forces, which naively scales as O(N²) for a system of N particles [38]. To make simulations of large systems feasible, this scaling must be reduced. This is achieved using neighbor list algorithms, principally the Verlet table (list) and the Linked-Cell (Cell-Linked List) methods [38] [39]. These algorithms efficiently identify particles within a specified cutoff distance, drastically reducing the number of pairwise interactions that need to be computed at each time step. This technical guide details the Verlet integration scheme, the core neighbor list algorithms, and their integration for achieving high computational efficiency, a critical consideration in fields ranging from material science to drug development.
The Verlet Integration Algorithm
The Verlet algorithm is a numerical method for integrating Newton's equations of motion, valued for its simplicity, stability, and time-reversibility [2].
Fundamental Formulation
The basic Störmer-Verlet algorithm is derived from Taylor expansions of the particle positions forward and backward in time [2] [40]: $$ \mathbf{r}(t+\Delta t) = \mathbf{r}(t) + \mathbf{v}(t)\Delta t + \frac{\mathbf{F}(t)}{2m}\Delta t^2 + \cdots $$ $$ \mathbf{r}(t-\Delta t) = \mathbf{r}(t) - \mathbf{v}(t)\Delta t + \frac{\mathbf{F}(t)}{2m}\Delta t^2 - \cdots $$
Adding these two equations eliminates the velocity term and yields the core Verlet position update formula: $$ \mathbf{r}(t+\Delta t) = 2\mathbf{r}(t) - \mathbf{r}(t-\Delta t) + \frac{\mathbf{F}(t)}{m}\Delta t^2 + \mathcal{O}(\Delta t^4) $$ This formulation provides excellent numerical stability and is symplectic, meaning it conserves the phase-space volume, which is crucial for long-term energy conservation in simulations [2].
Common Variants
While powerful, the basic Verlet algorithm does not explicitly handle velocities, which are needed for temperature computation and control. This led to the development of popular variants.
Table 1: Key Variants of the Verlet Algorithm
| Algorithm Variant | Key Features | Advantages | Disadvantages |
|---|---|---|---|
| Basic Verlet [2] [40] | - Positions updated using current and previous positions.- Velocities calculated post-hoc. | - Computationally simple.- Time-reversible and symplectic. | - Velocities not directly available.- Not self-starting. |
| Velocity Verlet [4] [40] | - Synchronous update of positions and velocities.- Requires single force calculation per step. | - Positions, velocities, and accelerations are all directly and accurately available at time t.- Self-starting. | - Slightly more complex than basic Verlet. |
| Leap-Frog [4] [40] | - Velocities and positions updated at staggered times.- Velocities at half-steps: v(t+½Δt) = v(t-½Δt) + a(t)Δt. |
- Computationally efficient.- Good numerical stability. | - Velocities and positions are not synchronous, complicating energy calculation. |
The Velocity Verlet algorithm is often preferred and follows these steps [4]:
- Update velocities by a half-step using the current force:
v_i(t + ½Δt) = v_i(t) + (F_i(t) / (2m_i)) * Δt - Update positions to the next step:
r_i(t + Δt) = r_i(t) + v_i(t + ½Δt) * Δt - Compute new forces
F_i(t + Δt)based on the new positions. - Complete the velocity update:
v_i(t + Δt) = v_i(t + ½Δt) + (F_i(t + Δt) / (2m_i)) * Δt
Neighbor List Algorithms
Force calculation is the most time-consuming part of an MD simulation, often accounting for over 98% of the total computational effort [41]. Neighbor list algorithms optimize this by creating a list of particles within a given cutoff sphere, avoiding the need to check all N² pairs.
Verlet Table (List) Algorithm
The Verlet table method constructs a list for each particle that includes all neighboring particles within a "search radius," R_s, which is slightly larger than the potential cutoff distance, R_cut [38]. This buffer ensures the list remains valid for several time steps without needing a rebuild.
Experimental Protocol for Verlet List Implementation:
- Parameter Definition: Set the potential cutoff distance R_cut and the skin depth (R_s - R_cut). A larger skin depth reduces the frequency of list updates but increases the list size and the number of pairwise checks.
- List Construction: For every particle i, loop over all other particles j and calculate the distance r_ij. If r_ij ≤ R_s, add particle j to the neighbor list of i.
- Force Calculation: During subsequent MD steps, only particle pairs present in the list are evaluated for non-bonded interactions.
- List Update: Rebuild the neighbor lists periodically after a predetermined number of steps (the "update interval") or when a particle has moved more than
(R_s - R_cut)/2since the last build [38].
Linked-Cell List Algorithm
The linked-cell list method partitions the simulation box into a grid of 3D cells with an edge length L_c equal to or slightly larger than R_cut [38] [39]. Each particle is assigned to a cell based on its coordinates. The neighbors of a particle in a given cell are then found by searching only within that cell and its 26 adjacent cells (in 3D), reducing the search complexity to O(N).
Experimental Protocol for Linked-Cell List Implementation:
- Domain Decomposition: Divide the simulation box into cells of size L_c ≥ R_cut.
- Cell Population and Linking: Assign each particle to a cell. Create a linked list (or a series of lists) for each cell to track its resident particles.
- Neighbor Search: For a particle in cell (i, j, k), the search for interaction partners is confined to particles in cells (i±1, j±1, k±1).
- List Update: The cell list must be updated every time step as particles move, but this process is highly efficient [39].
Diagram 1: Neighbor list integration in the MD loop. The list is updated periodically, not at every step.
Comparative Analysis and Integration
The choice between the Verlet table and linked-cell methods significantly impacts simulation performance, especially as system size grows.
Performance and Efficiency
Table 2: Quantitative Comparison of Verlet Table vs. Linked-Cell List
| Characteristic | Verlet Table (VT) | Linked-Cell List (CLL) |
|---|---|---|
| Computational Scaling | O(N²) for list build, but build is infrequent. | O(N) for both list build and force calculation [41]. |
| Memory Overhead | High, as a list of neighbors must be stored for each particle. | Low, primarily requiring storage for the cell grid and particle-cell mappings [39]. |
| Efficiency in Dense Systems | Good, but the list contains all particles within R_s, including some beyond R_cut. | Excellent, as the search is spatially restricted to local cells, minimizing unnecessary checks [38]. |
| Update Frequency | Infrequent (every 10-20 steps) due to the skin buffer. | Typically updated every single step, but the update is very fast [38]. |
| Suitability for Parallelism | Moderate. | High. The CLL is more suitable for modern multi-core and SIMD processors, with one study showing a 40% performance gain over VT in parallel implementations [39]. |
The linked-cell method is generally superior for large-scale simulations (N > 10,000) and is the algorithm of choice in modern parallel MD codes [39] [41]. Its regular data structure is more amenable to vectorization and parallel decomposition. However, for smaller systems or those with very disparate particle sizes, the Verlet list can remain competitive.
Advanced Application: Three-Body Interactions
The efficiency of these algorithms extends beyond simple pair potentials. For computing complex interactions like three-body forces (e.g., the Axilrod-Teller-Muto potential), the linked-cell structure is foundational [42]. The traversal for triplets (i, j, k) involves checking combinations of particles from three nearby cells. Furthermore, different truncation conditions can be applied once a triplet is identified, impacting both workload and accuracy [42]:
- Pairwise Cutoff: The interaction is computed only if all three pairwise distances
(r_ij, r_ik, r_jk)are less than the cutoff radius r_c. This is more restrictive. - Product Cutoff: The interaction is computed if the product of the three distances is less than the cube of the cutoff radius
(r_ij * r_ik * r_jk < r_c³). This includes a larger set of triplets and thus increases computational load [42].
Diagram 2: Linked-cell method for three-body interactions. Particle i's triplets are formed with particles j and k found in its own and adjacent cells.
The Scientist's Toolkit: Essential Research Reagents and Computational Components
Table 3: Key Components for MD Simulations with Neighbor Lists
| Item | Function/Description | Example/Note |
|---|---|---|
| Interaction Potential | Defines the forces between particles. | Lennard-Jones for simple fluids [5]; Axilrod-Teller-Muto for three-body interactions [42]. |
| Integrator | Numerically solves equations of motion. | Velocity Verlet; Leap-Frog [4] [40]. |
| Neighbor List Algorithm | Manages efficient force calculations. | Verlet Table; Linked-Cell List [38]. |
| Cutoff Radius (R_cut) | Distance beyond which interactions are truncated. | Must be chosen carefully to balance accuracy and speed. |
| Time Step (Δt) | The discrete time interval for integration. | Typically 1-2 femtoseconds (10â»Â¹âµ s) for atomic systems [40]. |
| Domain Decomposition | For parallel computing, divides the simulation box among processors. | Each processor handles a geometric domain of cells, minimizing communication [41]. |
| 2,2,5-Trimethyl-1,3-dioxolan-4-one | 2,2,5-Trimethyl-1,3-dioxolan-4-one|CAS 4158-85-4 | 2,2,5-Trimethyl-1,3-dioxolan-4-one (Me3DOX) is a key monomer for isotactic PLA synthesis. This product is For Research Use Only. Not for human or veterinary use. |
The integration of the robust Verlet integration scheme with efficient neighbor list algorithms is a cornerstone of modern molecular dynamics. While the Verlet table provides a straightforward approach, the linked-cell list method offers superior scalability and performance for large-scale simulations, a characteristic critical for harnessing the power of contemporary parallel computing architectures. The choice of algorithm directly influences the feasibility and efficiency of MD applications, from modeling simple liquids to capturing complex many-body interactions in drug design and materials science. As system size and physical complexity grow, the optimized integration of these core components remains an active and vital area of research and development.
Optimizing Verlet Simulations: Time Steps, Constraints, and Performance Tuning
The Verlet algorithm is a cornerstone of molecular dynamics (MD) simulations, a numerical method for integrating Newton's equations of motion to compute particle trajectories over time [2]. Its significance stems from its favorable properties: it is time-reversible, energy-conserving, and provides good numerical stability with relatively low computational cost compared to simpler methods like Euler integration [2] [3]. The choice of the integration time step (δt) is a critical parameter in any MD simulation based on the Verlet algorithm. An overly small time step wastes computational resources, while a too-large one leads to instability and inaccurate results [14] [3]. This guide provides an in-depth analysis of the stability limits of the Verlet algorithm and offers practical guidelines for selecting a time step in the 1-5 femtosecond (fs) range, a common interval for biomolecular simulations.
The Verlet Algorithm and its Variants
Core Algorithm
For a second-order differential equation of the type ( \ddot{\mathbf{x}}(t) = \mathbf{A}(\mathbf{x}(t)) ), the basic Størmer-Verlet algorithm updates particle positions using the following iteration [2]: [ \mathbf{x}{n+1} = 2\mathbf{x}{n} - \mathbf{x}{n-1} + \mathbf{a}{n} \Delta t^2 ] where ( \mathbf{a}{n} = \mathbf{A}(\mathbf{x}{n}) ) is the acceleration at time step ( n ). A notable feature of this formulation is that it does not explicitly use velocities to compute the new positions.
The Velocity Verlet Variant
The Velocity Verlet algorithm is a widely used variant that explicitly incorporates velocities, making it more convenient for MD simulations. It is mathematically equivalent to the original Verlet algorithm but calculates positions, velocities, and accelerations at the same time point [3]. The procedure for one time step is [3]:
- Use ( \mathbf{r}, \mathbf{v},\mathbf{a} ) at time ( t ) to compute the new position: ( \qquad\mathbf{r}(t+\delta{t})=\mathbf{r}(t)+\mathbf{v}(t)\delta{t}+\frac{1}{2}\mathbf{a}(t)\delta{t}^2 )
- Derive the acceleration ( \mathbf{a}(t+\delta{t}) ) from the interaction potential using the new positions ( \mathbf{r}(t+\delta{t}) ).
- Use both ( \mathbf{a}(t) ) and ( \mathbf{a}(t+\delta{t}) ) to compute the new velocity: ( \quad\mathbf{v}(t+\delta{t})=\mathbf{v}(t)+\frac{1}{2}(\mathbf{a}(t)+\mathbf{a}(t+\delta{t}))\delta{t} )
Due to its simplicity and stability, the Velocity Verlet algorithm has become the most widely used integrator in MD simulations [3].
Theoretical Stability Limits
The stability of the Verlet integrator is not unconditional; it is governed by the highest frequency motions present in the system. The following criteria define its theoretical stability limits.
General Stability Criterion for Harmonic Oscillators
For simple harmonic oscillators with a force constant ( k ), a stability analysis for the leapfrog method (closely related to Velocity Verlet) shows that the condition for stability is [43]: [ \sqrt{k} \epsilon < 2 ] where ( \epsilon ) is the time step. If this condition is violated, the integration becomes unstable. For a mass-spring system, the stiffest spring therefore determines the maximum stable time step [43].
Relationship to Vibration Frequency
A more general formulation states that Verlet-family integrators are stable for time steps that satisfy [3]: [ \delta{t}\leq\frac{1}{\pi f} ] where ( f ) is the highest oscillation frequency present in the system. This relationship directly links the fastest physical process in the simulated system to the maximum permissible numerical time step.
Table 1: Stability Limits for Common Molecular Motions
| Molecular Motion | Approximate Oscillation Period | Theoretical Maximum δt (fs) | Key Involved Atoms |
|---|---|---|---|
| Bond vibration with hydrogen [3] | 10 fs | 3.2 fs | C-H, O-H, N-H |
| Bond vibration of heavy atoms [3] | 20 fs | 6.4 fs | C-C, C-N |
| Angle vibration involving hydrogen [3] | 20 fs | 6.4 fs | H-C-H, H-O-C |
Practical Time Step Guidelines (1 fs to 5 fs)
In practice, time steps are chosen to be significantly smaller than the theoretical maximum to ensure accuracy and numerical stability. The following guidelines are standard for biomolecular simulations.
Standard Time Steps for Different System Types
The fastest motions in a molecular system are typically the stretching of bonds involving hydrogen atoms, which have a period of about 10 fs [3]. This limits the stable time step to less than approximately 3.2 fs. Consequently, a time step of 1 fs or 2 fs is often recommended to reliably describe this motion without constraints [3].
Table 2: Practical Time Step Selection Guide
| Time Step (fs) | Typical Use Case | Required System Constraints | Stability and Efficiency |
|---|---|---|---|
| 1 | Default, high-accuracy simulations [3] | None | High stability, lower efficiency |
| 2 | Common practice with constraints [3] | Bonds with H-atoms | Good stability, balanced efficiency |
| 4 | Hydrogen Mass Repartitioning (HMR) [3] | HMR technique allows larger δt | Increased efficiency, good stability |
| 4-5 | Advanced use with multiple constraints | All bonds + angles involving H [3] | Highest efficiency, requires careful setup |
Techniques for Enabling Larger Time Steps
To accelerate simulations beyond the 2 fs limit, several techniques are employed:
- Constraining Bonds with Hydrogen Atoms: Applying algorithms like SHAKE, LINCS, or SETTLE to fix the length of bonds involving hydrogen atoms allows the time step to be safely increased to 2 fs [3].
- Constraining All Bonds and Angles: Further increasing the time step to 4 fs requires constraining not only all bonds but also angles that involve hydrogen atoms [3].
- Hydrogen Mass Repartitioning (HMR): This technique allows for a time step of 4 fs by artificially increasing the mass of hydrogen atoms and decreasing the mass of bonded heavy atoms, thereby slowing down the highest frequency motions without significantly altering the system's dynamics [3].
Protocols for Determining System-Specific Time Steps
Workflow for Stability Assessment
The following diagram outlines a general workflow for determining the optimal time step for a specific molecular system.
Diagram Title: Workflow for Determining a Stable Time Step
Experimental Validation Protocol
Before commencing a production simulation, it is crucial to validate the stability of the chosen time step through a short test.
- System Preparation: Solvate your molecule of interest (e.g., a protein-ligand complex) in a water box and add necessary ions.
- Energy Minimization: Perform energy minimization to remove any bad contacts and prepare the system for dynamics.
- Equilibration: Carry out a short equilibration in the NVT ensemble, followed by a longer equilibration in the NPT ensemble to stabilize the system's temperature and density.
- NVE Stability Test:
- Switch to the NVE (microcanonical) ensemble using the Velocity Verlet integrator [14].
- Run a short simulation (e.g., 10-50 ps) using the candidate time step (e.g., 2 fs, 4 fs).
- Monitor the total energy of the system throughout the simulation. A stable time step will show no significant drift in the total energy over time [14] [3]. A clear upward drift indicates an unstable time step that must be reduced.
The Scientist's Toolkit: Key Reagents and Computational Methods
Table 3: Essential Tools for MD Simulations and Time Step Optimization
| Tool / Method | Function in Time Step Determination | Example Software |
|---|---|---|
| Velocity Verlet Integrator | Core algorithm for numerically integrating equations of motion; its stability dictates time step choice [3]. | LAMMPS, GROMACS (md-vv), NAMD [3] |
| Constraint Algorithms (SHAKE, LINCS, SETTLE) | Fix the length of specified bonds (and angles), removing high-frequency motions and enabling larger time steps [3]. | GROMACS, NAMD, AMBER |
| Hydrogen Mass Repartitioning (HMR) | Technique that modifies atomic masses to slow down high-frequency vibrations, allowing for a ~4 fs time step [3]. | GROMACS, AMBER, CHARMM |
| Multiple Time Stepping (MTS) | Accelerates simulation by computing slow forces (e.g., long-range electrostatics) less frequently than fast forces (bonded) [3]. | NAMD, GROMACS, LAMMPS |
| Molecular Dynamics Engines | Software packages that implement integration algorithms, constraint methods, and analysis tools. | GROMACS, NAMD, LAMMPS, AMBER [3] |
In molecular dynamics (MD) simulations, the presence of atoms with high vibrational frequencies, particularly hydrogen, presents a significant computational challenge. The fast motion of these atoms necessitates exceptionally small integration time steps to maintain numerical stability, dramatically increasing the computational cost of simulations. Within the context of Verlet integration algorithms—the fundamental framework for MD—constraint algorithms such as SHAKE and LINCS provide an elegant solution by effectively removing these highest frequency degrees of freedom. This technical guide examines the theoretical foundation, implementation methodologies, and practical considerations for constraining bonds involving hydrogen atoms, enabling researchers to perform longer, more stable simulations of biomolecular systems relevant to drug development.
The Verlet algorithm and its variants (Velocity-Verlet, Leap-Frog) form the mathematical backbone of time propagation in MD simulations [4]. These symplectic integrators conserve phase-space volume and ensure long-term stability, but their effectiveness depends critically on the chosen time step. Without constraints, the rapid bond vibrations of hydrogen atoms would limit time steps to approximately 0.5 femtoseconds, making simulations of biologically relevant timescales computationally prohibitive. By constraining these bonds to fixed lengths, algorithms like SHAKE allow time steps of 2 femtoseconds or more, accelerating research in pharmaceutical development and molecular design.
Mathematical Foundation: Constraint Algorithms in the Verlet Framework
The Verlet Integration Scheme
The standard Verlet algorithm calculates particle trajectories using the following position update formula [4]:
$$\mathbf{r}{i}(t+\Delta t) = 2\mathbf{r}{i}(t)-\mathbf{r}{i}(t-\Delta t)+\frac{\mathbf{F}{i}}{2m}(t)\Delta t^{2}+\mathcal{O}(\Delta t ^{3})$$
This formulation, while time-reversible and symplectic, does not explicitly include velocities. The Velocity-Verlet variant addresses this limitation and is decomposed into the following steps [4]:
- $\mathbf{v}{i}(t + \frac{1}{2}\Delta t)=\mathbf{v}{i}(t)+\frac{\mathbf{F}{i}(t)}{2m{i}}\Delta t$
- $\mathbf{r}{i}(t + \Delta t) = \mathbf{r}{i}(t) + \mathbf{v}_{i}(t + \frac{1}{2}\Delta t)\Delta t$
- Compute forces $\mathbf{F}_{i}(t)$ from the potential energy function
- $\mathbf{v}{i}(t + \Delta t)=\mathbf{v}{i}(t+\frac{1}{2}\Delta t)+\frac{\mathbf{F}{i}(t+\Delta t)}{2m{i}}\Delta t$
The Leap-Frog algorithm, another Verlet variant, implements time propagation as follows [4]:
- Compute forces $\mathbf{F}_{i}(t)$
- $\mathbf{v}{i}(t + \frac{1}{2}\Delta t)=\mathbf{v}{i}(t- \frac{1}{2}\Delta t)+\frac{\mathbf{F}{i}(t)}{m{i}}\Delta t$
- $\mathbf{r}{i}(t + \Delta t) = \mathbf{r}{i}(t) + \mathbf{v}_{i}(t + \frac{1}{2}\Delta t)\Delta t$
In this formulation, velocity and position vectors are asynchronous in time, but the algorithm remains computationally efficient and numerically stable.
The SHAKE Algorithm
The SHAKE algorithm, developed by Ryckaert et al., is a two-stage iterative procedure based on the Verlet integration scheme [44]. It solves for constraint forces that maintain fixed bond lengths despite the unconstrained forces acting on atoms. For a system with holonomic constraints, the equations of motion must fulfill $K$ constraints expressed as [45]:
$$\sigmak(\mathbf{r}1 \ldots \mathbf{r}_N) = 0; \;\; k=1 \ldots K$$
The constraint force $\mathbf{G}_i$ on atom $i$ is derived from Lagrange multipliers [45]:
$$\mathbf{G}i = -\sum{k=1}^K \lambdak \frac{\partial \sigmak}{\partial \mathbf{r}_i}$$
The SHAKE algorithm operates through the following iterative process [44]:
- Unconstrained Move: All atoms are moved using the Verlet algorithm, assuming no rigid bond forces
- Constraint Calculation: The deviation in each bond length is used to calculate corresponding constraint forces
- Iterative Correction: Steps 2 is repeated for all bonds until the largest deviation satisfies the convergence tolerance
For velocity Verlet integrators, an additional step called RATTLE constrains velocities by removing components parallel to bond vectors [45].
The LINCS Algorithm
The LINCS (Linear Constraint Solver) algorithm provides a non-iterative alternative to SHAKE, particularly efficient for bond constraints and isolated angle constraints [45]. LINCS operates in two distinct steps:
- First Projection: Sets the projections of new bonds on old bonds to zero
- Rotation Correction: Applies a correction for bond lengthening due to rotation
The mathematical formulation for the first step is [45]:
$$\mathbf{r}{n+1}=(\mathbf{I}-\mathbf{T}n \mathbf{B}n) \mathbf{r}{n+1}^{unc} + {\mathbf{T}}n \mathbf{d} = \mathbf{r}{n+1}^{unc} - {{\mathbf{M}}^{-1}}\mathbf{B}n ({\mathbf{B}}n {{\mathbf{M}}^{-1}}{\mathbf{B}}n^T)^{-1} ({\mathbf{B}}n \mathbf{r}_{n+1}^{unc} - \mathbf{d})$$
where ${\mathbf{T}}= {{\mathbf{M}}^{-1}}{\mathbf{B}}^T ({\mathbf{B}}{{\mathbf{M}}^{-1}}{\mathbf{B}}^T)^{-1}$ and $\mathbf{B}$ is the gradient matrix of constraint equations.
Table 1: Key Characteristics of Constraint Algorithms
| Algorithm | Mathematical Approach | Iteration Required | Computational Efficiency | Supported Constraints |
|---|---|---|---|---|
| SHAKE | Iterative Lagrange multipliers | Yes | Moderate | Bonds, angles |
| LINCS | Matrix inversion + projection | No | High | Bonds, isolated angles |
| SETTLE | Analytical solution | No | Very High | Rigid water molecules |
Methodologies: Implementing Constraints in Molecular Dynamics
System Preparation and Parameterization
Successful implementation of bond constraints begins with careful system preparation. For biomolecular simulations, researchers must:
- Identify Constrainable Bonds: Select all bonds involving hydrogen atoms, which typically exhibit the highest frequency vibrations
- Choose Appropriate Constraints: Decide whether to constrain only bond lengths or include angles involving hydrogen atoms
- Select Algorithm Parameters: Determine tolerance levels (typically $10^{-4}$ to $10^{-8}$ for SHAKE) and iteration limits [44]
For specific molecular systems like water, specialized algorithms such as SETTLE provide optimal performance. SETTLE uses an analytical solution for rigid water molecules, completely avoiding iterative processes and reducing rounding errors, which is particularly beneficial for large systems [45].
Integration with Force Calculations
Constraint algorithms must be properly integrated with force calculation procedures:
- Force Computation: Calculate unconstrained forces using empirical potential energy functions (e.g., Class I, II, or III force fields) [46]
- Position Update: Apply the Verlet algorithm to update atomic positions without constraints
- Constraint Application: Implement SHAKE or LINCS to correct positions preserving bond lengths
- Velocity Correction (for velocity Verlet): Apply RATTLE to ensure velocities remain perpendicular to constraint bonds
Table 2: Force Field Parameters for Hydrogen-Containing Bonds
| Bond Type | Equilibrium Length (Å) | Force Constant (kJ/mol/Ų) | Recommended Constraint Tolerance |
|---|---|---|---|
| O-H (water) | 0.9572 | 5020.8 | $10^{-6}$ |
| N-H (amide) | 1.0100 | 4340.0 | $10^{-5}$ |
| C-H (aliphatic) | 1.0900 | 2845.0 | $10^{-5}$ |
| S-H (thiol) | 1.3360 | 2070.0 | $10^{-5}$ |
Thermodynamic Consistency
Proper implementation of constraints must account for thermodynamic contributions:
Virial Calculation: Constraint forces contribute to the system virial, calculated for each constrained bond as [44]:
$$W = \mathbf{G}{ij} \cdot \mathbf{r}{ij}$$
Stress Tensor: The constraint contribution to the atomic stress tensor is [44]:
$$\sigma^{\alpha\beta} = G{ij}^{\alpha}r{ij}^{\beta}$$
where $\alpha$ and $\beta$ indicate components
Energy Conservation: While constraint forces perform no net work, they indirectly affect the system's kinetic and potential energy distribution
Comparative Analysis: SHAKE vs. LINCS Performance
Numerical Stability and Convergence
SHAKE's iterative approach provides robust performance across diverse molecular systems but may encounter convergence issues with complex constraint networks or large time steps. The algorithm continues until all constraints satisfy the specified relative tolerance, with error handling for excessive iterations [45].
LINCS employs a power series expansion to invert the constraint coupling matrix:
$$(\mathbf{I}-\mathbf{A}n)^{-1}= \mathbf{I} + \mathbf{A}n + \mathbf{A}n^2 + \mathbf{A}n^3 + \ldots$$
This expansion is valid when the absolute values of all eigenvalues of $\mathbf{A}_n$ are smaller than one, which generally holds for molecules with only bond constraints but may fail for systems with coupled angle constraints [45].
Computational Efficiency
For large biomolecular systems, LINCS typically outperforms SHAKE due to its non-iterative nature and better parallelization characteristics. The P-LINCS (Parallel LINCS) algorithm further enhances performance for distributed computing environments [45]. SHAKE remains valuable for systems with complex constraint networks where LINCS might encounter numerical instability.
Table 3: Performance Comparison of Constraint Algorithms
| Performance Metric | SHAKE | LINCS | SETTLE |
|---|---|---|---|
| Scalability | Moderate | High | Limited to water |
| Memory Requirements | Low | Moderate | Low |
| Parallel Efficiency | Moderate | High | High |
| Tolerance Control | Direct | Indirect via expansion order | Fixed |
Research Reagent Solutions: Essential Tools for Constrained MD
Table 4: Essential Software Tools for Constrained Molecular Dynamics
| Tool Name | Function | Implementation |
|---|---|---|
| GROMACS | MD simulation package | LINCS (default) and SHAKE |
| DL_POLY | MD simulation package | SHAKE and RD_SHAKE for parallel computing [44] |
| VASP | Ab initio MD simulation | Velocity-Verlet with constraints [4] |
| AMBER | Biomolecular simulation | SHAKE with support for hydrogen constraints |
| CHARMM | Biomolecular simulation | SHAKE with extended constraint options |
Advanced Applications in Drug Discovery
Constrained MD simulations have become indispensable in modern drug discovery, enabling:
- Enhanced Sampling: By removing hydrogen-related vibrations, constraints enable larger time steps and longer simulation timescales for studying drug-target interactions
- Membrane Protein Simulations: Constrained bonds maintain structural integrity of membrane-embedded drug targets during assembly and screening
- Free Energy Calculations: Alchemical transformation pathways benefit from constraints that maintain molecular geometry while perturbing non-essential interactions
Recent advances in machine learning potentials combine constraint algorithms with neural network force fields, improving simulation accuracy while maintaining numerical stability [47]. For example, deep learning architectures like MolFG integrate transformer embedding with graph neural networks to enhance molecular property prediction while leveraging constrained dynamics for structure generation [47].
Workflow Visualization: Constrained MD Implementation
Constrained MD Workflow: Integration of bond constraints within a Verlet-based molecular dynamics simulation.
Constraint algorithms for bonds involving hydrogen atoms represent a crucial enabling technology for molecular dynamics simulations in pharmaceutical research and drug development. By effectively removing the highest frequency vibrations, these algorithms allow for significantly larger time steps while maintaining numerical stability within the Verlet integration framework. The continued development of specialized constraint methods like LINCS and SETTLE, coupled with advances in machine learning potentials, promises further enhancements in simulation efficiency and accuracy. As MD simulations continue to address increasingly complex biological questions—from viral mechanisms to drug-receptor interactions—robust constraint handling will remain fundamental to extracting meaningful insights from computational experiments.
In molecular dynamics (MD) simulations, the maximum length of the time step for integrating Newton's equations of motion is restricted by the frequency of the fastest motions in the system, typically bond vibrations involving hydrogen atoms [48]. Constraint algorithms are mathematical methods that impose holonomic (geometric) constraints on selected degrees of freedom, most commonly bond lengths and angles, effectively removing these rapid vibrational modes from the dynamics [49]. This approach is fundamentally linked to the Verlet family of integration algorithms, which form the foundation for most MD simulations [3]. Within this numerical integration framework, constraints allow for significantly longer time steps—from 1 fs to 2-4 fs or more—by eliminating the highest frequency bond vibrations that would otherwise limit numerical stability [3] [50]. This can reduce required computer time by a factor of three or more, making constraints a crucial performance-enabling technique for simulating biological macromolecules and other complex systems [48].
The core mathematical challenge addressed by constraint algorithms is solving a set of coupled equations of motion subject to time-independent algebraic constraint equations [49]. For a system of N particles with positions represented by vector r, the Newtonian motion must satisfy both the standard equations of motion and M constraint equations of the form gâ±¼(r) = 0 (e.g., |râ‚ - râ‚‚| - d = 0 for a bond length constraint) [49] [51]. This is achieved through the introduction of Lagrange multipliers, which represent constraint forces that work to maintain the specified molecular geometry throughout the simulation [49].
Mathematical Foundation of Constraint Algorithms
The Lagrange Multiplier Formalism
The foundation for constraint algorithms in MD simulations lies in the method of Lagrange multipliers, which provides a mathematical framework for incorporating constraints directly into the equations of motion [49]. For a system subject to K holonomic constraints:
σₖ(râ‚...r_N) = 0; k=1...K
The forces in the system are derived from:
- ∂/∂ráµ¢ [ V + ∑ₖ₌â‚á´· λₖσₖ ]
where λₖ are the Lagrange multipliers that must be solved to fulfill the constraint equations [51]. The second term in this expression determines the constraint forces Gᵢ:
Gáµ¢ = -∑ₖ₌â‚á´· λₖ ∂σₖ/∂ráµ¢
In the context of Verlet integration algorithms, the displacement due to these constraint forces is proportional to (Gᵢ/mᵢ)(Δt)² [51]. Solving for the Lagrange multipliers—and consequently the constraint forces—requires solving a set of coupled equations of the second degree, which is the core computational challenge addressed by the various constraint algorithms [49].
Integration with Verlet Algorithms
Constraint algorithms are typically applied after an unconstrained update in the numerical integration scheme [51]. In the Velocity Verlet algorithm, for instance, the standard sequence is:
- Update positions and calculate velocities at half time steps
- Compute forces based on new positions
- Complete velocity updates
Constraint algorithms correct the positions (and velocities, if necessary) after the unconstrained position update to satisfy all specified bond constraints before force calculation [3]. For velocity Verlet, an additional round of constraining must be done to constrain the velocities, removing any component of the velocity parallel to the bond vector—a step implemented in the RATTLE algorithm [51].
Table 1: Time Step Limitations in Molecular Dynamics Simulations
| Motion Type | Approximate Oscillation Period | Stable Time Step Limit | Primary Constraint Method |
|---|---|---|---|
| Bond vibrations with H | 10 fs | 1-2 fs | Bond length constraints |
| Bond angles with H | 20 fs | 2-4 fs | Bond angle constraints |
| Bonds (heavy atoms) | 20 fs | 2-4 fs | Bond length constraints |
| Bond angles (heavy atoms) | 40 fs | 4-8 fs | Bond angle constraints |
| Torsional vibrations | 30-80 fs | 4-8 fs | Dihedral constraints (rare) |
Core Constraint Algorithms
SHAKE Algorithm
The SHAKE algorithm, one of the oldest and most robust constraint methods, solves constraint equations iteratively using the Gauss-Seidel method [48] [50]. After an unconstrained position update, SHAKE iteratively adjusts atom positions to satisfy all bond length constraints within a specified relative tolerance [51]. The algorithm requires continued iterations until all constraints are satisfied within the given tolerance, with error handling for cases where constraints cannot be satisfied due to excessively large forces or displacements [51].
The mathematical operation of SHAKE can be represented as:
SHAKE(r' → r''; r)
where r' represents the unconstrained coordinates, r'' represents the constrained coordinates that fulfill all distance constraints, and r represents the reference coordinates [51]. For velocity Verlet integration, the RATTLE extension performs a similar corrective procedure for velocities [51].
Key advantages of SHAKE include its simplicity, robustness, and ability to handle both bond and angle constraints [50]. Its primary limitations are slower performance compared to more modern algorithms, difficulty with parallelization, and potential convergence issues when constraints are highly coupled or displacements are large [3] [51].
LINCS Algorithm
The Linear Constraint Solver (LINCS) is a non-iterative algorithm that resets bonds to their correct lengths after an unconstrained update in exactly two steps [51] [45]. Unlike SHAKE, LINCS uses matrix operations without requiring full matrix-matrix multiplications [51]. The algorithm works by:
- Setting projections of new bonds on old bonds to zero
- Correcting for bond lengthening due to rotation
The mathematical formulation of LINCS defines how constrained coordinates rₙ₊₠are related to unconstrained coordinates rₙ₊â‚ᵘâ¿á¶œ through the expression:
rₙ₊₠= rₙ₊â‚ᵘâ¿á¶œ - Mâ»Â¹Bâ‚™(Bâ‚™Mâ»Â¹Bₙᵀ)â»Â¹(Bâ‚™rₙ₊â‚ᵘâ¿á¶œ - d)
where B is the gradient matrix of constraint equations, M is the diagonal mass matrix, and d is the constraint distance vector [51] [45].
To efficiently invert the constraint coupling matrix Bâ‚™Mâ»Â¹Bₙᵀ, LINCS employs a power series expansion:
(I - Aâ‚™)â»Â¹ = I + Aâ‚™ + Aₙ² + Aₙ³ + ...
This inversion is valid only when the absolute values of all eigenvalues of Aâ‚™ are smaller than one, which is generally true for molecules with only bond constraints but can become problematic for angle-constrained molecules [51] [45].
Key advantages of LINCS include its speed (3-4 times faster than SHAKE), easy parallelization, and particular stability in Brownian dynamics simulations [51]. Its main limitation is that it can only be used with bond constraints and isolated angle constraints, not with coupled angle constraints [51] [45].
SETTLE Algorithm
For rigid water molecules, which often constitute more than 80% of simulation systems, the SETTLE algorithm provides an analytical solution for applying constraints [51] [45]. SETTLE is a specialized algorithm that completely avoids iterative procedures, instead using direct mathematical computation to reset water molecules to their correct geometry [51].
The GROMACS implementation modifies the original SETTLE algorithm to avoid calculating the center of mass of water molecules, reducing both computational operations and rounding errors [45]. This modification changes the error dependence from quadratic to linear with respect to system size, enabling accurate integration of larger systems in single precision [45].
Figure 1: Constraint Algorithm Workflow - Relationship between unconstrained coordinates and constraint methods in MD integration.
Comparative Analysis of Algorithm Performance
Computational Efficiency and Accuracy
The choice between SHAKE, LINCS, and SETTLE involves important trade-offs between computational efficiency, accuracy, and applicability to different molecular systems [51]. SETTLE is unequivocally the most efficient method for water molecules, but for general biomolecular systems, the choice between SHAKE and LINCS depends on specific simulation requirements.
Table 2: Comparative Analysis of Constraint Algorithms
| Algorithm | Methodology | Supported Constraints | Parallelization | Relative Speed | Key Applications |
|---|---|---|---|---|---|
| SHAKE | Iterative (Gauss-Seidel) | Bonds, angles, dihedrals | Difficult | 1.0x (reference) | General purpose, robust error detection |
| LINCS | Non-iterative (matrix) | Bonds, isolated angles | Easy (P-LINCS) | 3-4x faster than SHAKE | Large systems, Brownian dynamics |
| SETTLE | Analytical solution | Complete molecular geometry | Native | Fastest for water | Water and small rigid molecules |
Practical Implementation Considerations
In practical MD simulations, the choice of constraint algorithm significantly impacts both performance and accuracy [3]. For bond constraints only, LINCS generally provides superior performance and is the default in GROMACS [51] [45]. However, for angle constraints or complex constraint networks, SHAKE may be more appropriate despite its slower performance [51] [50].
The accuracy of constraint satisfaction also varies between algorithms. LINCS typically maintains relative constraint deviations below 0.0001 for every constraint with a fourth-order expansion in MD simulations [51]. SHAKE accuracy depends on the specified tolerance but may require more iterations to achieve similar precision for complex systems [51].
Advanced Applications: Bond-Angle Constraints
Theoretical Framework for Angle Constraints
While bond-length constraints are standard in MD simulations, constraining bond angles presents additional challenges [52] [50]. Bond-angle constraints are implemented using extensions of the same Lagrange multiplier formalism, with algorithms such as SHAKEBAC for bond angles and SHAKEDAC for dihedral angles [50].
The mathematical framework for angle constraints follows the same principle as bond constraints but involves more complex geometry. For a bond angle θ formed by atoms i-j-k, the constraint equation is typically expressed as a function of the dot product of the vectors connecting the atoms, requiring solution of more complex coupled equations [52].
Performance Implications of Angle Constraints
Constraining bond angles involving hydrogen atoms can potentially increase the simulation time step from 2 fs to 4 fs [3] [50]. However, extensive studies on proteins have shown that constraining bond angles reduces molecular flexibility and entropy by approximately half, and can significantly alter the dynamics of the remaining degrees of freedom [50].
Unlike bond-length constraints, angle constraints exhibit stronger coupling to other molecular motions and more significant metric-tensor effects, which can introduce artifacts in the simulation [50]. For this reason, bond-angle constraints are typically applied only to angles involving hydrogen atoms, while angles between heavy atoms are left unconstrained to preserve more of the natural dynamics of the system [50].
Free Energy Considerations and Correction Methods
Thermodynamic Consequences of Constraints
Imposing constraints affects the phase space accessible to the molecular system, which has direct implications for thermodynamic properties and free energy calculations [48]. The free energy cost of applying constraints must be accounted for in rigorous free energy simulations, with deviations of 0.2-0.5 kcal/mol observed in solvation free energy calculations when bond-length constraints are employed [48].
For a simple harmonic oscillator with coordinate q and force constant K, the free energy of constraining the oscillator at position Δq_const is given by:
ΔG_constÊ°áµ’ = U(Δq_const) - Uâ‚€ + βâ»Â¹ln√(2Ï€/βK)
where the last term represents the entropic contribution from restricting the vibrational degree of freedom [48].
Constraint Correction Methods
For accurate free energy calculations, several methods have been developed to correct for the effects of constraints in post-processing [48]. These approaches typically involve:
- Explicit calculation of free energy costs for adding/removing constraints as additional steps in the free energy cycle
- Use of gradient calculations combined with normal mode analysis to approximate contributions to the partition function from constrained degrees of freedom
- Consideration of enthalpic, vibrational entropic, and Jacobian free energy terms
These correction methods are particularly valuable in multi-scale QM/MM simulations and other applications where constraints are used to increase phase space overlap between distinct energy surfaces [48].
Figure 2: Free Energy Correction Cycle - Incorporating constraint effects in free energy calculations.
Experimental Protocols and Implementation
Practical Implementation in MD Packages
The implementation of constraint algorithms varies across MD software packages. In GROMACS, multiple integration algorithms and constraint methods are specified in the run parameter file [3]:
In NAMD, the only available integration method is Verlet, with constraint options specified differently [3]. The time parameters in NAMD are typically defined as:
Protocol for Constraint Algorithm Selection
Selecting the appropriate constraint algorithm requires consideration of multiple factors:
Determine the required constraints: For pure bond constraints, LINCS is typically optimal. For angle constraints or complex molecular topologies, SHAKE may be necessary [51] [50].
Assess parallelization requirements: For simulations running on multiple processors, P-LINCS offers superior parallel scaling compared to SHAKE [51].
Consider system composition: For systems with significant water content, SETTLE should be used for water molecules regardless of the constraint algorithm for other molecules [51] [45].
Evaluate accuracy requirements: For energy minimization, higher-order expansions or tighter tolerances may be necessary compared to production MD [51].
Table 3: Research Reagent Solutions for Constrained MD Simulations
| Tool/Algorithm | Primary Function | Implementation Considerations | Typical Parameters |
|---|---|---|---|
| SHAKE | Iterative constraint satisfaction | Robust but slower, suitable for complex constraints | Relative tolerance: 10â»â´ to 10â»â¶ |
| LINCS | Matrix-based constraint solver | Fast parallelizable, bonds only | Expansion order: 4-8, 1 iteration for MD |
| SETTLE | Analytical water constraints | Optimal for water molecules | No parameters needed |
| RATTLE | Velocity constraint for Verlet | Required for velocity Verlet | Same tolerance as SHAKE |
| P-LINCS | Parallel LINCS implementation | Essential for multi-processor runs | Same as LINCS with parallel communication |
Constraint algorithms including SHAKE, LINCS, and SETTLE represent essential components of modern molecular dynamics simulations, enabling practical simulation timescales by allowing longer integration time steps. These algorithms, tightly integrated with Verlet integration schemes, maintain molecular geometry through Lagrange multipliers that enforce holonomic constraints on bond lengths and angles.
The continuing evolution of constraint algorithms focuses on improving parallel efficiency, accuracy for complex constraints, and integration with advanced sampling methods. Recent developments in angle constraint methods and free energy corrections highlight the growing sophistication of these fundamental computational tools, supporting more accurate and efficient simulations of biological macromolecules and materials systems.
As MD simulations continue to address increasingly complex scientific questions, constraint algorithms will remain indispensable for balancing computational efficiency with physical accuracy, enabling researchers to extract meaningful thermodynamic and kinetic information from molecular simulations.
Hydrogen Mass Repartitioning for Larger Time Steps (4 fs)
In molecular dynamics (MD) research, the Verlet algorithm and its variants, such as the velocity Verlet algorithm, form the cornerstone of numerical integration for Newton's equations of motion [53]. These symplectic integrators are prized for their numerical stability and energy conservation properties over long simulation timescales. The core of the Verlet algorithm involves calculating particle positions at time ( t + \Delta t ) using the current positions, forces, and positions from the previous time step [53]. However, the stability and accuracy of this algorithm, and MD simulations in general, are critically dependent on the size of the integration time step (( \Delta t )).
The choice of time step represents a fundamental compromise in MD. A shorter time step yields greater accuracy but increases the computational cost required to simulate a given physical timeframe. The time step is intrinsically limited by the highest frequency vibrations in the system, which typically involve bonds to hydrogen atoms due to their low mass. These X-H bond vibrations have periods on the order of 10 femtoseconds (fs) [53] [54]. The Nyquist theorem from signal processing dictates that to accurately capture these motions, the time step must be smaller than half the period of this fastest vibration—in practice, often 1/10 of the period or less [55]. This theorem historically limited time steps to 1-2 fs in all-atom MD simulations [56] [57] [54].
Hydrogen Mass Repartitioning (HMR) is a technique that directly addresses this limitation, enabling a larger integration time step while maintaining simulation stability. By strategically redistuting mass within molecules, HMR slows the fastest vibrational frequencies, allowing the Verlet integrator to propagate the system forward with a time step of up to 4 fs or more without significant loss of accuracy. This provides a potential performance improvement of close to a factor two [58], making it a highly valuable tool for researchers and drug development professionals seeking to access longer biological timescales.
The Principle and Implementation of Hydrogen Mass Repartitioning
Fundamental Concept
Hydrogen Mass Repartitioning is a simple yet powerful concept. The method involves increasing the mass of hydrogen atoms (typically to ~3.0–3.2 atomic mass units) and decreasing the mass of the heavy atoms to which they are bonded by a corresponding amount [56] [57] [54]. This redistribution is done in a way that preserves the total mass of the molecule [59]. The underlying physical principle is that the frequency of a harmonic vibration is inversely proportional to the square root of the reduced mass (( \omega \propto 1/\sqrt{\mu} )). By increasing the mass of the lightest atoms, HMR reduces the frequency of the fastest bond vibrations, which are the primary factor limiting the integration time step.
An alternative approach, Hydrogen Isotope Exchange (HIE), directly increases hydrogen masses without subtracting from the parent atom, effectively simulating a deuteration process [54]. However, HMR is generally preferred as it avoids altering the total molecular mass, which can affect diffusion and other kinetic properties [57].
Implementation Workflows
The implementation of HMR varies across different MD software packages. The general workflow and package-specific commands are detailed below.
Implementation in GROMACS
Recent versions of GROMACS (2024.2 and later) offer a highly flexible implementation of HMR directly through the grompp preprocessor using the mass-repartition-factor mdp option [60] [58]. This method modifies the masses on the fly during the creation of the run input file (.tpr), leaving the original topology unchanged. A typical setup in the mdp file is:
This setup repartitions masses by a factor designed to triple the mass of hydrogen atoms [60]. After modifying the mdp file, the simulation is prepared and run using standard commands (grompp followed by mdrun).
Implementation in AMBER
In AMBER, HMR is implemented using the parmed tool to modify the topology file (.parm7). The process is as follows [59]:
The hmassrepartition command by default triples the mass of all non-water hydrogens and adjusts the parent atom masses accordingly [59]. Subsequent equilibration and production MD use this modified topology file.
Associated Parameter Adjustments
When increasing the time step to 4 fs with HMR, other simulation parameters may require adjustment to maintain stability:
- Constraint Algorithm: Bonds involving hydrogen must remain constrained, typically using LINCS (in GROMACS) or SHAKE (in AMBER) algorithms with
constraints = h-bonds[60]. - LINCS Parameters: For improved stability, increasing
lincs-order(e.g., to 4) andlincs-iter(e.g., to 2) can be beneficial [60]. - Output Frequency: While not affecting the simulation physics, parameters like
nstcalcenergy(the frequency of energy calculation) can be adjusted for performance. Since the time per step is doubled, settingnstcalcenergy = 50withdt = 4 fsprovides energy output at the same physical time interval asnstcalcenergy = 100withdt = 2 fs[60]. - Thermostat and Barostat Coupling: The coupling constants (
tau-t,tau-p) are defined in physical time units (ps) and generally do not need adjustment when changing the time step [60].
Validation and Impact on Simulation Properties
Performance and Stability Considerations
The primary motivation for HMR is a significant reduction in computational cost. By allowing a time step of 4 fs instead of 2 fs, HMR can nearly double simulation speed [58]. However, stability must be carefully verified.
The optimal mass repartitioning factor is system-dependent. While a factor of 3 is common and often allows a 4 fs time step [60] [59], some systems, particularly those with methyl groups, might require a smaller factor or a more conservative time step (e.g., 3 fs) [60]. During setup, gmx grompp may issue a note if the estimated oscillational period of a bond is less than 10 times the time step [60]. This is often acceptable, but if followed by LINCS warnings during the simulation, it indicates instability. Mitigation strategies include reducing the time step to 3 fs, increasing the mass factor (if possible), or adjusting LINCS parameters [60].
Table 1: Summary of Validated Time Steps with HMR from Literature
| System Type | Force Field | HMR Factor | Stable Time Step (fs) | Key Structural Properties Maintained | Citation |
|---|---|---|---|---|---|
| Membrane Proteins | AMBER | 3 | 4 (3 if unstable) | Membrane structure, protein conformations | [60] |
| Pure Lipid Bilayers | CHARMM36 | Not specified | 4 | Area per lipid, order parameters, electron density profiles | [56] [57] |
| Cyclic Peptide Nanotubes | GROMOS54a7 | HMR & HIE | 5-7 | Pore diameter, H-bonding, peptide stacking | [54] |
Effects on Structural and Kinetic Properties
Extensive validation studies have demonstrated that HMR, when applied correctly, preserves key structural properties of biological systems, though some kinetic properties may be affected.
Table 2: Impact of HMR (with 4 fs time step) on Simulation Observables
| Observable Category | Specific Property | Impact of HMR | Notes |
|---|---|---|---|
| Structural Properties | Area per lipid (APL) | No significant difference | Validated in membrane systems [56] [57] |
| Electron density profile | No significant difference | Validated in membrane systems [56] [57] | |
| Deuterium order parameters | No significant difference | Validated in membrane systems [56] [57] | |
| Protein/Peptide conformation | Reproduces non-HMR conformations | Consistent with standard simulations [57] [54] | |
| Kinetic Properties | Lipid diffusion constant | Differences observed | Expected due to altered collision dynamics [56] [57] |
| Hydrogen-bond lifetimes | Little dependence | Similar dynamics observed [57] | |
| Viscosity of water | Increased if HMR applied to water | HMR should not be applied to water [57] |
For membrane systems—highly relevant to drug development—HMR with a 4 fs time step and a standard 12 Å cutoff produces structural results in excellent agreement with reference 2 fs simulations [56] [57]. This includes the conductance of porins, peptide partitioning, and membrane mixing processes [57]. Similarly, for complex systems like cyclic peptide nanotubes, HMR allowed time steps of 5-7 fs without serious alterations to structural and dynamic nanopore properties [54].
Table 3: Key Research Reagents and Computational Tools for HMR Simulations
| Item / Resource | Function / Role | Implementation Example |
|---|---|---|
| MD Software with HMR | Provides the computational engine to perform simulations with modified masses. | GROMACS (≥2024.2), AMBER, NAMD, ACEMD [58] [57] |
| Topology Modifier | Tool to modify atomic masses in the system topology. | grompp (GROMACS), parmed (AMBER) [60] [59] |
| Force Field | Defines the potential energy function and standard parameters for the system. | AMBER, CHARMM, OPLS, GROMOS [60] [54] |
| Constraint Algorithm | Algorithm to holonomically constrain the fastest bond vibrations. | LINCS (GROMACS), SHAKE (AMBER) [60] [54] |
| System Builder | Web-based or software tool to generate initial structures and topologies for complex systems. | CHARMM-GUI [60] [57] |
Hydrogen Mass Repartitioning is a robust and well-validated technique that integrates seamlessly with the core Verlet integration algorithms of molecular dynamics. By strategically altering atomic masses to slow the highest frequency vibrations, HMR enables a safe increase of the time step to 4 fs, nearly doubling computational throughput. For researchers and drug developers, this translates to the ability to simulate longer timescales or achieve better sampling within the same computational budget.
Critically, when implemented with a standard cutoff and without applying the mass change to water, HMR has been shown to preserve the structural integrity of a wide range of systems, from soluble proteins to complex membrane environments. While some kinetic properties like diffusion constants may be affected, the structural accuracy essential for most mechanistic studies is maintained. As MD continues to push the boundaries of simulated timescales and system complexity, HMR stands as a key optimization method within the researcher's toolkit, grounded in a solid theoretical foundation and extensive empirical validation.
Addressing Numerical Precision and Round-off Error
Within molecular dynamics (MD) research, the Verlet algorithm is a cornerstone numerical method for integrating Newton's equations of motion. Its popularity stems from its time-reversibility and symplectic nature, which contribute to excellent long-term energy conservation in simulations of physical systems [2]. However, like all numerical techniques, it is susceptible to errors introduced by the finite precision of computer arithmetic. This guide details the nature of these numerical precision challenges, specifically round-off errors, within the context of the Verlet algorithm and its variants. It provides a comprehensive overview of strategies to identify, quantify, and mitigate these errors, ensuring the reliability of MD simulations for applications such as drug development and materials science.
The core problem is that the standard Verlet algorithm, while elegant, performs operations that can lead to a significant loss of numerical precision. Understanding and addressing this vulnerability is crucial for researchers who require high-fidelity trajectories and accurate energy conservation over millions of simulation time steps.
The Verlet Algorithm and Its Numerical Vulnerabilities
Core Algorithm Formulation
The basic Størmer–Verlet algorithm is designed to solve Newton's second law, (\ddot{\mathbf{x}}(t) = \mathbf{A}(\mathbf{x}(t))), where (\mathbf{A}) is the acceleration, often computed as force divided by mass [2]. It does so without explicitly storing velocities at each time step. The update rule for positions is derived from central difference approximation and is given by:
[ \mathbf{x}{n+1} = 2\mathbf{x}{n} - \mathbf{x}{n-1} + \mathbf{a}n \Delta t^2 ]
where (\mathbf{x}{n+1}) is the new position, (\mathbf{x}{n}) is the current position, (\mathbf{x}{n-1}) is the previous position, (\mathbf{a}n) is the acceleration at the current time, and (\Delta t) is the time step [2] [8]. This formulation is second-order accurate in time and provides good numerical stability for a wide range of physical systems.
The primary numerical vulnerability of the standard Verlet algorithm lies in the calculation of the position update. The operation (2\mathbf{x}{n} - \mathbf{x}{n-1}) involves the difference of two numbers of similar magnitude, which is a well-known source of catastrophic cancellation in finite-precision arithmetic [12] [8]. This subtraction amplifies relative errors, leading to a gradual loss of precision in the particle trajectories over many time steps.
Furthermore, the algorithm is not self-starting [12] [8]. The initial step requires the position at a previous time (\mathbf{x}_{n-1}), which is typically generated using a different method, such as a simple Taylor expansion. Any error introduced in this initialization phase propagates through all subsequent iterations, potentially compounding the overall numerical inaccuracy.
Table 1: Key Numerical Characteristics of Verlet Algorithm Variants
| Algorithm Variant | Position Error | Velocity Error | Self-Starting | Primary Round-off Error Source |
|---|---|---|---|---|
| Basic Verlet | (O(\Delta t^4)) | (O(\Delta t^2)) [12] | No [8] | Position update: (2xn - x{n-1}) [12] |
| Velocity Verlet | (O(\Delta t^4)) [2] | (O(\Delta t^4)) [2] | Yes [8] | Accumulation in multi-step velocity updates |
| Stormer-Verlet | (O(\Delta t^4)) | (O(\Delta t^2)) [23] | No | Velocity calculation: (\frac{x{n+1} - x{n-1}}{2\Delta t}) [23] |
Optimized Verlet-like Algorithms for Enhanced Precision
To overcome the limitations of the basic Verlet algorithm, several optimized variants have been developed. These algorithms reformulate the integration steps to minimize operations that induce significant round-off errors.
Velocity Verlet Algorithm
The Velocity Verlet algorithm is a mathematically equivalent reformulation that explicitly stores and updates velocities at the same time as positions, making it self-starting and reducing round-off errors [8]. The update sequence is:
[ \begin{aligned} \mathbf{r}(t + \Delta t) &= \mathbf{r}(t) + \mathbf{v}(t) \Delta t + \frac{1}{2} \mathbf{a}(t) \Delta t^2 \ \mathbf{v}(t + \Delta t/2) &= \mathbf{v}(t) + \frac{1}{2} \mathbf{a}(t) \Delta t \ \mathbf{a}(t + \Delta t) &= -\frac{1}{m} \nabla V(\mathbf{r}(t + \Delta t)) \ \mathbf{v}(t + \Delta t) &= \mathbf{v}(t + \Delta t/2) + \frac{1}{2} \mathbf{a}(t + \Delta t) \Delta t \end{aligned} ]
This scheme avoids the direct subtraction of similar-magnitude position vectors, which is the main source of round-off error in the basic Verlet algorithm [8]. It requires (9N) memory locations to store positions, velocities, and accelerations for (N) particles but does not require storing values at two different times for any single quantity [8].
Recent Advancements and Optimized Algorithms
Research into optimized Verlet-like algorithms continues. A 2001 study introduced a family of Verlet-like algorithms derived using an extended decomposition scheme with a free parameter [20]. By optimizing this parameter, the authors developed algorithms that are more efficient and accurate than the original Verlet versions while remaining symplectic and time-reversible [20].
A very recent 2024 study highlights an Improved Velocity-Verlet Algorithm developed specifically for the Discrete Element Method, which is also relevant to MD [26]. This improvement corrects for unphysical results that can arise in mixtures with large particle size ratios, ensuring physically accurate outcomes by synchronizing the calculation of variables to consistent time steps [26].
Experimental Protocols for Quantifying Numerical Error
To validate the precision and stability of an integration scheme in practice, specific experimental protocols must be followed. These tests are essential for any researcher implementing or selecting an algorithm for production simulations.
Protocol for Energy Conservation Testing
Objective: To quantify the long-term stability and energy drift of the integrator in a conservative system.
- System Setup: Initialize a system with a well-defined potential energy function (e.g., Lennard-Jones fluid, harmonic oscillators).
- Simulation Parameters: Run a simulation over a sufficiently long time (e.g., (10^6) to (10^9) steps) at a constant time step (\Delta t).
- Data Collection: At every time step, or at regular intervals, record the total energy (E = K + V) (kinetic + potential).
- Analysis: Plot the total energy as a function of time. A high-quality, low-round-off-error integrator will show no discernible drift in the total energy over time, with only small fluctuations around the mean value [8]. The magnitude of these fluctuations and any linear drift trend should be quantified.
Protocol for Round-off Error Propagation Analysis
Objective: To directly measure the accumulation of round-off error in the integration trajectory.
- Reference Trajectory: Generate a simulation trajectory using a very small time step and high-precision arithmetic (e.g., quad-precision) to serve as a reference.
- Test Trajectory: Run the same simulation with the same initial conditions using the production integrator and standard double-precision arithmetic.
- Error Calculation: At regular intervals, calculate the root-mean-square deviation (RMSD) between the positions (and velocities, if applicable) of the test trajectory and the reference trajectory.
- Analysis: Plot the RMSD as a function of time. The growth rate of this error provides a direct measure of the algorithm's susceptibility to numerical precision loss.
Research Reagent Solutions: Essential Computational Tools
Table 2: Key Software and Numerical "Reagents" for Verlet-based MD
| Research Reagent | Function/Brief Explanation | Relevance to Precision |
|---|---|---|
| LAMMPS | A widely used open-source MD simulator [18] [26] | Provides tested, optimized implementations of Verlet algorithms; the improved 2024 Velocity-Verlet was implemented within it [26]. |
| High-Precision Arithmetic Libraries | Software libraries that enable computations beyond standard 64-bit double-precision. | Allows for the generation of reference trajectories and direct quantification of round-off error. |
| ProtoMol | An object-oriented framework for molecular dynamics [21] | Used as a testbed for implementing and evaluating new, stable integrators like the mollified impulse method. |
| Numerical Error Analysis Tools | Custom scripts or software to compute energy drift, trajectory RMSD, etc. | Essential for conducting the experimental protocols outlined in Section 4 to validate simulation accuracy. |
Workflow and Algorithmic Relationships
The following diagram illustrates the logical workflow for selecting and validating a Verlet integration scheme with a focus on managing numerical precision, summarizing the key concepts and relationships discussed in this guide.
Within the field of molecular dynamics (MD) research, the Verlet algorithm stands as a cornerstone numerical method for integrating Newton's equations of motion. Originally designed for calculating particle trajectories in molecular dynamics simulations, this algorithm provides good numerical stability, time reversibility, and preservation of the symplectic form on phase space at no significant additional computational cost over simpler methods like Euler integration [2]. The core Verlet integrator solves second-order differential equations of the type x¨(t)=A(x(t)) without explicitly calculating velocities, using the position update formula x_{n+1} = 2x_n - x_{n-1} + a_nΔt² [2]. Despite its mathematical elegance and favorable properties, the basic Verlet algorithm suffers from several practical limitations, including not being self-starting and potential roundoff errors, which have motivated numerous extensions and optimizations [12]. This technical guide explores the theoretical foundations, optimization methodologies, and practical implementations of Verlet-like algorithms, framed within the context of contemporary molecular dynamics research for drug development and scientific computing applications.
Theoretical Foundations of the Verlet Algorithm
Mathematical Derivation and Core Principles
The Verlet algorithm derives from a central difference approximation to the second derivative in Newton's equations of motion. For a conservative physical system described by Mx¨(t) = F(x(t)) = -∇V(x(t)), where M represents mass, F is force, and V denotes potential energy, the Verlet method discretizes time with step Δt and approximates acceleration using the position history [2]. The mathematical foundation emerges from Taylor expansions of position:
Adding these two expansions eliminates the odd-degree terms, including the velocity term, yielding the fundamental Verlet position update equation:
This formulation achieves third-order local accuracy for positions while using only one force evaluation per time step [2]. The global error for position is of order Δt³, while velocity calculations yield second-order accuracy [12]. The algorithm's time symmetry inherently reduces local discretization errors by eliminating odd-degree terms in the Taylor expansion, contributing to its remarkable numerical stability for long-time simulations [2].
Velocity Handling and Algorithmic Variants
The basic Størmer-Verlet algorithm does not explicitly calculate velocities, which presents challenges for molecular dynamics simulations where kinetic energy measurements are essential. Several mathematically equivalent variants address this limitation:
The Original Verlet Algorithm: Uses the position update
x_{n+1} = 2x_n - x_{n-1} + a_nΔt²without explicit velocity computation [2]. This approach suffers from numerical precision issues due to calculating velocities as differences between positions of similar magnitude [12].Velocity Verlet Algorithm: Provides self-starting capability and minimizes roundoff errors by explicitly tracking velocities [15] [12]. The algorithm follows a sequential update process:
This formulation uses current acceleration a(t) and the subsequent acceleration a(t+Δt) after position update, making it self-starting and reducing numerical precision loss [15].
- Leap-Frog Algorithm: Interleaves position and velocity updates at half-time steps, expressed as
v(t+Δt/2) = v(t-Δt/2) + a(t)Δtandx(t+Δt) = x(t) + v(t+Δt/2)Δt[11]. This approach provides better energy conservation properties for some systems.
The following diagram illustrates the logical relationships and computational workflow between these primary Verlet algorithm variants:
Optimization Strategies for Verlet-like Algorithms
Extended Decomposition with Free Parameters
Modern optimization approaches for Verlet algorithms involve extending the basic decomposition scheme through the introduction of free parameters that minimize the influence of truncated terms. Omelyan et al. proposed explicit velocity- and position-Verlet-like algorithms derived from an extended decomposition framework containing a free parameter [20]. When this parameter equals zero, the algorithms reduce to the original Verlet formulations. However, by optimizing the nonzero value of this parameter to minimize truncation error effects, the resulting algorithms demonstrate improved efficiency and accuracy while maintaining the symplectic and time-reversible properties essential for molecular dynamics simulations [20] [61].
The mathematical foundation for these optimized algorithms rests on a more general decomposition of the classical time-evolution operator. By introducing a parameter that controls the splitting between position and velocity updates, the method achieves a more balanced error distribution. The optimization process involves minimizing the expectation value of the squared error over a representative ensemble of molecular configurations, leading to algorithms that outperform the standard Verlet method at identical computational cost [20]. This approach represents a significant advancement in molecular dynamics integration techniques, particularly for simulations of Lennard-Jones fluids and other many-body systems where accuracy and stability are paramount.
Force-Gradient and Higher-Order Extensions
For systems requiring exceptionally high precision, force-gradient algorithms extend the Verlet method by incorporating higher-order derivatives of the force field. These advanced algorithms demonstrate particularly dramatic improvements for molecular, quantum, and celestial mechanics simulations [61]. The construction of these methods involves composing the basic Verlet integrator with force-gradient corrections, resulting in algorithms that can reach orders of 4 to 16 while maintaining favorable stability properties [61].
The efficiency gains from these high-order force-gradient algorithms are substantial, with fourth-order-based implementations outperforming standard methods by factors of 5 to 1000, depending on the required precision [61]. The implementation follows a multi-stage composition scheme where each stage incorporates corrected force evaluations based on gradient information. While computationally more expensive per time step, these methods enable significantly larger time steps while maintaining accuracy, resulting in net performance improvements for high-precision applications in drug development and materials science.
Comparative Analysis of Numerical Integrators
Accuracy and Stability Performance Metrics
The performance of integration algorithms can be evaluated across three primary criteria: computational performance (time per integration step), numerical stability (resistance to error accumulation with stiff forces), and accuracy (deviation from analytical solutions) [62]. Different Verlet formulations excel in different scenarios, making algorithm selection dependent on the specific physical system being simulated.
Table 1: Integrator Performance Comparison Across System Types
| Method | Accuracy Rank | Critically-Damped Stability | Over-Damped Stability | Under-Damped Stability | Force Evaluations per Step |
|---|---|---|---|---|---|
| Euler | 5 | 1 | 1 (tie) | 6 | 1 |
| SUVAT | 4 | 5 | 2 | 5 | 1 |
| Newton-Stormer-Verlet (NSV) | 6 | 6 | 5 | 2 | 1 |
| Modified Verlet | 3 | 3 | 4 | 1 | 1 |
| Improved Euler | 2 | 4 | 6 | 3 | 2 |
| Runge-Kutta 4 (RK4) | 1 | 2 | 3 | 4 | 4 |
Note: Accuracy and stability rankings are on a scale of 1 (best) to 6 (worst) based on tested performance across damping conditions. Data compiled from [62].
The ranking data reveals that no single algorithm performs optimally across all conditions. The NSV (Newton-Stormer-Verlet) and Modified Verlet algorithms demonstrate exceptional stability for under-damped systems, making them ideal for orbital mechanics and molecular dynamics where energy conservation is crucial [62]. Conversely, for overdamped systems with significant friction (common in tire simulations or protein folding), basic Euler and SUVAT methods show superior stability despite lower accuracy [62].
Discretization Error and Time-Step Selection
All numerical integrators accumulate discretization error due to Taylor series truncation, a phenomenon particularly relevant for Verlet algorithms where error terms grow exponentially with time-step size [15]. The following relationship illustrates how time-step selection dramatically impacts simulation accuracy:
As demonstrated in molecular dynamics simulations of spring-mass systems, larger time-steps (e.g., dt = 0.02) lead to visible energy non-conservation and divergent particle trajectories, while smaller time-steps (e.g., dt = 0.005) better preserve total system energy at the cost of increased computation [15]. This fundamental tradeoff between computational efficiency and physical accuracy represents a critical consideration when implementing Verlet algorithms for production molecular dynamics simulations in drug development.
Implementation Considerations for Molecular Dynamics
Molecular Dynamics Workflow and Integration
The integration of Verlet algorithms into complete molecular dynamics simulations requires careful attention to workflow sequencing and auxiliary computations. The global MD algorithm implemented in packages like GROMACS follows a structured approach [11]:
- Input Initial Conditions: Define potential interactions, initial positions, and initial velocities for all atoms
- Compute Forces: Calculate forces on atoms based on non-bonded interactions, bonded interactions, and external forces
- Update Configuration: Numerically solve Newton's equations of motion using the Verlet integrator
- Output Step: Write positions, velocities, energies, and other observables to trajectory files
The following workflow diagram illustrates this process with the Velocity Verlet integration steps expanded:
The velocity Verlet algorithm fits naturally within this framework, with position updates preceding force calculations, which then enable velocity updates [15] [11]. This sequencing ensures that all necessary information is available at each stage while maintaining the algorithm's self-starting property and minimizing numerical error.
Neighbor Searching and Performance Optimization
In production molecular dynamics codes, the Verlet algorithm couples with efficient neighbor searching techniques to handle non-bonded interactions. GROMACS implements an O(N) neighbor search algorithm that uses pair lists with a Verlet buffer scheme [11]. The pair list cut-off exceeds the interaction cut-off (râ„“ = rc + r_b) to create a buffer zone that accounts for atomic displacement between list updates, typically every 10-20 time steps.
The performance impact of this approach is significant, as pair list generation constitutes one of the more computationally expensive components of molecular dynamics simulations. By using cluster-based pair lists (grouping 4 or 8 particles) and dynamic list pruning, modern implementations maintain computational efficiency while preserving accuracy [11]. The buffer size can be determined automatically based on a tolerable energy drift threshold, typically set at 0.005 kJ/mol/ps per particle, though practical implementations often achieve an order of magnitude better performance [11].
Table 2: Research Reagent Solutions for Molecular Dynamics Implementation
| Component | Function | Implementation Example |
|---|---|---|
| Potential Interaction V | Defines force field between atoms | Lennard-Jones potential, AMBER, CHARMM force fields |
| Neighbor List | Identifies atom pairs within interaction range | Verlet buffer scheme with cluster pairs (4-8 atoms) |
| Periodic Boundary Conditions | Eliminates surface effects | Triclinic box transformation to rectangular coordinates |
| Thermostat | Maintains constant temperature | Maxwell-Boltzmann velocity distribution with COM correction |
| Initial Velocity Generator | Provides realistic starting velocities | Gaussian random numbers with standard deviation √(kT/m_i) |
| Integration Time Step (Δt) | Balances accuracy and computational cost | Typically 1-2 fs for molecular systems with Verlet |
| Force Calculation Kernel | Computes non-bonded and bonded interactions | NxM kernels optimized for SIMD/SIMT parallel processing |
These "research reagents" form the essential toolkit for implementing Verlet algorithms in production molecular dynamics environments, particularly in drug development where accurate sampling of conformational spaces is critical for binding affinity calculations and thermodynamic profiling.
Application in Drug Development and Research
Enhanced Sampling and Free Energy Calculations
Optimized Verlet-like algorithms enable more efficient free energy calculations through enhanced sampling techniques, a crucial capability in computer-aided drug design. The improved stability and accuracy of parameter-optimized Verlet algorithms facilitate longer time steps without sacrificing physical fidelity, directly accelerating molecular dynamics simulations used to compute binding free energies in protein-ligand systems [20]. These calculations form the basis for rational drug design, where small differences in binding affinity (1-2 kcal/mol) can separate effective pharmaceuticals from inactive compounds.
The numerical stability of symplectic Verlet integrators proves particularly valuable for Hamiltonian replica exchange methods, where multiple simulations at different temperatures exchange configurations to accelerate barrier crossing. The time-reversible and volume-preserving properties of optimized Verlet algorithms ensure detailed balance in these exchange attempts, maintaining correct thermodynamic sampling while dramatically improving conformational exploration compared to standard dynamics [61].
Force Field Compatibility and Scalability
Modern optimized Verlet algorithms demonstrate exceptional compatibility with advanced force fields used in drug development, including polarizable models and coarse-grained representations. The extended decomposition approaches maintain their stability advantages even with complex potential energy functions that include explicit polarization, van der Waals interactions, and sophisticated solvent models [20]. This compatibility ensures that accuracy improvements translate directly to biologically relevant systems with heterogeneous molecular environments.
For large-scale biomolecular simulations encompassing millions of atoms, the parallel scalability of Verlet-based integrators becomes essential. The local nature of the Verlet update (each position depends only on immediate neighbors) enables efficient domain decomposition across distributed computing resources [11]. This property, combined with the improved accuracy of optimized Verlet-like algorithms, supports the simulation of entire viral capsids, ribosomes, and membrane complexes at biologically relevant timescales - capabilities directly applicable to structure-based drug design and mechanistic studies of drug action.
The Verlet algorithm has evolved significantly from its original formulation into a family of optimized integration methods that balance numerical stability, computational efficiency, and implementation practicality. Through the introduction of free parameters in decomposition schemes, force-gradient corrections, and sophisticated neighbor-list management, modern Verlet-like algorithms deliver enhanced performance for molecular dynamics simulations in drug development and materials science. The continued refinement of these integration techniques, coupled with advancements in force field accuracy and high-performance computing, promises to further expand the scope and fidelity of molecular simulations in pharmaceutical research. As molecular dynamics approaches increasingly integrate with machine learning and automated experimentation, optimized Verlet algorithms will remain fundamental components in the computational toolkit for understanding molecular interactions and accelerating therapeutic discovery.
Verlet vs. Other Integrators: Accuracy, Stability, and Selection Criteria
In molecular dynamics (MD) research, the Verlet algorithm serves as a cornerstone numerical method for integrating Newton's equations of motion. Its significance stems from its ability to provide stable, long-time evolution for particle trajectories while preserving fundamental geometric properties of the true physical system [2]. However, like all numerical methods, it introduces discretization errors and, over sufficiently long timescales, can exhibit unphysical energy drift, potentially compromising simulation validity [63].
This technical guide examines the theoretical foundations for quantifying these numerical artifacts, framing the discussion within the context of the Verlet algorithm's properties. We explore the mathematical formalism of discretization errors through backward error analysis, establish methodologies for measuring their practical impact, and investigate the stochastic nature of energy drift. The insights presented herein provide researchers, particularly in computational drug development, with the framework necessary to critically assess and improve the numerical accuracy of their simulations.
The Verlet Algorithm: Foundations and Formulations
The Verlet algorithm, in its various forms, is a numerical integration scheme prized for its simplicity, computational efficiency, and desirable numerical properties, including time-reversibility and symplecticity [2]. These characteristics make it the integrator of choice for the vast majority of molecular dynamics simulations in materials science and biomolecular modeling [64].
Core Algorithmic Forms
The basic Störmer-Verlet scheme, derived from a central difference approximation to the second derivative, calculates a new position using the current and previous positions along with the current acceleration [2]. For a second-order differential equation of the form (\ddot{\mathbf{x}}(t) = \mathbf{A}(\mathbf{x}(t))), the iteration proceeds as:
- Initialization: Set (\mathbf{x}1 = \mathbf{x}0 + \mathbf{v}0 \Delta t + \frac{1}{2}\mathbf{A}(\mathbf{x}0) \Delta t^2).
- Iteration: For (n = 1, 2, \ldots), compute (\mathbf{x}{n+1} = 2\mathbf{x}n - \mathbf{x}{n-1} + \mathbf{A}(\mathbf{x}n) \Delta t^2).
This formulation, while explicit, does not directly include velocities. The velocity-Verlet algorithm, a functionally equivalent but more commonly implemented variant, explicitly calculates velocities at the same time step as positions and is summarized in the workflow diagram below.
Numerical Properties
The Verlet algorithm's superiority over simpler methods like Euler integration lies in its higher-order accuracy and structure-preserving nature. By being time-reversible and symplectic, it conserves the symplectic form on phase space, which is a fundamental property of Hamiltonian mechanics [2]. This leads to excellent long-term energy conservation behavior, meaning that while the energy in the numerical solution may fluctuate, it does not exhibit a systematic drift for a sufficiently small time step and stable system. Furthermore, its simple computational structure makes it highly efficient, with a cost per time step that scales linearly with the number of particles, (O(N)) [64].
Discretization Error: Theory and Quantification
Discretization error is the inherent inaccuracy introduced when a continuous differential equation is solved by a discrete numerical approximation. In MD, this means the computed trajectory diverges from the exact solution of Newton's equations. The choice of time step ((\Delta t)) is critical: a step too large causes instability, while a step too small wastes computational resources [65].
Backward Error Analysis
A powerful framework for understanding discretization errors is backward error analysis. Within this framework, the numerical solution produced by an integrator of order (r) (like Verlet, where (r=2)) is interpreted not as an approximate solution to the original system, but as an exact solution to a "shadow" or modified Hamiltonian system [65].
Formally, the modified flow (\tilde{\mathbf{F}}(\mathbf{z}; h)) and its corresponding modified Hamiltonian (\tilde{H}(\mathbf{z}; h)) for a symplectic method like Verlet can be expressed as power series in the step size (h):
[ \tilde{\mathbf{F}}(\mathbf{z}; h) = \mathbf{F}(\mathbf{z}) + h^r \mathbf{F}^{[r]}(\mathbf{z}) + h^{r+1} \mathbf{F}^{[r+1]}(\mathbf{z}) + \cdots ]
[ \tilde{H}(\mathbf{z}; h) = H(\mathbf{z}) + h^r H^{[r]}(\mathbf{z}) + h^{r+1} H^{[r+1]}(\mathbf{z}) + \cdots ]
The terms (H^{[j]}(\mathbf{z})) are corrections that depend on the original Hamiltonian and its derivatives. This explains why the energy of the numerical solution is nearly conserved for exponentially long times: it is closely tracking the energy of the shadow Hamiltonian [65]. The artifacts observed in simulations at large time steps—such as deviations in kinetic temperature or non-uniform pressure profiles—are therefore properties of the shadow system, not the original one [65].
Measuring Errors in Observed Quantities
The effect of discretization error on a measured observable (A) (e.g., temperature, pressure, potential energy) is a systematic bias. The phase space average (\langle A \rangleh) computed from a simulation with step size (h) relates to the true average (\langle A \rangle0) as [65]:
[ \langle A \rangle0 = \langle A \rangleh + h^r \langle A^{[r]} \rangleh + h^{r+1} \langle A^{[r+1]} \rangleh + \cdots ]
This power-series dependence means that the computed average depends on the step size. For a second-order Verlet integrator, the leading error term typically scales with (h^2). The table below summarizes the discretization errors for key properties observed in a TIP4P water simulation coupled to different thermostats.
Table 1: Discretization errors for key properties in a TIP4P water simulation with different thermostats. Error magnitudes are categorized as strong, moderate, or weak relative scaling with time step [65].
| Measured Quantity | NVE Ensemble (Microcanonical) | Nosé-Hoover Thermostat (Canonical) | Langevin Thermostat (Stochastic) |
|---|---|---|---|
| Kinetic Temperature (Translational) | Strong (h^2) dependence | Strong (h^2) dependence | Strong (h^2) dependence |
| Kinetic Temperature (Rotational) | Strong (h^2) dependence | Strong (h^2) dependence | Strong (h^2) dependence |
| Configurational Temperature | Weak (h^2) dependence | Weak (h^2) dependence | Weak (h^2) dependence |
| Potential Energy | Moderate (h^2) dependence | Moderate (h^2) dependence | Moderate (h^2) dependence |
| Pressure | Moderate (h^2) dependence | Moderate (h^2) dependence | Moderate (h^2) dependence |
| Radial Distribution Function | Minimal (h^2) dependence | Minimal (h^2) dependence | Minimal (h^2) dependence |
Energy Drift: Characterization and Analysis
In addition to the systematic bias from discretization error, another key indicator of numerical accuracy in microcanonical (NVE) simulations is energy drift, defined as a long-term, non-periodic change in the total energy of the system.
The Phenomenon of Energy Drift
For a symplectic integrator like Verlet applied to a stable Hamiltonian system, the total energy should oscillate around a stable mean without a secular drift. However, in practice, over very long simulation times, a slow drift can be observed. This drift is an unphysical artifact of numerical integration and is distinct from the small, bounded fluctuations expected from a symplectic integrator [63].
Stochastic Modeling of Energy Error
Research suggests that when a system is started from a random initial configuration, the error in energy from the numerical solution can be effectively modeled as a continuous-time stochastic process. Studies indicate that this error can be represented as geometric Brownian motion [63]. This model implies that the energy error behaves like a random walk with a drift component, providing a statistical framework for analyzing and predicting the long-term behavior of energy in MD simulations, rather than treating it as a purely deterministic error.
Experimental Protocols for Error Quantification
Establishing robust protocols for quantifying discretization error and energy drift is essential for validating simulation parameters and ensuring result reliability.
Protocol for Discretization Error Measurement
This protocol determines the systematic bias in measured observables due to the finite time step.
- System Preparation: Create an initial configuration of the system (e.g., a solvated protein or a box of water molecules) and equilibrate it thoroughly at the target state point (temperature, density) using a small time step (e.g., 1 fs) and a well-tested thermostat/barostat.
- Production Runs: Perform a set of independent production simulations from the same equilibrated starting point, varying only the integration time step (h). For example, use (h = 0.5, 1.0, 2.0, 4.0) fs. All other parameters (force field, cutoff schemes, thermostat/barostat settings) must remain identical.
- For NVE ensembles, the initial conditions must be identical for all runs.
- For thermostated ensembles, ensure the thermostat seeds are different for each run to avoid spurious correlations, or use deterministic thermostats.
- Data Collection: For each run, calculate the average values of key observables (A(h)) over the production trajectory. Ensure each simulation is long enough to achieve a small statistical error relative to the expected discretization error.
- Error Analysis: Plot the average value of each observable (\langle A \rangleh) against (h^2). Fit a linear or quadratic model to the data points. The y-intercept of the fit provides an extrapolated estimate of the true value (\langle A \rangle0), free from the leading order discretization error.
Protocol for Energy Drift Measurement
This protocol quantifies the long-term stability of energy conservation in NVE simulations.
- Initialization: Start from a well-equilibrated configuration, ensuring the system's energy is stable. Precisely record the initial total energy (E_0).
- NVE Production Run: Switch to the NVE ensemble and run a very long simulation (orders of magnitude longer than the correlation times of the slowest processes) using the Verlet integrator. The time step should be chosen as large as possible while maintaining stability; typically 90-100% of the maximum stable step.
- Energy Monitoring: Output the total energy (E(t)) at regular intervals throughout the simulation.
- Drift Calculation: Compute the energy drift using a linear regression of (E(t)) against simulation time (t). The slope of the best-fit line, (\frac{dE}{dt}), is the energy drift rate. It is often reported in units of energy per (simulation time x particle), e.g., kJ/(mol·ns·atom).
Mitigation Strategies and Correction Techniques
Understanding errors is a prerequisite for correcting them. Several strategies can mitigate the impact of discretization errors and energy drift.
Choosing an Appropriate Time Step
The most straightforward mitigation is using a smaller time step. The optimal choice balances computational cost against acceptable error. For all-atom biomolecular simulations with explicit solvent and flexible bonds, 2 fs is a common maximum, constrained by the fastest hydrogen vibration frequencies. Using holonomic constraints (e.g., SHAKE or LINCS) for bonds involving hydrogen atoms allows time steps of 2 fs, or even 4 fs for rigid water models [64].
Error Correction and Extrapolation
As outlined in Section 5.1, one can run simulations at multiple time steps and extrapolate the results to (h = 0). This provides a corrected estimate of the observable with the leading-order discretization error removed [65].
Using Shadow Hamiltonians
For some integrators, it is possible to construct a modified or "shadow" Hamiltonian (\tilde{H}) that is conserved more accurately by the numerical trajectory than the true Hamiltonian (H). Monitoring (\tilde{H}) can provide a more sensitive measure of integration quality [63].
Table 2: Methods for correcting discretization errors in molecular dynamics simulations [65].
| Method | Underlying Principle | Implementation Complexity | Limitations |
|---|---|---|---|
| Time Step Extrapolation | Extrapolates results from multiple simulations at different step sizes to Δt = 0. | Medium | Computationally expensive; requires multiple simulations. |
| Weighted Thermostating | Adjusts coupling of thermostat to different degrees of freedom to cancel leading-order error. | Low to Medium | System-specific; may require derivation and tuning for different observables. |
| Backward Error Analysis Corrections | Derives and computes modified expressions for observables based on the shadow Hamiltonian. | High | Mathematically complex; expressions are system and integrator-specific. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential computational "reagents" and tools for analyzing numerical accuracy in molecular dynamics.
| Tool/Reagent | Function in Analysis |
|---|---|
| Verlet Integrator (e.g., velocity-Verlet) | The core algorithm whose accuracy is being assessed; provides the numerical trajectory. |
| Stochastic Thermostat (e.g., Langevin) | Controls temperature while allowing study of discretization error in a stabilized context. |
| Deterministic Thermostat (e.g., Nosé-Hoover) | Controls temperature via extended Lagrangian; its own discretization error can be studied. |
| Holonomic Constraint Algorithm (e.g., SHAKE, LINCS) | Constrains fastest bond vibrations, enabling larger time steps and reducing the highest frequency errors. |
| Modified Hamiltonian ((\tilde{H})) | A theoretically conserved quantity for the numerical method; its drift serves as a sensitive accuracy probe. |
| Multi-Timestep Integrator (RESPA) | Allows different forces to be evaluated at different frequencies, potentially reducing error for fast forces. |
The Verlet algorithm provides a robust foundation for molecular dynamics, but a critical understanding of its numerical artifacts is non-negotiable for producing reliable scientific results. Discretization error introduces a systematic, time-step-dependent bias into measured observables, which can be rigorously analyzed and corrected via multi-step extrapolation. Energy drift, conversely, signals a long-term instability in energy conservation, often modeled stochastically. For researchers in drug development, where accurate computation of binding affinities, conformational dynamics, and thermodynamic properties is paramount, a rigorous quantitative approach to assessing and mitigating these numerical inaccuracies is not merely a best practice—it is a fundamental requirement for validating simulation-based conclusions.
Within molecular dynamics (MD) research, the accurate and efficient integration of Newton's equations of motion is a foundational computational challenge. The core problem involves solving a system of second-order differential equations for a many-body system, typically expressed as ( mi \ddot{\mathbf{x}}i = \mathbf{F}i(\mathbf{x}) ), where ( mi ) is the mass of particle ( i ), ( \mathbf{x}i ) is its position, and ( \mathbf{F}i ) is the force acting upon it [2]. The choice of numerical integration algorithm profoundly impacts the simulation's fidelity, stability, and ability to correctly model physical conservation laws. While simpler approaches like Euler's method offer intuitive implementation, the Verlet algorithm and its variants have emerged as the predominant integrators in modern molecular dynamics due to their superior stability and conservation properties [66] [67]. This technical analysis examines the theoretical and practical advantages of the Verlet algorithm over Euler's method, focusing on the numerical stability, energy conservation, and time-reversibility that are essential for reliable MD simulations in fields like drug development and materials science.
Theoretical Foundations of the Algorithms
Euler's Method: Formulation and Limitations
Euler's method is a first-order numerical procedure for solving ordinary differential equations with a given initial value [68]. For the equations of motion in molecular dynamics, it is typically implemented in its forward (explicit) form.
Mathematical Formulation: For a differential equation of the form ( y' = f(t, y) ), the Euler method computes the next value in the sequence as: [ y{n+1} = yn + h f(tn, yn) ] where ( h ) is the step size. Applied to molecular dynamics, this translates to two coupled equations for position and velocity [66]: [ \begin{aligned} x{n+1} &= xn + h vn \ v{n+1} &= vn + h an = vn + h \frac{F(xn)}{m} \end{aligned} ]
Truncation Error Analysis: The local truncation error (LTE) of Euler's method is ( O(h^2) ), while the global error is ( O(h) ), classifying it as a first-order method [69]. This relatively large error term introduces energy drift in Hamiltonian systems, making it unsuitable for long-term molecular dynamics simulations.
Numerical Stability Constraints: A critical limitation of the explicit Euler method is its conditional stability. For the linear test problem ( \dot{y} = -ky ), numerical stability requires the step size to satisfy ( h < 2/k ) [69]. This stringent requirement can force impractically small time steps in molecular systems with stiff bond potentials, severely limiting computational efficiency.
Verlet Algorithm: Formulation and Variants
The Verlet algorithm is a second-order method that directly integrates Newton's equations of motion by leveraging the symmetry of central difference approximations.
Basic Størmer-Verlet Algorithm: The original Verlet algorithm calculates the new position using the current and previous positions [2]: [ x{n+1} = 2xn - x{n-1} + an h^2 ] where ( an = F(xn)/m ). Notably, this formulation does not explicitly include velocity, which must be calculated retrospectively as ( vn = (x{n+1} - x_{n-1})/(2h) ) [67].
Velocity-Verlet Algorithm: A more computationally convenient variant explicitly includes velocity updates [67] [70]: [ \begin{aligned} x{n+1} &= xn + h vn + \frac{h^2}{2} an \ v{n+1} &= vn + \frac{h}{2} (an + a{n+1}) \end{aligned} ] This formulation provides direct access to velocities for kinetic energy calculations and maintains the favorable numerical properties of the basic Verlet method.
Truncation Error Analysis: The Verlet algorithm has a local truncation error of ( O(h^4) ) and a global error of ( O(h^2) ), making it a second-order method [2]. The expansion errors cancel effectively due to the time-reversible structure of the algorithm, leading to better long-term stability.
Quantitative Comparison of Algorithm Performance
Mathematical Property Comparison
Table 1: Comparative analysis of mathematical properties between Euler and Verlet integration methods
| Property | Euler Method | Verlet Algorithm |
|---|---|---|
| Order of Accuracy | First-order (( O(h) ) global error) [69] | Second-order (( O(h^2) ) global error) [2] |
| Stability Characteristics | Conditionally stable (requires ( h < 2/k ) for test equation) [69] | Unconditionally stable for simple harmonic motion [2] |
| Energy Conservation | Poor (significant energy drift) [67] | Excellent (bounded energy fluctuations) [67] |
| Time-Reversibility | No [71] | Yes [2] |
| Symplectic Property | Non-symplectic [71] | Symplectic [71] |
| Computational Cost per Step | Low (1 force evaluation) [66] | Moderate (1 force evaluation) [66] |
| Implementation Complexity | Low [66] | Moderate [66] |
Molecular Dynamics Application Performance
Table 2: Performance comparison in practical molecular dynamics simulations
| Performance Metric | Euler Method | Verlet Algorithm |
|---|---|---|
| Energy Drift in NVE Ensemble | Significant drift, unusable for production simulations [67] | Minimal drift, suitable for long simulations [67] |
| Orbital Stability (Celestial Example) | Earth spirals into sun with same timestep [67] | Stable orbits maintained over long periods [67] |
| Maximum Stable Timestep | Small (often < 0.5 fs for atomistic MD) | Larger (typically 1-2 fs for atomistic MD) [36] |
| Suitability for MD Simulations | Poor, not used in production MD [67] | Excellent, widely used in production MD [36] [2] |
The Symplectic Nature of Verlet Integration
A fundamental mathematical advantage of the Verlet algorithm lies in its symplectic property, which makes it particularly suited for Hamiltonian systems like molecular dynamics.
Symplectic Structure Preservation: A symplectic integrator preserves the symplectic form on phase space, meaning it conserves the area in the position-momentum phase space [71]. In molecular dynamics simulations, this mathematical property translates to superior energy conservation over long simulation times.
Time-Reversibility: The Verlet algorithm is time-reversible, a property inherent to the physical systems it models [2]. If the direction of time is reversed, the algorithm retraces its trajectory back to the initial conditions. This property is absent in Euler's method, which accumulates directional errors.
Phase Space Volume Conservation: In a symplectic method like Verlet, the mapping from one time step to the next conserves state-space volume [71]. This ensures that the simulation does not artificially contract or expand the phase space distribution, maintaining the statistical mechanical foundation of the molecular dynamics ensemble.
The impact of these properties is readily observable in practical simulations. For instance, when simulating the gravitational two-body problem (an analogue for molecular orbital interactions), Euler's method causes the earth to spiral into the sun, while Verlet integration maintains a stable orbit [67]. Similarly, in molecular dynamics, the total energy fluctuations with Verlet integration are typically two orders of magnitude smaller than those observed with Euler's method using the same time step [67].
Implementation Protocols for Molecular Dynamics
Velocity-Verlet Integration Protocol
The following protocol details the implementation of the velocity-Verlet algorithm for a standard molecular dynamics simulation:
Initialization:
- Set initial positions ( x0 ) and velocities ( v0 ) for all particles
- Choose an appropriate time step ( h ) (typically 1-2 femtoseconds for atomistic systems)
- Compute initial forces ( F0 = -\nabla V(x0) ) and accelerations ( a0 = F0/m )
Main Integration Loop (for each time step from ( n = 0 ) to ( N )):
Thermostat/Temperature Control (for NVT ensemble):
- Apply temperature scaling algorithms (Berendsen, Nosé-Hoover) to velocities if simulating canonical ensemble
Analysis:
- Monitor conservation of total energy, temperature, and pressure
- Analyze structural and dynamic properties from trajectory data
Parallelization Strategy for Large-Scale Systems
Modern molecular dynamics simulations of biologically relevant systems (proteins, nucleic acids) require parallel computing architectures for efficiency [36]:
- Domain Decomposition: The simulation box is divided into spatial domains, with each processor responsible for force calculations for particles within its domain
- Verlet List Construction: Each processor maintains a neighbor list for particles within its domain using the Verlet list algorithm, which tracks particles within a cutoff radius plus a skin distance
- Force Parallelization: Non-bonded forces are calculated in parallel, with processors communicating boundary particle information
- Synchronization Points: The algorithm requires synchronization after position updates and force calculations to ensure consistent evolution
This parallelization approach, combined with the numerical stability of the Verlet algorithm, enables simulations of millions of atoms over microsecond timescales [36].
Research Reagent Solutions: Computational Tools
Table 3: Essential software tools and algorithms for Verlet-based molecular dynamics research
| Tool/Algorithm | Type | Function in MD Research |
|---|---|---|
| Verlet Neighbor List | Algorithmic Optimization | Reduces computational cost of non-bonded interactions from ( O(N^2) ) to ( O(N) ) by maintaining list of nearby particles [36] |
| OpenMP | Parallel Programming API | Enables shared-memory parallelization of Verlet integration loops for multi-core processors [36] |
| GROMACS | MD Software Package | Highly optimized implementation of Verlet algorithms with GPU acceleration for biomolecular systems [36] |
| Lennard-Jones Potential | Force Field Component | Models van der Waals interactions in fluid and material systems; used to validate Verlet integrators [16] |
| Periodic Boundary Conditions | Simulation Methodology | Mimics bulk system behavior using repeating unit cells; requires special handling in Verlet integration [36] |
Visualization of Algorithm Workflows
Diagram 1: Comparative workflow of Euler vs. Velocity-Verlet integration algorithms. The Verlet method's symmetric force evaluation structure enables its superior conservation properties.
Diagram 2: Comparative stability behavior in orbital simulations. The Verlet algorithm maintains stable orbits with bounded energy fluctuations, while Euler's method exhibits energy drift and unstable trajectories [67].
The Verlet algorithm represents a fundamentally superior approach to numerical integration in molecular dynamics compared to Euler's method, particularly for simulations requiring long-term stability and faithful energy conservation. Its advantages stem from three interconnected mathematical properties: second-order accuracy with favorable error cancellation, symplectic structure preservation, and time-reversibility. These characteristics enable Verlet integration to maintain stable trajectories and bounded energy fluctuations in NVE ensemble simulations, whereas Euler's method exhibits significant energy drift even with small time steps. For molecular dynamics researchers investigating protein folding, drug-receptor interactions, or material properties, the Verlet algorithm provides the necessary numerical foundation for physically meaningful simulations. While modern molecular dynamics packages continue to employ optimized variants of the Verlet algorithm, understanding its core mathematical advantages over simpler methods like Euler's remains essential for critically evaluating simulation methodologies and interpreting computational results in pharmaceutical and materials research.
Within the field of molecular dynamics (MD) research, the Verlet algorithm stands as a cornerstone for numerical integration, prized for its ability to faithfully simulate the evolution of classical systems over long timescales [2] [72]. This technical guide examines the core characteristics of the Verlet integrator by contrasting it with the family of Runge-Kutta methods, focusing on the fundamental trade-off between long-term stability and short-term accuracy. For researchers and scientists in computational drug development, understanding this balance is not merely academic; it directly influences the reliability and computational cost of simulations probing protein dynamics, ligand binding, and other critical phenomena. The Verlet algorithm's design, which inherently preserves the symplectic form on phase space, makes it exceptionally suited for conserving energy in Hamiltonian systems, a property that underpins its superior long-term behavior [2].
Theoretical Foundations of the Algorithms
The Verlet and Runge-Kutta families of algorithms approach the problem of solving ordinary differential equations from fundamentally different philosophical and mathematical starting points. These foundational differences ultimately dictate their performance in practical applications.
Verlet Integration: A Symplectic Workhorse for Molecular Dynamics
Verlet integration is designed specifically to integrate Newton's equations of motion [2]. For a conservative physical system, this is expressed as: $$M\ddot{\mathbf{x}}(t) = F(\mathbf{x}(t)) = -\nabla V(\mathbf{x}(t)),$$ where (M) represents the mass matrix, (F) is the force, and (V) is the potential energy [2]. The algorithm operates by using a central difference approximation to the second derivative, leading to the core position-update formula: $$\mathbf{x}{n+1} = 2\mathbf{x}n - \mathbf{x}{n-1} + \mathbf{a}n \Delta t^2,$$ where (\mathbf{a}n = A(\mathbf{x}n)) is the acceleration at step (n) [2]. A key feature of this formulation is that it does not explicitly calculate velocities, though they can be derived from particle positions. The method is time-reversible and symplectic, meaning it preserves the geometric structure of phase space in Hamiltonian systems [2]. This property is crucial for the faithful long-term integration of conservative systems, as it leads to excellent energy conservation over millions of time steps.
Runge-Kutta Methods: Versatility through Intermediate Stages
Runge-Kutta (RK) methods form a family of iterative techniques for solving ordinary differential equations (ODEs) that extend the basic idea of Euler's method by using multiple derivative evaluations (stages) per step [73]. A general (s)-stage RK method computes the next value as: $$y{n+1} = yn + h\sum{i=1}^{s} biki,$$ where the intermediate stages are given by: $$ki = f(tn + cih, yn + h\sum{j=1}^{s} a{ij}kj).$$ The coefficients (a{ij}), (bi), and (c_i) are traditionally organized in a Butcher tableau [73]. These methods are classified by their order, which determines their local truncation error. Higher-order methods (e.g., the classic fourth-order RK, or RK4) achieve greater accuracy at the cost of more function evaluations per step. Unlike Verlet, standard RK methods are not generally symplectic, which affects their long-term energy conservation properties.
Quantitative Comparison of Algorithm Properties
The practical performance of numerical integrators can be quantified through several key metrics. The table below summarizes the fundamental characteristics of Verlet and Runge-Kutta methods.
Table 1: Core Algorithm Characteristics for Molecular Dynamics Simulations
| Property | Verlet Integration | Runge-Kutta Methods (Explicit) |
|---|---|---|
| Theoretical Basis | Central difference approximation for second-order ODEs [2] | Weighted average of intermediate slopes for first-order ODEs [73] |
| Typical Order | Second-order (local error (O(\Delta t^4))) [2] | Variable (commonly second to fourth-order, local error (O(h^{p+1}))) [73] |
| Symplectic Nature | Yes (preserves phase space structure) [2] | Not generally (except specialized symplectic variants) |
| Time Reversibility | Yes [2] | No |
| Stability for Conservative Systems | Excellent long-term energy conservation [72] | Potential energy drift in long simulations [73] |
| Computational Cost per Step | Low (one force evaluation per step) | Higher (multiple function evaluations per step, e.g., four for RK4) [73] |
| Stability Limit (Harmonic Oscillator) | (h \leq 2/\omega) [72] | Smaller than Verlet for a given order (region is bounded in complex left-half plane) [73] |
| Primary Strength | Long-term stability and energy conservation | High short-term accuracy and ease of implementation |
The stability analysis, particularly for MD simulations, often uses the linear test equation (y' = \lambda y). The stability region for a method is the set of complex values (z = h\lambda) for which the numerical solution remains bounded [73]. For the Verlet algorithm applied to a harmonic oscillator with frequency (\omega), the exact stability criterion is (h \leq 2/\omega) [72]. This provides a clear, physically interpretable limit for MD practitioners.
Table 2: Performance in Molecular Dynamics Applications
| Aspect | Verlet Integration | Runge-Kutta Methods |
|---|---|---|
| Energy Conservation | Excellent (due to existence of a shadow Hamiltonian) [72] | Tends to drift over long simulations |
| Recommended Use Case | Long-term MD trajectories, structural stability analysis | Short, high-accuracy trajectory segments, non-Hamiltonian systems |
| Implementation Complexity | Low (straightforward position/force updates) | Moderate (requires managing intermediate stages) [73] |
| Adaptive Step Size | Not inherent (requires modification) | Naturally supported (e.g., via embedded methods) [73] |
| Performance on Stiff Systems | Poor without very small time steps | Better with implicit variants (DIRK, Radau methods) [73] |
Stability Analysis: A Deeper Dive
The long-term stability of the Verlet integrator in molecular dynamics simulations can be understood through the concept of a shadow Hamiltonian (\tilde{H}) [72]. For a discrete symplectic algorithm like Verlet, there exists a slightly different, conserved Hamiltonian (\tilde{H}) that remains very close to the true Hamiltonian (H). The stability of a simulation can be monitored through the stability of this shadow energy. Simulations become unstable when the time increment (h) exceeds a threshold where the shadow energy is no longer conserved [72].
For a single harmonic mode with frequency (\omega), the stability limit is given exactly by (h \leq 2/\omega) [72]. In complex molecular systems with a spectrum of frequencies, the instantaneous highest frequency determines the stability limit. This explains why the hydrogen vibration, being the highest frequency mode in a typical biomolecular system, often dictates the maximum allowable time step. The relationship between the simulation time step and the stability governed by the highest frequency mode in the system is illustrated below.
Diagram 1: Stability determination workflow for MD simulations. The highest frequency mode dictates the maximum stable time step.
Runge-Kutta methods have different stability properties. Their stability region is derived from applying the method to the linear test equation (y' = \lambda y) [73]. The stability function (R(z)) is determined from the Butcher tableau via (R(z) = 1 + zb^T(I - zA)^{-1}e) [73]. The method is stable for step sizes where (|R(z)| \leq 1). While higher-order explicit RK methods can offer greater accuracy, their stability regions are often more restrictive than Verlet's for a given problem. Implicit RK methods can have superior stability properties (some are A-stable, meaning stable for all (h) when (Re(\lambda) < 0)), but this comes at the cost of solving nonlinear equations at each step [73].
Experimental Protocols and Validation
To quantitatively compare the stability and accuracy of Verlet and Runge-Kutta methods in a molecular dynamics context, specific experimental protocols can be employed. The following methodology outlines a robust approach for such a comparison.
Protocol for Stability and Accuracy Assessment
Test System Definition: Implement a standard benchmark system, such as a Lennard-Jones fluid or the viscous Kob-Andersen system, in a controlled simulation environment [72].
Algorithm Implementation:
- Implement the basic Størmer-Verlet algorithm as defined by the update rule: (x{n+1} = 2xn - x{n-1} + an \Delta t^2) [2].
- Implement a fourth-order Runge-Kutta (RK4) method, defined by the update: (y{n+1} = yn + \frac{h}{6}(k1 + 2k2 + 2k3 + k4)), where (k1) to (k4) are the standard intermediate stage calculations [73].
Stability Limit Determination:
- For a range of time steps (h), compute the shadow energy (for Verlet) or total energy (for RK4) over a long simulation (e.g., (10^6) steps).
- The stability limit is identified as the largest (h) for which the energy remains bounded and does not diverge [72].
- Compare the empirical stability limit with the theoretical prediction for a harmonic oscillator, (h \leq 2/\omega), where (\omega) is estimated from the highest frequency mode in the system [72].
Accuracy Quantification:
- For a short simulation where the exact solution is known (or can be accurately approximated by a high-order method with a very small time step), compute the global error for both methods.
- Plot the global error against the time step (h) on a log-log scale to verify the theoretical order of accuracy of each method.
Performance Benchmarking:
- For a given level of accuracy, compare the computational cost (CPU time) required by each method to integrate the equations of motion over a fixed physical time.
- For a fixed computational budget, compare the accuracy achieved by each method over a long simulation time, monitoring energy drift as a key metric.
Diagram 2: Experimental workflow for comparing Verlet and Runge-Kutta methods, highlighting key tests and metrics.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Computational Tools for Method Comparison Studies
| Item/Solution | Function in Analysis |
|---|---|
| Lennard-Jones Potential | A standard model potential for noble gases and simple fluids, used as a benchmark system for testing MD algorithms [72]. |
| Kob-Andersen Model | A classical model for a viscous liquid mixture, useful for testing algorithm performance in dense, glass-forming systems [72]. |
| Butcher Tableau | A standardized table of coefficients ((a{ij}), (bi), (c_i)) that completely defines a specific Runge-Kutta method [73]. |
| Shadow Hamiltonian ((\tilde{H})) | A conserved quantity for symplectic integrators like Verlet, used to monitor the stability and quality of the numerical integration [72]. |
| Stability Function (R(z)) | A complex function whose magnitude determines the stability region of a Runge-Kutta method for the linear test equation [73]. |
| Harmonic Oscillator Model | A simple system with a known analytical solution, used for deriving exact stability criteria (e.g., (h \leq 2/\omega)) [72]. |
The choice between the Verlet algorithm and Runge-Kutta methods is not a matter of identifying a universally superior technique, but rather of selecting the right tool for a specific scientific problem. For long-term molecular dynamics simulations where energy conservation and time-reversibility are critical for physical fidelity, the Verlet integrator is overwhelmingly the preferred choice [2] [72]. Its symplectic nature, computational efficiency, and existence of a shadow Hamiltonian make it uniquely suited for simulating the thermodynamic and dynamic properties of classical systems over extended timescales.
In contrast, Runge-Kutta methods, particularly higher-order explicit variants, excel in scenarios demanding high short-term accuracy for a given step size, for non-Hamiltonian systems, or when adaptive step-size control is highly beneficial [73]. The decision framework for researchers thus hinges on the core requirements of their simulation: when long-term stability and faithful energy conservation are the overriding concerns, Verlet is the definitive algorithm; when maximum accuracy for a single, short trajectory segment is the priority, a high-order Runge-Kutta method may be preferable. Understanding this fundamental trade-off empowers computational scientists in drug development and materials research to make informed choices that enhance the reliability and predictive power of their molecular simulations.
Molecular Dynamics (MD) simulation is a cornerstone computational technique in biophysics and drug development, enabling the study of biomolecular systems at atomic resolution. The Verlet integration algorithm is a foundational numerical method used to solve Newton's equations of motion in these simulations [2]. Its significance stems from its numerical stability, time-reversibility, and symplectic properties (meaning it conserves the geometric structure of phase space), which make it particularly suited for long-time MD simulations [2]. For researchers and drug development professionals, understanding the performance characteristics—computational cost and memory usage—of this algorithm and its implementations is crucial for planning and interpreting large-scale simulations, such as those used in virtual drug screening and protein folding studies.
Core Algorithm and Computational Workflow
The Verlet Integration Scheme
The Verlet algorithm integrates Newton's equations of motion for a system of N particles. For a conservative system, this is expressed as ( M\ddot{\mathbf{x}}(t) = -\nabla V(\mathbf{x}(t)) ), where ( M ) is the mass matrix, ( \mathbf{x} ) represents atomic positions, and ( V ) is the potential energy [2]. The most common form, the Störmer-Verlet method, calculates new positions using the recurrence relation: [ \mathbf{x}{n+1} = 2\mathbf{x}n - \mathbf{x}{n-1} + \mathbf{a}n \Delta t^2 ] where ( \mathbf{a}n = \mathbf{A}(\mathbf{x}n) ) is the acceleration at time step ( n ), and ( \Delta t ) is the integration time step [2]. A key characteristic of this basic formulation is that velocities are not explicitly computed, though they can be derived as ( \mathbf{v}n = (\mathbf{x}{n+1} - \mathbf{x}_{n-1}) / (2\Delta t) ).
Variants and Implementation in MD Software
Several variants of the basic Verlet algorithm have been developed to address specific needs. The leap-frog and velocity Verlet integrators are two popular implementations that explicitly track atomic velocities. In modern MD software packages like GROMACS, these are implemented as integrator=md (leap-frog) and integrator=md-vv (velocity Verlet) [74]. The velocity Verlet algorithm is noted for its improved accuracy in constant-temperature and constant-pressure simulations (e.g., with Nosé-Hoover or Parrinello-Rahman coupling) and provides more accurate velocity estimates, albeit at a slightly higher computational cost compared to the leap-frog method [74].
Table 1: Key Integrator Variants in GROMACS
| Integrator | Algorithm Type | Key Features | Performance Considerations |
|---|---|---|---|
md |
Leap-frog Verlet | Computationally efficient, good stability [74]. | Lower computational cost; sufficient for most production simulations [74]. |
md-vv |
Velocity Verlet | More accurate velocity integration, time-reversible [74]. | Higher computational cost; better for advanced thermostats/barostats [74]. |
sd |
Stochastic Dynamics | Leap-frog with friction and noise term for temperature coupling [74]. | Accurate and efficient for solvated systems; constraints require extra force calculations [74]. |
Performance Benchmarking and Computational Cost
Profiling of MD Simulation Components
In a GPU-optimized MD implementation, performance analysis reveals that force calculations and the evaluation of neighbor lists (including pair lists) constitute the majority of the total execution time [75]. This highlights these components as the primary targets for optimization efforts.
Benchmarking Data and Speedup
Performance gains are most pronounced when leveraging specialized hardware like Graphics Processing Units. One study demonstrated that a GPU-optimized MD code incorporating a parallel Verlet neighbor list algorithm achieved significant speedups compared to a CPU-optimized version. The speedup was found to be system-dependent (N-dependent), reaching approximately 30x for a large system like the full 70S ribosome (10,219 beads) [75]. The performance improvement was even more dramatic for specific subroutines, with the pair list and neighbor list evaluations showing speedups of about 25x and 55x, respectively [75].
Table 2: Performance Benchmark of a GPU-Optimized MD Implementation
| System Component | Speedup Factor (GPU vs. CPU) | Notes |
|---|---|---|
| Overall Simulation (70S Ribosome) | ~30x | Performance is N-dependent; system with 10,219 beads [75]. |
| Pair List Evaluation | ~25x | Critical for non-bonded force calculations [75]. |
| Neighbor List Evaluation | ~55x | Parallel Verlet algorithm shows extreme optimization potential [75]. |
Case Study: Large-Scale Datasets and Resource Implications
The computational cost of generating MD data for machine learning is substantial. The creation of the PLAS-20k dataset, for instance, required 97,500 independent MD simulations on a total of 19,500 distinct protein-ligand complexes to calculate binding affinities using the MMPBSA method [76]. Similarly, the mdCATH dataset represents over 62 milliseconds of accumulated simulation time for 5,398 protein domains, simulated at multiple temperatures and replicates [77]. These examples underscore the immense computational resources required for generating large-scale, statistically robust MD datasets, reinforcing the need for highly efficient algorithms and high-performance computing infrastructure in modern computational biophysics and drug discovery.
Memory Usage and Management
Critical Memory Structures
The Verlet neighbor list is a central data structure that dramatically influences performance and memory usage. It optimizes computation by maintaining a list of atom pairs within a specified cutoff radius, which is updated periodically rather than at every time step. The memory required for this list scales with the number of particles (N) and the density of the system.
Trajectory Storage and Data Volumes
In production MD simulations, storing trajectory data (atomic coordinates and velocities) constitutes a significant memory and storage burden. For example, the mdCATH dataset records atomic coordinates and forces every 1 nanosecond [77]. The total data volume generated by such large-scale efforts can easily reach terabytes. To manage this, efficient data formats like HDF5 are often employed. The mdCATH dataset, for instance, uses HDF5 to store trajectories, topology information (psf strings), and precomputed metadata (e.g., RMSD, RMSF) for efficient storage and random access [77].
Experimental Protocols for Performance Evaluation
Standard MD Simulation Protocol
A typical MD protocol for benchmarking, as used in generating the PLAS-20k dataset, involves several stages [76]:
- System Preparation: The protein-ligand complex is solvated in an explicit water box (e.g., TIP3P) with ions added for neutrality. Force fields like AMBER ff14SB for proteins and GAFF2 for ligands are applied [76].
- Energy Minimization: The system undergoes minimization (e.g., 1000-2000 steps) using algorithms like L-BFGS or steepest descent to remove steric clashes [76].
- Equilibration: The system is gradually heated to the target temperature (e.g., 300 K) and equilibrated with positional restraints on the protein backbone, first in the NVT ensemble and then in the NPT ensemble to achieve the correct density [76].
- Production Run: The final, unrestrained simulation is performed in the NPT ensemble for data collection. Integration is typically performed with a Verlet-type integrator (e.g.,
mdormd-vvin GROMACS) with a time step of 2 femtoseconds [76].
Workflow for MD Simulation and Performance Profiling
The following diagram illustrates the core workflow of an MD simulation, highlighting the integration of the Verlet algorithm and key performance-intensive steps:
Table 3: Key Software, Datasets, and Computational Resources for MD Research
| Resource | Type | Function and Application |
|---|---|---|
| GROMACS | Software Suite | High-performance MD simulation package offering multiple Verlet integrators (md, md-vv) and extensive benchmarking capabilities [74]. |
| OpenMM | Software Suite | Open library for MD simulations with GPU acceleration, used for high-throughput calculations in datasets like PLAS-5k/20k [76]. |
| AMBER ff14SB | Force Field | Provides parameters for simulating proteins, defining potential energy terms for force calculations [76]. |
| CHARMM22* | Force Field | State-of-the-art force field for all-atom protein simulations, used in the mdCATH dataset [77]. |
| PLAS-20k | Dataset | MD trajectory dataset for 19,500 protein-ligand complexes; benchmark for binding affinity prediction and ML model training [76]. |
| mdCATH | Dataset | Large-scale MD dataset of 5,398 protein domains; includes forces for training neural network potentials [77]. |
| HDF5 Format | Data Format | Efficient binary format for storing and managing large MD trajectories and associated data [77]. |
| GPUGRID.net | Compute Resource | Distributed computing network used for generating large-scale MD datasets like mdCATH [77]. |
In molecular dynamics (MD) research, the core task is to simulate the physical motions of atoms and molecules over time by numerically solving their equations of motion. [37] The choice of algorithm to integrate these equations is foundational, directly impacting the simulation's stability, accuracy, and conservation of physical quantities. The Verlet algorithm and its variants stand as the most widely used integration schemes in MD. Their selection, however, is not one-size-fits-all; it is profoundly influenced by the target statistical ensemble—a set of systems used to represent the thermodynamic state of the simulated molecular system. [78] This guide provides an in-depth examination of how the standard Verlet algorithm is ideally suited for the microcanonical (NVE) ensemble and how extended algorithms are employed to simulate other critical ensembles like the canonical (NVT) and isothermal-isobaric (NPT) ensembles, which are essential for mimicking realistic experimental conditions.
Core Theory: Statistical Ensembles in Molecular Dynamics
A statistical ensemble defines the macroscopic state of a system by specifying which microscopic states (atomic positions and velocities) are possible and their probabilities. The choice of ensemble dictates which thermodynamic variables—such as energy (E), temperature (T), or pressure (P)—are held constant during a simulation. [78]
Common MD Ensembles include:
- NVE Ensemble (Microcanonical): A system with a constant Number of particles (N), constant Volume (V), and constant total Energy (E). It is obtained by solving Newton's equations without any temperature or pressure control. [78]
- NVT Ensemble (Canonical): A system with a constant Number of particles (N), constant Volume (V), and constant Temperature (T). This is the default ensemble in many MD software packages and is appropriate for conformational searches in vacuum or when pressure is not a significant factor. [78]
- NPT Ensemble (Isothermal-Isobaric): A system with a constant Number of particles (N), constant Pressure (P), and constant Temperature (T). This ensemble allows the simulation box size to change and is used when correct pressure, volume, and density are critical. [78]
Although averages of properties are consistent across ensembles representing the same thermodynamic state, the fluctuations of these properties differ. [78] The integrator must therefore be compatible with, or adapted for, the specific ensemble being simulated.
The Verlet Family of Integrators
The Verlet algorithm and its derivatives are the workhorses of MD integration due to their numerical stability, time-reversibility, and conservation of energy.
The Original Verlet and Leap-Frog Algorithms
The original Verlet algorithm calculates new atomic positions based on the current positions, the previous positions, and the current accelerations. A closely related and mathematically equivalent variant is the leap-frog algorithm, which is the default integrator in packages like GROMACS. [79] The leap-frog algorithm updates positions and velocities at staggered times:
- Velocities are calculated at time ( t + \frac{1}{2}\Delta t ).
- Positions are calculated at time ( t + \Delta t ) using these mid-step velocities.
- Forces and accelerations are then computed at the new positions ( t + \Delta t ).
- The velocities are updated again for the next half-step. [79]
A known limitation of the standard leap-frog/Verlet scheme is that the positions and velocities are half a timestep out of sync, which can contribute to small fluctuations in the total energy. [78]
The Velocity-Verlet Algorithm
The velocity-Verlet algorithm is a reformulation that provides a more straightforward implementation while remaining equivalent to the standard Verlet method. Its key advantage is that it explicitly calculates positions, velocities, and accelerations all at the same time point at the end of the integration step, which simplifies the handling of thermostats and barostats.
Table 1: Key Integrator Equations and Properties
| Integrator | Update Equations | Key Features | Primary Ensemble |
|---|---|---|---|
| Leap-Frog | ( v(t + \frac{1}{2}\Delta t) = v(t - \frac{1}{2}\Delta t) + \frac{F(t)}{m} \Delta t ) ( r(t + \Delta t) = r(t) + v(t + \frac{1}{2}\Delta t) \Delta t ) | Positions & velocities are out of sync; efficient and stable. | NVE (base) [79] |
| Velocity-Verlet | ( v(t + \frac{1}{2}\Delta t) = v(t) + \frac{F(t)}{2m} \Delta t ) ( r(t + \Delta t) = r(t) + v(t + \frac{1}{2}\Delta t) \Delta t ) ( v(t + \Delta t) = v(t + \frac{1}{2}\Delta t) + \frac{F(t + \Delta t)}{2m} \Delta t ) | All quantities at same time; easier coupling to thermostats. | NVE (base) [17] |
Ensemble-Specific Integration Methods
While the base Verlet algorithms are designed for NVE simulations, most practical MD simulations require controlling temperature or pressure, necessitating extended algorithms.
NVE Ensemble: The Domain of Standard Verlet
The standard Verlet, leap-frog, and velocity-Verlet algorithms are inherently suited for the NVE ensemble because they directly integrate Newton's equations of motion, which naturally conserve energy. [78] This makes them ideal for exploring the constant-energy surface of a system's conformational space during production runs after equilibation. However, even in NVE, small energy drifts can occur due to numerical rounding errors and the inherent phase difference in the leap-frog algorithm. [78] Consequently, constant-energy simulations are not recommended for the equilibration phase, as they lack a mechanism to drive the system toward a desired temperature. [78]
Extended Algorithms for NVT and NPT Ensembles
Simulating NVT or NPT ensembles requires modifying the equations of motion to include coupling to an external heat bath (thermostat) and/or pressure bath (barostat). The base Verlet algorithm serves as the core, but is extended to accommodate these couplings.
NVT (Canonical) Ensemble: Temperature control is typically achieved by a thermostat that scales velocities or acts as a frictional force. In the initialization stage, direct velocity scaling can be used, while during data collection, more sophisticated methods like temperature-bath coupling (e.g., Nosé-Hoover, Berendsen thermostats) are employed, which subtly modify the force calculations and integration steps to maintain a constant temperature. [78]
NPT (Isothermal-Isobaric) Ensemble: This ensemble requires control over both temperature and pressure. A barostat is introduced to adjust the volume of the simulation cell. This means the integration algorithm must not only update atomic positions and velocities but also the simulation box vectors. This introduces additional complexity, as the equations of motion are extended to include the volume as a dynamic variable. [78]
Table 2: Summary of Common Ensembles and Control Methods
| Ensemble | Constant Quantities | Primary Application | Common Control Methods |
|---|---|---|---|
| NVE | Number, Volume, Energy | Exploring constant-energy surfaces; production runs after equilibration. | Newton's equations (No external control). [78] |
| NVT | Number, Volume, Temperature | Default for many simulations; conformational searches in vacuum. | Thermostats (e.g., velocity scaling, Nosé-Hoover, Berendsen). [78] |
| NPT | Number, Pressure, Temperature | Mimicking lab conditions; obtaining correct densities. | Thermostats + Barostats (e.g., Berendsen, Parrinello-Rahman). [78] |
The following diagram illustrates the logical workflow for selecting an appropriate integrator based on the target ensemble and simulation requirements.
Integrator selection is based on the target thermodynamic ensemble
Advanced Considerations and Recent Developments
Algorithmic Improvements and Pitfalls
Recent research highlights that subtle implementation details of the velocity-Verlet algorithm can lead to unphysical results in certain conditions. Studies in Discrete Element Method (DEM) simulations, which share algorithmic foundations with MD, have shown that the standard velocity-Verlet scheme can produce artificial particle trapping and oscillatory motion in systems with large particle size ratios (≥3:1). [17] [80] The root cause is the half-timestep phase difference between position and velocity calculations, which introduces inaccuracies in the tangential frictional force. An improved velocity-Verlet implementation has been proposed to correct these inaccuracies, ensuring physical accuracy even at extreme size ratios (up to 100:1). [80] This underscores the importance of rigorous validation, even for established algorithms.
The Rise of Machine-Learned Potentials and Hardware Acceleration
The field is undergoing a transformation with the integration of machine learning (ML). Traditional force fields are being supplemented or replaced by neural network potentials (NNPs) trained on massive quantum chemistry datasets, such as Meta's Open Molecules 2025 (OMol25). [81] These NNPs offer accuracy close to high-level quantum mechanics at a fraction of the computational cost, enabling simulations of larger systems and more complex chemistries. [81]
This shift demands new software interfaces. Frameworks like the ML-IAP-Kokkos interface in LAMMPS allow seamless integration of PyTorch-based NNPs, facilitating end-to-end GPU acceleration for scalable AI-driven MD simulations. [82] The choice of hardware, particularly GPUs, is critical for performance. Benchmarks show that while high-end data center GPUs like the H200 offer peak simulation speed, consumer-grade cards like the RTX 4090 and specialized GPUs like the L40S can provide exceptional cost-efficiency for traditional MD workloads. [83] [84]
Practical Protocols and the Scientist's Toolkit
Protocol: Setting Up an NPT Simulation with Thermostat/Barostat Coupling
- System Preparation: Construct the initial atomic coordinates, define the topology (bond connectivity, atom types), and assign force field parameters. Solvate the system in a water box if simulating in solution and add ions to neutralize the system's charge. [79]
- Energy Minimization: Run a steepest descent or conjugate gradient minimization to remove any bad contacts and relieve steric strain in the initial structure, bringing the system to a local energy minimum.
- Initial Equilibration (NVT): Perform a short MD simulation in the NVT ensemble. Initialize velocities from a Maxwell-Boltzmann distribution at the target temperature. [79] Use a thermostat (e.g., Berendsen or Nosé-Hoover) to stabilize the temperature. This step allows the solvent and ions to relax around the solute without pressurizing the box.
- Production Equilibration (NPT): Switch to the NPT ensemble. Continue the simulation using both a thermostat and a barostat (e.g., Parrinello-Rahman) to maintain the target temperature and pressure. This step allows the density of the system to converge to the correct value. Monitor the system's potential energy, temperature, pressure, and density until they stabilize.
- Production Run: Once equilibrated, begin the production simulation. Data from this phase is used for analysis. The integrator can remain in NPT or, for certain types of analysis, be switched back to NVE.
The Scientist's Toolkit: Essential Components for MD Simulations
Table 3: Key Software, Hardware, and Methodological Components
| Tool / Reagent | Function / Purpose | Example / Note |
|---|---|---|
| MD Engine | Software that performs the numerical integration and force calculation. | GROMACS, LAMMPS, AMBER, NAMD [83] [79] [82] |
| Integrator | Core algorithm that updates positions and velocities over time. | Verlet, Leap-Frog, Velocity-Verlet [79] |
| Thermostat | Algorithm to control and maintain the system temperature. | Nosé-Hoover, Berendsen [78] |
| Barostat | Algorithm to control and maintain the system pressure. | Parrinello-Rahman, Berendsen [78] |
| Force Field | Set of functions and parameters defining interatomic potentials. | CHARMM, AMBER; or Neural Network Potentials (NNPs) [81] [37] |
| Neural Network Potential (NNP) | ML-based model for highly accurate force/energy prediction. | Models trained on OMol25 (e.g., eSEN, UMA) [81] |
| GPU Accelerator | Hardware to drastically speed up force and integration calculations. | NVIDIA RTX 4090 (cost-effective), H200 (performance), L40S (balanced) [83] [84] |
Selecting the right integrator is a fundamental decision in molecular dynamics research. The Verlet algorithm, in its various forms, provides a robust and efficient foundation. For NVE simulations, the standard Verlet, leap-frog, or velocity-Verlet algorithms are the natural and correct choice, as they conserve energy. However, to simulate the experimentally relevant NVT and NPT ensembles, this core algorithm must be extended through coupling to thermostats and barostats. As the field advances with machine-learned potentials and increasingly complex applications, understanding these foundational integration methods and their appropriate application across different ensembles remains critical for generating accurate, reliable, and meaningful simulation data.
Molecular Dynamics (MD) simulation serves as a fundamental computational tool for exploring the temporal evolution of molecular systems, enabling researchers to study biological processes, material properties, and chemical reactions at atomic resolution. The core of any MD simulation lies in its integrator—the numerical algorithm that solves Newton's equations of motion to generate particle trajectories. Among available integrators, the Verlet algorithm and its variants have emerged as the dominant method in modern MD software due to their numerical stability, time-reversibility, and conservation properties. This technical guide examines the implementation specifics of the Verlet algorithm within three prominent MD packages: GROMACS, NAMD, and SIESTA, providing researchers with a practical framework for selecting and optimizing simulations for drug development and biomolecular research.
The Verlet algorithm, first used in 1791 by Delambre and rediscovered by Loup Verlet in the 1960s, has become the foundation of modern molecular dynamics due to its symplectic nature and minimal computational overhead. Its superiority over simpler methods like Euler integration stems from its time-reversibility and better energy conservation properties, making it particularly suitable for long-timescale simulations of biomolecular systems where numerical stability is paramount.
Theoretical Foundation of Verlet Integration
The Verlet algorithm operates on the principle of using current and previous positions along with current acceleration to propagate the system forward in time. The basic Störmer-Verlet algorithm derives from Taylor expansions of position forward and backward in time.
Mathematical Formulation
The position Taylor expansion forward in time is expressed as:
[ \mathbf{r}(t + \Delta t) = \mathbf{r}(t) + \Delta t \mathbf{v}(t) + \frac{1}{2} \Delta t^2 \mathbf{a}(t) + \frac{1}{6} \Delta t^3 \mathbf{b}(t) + \mathcal{O}(\Delta t^4) ]
The expansion backward in time is:
[ \mathbf{r}(t - \Delta t) = \mathbf{r}(t) - \Delta t \mathbf{v}(t) + \frac{1}{2} \Delta t^2 \mathbf{a}(t) - \frac{1}{6} \Delta t^3 \mathbf{b}(t) + \mathcal{O}(\Delta t^4) ]
Adding these two equations yields the primary Verlet position update formula:
[ \mathbf{r}(t + \Delta t) = 2\mathbf{r}(t) - \mathbf{r}(t - \Delta t) + \mathbf{a}(t) \Delta t^2 + \mathcal{O}(\Delta t^4) ]
where (\mathbf{a}(t) = \mathbf{F}(t)/m) represents acceleration computed from forces [2] [85]. This formulation provides fourth-order accuracy for positions while using only second-order calculations, making it computationally efficient.
Velocity Verlet Algorithm
The Velocity Verlet variant addresses the original algorithm's limitation by explicitly incorporating velocities:
[ \mathbf{r}(t + \Delta t) = \mathbf{r}(t) + \mathbf{v}(t) \Delta t + \frac{1}{2} \mathbf{a}(t) \Delta t^2 ]
[ \mathbf{v}(t + \Delta t) = \mathbf{v}(t) + \frac{\Delta t}{2} [\mathbf{a}(t) + \mathbf{a}(t + \Delta t)] ]
This formulation maintains time-reversibility and symplectic properties while providing explicit velocity values at each time step, which are essential for calculating kinetic energy and temperature [40] [85].
Leapfrog Verlet Algorithm
The leapfrog algorithm, another popular variant, computes velocities at half-time steps:
[ \mathbf{v}\left(t + \frac{1}{2}\Delta t\right) = \mathbf{v}\left(t - \frac{1}{2}\Delta t\right) + \Delta t \mathbf{a}(t) ]
[ \mathbf{r}(t + \Delta t) = \mathbf{r}(t) + \Delta t \mathbf{v}\left(t + \frac{1}{2}\Delta t\right) ]
This approach, used as the default integrator in GROMACS, offers numerical stability while minimizing memory requirements [11] [40].
Table 1: Comparison of Verlet Algorithm Variants
| Algorithm | Position Update | Velocity Update | Advantages | Disadvantages |
|---|---|---|---|---|
| Basic Verlet | r(t+Δt) = 2r(t) - r(t-Δt) + a(t)Δt² |
v(t) = [r(t+Δt) - r(t-Δt)]/(2Δt) |
Simple, memory efficient | Velocities not directly available |
| Velocity Verlet | r(t+Δt) = r(t) + v(t)Δt + ½a(t)Δt² |
v(t+Δt) = v(t) + ½[a(t) + a(t+Δt)]Δt |
All quantities synchronized | Requires two force evaluations per step |
| Leapfrog | r(t+Δt) = r(t) + v(t+½Δt)Δt |
v(t+½Δt) = v(t-½Δt) + a(t)Δt |
Numerically stable, efficient | Positions and velocities not synchronized |
Verlet Implementation in GROMACS
Core Integration Scheme
GROMACS employs the leapfrog Verlet algorithm as its default integrator for production MD simulations. The implementation uses the following discrete equations:
[ \mathbf{v}i\left(t + \frac{1}{2}\Delta t\right) = \mathbf{v}i\left(t - \frac{1}{2}\Delta t\right) + \Delta t \frac{\mathbf{F}i(t)}{mi} ]
[ \mathbf{r}i(t + \Delta t) = \mathbf{r}i(t) + \Delta t \mathbf{v}_i\left(t + \frac{1}{2}\Delta t\right) ]
where (\mathbf{F}i(t) = -\partial V/\partial \mathbf{r}i) represents the force on particle (i) computed from the potential energy function (V) [11]. This formulation provides excellent numerical stability with minimal computational overhead, enabling the femtosecond-scale time steps typical of atomistic simulations.
Neighbor Searching and Pair Lists
A critical implementation aspect in GROMACS is the efficient handling of non-bonded interactions through buffered Verlet lists. The algorithm uses a cluster-based approach where particles are grouped into clusters of 4 or 8 atoms, significantly reducing the overhead of neighbor list construction. The pair list is updated every nstlist steps (typically 10-20) using a buffered cut-off scheme where the list cut-off radius (r\ell) exceeds the interaction cut-off (rc) by a buffer size (r_b) [11]:
[ r\ell = rc + r_b ]
The buffer size is automatically determined based on the user-defined energy drift tolerance (verlet-buffer-tolerance) and accounts for particle diffusion over the lifetime of the pair list. This approach balances computational expense with numerical accuracy, minimizing unwanted energy drift while maintaining high performance [11] [86].
Table 2: GROMACS Verlet Scheme Performance Characteristics
| System Type | Group Scheme (ns/day) | Verlet Scheme (ns/day) | Verlet on GPU (ns/day) | Key Optimization |
|---|---|---|---|---|
| TIP3P Water | 208 (unbuffered) | 170 | 450 | Cluster-based pair lists |
| TIP3P Water | 116 (buffered) | 162 | 450 | Spatial clustering |
| united-atom | 129 (unbuffered) | 162 | 450 | NxM non-bonded kernels |
Advanced Features
GROMACS implements dynamic pair list pruning, where the list is periodically refined without full reconstruction. This approach, which can be overlapped with integration on CPU-GPU systems, significantly reduces the number of particle pairs evaluated in the force computation kernel. The non-bonded kernels are optimized for SIMD and SIMT architectures, processing multiple interactions simultaneously on modern CPUs and GPUs [11].
The implementation also includes automatic center-of-mass motion removal to prevent kinetic energy drift. Without this correction, numerical errors in the integration algorithm could lead to developing center-of-mass motion over long simulations, particularly when temperature coupling is employed [11].
Verlet Implementation in NAMD
Velocity Verlet with Multiple Time Stepping
NAMD utilizes the velocity form of the Verlet algorithm as its core integration method, enhanced with multiple time stepping (MTS) for computational efficiency. The implementation follows:
[ \mathbf{r}(t + \Delta t) = \mathbf{r}(t) + \mathbf{v}(t) \Delta t + \frac{1}{2} \mathbf{a}(t) \Delta t^2 ]
[ \mathbf{v}(t + \Delta t) = \mathbf{v}(t) + \frac{\Delta t}{2} [\mathbf{a}(t) + \mathbf{a}(t + \Delta t)] ]
NAMD extends this basic scheme through the MTSAlgorithm parameter, which implements different approaches for handling long-range forces [87]:
- naive: Uses constant extrapolation of long-range forces from the most recent cycle boundary
- verletI: Applies long-range forces as impulses only on cycle boundaries
- verletX: Employs linear extrapolation from previous two cycle boundaries
This multi-time stepping allows NAMD to evaluate computationally expensive long-range electrostatic forces less frequently than short-range interactions, significantly accelerating simulations of large biomolecular systems.
Extended Dynamics
Recent versions of NAMD have incorporated extensions to the velocity Verlet algorithm to handle external fields, including magnetic forces. This implementation modifies the force calculation to include Lorentz forces on charged particles while maintaining the numerical stability of the core integrator [9]. The magnetic field implementation has been validated through simulations of water properties and charged particle trajectories under magnetic influence, demonstrating the flexibility of the Verlet framework for specialized applications.
Implementation in SIESTA
Based on available documentation, SIESTA employs the Verlet algorithm for integrating equations of motion in its molecular dynamics simulations, though specific implementation details differ from GROMACS and NAMD due to its focus on ab initio and density functional theory (DFT) methods.
Integration Scheme and Parameters
SIESTA utilizes the standard velocity Verlet algorithm to propagate nuclear coordinates while electrons are treated with DFT. The time step is typically set to 1-2 fs for systems involving hydrogen atoms, consistent with other MD packages. SIESTA includes options for various thermostats and barostats to simulate different ensembles (NVT, NPT), maintaining the time-reversibility of the Verlet core while enabling temperature and pressure control.
Practical Implementation and Protocols
Initialization and System Setup
Proper initialization is critical for stable Verlet integration across all packages. The general protocol involves:
- System Construction: Build molecular topology using appropriate force field parameters (AMBER, CHARMM, OPLS)
- Solvation: Immerse the system in explicit solvent (typically TIP3P, SPC, or SPC/E water models) with sufficient padding from periodic boundaries
- Energy Minimization: Perform steepest descent or conjugate gradient minimization to remove bad contacts and high-energy configurations
- Velocity Initialization: Generate initial velocities from a Maxwell-Boltzmann distribution corresponding to the target temperature:
[ p(vi) = \sqrt{\frac{mi}{2 \pi kT}}\exp\left(-\frac{mi vi^2}{2kT}\right) ]
- Equilibration: Conduct gradual heating and density equilibration in NVT and NPT ensembles before production dynamics [11] [22]
Parameter Selection Guidelines
Table 3: Optimal Verlet Integration Parameters for Biomolecular Systems
| Parameter | Recommended Value | Rationale | Packages |
|---|---|---|---|
| Time Step | 1-2 fs | Limits discretization error for bond vibrations | All |
| Pair List Update | 10-20 steps | Balances neighbor search overhead with list accuracy | GROMACS, NAMD |
| Buffer Tolerance | 0.005 kJ/mol/ps/particle | Controls energy drift from particle diffusion | GROMACS |
| Coulomb Cut-off | 1.0-1.2 nm | Minimizes truncation artifacts | All |
| VDW Cut-off | 1.0-1.2 nm | Matches electrostatic cut-off | All |
Workflow Visualization
The Scientist's Toolkit
Table 4: Essential Components for Verlet-Based MD Simulations
| Component | Function | Examples/Alternatives |
|---|---|---|
| Force Fields | Defines potential energy function | AMBER, CHARMM, OPLS, GROMOS |
| Water Models | Solvent representation | TIP3P, SPC/E, TIP4P |
| Thermostats | Temperature control | Nosé-Hoover, Berendsen, v-rescale |
| Barostats | Pressure control | Parrinello-Rahman, Berendsen |
| Hardware | Computational acceleration | GPU clusters, SIMD processors |
| Analysis Tools | Trajectory processing | VMD, PyMOL, MDAnalysis, GROMACS tools |
The Verlet algorithm and its variants form the computational backbone of modern molecular dynamics across major simulation packages, each implementing the core mathematics with package-specific optimizations. GROMACS excels with its highly optimized leapfrog implementation and cluster-based neighbor searching, NAMD offers sophisticated multiple time stepping for large biomolecular systems, and SIESTA extends the method to ab initio molecular dynamics. Understanding these implementation differences enables researchers to select appropriate packages and parameters for specific scientific questions, particularly in drug development where simulation accuracy directly impacts predictive capability. As MD simulations continue to bridge temporal and spatial scales, the Verlet algorithm remains fundamental to extracting biologically and physically meaningful insights from atomic-level simulations.
Conclusion
The Verlet algorithm remains a fundamental pillar of Molecular Dynamics due to its simplicity, numerical stability, and crucial symplectic properties that ensure long-term energy conservation. Its various forms, particularly the Velocity Verlet algorithm, provide a robust framework for simulating biomolecular systems. For researchers in drug development, mastering this integrator—from its foundational theory to practical optimization strategies—is key to running efficient and physically meaningful simulations. Future directions involve continued development of optimized Verlet-like algorithms and their adept application in complex biological processes, such as protein-ligand binding and allosteric regulation, promising deeper insights and accelerating computer-aided drug discovery.
References
- 1. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development [https://www.mdpi.com/2227-9717/9/1/71]
- 2. Verlet integration - Wikipedia [https://en.wikipedia.org/wiki/Verlet_integration]
- 3. Advancing Simulation in Time – Practical considerations for Molecular Dynamics [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/03-Solving_Equations_of_Motion/index.html]
- 4. Time-propagation algorithms in molecular - VASP Wiki dynamics [https://www.vasp.at/wiki/Time-propagation_algorithms_in_molecular_dynamics]
- 5. GitHub - ansavin/ molecular - dynamics : Molecular ... dynamics [https://github.com/ansavin/molecular-dynamics]
- 6. - Drug Design Org Molecular Dynamics [https://www.drugdesign.org/chapters/molecular-dynamics/]
- 7. Democritus: Integration Algorithms [https://people.bath.ac.uk/chsscp/teach/md.bho/Theory/verlet.html]
- 8. CoCalc -- 02_02_ Verlet .ipynb [https://cocalc.app/github/afeiguin/comp-phys/blob/master/02_02_Verlet.ipynb]
- 9. Implementation of magnetic field force in molecular dynamics algorithm: NAMD source code version 2.12 - PubMed [https://pubmed.ncbi.nlm.nih.gov/32314035/]
- 10. Machine learning analysis of molecular dynamics properties influencing drug solubility | Scientific Reports [https://www.nature.com/articles/s41598-025-11392-1]
- 11. - GROMACS Molecular .1 documentation Dynamics 2025 [https://manual.gromacs.org/documentation/2025.1/reference-manual/algorithms/molecular-dynamics.html]
- 12. Computational Methods of Physics [https://www.physics.udel.edu/~bnikolic/teaching/phys660/numerical_ode/node5.html]
- 13. CECAM - When data science meets molecular dynamicsWhen data science meets molecular dynamics [https://www.cecam.org/workshop-details/when-data-science-meets-molecular-dynamics-1386]
- 14. — ASE documentation Molecular dynamics [https://ase-lib.org/ase/md.html]
- 15. Molecular Dynamics: Velocity Verlet [https://plainenglish.io/blog/molecular-dynamics-velocity-verlet-integration-with-python-5ae66b63a8fd]
- 16. Optimized Verlet-like algorithms for molecular dynamics simulations - PubMed [https://pubmed.ncbi.nlm.nih.gov/12059749/]
- 17. Improved velocity-Verlet algorithm for the discrete element method - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S001046552500027X]
- 18. integration | 555 Publications | 6608 Citations | Top Authors Verlet [https://scispace.com/topics/verlet-integration-gxix74o1]
- 19. ab initio MD: Velocity Verlet Algorithm [https://education.molssi.org/ab-initio-md/06-Velocity-Verlet/index.html]
- 20. Optimized Verlet -like algorithms for molecular simulations dynamics [https://arxiv.org/abs/cond-mat/0110438]
- 21. Overcoming Instabilities in Verlet -I/r-RESPA with the... | SpringerLink [https://link.springer.com/chapter/10.1007/978-3-642-56080-4_7]
- 22. A brief overview on molecular dynamics simulation of biomolecular... [https://ijpsr.com/bft-article/a-brief-overview-on-molecular-dynamics-simulation-of-biomolecular-system-procedure-algorithms-and-applications/?view=fulltext]
- 23. Integration · Arcane Verlet Archive Algorithm [https://www.algorithm-archive.org/contents/verlet_integration/verlet_integration.html]
- 24. Trotter derived algorithms for molecular dynamics with constraints: Velocity Verlet revisited - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0021999106003603]
- 25. - GROMACS Molecular .0-beta documentation Dynamics 2025 [https://manual.gromacs.org/2025.0-beta/reference-manual/algorithms/molecular-dynamics.html]
- 26. [2410.14798] Improved Velocity-Verlet Algorithm for the Discrete Element Method [https://arxiv.org/abs/2410.14798]
- 27. On the calculation of velocity-dependent properties in molecular dynamics simulations using the leapfrog integration algorithm - PubMed [https://pubmed.ncbi.nlm.nih.gov/18020625/]
- 28. - Temperature Dynamics [https://www.uoxray.uoregon.edu/local/manuals/biosym/discovery/General/Dynamics/Temperature.html]
- 29. - GROMACS Molecular .1 documentation Dynamics 2025 [https://manual.gromacs.org/2025.1/reference-manual/algorithms/molecular-dynamics.html]
- 30. Verlet [https://www.tu-chemnitz.de/physik/THUS/tmp/OSP/org/opensourcephysics/numerics/Verlet.html]
- 31. Welcome to GROMACS — GROMACS webpage https://www.gromacs.org documentation [https://www.gromacs.org/]
- 32. Comparison of software for molecular mechanics modeling - Wikipedia [https://en.wikipedia.org/wiki/Comparison_of_software_for_molecular_mechanics_modeling]
- 33. 20 Best Software for Molecular Modeling and Simulations in Academia 2025 [https://www.scijournal.org/articles/best-software-for-molecular-modeling-and-simulations]
- 34. | Molecular of Scientific Computing... | Fiveable dynamics Applications [https://fiveable.me/applications-of-scientific-computing/unit-10/molecular-dynamics/study-guide/ggnes8BdlCwCPflM]
- 35. LAMMPS Molecular Dynamics Simulator [https://www.lammps.org/]
- 36. Parallelization of Molecular Simulations Using Dynamics ... Verlet [https://link.springer.com/chapter/10.1007/978-981-99-8129-8_22]
- 37. : Survey of Methods for Simulating the Activity of... Molecular Dynamics [https://pmc.ncbi.nlm.nih.gov/articles/PMC2547409/]
- 38. Comparison research on the neighbor list algorithms: Verlet table and linked-cell - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0010465510001827]
- 39. Comparison of the Verlet and cell-linked list table for... algorithms [https://en.num-meth.ru/index.php/journal/article/view/402]
- 40. : Equations of motion - Compchems Molecular Dynamics [https://www.compchems.com/molecular-dynamics-equations-of-motion/]
- 41. (PDF) A Linked - Cell Domain Decomposition Method for Molecular ... [https://www.academia.edu/54423477/A_Linked_Cell_Domain_Decomposition_Method_for_Molecular_Dynamics_Simulation_on_a_Scalable_Multiprocessor]
- 42. Traversal Linked for Three-Body Interactions in... Cell Algorithms [https://arxiv.org/html/2510.21230v1]
- 43. stability - Stable time step limits for Velocity-Verlet integration - Computational Science Stack Exchange [https://scicomp.stackexchange.com/questions/10329/stable-time-step-limits-for-velocity-verlet-integration]
- 44. Bond Constraints [https://wanglab.hosted.uark.edu/DLPOLY2/node71.html]
- 45. - GROMACS 2025.3 documentation Constraint algorithms [https://manual.gromacs.org/current/reference-manual/algorithms/constraint-algorithms.html]
- 46. Practical considerations for Molecular Dynamics [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/aio/index.html]
- 47. Molecular representation contrastive learning via transformer embedding to graph neural networks - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1568494624007440]
- 48. Correcting for the free energy costs of bond or angle constraints in molecular dynamics simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4339525/]
- 49. Constraint (computational chemistry) - Wikipedia [https://en.wikipedia.org/wiki/Constraint_(computational_chemistry)]
- 50. On the use of intraâ€molecular distance and angle constraints to lengthen the time step in molecular and stochastic dynamics simulations of proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9293444/]
- 51. Constraint algorithms — GROMACS 2020-beta2 documentation [https://manual.gromacs.org/documentation/2020-beta2/reference-manual/algorithms/constraint-algorithms.html]
- 52. A method to apply bond-angle constraints in molecular dynamics simulations - PubMed [https://pubmed.ncbi.nlm.nih.gov/33351979/]
- 53. Lecture5 | PPTX [https://www.slideshare.net/slideshow/lecture5-46436732/46436732]
- 54. Molecular Dynamics Simulations of Transmembrane Cyclic Peptide Nanotubes Using Classical Force Fields, Hydrogen Mass Repartitioning, and Hydrogen Isotope Exchange Methods: A Critical Comparison [https://www.mdpi.com/1422-0067/23/6/3158]
- 55. numerical convergence - How should one choose the time step in a molecular dynamics integration? - Matter Modeling Stack Exchange [https://mattermodeling.stackexchange.com/questions/498/how-should-one-choose-the-time-step-in-a-molecular-dynamics-integration]
- 56. Accelerating Membrane Simulations with Hydrogen ... Mass [https://pubmed.ncbi.nlm.nih.gov/31265271/]
- 57. Accelerating membrane simulations with Hydrogen Mass Repartitioning - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7271963/]
- 58. Performance improvements - GROMACS 2024.3 documentation [https://manual.gromacs.org/2024.3/release-notes/2024/major/performance.html]
- 59. Hydrogen Mass Repartitioning [https://ambermd.org/tutorials/basic/tutorial12/index.php]
- 60. How does hydrogen with an... - GROMACS forums mass repartitioning [https://gromacs.bioexcel.eu/t/how-does-hydrogen-mass-repartitioning-with-an-increased-time-step-of-4-fs-affect-other-mdp-options/9173]
- 61. Reinhard Folk | Johannes Kepler University of Linz | 150 Publications [https://scispace.com/authors/reinhard-folk-4i1stgkah5]
- 62. Numerical Integration - VDrift [http://wiki.vdrift.net/index.php?title=Numerical_Integration]
- 63. Energy drift in molecular dynamics simulations [https://link.springer.com/article/10.1007/s10543-007-0134-z]
- 64. Best Practices for Foundations in Molecular Simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6884151/]
- 65. Discretization errors in molecular dynamics simulations ... [https://www.sciencedirect.com/science/article/abs/pii/S0021999110004985]
- 66. CLASSICAL MECHANICS AND NUMERICAL METHODS | Medium [https://medium.com/@sarthak.dangre20/classical-mechanics-and-numerical-methods-4c5b8f12c445]
- 67. Lecture notes for AP3082 Computational Physics [https://compphys.quantumtinkerer.tudelft.nl/proj1-moldyn-week3/]
- 68. Euler method - Wikipedia [https://en.wikipedia.org/wiki/Euler_method]
- 69. Forward and Backward Euler Methods [https://web.mit.edu/10.001/Web/Course_Notes/Differential_Equations_Notes/node3.html]
- 70. Integration | thammin/unity-spring | DeepWiki Verlet [https://deepwiki.com/thammin/unity-spring/4.4-verlet-integration]
- 71. physics - Why is Verlet integration better than Euler ... - Stack Overflow [https://stackoverflow.com/questions/2769466/why-is-verlet-integration-better-than-euler-integration]
- 72. of Stability simulations of classical systems molecular dynamics [https://pubmed.ncbi.nlm.nih.gov/23231212/]
- 73. - Runge : Theory and Derivation | Numerical... | Fiveable Kutta Methods [https://fiveable.me/numerical-analysis-i/unit-16/runge-kutta-methods-theory-derivation/study-guide/0zHp61d42mG6nZ2t]
- 74. parameters (.mdp options) - GROMACS... Molecular dynamics [https://manual.gromacs.org/2025.0-beta/user-guide/mdp-options.html]
- 75. (PDF) Performance Analyses of a Parallel Verlet Neighbor List... [https://www.academia.edu/36917680/Performance_Analyses_of_a_Parallel_Verlet_Neighbor_List_Algorithm_for_GPU_Optimized_MD_Simulations]
- 76. PLAS-20k: Extended Dataset of Protein-Ligand Affinities from MD Simulations for Machine Learning Applications | Scientific Data [https://www.nature.com/articles/s41597-023-02872-y]
- 77. mdCATH: A Large-Scale MD Dataset for Data-Driven Computational Biophysics | Scientific Data [https://www.nature.com/articles/s41597-024-04140-z]
- 78. Molecular Dynamics - Statistical Ensembles [https://www.uoxray.uoregon.edu/local/manuals/biosym/discovery/General/Dynamics/Ensembles.html]
- 79. Molecular Dynamics - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/current/reference-manual/algorithms/molecular-dynamics.html]
- 80. Improved Velocity-Verlet Algorithm for the Discrete Element ... [https://arxiv.org/html/2410.14798v1]
- 81. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 82. Enabling Scalable AI-Driven Molecular Dynamics ... [https://developer.nvidia.com/blog/enabling-scalable-ai-driven-molecular-dynamics-simulations/]
- 83. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOorR1PWHOY_j0Dtm9yrzO_DyKoSYOewdpms8mC2aNxB1f_pvQbsI]
- 84. Benchmarking GPUs for MD Simulations [https://www.linkedin.com/pulse/benchmarking-gpus-md-simulations-speed-cost-insights-barbosa-farias-ynoke]
- 85. Molecular Simulation/Molecular Dynamics - Wikibooks, open books for... [https://en.wikibooks.org/wiki/Molecular_Simulation/Molecular_Dynamics]
- 86. Non-bonded cut-off schemes — GROMACS 5.1.1 documentation [https://manual.gromacs.org/5.1.1/user-guide/cutoff-schemes.html]
- 87. Verlet method [https://www.ks.uiuc.edu/Research//namd/1.5/ug/node60.html]